

Abstract
Because outbreaks of severe acute respiratory syndrome coronavirus (SARS-CoV) might reemerge, identifying antiviral compounds is of key importance. Previously, we showed that the cellular factor TNF-α converting enzyme (TACE), activated by the spike protein of SARS-CoV (SARS-S protein), was positively involved in viral entry, implying that TACE is a possible target for developing antiviral compounds. To demonstrate this possibility, we here tested the effects of TACE inhibitors on viral entry. In vitro and in vivo data revealed that the TACE inhibitor TAPI-2 attenuated entry of both pseudotyped virus expressing the SARS-S protein in a lentiviral vector backbone and infectious SARS-CoV. TAPI-2 blocked both the SARS-S protein-induced shedding of angiotensin-converting enzyme 2 (ACE2), a receptor of SARS-CoV, and TNF-α production in lung tissues. Since the downregulation of ACE2 by SARS-S protein was proposed as an etiological event in the severe clinical manifestations, our data suggest that TACE antagonists block SARS-CoV infection and also attenuate its severe clinical outcome.
Keywords: SARS-CoV, TACE, ACE2, Shedding
During 2002–2003, an outbreak of severe acute respiratory syndrome coronavirus (SARS-CoV) infection spreads quickly from South China to more than 25 countries worldwide (WHO; http://www.who.int/csr/don/2003_04_17/en/). About 8000 individuals were infected and the mortality rate was 10% (Ksiazek et al., 2003). In 2003, angiotensin-converting enzyme 2 (ACE2) was identified as a cellular receptor of SARS-CoV (Li et al., 2003), and the binding of SARS-CoV spike protein (SARS-S protein) and ACE2 was clearly shown to be a required initial step for viral entry (Li et al., 2003, Wong et al., 2004). Based on an analysis of the molecular interaction between ACE2 and SARS-S protein and the subsequent process of viral entry (Kuhn et al., 2007), several targets are now available for developing antiviral compounds (Liu et al., 2004, Simmons et al., 2005, Sui et al., 2004). Since recurrent outbreaks of SARS-CoV can reemerge, developing compounds that will prevent SARS-CoV are critically important.
Previously, we found that TNF-α converting enzyme (TACE) was required for viral entry (Haga et al., 2008). TACE, originally identified as a metalloprotease required for processing the membrane form of TNF-α and its release as a soluble factor (Black et al., 1997), is involved in the cleavage and release of the ACE2 ectodomain (Lambert et al., 2005). TACE-dependent shedding of ACE2 is completely dependent on the binding of SARS-S protein to ACE2 (Haga et al., 2008). In addition, siRNA experiments demonstrated that TACE was required for efficient viral entry (Haga et al., 2008), implying that TACE might be a target for antiviral compounds against SARS-CoV. Since the downregulation of ACE2 caused by the SARS-S protein was postulated as being correlated with the severe respiratory failure (Kuba et al., 2005), TACE inhibitors might also attenuate the tissue damage caused by the viral infection. Therefore, we analyzed the antiviral activities of TAPI-0 and TAPI-2, hydroxamate-based TACE inhibitors (Mohler et al., 1994, Black et al., 1997).
First, we studied the effects of TACE inhibitors on ACE2 shedding after the binding of the SARS-S protein. Western blot analysis detected the shed form of the ACE2 ectodomain, which was about 80 kDa in size, in culture supernatants after treatment with the SARS-S protein (Fig. 1A, lanes 1–3). When cells were pretreated with 100 nM TAPI-0 (Fig. 1A, lanes 4 and 5) or 200 nM TAPI-2 (lanes 6 and 7) for 1 h, the SARS-S-induced ACE2 shedding was attenuated significantly. Since a soluble form of ACE2 released from the cell membrane has catalytic activity (Tipnis et al., 2000), we measured the carboxy-monopeptidase activity in the culture supernatants. Although the ACE2 activity increased after the SARS-S protein treatment (Fig. 1B–E), pretreatment with TAPI-0 and TAPI-2 decreased the activity to 40% (p < 0.01; Fig. 1B and C) and 80% (p < 0.05; Fig. 1 and 1) of the control, respectively.
Fig. 1.
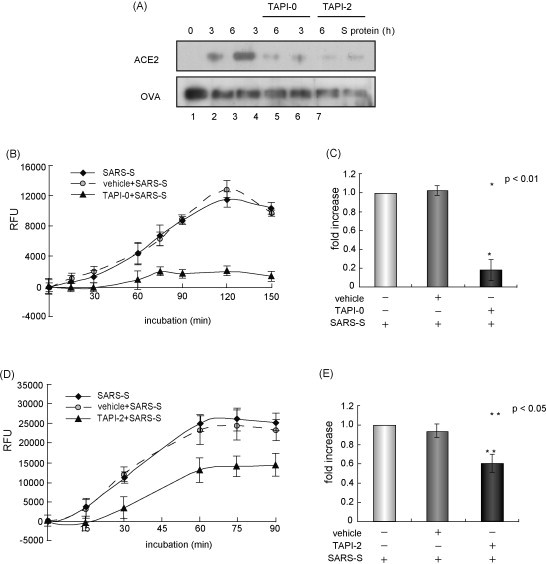
ACE2 shedding is inhibited by TACE inhibitors. (A) TACE inhibitors blocked SARS-S protein-induced processing of ACE2. Vero E6 cells were treated with two TACE inhibitors, TAPI-0 (100 nM, BIOMOL International, Plymouth Meeting, PA) or TAPI-2 (200 nM, BIOMOL International) for 1 h, and 100 μg/ml SARS-S protein (Haga et al., 2008) was added for 3 and 6 h. The proteins present in the culture supernatants were recovered by coprecipitation with ovalbumin (OVA) in trichloroacetic acid, and subjected to Western blot analysis, as previously described (Haga et al., 2008). Lane 1, control without the addition of SARS-S protein; lanes 2 and 3, saline with SARS-S protein; lanes 4 and 5, TAPI-0 with SARS-S protein; lanes 6 and 7, TAPI-2 with SARS-S protein. The culture supernatants were collected after 3 h (lanes 2, 4 and 6) or 6 h (lanes 3, 5 and 7). (B and C) TAPI-0 inhibits the release of ACE2 activity into the culture supernatants. The ACE2 activity was measured in the supernatants of Vero E6 cells cultured with or without 100 nM TAPI-0. The horizontal axis shows the incubation time of a fluorescence substrate of ACE2 (Mca-Y-V-A-D-A-P-K-Dnp-OH, amino acid depicted by single letters; R&D Systems, Minneapolis, MN) (B). The relative rate of inhibition calculated based on the data shown in (B) at 90 min (C). (D and E) TAPI-2 inhibits the release of ACE2 activity into the culture supernatants. The experiment was carried out by the same procedures as described in (B) and (C). TAPI-2 was used at a concentration of 200 nM.
To further show that the SARS-S protein induced TACE activation, we measured the TACE activity in cellular extracts. Time-course analysis of the TACE activity revealed that the addition of the SARS-S protein gradually increased the TACE activity in the cellular extracts (Fig. 2A). Six hours after adding the SARS-S protein, the TACE activity had increased significantly (p < 0.05; Fig. 2B). However, the addition of TAPI-2 blocked the increase in TACE activity upregulated by the SARS-S protein (Figs. 2A and B; p < 0.01). These data indicated that the shedding of the ACE2 ectodomain induced by SARS-S protein was coupled with the TACE activity, as previously proposed (Haga et al., 2008).
Fig. 2.
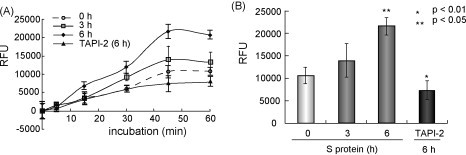
TACE inhibitors block the SARS-S protein-induced TACE activity. HuH-7 cells were treated with TAPI-2 (200 nM) for 1 h and then SARS-S protein (100 μg/ml) was added. After 3 or 6 h, cellular extracts were prepared and incubated with a fluorescence substrate of TACE (Mca-P-L-A-Q-A-V-Dpa-R-S-S-S-R-NH2, amino acid depicted by single letters; BIOMOL International, Plymouth Meeting, PA). The TACE activity was measured every 15 min after adding the substrate. (A) Time course of the TACE activity. The fluorescent intensity of cleaved substrate was monitored in extracts of cells treated with or without TAPI-2. After 45 min, a difference was detected in the TACE activity of the control sample and the sample treated with TAPI-2. (B) TAPI-2 blocked the TACE activity. TACE activity was detected 6 h after adding SARS-S protein, and this was blocked completely by pretreatment with TAPI-2 (p < 0.01).
Next, we examined the effects of TACE inhibitors on the viral infection. We first used a pseudotyped virus that expressed SARS-S protein in the core structure of a lentiviral vector (SARS-S virus), and measured intracellular p24, a gag protein of HIV-1, in the tested cells. In our previous observation, results obtained by measuring p24 were consistent with those measured using real-time RT-PCR of SARS-CoV mRNA (Haga et al., 2008). A plasmid expressing ACE2 was introduced into HEK293T cells and cell extracts were prepared after 4 h of viral infection (Fig. 3A). Treatment with two TACE inhibitors, TAPI-0 (100 nM) and TAPI-2 (100 and 200 nM), 1 h before infection with SARS-S virus, attenuated viral entry significantly (p < 0.01 for TAPI-0 and p < 0.05 for TAPI-2, shown in Fig. 3A and B, respectively). We next examined the inhibitory effects of TAPI-2 on viral entry of the infectious Frankfurt strain of SARS-CoV, and detected the marked reduction of the viral infection by the compound (Fig. 3C).
Fig. 3.
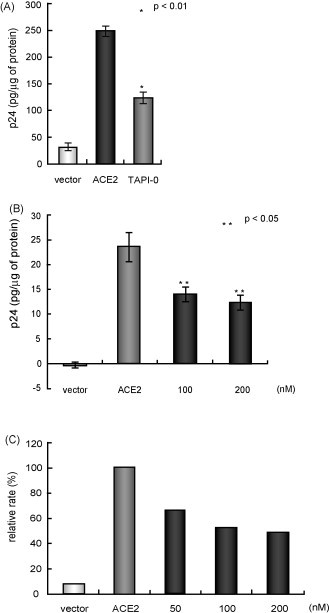
Effects of TACE inhibitors on viral entry in vitro. (A and B) TACE inhibitors suppressed SARS-S protein-dependent viral entry. Plasmid DNA encoding ACE2 was introduced into HEK293T cells, and then the cells were infected with the virus. The cells were treated with TAPI-0 (A) or TAPI-2 (B) for 1 h and then infected with the SARS-S pseudotyped lentivirus (Invitrogen, Carlsbad, CA) for 4 h. The intracellular p24 was measured using a Retro-Tek HIV-1 p24 ELISA kit (ZeptoMetrix, Buffalo, NY) according to the manufacturer's instructions. (C) TAPI-2 blocked the entry of infectious SARS-CoV. The viral infection efficiency was measured using real-time RT-PCR of viral mRNA, as described (Haga et al., 2008). Test samples were harvested and analyzed 4 h after infection with SARS-CoV. The data were normalized using 18S ribosomal RNA. In this experiment, TAPI-2 was used because TAPI-2 is water soluble, whereas TAPI-0 is water insoluble. TAPI-2 did not require control samples treated with of dimethylsulfoxide (DMSO), a solvent of TAPI-0.
The in vivo effects of TACE inhibitors on viral entry were further examined by using the SARS-S pseudotyped virus. After 4 h of intratracheal administration of 25 μl of viral solution (4 μg/ml p24), lung tissues were refluxed with 50 ml of phosphate-based buffer with 50 μg/ml of trypsin and excised. To measure intracellular p24, proteins of the lung tissues were extracted using a homogenizer and subjected to an enzyme-linked immunosorbent assay (ELISA). As shown in Fig. 4A, pretreatment of TAPI-0 and TAPI-2 at 2 h before the viral challenge significantly decreased the intracellular p24 (left panel for TAPI-0 and right panel for TAPI-2, respectively). Moreover, the TAPI-2 treatment also reduced the amount of soluble TNF-α content generated by the viral infection (p < 0.05; Fig. 4B).
Fig. 4.
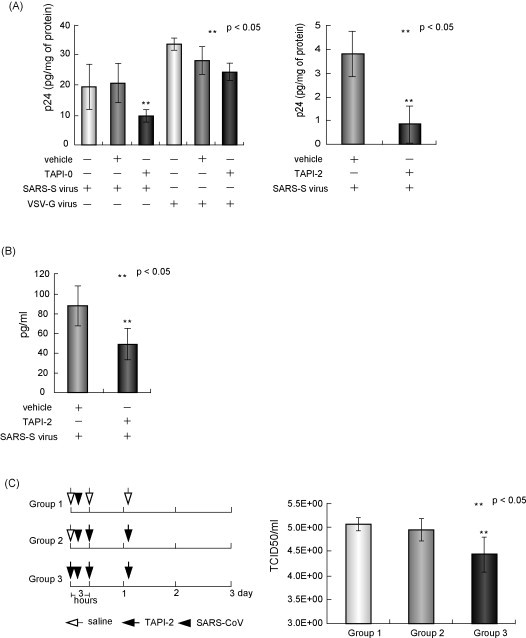
TACE inhibitors attenuated SARS-S protein-dependent viral entry in vivo. (A) Inhibitory effects of TAPI-0 and TAPI-2 on viral entry. Left panel: male C57BL/6j mice (5–7-week old, A, left panel) or female Balb/c mice (28–32-week old) were inoculated intratracheally with 25 μl of a solution of the SARS-S pseudotyped lentivirus (4 μg/ml p24). To test the effects of TAPI-0, 25 μl of 0.3 μM of the compound dissolved in 0.0075% of DMSO or the same amount of diluted DMSO was administered intratracheally 2 h before the viral challenge (n = 3 per group, respectively). As additional control, the effects on the viral entry of VSV-G pseudotyped lentivirus were also examined. After 4 h, lung tissues were first refluxed with 50 ml of phosphate-based buffer, 50 ml of phosphate-based buffer containing 500 μg/ml collagenase, and 50 ml of phosphate-buffered saline containing 50 μg/ml trypsin, and then extracted. Extracted p24 was measured using a p24 ELISA kit. Right panel: For testing the TAPI-2, 25 μl of 2 μM of the compound in water or the same volume of distilled water was administered by the same method (right panel, n = 6 per group). We measured intracellular p24 by the similar procedures. (B) Effect of TAPI-2 on the production of TNF-α. After viral infection with TAPI-2 or water as control, lung tissues, prepared without the refluxing using collagenase and trypsin, were subjected to the analysis of ELISA of TNF-α (n = 4 per group) (C) Effect of TAPI-2 on the viral infection of the infectious SARS-CoV. Left panel: Protocol for SARS-CoV inoculation and TAPI-2 treatment. In this experiment, we used TAPI-2 because it is water soluble. Although TAPI-0 seemed to be more effective than TAPI-2, TAPI-2 was better for in vivo experiments, because it was not necessary to include additional control groups treated with DMSO, a solvent of TAPI-0. Seven-week-old female Balb/c mice were pretreated with 20 μl of 2 μM of TAPI-2 or saline via intranasal injection under anesthesia. Soon afterward, these mice were infected with 20 μl of 106.0 TCID50 mouse-passaged Frankfurt isolate of SARS-CoV (Nagata et al., 2008) via intranasal inoculation. Three hours and 1 day after virus inoculation, animals were treated with 20 μl of TAPI-2 or saline via intranasal injection under anesthesia. Right panel: Virus titers in the lung lavage fluids 3 days after inoculation with saline or TAPI-2 (n = 4 per group). Balb/c mice on the virus titers in the lung lavage fluids after inoculation. The detection limit was 101.5 CCID50/ml of fluid.
Next, we tested the antiviral effects of TAPI-2 on infectious SARS-CoV challenged on adult female Balb/c mice. These animal experiments were approved by the Animal Care and Use Committee of the National Institute of Infectious Diseases, Tokyo, Japan, and work with infectious SARS-CoV was performed under biosafety level 3 conditions. The experimental protocol is shown in Fig. 4C (left panel). In this experiment, we used TAPI-2 instead of TAPI-0 because TAPI-2 is water soluble (see also legends for Fig. 4C). In group 2, TAPI-2 was administered twice after the viral challenge. In group 3, TAPI-2 was administered totally three times with an additional pretreatment just before the viral inoculation. As control, saline was administered under the same conditions (group 1). On day 3 after the viral challenge, lung lavage was performed and viral titers were measured. As shown in Fig. 4C (right panel), the triple administration of TAPI-2 significantly reduced the viral titer (p < 0.05).
Here, we showed evidence that TACE is a target candidate of anti-SARS-CoV agents. Our data revealed that nM levels of TAPIs reproducibly inhibited the viral entry in vitro. Additionally, TAPI-2 partially blocked the viral infection of SARS-CoV in vivo. Previously, we reported that TACE is a cellular factor involved in an efficient virus entry (Haga et al., 2008). Experiments using a cell line from embryonal fibroblasts of TACE-knockout mice revealed that infection by the SARS-S virus was strictly dependent on the exogenously transduced TACE cDNA. In addition, TACE siRNA attenuated the entry of both SARS-S pseudotyped virus and infectious SARS-CoV. Our current data are consistent with the previous report.
Although TAPI-2 effectively suppressed viral entry of the SARS-S pseudotyped virus (Fig. 4A, right panel) and infectious SARS-CoV (Fig. 3C) in vitro, its inhibitory effect was not drastic in vivo (Fig. 4C). As one possible explanation, TACE might not be a single target for complete suppression of viral infection. Work done by independent research groups has shown that SARS-CoV uses DC-SIGN (dendritic cell-specific ICAM-3 grabbing nonintegrin) and L (liver/lymph node-specific)-SIGN (DC/L-SIGN) as additional cellular receptors (Han et al., 2007, Jeffers et al., 2004, Shih et al., 2006). It was further proposed that the mode of the viral entry via these molecules was different from that of ACE2 (Han et al., 2007). These observations lend support to the notion that the combined administration of antagonists against TACE and DC/L-SIGN would lead to more remarkable inhibition on the viral infection of SARS-CoV. Another possibility is that complete blockage of virus entry might be required for efficient suppression of viral production. It seems that viral replication of SARS-CoV more crucially determines the whole amount of the virus produced in vivo. Even a single particle of virus, once infected, can vigorously replicate in vivo and overcome the subtle inhibitory effects of the compounds on viral entry, implicating the importance to develop more potent antagonists. Recently, Lu et al. (2008) identified a specific inhibitor of TACE having an IC50 of 1 nM against porcine TACE. It is plausible to speculate that such compound would suppress SARS-CoV infection more effectively than currently used TACE antagonists. The IC50 of TAPI-0, which inhibited viral entry more efficiently than TAPI-2, is about 100 nM.
Our data revealed that anti-TACE compounds also attenuated the production of TNF-α after viral infection, suggesting that the compounds might effectively protect patients from clinical manifestations of the disease. TACE, a member of the ADAM (a disintegrin and metalloprotease) family involved in a variety of biological functions, is a target for drug development in various diseases (Seals and Courtneidge, 2003).
Acknowledgment
This work was supported by Grants-in Aid for Research from the National Institute of Biomedical Innovation, and partly from the Naito Foundation.
References
- Black R.A., Rauch C.T., Kozlosky C.J., Peschon J.J., Slack J.L., Wolfson M.F., Castner B.J., Stocking K.L., Reddy P., Srinivasan S., Nelson N., Boiani N., Schooley K.A., Gerhart M., Davis R., Fitzner J.N., Johnson R.S., Paxton R.J., March C.J., Cerretti D.P. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Haga S., Yamamoto N., Nakai-Murakami C., Osawa Y., Tokunaga K., Sata T., Yamamoto N., Sasazuki T., Ishizaka Y. Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7809–7814. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D.P., Lohani M., Cho M.W. Specific asparagine-linked glycosylation sites are critical for DC-SIGN- and L-SIGN-mediated severe acute respiratory syndrome coronavirus entry. J. Virol. 2007;81:12029–12039. doi: 10.1128/JVI.00315-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers S.A., Tusell S.M., Gillim-Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Jr., Thackray L.B., Young M.D., Mason R.J., Ambrosino D.M., Wentworth D.E., Demartini J.C., Holmes K.V. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J., SARS Working Group A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W., Bao L., Zhang B., Liu G., Wang Z., Chappell M., Liu Y., Zheng D., Leibbrandt A., Wada T., Slutsky A.S., Liu D., Qin C., Jiang C., Penninger J.M. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn J.H., Li W., Radoshitzky S.R., Choe H., Farzan M. Severe acute respiratory syndrome coronavirus entry as a target of antiviral therapies. Antivir. Ther. 2007;12:639–650. [PubMed] [Google Scholar]
- Lambert D.W., Yarski M., Warner F.J., Thornhill P., Parkin E.T., Smith A.I., Hooper N.M., Turner A.J. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2) J. Biol. Chem. 2005;280:30113–30119. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C.R., Xiong H., Farmar J., Debnath A.K., Tien P., Jiang S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Ott G.R., Anand R., Liu R.Q., Covington M.B., Vaddi K., Qian M., Newton R.C., Christ D.D., Trzaskos J., Duan J.J. Potent, selective, orally bioavailable inhibitors of tumor necrosis factor-α converting enzyme (TACE): discovery of indole, benzofuran, imidazopyridine and pyrazolopyridine P1’ substituents. Bioorg. Med. Chem. Lett. 2008;18:1958–1962. doi: 10.1016/j.bmcl.2008.01.120. [DOI] [PubMed] [Google Scholar]
- Mohler K.M., Sleath P.R., Fitzner J.N., Cerretti D.P., Alderson M., Kerwar S.S., Torrance D.S., Otten-Evans C., Greenstreet T., Weerawarna K., Kronheim S.R., Petersen M., Gerhart M., Kozlosky C.J., March C.J., Black R.A. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature. 1994;370:218–220. doi: 10.1038/370218a0. [DOI] [PubMed] [Google Scholar]
- Nagata N., Iwata N., Hasegawa H., Fukushi S., Harashima A., Sato Y., Saijo M., Taguchi F., Morikawa S., Sata T. Mouse-passaged severe acute respiratory syndrome-associated coronavirus leads to lethal pulmonary edema and diffuse alveolar damage in adult but not young mice. Am. J. Pathol. 2008;172:1625–1637. doi: 10.2353/ajpath.2008.071060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals D.F., Courtneidge S.A. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17:7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- Shih Y.P., Chen C.Y., Liu S.J., Chen K.H., Lee Y.M., Chao Y.C., Chen Y.M. Identifying epitopes responsible for neutralizing antibody and DC-SIGN binding on the spike glycoprotein of the severe acute respiratory syndrome coronavirus. J. Virol. 2006;80:10315–10324. doi: 10.1128/JVI.01138-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui J., Li W., Murakami A., Tamin A., Matthews L.J., Wong S.K., Moore M.J., Tallarico A.S., Olurinde M., Choe H., Anderson L.J., Bellini W.J., Farzan M., Marasco W.A. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. U.S.A. 2004;101:2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- WHO, website, http://www.who.int/csr/don/2003_04_17/en/.
- Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]