

Abstract
Life-threatening RNA viruses emerge regularly, and often in an unpredictable manner. Yet, the very few drugs available against known RNA viruses have sometimes required decades of research for development. Can we generate preparedness for outbreaks of the, as yet, unknown viruses? The VIZIER (VIral enZymes InvolvEd in Replication) (http://www.vizier-europe.org/) project has been set-up to develop the scientific foundations for countering this challenge to society. VIZIER studies the most conserved viral enzymes (that of the replication machinery, or replicases) that constitute attractive targets for drug-design. The aim of VIZIER is to determine as many replicase crystal structures as possible from a carefully selected list of viruses in order to comprehensively cover the diversity of the RNA virus universe, and generate critical knowledge that could be efficiently utilized to jump-start research on any emerging RNA virus. VIZIER is a multidisciplinary project involving (i) bioinformatics to define functional domains, (ii) viral genomics to increase the number of characterized viral genomes and prepare defined targets, (iii) proteomics to express, purify, and characterize targets, (iv) structural biology to solve their crystal structures, and (v) pre-lead discovery to propose active scaffolds of antiviral molecules.
Keywords: RNA virus, Genomics, Crystal structure, Replicase, Antivirals, Drug-design
1. Introduction
1.1. World epidemic context
Since the discovery of the deadly Ebola virus (filovirus) in 1976 a series of previously unknown pathogenic (emerging) viruses have been discovered, with greater human communication and mobility contributing to awareness and spread, respectively. The World Health Organization (WHO) estimates that the human immunodeficiency virus (HIV), since its discovery in 1983, has killed more than 25 million people and that about 40 million people are today infected with the virus. In 1993, a then unknown lethal hantavirus the Sin Nombre virus, emerged in the Western USA. Shortly thereafter, two unknown paramyxoviruses, both causing lethal diseases, emerged: Nipah virus in Malaysia and Hendra virus in Australia. Before 2002, human coronaviruses were known to cause only mild upper respiratory tract infections. However, the severe acute respiratory syndrome coronavirus (SARS CoV), which appeared in 2002, was highly pathogenic and had a high fatality rate. The avian influenza virus H5N1 strain was first isolated from humans in 1996 in Hong Kong and has caused, since 2003, about 200 casualties. H5N1, as well as other avian influenza viruses, have the potential to develop, either by genetic drift or recombination with other influenza virus strains, into viruses that are highly pathogenic to humans and which have the potential of causing a pandemic. Several viruses have, in recent years, widely expanded their territory, causing the death of an increasing number of people. One such example is the West Nile flavivirus, that was introduced into the USA in 1999, and has since then become endemic in the entire USA and parts of Canada with extensions into Latin America. In the period 2005/2006, the Chikungunya alphavirus resulted in more than 500 000 cases in the islands of the Indian Ocean, probably more than 1.5 million cases in India and is currently sweeping through large parts of southeast Asia, central Africa, and recently Italy. Since the 1970s the number of people infected with the dengue virus (a flavivirus) has been dramatically increasing. In 1975, the WHO surveillance network reported 10 dengue endemic countries and ∼60 000 dengue cases. In 2005, the WHO reported 65 dengue-infested countries, with 1 million reported cases and an estimate of 50 millions actual cases/year.
On the basis of this large, but non-exhaustive list, it is evident that almost all newly emerging, human pathogenic viruses belong to the group of RNA viruses. The RNA-dependent RNA polymerases of RNA viruses have no proofreading capability. As a consequence, these viruses have a very high mutation rate on average about one mutation/virus/replication cycle, allowing fast adaptation to new hosts and environments. Another remarkable observation is that almost all emerging viral infections are zoonotic in nature. The natural hosts for Ebola, HIV, Nipah, Hendra, SARS, H5N1, Sin Nombre, are animals such as bats, monkeys and birds, in which they often have no strong pathogenic effect. An enormous number of yet undiscovered viruses exist in vertebrates. One can state with a high degree of certainty that novel, sometimes highly lethal RNA viruses, will emerge in the future from this large natural genetic pool. Human activities such as deforestation and international travel as well as the results of climate change may facilitate the emergence of such viruses. Other RNA viruses that are known to be pathogenic to man may emerge in regions where they are not yet present, as examplified by the dengue virus in recent years. In addition, changes in the fragile balance of specific ecological niches, may favour emergence of novel pathogenic agents.
1.2. How ready are we to face epidemics?
Fear that H5N1 influenza might develop into an epidemic like that of 1918 fuelled a global effort to be prepared for a future pandemic. A human population of more than 6 billion people must also be prepared for the inevitable emergence of new, highly contagious and lethal viral infections of other origins. The introduction of HIV, the Ebola virus, Sin Nombre virus, Nipah, Hendra, SARS CoV, and many other novel viruses, in the human population was not, and could not have been predicted. Likewise, we are today unable to predict which virus may emerge tomorrow. Preparedness to meet such a threat of an emerging virus includes the ability to rapidly characterize the virus and to be able to take the necessary measures for control. The latter may include vector control, development of vaccines as well as the development of selective antiviral drugs. The former two strategies are most appropriate when a threat has been recognised (i.e. effective in prevention of infection) whilst the latter are essential for quick response to an unknown threat. Today antiviral drugs are only available for the treatment of infections with herpesviruses, the hepatitis B and C viruses, HIV and influenza viruses. In addition Ribavirin has been approved and has shown clinical benefit for the treatment of Lassa virus infections. No specific therapy is available for the treatment of other viral infections. In fact, a crucial statement has to be made here. The drugs that are available against these viruses, have required decades of scientific effort both in the academic and industrial sectors for their development.
1.3. Should we start the chess game?
Why then should not we start now and have these decades of anticipative research already in place whenever our next viral challenger comes? For most of the RNA viruses, very fundamental insights into potential molecular targets for therapy are lacking. When the SARS coronavirus (SARS-CoV) was identified as the causative agent of the SARS epidemic, research on anti-coronavirus drugs was in its infancy and no knowledge was available as to how to selectively inhibit the replication of a coronavirus. Since then, many important advances have been made, although a search for antivirals against SARS-CoV has yet to produce drugs approved for human treatment. Sadly, one can state with near certainty that no antiviral drugs will be available for the treatment of infections of the next newly emerging virus.
Let us examine the concept. For HIV there are now more than 20 drugs available. If another deadly retrovirus should emerge now, would not the two decades of HIV research make a big difference? The answer is obvious.
2. The birth of viral structural genomics
It became quickly apparent that Structural Genomics projects are adaptable to a pan-viral approach and could be useful to develop the anticipative concept.
SPINE (Structural Proteomics in Europe, an EU integrated project coordinated by D.I. Stuart, Oxford, UK) was the prototype of a large-scale, European structural proteomics project launched by the European Union in 2001. Amongst the eukaryotic targets, SPINE contained one workpackage dedicated to viral proteins. At that time, the frustration of virologists facing large-scale genomic projects originated from the small genome size of viruses. It was obvious that viruses of the Herpesviridae or Poxviridae could present respectable targets. The true innovation, however, came with the proposition that studying large families of related viruses could in fact address the well-known problem of poor crystallization success rate. For example, the structure of the first flavivirus polymerase, even from an obscure virus, would immediately have an impact on the whole flavivirus field. Additionally molecular replacement techniques would then considerably speed up the structure solution of more medically important flavivirus polymerases once crystals became available. We also hoped that a gallery of structures from similar viruses would uncover small, as yet-unknown differences that could translate into larger in vivo differences, impacting for example on pathogenic properties, host range and mechanistics peculiarities. In fact, this approach had been met with partial success for RNA viruses. The main difficulties originated from the shortage of properly defined viral targets. In other words, at the time, it was a bit ambitious to start structural proteomics without addressing viral genomics. VIZIER, which started as an European FP6 Integrated Project in October 2004 with a 13 million € budget, specifically addressed this limitation by providing a comprehensive structural characterization of a diverse set of RNA viruses. In the meantime, a part of the efforts in the SPINE project were devoted to method developments, and technical progress implemented in the individual laboratories were combined. This study (published as special issue in Acta Crystallogaphica Section D, Volume 62, Part 10, October 2006) provided strategic information needed to set up a pipeline adapted for Structural and Functional Genomics of viral proteins involved in replication.
3. VIZIER: viral enzymes involved in replication (http://www.vizier-europe.org/)
The overall VIZIER organization is shown in Fig. 1 . The bioinformatics and virology represent the genomics core, namely data gathering and analysis, and protein production/crystallization facilities represent structural genomics aspects. The validation section aims at discovering novel enzyme activities as well as potential lead compounds for drug discovery, that are characterized in close collaboration with all other sections of the project. The flow of information, material, and activities is depicted in Fig. 2 . Thus VIZIER is organized into a pipeline containing six thematic sections.
Fig. 1.
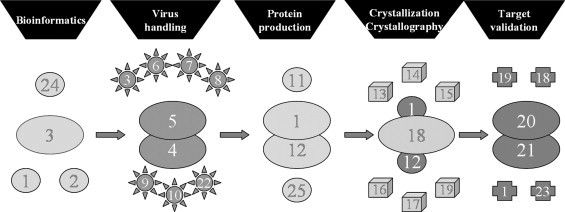
The pipeline organization of the VIZIER project, with its core (large symbols) and satellite labs (smaller symbols) represented by the numbers 1–25; The numbers 1–25 correspond to the a–z author laboratory addresses, respectively.
Fig. 2.
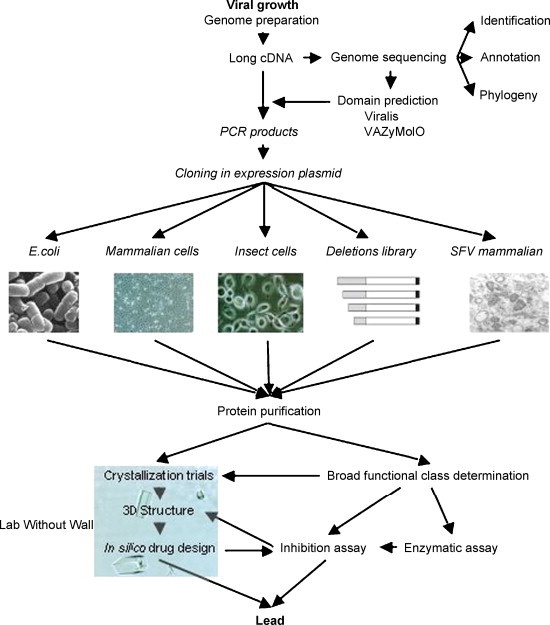
The flow of information, material, and activities within the VIZIER pipeline. Viralis: Virus Alignment Suite; VaZyMolO: Viral Enzymes Modular Organization; SFV: Semliki Forest Virus.
3.1. Section 1: bioinformatics (Head: A.E. Gorbalenya, Leiden)
RNA viruses commonly produce replicative proteins as large molecular weight polyproteins that may be autocatalytically processed to mature products. Because of their size and complex organization, the production and crystallization of recombinant full-length viral proteins is notoriously difficult and usually associated with low expression levels, protein degradation and/or insolubility. To ensure a high success rate at the production, purification and crystallization steps, expression of each domain individually as well as combinations of domains was adopted as a main VIZIER strategy. Its success relies on a reliable dissection of the domain organization of viral polyproteins that is accomplished through the use of two complementary approaches utilizing, respectively, the VirAliS (Virus Alignment Suite) (Gorbalenya et al., unpublished) and VaZyMolO (Viral Enzymes Modular Organization) (Ferron et al., 2005) software platforms developed and advanced by researchers of LUMC/MSU (Viralis) and AFMB (VaZyMolO). The partners in the VIZIER consortium communicate with these platforms through dedicated web servers. Viralis provides a state-of-the-art multi-modular environment for expert analysis of RNA virus genomes using multiple alignments and phylogenetic analysis. A recent study of the origin and evolution of poliovirus (Jiang et al., 2007) is a good example. It has been used for delineation of a large variety of domains also including those that are functionally poorly characterized and those that are hard to express in soluble form. The VaZyMolO platform allows the VIZIER partners to make, or refine, their own domain predictions, using different bioinformatic tools (BLAST, HCA plot, etc.) and to access information on previously made predictions using old and new RNA virus sequences. It will be interesting to compare the crystallization success rate for domains predicted using Viralis and VaZyMolO.
Domain designs are converted into polymerase chain reaction (PCR) products by Section 2 partners and are then submitted for further target processing. Information about targets and their processing, from the cloning to the protein purification, is stored and updated in the central VIZIER target relational database (http://www.vizier-europe.org/). The web site also hosts a public domain containing general data on RNA viruses. The structural information, and the progress to structure solution, is stored in an adapted version of Xtrack (Harris and Jones, 2002). The functional aspects (e.g. protocols, lists of antiviral compounds) are stored in a database called Drugbase (Leuven). Although all these databases are maintained in different places (Leiden, Marseilles, Uppsala and Leuven), they are linked to the VIZIER website so that a VIZIER user can request technical data on each step of the process for any target.
3.2. Section 2: virology (Head: X. de Lamballerie, Marseille)
The main lesson learnt from the SPINE project was the difficulty in feeding the production pipeline with a sufficient number of RNA virus isolates. An incredible discrepancy was also observed between published viral sequences and the actual sequences of produced viral proteins. A cruel fact is that many virology laboratories work with viruses which differ appreciably in sequence from the native isolates. Likewise, there were large virus families for which complete genome sequences were quite fragmentary (see the flavivirus example below). As a consequence, VIZIER is first a viral genomics program, upon which structural genomics and early lead discovery work is superimposed. The virology section is involved in the genomics part of the project by (i) implementing viral growth and virus storage under appropriate bio-safety level conditions, (ii) sequencing the viral genomes and depositing sequences in public databases, and (iii) analyzing the sequence data for the purpose of virus identification as well as for evolution/phylogeny studies. No attempt has been made to collect and centralize biological material. Dangerous viruses are cultivated under appropriate safety conditions by laboratories already experienced with this task. Initially the tasks of the Section had to span about 145 viruses and this ambitious goal required the design of protocols adapted for large-scale genome sequencing (Emonet et al., 2007). This procedure, by generating long cDNAs, enabled easy subsequent one-step amplifications of domains designed by Section 1 for protein domain expression.
3.3. Section 3: protein production (Head: B. Coutard, Marseille)
Once the PCR products are generated, they are cloned into expression plasmids compatible for several expression systems without the need for subsequent recloning steps (pDEST14 from Invitrogen and/or the pOPIN series (Berrow et al., 2007)). All the constructs, fused to a hexa-histidine tag, are first tested in a robust E. coli expression screening that was set up according to the lessons learnt in the SPINE project by the Marseille and Oxford Laboratories (Berrow et al., 2006, Alzari et al., 2006). When proteins are expressed in the soluble fraction, they are purified through a two-stage purification protocol (immobilized metal affinity chromatography followed by size exclusion chromatography) on automated fast protein liquid chromatography systems (Äkta Xpress from GE Healthcare) in order to recover pure protein suitable for initial crystallization screenings and protein characterization. When expression in E. coli fails, additional strategies are applied: platforms are also available for expression in mammalian and insect cells (in the Oxford group) and Semliki Forest virus (in the Lausanne group (Hassaine et al., 2006)). Another one is devoted to the use deletion libraries (the Stockholm group (Cornvik et al., 2005)) that can provide not only soluble proteins but also new domain designs. The concept of Section 3 initially included preliminary crystallization screening, but due to protein instability during protein transit, most of the crystallization efforts are, in practice, carried out by the partner involved in the structure determination.
3.4. Section 4: structure determination (Head: T.A. Jones, Uppsala)
Crystal production (and increasingly rescue strategies for high value targets) is performed in the Section 4 Laboratories where implementation of standard protocols and automated platforms guarantees reproducible results and requires less protein compared to classical manual crystallogenesis (Sulzenbacher et al., 2002, Fogg et al., 2006). Crystals are then exposed to X-rays at the ESRF (Grenoble) or the EMBL (DESY, Hamburg) for data collection. The crystal structures can then be solved by conventional techniques (heavy atoms derivatisation, usually involving the production of selenomethionine containing protein), or molecular replacement when appropriate. In a second step, co-crystallization experiments are performed with putative binding molecules according to functional data provided by the Section 5 Laboratories. Section 4 was designed according to the “Lab Without Walls” concept, where all the crystallization and structural data can be shared in order to promote synergy between partners. This synergy also involves Global Phasing, Inc., which develops and distributes beta-versions of crystallographic software useful for “difficult” structures. Once a structure is solved in one virus family, other groups in the consortium can use the experimental data (for example, domain design, crystallization procedures and possible binding partners) to speed up the structure solution of homologues.
To take one example: a large number of flavivirus methyltransferases were solved on the basis of the structural data provided by the dengue virus construct (Egloff et al., 2002). The structure revealed an unfolded C-terminal domain that also appeared to be cleaved off when producing the full-length NS5 protein of a TBE flavivirus. These data suggested that at least two constructs should be tested for the other flavivirus methyltransferases (with and without the flexible C-terminal domain). This strategy is validated since the Meaban virus domain was solved with the smaller construct whereas the Yokose virus one was solved with the larger (Mastrangelo et al., 2006a). Moreover, the substrates provided by Section 5 activities (Luzhkov et al., 2007, Peyrane et al., 2007) are available for co-crystallization experiments with all flavivirus methyltransferase constructs. It is also important to note that the compounds screened against the dengue methyltransferase structure in silico may also be suitable to evaluate the relative efficiency for all the viral methyltransferases, not only the flavivirus ones.
3.5. Section 5: validation (Head: J. Neyts, Leuven)
The validation section aims at deciphering the functional aspects of the purified domains. The predicted functions are first tested in vitro, in different specialized platforms (the RNA-dependent RNA polymerases in Marseille and Dresden, the proteases in Lubeck and Dresden, the capping enzymes in Marseille and the helicases in Dresden and Milano). When no putative function is available for a target protein, a first generic screening is performed (RNA and nucleotide binding) before being refined on the corresponding platform. Once the enzyme is well characterized, compounds from the drugbase can be tested by in vitro inhibition assays before in cellulo evaluation of the effect on the replicative system. In a reverse process, data mining of old active compounds can generate ligands with either interesting antiviral properties (De Palma et al., 2007) on novel infected cell systems, or for use in crystallographic studies. It is a commonly held view that inhibitors or binding partners of proteins help crystallization by stabilizing the protein (Vedadi et al., 2006).
3.6. Section 6: training, dissemination, and the industrial platform (Head: P.A. Tucker, Hamburg)
The dissemination of knowledge generated within the VIZIER project is one of the missions encouraged and valued by the European Commission. Different tools such as workshops, international congresses, promotional videos and a website are used to distribute information.
The VIZIER project has rapidly generated a need for intra- and extra-consortium vehicles aimed at optimizing data/results sharing, but communication to the general public is also of great importance given the relevance of the project to human health and the need to justify the use of public money. A video describing VIZIER, targeted at the general public, is available online (a longer version is available from the Section Head upon request), and the open access part of the website has been radically redesigned to address the increasing public awareness and interest in emerging viruses. The access is easy (as of September 2007, the site had third position when VIZIER is used a request on the Google search engine in France). The site is visited by more than 200 persons each day. Different links are organized around RNA viruses, and about viral diseases. A specific agreement has been signed to enable the display of WHO information. Detailed topical information can be found concerning VIZIER and the ongoing research both within VIZIER and the wider community of virologists. The VIZIER website provides two types of protected access. The first one provides access to technical data that is shared by VIZIER scientists. The second one is an entry to the VIZIER industrial platform (VIP). The VIP has been created for project promotion to the industrial sector and to allow the members of the VIP to access potential commercially exploitable products of the consortiums research. This VIP is accessible to companies having a membership agreement with the consortium where members have privileged access to information such as know-how, materials and techniques, as well as to unpublished scientific results. A unique and fundamental feature of the VIP is to facilitate the contractual procedures between a potential member and any of the VIZIER laboratories. An agreement has been signed within the consortium to delegate, where desired, the first negotiation steps to the VIP manager. Intellectual property rules has been established and agreed upon by all VIZIER partners. When partners decide to patent a discovery, a procedure for the partition of royalties between the partners (the “partition owners”) is organized by the VIP manager together with the different technology transfer departments of the institutes involved in the patenting procedure. The overall objective of the VIP is to produce conditions for an active collaboration between VIZIER scientists and the corporate world. It is also to increase and facilitate the collaboration with the industrial sector and even possibly to extend, beyond the funding period, certain aspects of the project by generating additional funds.
4. Flaviviruses: from proof-of-concept to structure-based drug-design
Flaviviruses served as an interesting test-case for the functional domain approach. In 2002, the crystal structure of the dengue virus N-terminal methyltransferase (MTase) domain of the NS5 protein was determined, and shown to be functional (Egloff et al., 2002). The complementary C-terminal RNA-dependent RNA polymerase (RdRp) domain was subsequently shown to be functional as well. Given this information, coupled with the large number of human pathogens within the genus, it is thus not surprising that flaviviruses were early targets of the VIZIER project. Progress within this important viral genus illustrates the exciting merging of traditional virology and genomics, with structural and functional proteomics. As described in Fig. 3 , VIZIER contributed to the sequencing of 34 flaviviruses, amongst them the N’Goye virus, which was previously totally unknown (Grard et al., 2006). Ten RdRps, nineteen MTases, nine helicases and four proteases were produced and purified, leading, so far, to thirteen new structures.
Fig. 3.
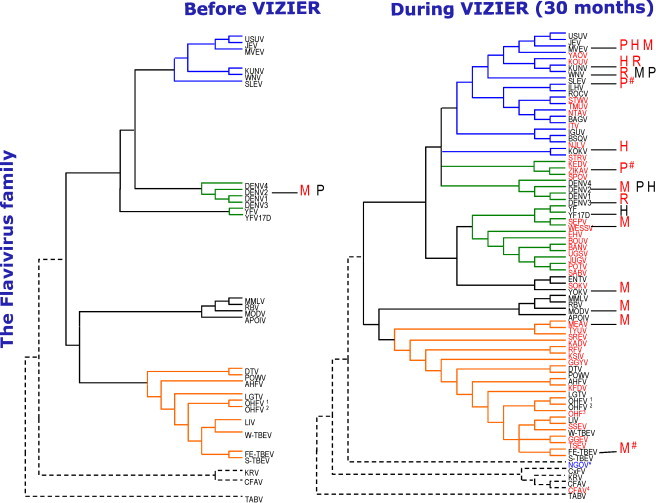
Phylogentic tree and crystal structures available before the start of the VIZIER project and now. The VIZIER contribution is shown in red (letters beside tree), with other contributions in blackI. P: Protease (Nt-NS3), H: Helicase (Ct-NS3), M: Mtase (Nt-NS5), R: RNA polymerase (Ct-NS5), #: crystals only. Only complete genome sequences are represented. In red (tree): sequences determined as part of the VIZIER project. NGOV is a new virus discovered and sequenced in VIZIER (Grard et al., 2006). This figure was prepared by Gilda Grard (Unité des Virus Emergents, Marseille).
When feasible, molecular replacement techniques allow not only a remarkable acceleration of 3D structure phylum coverage, but also foster collaboration between project partners. The structure solution of the West Nile virus polymerase domain speeded-up that of the dengue virus domain, and it is obvious that these atomic coordinates, much like that of the MTase NS5 domain, will remain useful for the whole flavivirus genus.
5. Applications in drug-design
5.1. Flavivirus MTase domain
Due to the early structure determination of the flavivirus MTase domain, this was used to perform virtual screening studies aiming at finding inhibitors (Luzhkov et al., 2007). The virtual high-throughput screening focused on the S-adenosyl-l-methionine (SAM, which is the methyl donor for the methyltransfer) binding site based on the structure of the dengue virus NS5 MTase domain (NS5MTaseDV) in complex with GTP analogs and S-adenosyl-homocysteine (SAH) as shown in Fig. 4A (Egloff et al., 2002). The procedure started with a pharmacophore and a 2D similarity search of a database of 2.1 million commercially available compounds. After docking a set of selected ligands, 17 top-ranking compounds, representing different scaffolds, were purchased. Inhibitory activity was tested on recombinant NS5MTaseDV using the capped RNA substrate 7MeGpppAC5 to measure (adenosine-2′O) methyltransferase activity (Peyrane et al., 2007). Finally, we identified a novel inhibitor with a previously unknown scaffold (see Fig. 4B) showing moderate inhibitory activity (see Fig. 4C). In addition another SAH analog, sinefungin, was identified as a potent inhibitor (in the submicromolar range), as were other SAH analogs which proved to be moderate inhibitors (unpublished results). Although capping might not be a primary target, because there is only a single RNA capping event, during replication of the Flavivirus genome, these compounds deserve further attention since they could be combined with other anti-viral molecules.
Fig. 4.
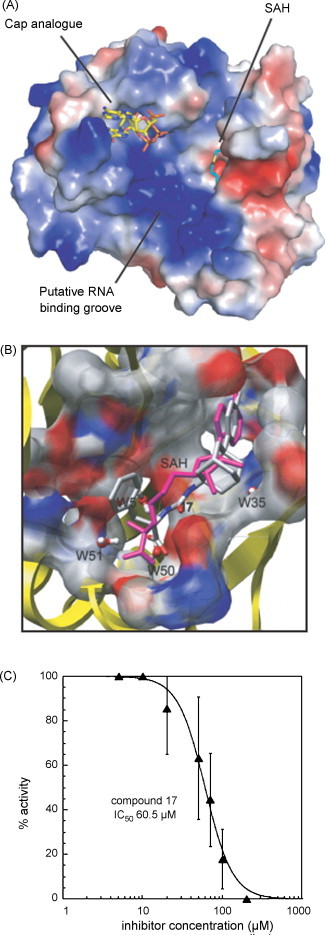
Discovery of a moderate flavivirus MTase inhibitor by virtual screening. (A) Electrostatic surface representation of the dengue virus NS5 MTase domain in complex with a cap analog depicting the cap-binding site during 2′O-methylation and SAH the co-product of the methyltransfer. The blue (positively charged) zone is thought to accommodate the RNA substrate. (B) Close-up of the SAM/SAH binding site showing a superposition of the docked inhibitor with an adamantane scaffold (coloured according to atom type) and SAH (pink). (C) Inhibition of the 2′OMTase activity of NS5MTaseDV by compound 17.
5.2. Flavivirus RdRp domain
The RdRp domain is a good example of the viral structural genomics approach. Several flavivirus NS5 genes had their MTase N-terminal domains removed, but the largest part of the challenge was to find a soluble RdRp domain construct that was enzymatically active, but that would crystallize easily. Up to 70 constructs were designed, and of these a single (a.a. 273–900 of NS5) enzymatically active one could be crystallized which diffracted to 3.0 Å resolution. Unfortunately it did not allow phase determination either using selenomethionine-substituted protein or more conventional heavy atom methods. Another construct (a.a. 317–900) gave crystals of an inactive enzyme, whose structure was solved at 2.4 Å, and used as a model to solve the original, enzymatically active protein (Malet et al., 2007). Both the construct length, and later, the coordinates were made available to the Lescar group, which expedited the crystal structure solution of the dengue virus RdRp domain in complex with a nucleotide at 1.8 Å resolution (Yap et al., 2007). We fully anticipate that these two first flavivirus polymerase structures will speed up crystal structure solution of other flavivirus polymerases, such as those of the BSL-4 hemorrhagic viruses, which could, then, be removed from the category of neglected, but deadly, pathogens. Led by the Novartis Institute of Tropical Diseases (NITD) in Singapore, there is currently a very active research programme merging high-throughput screening results (i.e. hits) with crystal soaking experiments in order to drive forward the structure-based drug-design efforts on flavivirus RdRps.
5.3. Other flavivirus targets: the NS3 helicase
The first flavivirus helicase crystal structure, that from yellow fever virus, was determined by Wu et al. (2005) followed closely by that from dengue virus (Xu et al., 2005, Sampath et al., 2006). In turn, these structures considerably help in the aims of the VIZIER consortium to cover structures encompassing the genomic tree of the flavivirus genus. The structures of West Nile (Kunjin) (Mastrangelo et al., 2006b), Kokobera (De Colibus et al., 2007), and Murray Valley Encephalitis virus (Mancini et al., unpublished) helicases have been recently determined. In summary these results now make structure-based drug-design of inhibitors possible for the four enzymatic activities carried by the flavivirus NS3 and NS5 proteins.
6. Beyond viral structural genomics
As addressed by the VIZIER project, viral structural genomics looks exciting for the crystallographer at an early stage of study in a given virus family, where no structural data yet exists. The hunger for novel structures is often in contradiction with the crystal structure determination of many similar structures (for example using molecular replacement techniques) inside a given virus family. But it is imperative to realize that even if VIZIER did not contribute to many original folds yet, this potential disenchantment on the part of structural biologists is not shared with virologists, who foresee connection of long-known virus-specific data (e.g. host range and specificity, disease specificity, geographical distribution, to name but a few) with new structural data. We realize that an understanding of systems biology has implications and applications in host–pathogen interactions. Besides the obvious desire to be prepared to tackle an emerging disease, the information content of a large number of crystal structures within a virus family cannot be currently estimated. Likewise, the anticipated crystal structures of complexes of replicases with substrates or, perhaps more importantly, protein partners will surely benefit from the wealth of structures determined both within VIZIER project and elsewhere. Before starting the flavivirus polymerase project, it was not possible to predict which flavivirus polymerase domain would yield a crystal structure. The same difficulty will apply to the flavivirus NS3-NS5 complexes, but the availability of many helicase and polymerase structures is likely to speed up exciting crystallographic work on these complexes.
It is possible that homologous domains in different virus families have different functions. The macrodomain (or X domain) may be such an example. In addition to archae, eukarya and prokarya, this domain is present in coronaviruses, alphaviruses, rubella virus and hepatitis E virus. However, since its associated phosphatase activity and ribose-binding activity differ between these phyla, why should we a priori consider that the function would remain conserved? The crystal structure of macrodomains, coupled with functional studies (e.g. Egloff et al., 2006) will undoubtedly help to find new and unexpected functions across different virus families.
Tackling several targets in parallel may provide research incentives on viruses presently unaddressed by the drug-design community. The project, after 30 months, has resulted in more than 1800 viral cDNAs being introduced into the protein production pipeline, and this has resulted in about 50 original solved apo- and binary-complex crystal structures for alphaviruses, rhabdoviruses, caliciviruses, coronaviruses, reoviruses, and picornaviruses (a full account will be reported elsewhere). In the latter genus, the simultaneous structure determination of the Coxsackie B3 polymerase (Jabafi et al., 2007; Gruez et al., unpublished), the 3A protease (Hilgenfeld et al., unpublished), and the discovery of potent inhibitors of the putative 2C helicase (De Palma et al., unpublished) provides a unique opportunity to comparatively evaluate the efficacy of targeting different components of the replicon, and perhaps simultaneous developments will lead to potent drug combinations. It may well be that these data can be “translated” to the poliovirus field, where antiviral molecules may help complete the eradication campaign.
Finally, the VIZIER project is a first step towards integrating the study of viral replicases into their cellular context. An unravelling of the complexity of the interactome existing around viral replicases will benefit from these structural data and allow future systems biology approaches. Detailed structural, functional, temporal, and interaction studies of replicases with their ligands, substrates, and viral/cellular partners will undoubtedly extend our knowledge beyond the antiviral field.
Acknowledgements
We thank Ségolène Arnoux, Julie Caprili, and Tania Langon, from ALMA Consulting Group (http://www.almacg.com), for administrative management of the project, Fabrice Malergue for the VIZIER Industrial Platform (VIP), as well as many colleagues, labmates, and partners, too numerous to name, for their enthusiasm and commitment to the VIZIER project. This work was supported by the VIZIER integrated project (LSHG-CT-2004-511960) of the European Union 6th Framework Programme (FP6)
References
- Alzari P.M., Berglund H., Berrow N.S., Blagova E., Busso D., Cambillau C., Campanacci V., Christodoulou E., Eiler S., Fogg M.J., Folkers G., Geerlof A., Hart D., Haouz A., Herman M.D., Macieira S., Nordlund P., Perrakis A., Quevillon-Cheruel S., Tarandeau F., van Tilbeurgh H., Unger T., Luna-Vargas M.P., Velarde M., Willmanns M., Owens R.J. Implementation of semi-automated cloning and prokaryotic expression screening: the impact of SPINE. Acta Crystallogr. D: Biol. Crystallogr. 2006;62:1103–1113. doi: 10.1107/S0907444906029775. [DOI] [PubMed] [Google Scholar]
- Berrow N.S., Bussow K., Coutard B., Diprose J., Ekberg M., Folkers G.E., Levy N., Lieu V., Owens R.J., Peleg Y., Pinaglia C., Quevillon-Cheruel S., Salim L., Scheich C., Vincentelli R., Busso D. Recombinant protein expression and solubility screening in Escherichia coli: a comparative study. Acta Crystallogr. D: Biol. Crystallogr. 2006;62:1218–1226. doi: 10.1107/S0907444906031337. [DOI] [PubMed] [Google Scholar]
- Berrow N.S., Alderton D., Sainsbury S., Nettleship J., Assenberg R., Rahman N., Stuart D.I., Owens R.J. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 2007;35:e45. doi: 10.1093/nar/gkm047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornvik T., Dahlroth S.L., Magnusdottir A., Herman M.D., Knaust R., Ekberg M., Nordlund P. Colony filtration blot: a new screening method for soluble protein expression in Escherichia coli. Nat. Methods. 2005;2:507–509. doi: 10.1038/nmeth767. [DOI] [PubMed] [Google Scholar]
- De Colibus L., Speroni S., Coutard B., Forrester N.L., Gould E., Canard B., Mattevi A. Purification and crystallization of Kokobera virus helicase. Acta Crystallograph. Sect. F: Struct. Biol. Cryst. Commun. 2007;63:193–195. doi: 10.1107/S1744309107005283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma A.M., Heggermont W., Leyssen P., Purstinger G., Wimmer E., De Clercq E., Rao A., Monforte A.M., Chimirri A., Neyts J. Anti-enterovirus activity and structure-activity relationship of a series of 2,6-dihalophenyl-substituted 1H,3H-thiazolo[3,4-a]benzimidazoles. Biochem. Biophys. Res. Commun. 2007;353:628–632. doi: 10.1016/j.bbrc.2006.12.063. [DOI] [PubMed] [Google Scholar]
- Egloff M.P., Benarroch D., Selisko B., Romette J.L., Canard B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M.P., Malet H., Putics A., Heinonen M., Dutartre H., Frangeul A., Gruez A., Campanacci V., Cambillau C., Ziebuhr J., Ahola T., Canard B. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macrodomains. J. Virol. 2006;80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emonet S.F., Grard G., Brisbarre N.M., Moureau G.N., Temmam S., Charrel R.N., de Lamballerie X. Long PCR product sequencing (LoPPS): a shotgun-based approach to sequence long PCR products. Nat. Protoc. 2007;2:340–346. doi: 10.1038/nprot.2006.453. [DOI] [PubMed] [Google Scholar]
- Ferron F., Rancurel C., Longhi S., Cambillau C., Henrissat B., Canard B. VaZyMolO: a tool to define and classify modularity in viral proteins. J. Gen. Virol. 2005;86:743–749. doi: 10.1099/vir.0.80590-0. [DOI] [PubMed] [Google Scholar]
- Fogg M.J., Alzari P., Bahar M., Bertini I., Betton J.M., Burmeister W.P., Cambillau C., Canard B., Corrondo M.A., Coll M., Daenke S., Dym O., Egloff M.P., Enguita F.J., Geerlof A., Haouz A., Jones T.A., Ma Q., Manicka S.N., Migliardi M., Nordlund P., Owens R.J., Peleg Y., Schneider G., Schnell R., Stuart D.I., Tarbouriech N., Unge T., Wilkinson A.J., Wilmanns M., Wilson K.S., Zimhony O., Grimes J.M. Application of the use of high-throughput technologies to the determination of protein structures of bacterial and viral pathogens. Acta Crystallogr. D: Biol. Crystallogr. 2006;62:1196–1207. doi: 10.1107/S0907444906030915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grard G., Lemasson J.J., Sylla M., Dubot A., Cook S., Molez J.F., Pourrut X., Charrel R., Gonzalez J.P., Munderloh U., Holmes E.C., de Lamballerie X. Ngoye virus: a novel evolutionary lineage within the genus flavivirus. J. Gen. Virol. 2006;87:3273–3277. doi: 10.1099/vir.0.82071-0. [DOI] [PubMed] [Google Scholar]
- Harris M., Jones T.A. Xtrack—a web-based crystallographic notebook. Acta Crystallogr. D: Biol Crystallogr. 2002;58:1889–1891. doi: 10.1107/s0907444902012696. [DOI] [PubMed] [Google Scholar]
- Hassaine G., Wagner R., Kempf J., Cherouati N., Hassaine N., Prual C., Andre N., Reinhart C., Pattus F., Lundstrom K. Semliki Forest virus vectors for overexpression of 101 G protein-coupled receptors in mammalian host cells. Protein Expr. Purif. 2006;45:343–351. doi: 10.1016/j.pep.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Jabafi I., Selisko B., Coutard B., De Palma A.M., Neyts J., Egloff M.P., Grisel S., Dalle K., Campanacci V., Spinelli S., Cambillau C., Canard B., Gruez A. Improved crystallization of the coxsackievirus B3 RNA-dependent RNA polymerase. Acta Crystallograph. Sect. F: Struct. Biol. Cryst. Commun. 2007;63:495–498. doi: 10.1107/S1744309107020416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P., Faase J.A., Toyoda H., Paul A., Wimmer E., Gorbalenya A.E. Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc. Natl. Acad. Sci. U.S.A. 2007;104:9457–9462. doi: 10.1073/pnas.0700451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzhkov V.B., Selisko B., Nordqvist A., Peyrane F., Decroly E., Alvarez K., Karlen A., Canard B., Qvist J. Virtual screening and bioassay study of novel inhibitors for dengue virus mRNA cap (nucleoside-2’O)-methyltransferase. Bioorg Med Chem. 2007;15:7795–7802. doi: 10.1016/j.bmc.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Malet H., Egloff M.P., Selisko B., Butcher R.E., Wright P.J., Roberts M., Gruez A., Sulzenbacher G., Vonrhein C., Bricogne G., Mackenzie J.M., Khromykh A.A., Davidson A.D., Canard B. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J. Biol. Chem. 2007;282:10678–10689. doi: 10.1074/jbc.M607273200. [DOI] [PubMed] [Google Scholar]
- Mastrangelo E., Bollati M., Milani M., de Lamballerie X., Brisbarre N., Dalle K., Lantez V., Egloff M.P., Coutard B., Canard B., Gould E., Forrester N., Bolognesi M. Preliminary characterization of (nucleoside-2′-O-)-methyltransferase crystals from Meaban and Yokose flaviviruses. Acta Crystallograph. Sect. F: Struct. Biol. Cryst. Commun. 2006;62:768–770. doi: 10.1107/S1744309106025553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrangelo E., Bollati M., Milani M., Brisbarre N., de Lamballerie X., Coutard B., Canard B., Khromykh A., Bolognesi M. Preliminary crystallographic characterization of an RNA helicase from Kunjin virus. Acta Crystallograph. Sect. F: Struct. Biol. Cryst. Commun. 2006;62:876–879. doi: 10.1107/S1744309106028776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrane F., Selisko B., Decroly E., Vasseur J.J., Benarroch D., Canard B., Alvarez K. High-yield production of short GpppA- and 7MeGpppA-capped RNAs and HPLC-monitoring of methyltransfer reactions at the guanine-N7 and adenosine-2′O positions. Nucleic Acids Res. 2007;35:e26. doi: 10.1093/nar/gkl1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath A., Xu T., Chao A., Luo D., Lescar J., Vasudevan S.G. Structure-based mutational analysis of the NS3 helicase from dengue virus. J. Virol. 2006;80:6686–6690. doi: 10.1128/JVI.02215-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzenbacher G., Gruez A., Roig-Zamboni V., Spinelli S., Valencia C., Pagot F., Vincentelli R., Bignon C., Salomoni A., Grisel S., Maurin D., Huyghe C., Johansson K., Grassick A., Roussel A., Bourne Y., Perrier S., Miallau L., Cantau P., Blanc E., Genevois M., Grossi A., Zenatti A., Campanacci V., Cambillau C. A medium-throughput crystallization approach. Acta Crystallogr. D: Biol. Crystallogr. 2002;58:2109–2115. doi: 10.1107/s0907444902013938. [DOI] [PubMed] [Google Scholar]
- Vedadi M., Niesen F.H., Allali-Hassani A., Fedorov O.Y., Finerty, Wasney G.A., Yeung R., Arrowsmith C., Ball L.J., Berglund H., Hui R., Marsden B.D., Nordlund P., Sundstrom M., Weigelt J., Edwards A.M. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U.S.A. 2006;103:15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Bera A.K., Kuhn R.J., Smith J.L. Structure of the flavivirus helicase: implications for catalytic activity, protein interactions, and proteolytic processing. J. Virol. 2005;79:10268–10277. doi: 10.1128/JVI.79.16.10268-10277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T., Sampath A., Chao A., Wen D., Nanao M., Chene P., Vasudevan S.G., Lescar J. Structure of the Dengue virus helicase/nucleoside triphosphatase catalytic domain at a resolution of 2.4 A. J Virol. 2005;79:10278–10288. doi: 10.1128/JVI.79.16.10278-10288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap T.L., Xu T., Chen Y.L., Malet H., Egloff M.P., Canard B., Vasudevan S.G., Lescar J. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 2007;81:4753–4765. doi: 10.1128/JVI.02283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]