

Abstract
Human metapneumovirus (hMPV), a newly discovered virus of the family Paramyxoviridae, has been associated with upper and lower respiratory tract infections in different age groups in many countries. The putative attachment (G) glycoprotein of this virus was previously reported to have shown more extensive nucleotide and deduced amino acid sequence polymorphism than any other genomic regions of this virus, leading to four sub-lineages. Using a maximum likelihood-based codon substitution model of sequence evolution, here we report that sequences of extracellular domain of 8 amino acid sites in lineage 1a, and 3 amino acid sites each in lineage 1b, 2a, and 2b have a higher rate of nonsynonymous substitutions (dN) than the synonymous substitutions (dS) with a posterior probability above 0.95, thus suggesting the evidence of adaptive evolution driven by Darwinian selection. Although it is unclear whether these amino acid adaptations are driven by differential immune pressure or some other factors, identification of these positively selected amino acid sites would help in better screening using epitope mapping technology to identify and localize the sites that can be recognized by the immune system. We also observed surprisingly higher nucleotide substitution rates per site, per year for each lineage of hMPV than the rates that were previously reported for the human respiratory syncytial virus, suggesting rapid evolutionary dynamics of hMPV.
Keywords: Human metapneumovirus, Attachment glycoprotein, Phylogeny, Adaptive evolution, Substitution rate, Evolutionary dynamics
Human metapneumovirus (hMPV) of the family Paramyxoviridae and subfamily Pneumoviridae was first discovered in The Netherlands from infants and children suffering from acute respiratory tract disease (van den Hoogen et al., 2001). Since then considerable progress has been made in identification and characterization (Cote et al., 2003, Mackay et al., 2003, Ebihara et al., 2004, Maertzdorf et al., 2004, Skiadopoulos et al., 2004, Hamelin and Boivin, 2005, Leung et al., 2005, Gray et al., 2006a, Gray et al., 2006b, Ulbrand et al., 2006, van den Hoogen, 2007) as well as in understanding its genetic diversity (Bastien et al., 2003, Bastien et al., 2004, Biacchesi et al., 2003, Ishiguro et al., 2004, Peret et al., 2004, Carr et al., 2005, Ludewick et al., 2005, Galiano et al., 2006, Boivin et al., 2007). To date this virus has been identified in many countries from different age groups and reported to cause upper respiratory tract infections, flu-like infections, and has also been associated with lower respiratory tract infections (van den Hoogen et al., 2001, Stockton et al., 2002, Biacchesi et al., 2003, Bastien et al., 2003, Bastien et al., 2004, Ishiguro et al., 2004, Peret et al., 2004, Carr et al., 2005, Ludewick et al., 2005, Fouchier et al., 2005, Regev et al., 2006, Galiano et al., 2006, Kahn, 2006, Gray et al., 2006a, Gray et al., 2006b), a pattern similar to that reported for human respiratory syncytial virus (HRSV). Although comparative genome mapping analyses suggested that this virus has structural and functional similarities with HRSV (Kahn, 2006), recent studies reported that the attachment (G) glycoprotein of these paramimyxoviruses exhibit extensive nucleotide and amino acid variation, with most differences located in the extracellular domain (Peret et al., 2004, Kahn, 2006). Therefore, G-protein has been widely used to infer evolutionary relationships among the isolates from different geographic regions (e.g., Peret et al., 2004, Ishiguro et al., 2004). Although phylogenetic analyses of hMPVs from the complete nucleotide coding sequences revealed the existence of two major lineages of hMPVs (Ishiguro et al., 2004), recent analyses based on G-protein phylogeny revealed the existence of two minor sub-groups within each major lineage (Peret et al., 2004). Despite the knowledge of identification and characterization of hMPVs, the possible mechanism by which hMPV G-proteins have evolved is poorly understood.
Earlier studies on the molecular evolution of HRSV G-protein reported that certain amino acid sites that correspond to sites of O-glycosylation, or amino acid sites that were previously described as monoclonal antibody-induced in vitro escape mutants, are under positive selection and thus showed strong association between these positively selected sites and the mapped neutralizing epitopes (Zlateva et al., 2004). Recently, Zhang et al. (2006) also reported that certain amino acid sites in severe acute respiratory syndrome (SARS) coronavirus (CoV) are evolved by positive Darwinian selection. These lines of evidence suggest an interesting evolutionary pattern of the respiratory viruses. At the genomic level, whether a gene, or a particular amino acid within a gene, is under relaxed selection or remains functionally constrained throughout evolution can be detected by comparing the rate of nonsynonymous nucleotide substitutions per nonsynonymous site (dN) with that of synonymous substitutions per synonymous site (dS) (Hughes and Nei, 1989). If dN/dS (hereafter referred as ω) is greater than one, then positive selection is said to be operating. Alternatively, if ω < 1, the gene is under strong purifying selection and presumed to be functionally constrained.
Identifying genes that have evolved by adaptation is central to understanding molecular evolution. However, not all amino acid differences observed among the closely related sequences from ecologically/geographically isolated strains are adaptive (e.g., Zlateva et al., 2004). Therefore, analyzing patterns of amino acid substitutions would provide insight into understanding protein adaptation by identifying candidate codon sites on which positive selection has been operating. Identifying the positively selected amino acid sites would also help in further immunization studies. Maximum likelihood (ML)-based codon substitution models, which account for variable ω ratios among codon sites and detect codon sites that are subjected to positive selection (Yang et al., 2000), have been widely used in detecting positive selection in a number of respiratory viral groups (e.g., Zlateva et al., 2004, Zhang et al., 2006). Here we used Yang et al's (2000) ML codon substitution models to test whether there was evidence at the nucleotide sequence level that a subset of amino acid sites in G-protein of hMPV sequences that represent each subgroup has been under positive selection. In addition, we used a Bayesian MCMC approach implemented in BEAST version 1.4.4 (Drummond and Rambaut, 2006) that utilize the number and temporal distribution of genetic differences among viruses sampled at different times (Drummond et al., 2002, Drummond et al., 2006) to estimate the evolutionary change for each lineage.
A total of 144 published unique nucleotide coding sequences of G-protein representing four sub-lineages (1a = 46, 1b = 40, 2a = 38, 2b = 20) were retrieved from GenBank (Table 1 ). Sequences were aligned using Mesquite version 1.2 (Maddison and Maddison, 2006), DAMBE version 4.5.2 (Xia, 2000, Xia and Xie, 2001), and BioEdit version 7.0.5.3 (Hall, 1999) software packages. To infer phylogenetic relationship among these strains of hMPVs, we reconstructed a neighbor joining tree from their predicted amino acid sequence data with p-distance implemented in MEGA version 3.1 (Kumar et al., 2004). Using the same program, nodal supports were estimated with 10,000 nonparametric bootstrap replicates. For selection analyses, we reconstructed unrooted ML trees for each lineage from their respective nucleotide sequence data using the appropriate nucleotide substitution model identified by the hierarchical likelihood ratio test implemented in Modeltest version 3.5 (Posada and Crandall, 1998). PHYML version 2.4.4 (Guindon and Gascuel, 2003) was used to conduct ML analyses.
Table 1.
GenBank accession number, strain name, country of origin, and the year of isolation of 144 unique hMPV G-protein sequences used in the study
| GenBank No. | Strain name | Country of origin | Year of Isolation | Source | Group |
|---|---|---|---|---|---|
| AF371337 | 00-1 | The Netherlands | van den Hoogen et al. (2001) | 1a | |
| AY296015 | FL/4/01 | The Netherlands | 2001 | van den Hoogen et al. (2001) | 1a |
| AY296016 | FL/3/01 | The Netherlands | 2001 | van den Hoogen et al. (2001) | 1a |
| AY296017 | FL/8/01 | The Netherlands | 2001 | van den Hoogen et al. (2001) | 1a |
| AY296018 | FL/10/01 | The Netherlands | 2001 | van den Hoogen et al. (2001) | 1a |
| AY296019 | NL/10/01 | The Netherlands | 2001 | van den Hoogen et al. (2001) | 1a |
| AY296020 | NL/2/02 | The Netherlands | 2002 | van den Hoogen et al. (2001) | 1a |
| AY327802 | 201-7182 | Australia | GenBank | 1a | |
| AY327803 | 201-4199 | Australia | – | GenBank | 1a |
| AY327804 | Q01-6410 | Australia | – | GenBank | 1a |
| AY327805 | Q01-7262 | Australia | – | GenBank | 1a |
| AY327806 | Q01-6346 | Australia | – | GenBank | 1a |
| AY327807 | Q01-7292 | Australia | – | GenBank | 1a |
| AY327808 | Q01-7252A | Australia | – | GenBank | 1a |
| AY327809 | Q01-7292 | Australia | – | GenBank | 1a |
| AY327810 | Q016297 | Australia | – | GenBank | 1a |
| AY485232 | hMPV13-2000 | Canada | 2000 | Peret et al. (2004) | 1a |
| AY485235 | hMP V193-2002 | Canada | 2002 | Peret et al. (2004) | 1a |
| AY485236 | hMPV22-2001 | Canada | 2001 | Peret et al. (2004) | 1a |
| AY485238 | hMPV23-2001 | Canada | 2001 | Peret et al. (2004) | 1a |
| AY485251 | hMPV81-1999 | Canada | 1999 | Peret et al. (2004) | 1a |
| AY485254 | hMPV86316-2002 | Canada | 2002 | Peret et al. (2004) | 1a |
| AY485255 | hMPV88448-2002 | Canada | 2002 | Peret et al. (2004) | 1a |
| AY485256 | hMPV88470-2002 | Canada | 2002 | Peret et al. (2004) | 1a |
| AY530092 | JPS03-180 | Japan | 2003 | Ishiguro et al. (2004) | 1a |
| AY574225 | CAN34-02 | Canada | 2002 | Ishiguro et al. (2004) | 1a |
| AY574226 | CAN40-02 | Canada | 2002 | Ishiguro et al. (2004) | 1a |
| AY574228 | CAN97-02 | Canada | 2002 | Ishiguro et al. (2004) | 1a |
| AY574231 | CAN187-02 | Canada | 2002 | Ishiguro et al. (2004) | 1a |
| AY574237 | CAN216-02 | Canada | 2002 | Ishiguro et al. (2004) | 1a |
| AY574243 | CAN464-02 | Canada | 2002 | Ishiguro et al. (2004) | 1a |
| AY574244 | CAN532-02 | Canada | 2002 | Ishiguro et al. (2004) | 1a |
| AY848881 | RSA/39/01 | South Africa | 2001 | Ludewick et al. (2005) | 1a |
| AY848882 | RSA/1/02 | South Africa | 2002 | Ludewick et al. (2005) | 1a |
| AY848885 | RSA/4/02 | South Africa | 2002 | Ludewick et al. (2005) | 1a |
| AY848887 | RSA/17/02 | South Africa | 2002 | Ludewick et al. (2005) | 1a |
| AY848889 | RSA/31/01 | South Africa | 2001 | Ludewick et al. (2005) | 1a |
| AY848890 | RSA/33/01 | South Africa | 2001 | Ludewick et al. (2005) | 1a |
| AY848893 | RSA/8/02 | South Africa | 2002 | Ludewick et al. (2005) | 1a |
| AY848896 | RSA/3/02 | South Africa | 2002 | Ludewick et al. (2005) | 1a |
| AY848897 | RSA/10/02 | South Africa | 2002 | Ludewick et al. (2005) | 1a |
| AY848901 | RSA/14/02 | South Africa | 2002 | Ludewick et al. (2005) | 1a |
| AY848903 | RSA/34/01 | South Africa | 2001 | Ludewick et al. (2005) | 1a |
| DQ312444 | IA3-2002 | USA | 2002 | Gray et al., 2006a, Gray et al., 2006b | 1a |
| DQ362949 | Arg/1/03 | Argentina | 2003 | Galiano et al. (2006) | 1a |
| DQ362950 | Arg/2/02 | Argentina | 2002 | Galiano et al. (2006) | 1a |
| AY296021 | NL/17/00 | The Netherlands | 2000 | van den Hoogen et al. (2004) | 1b |
| AY296022 | NL/1/81 | The Netherlands | 1981 | van den Hoogen et al. (2004) | 1b |
| AY296023 | NL/1/93 | The Netherlands | 1993 | van den Hoogen et al. (2004) | 1b |
| AY296025 | NL/3/93 | The Netherlands | 1993 | van den Hoogen et al. (2004) | 1b |
| AY296026 | NL/1/95 | The Netherlands | 1995 | van den Hoogen et al. (2004) | 1b |
| AY296028 | NL/13/96 | The Netherlands | 1996 | van den Hoogen et al. (2004) | 1b |
| AY296029 | NL/22/01 | The Netherlands | 2001 | van den Hoogen et al. (2004) | 1b |
| AY296030 | NL/24/01 | The Netherlands | 2001 | van den Hoogen et al. (2004) | 1b |
| AY296032 | NL/29/01 | The Netherlands | 2001 | van den Hoogen et al. (2004) | 1b |
| AY296033 | NL/302 | The Netherlands | 2002 | van den Hoogen et al. (2004) | 1b |
| AY485234 | hMPV17-2000 | Canada | 2000 | Peret et al. (2004) | 1b |
| AY485250 | hMPV80-1999 | Canada | 1999 | Peret et al. (2004) | 1b |
| AY530090 | JPS03-176 | Japan | 2003 | Ishiguro et al. (2004) | 1b |
| AY530091 | JPS03-178 | Japan | 2003 | Ishiguro et al. (2004) | 1b |
| AY530093 | JPS03-187 | Japan | 2003 | Ishiguro et al. (2004) | 1b |
| AY530095 | JPS03-240 | Japan | 2003 | Ishiguro et al. (2004) | 1b |
| AY574227 | CAN58-02 | Canada | 2002 | Bastien et al. (2004) | 1b |
| AY574229 | CAN164-02 | Canada | 2002 | Bastien et al. (2004) | 1b |
| AY574230 | CAN182-02 | Canada | 2002 | Bastien et al. (2004) | 1b |
| AY574234 | CAN197-02 | Canada | 2002 | Bastien et al. (2004) | 1b |
| AY574235 | CAN208-02 | Canada | 2002 | Bastien et al. (2004) | 1b |
| AY574236 | CAN215-02 | Canada | 2002 | Bastien et al. (2004) | 1b |
| AY574241 | CAN348-02 | Canada | 2002 | Bastien et al. (2004) | 1b |
| AY848910 | RSA/27/00 | South Africa | 2000 | Ludewick et al. (2005) | 1b |
| AY848911 | RSA/7/00 | South Africa | 2000 | Ludewick et al. (2005) | 1b |
| AY848912 | RSA/26/00 | South Africa | 2000 | Ludewick et al. (2005) | 1b |
| AY848914 | RSA/7/01 | South Africa | 2000 | Ludewick et al. (2005) | 1b |
| AY848915 | RSA/20/00 | South Africa | 2000 | Ludewick et al. (2005) | 1b |
| AY848916 | RS A/20/01 | South Africa | 2001 | Ludewick et al. (2005) | 1b |
| AY848917 | RSA/49/00 | South Africa | 2000 | Ludewick et al. (2005) | 1b |
| AY848919 | RSA/44/00 | South Africa | 2000 | Ludewick et al. (2005) | 1b |
| DQ270215 | BJ1819 | China | 2000 | GenBank | 1b |
| DQ312449 | IA-8-2003 | USA | 2003 | Gray et al. (2006a) | 1b |
| DQ270217 | BJ1824 | China | – | GenBank | 1b |
| DQ312458 | IA-17-2003 | USA | 2003 | Gray et al. (2006a) | 1b |
| DQ312462 | IA21-2004 | USA | 2004 | Gray et al. (2006a) | 1b |
| DQ312463 | IA22-2004 | USA | 2004 | Gray et al. (2006a) | 1b |
| DQ312464 | IA23-2004 | USA | 2004 | Gray et al. (2006a) | 1b |
| DQ362952 | Arg/3/00 | Argentina | 2000 | Galiano et al. (2006) | 1b |
| NC_004148 | CAN97-83 | Canada | 1997 | Biacchesi et al. (2003) | 1b |
| AY296040 | NL/1/94 | The Netherlands | 1994 | van den Hoogen et al. (2004) | 2a |
| AY296041 | NL/1/82 | The Netherlands | 1982 | van den Hoogen et al. (2004) | 2a |
| AY296042 | NL/1/96 | The Netherlands | 1996 | van den Hoogen et al. (2004) | 2a |
| AY296044 | NL/9/00 | The Netherlands | 2000 | van den Hoogen et al. (2004) | 2a |
| AY296045 | NL/3/01 | The Netherlands | 2001 | van den Hoogen et al. (2004) | 2a |
| AY296046 | NL/4/01 | The Netherlands | 2001 | van den Hoogen et al. (2004) | 2a |
| AY296047 | UK/5/01 | UK | 2001 | van den Hoogen et al. (2004) | 2a |
| AY297748 | CAN98-75 | Canada | 1998 | Biacchesi et al. (2003) | 2a |
| AY485243 | hMPV73-1998 | Canada | 1998 | Peret et al. (2004) | 2a |
| AY485244 | hMPV74-1998 | Canada | 1998 | Peret et al. (2004) | 2a |
| AY485245 | hMPV75-1998 | Canada | 1998 | Peret et al. (2004) | 2a |
| AY485246 | hMPV76-1998 | Canada | 1998 | Peret et al. (2004) | 2a |
| AY485247 | hMPV77-1998 | Canada | 1998 | Peret et al. (2004) | 2a |
| AY485248 | hMPV78-1998 | Canada | 1998 | Peret et al. (2004) | 2a |
| AY485249 | hMPV79-1998 | Canada | 1998 | Peret et al. (2004) | 2a |
| DQ270219 | BJ1921 | China | – | GenBank | 2a |
| DQ270220 | BJ2034 | China | – | GenBank | 2a |
| DQ270221 | BJ4879 | China | – | GenBank | 2a |
| DQ270222 | BJ4944 | China | – | GenBank | 2a |
| DQ270223 | BJ5128 | China | – | GenBank | 2a |
| DQ270224 | BJ5129 | China | – | GenBank | 2a |
| DQ312443 | IA2-2002 | USA | 2002 | Gray et al. (2006a) | 2a |
| DQ312457 | IA16-2003 | USA | 2003 | Gray et al. (2006a) | 2a |
| DQ312460 | IA19-2003 | USA | 2003 | Gray et al. (2006a) | 2a |
| DQ393715 | Peru1-2002 | USA | 2002 | Gray et al. (2006b) | 2a |
| DQ843658 | BJ1816 | China | – | GenBank | 2a |
| AY848861 | RSA/4/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848862 | RSA/71/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848864 | RSA/37/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848865 | RSA/16/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848866 | RSA/12/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848868 | RSA/29/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848869 | RSA/58/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848875 | RSA/54/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848878 | RSA/23/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848879 | RSA/90/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| AY848880 | RSA/93/00 | South Africa | 2000 | Ludewick et al. (2005) | 2a |
| DQ312453 | IA12-2003 | USA | 2003 | Gray et al. (2006a) | 2a |
| AY296034 | NL/1/99 | The Netherlands | 1999 | van den Hoogen et al. (2004) | 2b |
| AY296035 | NL/11/00 | The Netherlands | 2000 | van den Hoogen et al. (2004) | 2b |
| AY296036 | NL/12/00 | The Netherlands | 2000 | van den Hoogen et al. (2004) | 2b |
| AY296037 | NL/5/01 | The Netherlands | 2001 | van den Hoogen et al. (2004) | 2b |
| AY296038 | NL/9/01 | The Netherlands | 2001 | van den Hoogen et al. (2004) | 2b |
| AY296039 | NL/21/01 | The Netherlands | 2001 | van den Hoogen et al. (2004) | 2b |
| AY485242 | hMPV33-2001 | Canada | 2001 | Peret et al. (2004) | 2b |
| AY485252 | hMPV82-1997 | Canada | 1997 | Peret et al. (2004) | 2b |
| AY530089 | JPS02-76 | Japan | 2002 | Ishiguro et al. (2004) | 2b |
| DQ312445 | IA4-2002 | USA | 2002 | Gray et al. (2006a) | 2b |
| DQ312446 | IA5-2002 | USA | 2002 | Gray et al. (2006a) | 2b |
| DQ312448 | IA7-2003 | USA | 2003 | Gray et al. (2006a) | 2b |
| DQ312454 | IA13-2003 | USA | 2003 | Gray et al. (2006a) | 2b |
| DQ312455 | IA14-2003 | USA | 2003 | Gray et al. (2006a) | 2b |
| DQ312461 | IA20-2003 | USA | 2003 | Gray et al. (2006a) | 2b |
| DQ393716 | Peru2-2002 | Peru | 2002 | Gray et al. (2006b) | 2b |
| DQ393717 | Peru3-2003 | Peru | 2003 | Gray et al. (2006b) | 2b |
| DQ393718 | Peru4-2003 | Peru | 2003 | Gray et al. (2006b) | 2b |
| DQ393719 | Peru5-2003 | Peru | 2003 | Gray et al. (2006b) | 2b |
| AY530094 | JPS03-194 | Japan | 2003 | Ishiguro et al. (2004) | 2b |
Overall substitution rate (nucleotide substitutions per site per year) of each lineage was estimated using the Bayesian skyline model, with both relaxed (variable) molecular clock (with uncorrelated lognormal model) and strict clock implemented in the BEAST version 1.4.4 (Drummond and Rambaut, 2006). This model employs a Bayesian MCMC approach and utilize the number and temporal distribution of genetic differences among viruses sampled at different times (Drummond et al., 2002, Drummond et al., 2006). Bayesian skyline plots with 10 grouped intervals were reconstructed to infer demographic history (Drummond et al., 2005). Phylogenies were evaluated using a chain length of 30 million states under the HKY85 + Γ4 substitution model and with uncertainty in the data reflected in the 95% high-probability density (HPD) intervals. Convergence of trees was checked using Tracer version 1.3 (Rambaut and Drummond, 2006).
To determine the synonymous and nonsynonymous sequence divergence distribution pattern across the entire coding region of each lineage (Fig. 1 ), we used a sliding window approach (window size = 6, step = 1) implemented in DNAsp version 4.0 (Rozas et al., 2003).
Fig. 1.
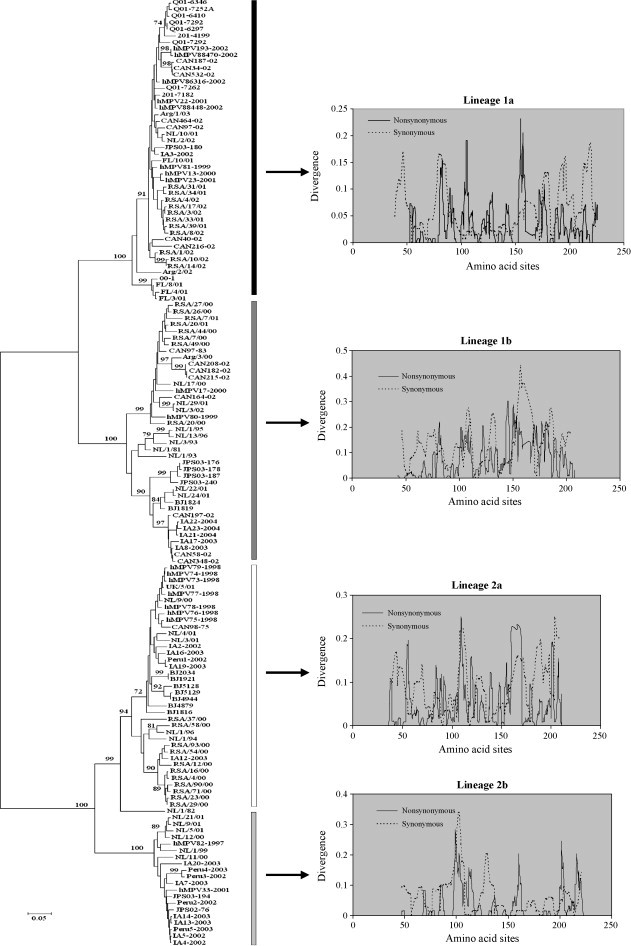
NJ tree inferred from 144 amino acid sequences of human metapneumovirus G glycoprotein representing four lineages. Nodal support is mentioned at the base of the node. The sliding window analyses of respective lineages show the synonymous and nonsynonymous divergence.
To assess whether positive selection is operating in any codon sites, we used the alignment and ML trees of respective lineages as input for the CODEML program of PAML version 3.15 (Yang, 1997). The PAML program incorporates six different codon substitution models that account for variable ω for each codon site. The six codon substitution models are: M0 (one-ratio), M1a (nearly neutral), M2a (positive selection), M7 (β distribution; 0 ≤ ω ≤ 1), M8 (β + ω > 1: continuous) (Yang et al., 2000), and M8a (β + ω = 1) (Swanson et al., 2003). The M0 model estimates overall ω for the data. The M1a model estimates a single parameter, p 0, with ω 0 = 0, and the remaining sites with frequency p 1 (p 1 = 1 − p 0) assuming ω 1 = 1. The M2a model adds a class of positively selected sites with frequency p 2 (where p 2 = 1 − p 1 − p 0) with ω 2 estimated from the data. In the M7 model, ω follows a beta distribution and is allowed to vary between 0 and 1, and two parameters (p and q) of the beta distribution are estimated from the data. In the M8 model, a proportion, p 0 , of sites have ω drawn from the beta distribution and the remaining sites with proportion p 1 are positively selected (ω 1 > 1). The LRTs between nested models were conducted by comparing twice the difference in log-likelihood values (2lnΔl) against a χ 2-distribution with degrees of freedom equal to the difference in the number of parameters between models (Yang, 1997). Three LRTs were conducted. The first comparison was made between M1a, which allows for two site classes (0 < ω < 1, ω = 1), and M2a, which allows three site classes (0 < ω < 1, ω = 1 or ω > 1). The second comparison was between M7 and M8, and the last comparison was between M8 and M8a, in which ω for M8a was constrained to 1. In all LRTs good evidence for positive selection is found if the LRT indicates that models that allow for selection (i.e. M2a and M8; alternative models) are significantly better than their respective null models (M1a, M7 and M8a) (Yang, 1997). Posterior probabilities of the inferred positively selected sites were estimated by the Bayes empirical Bayes (BEB) approach that takes sampling errors into account (Yang et al., 2005).
Consistent with earlier studies (Peret et al., 2004), G-protein based phylogeny in the present study has also revealed the existence of multiple lineages of this virus (Fig. 1). All four lineages showed some degree of spatial structure; however, few strains in each lineage did not show any spatial structure, indicating extensive viral gene flow across the regions in a given epidemic season. Relatively weak temporal structure across the regions further suggested that either certain strains can remain stable for more than one epidemic season (e.g., HRSV, Zlateva et al., 2004, Zlateva et al., 2005), or mutations might not have occurred in a linear fashion with the preservation of changes in the circulating viral strains. Thus, virus genotypes would frequently appear and disappear along with new mutations in the populations. However, HRSV (Zlateva et al., 2004, Zlateva et al., 2005) showed a strong correlation between the accumulation of genetic divergence and the isolation date of the sequences. Based on the relaxed clock assumption, the evolutionary rate of each major lineage of hMPVs (1 and 2; Table 2 ) are 5.18 × 10−3 and 6.49 × 10−3 substitutions/site/year, respectively. Although these rates are compatible with the substitution rates reported for influenza viruses (Chen and Holmes, 2006), these rates are higher than the estimates of HRSV (HRSV A: 1.83 × 10−3, Zlateva et al., 2004; HRSV B: 1.95 × 10−3, Zlateva et al., 2005; HRSV-BA: 3 × 10−3 substitutions/site/year, Trento et al., 2006; HRSV-A: 2.6 × 10−3, HRSV-B: 3.5 × 10−3, Matheson et al., 2006) and other paramyxoviruses (e.g., measles: Woelk et al., 2002). These discrepancies in the evolutionary rates could be associated with the differential selective pressures targeting different genomic regions. For example, the presence of a greater number of adaptively evolved amino acid sites in the gene can cause an accelerated rate of evolution. As a consequence, the overall evolutionary rate is expected to be higher (Trento et al., 2006). Both major lineages of hMPVs showed interesting population dynamics (Fig. 2 ). The times to the most recent common ancestor for lineage 1 and 2 are 49.452 (29.08–70.8) and 26.091 (21–36.651) years, respectively. While the population size of lineage 1 recently declined, the lineage 2 population size did not show any declining trend. This contrast in the population size could be associated with fitness of the virus.
Table 2.
Mean nucleotide substitution rates (95% HPD interval in parenthesis) in hMPV G-gene estimated using Bayesian MCMC approach, with both relaxed and strict clock
| Lineage | Relaxed clock |
Strict clock |
||
|---|---|---|---|---|
| Substitution rate (×10−3 substitutions/site/year) | Likelihood score | Substitution rate (×10−3 substitutions/site/year) | Likelihood score | |
| 1a | 4.58 (2.400–7.048) | −2250.481 | 4.152 (2.235–6.196) | −2256.156 |
| 1b | 5.344 (3.995–6.898) | −2946.824 | 4.817 (3.809–5.889) | −2975.021 |
| 2a | 6.139 (4.318–7.825) | −2530.280 | 5.275 (3.733–6.798) | −2556.508 |
| 2b | 7.865 (4.060–11.63) | −1840.066 | 3.795 (2.687–7.625) | −1868.507 |
| 1(a + b) | 5.182 (3.761–6.781) | −4689.161 | 4.621 (3.639–5.647) | −4702.717 |
| 2(a + b) | 6.494 (4.599–8.438) | −3783.320 | 4.770 (3.555–6.012) | −3833.563 |
Estimates with relaxed clock are better fit to the data.
Fig. 2.
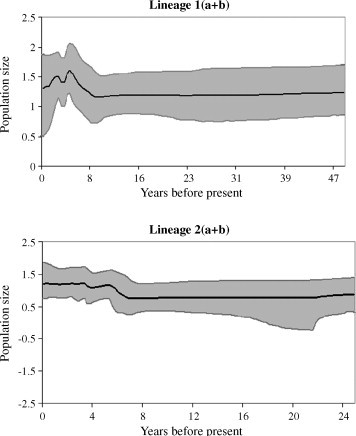
Skyline plots estimated from Bayesian MCMC analyses of hMPV G-protein sequences belong to lineage 1(a + b) and lineage 2(a + b). Population size (in Y-axis) is expressed in logarithmic scale. The solid line shows the median estimate of population size (Ne × g) throughout the given time period. The grey area gives the 95% HPD interval of these estimates.
Despite the weak temporal and spatial structure, viral strains belonging to lineage 1a (Australia, Canada, The Netherlands, South Africa, USA, Argentina, and Japan) and 1b (Canada, The Netherlands, South Africa, USA, Japan, China, and Argentina) have a wider geographic spread than the strains belonging to lineage 2a (Canada, UK, The Netherlands, USA, China, and South Africa) and 2b (The Netherlands, Canada, USA, Peru, and Japan), indicating that fitness of the viral strains might have played a crucial role in the uneven distribution across the wide geographic regions. The extensive polymorphisms of the hMPV G-gene may have resulted from mutations occurring during virus propagation in cell culture; however, Peret et al. (2004) reported identical sequences of the same viral strain after multiple passages, and thus, the observed variation in the G-gene of hMPVs due to multiple passages is more unlikely. However, it is unclear whether the hMPV G-gene experienced differential selection pressures, or all the deduced amino acid sites evolved due to stochastic mutational processes? Sliding window analyses of each lineage revealed that in the majority of regions synonymous divergence exceeds the corresponding nonsynonymous divergence, thus suggesting that the G-gene of hMPV is influenced by purifying selection (Fig. 1). However, a few coding regions in all the lineages showed relatively higher nonsynonymous divergence than synonymous divergence, therefore indicating the pervasive role of positive selection in certain amino acid sites. To identify the codon sites that are positively selected, we performed ML-based codon substitution analyses. Consistent with sliding window results, the M0 model revealed that the average ω for each lineage is less than one (Table 3 ), thus suggesting each lineage experienced purifying selection. However, comparison of the models that assume positive selection (M2a, M8) with the models (M1a, M7, and M8a) that assume no positive selection, detected approximately 6%, 1.3%, 7.3%, and 3% positively selected codons in lineage 1a, 1b, 2a, and 2b, respectively (Table 3). There are eight positively selected sites (site 93, 105, 106, 154, 158, 171, 173, and 188) with posterior probability ≥0.95 within lineage 1a, whereas lineage 1b (site 146, 183, and 196) and lineage 2a (85, 232, and 239) each have three positively selected sites with posterior probability ≥0.95. Lineage 2b has only two positively selected sites (site 100 and 105) with posterior probability ≥0.95. Except site 105, which is positively selected in lineage 1a and 2b, none of the positively selected sites are overlapping among the lineages. It is unclear whether these positively selected sites are associated with the fitness of this virus. Research with monoclonal antibodies has shown that the hMPV F-protein carries neutralizing epitopes (Skiadopoulos et al., 2004, Ulbrand et al., 2006); therefore, antigenic variation due to immune selection in the hMPV F-protein is more likely. Although, the overall excess of synonymous substitutions at the hMPV G-protein indicates that host immune selection might not be the dominant selective force, the findings of several hotspots of nonsynonymous substitutions in this protein suggests that host immune selection might also play a role in maintaining diversity. Recent study has shown that a majority of the neutralizing epitopes in the HRSV G-gene is strongly associated with positively selected sites, and some of the positively selected sites correspond to the sites of O-glycosylation (Zlateva et al., 2004). Like HRSV, although all the positively selected codons of hMPV G-gene are located in the extracellular domain and some of them correspond to sites of O-glycosylation, the putative role is still unclear for these positively selected sites, as is whether some of these positively selected sites are associated with the region of antigenic determinants. We intended to map these positively selected sites with the HRSV G-protein to see whether the same sites were also positively selected in HRSV (Zlateva et al., 2004, Zlateva et al., 2005); however, the predicted G-gene amino acid sequences of the two viruses could not be aligned (van den Hoogen et al., 2002, Kahn, 2006). Although a vast majority of codon sites (>95% in most cases) are shown to have been under purifying selection, significantly higher ω values (>1) at certain codon sites (Table 3) indicate the hMPV G-gene is under positive selection. Identification of these positively selected amino acid sites would help in better screening using epitope mapping technology to identify and localize the sites that can be recognized by the immune system. Knowledge of sites that have adaptively evolved can greatly cut the cost of these screening processes and thereby help in developing better immunization techniques (Mes and van Putten, 2007).
Table 3.
Test for variable selection pressures on different codons based on ML-based codon substitution models of Yang et al. (2000)
| Model | Free parameters | Parameter estimates | Likelihood scores | Model comparison (2Δl, d.f., p) | Positively selected sites | ω ± S.E. |
|---|---|---|---|---|---|---|
| Lineage 1a | ||||||
| M0: One-ratio | 1 | ω = 0.6152 | −2510.069374 | None | ||
| M1a: Nearly neutral | 1 | ω0 = 0.1, ω1 = 1, (p0 = 0.62, p1 = 0.38) | −2473.399503 | Not allowed | ||
| M2a: Positive selection | 3 | ω0 = 0, ω1 = 1, ω2 = 7.31; (p0 = 0.62, p1 = 0.32, p2 = 0.06) | −2444.710131 | (M1a vs. M2a), 57.378744, d.f. = 2, p = 0.0000 | 93-H(0.989) | 7.523 ± 1.614 |
| 105-Y (0.987) | 7.504 ± 1.640 | |||||
| 106-F (1.000) | 7.595 ± 1.464 | |||||
| 143-K (0.748) | 5.832 ± 3.077 | |||||
| 154-P (1.000) | 7.594 ± 1.466 | |||||
| 155-R(0.667) | 5.263 ± 3.239 | |||||
| 158-S (0.980) | 7.456 ± 1.718 | |||||
| 171-R(0.958) | 7.323 ± 1.953 | |||||
| 173-T (0.971) | 7.380 ± 1.805 | |||||
| 176-T (0.583) | 4.674 ± 3.305 | |||||
| 188-T (0.973) | 7.393 ± 1.788 | |||||
| M7: β | 2 | p = 0.1085, q = 0.1183 | −2474.985643 | Not allowed | ||
| M8: β + ωs > 1 | 4 | p0 = 0.94, p1 = 0.06, p = 0.36716, q = 0.47347, ω = 6.83 | −2444.453952 | (M7 vs. M8), 61.063382, d.f. = 2, p = 0.0000 | 93-H (0.993) | 7.265 ± 1.428 |
| 105Y (0.993) | 7.264 ± 1.426 | |||||
| 106-F (1.000) | 7.307 ± 1.332 | |||||
| 143-K (0.814) | 6.055 ± 2.779 | |||||
| 154-P (1.000) | 7.306 ± 1.332 | |||||
| 155-R (0.744) | 5.575 ± 3.020 | |||||
| 156-T (0.649) | 4.762 ± 2.999 | |||||
| 158-S (0.990) | 7.242 ± 1.468 | |||||
| 171-R(0.970) | 7.119 ± 1.718 | |||||
| 173-T (0.989) | 7.228 ± 1.480 | |||||
| 176-T (0.664) | 5.028 ± 3.191 | |||||
| 188-T (0.991) | 7.239 ± 1.460 | |||||
| M8a: β + ωs = 1 | 3 | p0 = 0.62, p1 = 0.38, p = 11.37, q = 99, ω = 1 | −2473.400411 | (M8 vs. M8a), 57.892918, d.f. = 1, p = 0.0000 | Not allowed | |
| Lineage 1b | ||||||
| M0: One-ratio | 1 | ω = 0.4649 | −3088.137934 | None | ||
| M1a: Nearly neutral | 1 | ω0 = 0.166, ω1 = 1, (p0 = 0.72, p1 = 0.28) | −3048.588195 | Not allowed | ||
| M2a: Positive selection | 3 | ω0 = 0.179, ω1 = 1, ω2 = 9.729; (p0 = 0.696, p1 = 0.289, p2 = 0.013) | −3028.791341 | (M1a vs. M2a), 39.593708, d.f. = 2, p = 0.0000 | 146-P (1.00) | 8.326 ± 1.713 |
| 183-F (1.00) | 8.325 ± 1.714 | |||||
| 196-L (0.999) | 8.316 ± 1.732 | |||||
| M7: β | 2 | p = 0.393, q = 0.546 | −3054.125576 | Not allowed | ||
| M8: β + ωs > 1 | 4 | p0 = 0.89, p1 = 0.11p = 1.777, q = 4.03, ω = 2.84 | −3034.377594 | (M7 vs. M8), 39.495964, d.f. = 2, p = 0.0000 | 146-P (1.000) | 5.183 ± 1.880 |
| 157-F (0.718) | 3.601 ± 2.137 | |||||
| 183-F (1.000) | 5.183 ± 1.880 | |||||
| 196-L (0.999) | 5.181 ± 1.882 | |||||
| 199-S (0.573) | 2.935 ± 2.110 | |||||
| M8a: β + ωs = 1 | 3 | p0 = 0.72, p1 = 0.28, p = 19.98, q = 99, ω = 1 | −3048.6126 | (M8 vs. M8a), 28.470012, d.f. = 1, p = 0.0000 | Not allowed | |
| Lineage 2a | ||||||
| M0: One-ratio | 1 | ω = 0.6898 | −2927.296491 | None | ||
| M1a: Nearly neutral | 1 | ω0 = 0.248, ω1 = 1, (p0 = 0.565, p1 = 0.435) | −2913.654666 | Not allowed | ||
| M2a: Positive selection | 3 | ω0 = 0.382, ω2 = 4.487; (p0 = 0.69, p1 = 0.23, p2 = 0.073) | −2898.698295 | (Mla vs. M2a), 29.912742, d.f. = 2, p = 0.0000 | 85-L (1.000) | 5.340 ± 1.570 |
| 93-Q (0.888) | 4.839 ± 2.038 | |||||
| 105-L (0.878) | 4.694 ± 1.966 | |||||
| 109-S(0.913) | 4.898 ± 1.899 | |||||
| 113-L (0.732) | 3.959 ± 2.193 | |||||
| 121-P (0.510) | 2.849 ± 2.078 | |||||
| 180-L (0.535) | 3.024 ± 2.228 | |||||
| 202-S (0.508) | 2.890 ± 2.196 | |||||
| 232-Y (0.989) | 5.295 ± 1.627 | |||||
| 239-P (0.975) | 5.226 ± 1.690 | |||||
| M7: β | 2 | p = 0.606, q = 0.379 | −2917.092133 | Not allowed | ||
| M8: β + ωs > 1 | 4 | p0 = 0.89, p1 = 0.11, p = 28.418, q = 31.77, ω = 3.65 | −2898.947133 | (M7 vs. M8), 36.29, d.f. = 2, p = 0.0000 | 85-L (1.000) | 5.381 ± 1.324 |
| 93-Q (0.900) | 4.945 ± 1.872 | |||||
| 105-L (0.920) | 4.988 ± 1.751 | |||||
| 109-S (0.939) | 5.093 ± 1.673 | |||||
| 113-L (0.777) | 4.282 ± 2.179 | |||||
| 121-P (0.528) | 3.028 ± 2.275 | |||||
| 180-L (0.546) | 3.173 ± 2.375 | |||||
| 202-S (0.519) | 3.038 ± 2.358 | |||||
| 232-Y (0.992) | 5.351 ± 1.376 | |||||
| 239-P (0.983) | 5.306 ± 1.439 | |||||
| M8a: β + ωs = 1 | 3 | p0 = 0.57, p1 = 0.43, p = 33.25, q = 99, ω = 1 | −2913.719868 | (M8 vs. M8a), 29.54547 d.f. = 1, p = 0.0000 | Not allowed | |
| Lineage 2b | ||||||
| M0: One-ratio | 1 | ω = 0.7065 | −1855.459267 | None | ||
| M1a: Nearly neutral | 1 | ω0 = 0, ω1 = 1, (p0 = 0.49, p1 = 0.51) | 840.547215 | Not allowed | ||
| M2a: Positive selection | 3 | ω0 = 0, ω1 = 1, ω2 = 10.0195; (p0 = 0.45, p1 = 0.52, p2 = 0.03) | −1829.903873 | (M1a vs. M2a), 21.286684, d.f. = 2, p = 0.00002 | 100-E (0.999) | 7.607 ± 2.041 |
| 105-P (0.971) | 7.432 ± 2.288 | |||||
| 109-P (0.911) | 6.994 ± 2.703 | |||||
| 213-R (0.682) | 5.477 ± 3.516 | |||||
| M7: β | 2 | p = 0.00517, q = 0.005 | −1840.570751 | Not allowed | ||
| M8: β + ωs > 1 | 4 | p0 = 0.97, p1 = 0.03, p = 0.005, q = 0.005, ω = 9.6 | 830.002479 | (M7 vs. M8), 21.136554, d.f. = 2, p = 0.00003 | 100-E (1.000) | 6.823 ± 2.093 |
| 105-P (0.985) | 6.745 ± 2.196 | |||||
| 109-P (0.958) | 6.561 ± 2.375 | |||||
| 114-Y (0.515) | 3.679 ± 3.226 | |||||
| 116-G (0.572) | 4.071 ± 3.293 | |||||
| 162-E (0.606) | 4.080 ± 3.052 | |||||
| 201-T (0.500) | 3.579 ± 3.206 | |||||
| 213-R (0.770) | 5.424 ± 3.159 | |||||
| 220-P (0.629) | 4.385 ± 3.236 | |||||
| M8a: β + ωs = 1 | 3 | p0 = 0.49, p1 = 0.51, p = 0.005, q = 2.785, ω = 1 | 840.547213 | (M8 vs. M8a), 21.089468, d.f. = 1, p = 0.0000 | Not allowed | |
Null models (M1a, M7, and M8a) are compared with their respective alternative models (M2a, M8) that allow ω > 1. Proportion of positively selected sites and their corresponding ω-values in M2a and M8 models are in bold. The posterior probability of each positively selected amino acid site is in parenthesis. Posterior probabilities are estimated based on Bayes Empirical bayes analyses (Yang et al., 2005).
Acknowledgements
We thank the University of Tulsa for providing facilities to carry out this work and Dr. Peggy S.M. Hill for editing and comments for improving the manuscript. We thank two anonymous reviewers for the helpful comments on the earlier version of the manuscript.
References
- Bastien N., Liu L., Ward D., Taylor T., Li Y. Genetic variability of the G glycoprotein gene of human metapneumovirus. J. Clin. Microbiol. 2004;42:3532–3537. doi: 10.1128/JCM.42.8.3532-3537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien N., Ward D., Caeseele P.V., Brandt K., Lee S.H.S., McNabb G., Klisko B., Chan E., Li Y. Human metapneumovirus infection in the Canadian population. J. Clin. Microbiol. 2003;41:4642–4646. doi: 10.1128/JCM.41.10.4642-4646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacchesi S., Skiadopoulos M.H., Boivin G., Hanson C.T., Murphy B.R., Collins P.L., Buchholz U.J. Genetic diversity between human metapneumovirus subgroups. Virology. 2003;315:1–9. doi: 10.1016/s0042-6822(03)00528-2. [DOI] [PubMed] [Google Scholar]
- Boivin G., De Serres G., Hamelin M.E., Cote S., Argouin M., Tremblay G., Maranda-Aubut R., Sauvageau C., Ouakki M., Boulianne N., Couture C. An outbreak of severe respiratory tract infection due to human metapneumovirus in a long-term care facility. Clin. Infect. Dis. 2007;44:1152–1158. doi: 10.1086/513204. [DOI] [PubMed] [Google Scholar]
- Carr M.J., McCormack G.P., Crowley B. Human metapneumovirus-associated respiratory tract infections in the republic of Ireland during the influenza season of 2003–2004. Clin. Microbiol. Infect. 2005;11:366–371. doi: 10.1111/j.1469-0691.2005.01129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Holmes E.C. Avian influenza virus exhibits rapid evolutionary dynamics. Mol. Biol. Evol. 2006;23:2336–2341. doi: 10.1093/molbev/msl102. [DOI] [PubMed] [Google Scholar]
- Cote S., Abed Y., Boivin G. Comparative evaluation of real-time PCR assays for detection of the human metapneumovirus. J. Clin. Microbiol. 2003;41:3631–3635. doi: 10.1128/JCM.41.8.3631-3635.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A.J., Rambaut, A., 2006. BEAST version 1.4.4. Available from http://beast.bio.ed.ac.uk/.
- Drummond A.J., Ho S.Y.W., Phillips M.J., Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Nicholls G.K., Rodrigo A.G., Solomon W. Estimating mutation parameters, population history, and genealogy simultaneously from temporally spaced sequence data. Genetics. 2002;161:1307–1320. doi: 10.1093/genetics/161.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A., Shapiro B., Pybus O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- Ebihara T., Endo R., Kikuta H., Ishiguro N., Ishiko H., Kobayashi K. Comparison of the seroprevalence of human metapneumovirus and human respiratory syncytial virus. J. Med. Virol. 2004;72:304–306. doi: 10.1002/jmv.10572. [DOI] [PubMed] [Google Scholar]
- Fouchier R.A.M., Rimmelzawaan G.F., Kuiken T., Osterhaus A.D.M.E. Newer respiratory virus infections: human metapneumovirus, avian influenza, and coronaviruses. Curr. Opin. Infect. Dis. 2005;18:141–146. doi: 10.1097/01.qco.0000160903.56566.84. [DOI] [PubMed] [Google Scholar]
- Galiano M., Trento A., Ver L., Carballal G., Videla C. Genetic heterogeneity of G and F protein genes from Argentinean human metapneumovirus strains. J. Med. Virol. 2006;78:631–637. doi: 10.1002/jmv.20586. [DOI] [PubMed] [Google Scholar]
- Gray G., Capuano A., Setterquist S., Sanchez J., Neville J., Olson J. Human metapneumovirus, Peru. Emerg. Infect. Dis. 2006;12:347–350. doi: 10.3201/eid1202.051133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray G.C., Capuano A.W., Setterquist S.F., Erdman D.D., Nobbs N.D., Abed Y., Doern G.V., Starks S.E., Boivin G. Multi-year study of human metapneumovirus infection at a large US midwestern medical referral center. J. Clin. Virol. 2006;37:269–276. doi: 10.1016/j.jcv.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hall T.A. BioEdit: a user friendly biological sequence alignment editor and analyses program for windows 95/98/N. J. Nucl. Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hamelin M.E., Boivin G. Development and validation of an enzyme-linked immunosorbent assay for human metapneumovirus serology based on a recombinant viral protein. Clin. Diagn. Lab. Immunol. 2005;12:249–253. doi: 10.1128/CDLI.12.2.249-253.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A.L., Nei M. Nucleotide substitution at major histocompatibility complex class II loci: evidence for overdominant selection. Proc. Natl. Acad. Sci. U.S.A. 1989;86:958–962. doi: 10.1073/pnas.86.3.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro N., Ebihara T., Endo R., Ma X., Kikuta H., Ishiko H., Kobayashi K. High genetic diversity of the attachment (G) protein of human metapneumovirus. J. Clin. Microbiol. 2004;42:3406–3414. doi: 10.1128/JCM.42.8.3406-3414.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn J.S. Epidemiology of human metapneumovirus. Clin. Microbiol. Rev. 2006;19:546–557. doi: 10.1128/CMR.00014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Tamura K., Nei M. MEGA3, 1: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Leung J., Esper F., Weibel C., Kahn J.S. Seroepidemiology of human metapneumovirus (hMPV) on the basis of a novel enzyme-linked immunosorbent assay utilizing hMPV fusion protein expressed in recombinant vesicular stomatitis virus. J. Clin. Microbiol. 2005;43:1213–1219. doi: 10.1128/JCM.43.3.1213-1219.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewick H.P., Abed Y., van Niekerk N., Boivin G., Klugman K.P., Madhi S.A. Human metapneumovirus genetic variability, South Africa. Emerg. Infect. Dis. 2005;11:1074–1078. doi: 10.3201/eid1107.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay I.M., Jacob K.C., Woolhouse D., Waller K., Syrmis M.W., Whiley D.M. Molecular assays for detection of human metapneumovirus. J. Clin. Microbiol. 2003;41:100–105. doi: 10.1128/JCM.41.1.100-105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison, Y., Maddison, D., 2006. Mesquite: a modular system for evolutionary analysis. Version 1.12. http://mesquiteproject.org.
- Maertzdorf J., Wang C.K., Brown J.B., Quinto J.D., Chu M., de Graaf M. Real-time reverse transcriptase PCR assay for detection of human metapneumoviruses from all known genetic lineages. J. Clin. Microbiol. 2004;42:981–986. doi: 10.1128/JCM.42.3.981-986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson J.W., Rich F.J., Cohet C., Grimwood K., Huang Q.S., Penny D., Hendy M.D., Kirman J.R. Distinct patterns of evolution between respiratory syncytial virus subgroups A and B from New Zealland isolates collected over thirty-seven years. J. Med. Virol. 2006;78:1354–1364. doi: 10.1002/jmv.20702. [DOI] [PubMed] [Google Scholar]
- Mes T.H.M., van Putten J.P.M. Positively selected codons in immune-exposed loops of the vaccine candidate OMP-P1 of Haemophilus influenzae. J. Mol. Evol. 2007;64:411–422. doi: 10.1007/s00239-006-0021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peret T.C., Abed Y., Anderson L.J., Erdman D.D., Boivin G. Sequence polymorphism of the predicted human metapneumovirus G glycoprotein. J. Gen. Virol. 2004;85:679–686. doi: 10.1099/vir.0.19504-0. [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K.A. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Rambaut, A., Drummond, A.J., 2006. Tracer version 1.3. Available from http://tree.bio.ed.ac.uk/software/tracer/.
- Regev L., Hindiyeh M., Shulman L.M., Barak A., Levy V., Azar R., Shalev Y., Grossman Z., Mendelson E.J. Characterization of human metapneumovirus infections in Israel. Clin. Microbiol. 2006;44:1484–1489. doi: 10.1128/JCM.44.4.1484-1489.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J., Sánchez-DelBarrio J.C., Messeguer X., Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Skiadopoulos M.H., Biacchesi S., Buchholz U.J., Riggs J.M., Surman S.R., Carambot E.A., McAuliffe J.M., Elkins W.R., Claire M.S., Collins P.L., Murphy B.R. The two major human metapneumovirus genetic lineages are highly related antigenically, and the fusion (F) protein is a major contributor to this antigenic relatedness. J. Virol. 2004;78:6927–6937. doi: 10.1128/JVI.78.13.6927-6937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockton J., Stephenson I., Fleming D., Zambon M. Human metapneumovirus as a cause of community-acquired respiratory illness. Emerg. Infect. Dis. 2002;8:897–901. doi: 10.3201/eid0809.020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson W.J., Nielsen R., Yang Q. Pervasive adaptive evolution in mammalian fertilization proteins. Mol. Biol. Evol. 2003;20:18–20. doi: 10.1093/oxfordjournals.molbev.a004233. [DOI] [PubMed] [Google Scholar]
- Trento A., Viegas M., Galiano M., Videla C., Carballal G., Mistchenko A.S., Melero J.A. Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J. Virol. 2006;80:975–984. doi: 10.1128/JVI.80.2.975-984.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrand N.D., Ji H., Patel N.K., Riggs J.M., Brewah Y.A., Ready S., Donacki N.E., Folliot K., Barnes A.S., Senthil K., Wilson S., Chen M., Clarke L., MacPhail M., Li J., Woods R.M., Coelingh K., Reed J.L., McCarthy M.P., Pfarr D.S., Osterhaus A.D.M.E., Fouchier R.A.M., Kiener P.A., Suzich J.A.A. Isolation and characterization of monoclonal antibodies which neutralize human metapneumovirus in in vitro and in vivo. J. Virol. 2006;80:799–7806. doi: 10.1128/JVI.00318-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogen B.G. Respiratory tract infection due to human metapneumovirus among elderly patients. Clin. Infect. Dis. 2007;44:1159–1160. doi: 10.1086/513295. [DOI] [PubMed] [Google Scholar]
- van den Hoogen B.G., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Analysis of the genomic sequence of a human metapneumovirus. Virology. 2002;295:119–132. doi: 10.1006/viro.2001.1355. [DOI] [PubMed] [Google Scholar]
- van den Hoogen B.G., de Jong J.C., Groen J., Kuiken T., de Groot R., Fouchier R.A., Osterhaus A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001;7:719–724. doi: 10.1038/89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogen B.G., Herfst S., Sprong L., Cane P.A., Forleo-Neto E., de Swart R.L., Osterhaus A.D.M.E., Fouchier R.A.M. Antigenic and genetic variability of human metapneumoviruses. Emerg. Infect. Dis. 2004;10(4):658–666. doi: 10.3201/eid1004.030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woelk C.H., Pybus O.G., Jin L., Brown D.W.G., Holmes E.C. Increased positive selection pressure in persistent (SSPE) versus acute measles virus infections. J. Gen. Virol. 2002;83:1419–1430. doi: 10.1099/0022-1317-83-6-1419. [DOI] [PubMed] [Google Scholar]
- Xia, X., 2000. Data analysis in molecular biology and evolution. pp. 276.
- Xia X., Xie Z. DAMBE: Data analysis in molecular biology and evolution. J. Heredity. 2001;92:371–373. doi: 10.1093/jhered/92.4.371. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z., Nielsen R., Goldman N., Pederson A.M. Codon-substitution models for heterogeneous selection pressures at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Wong W.S., Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Zhang C.-Y., Wei J.-F., He S.-H. Adaptive evolution of the spike gene of SARS coronavirus: changes in positively selected sites in different epidemic groups. BMC Microbiol. 2006;6:88. doi: 10.1186/1471-2180-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlateva K.T., Lemey P., Moës E., Vandamme A.-M., Ranst M.V. Genetic variability and molecular evolution of the human respiratory syncytial virus subgroup B attachment G protein. J. Virol. 2005;79:9157–9167. doi: 10.1128/JVI.79.14.9157-9167.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlateva K.T., Lemey P., Vandamme A.-M., Ranst M.V. Molecular evolution and circulation patterns of human respiratory syncytial virus subgroup A: Positively selected sites in the attachment G glycoprotein. J. Virol. 2004;78:4675–4683. doi: 10.1128/JVI.78.9.4675-4683.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]