

Abstract
There is a continuing threat that the highly pathogenic avian influenza virus will cause future influenza pandemics. In this study, we screened a library of compounds that are biologically active and structurally diverse for inhibitory activity against influenza neuraminidase (NA). We found that aurintricarboxylic acid (ATA) is a potent inhibitor of NA activity of both group-1 and group-2 influenza viruses with IC50s (effective concentration to inhibit NA activity by 50%) values at low micromolar concentrations. ATA was equally potent in inhibiting the NA activity derived from wild-type NA and its H274Y mutant which renders NA resistance to inhibition by oseltamivir. Although ATA is structurally distinct from sialic acid, molecular modeling experiments suggested that ATA binds to NA at the enzyme’s substrate binding site. These results indicate that ATA may be a good starting material for the design of a novel class of NA inhibitors for the treatment influenza viruses.
Keywords: Aurintricarboxylic acid, Influenza, Neuraminidase, Antivirals
1. Introduction
Influenza virus causes severe epidemics of respiratory illness each year. In recent years, frequent outbreaks of H5N1 subtype avian influenza infections in humans have caused serious concerns (Chang et al., 2006). Although vaccination is considered the first line of defense, current strategies for vaccine design and manufacturing may not be sufficient to combat an influenza pandemic. Neuraminidase (NA) inhibitors (NAIs) are effective therapeutic agents against influenza viruses by targeting the viral NA protein (von Itzstein et al., 1993, Li et al., 1998, Moscona, 2005). The advent of zanamivir (Relenza) and oseltamivir (Tamiflu) has clearly established that NA is a valid drug target for development of anti-influenza therapeutics (Klumpp and Graves, 2006, Garman and Laver, 2004). Although these agents are effective as prophylactics and treatment of influenza virus infection, alternative treatments are needed as drug-resistant strains have already emerged in some patients who received oseltamivir treatment (de Jong et al., 2005, Carr et al., 2002, Gubareva et al., 2001, Monto et al., 2006, Yen et al., 2007).
Influenza virus NA is essential for virus replication by facilitating the release of progeny virions from infected cells (Seto and Rott, 1966, Palese et al., 1974). NA is also important for the trafficking of virus in the mucus layer of the respiratory tract so that the viruses can get access to the underlying epithelial cells (Klenk and Rott, 1988). NA is present on the surface of viral envelope and is composed of a tetramer of identical 60-kDa glycosylated subunits. X-ray crystallographic studies have revealed the structure of the head of the tetrameric NA (Colman et al., 1983). NAs from influenza A viruses are classified into two genetically distinct groups: group-2 includes the N2 and N9 subtypes that have been used for structure-based drug design (Varghese et al., 1983, von Itzstein et al., 1993) and group-1 includes the N1 subtype of the H5N1 or H1N1 strains. The structure of group-1 H5N1 avian influenza NA was recently solved, and it has suggested new opportunities for drug design because the discovery of a novel cavity adjacent to the active site that would close upon substrate binding (Russell et al., 2006).
In this study, we screened a compound library for inhibition of the NA activity derived from influenza A/Udorn/72 (H3N2) virus and identified aurintricarboxylic acid (ATA) as a candidate inhibitor. ATA has previously been shown to interfere with a variety of cellular processes. It was described as a general inhibitor of nucleases (Hallick et al., 1977). ATA has also been shown to protect various cell types from apoptotic cell death which was induced by a variety of factors (Roberts-Lewis et al., 1993, Benchokroun et al., 1995, Andrew et al., 1999, Tsi et al., 2002, Marchisio et al., 2003). There were also reports to indicate that ATA could inhibit various types of kinases involved in cellular singaling pathways, e.g., I kappa B kinase (IKK), extracellular signal-regulated kinase (ERK) (Tsi et al., 2002, Myskiw et al., 2007). ATA was also shown to inhibit cytokine-induced JAK-STAT signaling pathway (Chen et al., 2002). Further, ATA has been described as a insulin-like growth factor I (IGF-I) mimetic and could promote proliferation of cells in serum-free culture medium presumably through activation of the insulin-like growth factor 1 signaling pathway (Beery et al., 2001).
In addition to the aforementioned cellular processes that ATA may be interfering, ATA has also been reported to inhibit the replication of different kinds of viruses including human immunodeficiency virus (HIV) (Schols et al., 1989), SARS-CoV (Yap et al., 2005, He et al., 2004), and vaccinia virus (Myskiw et al., 2007), etc. In this study, we discovered that ATA also inhibits the function of the NA encoded by influenza viruses. We also found that ATA is equally effective in inhibiting the wild-type NA and the oseltamivir-resistant NA with a H274Y mutation (NAH274Y) (de Jong et al., 2005). Molecular modeling experiments indicate that ATA binds to the same binding site as the substrate does.
2. Materials and methods
2.1. Drugs and reagents
The test compounds were mainly from The Spectrum Collection (MicroSource Discovery Systems, Inc., Gaylordsville, CT, USA) containing 2000 biologically active and structurally diverse compounds. In addition, several in-house collected compounds including ATA were also included for screening as NA inhibitors. ATA was purchased from Sigma–Aldrich (St. Louis, MO, USA). It was dissolved in dimethyl sulfoxide (DMSO) as a 10 mM stock stock solution and stored at −20 °C. Oseltamivir carboxylate (GS4071) was synthesized by Dr. K.-S. Shia at the National Health Research Institutes (NHRI) of Taiwan. The chemiluminescent substrate sodium (2-chloro-5-(4-methoxyspiro{1,2-dioxetane-3,2′-(5-chloro) tricycle [3.3.1.1]decan}-4-yl-phenyl 5-acetamido-3,5-dideoxy-α-d-glycero-d-galacto-2-nonulopyranoside)onate) and the NA released the product 1,2-dioxetane utilized in the NA high-throughput screening assay were indicated in the NA-Star ® Kit (Applied Biosystems, Foster City, CA, USA). The fluorogenic substrate 2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid (MU-NANA) and the product 4-methylumbelliferyl (4-MU) utilized in the NA assay were obtained from Sigma–Aldrich.
2.2. Viruses and cells
The influenza strains A/WSN/33 (H1N1), A/Udorn/72 (H3N2) (Shih et al., 1998) and the H5N1 influenza virus reassortant strain NIBRG-14 (Wood and Robertson, 2007) were utilized in this study. Madin-Darby canine kidney (MDCK) cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum. DMEM containing 2 μg/ml TPCK (tolylsulfonyl phenylalanyl chloromethyl ketone)-trypsin was used as an infection medium for MDCK cells.
2.3. Site-directed mutagenesis of the NA gene
pGEM-T Easy plasmids (Promega, Madison, WI, USA) bearing the NA gene fragment from influenza virus strains A/WSN/33 and NIBRG-14 were used as targets for site-directed PCR mutagenesis of NA. To generate site-specific mutations, two primer sets were designed:
-
•
WSN H274Y-F (5′-AAT TCT TAC TAC GAG GAA TGT TCC TGT TAC-3′).
-
•
WSN H274Y-R (5′-CTC GTA GTA AGA ATT AGG TGC ATT CAA CTC-3′).
-
•
VN H274Y-F (5′-AAT TAT GCC TAT GAG GAA TGC TCC TGT TAT-3′).
-
•
VN H274Y-R (5′-CTC ATA GGC ATA ATT AGG ATC CAA TTC-3′).
Mutation sites were created by PCR using 0.4 mM dNTP, 0.3 μM primers, 2 ng template DNA and an appropriate amount of Turbo Pfu DNA polymerase and buffer (Stratagene, La Jolla, CA, USA). The thermal cycling profile was 95 °C for 5 min followed by 20 cycles of 94 °C for 30 s, 50 °C for 1 min, and 68 °C for 10 min, with final extension of 68 °C for 10 min. After PCR amplification, DpnI was added to digest the plasmid template. The amplified DpnI-insensitive mutated plasmids were introduced into E. coli for selection of the designed mutant sequences.
2.4. NA production in insect cells
The NA gene for strain A/WSN/33 (H1N1) was obtained from pPOLI-N1-R (Fodor et al., 2002) by PCR with primers carrying sites for EcoRI and XhoI, N1Bac-F: 5′-CCG CTC GAG CGG CGC CAC CAT GAA TCC AAA CCA GAA AAT-3′ and N1Bac-R: 5′-CCG GAA TTC CGG CTA CTT GTC AAT GGT GAA CGG CAA-3′ C. The NIBRG-14 (H5N1) subtype NA gene was retrieved from a cDNA library established by reverse transcription with primer Uni12: 5′-AGC AAA AGC AGG-3′ followed by PCR amplification with primers N1Bac-F and N1Bac-R.
The PCR-amplified NA inserts were cloned into pGEM-T Easy Vectors (Promega). Then, NA genes were cut out of the pGEM-T plasmid by digestion with EcoRI and XhoI restriction enzymes. The EcoRI–XhoI fragments were then introduced into the modified transfer vector pBacPAK8 (Clontech, Palo Alto, CA, USA), which carries an Enhanced Green Fluorescent Protein gene under the weak metallothionein promoter as a selection marker (Chen et al., 2004). These constructs were co-transfected with linear BacPAK8 viral DNA into Sf9 insect cells. In the Sf9 system, successful homologous recombination produces infectious baculovirus expressing NA proteins. To select single clones of recombinant baculoviruses, the culture medium from baculovirus-infected insect cell cultures was harvested and serially diluted to infect Sf9 cells in 96-well plates. Subsequently, selected virus clones were amplified in a 6-well plate. Culture media of infected cells were then collected and stored as virus stocks for the production of NA to be utilized in the NA enzymatic assay. Recombinant baculoviruses, i.e., Bac-NAWT(WSN), Bac-NAH274Y(WSN), Bac-NAWT(H5N1), Bac-NAH274Y(H5N1), were generated to express the wild-type and H274Y mutants of NA originating from influenza A/WSN/33 (H1N1) and NIBRG-14 (H5N1). Although the His is not exactly located at 274 in the NAWT(WSN) or NAWT(H5N1), the His to Phe point mutation was referred to as H274Y in this study because “H274Y” has been utilized in the literature to designate this specific amino acid change responsible for Tamiflu resistance (Zürcher et al., 2006, Abed et al., 2006, Baz et al., 2007).
2.5. Neuraminidase inhibition assay
The large-scale influenza virus suspension was prepared from MDCK cells infected with influenza virus. To inactivate viral infectivity, virus suspensions were treated with formaldehyde at a final concentration of 0.01% at 37 °C for 30 min. We demonstrated that such preparations were safe for handling on the bench because the viral titer is under the detection limit without decreasing the NA activity (results not shown). Aliquots of the inactivated virus supernatants were stored at −80 °C. The NA enzymatic activity was measured using the fluorogenic substrate MU-NANA (Potier et al., 1979). The NA activity assay was conducted in 96-well plates containing 10 μl diluted virus supernatant (containing active influenza NA) and 100 μM MU-NANA substrate per well in buffer (32.5 mM 2-(N-morpholino)-ethanesulfonic acid (Wood and Robertson, 2007), 4 mM CaCl2 at pH 6.5). For compounds screening or for evaluating the inhibitory activity of ATA, the inactivated virus supernatants were pre-incubated with the test compounds for 30 min at 30 °C. The enzymatic reactions were then carried out for 1 h at 37 °C and then terminated by 150 μl of the stop solution (25% ethanol, 0.1 M glycine, pH 10.7), and fluorescence intensity of the product 4-MU was measured using a Fluoroskan spectrofluorometer (Labsystems, Helsinki, Finland) with excitation and emission wavelengths of 330 and 445 nm, respectively. The IC50 for reducing NA activity was then determined. Free 4-MU was used to generate a standard curve to assess NA activity.
2.6. CPE inhibition test
To examine if the initial hit compounds identified from the NA inhibition assay can inhibit the cytopathic effect (CPE) of cells infected by influenza viruses, 96-well tissue culture plates were seeded with 200 μl of 1 × 105 MDCK cells/ml in DMEM with 10% FBS. After cells were incubated 18–24 h at 37 °C, virus at a multiplicity of infectivity (MOI) of 0.2 per cell was mixed with different concentrations of compounds and were then added to the cells. After 1 h viral adsorption, the infected cells were overlaid with 50 μl DMEM and 0.5% DMSO and incubated at 37 °C for 72 h. At the end of the incubation, cells were fixed with 100 μl of 10% formaldehyde for 1 h at room temperature. After removal of the formaldehyde, the cells were stained with 0.1% crystal violet for 15 min at room temperature. The plates were washed and dried, and the intensity of crystal violet staining for each well was measured at 570 nm. The concentration required for a test compound to reduce the CPE of the virus by 50% (IC50) was determined. Oseltamivir carboxylate (GS4071) was used as a reference control in the CPE inhibition test.
2.7. Cytotoxicity assay
Cell viability was determined by MTS assay (Cory et al., 1991). MDCK cells were grown (7000 cells/well) in 96-well plate for 24 h. The medium was replaced with that containing serial diluted compound and the cells were further incubated for 72 h. The culture medium were removed and added 100 μl including MTS and PMS mixture solution, the plate was incubated for 30 min. MTS and PMS are purchased from sigma and prepared in PBS (phosphate-buffered saline). To identify a 96-well microtiter plate, 2 ml reagent containing both MTS and PMS at the ratio of 20:1 was mixed immediately with 8 ml serum-free DMEM. Each drug concentration was performed with four repeats. The optical density was measured at OD490 nm in ELISA reader.
2.8. Plaque assay
Plaque assays were used to determine the effect of ATA on influenza virus replication. The plaque assay was performed as described (Sidwell and Smee, 2002) in 6-well plates containing confluent MDCK monolayer cells. GS4071 was used as a reference control in this experiment. The cells were inoculated with influenza A/WSN/33 (H1N1), A/Udorn/72 (H3N2), or NIBRG-14 (H5N1) virus at the titer of approximately 50–100 plaque forming units (PFU) per well. After 1 h incubation at 35 °C, the infection media were aspirated off and each well was then covered with 3 ml agar overlay media containing various concentrations of compounds and incubated for 2 days at 35 °C in a CO2 incubator. The cells were then fixed by the addition of 1 ml 10% formaldehyde for 1 h at room temperature. After the removal of formaldehyde, the cells were stained with 0.5% crystal violet for 15 min at room temperature. The plates were washed and dried, and the number of plaques was counted for each well. The concentration required for ATA to reduce the number of plaques by 50% (EC50) was then determined.
2.9. Virus yield reduction assay
Vero cells were infected with influenza virus A at an MOI of 0.0001 and various concentrations of compounds were added to the cell culture media. After 48 h, the culture media were collected and then subjected to virus titration by plaque forming assay as described.
2.10. Molecular modeling
The docking of ATA into the binding site of the H5N1 avian influenza NA protein (Russell et al., 2006) was explored using GEMDOCK (Yang and Chen, 2004, Yang and Shen, 2005, Yang et al., 2007) and GOLD (Jones et al., 1997) softwares, which have been shown to be powerful tools for molecular recognition and virtual screening (Thomsen and Christensen, 2006, Knox et al., 2007). To validate the molecular modeling programs, we first evaluated the docking accuracies of GEMDOCK and GOLD by docking two known NA inhibitors, GS4071 (oseltamivir carboxylate) and zanamivir, into the binding site. The 3D structure of ATA was prepared by CORINA (Sadowski et al., 1994) and the structures of the GS4071 and zanamivir were extracted from the crystal structures for N1 NA (PDB code 2HU4 (Russell et al., 2006)) and N8 NA (PDB code 2HTQ (Russell et al., 2006)), respectively, in the Protein Data Bank (PDB). The binding pocket (called NA2HU4) of the H5N1 avian influenza NA (PDB code 2HU4 (Russell et al., 2006)) was defined to include the amino acid residues within a 10 Å radius sphere centered around the binding site of GS4071. The coordinates of the atoms in the binding pocket were obtained from the PDB.
3. Results
3.1. ATA inhibits the NA and replication of influenza virus
The Microsource Spectrum Collection compounds and several in-house collected compounds were tested for inhibition of NA activity using the chemiluminescent NA-Star ® assay kit. In this assay, a decrease in chemiluminescence would indicate inhibition of the release of 1,2-dioxetane by enzymatic hydrolysis of NA-Star ® mediated by NA (inactivated viral suspensions of influenza A/Udorn/72 (H3N2)). The compounds were tested at 50 μM in the NA activity assay, where GS4071 was employed as a positive control. In this screening assay, NA derived from the NIBRG-14 strain was untilized. There were 24 initial hits, including ATA, displaying inhibition of >50% of chemiluminescence signal (Table 1 ). Since we wished to discover compounds that are active in inhibiting influenza virus replication, the initial hits from the NA inhibition assay were subject to CPE inhibition assay. ATA and periplocymarin were the only two compounds that at 10 μM could inhibit >50% of CPE without causing apparent cytotoxicity (Table 1). The IC50 of ATA in the CPE inhibition assay was 9.4 μM and the selectivity index (CC50/IC50) for ATA was approximate 10.2.
Table 1.
Anti-influenza virus agents identified by high-throughput screening.
| Chemical name | NA activity inhibition at 50 μM (%) | CPE inhibition IC50 (<10 μM) | Cytotoxicity assay CC50 (μM) |
|---|---|---|---|
| Theaflavin digallate | 61.5 | – | N.D. |
| Isoliquiritigenin | 62.6 | – | N.D. |
| Amprolium | 95.0 | – | N.D. |
| 2′2′-Bisepigallocatechin digallate | 60.6 | – | >50.0 |
| Periplocymarin | 75.2 | 0.3 | <50.0 |
| Citromycetin | 46.8 | – | N.D. |
| Mecysteine hydrochloride | 51.3 | – | N.D. |
| Ketoconazole | 72.2 | – | N.D. |
| Miconazole nitrate | 100.0 | – | N.D. |
| Piperazine | 100.0 | – | N.D. |
| Sulconazole nitrate | 99.6 | – | N.D. |
| Econazole nitrate | 96.4 | – | N.D. |
| Clotrimazole | 94.5 | – | N.D. |
| Cyproterone | 55.7 | – | N.D. |
| Myricetin | 61.6 | – | N.D. |
| Diosmetin | 59.0 | – | N.D. |
| Sennoside A | 61.9 | – | N.D. |
| Tetrachloroisophthalonitrile | 76.9 | – | N.D. |
| Meloxicam | 62.1 | – | N.D. |
| Bisanhydrorutilantinone | 71.9 | – | N.D. |
| Ethyl1-benzyl-3-hydroxy-2-oxo(5H)pyrrole-4-carboxylate | 71.9 | – | N.D. |
| Benzyl isothiocyanate | 75.5 | – | N.D. |
| Ascorbic acid acetonide | 83.5 | – | N.D. |
| Aurintricarboxylic acid (ATA) | 100.0 | 9.4 | 95.5 |
| GS4071a | 100.0 | 0.1 | >25.0 |
N.D.: Not-determined. Symbol (–) means the value of IC50 was above than 10 μM.
GS4071 was included as a positive control.
Further analysis revealed that ATA inhibited influenza virus NAs in a dose-dependent manner (Fig. 1 ). The IC50 values for ATA were 3.3 μM, 13.8 μM, and 3.3 μM for NA in inactivated viral suspensions of influenza A/WSN/33 (H1N1), A/Udorn/72 (H3N2), and NIBRG-14 (H5N1) by using fluorogenic substrate-Mu-NANA, respectively. Thus, ATA was a potent inhibitor of human influenza virus NAs of both group-1 and -2 NAs (Russell et al., 2006). Control experiments determined that ATA, up to 1.18 mM, did not interfere with the fluorescence emission of 4-MU, the enzymatic product in the NA reaction (data not shown). Thus, ATA indeed inhibited the NA-mediated hydrolysis of MU-NANA. The IC50 of GS4071 was less than 0.1 nM for all the 3 kinds of influenza A viruses tested (not shown).
Fig. 1.

Inhibition of NAs derived from A/WSN/33 (H1N1), A/Udorn/72 (H3N2), and NIBRG-14 (H5N1) by ATA. Formaldehyde-treated virus suspensions were used in the MU-NANA fluorogenic substrate method with various concentrations of ATA. The squares, triangles, and circles represent A/WSN/33 (H1N1), A/Udorn/72 (H3N2), and NIBRG-14 (H5N1) NA, respectively.
The effects of ATA on influenza virus replication were further evaluated using plaque reduction and yield reduction assays. ATA at various concentrations was evaluated in virus plaque reduction assays for influenza A/WSN/33 (H1N1), A/Udorn/72 (H3N2), and the reassortant NIBRG-14 (H5N1). ATA, in a dose-dependent manner, reduced the plaque formation units (PFU) caused by infection of MDCK cells with different strains of influenza virus (Fig. 2A). The EC50 values for viral plaque formation were estimated to be 4.1 μM, 6.3 μM, and 5.4 μM for influenza A/WSN/33 (H1N1), A/Udorn/72 (H3N2), and NIBRG-14 (H5N1), respectively. The EC50 of GS4071 was approximately 40 nM for all the 3 kinds of influenza A viruses tested (not shown).
Fig. 2.

Inhibition of influenza virus plaque formation units by ATA (A) and reduction in viral yields from infected cells treated with ATA (B) and GS4071 (C) at different concentrations. (A) Approximately 50–100 PFU/well of three different subtypes of influenza A virus, A/WSN/33 (H1N1), A/Udorn/72 (H3N2), or NIBRG-14 (H5N1), was used to infect MDCK cells in 6-well plates. After the viral adsorption stage, 3 ml of agar overlay media containing various concentrations of ATA was added to the cells. The concentration of ATA is indicated at the top. MDCK cells were infected with MOI 0.001 A/WSN/33 (H1N1) and various concentrations of ATA (B) and GS4071 (C) were added to the infected cells at the adsorption stage of A/WSN/33 replication cycle. After 48 post-infection hours, the culture supernatant were collected for virus titration by plaque forming assay.
To evaluate the effect of ATA in the yield of progeny virus assay, various concentrations of ATA were added to infected MDCK cells by influenza A/WSN/33 with an MOI of 0.0001. After 48 h post-infection, the culture supernatants were collected for virus yield determination by plaque forming assay. As shown in Fig. 2B, at 24 μM ATA seemed to have only marginal activity for reducing the yield of influenza progeny virus from infected cells. However, at 48 μM, ATA exhibited strong antiviral activity to reduce the viral yield by >1000 fold. As a reference control, GS4071 was only effective to reduce the viral yield when cells were treated by GS4071 at 10 μM (Fig. 2C). At lower concentrations of GS4071, no reduction in the yield of progeny virus could be observed.
3.2. ATA inhibited the wild-type NA and H274Y oseltamivir-resistant NA
We were interested in examining whether ATA is also effective in inhibiting the oseltamivir-resistant of H5N1 and H1N1 NAs due to a H274Y mutation in NAs (Wang et al., 2002). For safety reasons, we intended to avoid generating and manipulating drug-resistant influenza virus mutants. Therefore, insect cell protein expression technology was employed to express several variant NAs for the study of their sensitivity to ATA. Both cell lysates and cell culture supernatants of insect cells infected with Bac-NAWT(WSN), Bac-NAH274Y(WSN), Bac-NAWT(H5N1), or Bac-NAH274Y(H5N1) had NA activity (data not shown). That the cell culture supernatants also contained substantial NA activity may be due to the fact that NA is likely to be embedded in the envelope of budded baculovirus particles as suggested by Mather et al. (1992). The sensitivity of these NA variants to ATA was tested using cell lysates prepared from insect cells infected by baculoviruses. In our assay, the H274Y mutation in NA of the H1N1 and H5N1 subtypes, i.e., NAH274Y(WSN)and NAH274(H5N1), resulted in resistance to oseltamivir carboxylate (GS4071), the active ingredient of oseltamivir (Mendel et al., 1998), as indicated by a 191- and 885-fold increase in the IC50 values, respectively (not shown). Such results are largely consistent with a previous report indicating that the H274Y mutation in NAWT(WSN) conferred a 754-fold increase in the IC50 of oseltamivir (Abed et al., 2006). The IC50 values of ATA in inhibiting NA activity were estimated to be 8.7 μM, 18.4 μM, 5.4 μM and 2.3 μM for NAWT(WSN), NAH274Y(WSN), NAWT(H5N1), and NAH274Y(H5N1), respectively (Fig. 3 ). Thus, ATA remained equally potent in inhibiting the activity of the H274Y mutant forms and the wild-type NAs derived from either the H5N1 or the H1N1 influenza subtypes.
Fig. 3.

Effects of ATA on wild-type and mutant H274Y NA activity. Insect cells were infected with Bac-NAWT(WSN), Bac-NAH274Y(WSN), Bac-NAWT(H5N1), or Bac-NAH274Y(H5N1) for 72 h. The NA enzymatic activity of cell lysates containing 2 μg protein treated with the indicated concentrations of ATA was detected by the MU-NANA fluorogenic substrate method. (A) The NAWT and NAH274Y were derived from influenza A/WSN/33 (H1N1). (B) The NAWT and NAH274Y were derived from influenza NIBRG-14 (H5N1).
3.3. Molecular modeling
The GEMDOCK and GOLD were employed to predict the docked conformations of ATA, GS4071, and zanamivir, respectively, in the binding cavity of NA2HU4 based on calculated binding energies. Fig. 4 shows that the docked conformations of two known compounds (GS4071 and zanamivir) with the lowest scoring values were compared with the crystal structures based on the root-mean-square deviation (RMSD) of heavy atoms. The average RMSDs from GEMDOCK and GOLD were less than 1.2 Å and 1.5 Å for GS4071 and zanamivir, respectively. Thus, the derived GEMDOCK and GOLD parameters were adapted to predict the docked conformation of ATA in the binding site.
Fig. 4.
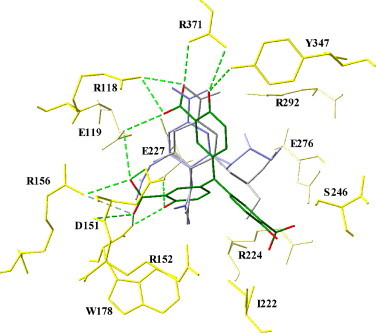
Docked conformations of aurintricarboxylic acid (ATA) (green), GS4071 (gray), and zanamivir (blue) in H5N1 avian influenza neuraminidase. The docked conformation of ATA is obtained by using GEMDOCK and the binding pocket of N1 neuraminidase is derived from Protein Data Bank (PDB code 2HU4). The main contact residues (yellow) of N1 neuraminidase are labeled and hydrogen bonds (dash with green line) between ATA (green) and the neuraminidase are indicated. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
The docked conformation (Fig. 4, Fig. 5 ) and the hydrogen bond network of ATA are slightly different from those of GS4071 and of zanamivir. To analyze the binding mechanism of ATA to NA2HU4, we divided the binding site of NA2HU4 into several subsites, S1 (R292, Y347, R371 and R118), S2-S3 (E119, R156, D151, R152, W178 and E227) and S4-S5 (I222, R224, S246 and E276) (Stoll et al., 2003, Kati et al., 2002). GS4071 and zanamivir inhibits NA by mimicking the electrostatic and hydrogen bond interactions of substrate binding at three subsites (S1, S2, and S3) and hydrophobic interactions of the S4–S5 subsites (Kim et al., 1997). The conformational analysis indicated that in the subsite S1, ATA forms a hydrogen bond network that is highly conserved for the known NAIs (Stoll et al., 2003, Kati et al., 2002, Russell et al., 2006) (Table 2 ). ATA also forms hydrogen bonds with Y347, R371, and R118 but not with the guanidine group of R292, which forms a hydrogen bond with GS4071 and zanamivir. An in vitro selection study demonstrated that the mutation R292K is the most common substitution in Group 2 NAs resistant to oseltamivir (Tai et al., 1998). The resistance results partially from the loss of a hydrogen bond from R292 to the carboxylate group of oseltamivir. This mutation would have little effect in Group 1 NAs because the carboxylate group forms an additional hydrogen bond with Y347 (Russell et al., 2006).
Fig. 5.

Computational conformation of aurintricarboxylic acid (ATA) in H5N1 avian influenza neuraminidase. The conformations of ATA and zanamivir are docked by using GEMDOCK, and the conformation of zanamivir is obtained from the X-ray structure (PDB code 2HU4). The main contact residues (yellow) are labeled and hydrogen bonds (dash with green line) between ATA and the neuraminidase are indicated. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Table 2.
The contact residues, hydrogen bonds, and close hydrophobic contacts between H5N1 avian influenza neuraminidase and docked conformations of three compounds.
| ATA (−99.78 kcal/mola) | Oseltamivir (−101.65 kcal/mola) | Zanamivir (−123.52 kcal/mola) | |
|---|---|---|---|
| S1 | |||
| Y347 | R347-O.hydroxylb-O.hydroxylc, 2.23d | R347-O.hydroxyl-O.carboxylic, 2.91 | – |
| R292 | – | R292-N.guanidino-O.carboxylic, 2.60 | R292-N.guanidino-O.carboxylic, 3.28 292-N.guanidino-O.carboxylic, 3.32 |
| R371 | R371-N.guanidino-O.hydroxyl, 3.25 R371-N.guanidino-O.carboxylic, 2.61 | R371-N.guanidino-O.carboxylic, 2.68 R371-N.guanidino-O.carboxylic, 2.83 | R371-N.guanidino-O.carboxylic, 2.80 R371-N.guanidino-O.carboxylic, 2.95 |
| R118 | R118-N.guanidino-O.carboxylic, 3.10 R118-N.guanidino-O.carboxylic, 2.89 | – | R118-N.guanidino-O.carboxylic, 2.87 R118-N.guanidino-O.carboxylic, 3.12 |
| S2–S3 | |||
| E119 | R119-O.carboxylic-O.carboxylic, 3.27 R119-O.carboxylic-O.carboxylic, 3.30 | R119-O.carboxylic-N.amine, 2.60 | R119-O.carboxylic-N.guanidino, 3.02 |
| D151 | D151-O.amide-O.hydroxyl, 2.99 D151-O.carboxylic-O.carboxylic, 2.92 | D151-O.carboxylic-N.amine, 2.56 D151-O.carboxylic-O.amide, 3.09 | D151-O.carboxylic-N.guanidino, 2.60 |
| R156 | R156-N.guanidino-O.carboxylic, 3.11 R156-N.guanidino-O.carboxylic, 3.34 | – | – |
| E227 | E227-O.carboxylic-O.hydroxyl, 2.60 | – | E227-O.carboxylic-N.guanidino, 2.96 |
| R152 | – | – | R152-N.guanidino-O.amide, 3.10 |
| W178 | W178-O.amide-O.hydroxyl, 2.51 W178-O.amide-O.carboxylic, 2.38 | – | W178-O.amide-N.guanidino, 3.10 W178-O.amide-N.guanidino, 3.23 |
| S4–S5 | |||
| I222 | These five residues form hydrophobic interactions with substitution 2-hydroxybenzoic acid group | These five residues form hydrophobic interactions with substitution 1-ethylpropyl group | These five residues form hydrophobic interactions with substitution 1,2,3-trihydroxypropyl group |
| R224 | |||
| S246 | |||
| E276 | E276-O.carboxylic-O.hydroxyl, 3.11 | ||
| E277 | E277-O.carboxylic-O.hydroxyl, 2.96 | ||
Docked energy.
The atom name of a residue in the neuraminidase.
The atom name of a compound.
The distance of a hydrogen bond between compounds and neuraminidase.
Results from GEMDOCK and GOLD suggest that ATA interacts in subsite S2 with the side chain of E119 and the backbone oxygen of D151 and, in subsite S3, with the carboxyl group of E227 and backbone oxygen of W178 through several hydrogen bond interactions (Fig. 4, Fig. 5 and Table 2). In addition, the carboxyl group of ATA appears to form new hydrogen bonds with the nitrogen atoms of R156 in a bidentate hydrogen bond donor–acceptor mode. On the other hand, the known NAIs GS4071 and zanamivir utilized the amino group to form strong charge–charge type hydrogen bond interactions with E119 and D151 (Kim et al., 1997). The 2-hydroxybenzoic acid group of ATA has steric interactions with the hydrocarbon chains of I222, R224, S246 and E276 that constitute a hydrophobic pocket in S4 and S5 and is related to the inhibition of NA by ATA (Russell et al., 2006, Kim et al., 1997). The H274 amino acid residue that is mutated in GS4017-resistant influenza strains is positioned in this pocket (Russell et al., 2006, Le et al., 2005). The H274Y mutation could perturb the rearrangement space of E276, leading to increased resistance to GS4071 (Varghese et al., 1998, Yen et al., 2005, Wang et al., 2002). Upon comparison of the orientations of the substitution groups of GS4071 and ATA in this pocket, we observed that ATA binding allows for more rearrangement space for E276.
4. Discussion
There is an urgent need for new anti-influenza therapeutics and the objective of this study was to identify novel NA inhibitors whose chemical structures are distinct from that of sialic acid. After screening approximately 2000 structurally diverse compounds, we found that ATA has potent inhibitory activity for NA. ATA has previously been found to inhibit influenza virus replication and the mechanism of action was attributed to the inhibition of influenza viral polymerases activity by ATA (Steward et al., 1977, Liao et al., 1975). In these previous studies, ATA was shown to inhibit viral polymerase in in vitro assays using purified influenza virus protein. However, we have tested the inhibitory activity of ATA against influenza viral polymerase using a cell-based assay and we found that ATA did not inhibit influenza polymerase. In this assay, we added ATA to human embryonic kidney 293 cells co-expressing influenza A/WSN/33 (H1N1) polymerase complex proteins including PA, PB1, PB2, and NP, along with a reporter plasmid, pPOLI-CAT-RT. The reporter plasmid, pPOLI-CAT-RT, contained a vRNA-like RNA encoding the reporter gene chloramphenicol acetyltransferase (CAT), in negative sense, flanked by the 5′ and 3′ noncoding regions of the NS vRNA segment of influenza virus (Pleschka et al., 1996). To our surprise, ATA did not inhibit RNA-dependent RNA polymerase (RdRp)-derived expression of CAT at a concentration up to 12 μM (not shown). At this concentration, viral plaque formation was clearly inhibited (Fig. 2A). Based on the cell-based RdRp assay, we concluded that the polymerase of influenza A/WSN/33 (H1N1) may not be the primary molecular target of ATA.
Our findings are reminiscent of previous studies on the inhibitory effects of ATA for HIV-1. ATA was found to inhibit HIV replication and the mechanism was proposed to be through the inhibition of the viral reverse transcriptase (RT) (Balzarini et al., 1986). However, later results indicated that ATA inhibits HIV replication primarily through the binding of virus to CD4 receptor on cell surface (Schols et al., 1989, De Clercq, 2005). In addition to influenza virus and HIV, ATA was recently reported to inhibit other viruses such as SARS-CoV and vaccinia virus (He et al., 2004, Myskiw et al., 2007). ATA is a very strong inhibitor of influenza virus compared to its anti-SARS-CoV activity; the EC50 of ATA for SARS-CoV was 0.2 mg/mL (He et al., 2004), whereas the EC50 of ATA for influenza virus in this study was in the low μM range. It is interesting to note that the EC50 of ATA, measured in plaque reduction assay, and IC50, measured in the NA inhibition assay, were in the same order of magnitude. However, the IC50 and EC50 of GS4071 were less than 0.1 nM and ∼40 nM, respectively, for influenza A/WSN/33 (H1N1), A/Udorn/72 (H3N2), and the reassortant NIBRG-14 (H5N1). Inasmuch as ATA also has a pleiotropic effect on cellular functions (Hallick et al., 1977, Roberts-Lewis et al., 1993, Benchokroun et al., 1995, Andrew et al., 1999, Tsi et al., 2002, Marchisio et al., 2003, Myskiw et al., 2007, Chen et al., 2002, Beery et al., 2001), NA may not be the sole target that ATA interferes to exert its strong antiviral actions. Our findings are congruent with a recent report indicating that ATA inhibits the replication of vaccinia virus through targeting both cellular and viral factors (Myskiw et al., 2007).
Finally, molecular modeling experiments predicted that ATA binds to the substrate binding site of NA. The molecular modeling experiments have been applied to examine the docking of ATA in its monomeric, dimeric, and trimeric froms since ATA was known to be present in multimeric forms (Cushman et al., 1991a, Cushman et al., 1991b). The docked conformations of ATA monomer, ATA dimer, ATA trimer, and an ATA polymer using GEMDOCK are distinctive (results not shown). We found that ATA dimer and ATA trimer lost some important hydrogen bond interactions, which are present upon binding of ATA monomer, GS4071 and zanamivir to NA (Table 2). Further, an ATA polymer, which consists of more than three ATAs, cannot be docked into the pocket. These observations imply that the ATA monomer might be the best form to inhibit the activities of NA among the ATA monomeric or polymeric forms. It is warranted to examine the exact binding mode of ATA and NA through crystallographic studies. An understanding of the exact binding modes ATA to NA would shed light on the design of a novel class of NAIs to combat influenza virus infections. The finding that ATA is a novel NAI shall prove to be important in the design of second-generation NAIs to block the replication of influenza viruses that are resistant to oseltamivir treatment.
Acknowledgements
This work was supported by the National Health Research Institutes (NHRI) and National Science Council (NSC) in Taiwan, Republic of China (grant no. NHRI-BP-097-PP-07 and NSC95-2745-B-400-002).
References
- Abed Y., Baz M., Boivin G. Impact of neuraminidase mutations conferring influenza resistance to neuraminidase inhibitors in the N1 and N2 genetic backgrounds. Antiviral Ther. 2006;11:971–976. [PubMed] [Google Scholar]
- Andrew D.J., Hay A.W., Evans S.W. Aurintricarboxylic acid inhibits apoptosis and supports proliferation in a haemopoietic growth-factor dependent myeloid cell line. Immunopharmacology. 1999;41:1–10. doi: 10.1016/s0162-3109(98)00049-6. [DOI] [PubMed] [Google Scholar]
- Balzarini J., Mitsuya H., De Clercq E., Broder S. Aurintricarboxylic acid and Evans Blue represent two different classes of anionic compounds which selectively inhibit the cytopathogenicity of human T-cell lymphotropic virus type III/lymphadenopathy-associated virus. Biochem. Biophys. Res. Commun. 1986;136:64–71. doi: 10.1016/0006-291x(86)90877-6. [DOI] [PubMed] [Google Scholar]
- Baz M., Abed Y., Boivin G. Characterization of drug-resistant recombinant influenza A/H1N1 viruses selected in vitro with peramivir and zanamivir. Antiviral Res. 2007;74:159–162. doi: 10.1016/j.antiviral.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Beery R., Haimsohn M., Wertheim N., Hemi R., Nir U., Karasik A., Kanety H., Geier A. Activation of the insulin-like growth factor 1 signaling pathway by the antiapoptotic agents aurintricarboxylic acid and evans blue. Endocrinology. 2001;142:3098–3107. doi: 10.1210/endo.142.7.8265. [DOI] [PubMed] [Google Scholar]
- Benchokroun Y., Couprie J., Larsen A.K. Aurintricarboxylic acid, a putative inhibitor of apoptosis, is a potent inhibitor of DNA topoisomerase II in vitro and in Chinese hamster fibrosarcoma cells. Biochem. Pharmacol. 1995;49:305–313. doi: 10.1016/0006-2952(94)00465-x. [DOI] [PubMed] [Google Scholar]
- Carr J., Ives J., Kelly L., Lambkin R., Oxford J., Mendel D., Tai L., Roberts N. Influenza virus carrying neuraminidase with reduced sensitivity to oseltamivir carboxylate has altered properties in vitro and is compromised for infectivity and replicative ability in vivo. Antiviral Res. 2002;54:79–88. doi: 10.1016/s0166-3542(01)00215-7. [DOI] [PubMed] [Google Scholar]
- Chang S.C., Cheng Y.Y., Shih S.R. Avian influenza virus: the threat of a pandemic. Chang Gung Med. J. 2006;29:130–134. [PubMed] [Google Scholar]
- Chen C.W., Chao Y., Chang Y.H., Hsu M.J., Lin W.W. Inhibition of cytokine-induced JAK-STAT signalling pathways by an endonuclease inhibitor aurintricarboxylic acid. Br. J. Pharmacol. 2002;137:1011–1020. doi: 10.1038/sj.bjp.0704955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.S., Chien C.H., Goparaju C.M., Hsu J.T., Liang P.H., Chen X. Purification and characterization of human prolyl dipeptidase DPP8 in Sf9 insect cells. Protein Exp. Purif. 2004;35:142–146. doi: 10.1016/j.pep.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Colman P.M., Varghese J.N., Laver W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature. 1983;303:41–44. doi: 10.1038/303041a0. [DOI] [PubMed] [Google Scholar]
- Cory A.H., Owen T.C., Barltrop J.A., Cory J.G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- Cushman M., Kanamathareddy S., De Clercq E., Schols D., Goldman M.E., Bowen J.A. Synthesis and anti-HIV activities of low molecular weight aurintricarboxylic acid fragments and related compounds. J. Med. Chem. 1991;34:337–342. doi: 10.1021/jm00105a053. [DOI] [PubMed] [Google Scholar]
- Cushman M., Wang P.L., Chang S.H., Wild C., De Clercq E., Schols D., Goldman M.E., Bowen J.A. Preparation and anti-HIV activities of aurintricarboxylic acid fractions and analogues: direct correlation of antiviral potency with molecular weight. J. Med. Chem. 1991;34:329–337. doi: 10.1021/jm00105a052. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Emerging anti-HIV drugs. Expert Opin. Emerg. Drugs. 2005;10:241–273. doi: 10.1517/14728214.10.2.241. [DOI] [PubMed] [Google Scholar]
- de Jong M.D., Tran T.T., Truong H.K., Vo M.H., Smith G.J., Nguyen V.C., Bach V.C., Phan T.Q., Do Q.H., Guan Y., Peiris J.S., Tran T.H., Farrar J. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N. Engl. J. Med. 2005;353:2667–2672. doi: 10.1056/NEJMoa054512. [DOI] [PubMed] [Google Scholar]
- Fodor E., Crow M., Mingay L.J., Deng T., Sharps J., Fechter P., Brownlee G.G. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 2002;76:8989–9001. doi: 10.1128/JVI.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garman E., Laver G. Controlling influenza by inhibiting the virus’s neuraminidase. Curr. Drug Targets. 2004;5:119–136. doi: 10.2174/1389450043490604. [DOI] [PubMed] [Google Scholar]
- Gubareva L.V., Kaiser L., Matrosovich M.N., Soo-Hoo Y., Hayden F.G. Selection of influenza virus mutants in experimentally infected volunteers treated with oseltamivir. J. Infect. Dis. 2001;183:523–531. doi: 10.1086/318537. [DOI] [PubMed] [Google Scholar]
- Hallick R.B., Chelm B.K., Gray P.W., Orozco E.M., Jr. Use of aurintricarboxylic acid as an inhibitor of nucleases during nucleic acid isolation. Nucleic Acids Res. 1977;4:3055–3064. doi: 10.1093/nar/4.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R., Adonov A., Traykova-Adonova M., Cao J., Cutts T., Grudesky E., Deschambaul Y., Berry J., Drebot M., Li X. Potent and selective inhibition of SARS coronavirus replication by aurintricarboxylic acid. Biochem. Biophys. Res. Commun. 2004;320:1199–1203. doi: 10.1016/j.bbrc.2004.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Kati W.M., Montgomery D., Carrick R., Gubareva L., Maring C., McDaniel K., Steffy K., Molla A., Hayden F., Kempf D., Kohlbrenner W. In vitro characterization of A-315675, a highly potent inhibitor of A and B strain influenza virus neuraminidases and influenza virus replication. Antimicrob. Agents Chemother. 2002;46:1014–1021. doi: 10.1128/AAC.46.4.1014-1021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.U., Lew W., Williams M.A., Liu H., Zhang L., Swaminathan S., Bischofberger N., Chen M.S., Mendel D.B., Tai C.Y., Laver W.G., Stevens R.C. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- Klenk H.D., Rott R. The molecular biology of influenza virus pathogenicity. Adv. Virus Res. 1988;34:247–281. doi: 10.1016/S0065-3527(08)60520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp K., Graves B.J. Optimization of small molecule drugs binding to highly polar target sites: lessons from the discovery and development of neuraminidase inhibitors. Curr. Top. Med. Chem. 2006;6:423–434. doi: 10.2174/156802606776743138. [DOI] [PubMed] [Google Scholar]
- Knox A.J., Meegan M.J., Sobolev V., Frost D., Zisterer D.M., Williams D.C., Lloyd D.G. Target specific virtual screening: optimization of an estrogen receptor screening platform. J. Med. Chem. 2007;50:5301–5310. doi: 10.1021/jm0700262. [DOI] [PubMed] [Google Scholar]
- Le Q.M., Kiso M., Someya K., Sakai Y.T., Nguyen T.H., Nguyen K.H., Pham N.D., Ngyen H.H., Yamada S., Muramoto Y., Horimoto T., Takada A., Goto H., Suzuki T., Suzuki Y., Kawaoka Y. Avian flu: isolation of drug-resistant H5N1 virus. Nature. 2005;437:1108. doi: 10.1038/4371108a. [DOI] [PubMed] [Google Scholar]
- Li W., Escarpe P.A., Eisenberg E.J., Cundy K.C., Sweet C., Jakeman K.J., Merson J., Lew W., Williams M., Zhang L., Kim C.U., Bischofberger N., Chen M.S., Mendel D.B. Identification of GS 4104 as an orally bioavailable prodrug of the influenza virus neuraminidase inhibitor GS 4071. Antimicrob. Agents Chemother. 1998;42:647–653. doi: 10.1128/aac.42.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L.L., Horwitz S.B., Huang M.T., Grollman A.P., Steward D., Martin J. Triphenylmethane dyes as inhibitors of reverse transcriptase, ribonucleic acid polymerase, and protein synthesis. Structure–activity relationships. J. Med. Chem. 1975;18:117–120. doi: 10.1021/jm00235a029. [DOI] [PubMed] [Google Scholar]
- Marchisio M., Brugnoli F., Santavenere E., Paludi M., Ciccocioppo F., Miscia S. Mitigation of tumor necrosis factor alpha cytotoxicity by aurintricarboxylic acid in human peripheral B lymphocytes. Biochem. Pharmacol. 2003;66:1973–1979. doi: 10.1016/s0006-2952(03)00583-5. [DOI] [PubMed] [Google Scholar]
- Mather K.A., White J.F., Hudson P.J., McKimm-Breschkin J.L. Expression of influenza neuraminidase in baculovirus-infected cells. Virus Res. 1992;26:127–139. doi: 10.1016/0168-1702(92)90152-y. [DOI] [PubMed] [Google Scholar]
- Mendel D.B., Tai C.Y., Escarpe P.A., Li W., Sidwell R.W., Huffman J.H., Sweet C., Jakeman K.J., Merson J., Lacy S.A., Lew W., Williams M.A., Zhang L., Chen M.S., Bischofberger N., Kim C.U. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob. Agents Chemother. 1998;42:640–646. doi: 10.1128/aac.42.3.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto A.S., McKimm-Breschkin J.L., Macken C., Hampson A.W., Hay A., Klimov A., Tashiro M., Webster R.G., Aymard M., Hayden F.G., Zambon M. Detection of influenza viruses resistant to neuraminidase inhibitors in global surveillance during the first 3 years of their use. Antimicrob. Agents Chemother. 2006;50:2395–2402. doi: 10.1128/AAC.01339-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J. Clin. Invest. 2005;115:1688–1698. doi: 10.1172/JCI25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myskiw C., Deschambault Y., Jefferies K., He R., Cao J. Aurintricarboxylic acid inhibits the early stage of vaccinia virus replication by targeting both cellular and viral factors. J. Virol. 2007;81:3027–3032. doi: 10.1128/JVI.02531-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P., Schulman J.L., Bodo G., Meindl P. Inhibition of influenza and parainfluenza virus replication in tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA) Virology. 1974;59:490–498. doi: 10.1016/0042-6822(74)90458-9. [DOI] [PubMed] [Google Scholar]
- Pleschka S., Jaskunas R., Engelhardt O.G., Zürcher T., Palese P., García-Sastre A. A plasmid-based reverse genetics system for influenza A virus. J. Virol. 1996;70:4188–4192. doi: 10.1128/jvi.70.6.4188-4192.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier M., Mameli L., Bélisle M., Dallaire L., Melançon S.B. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-alpha-d-N-acetylneuraminate) substrate. Anal. Biochem. 1979;94:287–296. doi: 10.1016/0003-2697(79)90362-2. [DOI] [PubMed] [Google Scholar]
- Roberts-Lewis J.M., Marcy V.R., Zhao Y., Vaught J.L., Siman R., Lewis M.E. Aurintricarboxylic acid protects hippocampal neurons from NMDA- and ischemia-induced toxicity in vivo. J. Neurochem. 1993;61:378–381. doi: 10.1111/j.1471-4159.1993.tb03583.x. [DOI] [PubMed] [Google Scholar]
- Russell R.J., Haire L.F., Stevens D.J., Collins P.J., Lin Y.P., Blackburn G.M., Hay A.J., Gamblin S.J., Skehel J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- Sadowski J., Gasteiger J., Klebe G. Comparison of automatic three-dimensional model builders using 639 X-ray structures. J. Chem. Inf. Comput. Sci. 1994;34:1000–1008. [Google Scholar]
- Schols D., Baba M., Pauwels R., Desmyter J., De Clercq E. Specific interaction of aurintricarboxylic acid with the human immunodeficiency virus/CD4 cell receptor. Proc. Natl. Acad. Sci. U.S.A. 1989;86:3322–3326. doi: 10.1073/pnas.86.9.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto J.T., Rott R. Functional significance of sialidose during influenza virus multiplication. Virology. 1966;30:731–737. doi: 10.1016/0042-6822(66)90178-4. [DOI] [PubMed] [Google Scholar]
- Shih S.R., Suen P.C., Chen Y.S., Chang S.C. A novel spliced transcript of influenza A/WSN/33 virus. Virus Genes. 1998;17:179–183. doi: 10.1023/a:1008024909222. [DOI] [PubMed] [Google Scholar]
- Sidwell R.W., Smee D.F. Peramivir (BCX-1812, RWJ-270201): potential new therapy for influenza. Expert Opin. Emerg. Drugs. 2002;11:859–869. doi: 10.1517/13543784.11.6.859. [DOI] [PubMed] [Google Scholar]
- Steward D.L., Martin J., Grollman A.P. Inhibition of influenza virus by triphenylmethane compounds. Ann. N. Y. Acad. Sci. 1977;284:638–649. doi: 10.1111/j.1749-6632.1977.tb21998.x. [DOI] [PubMed] [Google Scholar]
- Stoll V., Stewart K.D., Maring C.J., Muchmore S., Giranda V., Gu Y.G., Wang G., Chen Y., Sun M., Zhao C., Kennedy A.L., Madigan D.L., Xu Y., Saldivar A., Kati W., Laver G., Sowin T., Sham H.L., Greer J., Kempf D. Influenza neuraminidase inhibitors: structure-based design of a novel inhibitor series. Biochemistry. 2003;42:718–727. doi: 10.1021/bi0205449. [DOI] [PubMed] [Google Scholar]
- Tai C.Y., Escarpe P.A., Sidwell R.W., Williams M.A., Lew W., Wu H., Kim C.U., Mendel D.B. Characterization of human influenza virus variants selected in vitro in the presence of the neuraminidase inhibitor GS 4071. Antimicrob. Agents Chemother. 1998;42:3234–3241. doi: 10.1128/aac.42.12.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R., Christensen M.H. MolDock: a new technique for high-accuracy molecular docking. J. Med. Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- Tsi C.J., Chao Y., Chen C.W., Lin W.W. Aurintricarboxylic acid protects against cell death caused by lipopolysaccharide in macrophages by decreasing inducible nitric-oxide synthase induction via IkappaB kinase, extracellular signal-regulated kinase, and p38 mitogen-activated protein kinase inhibition. Mol. Pharmacol. 2002;62:90–101. doi: 10.1124/mol.62.1.90. [DOI] [PubMed] [Google Scholar]
- Varghese J.N., Laver W.G., Colman P.M. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature. 1983;303:35–40. doi: 10.1038/303035a0. [DOI] [PubMed] [Google Scholar]
- Varghese J.N., Smith P.W., Sollis S.L., Blick T.J., Sahasrabudhe A., McKimm-Breschkin J.L., Colman P.M. Drug design against a shifting target: a structural basis for resistance to inhibitors in a variant of influenza virus neuraminidase. Structure. 1998;6:735–746. doi: 10.1016/s0969-2126(98)00075-6. [DOI] [PubMed] [Google Scholar]
- von Itzstein M., Wu W.Y., Kok G.B., Pegg M.S., Dyason J.C., Jin B., Van Phan T., Smythe M.L., White H.F., Oliver S.W. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- Wang M.Z., Tai C.Y., Mendel D.B. Mechanism by which mutations at his274 alter sensitivity of influenza a virus n1 neuraminidase to oseltamivir carboxylate and zanamivir. Antimicrob. Agents Chemother. 2002;46:3809–3816. doi: 10.1128/AAC.46.12.3809-3816.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood J.M., Robertson J.S. Reference viruses for seasonal and pandemic influenza vaccine preparation. Influenza. 2007;1:5–9. doi: 10.1111/j.1750-2659.2006.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.-M., Chen C.-C. GEMDOCK: a generic evolutionary method for molecular docking. Proteins. 2004;55:288–304. doi: 10.1002/prot.20035. [DOI] [PubMed] [Google Scholar]
- Yang J.-M., Shen T.-W. A pharmacophore-based evolutionary approach for screening selective estrogen receptor modulators. Proteins. 2005;59:205–220. doi: 10.1002/prot.20387. [DOI] [PubMed] [Google Scholar]
- Yang J.M., Chen Y.F., Tu Y.Y., Yen K.R., Yang Y.L. Combinatorial computational approaches to identify tetracycline derivatives as flavivirus inhibitors. PLoS ONE. 2007:e428. doi: 10.1371/journal.pone.0000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap Y., Zhang X., Andonov A., He R. Structural analysis of inhibition mechanisms of aurintricarboxylic acid on SARS-CoV polymerase and other proteins. Comput. Biol. Chem. 2005;29:212–219. doi: 10.1016/j.compbiolchem.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen H.L., Herlocher L.M., Hoffmann E., Matrosovich M.N., Monto A.S., Webster R.G., Govorkova E.A. Neuraminidase inhibitor-resistant influenza viruses may differ substantially in fitness and transmissibility. Antimicrob. Agents Chemother. 2005;49:4075–4084. doi: 10.1128/AAC.49.10.4075-4084.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen H.L., Ilyushina N.A., Salomon R., Hoffmann E., Webster R.G., Govorkova E.A. Neuraminidase inhibitor-resistant recombinant A/Vietnam/1203/04 (H5N1) influenza viruses retain their replication efficiency and pathogenicity in vitro and in vivo. J. Virol. 2007;81:12418–12426. doi: 10.1128/JVI.01067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zürcher T., Yates P.J., Daly J., Sahasrabudhe A., Walters M., Dash L., Tisdale M., McKimm-Breschkin J.L. Mutations conferring zanamivir resistance in human influenza virus N2 neuraminidases compromise virus fitness and are not stably maintained in vitro. J. Antimicrob. Chemother. 2006;58:723–732. doi: 10.1093/jac/dkl321. [DOI] [PubMed] [Google Scholar]