

Abstract
Cardiometabolic disorders including obesity, diabetes and cardiovascular disease are among the most severe health problems worldwide. DPP4 enzymatic inhibitors were first developed as anti-diabetic reagents which preserve incretin hormones and promote post-prandial insulin secretion. It's been shown in animal studies that incretin-based therapy has a beneficial effect on cardiovascular disease. Recent studies demonstrated novel non-catalytic functions of DPP4 that may play a role in cardiometabolic disease. Although the role of DPP4 inhibition-mediated incretin effects has been well-reviewed, little information of its incretin-independent actions was introduced in cardiometabolic disease. In the current review, we will summarize the catalytic dependent and independent effects of DPP4 inhibition on cardiometabolic disease.
Keywords: DPP4, Incretin, GLP-1, Cardiometabolic disease, Diabetes
Highlights
-
•
Discuss the findings from recent large scale clinical trials (EXAMINE and SAVOR-TIMI 53)
-
•
Summarize the catalytic dependent and independent effects of DPP4 inhibition on cardiometabolic disease
-
•
Focus on recent evidence linking DPP4 inhibition therapy with cardiovascular disease
-
•
Provide mechanistic insights into the cardiovascular effect of DPP4
1. Introduction
Dipeptidyl peptidase-4 (DPP4), also known as CD26 or adenosine deaminase binding protein (ADBP), is a type-II integral membrane glycoprotein that has recently gained attention owing to its role in the catalytic degradation of incretins. The development of DPP4 inhibitors as a class of anti-diabetic medications has been widely accepted due to their ease of administration and lack of serious side-effects. It's been widely believed that DPP4 inhibition has beneficial effect on cardiovascular diseases [1], [2], [3], [4], [5], [6]. However, recently completed large-scale phase 3 and phase 4 clinical trials (EXAMINE and SAVOR-TIMI 53) showed no significant improvements in primary cardiovascular endpoints in patients treated with DPP4 enzymatic inhibitors compared to those with placebo [7], [8], calling into question the cardiovascular efficacy of these agents. However it must be noted that these trials tested whether enzymatic inhibition of DPP4 is beneficial in the short term, and the importance of any of other non-enzymatic effects was not tested.
In addition to catalytic actions, DPP4 also exerts catalytic independent functions by interacting with a number of ligands such as adenosine deaminase (ADA), fibronectin, caveolin-1, and Middle Eastern Respiratory Syndrome-Corona Virus spike protein. The cardiovascular effect of DPP4 was believed to be majorly mediated through an incretin-dependent mechanism. Recent advances in DPP4 non-catalytic activities suggest that the incretin-independent actions of DPP4 may also play an essential role in cardiometabolic disease. However, little information of its incretin-independent actions in cardiometabolic disease was introduced in previous reviews. In this review, we will review the importance of DPP4 catalytic-dependent versus -independent activities in the pathophysiology of cardiometabolic disorders. We will also provide recent clinical trial evidence that have tested its effects in cardiovascular disease.
2. Overview of dipeptidyl peptidases of S9B family
DPP4 belongs to S9B family which consists of several structurally homologous serine peptidases, such as quiescent cell proline dipeptidase (QPP, also called DPP2), fibroblast activation protein (FAP), DPP8, and DPP9 [9]. Proteins in S9B family are able to cleave N-terminal dipeptides from proteins containing proline or alanine in the penultimate position [10]. DPP4 was first identified in 1966 as a glycylproline naphthylamidase [11] and was subsequently purified from rat liver [12] and pig kidney [13].
2.1. Structural and cellular biology of DPP4
Human DPP4 is a 766 amino acid membrane glycoprotein encoded by the human Dpp4 gene localized to chromosome 2q24, adjacent to GLP-1-encoding gene [14]. Sequence comparison reveals a very high degree of sequence conservation. The overall sequence identity is 88% between human and porcine DPP4, and 83% between human and mouse [15]. DPP4 has a large extracellular domain and a short cytoplasmic (AA 1–6) and transmembrane domain (AA7–29). The extracellular domain contains a α/β-hydrolase domain (Gln508-Pro766) and an eight-blade β-propeller domain (Arg54-Asn497) (Fig. 1 ) [16]. The C terminal extracellular domain is responsible for the catalytic activity (catalytic triad: Asp708, His740, and Ser630) and binding to a number of proteins including ADA and matrix proteins [16], [17], [18]. DPP4 functions as a homodimer, relying on broad intermolecule contacts contributed by the hydrolase domain and the extended strands in blade IV of the β-propeller [19]. The catalytic activity of DPP4 depends upon its dimerization state [20], with residues 630, residues 708 and 740 playing a critical role in substrate cleavage [21], [22], [23], [24]. Glycosylation of DPP4 also appears to be important in determining catalytic activity. Glycosylation of Asn-281 in human DPP4 has been proposed to control its assembly [25]. DPP4 can assemble into tetramers on the cell surface and in the circulation by linkage of two homodimers.
Fig. 1.
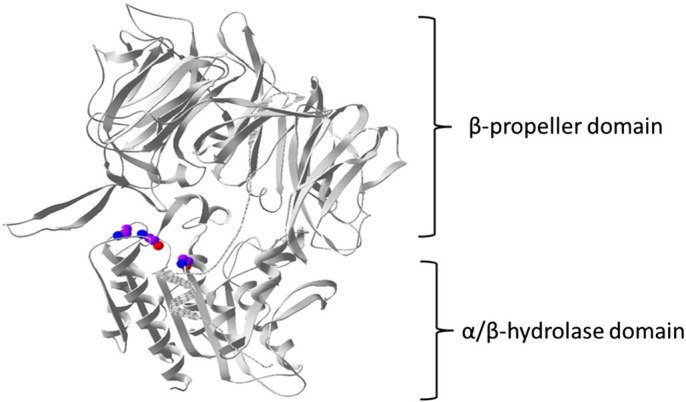
Structure of DPP4 Extracellular Domain: The extracellular portion of DPP4 is divided into an eight-bladed β-propeller (Arg54-Asn497) and a α/β-hydrolase domain (Gln508-Pro766). The backbone of catalytic triad (Ser630, Asp708, and Hsp740) is shown (Purple: C; Blue: N; Red: O).
2.2. Cellular and tissue distribution of DPP4
DPP4 is widely distributed throughout the body, with particularly high expression exocrine glands and absorptive epithelia. In humans, DPP4 is found at high levels in brush border of the small intestine, kidney proximal tubular cells and glomerular cells, hepatocytes, and activated leukocytes [26]. Significant amounts of DPP4 are also present in blood vessels, particularly in the endothelial cells. Among hematopoietic cells, DPP4 is expressed at the highest level on T cells especially CD4+ T cells, and at lower levels in monocytes and dendritic cells. DPP4 expression is increased as monocytes differentiate into macrophages as well as during T cell activation [27]. Indeed DPP4 was described as a T cell activation marker for a number of years. At organ level, DPP4 is highly expressed in solid organs, including kidney, spleen, lung, pancreas and prostate [20].
2.3. Soluble DPP4 in circulation
Soluble DPP4 is a homodimer with a molecular weight range of 210–290 kDa [28], but can form higher molecular weight assemblies migrating as 900-kDa complexes [29]. Soluble DPP4 lacks the cytoplasmic and transmembrane domain with preserved catalytic activity. The major cellular source of circulating DPP4 appears to be bone marrow cells and adipocytes [30], [31]. However, whether sDPP4 is cleaved from the membrane or is secreted is unclear. Studies investigating viral liver infection suggested that sDPP4 is shed from membrane bound DPP4 [32], while other studies suggest a secretory mechanism as evidenced by the fact that sDPP4 was detected in the lumen of secretory granules in pancreatic A cells and exocytic secretory lysosomes of natural killer cells [33], [34].
2.4. Other members of the DPP family
Other members of S9B family include FAP, DPP2, DPP8, and DPP9 [9]. FAP, also known as Seprase, is a 170 kDa membrane glycoprotein with dipeptidylpeptidase and type I collagenase activity [35]. It shares 54% sequence identity with DPP4 [36]. FAP is absent or produced at low levels in normal adult tissues under baseline conditions and is induced in response to inflammatory conditions and tumors. FAP expression levels correlate with prognosis in a number of malignancies and targeting FAP in epithelial-derived solid tumors has been shown to inhibit tumor growth [37]. FAP associates with thin-cap human coronary fibroatheromata and contributes to type I collagen breakdown in fibrous caps. The origin of FAP in plaque interestingly has been shown to derive from smooth muscle cells rather than macrophages and expression correlates with the degree of inflammation [38]. Plasma levels of FAP have been shown to inversely correlate with mortality in patients with ACS, pointing again to a divergent regulation of plasma vs. tissue levels of FAP [39].
DPP2 is widely distributed across a range of mammalian tissues. It is located in intracellular vesicles and can be secreted upon activation [40]. Interestingly it bears no significant nucleotide or amino acid homology with DPP4. Whereas the order of the catalytic amino acids, Ser-Asp-His, is identical in DPP4 and DPP2, the spacing of the catalytic triad is dissimilar, suggesting different substrate specificities. DPP2 knockout mice are embryonically lethal. Conditional knock-down of DPP2 in specific cell types such as neuroendocrine cells (expressing neurogenin 3, NGN3) in the pancreas as well as regions of the hypothalamus and brain stem displayed a phenotype which is in stark contrast to that of DPP4 knockout mice [41], [42]. NGN3-DPP2 knock-down mice had unaltered levels of active GLP-1 but presented with hyperinsulinemia, accompanied by marked impairment of glucose tolerance, insulin resistance, enhanced liver steatosis, and visceral obesity, whereas DPP4 knockout mice are resistant to diet-induced obesity and insulin resistance [41], [42]. These findings suggest that dipeptidases DPP2 and DPP4 may have distinct substrate specificities and opposing functional roles.
Human DPP8 and DPP9 share significant homology (62% identity over 820 amino acids), whereas DPP4 is less homologous, e.g. 27% identical amino acids over a span of 677 amino acids with DPP8 [43]. In contrast to DPP4, both DPP8 and DPP9 are expressed in cytoplasma. They share substrate specificity with DPP4 based on in vitro assays utilizing native peptides or peptide pseudosubstrates. However, their physiologically relevant natural substrates and functions in vivo were not known until recently [44]. Utilizing cytosol-wide analysis of proteome in human ovarian cancer lines stably transfected with DPP8 or 9 using terminal amine isotopic labeling of substrates approach, a recent study identified more than 29 candidate natural substrates and pathways affected by DPP8/DP9 overexpression. Cleavage of 14 substrates was investigated in vitro; 9/14 substrates for both DPP8 and DPP9 were confirmed by MALDI-TOF mass spectrometry, including two of high confidence, calreticulin and adenylate kinase 2. No unique cleavage was identified for DP8 or DP9 demonstrating similar enzyme specificity and large substrate overlap between these enzymes Adenylate kinase 2 plays key roles in cellular energy and nucleotide homeostasis [44]. While in vitro studies have demonstrated non-enzymatic roles for DPP8 and DPP9 in cell migration, proliferation, and apoptosis, the exact physiologic role of these dipeptidases and their in-vivo substrates remain poorly defined [44].
3. Alteration of DPP4 expression in cardiometabolic disorders
3.1. DPP4 expression in the setting of cardiometabolic syndrome
The alteration of DPP4 expression has been studied in a variety of disease conditions including cardiometabolic disease. It has been shown that both expression and catalytic activity of DPP4 were enhanced in diabetes. When in vitro exposed to high glucose, human glomerular endothelial cells displayed an enhanced expression and activity of DPP4 [45], [46]. In line with this finding, circulating DPP4 activity was reported to be increased in patients with type 2 diabetes and positively correlated with HbA1c levels [47], [48]. In diabetic patients, the increase of circulating DPP4 activity results in a reduction of plasma GLP-1 both at fasting and in response to the meal [49]. In addition to circulating DPP4 activity, expression of DPP4 on T cells was reported to increase in patients with T2DM [50]. However, there are also reports suggesting a decrease of circulating DPP4 activity in patients with T2DM [51]. This is probably because most patients included in this study were on anti-diabetic agents, while those medications were reported to reduce circulating DPP4. Several widely-used anti-diabetic agents such as metformin, thiazolidinedione, and pioglitazone were reported to reduce circulating DPP4 [52], [53], [54] and DPP4 on T cells [50].
An enhanced expression of hepatic DPP4 was also reported in non-alcoholic fatty liver disease and its expression may adversely affect glucose metabolism in this liver disease [55]. In vitro stimulation of HepG2 cells with high glucose increased the expression of DPP4, whereas insulin, fatty acids and cholesterol did not [55]. In addition to cardiometabolic diseases, an altered DPP4 expression has also been reported in many other diseases such as rheumatoid arthritis [56], [57], systemic lupus erythematosus [58], major depression [59], and allograft rejection [60].
3.2. Molecular mechanisms underlying regulation of DPP4 expression
The signaling pathways mediated by DPP4 are not completely understood and what is known may vary depending on the cell type, context and the microenvironment.
Hepatocyte nuclear factor (HNF)− 1α, − 1β, and signal transducer and activator of transcription-α (STAT1α) were identified as transcription factors for DPP4 expression [61], [62], [63]. The expression of DPP4 was reduced by 60–80% by mutations of HNF1 [62]. The promoter of DPP4 contains a consensus GAS (interferon gamma-activated sequence) motif at bp − 35 to − 27, which has been shown to be a binding motif by STAT1α. Administrations of both interferon and retinoic acid result in the tyrosine phosphorylation of STAT1α, and subsequent nuclear translocation and binding to the GAS motif resulting in DPP4 transcription [61].
The regulation of DPP4 expression is highly dependent on cell types. DPP4 expression on T cells increases during activation, independent of CD4 or CD8 phenotype. IL-12 enhances the translation but not transcription of DPP4 in activated lymphocytes, while TNFα decrease cell surface expression of DPP4, suggesting a regulatory role of IL-12 and TNFα in the translation and probably translocation of DDP4 toward the cell surface [64]. When stimulated by IL-12, IFN-γ-producing T cells expressed higher levels of DPP4 compared with IFN-γ negative cells [27]. Both IFN-γ and retinoic acid could increase DPP4 transcription in malignant B lymphocytes through STAT1α pathway [61]. IFN-γ also induces DPP4 expression on renal epithelial cells [65]. In contrast, DPP4 expression on natural killer cells is induced by IL-2, IL-12, or IL-15, but not IFN-γ [66]. The expression of DPP4 on gingival fibroblasts is shown to be regulated by IL-1α [67]. A recent study demonstrates that IL-1α also contributes to the upregulation of DPP4 expression on epithelial cells and stromal cells in wounded dermis [68].
4. Catalytic and non-catalytic function of DPP4
4.1. Catalytic function
As a member of dipeptidyl peptidase family proteins, DPP4 functions as an N-terminal dipeptidase with a preference for a proline or alanine residue at the penultimate position (i.e., X-Pro-peptide, X means any amino acid). DPP4 thus cleaves X-Pro or X-Ala dipeptides from the N-terminal end of a diverse range of proteins including glucagon-like peptide (GLP) − 1 and − 2, glucose-dependent insulinotropic peptide (GIP), neuropeptide Y (NPY), CCL5, and CXCL9 can be cleaved by DPP4. The currently available DPP4 inhibitors in clinic suppress the dipeptidase function and preserve the function of its substrates.
Substrates of DPP4 can be categorized into 3 groups: regulatory peptides, chomokines and cytokines, and neuropeptides [48]. The regulatory peptide substrates include GLP-1, GLP-2, GIP, gastrin-releasing peptide (GRP), and growth-hormone-releasing factor (GHRF). The active form of GLP-1, GLP-1(7–36), could be rapidly converted to an inactive form GLP-1(9–36). Similarly, GLP-2(1–33), GIP(1–42) and GHRF(1–29) can be inactivated by DPP4 and converted to GLP-2(3–33), GIP(3–42), and GHRF(3–29) respectively. The chemokines that can be degraded by DPP4 include regulated on activation normal T-cell expressed and presumably secreted (RANTES, also known as CCL5), macrophage derived chemokine (MDC, also known as CCL22), eotaxin (also known as CCL11), monokine induced by IFN-γ (Mig, also known as CXCL9), IFN-γ-induced protein-10 (IP-10, also known as CXCL10), interferon-inducible T-cell α chemoattractant (ITAC, also known as CXCL11), and stromal-cell-derived factor-1 (SDF-1, also known as CXCL12). DPP4 alters the specificity of MDC on recruited cell types, while cleavage of N-terminal dipeptides inactivates the chemotactic activity of the other chemokines. A recent study suggests DPP4 also truncates a number of cytokines including GM-CSF, G-CSF, IL-3, and Epo [69]. Genetic ablation of DPP4 or enzymatic inhibition DPP4 activities by inhibitors enhanced hematopoiesis and bone marrow engraftment in mice after irradiation or chemotherapy. More importantly, truncated GM-CSF could serve as an antagonist for full-length GM-CSF as truncated GM-CSF has higher affinity to its receptor but devoid of bioactivity [69]. The neuropeptide substrates of DPP4 include NPY, B-type natriuretic peptide (BNP), Peptide YY (PYY), and Substance P. Removal of N-terminal dipeptides by DPP4 alters the receptor subtype specificity of NPY and PYY, reduces activity of substance P and BNP.
4.2. Non-catalytic function
In addition to its well-known peptidase activity, DPP4 also possesses non-catalytic functions via interacting with a range of ligands including adenosine deaminase (ADA), caveolin-1, fibronectin, and CXCR4. By interacting with its ligands, DPP4 plays a role in a variety of processes such as immune regulatory function such as enhancing T-cell activation and functional modulation of antigen-presenting cells (APCs).
DPP4 is expressed on T cells and represents as a marker of T cell activation [70]. Upon activation, both the percentage of DPP4+ T cells and the number of molecules per cell are elevated [71]. DPP4 on the cell surface of T cells has been demonstrated to be able to directly provide co-stimulatory signal by binding to ADA [70], [72], [73], [74], [75], [76]. The engagement of ADA by DPP4 enhances T-cell activation by activating CD45 [77]. Activation of DPP4 by anti-DPP4 antibodies prolonged and enhanced tyrosine phosphorylation of a variety of proteins activated in response to TCR/CD3 engagement [78]. In addition, ADA-independent non-catalytic activity of DPP4 also plays a role in T cell activation. Yu et al. reported that phytohaemagglutinin (PHA)- and Herpes simplex virus antigen (HSV Ag)-induced T-cell proliferation can be enhanced by soluble DPP4, although DPP4 showed no effect on anti-CD3-induced T cell proliferation. Moreover, both enzyme inactive mutant and ADA non-binding mutant promoted HSV Ag-induced T cell proliferation, indicating this effect of soluble DPP4 is mechanistically independent of both the enzymatic activity and ADA-binding capability of DPP4 [79].
Recent findings showed that DPP4 in a number of species including human, bat, macaque, horse, rabbit, and camel is able to bind spike protein on Middle Eastern Respiratory Syndrome-Corona Virus (MERS-CoV) and mediates its entrance into host cells [69], [80]. More interestingly, there is an overlap between virus-DPP4 and ADA-DPP4 binding interface, indicating a possible manipulation of the host immune response by MERS-CoV through competition for the ADA-recognition site [81].
5. DPP4 and diabetes
5.1. Incretin-dependent function and diabetes
5.1.1. Incretin system
Orally introduced glucose induces a greater insulin response than intravenously administered glucose, a concept known as incretin effect [82], [83]. Incretin hormones include glucagons-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). GLP-1 and GIP are produced by enteroendocrine L cells and K cells, respectively.
As the major types of incretin peptides, one of the primary functions for GLP-1 and GIP is to promote postprandial insulin secretion (Fig. 2 ). Both GLP-1 and GIP increase the transcription and biosynthesis of insulin, promote β-cell proliferation, and suppress β-cell death [84], [85], [86]. GLP-1 is also shown to be able to enhance glucose sensitivity of β-cell and induce glucose competence in previously unresponsive β-cells [87], [88]. Clinical studies confirmed the blood glucose lowering effect of exogenous administration of GLP-1 [89], [90]. In addition to pancreas, GLP-1 also reduces blood sugar by acting on many other organs including liver, brain, adipose tissue, and intestine. GLP-1 decreases glycemia in subjects during a pancreatic clamp, a condition with fixed insulin and glucagons level, suggesting that GLP-1 can suppress glucose production independent of islet hormones [91]. GLP-1 diminishes hepatic glucose production, which also contributes to its anti-hyperglyciemia effect [92]. GLP-1 also acts as a neuropeptide to reduce food intake by inhibiting appetite and regulates muscle glucose utilization and insulin secretion by acting on brain GLP-1 receptor (GLP-1R) [93], [94], [95]. Furthermore, GLP-1 also suppresses gastric emptying, inhibits adipose inflammation and promotes adipocyte formation by acting on digestive duct and adipose tissue [96], [97], [98].
Fig. 2.
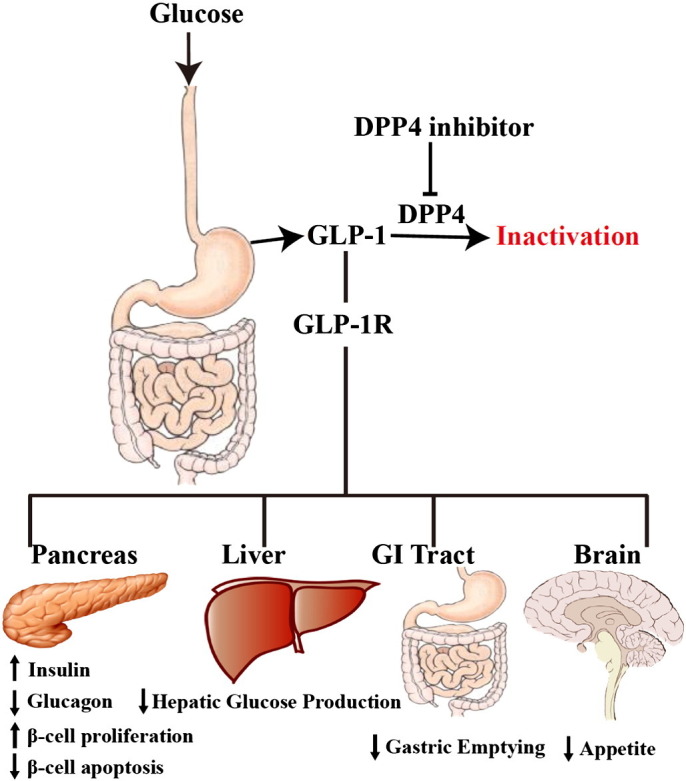
Regulatory function of GLP-1 on glucose metabolism.
In response to meal intake, GLP-1 is released by L cells distributed throughout the gastrointestinal tract (GI Tract). GLP-1 acts on GLP-1 receptor (GLP-1R) that is expressed in various organs and mediates multiple processes to maintain glucose homeostasis.
Despite plenty benefits, GLP-1/GIP treatment is limited by its short half-life. The half-life of GLP-1 is less than 2 min, while the half-life of GIP is less than 2 min in rodents and 7 min in human [84], [99], [100]. GLP-1(7–36) and GIP(1–42), the active forms of GLP-1 and GIP, are converted into inactive forms: GLP-1(9–36) and GIP(3–42) by a widely expressed enzyme, DPP4. The degradation of GLP-1 and GIP by DPP4 was first reported in early 1990s [101]. An enzymatic removal of N-terminal dipeptides His7-Ala8 from GLP-1 and Tyr1-Ala2 from GIP in plasma was observed in an in vitro experiment, using DPP4 purified from human placenta. Subsequent studies confirmed the metabolism of GLP-1 and GIP mediated by DPP4 did indeed occur in vivo [99], [102]. Accordingly, DPP4 inhibitors emerged as a novel therapeutic approach for type 2 diabetes in clinic [103].
5.1.2. DPP4 inhibitors and diabetes
Incretin response is found impaired in patients with type 2 diabetes and DPP4 enzymatic activity correlates with the degree of glucose homeostasis, suggesting that DPP4 mediated incretin degradation is involved in the pathogenesis of type 2 diabetes. A significant correlation between DPP4 and HbA1c was observed in Type 2 diabetic subjects who also demonstrated higher DPP4 activity than controls or those with impaired glucose tolerance [47].
DPP4 inhibitors are a class of newly available oral agents for type 2 diabetes, either as monotherapy or in combination with oral medications such as metformin, glitazones, and sulphonylureas [104]. Treatment with DPP4 inhibitors is usually accompanied with improvement in β cell function: improved homeostasis model assessment beta cell function index (HOMA-β) and fasting proinsulin:insulin ratio [105]. The therapeutic effects of DPP4 inhibitors are based on the ability of DPP4 to degrade the incretin peptides. Inhibition of DPP4 conversely increases the half-life of incretins and contributes to glucose lowering in the post-prandial state [104]. DPP4 inhibitors are only modestly effective as sole glucose-lowering drugs, with a 0.6–0.8% lowering of HbA1C in patients with a starting level around 8% [106]. However, they are being increasingly used in clinic due to the safety of these drugs. The side effect incidence is very low with these agents [105], [107]. The most reported side effects of DPP4 inhibitors in clinical trials include nasopharyngitis, upper respiratory tract infection, urinary tract infection and headache [108]. There are currently several DPP4 inhibitors approved or being approved by FDA or EU, which may be broadly divided into two classes based on structure: DPP4 dipeptide structure mimics and non-peptidomimetics. The first class includes Sitagliptin (β-amino acid-based), vildagliptin, and saxagliptin, whereas the non-peptidomimetics includes alogliptin (modified pyrimidinedione) and linagliptin (xanthine-based). The approved DPP4 inhibitors available in the market include: sitagliptin (approved by FDA in 2006), vildagliptin (approved by EU in 2007), saxagliptin (approved by FDA in 2009), linagliptin (approved by FDA in 2011), teneligliptin (approved in Japan 2012), anagliptin (approved in Japan 2012), and alogliptin (approved by FDA in 2013). In addition to those approved drugs, more DPP4 inhibitors including dutogliptin [109] and gemigliptin are under development and awaiting FDA approval.
5.2. Incretin-independent function and diabetes
It has been shown that inflammation is a major contributor to insulin resistance and T2DM [110], [111], [112]. Both innate immunity and adaptive immunity are responsible for the development of metaflammation (a metabolic inflammatory state) [110], [111], [112], [113], [114], [115]. Given the role of DPP4 in regulating T cell activation, DPP4 may be an important regulator of metaflammation. In addition, the binding of ADA to membrane-bound DPP4 may also promote T cell activation by clearing peri-cellular adenosine. Extracellular adenosine is generated from ATP by cell surface CD39 [116] and CD73. Excessive amount of adenosine suppresses T cell proliferation. Adenosine is degradated by ADA and generates to inosine, a nontoxic product [117]. Loss-of-function mutations in ADA could cause severe combined immunodeficiency (SCID) in both human and mouse by resulting in the accumulation of adenosine [118], [119], [120]. Jurkat cells with a DPP4 mutant devoid of ADA binding activity are vulnerable to inhibitory effect on proliferation [121], indicating that DPP4 on immune cells facilitates adenosine clearance by binding to ADA (Fig. 3 ). There are two isoforms of ADA, ADA1 and ADA2. ADA1 is ubiquitously expressed and present in cytosol as well as on the cell membrane. The primary role of ADA1 is to degrade adenosine and deoxyadenosine [122]. ADA2 is the predominant isoform found in plasma. Unlike ADA1, ADA2 possesses a much higher costimulatory activity than ADA1 [123] and has a much lower capacity to catalyze adenosine deamination [124].
Fig. 3.

ADA and its role in ATP metabolism.
ATP or ADP is converted into AMP by membrane-bound CD39. AMP is further catalyzed by CD73 and produce adenosine. Adenosine is then degraded by ADA bound to membrane-anchoring DPP4.
DPP4 on T cells may also interact with caveolin-1 present on APCs and initiates a signaling cascade in antigen loaded APCs, resulting in their activation [125], [126]. Upon binding to DPP4, caveolin-1 is phosphorylated, resulting in the phosphorylation of IRAK-1 and eventual NF-kappa B (NFkB) activation [127], [128]. Activated NFkB in turn upregulates CD86 [126]. The interaction between DPP4 and caveolin-1 has been reported to be involved in the pathogenesis of arthritis [129]. DPP4 has been reported to bind multiple components of extracellular matrix such as collagen, fibronectin, and the HIV-1 Tat protein [9], [130]. Interactions with these matrix components may play a role in sequestration of DPP4 and allow additional functions such as matrix remodeling, metastasis and chemotaxis.
Our recent work suggests that DPP4 expressed on APCs (including macrophage and dendritic cell) also promotes metaflammation by interacting with ADA [27]. DPP4 levels in adipose tissue inflammatory cells were higher than their counterparts in the circulation. DPP4 expression in visceral adipose tissue increased in obese patients with a positive correlation between DPP4 levels and the degree of insulin resistance (HOMA-IR). Further evidence suggests that APC-expressing DPP4 promotes T cell proliferation and activation independent of its catalytic activity as catalytic inhibition of DPP4 or addition of exogenous soluble DPP4 did not have such effect. In vitro co-culture experiments suggest that DPP4 on APCs is able to anchor ADA on the cell surface and modulates peri-cellular microenvironment through clearing adenosine [27]. These results suggested an involvement of DPP4 non-catalytic function in adipose inflammation and diabetes.
6. Catalytic-dependent and -independent protective effects of DPP4 inhibition on cardiovascular system
Recently, we and others demonstrated that DPP4 inhibitors confer cardioprotective effects in addition to their anti-glycemia effect. These may include both incretin-dependent and -independent effects.
6.1. Incretin-dependent effect of DPP4 inhibition on cardiovascular disease
By including 18 randomized clinical trials with 4988 patients on DPP4 inhibition therapy and 3546 patients on control treatment (other diabetic treatments or placebo), a recent meta-analysis demonstrated that DPP4 inhibitors are safe from cardiovascular standpoint and have beneficial effects on decreasing adverse cardiovascular events compared to other diabetic medications or placebo [4]. Studies indicate that agonists of GLP-1, a substrate of DPP4, improve the outcome of cardiovascular disease. It has been suggested for some time that receptor for GLP-1 (GLP-1R) is expressed in cardiovascular cells including endothelial cells, cardiomyocytes and coronary smooth muscle cells [131]. GLP-1R belongs to G protein-coupled receptors. The engagement of GLP-1R leads to the activation of adenylate cyclase through stimulatory Gs subunit and subsequent accumulation of cAMP in classically responsive cells such as pancreatic β cells [132], [133]. For example, in β cell, the activation of GLP-1 activates PKA, which subsequently reduces Foxo1 and results in an increase of Foxa2. Foxa2 then increases PDX-1, an transcription factor for insulin [134], [135]. Via a cAMP-dependent pathway, GLP-1R signaling may also induce the activation of PI3K which further increases the expression of Bcl-2 and Bcl-xL, two antiapoptotic proteins [136].
There is compelling evidence indicating that GLP-1 signaling is involved in the cardioprotective effects of DPP4 inhibition. For example, it has been shown that acute infusion of GLP-1 improves endothelial dysfunction in patients with type 2 diabetes [137], [138]. Exendin-4, a GLP-1R agonist, was also shown to stimulate proliferation of human coronary artery endothelial cells through eNOS-, PKA-, and PI3K/Akt-dependent pathways [139]. Both DPP4 inhibitor and GLP-1 can increase endothelial progenitor cells (EPCs), suggesting a role of GLP-1-dependent pathway in the development of EPCs [140], [141]. Moreover, GLP-1 can increase left ventricular developed pressure and coronary flow in isolated mouse hearts [142]. Further studies confirmed that the protective effect of GLP-1 on endothelial cells is mediated through increasing nitric oxide (NO) production [143]. Those effects could be mediated by in both GLP-1R-dependent and -independent pathways [142].
There are several large scale phase 3 or phase 4 clinical trials to assess the cardiovascular effect of DPP4 inhibitors. Those trials include EXAMINE (EXamination of cArdiovascular outcoMes with alogliptIN versus standard of carE in patients with type 2 diabetes mellitus and acute coronary syndrome) on alogliptin, TECOS (Trial Evaluating Cardiovascular Outcomes With Sitagliptin) on sitagliptin, the SAVOR-TIMI 53(Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus-Thrombolysis in Myocardial Infarction 53 trial) on saxagliptin, and CAROLINA (Cardiovascular Outcome Study of Linagliptin Versus Glimepiride in Patients With Type 2 Diabetes) on linagliptin. Among those trials, EXAMINE and SAVOR-TIMI 53 were recently completed. In the EXAMINE study, 5380 diabetic patients with a recent (< 90 days) myocardial infarction or unstable angina requiring hospitalization were randomized to the treatment of alogliptin or placebo. The patients were followed for a period up to 40 months (a median of 18 months). Alogliptin treatment did not improve the outcome of primary endpoints including death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke [8]. A total of 16,492 diabetic subjects with a history of MI, or documented atherosclerosis, or at least one of hypertension, smoking, or dyslipidemia and HbA1c 6.5–12% were recruited in the trial of SAVOR-TIMI 53 to evaluate the effect of saxagliptin on cardiovascular outcomes (a composite of cardiovascular death, nonfatal myocardial infarction or nonfatal ischemic stroke). The patients were randomized to saxagliptin group or placebo group. Additional anti-diabetic agents were prescribed throughout the study. Median follow-up time was 2.1 years and maximum was 2.9 years. No improvements in cardiovascular outcomes were observed in saxagliptin treatment group compared to placebo-treated patients. However, hospitalization rate for heart failure was higher in saxagliptin-treated subjects (3.5% vs. 2.8%; hazard ratio, 1.27; 95% CI, 1.07 to 1.51; P = 0.007) [7]. No differences in multiple safety endpoints (such as pancreatitis, cancer, and hypoglycemia) were observed across groups in both trials. These trials suggest that catalytic inhibition of DPP4 is basically safe from cardiovascular standpoint but also does not improve cardiovascular endpoints. There are several reasons for this discrepancy between these clinical trial results and previous reports that DPP4 inhibition possessed beneficial cardiovascular effect. One of the possible reasons is that the follow-up period of those trials is not long enough to observe the difference between treatment group and placebo group (Median follow-up periods were 1.5 years and 2.1 years for EXAMINE and SAVOR-TIMI 53 respectively). Another possible reason is that patients in both groups were receiving other anti-diabetic treatments throughout the study. The possible reason for this neutral effect on cardiovascular endpoints may involve other substrates of DPP4. As mentioned previously, the substrates of DPP4 include a variety of peptides. Those treatments might interfere with or cover the cardiovascular effects of DPP4 inhibition. Many of these substrates, such as SDF-1 and NPY, are implicated in diabetes and cardiovascular disease. However, it is also possible that catalytic inhibition alone is insufficient to improve cardiovascular outcomes.
6.2. Incretin-independent effect of DPP4 inhibition on cardiovascular disease
DPP4 is expressed in endothelial cells may mediate cardiovascular effects in both GLP-1-dependent or -independent manner, although the functional significance of these effects has not been fully elucidated. By incubating isolated aorta rings with DPP4 enzymatic inhibitor, we provided direct evidence showing DPP4 inhibition relaxes aorta through a GLP-1-independent pathway. Alogliptin, a DPP4 inhibitor, but not GLP-1 induces eNOS and Akt phosphorylation (Ser1177 and Ser473 respectively) paralleled by a rapid increase in nitric oxide [144]. Inhibition of Src kinase decreased eNOS and Akt phosphorylation in response to alogliptin, in contrast to a lack of any effect on insulin mediated activation of the eNOS-Akt, suggesting that alogliptin mediates vasodilation through Src kinase mediated effects on eNOS-Akt. Whether DPP4 exerts effects directly on eNOS function and the mechanisms by which it exerts these effects remain to be seen [144].
DPP4 has also been shown to assist reconstruction of vasculature by promoting EPCs in a GLP-1-independent pathway. SDF-1α, a well-studied substrate of DPP4, is a regulator of EPCs. By comparing four weeks of sitagliptin versus no additional treatment added to baseline metformin and/or sulfonylurea therapy in 32 diabetic patients, Fadini and coworkers demonstrated patients with DPP4 inhibition had a 2-fold increase of EPCs, 50% increase of SDF-1α [145]. Animal hindlimb ischemia and myocardial infarction studies confirmed the promotive effect of DPP4 inhibition on vasculogenesis through SDF-1 [140], [146]. SDF-1 also mediates the homing and engraftment of progenitor cells in transplantation. Enzymatic inhibition or deletion of DPP4 greatly promotes the homing and engraftment of both mouse and human hematopoietic progenitor cells via a mechanism involving SDF-1 [147], [148], [149], [150]. Further study suggests that truncation of colony-stimulating factors (CSF) such as GM-CSF, G-CSF, erythropoietin, and IL-3 is also involved in DPP4-mediated regulation of progenitor cell homing and engraftment [69]. However, degradation of another substrate NPY(1–36) by DPP4 has been suggested to promote angiogenesis by enhancing the production of NPY(3–36). Both NPY(1–36) and NPY(3–36) have been shown to promote human umbilical endothelial cell migration in an endothelial wound assay [151]. Inhibition of DPP4 activity by a neutralizing antibody suppresses angiogenic activity of NPY(1–36) but not NPY(3–36), suggesting that the production of NPY(3–36) is required for angiogenic activity of NPY [151].
Another important mechanism by which DPP4 inhibition proffer cardiovascular benefits is through a reduction in inflammation. Inflammation plays an etiological role in the development and maintenance of cardiovascular disease [152]. DPP4 is also known as the T-cell antigen CD26. In addition to cardiovascular cells, DPP4 is also highly expressed on many inflammatory cells including T cells and monocytes, and regulates their biological processes and functions [48]. Both enzymatic and non-enzymatic functions of DPP4 play an important role in T cell activation. T cell activation can be blocked by enzymatic inhibition of DPP4 [153], [154]. Suppression of dipeptidase activity of DPP4 resulted in a reduced production of cytokines including IL-2, IL-10, IL-12, and IFN-γ by peripheral blood mononuclear cells and T cells [153], [155], [156]. TGF-β1, an immunosuppressive cytokine, was shown to be up-regulated by DPP4 inhibition [155], [157], [158]. T cells transfected with mutant DPP4 devoid of enzymatic activity displayed reduced activation compared with that of transfected with wild-type DPP4 [159]. Consistent with that, the addition of soluble DPP4 promoted the recall antigen-induced proliferation of peripheral blood lymphocytes, while soluble DPP4 mutant without enzymatic activity did not, providing a direct evidence that enzymatic activity of DPP4 is involved in the T cell activation [160]. Other than its enzymatic activity, the non-enzymatic function of DPP4 is also important for T cell activation. DPP4 has been shown to be able to directly bind to several extracellular molecules such as ADA (adenosine deaminase), collagen and fibronectin [48]. Engagement of DPP4 by ADA or antibodies induces co-stimulatory signal and activates T-cell through interacting with CD45, a membrane-linked tyrosine phosphatase [77]. Following the activation of T cells, both the percentage of DPP4+ cells and the number of molecules per cell are elevated [71]. DPP4-induced T-cell activation requires TCR/CD3 complex. DPP4 engagement can induce cytotoxicity only in CD3-expressing clones but not CD3-negative clones [161]. Furthermore, down-regulation of CD3 by antibody pre-incubation markedly reduced DPP4-mediated T-cell proliferation [72]. The activation of DPP4 enhances the tyrosine phosphorylation of p56lck and ζ chain in CD3 and induces IL-2 production [77], [162]. In addition, the phosphorylation of a range of intracellular proteins which are involved in the TCR/CD3 signaling, such as p56lck, p59fyn, ZAP-70, MAP kinase, c-Cbl, and phospholipase Cγ was increased and prolonged by the cross-linking of DPP4 [78].
Other than T-cell, DPP4 also regulates the functionality of antigen-presenting cells (APCs) including macrophages and dendritic cells. Soluble DPP4 has been reported to be able to upregulate the expression of CD86 on APCs and thus enhance antigen presentation function [125]. Additional studies demonstrated that DPP4 directly binds caveolin-1 on the cell surface of APCs. The interaction between DPP4 and Caveolin-1 expressed on APCs mediates a signaling cascade in antigen loaded APCs, resulting in their activation [125], [126]. The binding of DPP4 to caveolin-1 induces its phosphorylation, which subsequently activates IRAK-1 and NFκB [126]. Recently, we showed that DPP4 on macrophages and dendritic cells is important for their inflammatory function. By binding ADA, APC-expressing DPP4 provides a microenvironment suitable for T cell proliferation and enhances visceral adipose inflammation [27]. In addition to ADA and caveolin-1, DPP4 has been reported to bind multiple extracellular matrix components including collagen and fibronectin [24], [74], [163]. The interaction between DPP4 and the matrix may play a role in matrix remodeling, metastasis and chemotaxis.
7. Conclusion
The development and application of specific DPP4 inhibitors in clinic reemphasized the importance of DPP4 in both physiological and pathological processes. Recent studies suggest an implication of DPP4 in cardiovascular disease and the non-catalytic activity of DPP4 may also play a role in metabolic dysfunction. However, the details of the involvement in various biological processes and diseases are not fully elucidated. Further studies are required to explore these implications.
Sources of funding
This work was supported by grants from AHA (15SDG25700381 and 13POST17210033) and NSFC (81101553/H1604) to Dr. Zhong.
Disclosures
The authors declare no conflict of interests in this paper.
Contributor Information
Sanjay Rajagopalan, Email: srajagopalan@medicine.umaryland.edu.
Jixin Zhong, Email: jzhong@medicine.umaryland.edu.
References
- 1.Kwok A.J., Mashar M., Khavandi K., Sabir I. DPP-IV inhibitors: beyond glycaemic control? Trends Cardiovasc. Med. 2014;24:157–164. doi: 10.1016/j.tcm.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Sauve M., Ban K., Momen M.A., Zhou Y.Q., Henkelman R.M., Husain M. Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes after myocardial infarction in mice. Diabetes. 2010;59:1063–1073. doi: 10.2337/db09-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shigeta T., Aoyama M., Bando Y.K., Monji A., Mitsui T., Takatsu M. Dipeptidyl peptidase-4 modulates left ventricular dysfunction in chronic heart failure via angiogenesis-dependent and -independent actions. Circulation. 2012;126:1838–1851. doi: 10.1161/CIRCULATIONAHA.112.096479. [DOI] [PubMed] [Google Scholar]
- 4.Patil H.R., Al Badarin F.J., Al Shami H.A., Bhatti S.K., Lavie C.J., Bell D.S. Meta-analysis of effect of dipeptidyl peptidase-4 inhibitors on cardiovascular risk in type 2 diabetes mellitus. Am. J. Cardiol. 2012;110:826–833. doi: 10.1016/j.amjcard.2012.04.061. [DOI] [PubMed] [Google Scholar]
- 5.Read P.A., Khan F.Z., Heck P.M., Hoole S.P., Dutka D.P. DPP-4 inhibition by sitagliptin improves the myocardial response to dobutamine stress and mitigates stunning in a pilot study of patients with coronary artery disease. Circ. Cardiovasc. Imaging. 2010;3:195–201. doi: 10.1161/CIRCIMAGING.109.899377. [DOI] [PubMed] [Google Scholar]
- 6.Scheen A.J. Cardiovascular effects of gliptins. Nat. Rev. Cardiol. 2013;10:73–84. doi: 10.1038/nrcardio.2012.183. [DOI] [PubMed] [Google Scholar]
- 7.Scirica B.M., Bhatt D.L., Braunwald E., Steg P.G., Davidson J., Hirshberg B. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013;369:1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 8.White W.B., Cannon C.P., Heller S.R., Nissen S.E., Bergenstal R.M., Bakris G.L. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N. Engl. J. Med. 2013;369:1327–1335. doi: 10.1056/NEJMoa1305889. [DOI] [PubMed] [Google Scholar]
- 9.Zhong J., Rao X., Rajagopalan S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: potential implications in cardiovascular disease. Atherosclerosis. 2013;226:305–314. doi: 10.1016/j.atherosclerosis.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 10.Abbott C.A., Yu D.M., Woollatt E., Sutherland G.R., McCaughan G.W., Gorrell M.D. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur. J. Biochem. 2000;267:6140–6150. doi: 10.1046/j.1432-1327.2000.01617.x. [DOI] [PubMed] [Google Scholar]
- 11.Hopsu-Havu V.K., Glenner G.G. A new dipeptide naphthylamidase hydrolyzing glycyl-prolyl-beta-naphthylamide. Histochem. Histochem. Histochim. 1966;7:197–201. doi: 10.1007/BF00577838. [DOI] [PubMed] [Google Scholar]
- 12.Hopsu-Havu V.K., Sarimo S.R. Purification and characterization of an aminopeptidase hydrolyzing glycyl-proline-naphthylamide. Hoppe-Seyler's Z. Physiol. Chem. 1967;348:1540–1550. doi: 10.1515/bchm2.1967.348.1.1540. [DOI] [PubMed] [Google Scholar]
- 13.Hopsu-Havu V.K., Rintola P., Glenner G.G. A hog kidney aminopeptidase liberating N-terminal dipeptides. Partial purification and characteristics. Acta Chem. Scand. 1968;22:299–308. doi: 10.3891/acta.chem.scand.22-0299. [DOI] [PubMed] [Google Scholar]
- 14.Drucker D.J. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care. 2003;26:2929–2940. doi: 10.2337/diacare.26.10.2929. [DOI] [PubMed] [Google Scholar]
- 15.Abbott C.A., McCaughan G.W., Levy M.T., Church W.B., Gorrell M.D. Binding to human dipeptidyl peptidase IV by adenosine deaminase and antibodies that inhibit ligand binding involves overlapping, discontinuous sites on a predicted beta propeller domain. Eur. J. Biochem. 1999;266:798–810. doi: 10.1046/j.1432-1327.1999.00902.x. [DOI] [PubMed] [Google Scholar]
- 16.Richard E., Arredondo-Vega F.X., Santisteban I., Kelly S.J., Patel D.D., Hershfield M.S. The binding site of human adenosine deaminase for CD26/Dipeptidyl peptidase IV: the Arg142Gln mutation impairs binding to cd26 but does not cause immune deficiency. J. Exp. Med. 2000;192:1223–1236. doi: 10.1084/jem.192.9.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwaki-Egawa S., Watanabe Y., Kikuya Y., Fujimoto Y. Dipeptidyl peptidase IV from human serum: purification, characterization, and N-terminal amino acid sequence. J. Biochem. 1998;124:428–433. doi: 10.1093/oxfordjournals.jbchem.a022130. [DOI] [PubMed] [Google Scholar]
- 18.McCaughan G.W., Wickson J.E., Creswick P.F., Gorrell M.D. Identification of the bile canalicular cell surface molecule GP110 as the ectopeptidase dipeptidyl peptidase IV: an analysis by tissue distribution, purification and N-terminal amino acid sequence. Hepatology. 1990;11:534–544. doi: 10.1002/hep.1840110403. [DOI] [PubMed] [Google Scholar]
- 19.Rasmussen H.B., Branner S., Wiberg F.C., Wagtmann N. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat. Struct. Biol. 2003;10:19–25. doi: 10.1038/nsb882. [DOI] [PubMed] [Google Scholar]
- 20.Gorrell M.D. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin. Sci. (Lond.) 2005;108:277–292. doi: 10.1042/CS20040302. [DOI] [PubMed] [Google Scholar]
- 21.Ajami K., Abbott C.A., Obradovic M., Gysbers V., Kahne T., McCaughan G.W. Structural requirements for catalysis, expression, and dimerization in the CD26/DPIV gene family. Biochemistry. 2003;42:694–701. doi: 10.1021/bi026846s. [DOI] [PubMed] [Google Scholar]
- 22.David F., Bernard A.M., Pierres M., Marguet D. Identification of serine 624, aspartic acid 702, and histidine 734 as the catalytic triad residues of mouse dipeptidyl-peptidase IV (CD26). A member of a novel family of nonclassical serine hydrolases. J. Biol. Chem. 1993;268:17247–17252. [PubMed] [Google Scholar]
- 23.Dobers J., Zimmermann-Kordmann M., Leddermann M., Schewe T., Reutter W., Fan H. Expression, purification, and characterization of human dipeptidyl peptidase IV/CD26 in Sf9 insect cells. Protein Expr. Purif. 2002;25:527–532. doi: 10.1016/s1046-5928(02)00043-8. [DOI] [PubMed] [Google Scholar]
- 24.Fleischer B. CD26: a surface protease involved in T-cell activation. Immunol. Today. 1994;15:180–184. doi: 10.1016/0167-5699(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 25.Engel M., Hoffmann T., Wagner L., Wermann M., Heiser U., Kiefersauer R. The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc. Natl. Acad. Sci. U. S. A. 2003;100:5063–5068. doi: 10.1073/pnas.0230620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mentzel S., Dijkman H.B., Van Son J.P., Koene R.A., Assmann K.J. Organ distribution of aminopeptidase A and dipeptidyl peptidase IV in normal mice. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1996;44:445–461. doi: 10.1177/44.5.8627002. [DOI] [PubMed] [Google Scholar]
- 27.Zhong J., Rao X., Deiuliis J., Braunstein Z., Narula V., Hazey J. A potential role for dendritic cell/macrophage-expressing DPP4 in obesity-induced visceral inflammation. Diabetes. 2013;62:149–157. doi: 10.2337/db12-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duke-Cohan J.S., Morimoto C., Rocker J.A., Schlossman S.F. Serum high molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen DPPT-L released from activated T cells. J. Immunol. 1996;156:1714–1721. [PubMed] [Google Scholar]
- 29.Lambeir A.M., Diaz Pereira J.F., Chacon P., Vermeulen G., Heremans K., Devreese B. A prediction of DPP IV/CD26 domain structure from a physico-chemical investigation of dipeptidyl peptidase IV (CD26) from human seminal plasma. Biochim. Biophys. Acta. 1997;1340:215–226. doi: 10.1016/s0167-4838(97)00045-9. [DOI] [PubMed] [Google Scholar]
- 30.Lamers D., Famulla S., Wronkowitz N., Hartwig S., Lehr S., Ouwens D.M. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60:1917–1925. doi: 10.2337/db10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z., Grigo C., Steinbeck J., von Horsten S., Amann K., Daniel C. Soluble DPP4 originates in part from bone marrow cells and not from the kidney. Peptides. 2014;57:109–117. doi: 10.1016/j.peptides.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Andrieu T., Thibault V., Malet I., Laporte J., Bauvois B., Agut H. Similar increased serum dipeptidyl peptidase IV activity in chronic hepatitis C and other viral infections. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2003;27:59–68. doi: 10.1016/s1386-6532(02)00128-2. [DOI] [PubMed] [Google Scholar]
- 33.Grondin G., Hooper N.M., LeBel D. Specific localization of membrane dipeptidase and dipeptidyl peptidase IV in secretion granules of two different pancreatic islet cells. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1999;47:489–498. doi: 10.1177/002215549904700407. [DOI] [PubMed] [Google Scholar]
- 34.Topham N.J., Hewitt E.W. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128:7–15. doi: 10.1111/j.1365-2567.2009.03123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park J.E., Lenter M.C., Zimmermann R.N., Garin-Chesa P., Old L.J., Rettig W.J. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 1999;274:36505–36512. doi: 10.1074/jbc.274.51.36505. [DOI] [PubMed] [Google Scholar]
- 36.Stremenova J., Mares V., Lisa V., Hilser M., Krepela E., Vanickova Z. Expression of dipeptidyl peptidase-IV activity and/or structure homologs in human meningiomas. Int. J. Oncol. 2010;36:351–358. [PubMed] [Google Scholar]
- 37.Santos A.M., Jung J., Aziz N., Kissil J.L., Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Invest. 2009;119:3613–3625. doi: 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brokopp C.E., Schoenauer R., Richards P., Bauer S., Lohmann C., Emmert M.Y. Fibroblast activation protein is induced by inflammation and degrades type I collagen in thin-cap fibroatheromata. Eur. Heart J. 2011;32:2713–2722. doi: 10.1093/eurheartj/ehq519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tillmanns J., Widera C., Habbaba Y., Galuppo P., Kempf T., Wollert K.C. Circulating concentrations of fibroblast activation protein alpha in apparently healthy individuals and patients with acute coronary syndrome as assessed by sandwich ELISA. Int. J. Cardiol. 2013;168:3926–3931. doi: 10.1016/j.ijcard.2013.06.061. [DOI] [PubMed] [Google Scholar]
- 40.Pangalos M.N., Neefs J.M., Somers M., Verhasselt P., Bekkers M., van der Helm L. Isolation and expression of novel human glutamate carboxypeptidases with N-acetylated alpha-linked acidic dipeptidase and dipeptidyl peptidase IV activity. J. Biol. Chem. 1999;274:8470–8483. doi: 10.1074/jbc.274.13.8470. [DOI] [PubMed] [Google Scholar]
- 41.Marguet D., Baggio L., Kobayashi T., Bernard A.M., Pierres M., Nielsen P.F. Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6874–6879. doi: 10.1073/pnas.120069197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conarello S.L., Li Z., Ronan J., Roy R.S., Zhu L., Jiang G. Mice lacking dipeptidyl peptidase IV are protected against obesity and insulin resistance. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6825–6830. doi: 10.1073/pnas.0631828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Augustyns K., Bal G., Thonus G., Belyaev A., Zhang X.M., Bollaert W. The unique properties of dipeptidyl-peptidase IV (DPP IV/CD26) and the therapeutic potential of DPP IV inhibitors. Curr. Med. Chem. 1999;6:311–327. [PubMed] [Google Scholar]
- 44.Wilson C.H., Indarto D., Doucet A., Pogson L.D., Pitman M.R., McNicholas K. Identifying natural substrates for dipeptidyl peptidases 8 and 9 using terminal amine isotopic labeling of substrates (TAILS) reveals in vivo roles in cellular homeostasis and energy metabolism. J. Biol. Chem. 2013;288:13936–13949. doi: 10.1074/jbc.M112.445841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pala L., Mannucci E., Pezzatini A., Ciani S., Sardi J., Raimondi L. Dipeptidyl peptidase-IV expression and activity in human glomerular endothelial cells. Biochem. Biophys. Res. Commun. 2003;310:28–31. doi: 10.1016/j.bbrc.2003.08.111. [DOI] [PubMed] [Google Scholar]
- 46.Yu D.M., Wang X.M., McCaughan G.W., Gorrell M.D. Extraenzymatic functions of the dipeptidyl peptidase IV-related proteins DP8 and DP9 in cell adhesion, migration and apoptosis. FEBS J. 2006;273:2447–2460. doi: 10.1111/j.1742-4658.2006.05253.x. [DOI] [PubMed] [Google Scholar]
- 47.Mannucci E., Pala L., Ciani S., Bardini G., Pezzatini A., Sposato I. Hyperglycaemia increases dipeptidyl peptidase IV activity in diabetes mellitus. Diabetologia. 2005;48:1168–1172. doi: 10.1007/s00125-005-1749-8. [DOI] [PubMed] [Google Scholar]
- 48.Ryskjaer J., Deacon C.F., Carr R.D., Krarup T., Madsbad S., Holst J. Plasma dipeptidyl peptidase-IV activity in patients with type-2 diabetes mellitus correlates positively with HbAlc levels, but is not acutely affected by food intake. Eur. J. Endocrinol. 2006;155:485–493. doi: 10.1530/eje.1.02221. [DOI] [PubMed] [Google Scholar]
- 49.Lugari R., Dei Cas A., Ugolotti D., Barilli A.L., Camellini C., Ganzerla G.C. Glucagon-like peptide 1 (GLP-1) secretion and plasma dipeptidyl peptidase IV (DPP-IV) activity in morbidly obese patients undergoing biliopancreatic diversion. Horm. Metab. Res. 2004;36:111–115. doi: 10.1055/s-2004-814222. [DOI] [PubMed] [Google Scholar]
- 50.Lee S.A., Kim Y.R., Yang E.J., Kwon E.J., Kim S.H., Kang S.H. CD26/DPP4 levels in peripheral blood and T cells in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2013;98:2553–2561. doi: 10.1210/jc.2012-4288. [DOI] [PubMed] [Google Scholar]
- 51.McKillop A.M., Duffy N.A., Lindsay J.R., O'Harte F.P., Bell P.M., Flatt P.R. Decreased dipeptidyl peptidase-IV activity and glucagon-like peptide-1(7–36)amide degradation in type 2 diabetic subjects. Diabetes Res. Clin. Pract. 2008;79:79–85. doi: 10.1016/j.diabres.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Lenhard J.M., Croom D.K., Minnick D.T. Reduced serum dipeptidyl peptidase-IV after metformin and pioglitazone treatments. Biochem. Biophys. Res. Commun. 2004;324:92–97. doi: 10.1016/j.bbrc.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 53.Lindsay J.R., Duffy N.A., McKillop A.M., Ardill J., O'Harte F.P., Flatt P.R. Inhibition of dipeptidyl peptidase IV activity by oral metformin in type 2 diabetes. Diabet. Med. 2005;22:654–657. doi: 10.1111/j.1464-5491.2005.01461.x. [DOI] [PubMed] [Google Scholar]
- 54.Green B.D., Irwin N., Duffy N.A., Gault V.A., O'Harte F.P., Flatt P.R. Inhibition of dipeptidyl peptidase-IV activity by metformin enhances the antidiabetic effects of glucagon-like peptide-1. Eur. J. Pharmacol. 2006;547:192–199. doi: 10.1016/j.ejphar.2006.07.043. [DOI] [PubMed] [Google Scholar]
- 55.Miyazaki M., Kato M., Tanaka K., Tanaka M., Kohjima M., Nakamura K. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol. Med. Rep. 2012;5:729–733. doi: 10.3892/mmr.2011.707. [DOI] [PubMed] [Google Scholar]
- 56.Hildebrandt M., Reutter W., Arck P., Rose M., Klapp B.F. A guardian angel: the involvement of dipeptidyl peptidase IV in psychoneuroendocrine function, nutrition and immune defence. Clin. Sci. (Lond.) 2000;99:93–104. [PubMed] [Google Scholar]
- 57.Kamori M., Hagihara M., Nagatsu T., Iwata H., Miura T. Activities of dipeptidyl peptidase II, dipeptidyl peptidase IV, prolyl endopeptidase, and collagenase-like peptidase in synovial membrane from patients with rheumatoid arthritis and osteoarthritis. Biochem. Med. Metab. Biol. 1991;45:154–160. doi: 10.1016/0885-4505(91)90016-e. [DOI] [PubMed] [Google Scholar]
- 58.Stancikova M., Lojda Z., Lukac J., Ruzickova M. Dipeptidyl peptidase IV in patients with systemic lupus erythematosus. Clin. Exp. Rheumatol. 1992;10:381–385. [PubMed] [Google Scholar]
- 59.Maes M., De Meester I., Vanhoof G., Scharpe S., Bosmans E., Vandervorst C. Decreased serum dipeptidyl peptidase IV activity in major depression. Biol. Psychiatry. 1991;30:577–586. doi: 10.1016/0006-3223(91)90027-j. [DOI] [PubMed] [Google Scholar]
- 60.Korom S., De Meester I., Stadlbauer T.H., Chandraker A., Schaub M., Sayegh M.H. Inhibition of CD26/dipeptidyl peptidase IV activity in vivo prolongs cardiac allograft survival in rat recipients. Transplantation. 1997;63:1495–1500. doi: 10.1097/00007890-199705270-00021. [DOI] [PubMed] [Google Scholar]
- 61.Bauvois B., Djavaheri-Mergny M., Rouillard D., Dumont J., Wietzerbin J. Regulation of CD26/DPPIV gene expression by interferons and retinoic acid in tumor B cells. Oncogene. 2000;19:265–272. doi: 10.1038/sj.onc.1203292. [DOI] [PubMed] [Google Scholar]
- 62.D'Angelo A., Bluteau O., Garcia-Gonzalez M.A., Gresh L., Doyen A., Garbay S. Hepatocyte nuclear factor 1alpha and beta control terminal differentiation and cell fate commitment in the gut epithelium. Development. 2010;137:1573–1582. doi: 10.1242/dev.044420. [DOI] [PubMed] [Google Scholar]
- 63.Erickson R.H., Lai R.S., Kim Y.S. Role of hepatocyte nuclear factor 1alpha and 1beta in the transcriptional regulation of human dipeptidyl peptidase IV during differentiation of Caco-2 cells. Biochem. Biophys. Res. Commun. 2000;270:235–239. doi: 10.1006/bbrc.2000.2420. [DOI] [PubMed] [Google Scholar]
- 64.Salgado F.J., Vela E., Martin M., Franco R., Nogueira M., Cordero O.J. Mechanisms of CD26/dipeptidyl peptidase IV cytokine-dependent regulation on human activated lymphocytes. Cytokine. 2000;12:1136–1141. doi: 10.1006/cyto.1999.0643. [DOI] [PubMed] [Google Scholar]
- 65.Stefanovic V., Ardaillou N., Vlahovic P., Placier S., Ronco P., Ardaillou R. Interferon-gamma induces dipeptidylpeptidase IV expression in human glomerular epithelial cells. Immunology. 1993;80:465–470. [PMC free article] [PubMed] [Google Scholar]
- 66.Yamabe T., Takakura K., Sugie K., Kitaoka Y., Takeda S., Okubo Y. Induction of the 2B9 antigen/dipeptidyl peptidase IV/CD26 on human natural killer cells by IL-2, IL-12 or IL-15. Immunology. 1997;91:151–158. doi: 10.1046/j.1365-2567.1997.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nemoto E., Sugawara S., Takada H., Shoji S., Horiuch H. Increase of CD26/dipeptidyl peptidase IV expression on human gingival fibroblasts upon stimulation with cytokines and bacterial components. Infect. Immun. 1999;67:6225–6233. doi: 10.1128/iai.67.12.6225-6233.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arwert E.N., Mentink R.A., Driskell R.R., Hoste E., Goldie S.J., Quist S. Upregulation of CD26 expression in epithelial cells and stromal cells during wound-induced skin tumour formation. Oncogene. 2011;31(8):992–1000. doi: 10.1038/onc.2011.298. [DOI] [PubMed] [Google Scholar]
- 69.Broxmeyer H.E., Hoggatt J., O'Leary H.A., Mantel C., Chitteti B.R., Cooper S. Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nat. Med. 2012;18:1786–1796. doi: 10.1038/nm.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morimoto C., Torimoto Y., Levinson G., Rudd C.E., Schrieber M., Dang N.H. 1F7, a novel cell surface molecule, involved in helper function of CD4 cells. J. Immunol. 1989;143:3430–3439. [PubMed] [Google Scholar]
- 71.Martin M., Huguet J., Centelles J.J., Franco R. Expression of ecto-adenosine deaminase and CD26 in human T cells triggered by the TCR-CD3 complex. Possible role of adenosine deaminase as costimulatory molecule. J. Immunol. 1995;155:4630–4643. [PubMed] [Google Scholar]
- 72.Dang N.H., Torimoto Y., Sugita K., Daley J.F., Schow P., Prado C. Cell surface modulation of CD26 by anti-1F7 monoclonal antibody. Analysis of surface expression and human T cell activation. J. Immunol. 1990;145:3963–3971. [PubMed] [Google Scholar]
- 73.Dang N.H., Torimoto Y., Shimamura K., Tanaka T., Daley J.F., Schlossman S.F. 1F7 (CD26): a marker of thymic maturation involved in the differential regulation of the CD3 and CD2 pathways of human thymocyte activation. J. Immunol. 1991;147:2825–2832. [PubMed] [Google Scholar]
- 74.Morimoto C., Schlossman S.F. The structure and function of CD26 in the T-cell immune response. Immunol. Rev. 1998;161:55–70. doi: 10.1111/j.1600-065x.1998.tb01571.x. [DOI] [PubMed] [Google Scholar]
- 75.Pacheco R., Martinez-Navio J.M., Lejeune M., Climent N., Oliva H., Gatell J.M. CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9583–9588. doi: 10.1073/pnas.0501050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Meester I.A., Kestens L.L., Vanham G.L., Vanhoof G.C., Vingerhoets J.H., Gigase P.L. Costimulation of CD4 + and CD8 + T cells through CD26: the ADA-binding epitope is not essential for complete signaling. J. Leukoc. Biol. 1995;58:325–330. doi: 10.1002/jlb.58.3.325. [DOI] [PubMed] [Google Scholar]
- 77.Torimoto Y., Dang N.H., Vivier E., Tanaka T., Schlossman S.F., Morimoto C. Coassociation of CD26 (dipeptidyl peptidase IV) with CD45 on the surface of human T lymphocytes. J. Immunol. 1991;147:2514–2517. [PubMed] [Google Scholar]
- 78.Hegen M., Kameoka J., Dong R.P., Schlossman S.F., Morimoto C. Cross-linking of CD26 by antibody induces tyrosine phosphorylation and activation of mitogen-activated protein kinase. Immunology. 1997;90:257–264. doi: 10.1046/j.1365-2567.1997.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu D.M., Slaitini L., Gysbers V., Riekhoff A.G., Kahne T., Knott H.M. Soluble CD26/dipeptidyl peptidase IV enhances human lymphocyte proliferation in vitro independent of dipeptidyl peptidase enzyme activity and adenosine deaminase binding. Scand. J. Immunol. 2011;73:102–111. doi: 10.1111/j.1365-3083.2010.02488.x. [DOI] [PubMed] [Google Scholar]
- 80.Barlan A., Zhao J., Sarkar M.K., Li K., McCray P.B., Jr., Perlman S. Receptor variation and susceptibility to Middle East respiratory syndrome coronavirus infection. J. Virol. 2014;88:4953–4961. doi: 10.1128/JVI.00161-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu G., Hu Y., Wang Q., Qi J., Gao F., Li Y. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature. 2013;500:227–231. doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McIntyre N., Holdsworth C.D., Turner D.S. New interpretation of oral glucose tolerance. Lancet. 1964;2:20–21. doi: 10.1016/s0140-6736(64)90011-x. [DOI] [PubMed] [Google Scholar]
- 83.Plutzky J. The incretin axis in cardiovascular disease. Circulation. 2011;124:2285–2289. doi: 10.1161/CIRCULATIONAHA.111.064139. [DOI] [PubMed] [Google Scholar]
- 84.Baggio L.L., Drucker D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 85.Kim S.J., Winter K., Nian C., Tsuneoka M., Koda Y., McIntosh C.H. Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J. Biol. Chem. 2005;280:22297–22307. doi: 10.1074/jbc.M500540200. [DOI] [PubMed] [Google Scholar]
- 86.Wang Y., Montrose-Rafizadeh C., Adams L., Raygada M., Nadiv O., Egan J.M. GIP regulates glucose transporters, hexokinases, and glucose-induced insulin secretion in RIN 1046–38 cells. Mol. Cell. Endocrinol. 1996;116:81–87. doi: 10.1016/0303-7207(95)03701-2. [DOI] [PubMed] [Google Scholar]
- 87.Holz GGt, Kuhtreiber W.M., Habener J.F. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7–37) Nature. 1993;361:362–365. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rachman J., Gribble F.M., Barrow B.A., Levy J.C., Buchanan K.D., Turner R.C. Normalization of insulin responses to glucose by overnight infusion of glucagon-like peptide 1 (7–36) amide in patients with NIDDM. Diabetes. 1996;45:1524–1530. doi: 10.2337/diab.45.11.1524. [DOI] [PubMed] [Google Scholar]
- 89.Nauck M.A., Kleine N., Orskov C., Holst J.J., Willms B., Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1993;36:741–744. doi: 10.1007/BF00401145. [DOI] [PubMed] [Google Scholar]
- 90.Zander M., Madsbad S., Madsen J.L., Holst J.J. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- 91.Prigeon R.L., Quddusi S., Paty B., D'Alessio D.A. Suppression of glucose production by GLP-1 independent of islet hormones: a novel extrapancreatic effect. Am. J. Physiol. Endocrinol. Metab. 2003;285:E701–E707. doi: 10.1152/ajpendo.00024.2003. [DOI] [PubMed] [Google Scholar]
- 92.Larsson H., Holst J.J., Ahren B. Glucagon-like peptide-1 reduces hepatic glucose production indirectly through insulin and glucagon in humans. Acta Physiol. Scand. 1997;160:413–422. doi: 10.1046/j.1365-201X.1997.00161.x. [DOI] [PubMed] [Google Scholar]
- 93.Barrera J.G., Sandoval D.A., D'Alessio D.A., Seeley R.J. GLP-1 and energy balance: an integrated model of short-term and long-term control. Nat. Rev. Endocrinol. 2011;7:507–516. doi: 10.1038/nrendo.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Knauf C., Cani P.D., Perrin C., Iglesias M.A., Maury J.F., Bernard E. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J. Clin. Invest. 2005;115:3554–3563. doi: 10.1172/JCI25764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ussher J.R., Drucker D.J. Cardiovascular biology of the incretin system. Endocr. Rev. 2012;33:187–215. doi: 10.1210/er.2011-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Naslund E., Bogefors J., Skogar S., Gryback P., Jacobsson H., Holst J.J. GLP-1 slows solid gastric emptying and inhibits insulin, glucagon, and PYY release in humans. Am. J. Physiol. 1999;277:R910–R916. doi: 10.1152/ajpregu.1999.277.3.R910. [DOI] [PubMed] [Google Scholar]
- 97.Lee Y.S., Park M.S., Choung J.S., Kim S.S., Oh H.H., Choi C.S. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia. 2012;55:2456–2468. doi: 10.1007/s00125-012-2592-3. [DOI] [PubMed] [Google Scholar]
- 98.Challa T.D., Beaton N., Arnold M., Rudofsky G., Langhans W., Wolfrum C. Regulation of adipocyte formation by GLP-1/GLP-1R signaling. J. Biol. Chem. 2012;287:6421–6430. doi: 10.1074/jbc.M111.310342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kieffer T.J., McIntosh C.H., Pederson R.A. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology. 1995;136:3585–3596. doi: 10.1210/endo.136.8.7628397. [DOI] [PubMed] [Google Scholar]
- 100.Pridal L., Deacon C.F., Kirk O., Christensen J.V., Carr R.D., Holst J.J. Glucagon-like peptide-1(7–37) has a larger volume of distribution than glucagon-like peptide-1(7–36)amide in dogs and is degraded more quickly in vitro by dog plasma. Eur. J. Drug Metab. Pharmacokinet. 1996;21:51–59. doi: 10.1007/BF03190278. [DOI] [PubMed] [Google Scholar]
- 101.Mentlein R., Gallwitz B., Schmidt W.E. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur. J. Biochem. 1993;214:829–835. doi: 10.1111/j.1432-1033.1993.tb17986.x. [DOI] [PubMed] [Google Scholar]
- 102.Deacon C.F., Johnsen A.H., Holst J.J. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J. Clin. Endocrinol. Metab. 1995;80:952–957. doi: 10.1210/jcem.80.3.7883856. [DOI] [PubMed] [Google Scholar]
- 103.Yazbeck R., Howarth G.S., Abbott C.A. Dipeptidyl peptidase inhibitors, an emerging drug class for inflammatory disease? Trends Pharmacol. Sci. 2009;30:600–607. doi: 10.1016/j.tips.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 104.Drucker D.J., Nauck M.A. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 105.Raz I., Hanefeld M., Xu L., Caria C., Williams-Herman D., Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia. 2006;49:2564–2571. doi: 10.1007/s00125-006-0416-z. [DOI] [PubMed] [Google Scholar]
- 106.Inzucchi S.E., McGuire D.K. New drugs for the treatment of diabetes: part II: Incretin-based therapy and beyond. Circulation. 2008;117:574–584. doi: 10.1161/CIRCULATIONAHA.107.735795. [DOI] [PubMed] [Google Scholar]
- 107.Lambeir A.M., Scharpe S., De Meester I. DPP4 inhibitors for diabetes—what next? Biochem. Pharmacol. 2008;76:1637–1643. doi: 10.1016/j.bcp.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 108.Jose T., Inzucchi S.E. Cardiovascular effects of the DPP-4 inhibitors. Diabetes Vasc. Dis. Res. 2012;9:109–116. doi: 10.1177/1479164111436236. [DOI] [PubMed] [Google Scholar]
- 109.Pattzi H.M., Pitale S., Alpizar M., Bennett C., O'Farrell A.M., Li J. Dutogliptin, a selective DPP4 inhibitor, improves glycaemic control in patients with type 2 diabetes: a 12-week, double-blind, randomized, placebo-controlled, multicentre trial. Diabetes Obes. Metab. 2010;12:348–355. doi: 10.1111/j.1463-1326.2010.01195.x. [DOI] [PubMed] [Google Scholar]
- 110.Hotamisligil G.S. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 111.Despres J.P., Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- 112.Horng T., Hotamisligil G.S. Linking the inflammasome to obesity-related disease. Nat. Med. 2011;17:164–165. doi: 10.1038/nm0211-164. [DOI] [PubMed] [Google Scholar]
- 113.Feuerer M., Herrero L., Cipolletta D., Naaz A., Wong J., Nayer A. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nishimura S., Manabe I., Nagasaki M., Eto K., Yamashita H., Ohsugi M. CD8 + effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 115.Sell H., Habich C., Eckel J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012;8:709–716. doi: 10.1038/nrendo.2012.114. [DOI] [PubMed] [Google Scholar]
- 116.Deaglio S., Dwyer K.M., Gao W., Friedman D., Usheva A., Erat A. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Resta R., Yamashita Y., Thompson L.F. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol. Rev. 1998;161:95–109. doi: 10.1111/j.1600-065x.1998.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 118.Hershfield M.S. New insights into adenosine-receptor-mediated immunosuppression and the role of adenosine in causing the immunodeficiency associated with adenosine deaminase deficiency. Eur. J. Immunol. 2005;35:25–30. doi: 10.1002/eji.200425738. [DOI] [PubMed] [Google Scholar]
- 119.Mortellaro A., Hernandez R.J., Guerrini M.M., Carlucci F., Tabucchi A., Ponzoni M. Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood. 2006;108:2979–2988. doi: 10.1182/blood-2006-05-023507. [DOI] [PubMed] [Google Scholar]
- 120.Sanchez J.J., Monaghan G., Borsting C., Norbury G., Morling N., Gaspar H.B. Carrier frequency of a nonsense mutation in the adenosine deaminase (ADA) gene implies a high incidence of ADA-deficient severe combined immunodeficiency (SCID) in Somalia and a single, common haplotype indicates common ancestry. Ann. Hum. Genet. 2007;71:336–347. doi: 10.1111/j.1469-1809.2006.00338.x. [DOI] [PubMed] [Google Scholar]
- 121.Dong R.P., Tachibana K., Hegen M., Munakata Y., Cho D., Schlossman S.F. Determination of adenosine deaminase binding domain on CD26 and its immunoregulatory effect on T cell activation. J. Immunol. 1997;159:6070–6076. [PubMed] [Google Scholar]
- 122.Franco R., Pacheco R., Gatell J.M., Gallart T., Lluis C. Enzymatic and extraenzymatic role of adenosine deaminase 1 in T-cell-dendritic cell contacts and in alterations of the immune function. Crit. Rev. Immunol. 2007;27:495–509. doi: 10.1615/critrevimmunol.v27.i6.10. [DOI] [PubMed] [Google Scholar]
- 123.Zavialov A.V., Gracia E., Glaichenhaus N., Franco R., Lauvau G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J. Leukoc. Biol. 2010;88:279–290. doi: 10.1189/jlb.1109764. [DOI] [PubMed] [Google Scholar]
- 124.Zavialov A.V., Engstrom A. Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochem. J. 2005;391:51–57. doi: 10.1042/BJ20050683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ohnuma K., Munakata Y., Ishii T., Iwata S., Kobayashi S., Hosono O. Soluble CD26/dipeptidyl peptidase IV induces T cell proliferation through CD86 up-regulation on APCs. J. Immunol. 2001;167:6745–6755. doi: 10.4049/jimmunol.167.12.6745. [DOI] [PubMed] [Google Scholar]
- 126.Ohnuma K., Yamochi T., Uchiyama M., Nishibashi K., Yoshikawa N., Shimizu N. CD26 up-regulates expression of CD86 on antigen-presenting cells by means of caveolin-1. Proc. Natl. Acad. Sci. U. S. A. 2004;101:14186–14191. doi: 10.1073/pnas.0405266101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ohnuma K., Takahashi N., Yamochi T., Hosono O., Dang N.H., Morimoto C. Role of CD26/dipeptidyl peptidase IV in human T cell activation and function. Front. Biosci. 2008;13:2299–2310. doi: 10.2741/2844. [DOI] [PubMed] [Google Scholar]
- 128.Ohnuma K., Yamochi T., Uchiyama M., Nishibashi K., Iwata S., Hosono O. CD26 mediates dissociation of Tollip and IRAK-1 from caveolin-1 and induces upregulation of CD86 on antigen-presenting cells. Mol. Cell. Biol. 2005;25:7743–7757. doi: 10.1128/MCB.25.17.7743-7757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ohnuma K., Inoue H., Uchiyama M., Yamochi T., Hosono O., Dang N.H. T-cell activation via CD26 and caveolin-1 in rheumatoid synovium. Mod. Rheumatol. 2006;16:3–13. doi: 10.1007/s10165-005-0452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Loster K., Zeilinger K., Schuppan D., Reutter W. The cysteine-rich region of dipeptidyl peptidase IV (CD 26) is the collagen-binding site. Biochem. Biophys. Res. Commun. 1995;217:341–348. doi: 10.1006/bbrc.1995.2782. [DOI] [PubMed] [Google Scholar]
- 131.Bullock B.P., Heller R.S., Habener J.F. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology. 1996;137:2968–2978. doi: 10.1210/endo.137.7.8770921. [DOI] [PubMed] [Google Scholar]
- 132.Mayo K.E., Miller L.J., Bataille D., Dalle S., Goke B., Thorens B. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol. Rev. 2003;55:167–194. doi: 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- 133.Thorens B. Expression cloning of the pancreatic beta cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc. Natl. Acad. Sci. U. S. A. 1992;89:8641–8645. doi: 10.1073/pnas.89.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kitamura T., Nakae J., Kitamura Y., Kido Y., Biggs W.H., III, Wright C.V. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J. Clin. Invest. 2002;110:1839–1847. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Buteau J., Spatz M.L., Accili D. Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic beta-cell mass. Diabetes. 2006;55:1190–1196. doi: 10.2337/db05-0825. [DOI] [PubMed] [Google Scholar]
- 136.Hui H., Nourparvar A., Zhao X., Perfetti R. Glucagon-like peptide-1 inhibits apoptosis of insulin-secreting cells via a cyclic 5′-adenosine monophosphate-dependent protein kinase A- and a phosphatidylinositol 3-kinase-dependent pathway. Endocrinology. 2003;144:1444–1455. doi: 10.1210/en.2002-220897. [DOI] [PubMed] [Google Scholar]
- 137.Nystrom T., Gutniak M.K., Zhang Q., Zhang F., Holst J.J., Ahren B. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am. J. Physiol. Endocrinol. Metab. 2004;287:E1209–E1215. doi: 10.1152/ajpendo.00237.2004. [DOI] [PubMed] [Google Scholar]
- 138.Ceriello A., Esposito K., Testa R., Bonfigli A.R., Marra M., Giugliano D. The possible protective role of glucagon-like peptide 1 on endothelium during the meal and evidence for an “endothelial resistance” to glucagon-like peptide 1 in diabetes. Diabetes Care. 2011;34:697–702. doi: 10.2337/dc10-1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Erdogdu O., Nathanson D., Sjoholm A., Nystrom T., Zhang Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol. Cell. Endocrinol. 2010;325:26–35. doi: 10.1016/j.mce.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 140.Huang C.Y., Shih C.M., Tsao N.W., Lin Y.W., Huang P.H., Wu S.C. Dipeptidyl peptidase-4 inhibitor improves neovascularization by increasing circulating endothelial progenitor cells. Br. J. Pharmacol. 2012;167:1506–1519. doi: 10.1111/j.1476-5381.2012.02102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xiao-Yun X., Zhao-Hui M., Ke C., Hong-Hui H., Yan-Hong X. Glucagon-like peptide-1 improves proliferation and differentiation of endothelial progenitor cells via upregulating VEGF generation. Med. Sci. Monit. 2011;17:BR35–BR41. doi: 10.12659/MSM.881383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ban K., Noyan-Ashraf M.H., Hoefer J., Bolz S.S., Drucker D.J., Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117:2340–2350. doi: 10.1161/CIRCULATIONAHA.107.739938. [DOI] [PubMed] [Google Scholar]
- 143.Hattori Y., Jojima T., Tomizawa A., Satoh H., Hattori S., Kasai K. A glucagon-like peptide-1 (GLP-1) analogue, liraglutide, upregulates nitric oxide production and exerts anti-inflammatory action in endothelial cells. Diabetologia. 2010;53:2256–2263. doi: 10.1007/s00125-010-1831-8. [DOI] [PubMed] [Google Scholar]
- 144.Shah Z., Pineda C., Kampfrath T., Maiseyeu A., Ying Z., Racoma I. Acute DPP-4 inhibition modulates vascular tone through GLP-1 independent pathways. Vasc. Pharmacol. 2011;55:2–9. doi: 10.1016/j.vph.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fadini G.P., Boscaro E., Albiero M., Menegazzo L., Frison V., de Kreutzenberg S. The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal-derived factor-1alpha. Diabetes Care. 2010;33:1607–1609. doi: 10.2337/dc10-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hocher B., Sharkovska Y., Mark M., Klein T., Pfab T. The novel DPP-4 inhibitors linagliptin and BI 14361 reduce infarct size after myocardial ischemia/reperfusion in rats. Int. J. Cardiol. 2013;167:87–93. doi: 10.1016/j.ijcard.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 147.Christopherson K.W., II, Hangoc G., Mantel C.R., Broxmeyer H.E. Modulation of hematopoietic stem cell homing and engraftment by CD26. Science. 2004;305:1000–1003. doi: 10.1126/science.1097071. [DOI] [PubMed] [Google Scholar]
- 148.Christopherson K.W., II, Paganessi L.A., Napier S., Porecha N.K. CD26 inhibition on CD34 + or lineage- human umbilical cord blood donor hematopoietic stem cells/hematopoietic progenitor cells improves long-term engraftment into NOD/SCID/Beta2null immunodeficient mice. Stem Cells Dev. 2007;16:355–360. doi: 10.1089/scd.2007.9996. [DOI] [PubMed] [Google Scholar]
- 149.Christopherson K.W., II, Hangoc G., Broxmeyer H.E. Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1 alpha-mediated chemotaxis of human cord blood CD34 + progenitor cells. J. Immunol. 2002;169:7000–7008. doi: 10.4049/jimmunol.169.12.7000. [DOI] [PubMed] [Google Scholar]
- 150.Peled A., Petit I., Kollet O., Magid M., Ponomaryov T., Byk T. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283:845–848. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- 151.Ghersi G., Chen W., Lee E.W., Zukowska Z. Critical role of dipeptidyl peptidase IV in neuropeptide Y-mediated endothelial cell migration in response to wounding. Peptides. 2001;22:453–458. doi: 10.1016/s0196-9781(01)00340-0. [DOI] [PubMed] [Google Scholar]
- 152.Olinde K.D., O'Connell J.B. Inflammatory heart disease: pathogenesis, clinical manifestations, and treatment of myocarditis. Annu. Rev. Med. 1994;45:481–490. doi: 10.1146/annurev.med.45.1.481. [DOI] [PubMed] [Google Scholar]
- 153.Schon E., Jahn S., Kiessig S.T., Demuth H.U., Neubert K., Barth A. The role of dipeptidyl peptidase IV in human T lymphocyte activation. Inhibitors and antibodies against dipeptidyl peptidase IV suppress lymphocyte proliferation and immunoglobulin synthesis in vitro. Eur. J. Immunol. 1987;17:1821–1826. doi: 10.1002/eji.1830171222. [DOI] [PubMed] [Google Scholar]
- 154.Flentke G.R., Munoz E., Huber B.T., Plaut A.G., Kettner C.A., Bachovchin W.W. Inhibition of dipeptidyl aminopeptidase IV (DP-IV) by Xaa-boroPro dipeptides and use of these inhibitors to examine the role of DP-IV in T-cell function. Proc. Natl. Acad. Sci. U. S. A. 1991;88:1556–1559. doi: 10.1073/pnas.88.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Reinhold D., Bank U., Buhling F., Lendeckel U., Faust J., Neubert K. Inhibitors of dipeptidyl peptidase IV induce secretion of transforming growth factor-beta 1 in PWM-stimulated PBMC and T cells. Immunology. 1997;91:354–360. doi: 10.1046/j.1365-2567.1997.d01-2258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Reinhold D., Bank U., Buhling F., Neubert K., Mattern T., Ulmer A.J. Dipeptidyl peptidase IV (CD26) on human lymphocytes. Synthetic inhibitors of and antibodies against dipeptidyl peptidase IV suppress the proliferation of pokeweed mitogen-stimulated peripheral blood mononuclear cells, and IL-2 and IL-6 production. Immunobiology. 1993;188:403–414. doi: 10.1016/S0171-2985(11)80223-8. [DOI] [PubMed] [Google Scholar]
- 157.Reinhold D., Bank U., Buhling F., Tager M., Born I., Faust J. Inhibitors of dipeptidyl peptidase IV (DP IV, CD26) induces secretion of transforming growth factor-beta 1 (TGF-beta 1) in stimulated mouse splenocytes and thymocytes. Immunol. Lett. 1997;58:29–35. doi: 10.1016/s0165-2478(97)02716-8. [DOI] [PubMed] [Google Scholar]
- 158.Kahne T., Lendeckel U., Wrenger S., Neubert K., Ansorge S., Reinhold D. Dipeptidyl peptidase IV: a cell surface peptidase involved in regulating T cell growth (review) Int. J. Mol. Med. 1999;4:3–15. doi: 10.3892/ijmm.4.1.3. [DOI] [PubMed] [Google Scholar]
- 159.Tanaka T., Kameoka J., Yaron A., Schlossman S.F., Morimoto C. The costimulatory activity of the CD26 antigen requires dipeptidyl peptidase IV enzymatic activity. Proc. Natl. Acad. Sci. U. S. A. 1993;90:4586–4590. doi: 10.1073/pnas.90.10.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tanaka T., Duke-Cohan J.S., Kameoka J., Yaron A., Lee I., Schlossman S.F. Enhancement of antigen-induced T-cell proliferation by soluble CD26/dipeptidyl peptidase IV. Proc. Natl. Acad. Sci. U. S. A. 1994;91:3082–3086. doi: 10.1073/pnas.91.8.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fleischer B., Sturm E., De Vries J.E., Spits H. Triggering of cytotoxic T lymphocytes and NK cells via the Tp103 pathway is dependent on the expression of the T cell receptor/CD3 complex. J. Immunol. 1988;141:1103–1107. [PubMed] [Google Scholar]
- 162.Mittrucker H.W., Steeg C., Malissen B., Fleischer B. The cytoplasmic tail of the T cell receptor zeta chain is required for signaling via CD26. Eur. J. Immunol. 1995;25:295–297. doi: 10.1002/eji.1830250149. [DOI] [PubMed] [Google Scholar]
- 163.Dang N.H., Morimoto C. CD26: an expanding role in immune regulation and cancer. Histol. Histopathol. 2002;17:1213–1226. doi: 10.14670/HH-17.1213. [DOI] [PubMed] [Google Scholar]