

Abstract
Recent advances in cell reprogramming have enabled assessment of disease‐related cellular traits in patient‐derived somatic cells, thus providing a versatile platform for disease modeling and drug development. Given the limited access to vital human brain cells, this technology is especially relevant for neurodegenerative disorders such as Parkinson's disease (PD) as a tool to decipher underlying pathomechanisms. Importantly, recent progress in genome‐editing technologies has provided an ability to analyze isogenic induced pluripotent stem cell (iPSC) pairs that differ only in a single genetic change, thus allowing a thorough assessment of the molecular and cellular phenotypes that result from monogenetic risk factors. In this review, we summarize the current state of iPSC‐based modeling of PD with a focus on leucine‐rich repeat kinase 2 (LRRK2), one of the most prominent monogenetic risk factors for PD linked to both familial and idiopathic forms. The LRRK2 protein is a primarily cytosolic multi‐domain protein contributing to regulation of several pathways including autophagy, mitochondrial function, vesicle transport, nuclear architecture and cell morphology. We summarize iPSC‐based studies that contributed to improving our understanding of the function of LRRK2 and its variants in the context of PD etiopathology. These data, along with results obtained in our own studies, underscore the multifaceted role of LRRK2 in regulating cellular homeostasis on several levels, including proteostasis, mitochondrial dynamics and regulation of the cytoskeleton. Finally, we expound advantages and limitations of reprogramming technologies for disease modeling and drug development and provide an outlook on future challenges and expectations offered by this exciting technology.
Keywords: disease modeling, iPSC, LRRK2, mitophagy, Parkinson's disease
Reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) followed by differentiation has enabled comparison of pathophenotypes in affected cell types. Focusing on the Parkinson's disease‐linked leucine‐rich repeat kinase 2 (LRRK2), we summarize how iPSC models help revealing the role of LRRK2 variants for pathologically relevant pathways such as proteostasis, mitochondrial homeostasis and cytoskeleton dynamics. Future prospects and limitations of the technology are discussed.
Abbreviations
- ALDH
aldehyde dehydrogenase
- ANK
ankyrin
- ARM
armadillo
- ASCL1
achaete‐scute homolog 1
- ASK1
apoptosis signal‐regulating kinase 1
- ATG
autophagy‐related
- ATP
adenosine triphosphate
- Ca2+
calcium
- CaV channels
voltage‐gated Ca2+ channels
- c‐casp3
cleaved‐caspase 3
- CMA
chaperone‐mediated autophagy
- CNS
central nervous system
- COPII
coat protein complex II
- COR
C‐terminus of ROC
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
- c‐Myc
avian myelocytomatosis virus oncogene cellular homolog
- DA
dopamine
- DAT
dopamine transporter
- DRP1
dynamin‐related protein 1
- EM
electron microscopy
- ER
endoplasmic reticulum
- ERK
extracellular signal‐regulated kinase
- FACS
fluorescence‐activated cell sorting
- FAK
focal adhesion kinase 1
- FOXA2
forkhead box protein A2
- GA
Golgi apparatus
- GAK
cyclin‐G‐associated kinase
- GBA
glucosylceramidase beta
- GC
glucocerebrosidase
- GD
Gaucher's disease
- GSK3‐β
glycogen synthase kinase 3 beta
- GWAS
genome‐wide association study
- HD
Huntington's disease
- hESCs
human embryonic stem cells
- H2O2
hydrogen peroxide
- iDA
induced dopamine neurons
- IL
interleukin
- iMSN
induced medium spiny neurons
- iN
induced neuron
- IN‐1
LRRK2 kinase inhibitor‐1
- iNSCs
induced neural stem cells
- IFN
interferon
- iPD
idiopathic PD
- iPSC
induced pluripotent stem cell
- K‐ATP channel
ATP‐sensitive potassium channel
- Klf4
Kruppel‐like factor 4
- KO
knock out
- LAMP1
lysosomal‐associated membrane protein 1
- LAMP2A
lysosomal‐associated membrane protein 2A
- LAP2α
lamina‐associated polypeptide 2α
- LMX1A/B
LIM homeobox transcription factor 1 A and B
- LRR
leucine‐rich repeat
- LRRK2
leucine‐rich repeat kinase 2
- LRS
leucyl‐tRNA synthetase
- MACS
magnetic‐activated cell sorting
- mesDA neurons
mesencephalic dopaminergic neurons
- MEA
multi‐electrode array
- MFN1
mitofusin1
- MLOs
midbrain‐like organoids
- MPP+
1‐methyl‐4‐phenyl‐4‐dihydropyridine
- MPTP
1‐methyl‐4‐phenyl‐1‐2‐3‐6‐tetrahydropyridine
- mTOR
mammalian target of rapamycin
- MYD88
myeloid differentiation primary response protein 88
- MYT1L
myelin transcription factor 1 like
- NAC
N‐acetyl cysteine
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NAD
nicotinamide adenine dinucleotide
- NF‐κB
nuclear factor‐kappa B
- NFAT
nuclear factor of activated T cells
- NEUROD1
neurogenic differentiation 1
- NEUROG2
neurogenin 2
- NM
neuromelanin
- NPC
neural progenitor cell
- NRXN1
neurexin 1
- NSC
neural stem cell
- NURR1
nuclear receptor related‐1 protein
- OCR
oxygen consumption rate
- Oct4
octamer‐binding transcription factor 4
- OPA1
mitochondrial dynamin like GTPase
- OSKM
Oct3/4, Sox2 Klf4 and c‐Myc
- OTX2
orthodenticle homeobox 2
- PAK6
p21 RAC-activated kinase 6
- PAMPs
pathogen‐associated molecular patterns
- PD
Parkinson's disease
- PINK1
PTEN‐induced putative kinase 1
- POU3F2
POU class 3 homeobox 2
- PRKN
parkin RBR E3 ubiquitin protein ligase
- PSC
pluripotent stem cell
- Rab
Ras‐related in brain
- RILPL
Rab‐interacting lysosomal protein like
- ROC
Ras of complex proteins
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SEC16A
protein transport protein Sec 16A
- SHANK3
SH and multiple ankyrin repeat domains 3
- SHH
sonic hedgehog
- SN
substantia nigra
- SNCA
synuclein alpha
- SNP
single nucleotide polymorphism
- SNpc
substantia nigra pars compacta
- SNV
single nucleotide variant
- Sox2
sex‐determining region Y‐box 2
- SPN
spiny projection neurons
- SVE
synaptic vesicle endocytosis
- TALEN
transcription activator‐like effector nucleases
- TH
tyrosine hydroxylase
- TLR
Toll‐like receptor
- TNF
tumor necrosis factor
- ULK1
Unc‐51 like autophagy activating kinase 1
- VPS35
Retromer Complex Component
- VTA
ventral tegemental area
- ZFN
zinc‐finger nucleases
- α‐syn
α‐synuclein
- 6‐OHDA
6‐hydroxydopamine
1. THE ERA OF REPROGRAMMING TECHNOLOGIES
For centuries, developmental biologists considered cell fate decisions that is the development of specialized, differentiated cells from less differentiated precursors, to be unidirectional and irreversible. Only embryonic cells were believed to possess the capacity to give rise to all three germ layers (ectoderm, endoderm and mesoderm) and the more than 200 cell types that make up the human body. This paradigm was not only challenged but also negated with the advent of transcription factor‐based cell reprogramming only one decade ago (Takahashi & Yamanaka, 2006). With a number of elegant transfection experiments, the authors could demonstrate that ectopic expression of a defined set of transcription factors (octamer‐binding transcription factor 4 [Oct4], sex‐determining region Y‐box 2 [Sox2], Krüppel‐like factor 4 [Klf4], avian myelocytomatosis virus oncogene cellular homolog [c‐Myc], collectively referred to as ‘Yamanaka factors’ and commonly abbreviated with ‘OSKM’) is capable to rewire the gene regulatory networks that define a specific cell fate and to put adult mouse fibroblasts back into an embryonic‐like state. Only one year later, the same group proved general applicability of this approach by translating the experiments to human fibroblasts (Takahashi et al., 2007). These rewired cells (referred to as ‘induced pluripotent stem cells’, iPSCs) are highly similar to human embryonic stem cells (hESCs), can self‐renew while maintaining their identity as stem cells and are able to generate differentiated progeny of all germ layers, including cardiomyocytes, hepatocytes, blood cells, glial cells and neurons. Cells generated that way thus reflect the genotype of the donor and the identity of tissue‐specific cells, making them an attractive resource for studying patient‐, disease‐ and cell‐type specific traits (e.g. pathophenotypes, drug response patterns) in the cell culture dish. These salient findings, which together with Sir John B. Gurdon's description of reprogramming of frog cells by nuclear transfer were awarded with the Nobel prize in 2012, kicked off a new era for in vitro disease modeling and patient‐specific drug development.
Since the advent of iPSC technology 12 years ago, intense research in this field has provided a number of modifications and improvements to the initial methodology which relied on integrating retroviral vectors and co‐culture with murine feeder cells. Meanwhile, it has become a standard in the field to efficiently reprogram fibroblasts or blood cells under feeder‐free conditions with the help of non‐integrating vectors such as Sendai viruses, synthetic mRNAs or episomal plasmids. However, despite these methodological changes, the basic principle that is the forced expression of the OSKM reprogramming factors is still mainly applied (Takahashi & Yamanaka, 2016).
While reprogramming technologies in combination with directed differentiation into specific cell types are, in principle, applicable to any somatic tissue and cell type, it is the non‐regenerative tissues that have become a key focus of iPSC‐related activities. This is particularly true for the central nervous system (CNS) and its otherwise inaccessible cell types. Today, there is a huge number of protocols enabling the generation of various CNS cell types from iPSCs, typically based on the modulation of developmentally relevant signaling pathways by applying morphogens and/or small molecules (Tabar & Studer, 2014).
One key question related to in vitro disease modeling is which cells to use as appropriate control. While early studies employed iPSC derived from unrelated healthy donors or, if available, unaffected family members as donors for control cells, these two types of controls are compromised by the significant genomic variation between individuals. This variability may obscure phenotypic read‐outs and limit the system's capacity to fully depict effects caused by a specific mutation. Comparative studies encompassing iPSC lines from different donors and donor tissues revealed that variability derives largely from the genetic background rather than the source cell. Accordingly, iPSC lines generated from different donor tissues with the same genetic background showed a much higher similarity than iPSC lines from different donors (Kyttala et al., 2016; Rouhani et al., 2014). Initially, this interindividual variability necessitated an increase of the number of experiments and cell lines studied for each pathophenotype, thereby severely delaying or even precluding the establishment of reliable disease models.
Luckily, the vast and unexpected technological progress made during the last 5 years in a complementary field of biomedical research now appears as a ‘perfect match’ to address this important issue: The recent advances in genome‐editing technologies and, in particular, CRISPR/Cas9 technology (Cong et al., 2013; Jinek et al., 2012; Mali et al., 2013) have dramatically changed the armamentarium for in vitro disease modeling (Avior, Sagi, & Benvenisty, 2016) (see also the chapter by Thilo Kunath and colleagues in this Special Issue). These technologies enable the introduction of sequence‐specific DNA double‐strand breaks at single‐base resolution, thus allowing even multiple rounds of DNA editing and offering the (theoretical) possibility to create any desired genotype. In the context of iPSC‐mediated disease modeling, the genome‐editing strategy can start from either healthy control iPSCs by introducing the mutation(s) to be analyzed, or from patient cells and reverting existing aberrations (‘gene correction’). This ‘isogenic’ approach (meaning that besides the mutation in question the diseased and control cells in theory share the same genome) offers the unique possibility to analyze the effects of a single mutation within the same genetic background, thus eliminating phenotype variations originating from interindividual genetic variation.
It is important to note that even before the advent of the powerful CRISPR technology Liu et al. already generated isogenic PD pluripotent stem cells (PSCs) with or without LRRK2G2019S(see below), by either correcting diseased variants in PD iPSCs or knocking in the same pathogenic variant in human embryonic stem cells (hESCs) with adenovirus‐mediated homologous recombination (Liu et al., 2012). This study showed that neural stem cells (NSCs) with the LRRK2G2019S variant displayed progressive disorganization of the nuclear lamina and a decline in neuronal differentiation potential after extensive passaging. Interestingly, similar nuclear envelope abnormalities were also observed in hippocampal postmortem samples of LRRK2G2019S carriers and idiopathic PD patients (Liu et al., 2012), supporting the validity of this experimental approach.
It is expected that the experimental paradigm of using isogenic iPSC lines will form the basis for most if not all future disease modeling efforts (see Figure 1). We will demonstrate the applicability of this approach by providing exemplary data based on an isogenic disease model in the context of this article to visualize the nature of expected data sets.
Figure 1.
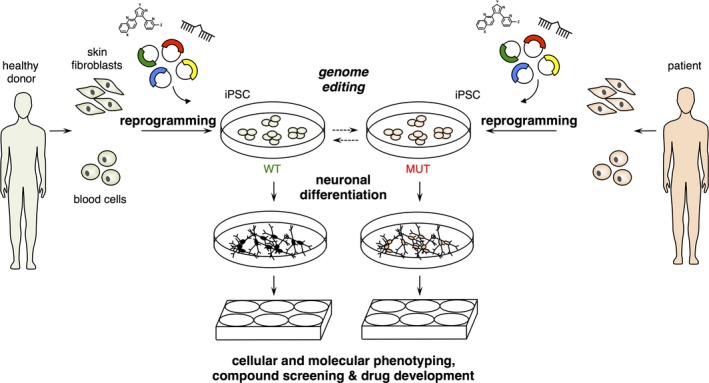
Reprogramming approaches in combination with genome editing facilitate disease modeling and drug discovery. Somatic cells (skin fibroblasts, blood cells) from healthy or diseased donors are reprogrammed into induced pluripotent stem cells (iPSCs) via various approaches, including ectopic expression of reprogramming factors, combinations of transcription factors and microRNAs or application of small molecules. The genome of resulting iPSCs can be edited to insert disease‐specific mutations into cells of healthy donors or to repair mutants in patient‐derived cells. Both approaches result in isogenic pairs of iPSCs which differ only by the respective disease variant, thereby eliminating phenotypic variability due to interindividual genetic differences. Optimized differentiation routines are next applied to generate the desired cell type(s) for in vitro disease modeling and drug discovery
2. THE ROLE OF LRRK2 IN THE PATHOGENESIS OF PARKINSON'S DISEASE
2.1. Introduction to LRRK2 protein structure and functions
The LRRK2 gene encodes a multi‐domain protein with a complex structure and highly pleiotropic functions (Figure 2). The central part of the protein contains the catalytic core with two distinct enzymatic activities: A Ras of complex proteins (ROC) GTPase domain with an adjacent C‐terminus of ROC (COR) domain, followed directly by a serine/threonine kinase domain. This catalytic core is surrounded by several modules of protein–protein interaction domains, including an armadillo (ARM)‐, an ankyrin (ANK)‐ and a leucine‐rich repeat (LRR)‐domain at the N‐terminus, and a WD40 domain at the C‐terminus (Figure 2a). LRRK2 exists as an almost inactive monomer in the cytosol, while the predominantly active dimer is membrane‐bound and exhibits a higher kinase activity compared to cytosolic LRRK2 (Berger, Smith, & LaVoie, 2010; Rosenbusch & Kortholt, 2016). The protein has been described to localize to a variety of subcellular compartments and organelles (Cho et al., 2014; Larsen, Hanss, & Krüger, 2018; Li, Tan, & Yu, 2014; Roosen & Cookson, 2016; Ryan, Hoek, Fon, & Wade‐Martins, 2015; Yang et al., 2014) and has been implicated in a plethora of different subcellular functions (Figure 2b). LRRK2's GTPase activity is considered to mediate its interaction with components of the cytoskeleton (such as tubulins and tau), thereby regulating stability of microtubules and thus directly impacting cell morphology and vesicle transport processes (Kawakami et al., 2014). In addition, LRRK2 has been shown to interact with and regulate the actin cytoskeleton regulators moesin, p21 (RAC)‐activated kinase 6 (PAK6) and focal adhesion kinase (FAK) (Civiero et al., 2015, 2017; Jaleel et al., 2007; Wallings, Manzoni, & Bandopadhyay, 2015). In this context, it is noteworthy that LRRK2's central catalytic core (comprising a ROC‐GTPase, a COR and a kinase domain) is reminiscent of the evolutionary conserved protein family of ROCO proteins that are reported to modulate cytoskeleton dynamics in eukaryotes (Civiero, Dihanich, Lewis, & Greggio, 2014; Lewis, 2009). For example, members of the ROCO family regulate chemotaxis and colony formation in the slime mold Dictyostelium discoideum, presumably via phosphorylation‐dependent regulation of myosin dynamics (Bosgraaf et al., 2005). The involvement of LRRK2 in regulating cytoskeletal stability (reviewed in Parisiadou & Cai, 2010) may thus point to an ancient and primordial aspect of this protein family's functionality.
Figure 2.
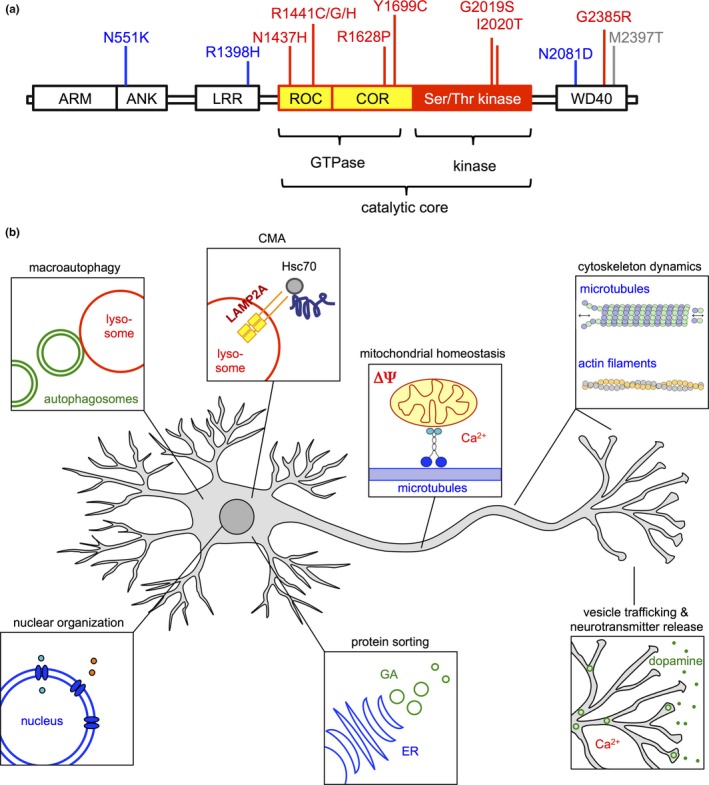
Leucine‐rich repeat kinase 2 protein structure and functions. (a) LRRK2 protein architecture and disease‐linked variants. Colored boxes, domains comprising the catalytic core; white boxes, protein–protein interaction domains, color‐coding of disease‐associated variants: red, Parkinson's disease; blue, Crohn disease; gray, leprosy. (b) Pleiotropic functions of LRRK2. The LRRK2 protein is contributing to the homeostasis and regulation of several cellular pathways and compartments including autophagy, mitochondrial function, vesicle transport and nuclear architecture. CMA, chaperone‐mediated autophagy; Ca2+, Calcium2+; ER, endoplasmatic reticulum; GA, Golgi apparatus; ΔΨ, mitochondrial membrane potential. See main text for details
LRRK2 is pivotally involved in regulating proteostasis by its effect on (at least) two major proteostatic mechanisms: on the one hand, it regulates macroautophagy by affecting the phosphorylation status of the key autophagy protein Beclin‐1 (Manzoni et al., 2016) and the mammalian target of rapamycin (mTOR) regulator Leucyl‐tRNA synthetase (LRS) (Ho et al., 2018). On the other hand, LRRK2 is under normal conditions degraded via both proteasomal and lysosomal pathways. The lysosomal degradation of LRRK2 is facilitated by the chaperone‐mediated autophagy (CMA) pathway that is via binding of chaperone heat shock cognate 71 kDa protein (Hsc70) to cargo protein and mediating its import into the lysosome via lysosomal‐associated membrane protein 2A (LAMP‐2A) (Kaushik & Cuervo, 2009). Importantly, aggregation of mutant LRRK2 protein with LAMP‐2A at the lysosomal membrane interferes with import and degradation of other CMA substrates (Orenstein et al., 2013).
A major body of evidence links LRRK2 function to mitochondrial functionality. LRRK2 interacts with dynamin‐related protein 1 (DRP1), which controls mitochondrial fission, and other mitochondrial control checkpoints such as mitochondrial dynamin‐like GTPase (OPA1) and mitofusin1 (MFN1) (Stafa et al., 2014; Wang et al., 2012). In addition, LRRK2 is directly involved in autophagy‐mediated turnover of mitochondria (‘mitophagy’) by enabling engulfment of damaged or aged mitochondria by autophagosomes via removing the protein motor anchor MIRO1 that orchestrates movement of mitochondria along the cytoskeleton via the MIRO1/MILTON complex (Hsieh et al., 2016).
LRRK2 is further involved in the control of vesicle endocytosis and intracellular transport processes by regulatory phosphorylation of various Ras‐related in brain (Rab) proteins (including Rab1a/b, 5, 7/L1, 8, 10, 12, 29, dependent on the experimental system chosen) (Purlyte et al., 2018; Steger et al., 2016). Interestingly, the LRRK2‐dependent phosphorylation of a subset of these bona fide LRRK2 substrates (i.e. Rab8, 10) has been reported to affect their interaction with primary regulators of ciliogenesis such as RILPL1 and RILPL2 (Rab‐interacting lysosomal protein like 1 and 2) (Dhekne et al., 2018). Given the prominent role of primary cilia as essential signal transducers of the sonic hedgehog (SHH) signaling pathway (Goetz & Anderson, 2010), these findings are linking LRRK2 kinase activity to regulation of cilia‐dependent developmental signaling. In addition, LRRK2 has been described to contribute to anterograde vesicle and protein transport by anchoring the endoplasmatic reticulum (ER) export factor SEC16A (SEC16 homolog A, endoplasmic reticulum export factor) to the ER exit site and thus affecting ER‐to‐Golgi shuttling of coat protein complex II (COPII) vesicles. In the context of neuronal function, this mechanism is discussed to be involved in the directed vesicle‐mediated transport of neurotransmitter receptors and, as a consequence, formation and function of synapses (Cho et al., 2014).
Several lines of evidence link LRRK2 function to regulation of intracellular calcium (Ca2+) homeostasis: LRRK2 induces the extracellular signal‐regulated kinase (ERK) 1/2‐dependent upregulation of mitochondrial calcium uptake proteins (Verma et al., 2017). Moreover, Ca2+ is mobilized from lysosomal storage by LRRK2 in a nicotinic acid adenine dinucleotide phosphate (NAADP)‐dependent manner (Gomez‐Suaga, Luzon‐Toro et al., 2012). Finally, LRRK2 augments Ca2+ influx via voltage‐gated Ca2+ (CaV) channels, although the underlying mechanisms are still unknown (Bedford, Sears, Perez‐Carrion, Piccoli, & Condliffe, 2016). The central role of Ca2+ ions in key cellular pathways (e.g. as second messenger in signal transduction pathways, as regulators of vesicle internalization and release) may provide a further explanation for the pleiotropic effects of LRRK2 encompassing lysosomal function, autophagy, mitochondrial metabolism as well as vesicle trafficking and neurotransmitter homeostasis (Gomez‐Suaga, Fdez, Blanca Ramirez, & Hilfiker, 2012; Li et al., 2014; Rassu et al., 2017).
Finally, LRRK2 function has also been implicated in regulation of the subcellular architecture: organization of the nuclear envelope is affected by phosphorylation status of lamins B1 and B2 which have been shown to represent potential substrates for LRRK2's kinase activity (Liu et al., 2012). Moreover, LRRK2 interacts with cyclin‐G‐associated kinase (GAK) and RAB29/ RAB7L1 that control morphology and function of the Golgi apparatus (GA) and vesicle trafficking through GA cisternae and the ER (Beilina et al., 2014; Liu et al., 2018; MacLeod et al., 2013).
2.2. LRRK2 variants and Parkinson's disease
Familial forms of Parkinson's disease are associated with variants in a number of well characterized risk genes, including, among others, SNCA (synuclein alpha), LRRK2 (leucine‐rich repeat kinase 2), GBA (glucosylceramidase beta), PINK1 (PTEN‐induced putative kinase 1), PRKN (parkin RBR E3 ubiquitin protein ligase, formerly known as PARK2), PARK7 (protein deglycase DJ‐1) and VPS35 (VPS35, retromer complex component). Among these genetic risk factors, the gene LRRK2 plays a prominent role as the LRRK2 locus harbors one of the most common polymorphisms associated with PD (G2019S, see below), which has been reported to be associated with up to 2% of sporadic cases and up to 6% of total familial cases (Bardien, Lesage, Brice, & Carr, 2011; Berg et al., 2005; Bonifati et al., 2002). While frequency estimates (especially of heterozygote alleles) derived from small‐scale clinical studies (in contrast to epidemiological studies) may sometimes be biased by clinical referral, large scale genome‐wide association studies (GWAS) and recent meta‐analyses on multi‐ethnic PD cohorts further emphasize the importance of individual LRRK2 variants and the associated pathways in a wide patient population. Interestingly, it was shown that distinct variants in LRRK2 can exert independent and in some cases even protective effects on the disease susceptibility (Foo et al., 2017; Ross et al., 2011). These data and the presence of enzymatically active domains (see below) make the LRRK2 protein a highly attractive target for PD therapy.
Interestingly, almost all variants associated with PD are clustered within the central catalytic core. The most frequent variation, affecting position 2019, results in an amino acid exchange from glycine to serine (G2019S) within a highly conserved DYG (aspartic acid/D, tyrosine/Y, glycine/G; or DFG [aspartic acid/D, phenylalanine/F, glycine/G] in most other kinases) motif in direct proximity to the activation loop of the kinase activity (Cookson, 2010; Kachergus et al., 2005; Mata, Wedemeyer, Farrer, Taylor, & Gallo, 2006). By today, several studies have reported that LRRK2G2019S increases the kinase activity of the protein (first demonstrated by Jaleel et al., 2007; reviewed by Price and colleagues (Price, Manzoni, Cookson, & Lewis, 2018)), while the effect of the I2020T variant (affecting the adjacent C‐terminal amino acid) on LRRK2 kinase activity has been discussed controversially (Gloeckner et al., 2006; Jaleel et al., 2007). Variants within or close to the ROC/COR domains (e.g. R1441C) have been shown to decrease the GTPase function (Lewis et al., 2007). A common dominator of these early studies is that they argue for a causative role of dysregulated enzymatic activity in PD etiopathology. Interestingly, elevated LRRK2 phosphorylation at Ser1292, that is the residue, that is estimated to be best suited to monitor LRRK2 kinase activity by autophosphorylation (Sheng et al., 2012), has recently also been shown in postmortem samples of substantia nigra tissue from patients with idiopathic PD (Di Maio et al., 2018). In this context, it is noteworthy that the activity of the kinase domain has also been linked to regulation of LRRK2 protein solubility and stability, suggesting that abundance of the solube protein—in addition to the level of its catalytic activity—may represent a causative factor for neuronal cell death (Skibinski, Nakamura, Cookson, & Finkbeiner, 2014; Skibinski et al., 2017).
2.3. Role of LRRK2 in immune disorders
It is noteworthy that LRRK2 variants are not only associated with PD, but also with susceptibility for immune‐related disorders, namely the chronic inflammatory bowel disease Crohn's disease and leprosy (Franke et al., 2010; Jostins et al., 2012; Marcinek et al., 2013; Witoelar et al., 2017) (Figure 2a). The respective variants for these immune disorders tend to localize outside of the catalytic core within the protein interaction modules. The C‐terminal WD40 domain harbors the M2397T variant associated with excessive inflammation in leprosy (Fava et al., 2016) and the N2081D risk variant for Crohn's disease (Hui et al., 2018). Interestingly, Parkinson's disease and Crohn’ s disease share a protective haplotype that involves N551K and R1398H located N‐terminally of the catalytic domains, suggesting shared aspects of pathology for both diseases. In addition to its role in neurons, LRRK2 plays an important role in various immune cells: LRRK2 is strongly expressed in dendritic cells, lymphocytes and macrophages and can be even further upregulated by interferon (IFN)‐γ signaling via activation of ERK5 (Kuss, Adamopoulou, & Kahle, 2014). Moreover, LRRK2 contributes to activation of microglia (the CNS counterpart of monocytes/macrophages) via regulating activity of nuclear factor‐kappa B (NF‐κB), a pivotal transcription factor involved in pro‐inflammatory signaling via induction of, for example, tumor necrosis factor (TNF)‐α and interleukin (IL)‐6 (Hirsch, Hunot, Damier, & Faucheux, 1998; McGeer, Itagaki, Boyes, & McGeer, 1988; Russo et al., 2015). While microglia have been found to be highly activated in PD patient samples irrespective of the LRRK2 status (Gerhard et al., 2006), it is important to note that their migration and activation status can be strongly mediated in a LRRK2‐dependent manner (Choi et al., 2015; Ma et al., 2016). In addition, LRRK2 has been shown to be an essential part of a protein complex that inhibits the activity of the immune‐regulatory transcription factor nuclear factor of activated T cells (NFAT), and this mechanism may have implications for the activation status of the immune system in multiple disease scenarios (Liu et al., 2011). Finally, LRRK2 activity is described to be stabilized by and act downstream of Toll‐like receptors (TLR)‐2 and ‐4 and their common adaptor myeloid differentiation primary response protein 88 (MYD88), placing LRRK2 function in the context of sensing of bacterial pathogen‐associated molecular patterns (PAMPs) (Dzamko et al., 2012). Given the well‐established role of inflammation in development of neurodegenerative disorders including PD (see below), it is tempting to speculate that the emerging immunomodulatory functions of LRRK2 (e.g. activation of microglia) may contribute to the neuroinflammatory conditions that shape PD etiopathology (Russo, Bubacco, & Greggio, 2014).
3. PD‐ASSOCIATED LRRK2 VARIANTS IN iPSC‐BASED DISEASE MODELING
In the following paragraphs, we aim at dissecting how iPSC‐based disease modeling was applied to study the involvement of LRRK2 in pathologically relevant pathophenotypes in the context of PD. It is important to note that salient findings about the diverse functions of LRRK2 and the role of its disease‐associated variants are based on the use of human cancer cell line models (such as HeLa or HEK 293 cells) or immortalized cells, and that these approaches represent valid systems to unravel general biological principles. Still, when it comes to the many peculiarities of neuronal cells and their various subtypes (see below), primary cells and iPSC‐based strategies are usually considered superior to demonstrate cell‐type specific effects. We included information about whether a given phenotype was found to be specific for LRRK2 or could also be shown in other models of familial PD. Where suitable we included own data generated with an isogenic pair of normal and G2019S iPSC lines (Liu et al., 2012) that were differentiated into mesencephalic dopamine (mesDA) neurons following a modified floor plate‐based differentiation protocol (Kriks et al., 2011; Ryan et al., 2013), (Figure 3a,b). This differentiation protocol takes into account the developmental origin of mesDA neurons from floor plate progenitor cells and thus recapitulates the development of this specific neuronal cell type in vitro (Blaess et al., 2011; Joksimovic et al., 2009; Ono et al., 2007). Compared to other approaches that involve a neural stem cell expansion phase, floor plate‐derived mesDA neurons are currently considered to be the most authentic population of in vitro generated midbrain dopamine‐like neurons (Barker, Parmar, Studer, & Takahashi, 2017). This notion is based on the in‐depth analysis of substantia nigra pars compacta (SNpc)‐specific marker expression profiles, the synthesis and release of dopamine and its metabolites, SNpc‐specific electrophysiological properties (e.g. spontaneous spiking at 1–3 Hz; ‘pacemaking activity’) and the ability to function as midbrain dopaminergic neurons in culture slices and upon transplantation in animal models (Kikuchi et al., 2017; Kirkeby et al., 2012; Kriks et al., 2011). We will refer to these iPSC‐derived mesDA neurons simply as ‘mesDA neurons’ in the following sections.
Figure 3.
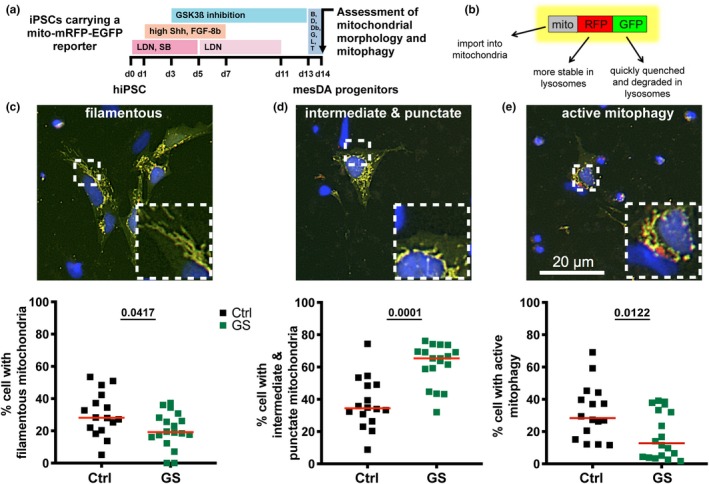
An isogenic iPSC‐based model of LRRK2G2019S‐linked PD reveals alterations in mitochondrial morphology and impaired mitophagy in mesDA progenitors. (a) Schematic overview of the experimental setup. Isogenic iPSCs expressing the mito‐RFP‐EGFP reporter were subjected to differentiation into floor plate‐derived mesDA neurons. Mitochondrial morphology and mitophagy were analyzed at day 14 (d14). LDN, LDN193189; SB, SB431542; B, BDNF; D, DAPT; Db, dbcAMP; G, GDNF; L, LAAP; T, TGFβ. (b) Illustration of the mitophagy reporter mito‐RFP‐EGFP, which is imported into the mitochondrial matrix upon translation and indicates mitophagy by a pH‐dependent color shift from yellow (RFP+EGFP) to red (RFP only) upon lysosomal delivery. (c–e) Quantification and exemplary images of mitochondrial morphologies in isogenic G2019S mesDA progenitors at d14. LRRK2G2019S cultures display reduced levels of filamentous mitochondria (c) and increased levels of intermediate and punctate mitochondria (d). (e) At d14, levels of active mitophagy are reduced in G2019S cultures. For each condition, 5–6 wells were quantified per experiment (n = 3). The percentages of cells containing mitochondria of a specific category per well are depicted as dot plots. Lines indicate the median. For details on the methodology, see Supporting Information
3.1. Pathogenic protein accumulation
The gene SNCA (previous gene symbols: PARK1, PARK4) encoding the protein α‐synuclein (α‐syn) is genetically linked to PD, and its accumulation in ‘Lewy bodies’ and ‘Lewy neurites’ represents a pathological hallmark of the disease in vivo (see also the chapter by Andrew West and colleagues, and the chapter by Patrik Brundin and Gerhard Coetzee). While α‐syn has been described to be a small soluble protein predominantly expressed in the brain, many questions regarding its exact physiological function as well as the mechanisms leading to α‐syn accumulation and finally neurodegeneration remain unanswered (Steiner, Quansah, & Brundin, 2018). Given its prominent role in the pathology of the disease, several groups investigated α‐syn protein levels and subsequent α‐syn accumulation in iPSC‐derived mesDA‐neuronal cultures. Significantly increased α‐syn protein levels were detected in iPSC‐derived LRRK2G2019S mesDA‐neurons by immunoblotting (Nguyen et al., 2011). Subsequently, this striking finding was verified by several independent studies comparing either cultures of patient lines and controls of different genetic background (Lopez de Maturana et al., 2016; Nguyen & Krainc, 2018), or isogenic iPSC pairs (Reinhardt, Schmid et al., 2013).
Importantly, accumulation of α‐syn does not represent a LRRK2‐specific pathophenotype as similar data have been generated for various different models of familial PD, including iPSC models for triplication of the SNCA gene (SNCAtrp) (Byers et al., 2011; Flierl et al., 2014; Mazzulli, Zunke, Isacson, Studer, & Krainc, 2016), the SNCAA53T point mutation (Kouroupi et al., 2017; Mazzulli, Zunke, Tsunemi et al., 2016; Ryan et al., 2013), mutations in GBA (Woodard et al., 2014), PRKN (Chang et al., 2016; Chung et al., 2016; Shaltouki et al., 2015) and PINK1 (Chung et al., 2016), in homozygous DJ‐1 mutant cells and in DJ‐1 knock‐out neurons (Burbulla et al., 2017) as well as in iPSC‐derived neurons from an idiopathic PD (iPD) patient (Mazzulli, Zunke, Isacson et al., 2016).
In addition, pronounced accumulation of tau (a protein family whose members mediate stability of microtubules) has been described as a pathological hallmark in postmortem samples obtained from carriers of LRRK2 variants, including G2019S and I2020T (Ohta et al., 2015; Rajput et al., 2006; Zimprich et al., 2004). It has been shown that wildtype LRRK2 as well as LRRK2G2019S can either directly or indirectly induce glycogen synthase kinase 3 beta (GSK‐3β)‐mediated tau phosphorylation in SH‐SY5Y cells overexpressing LRRK2 (Kawakami et al., 2014). Several groups analyzed tau and phospho‐tau (p‐tau) levels in iPSC‐derived mesDA neurons. A study by Reinhardt et al. revealed elevated levels of total‐tau and p‐tau in LRRK2G2019S mesDA‐neurons compared to isogenic controls (Reinhardt, Schmid et al., 2013), and a similar observation was made in sensory neurons carrying the G2019S variant (Schwab & Ebert, 2015). Interestingly, in the same study, the authors did not observe accumulation of tau or p‐tau in an SNCAtrp patient (Schwab & Ebert, 2015). Finally, mesDA neurons carrying the LRRK2I2020T variant showed accumulation of total‐tau as well as increased phosphorylation of tau (Ohta et al., 2015). It is intriguing that tau is cytotoxic when overexpressed not only in mesDA neurons but in almost all cell types examined, while α‐syn on its own does not convey a comparably strong cytotoxicity (Klein, Dayton, Henderson, & Petrucelli, 2006; Klein, Dayton, Lin, & Dickson, 2005). This toxic effect of tau has been functionally linked to impairment of axonal vesicle transport (Beevers et al., 2017; Wu et al., 2013) and can be rescued by parkin overexpression (Klein et al., 2006), thus arguing for crosstalk at the level of mitochondrial homeostasis and for a potential direct role of dysregulated tau protein levels in PD etiopathology.
As with accumulation of α‐syn, this effect is neither specific for LRRK2 nor restricted to PD. For example, Mazzulli and colleagues found a mild increase in tau levels in mesDA‐neuronal cultures derived from patients with GBAN370S/ins84GG‐associated Gaucher's disease (GD), a lysosomal storage disease clinically linked to PD (Mazzulli et al., 2011). These findings underscore the existence of shared underlying disease mechanisms for PD and GD and point to a potential involvement of a lysosomal degradation deficit as a common pathomechanism.
Taken together, accumulation of pathogenically linked proteins such as α‐syn and (hyperphosphorylated) tau represent highly reproducible phenotypes in iPSC‐based models of LRRK2‐associated PD and related models of neurodegeneration.
3.2. Impairment of the autophagy‐lysosomal system
Evidence for dysregulation of the autophagy–lysosomal system (e.g. accumulation of autophagosomes, alterations in lysosomal marker expression) has frequently been observed in postmortem samples of PD patients, suggesting a pathogenic role of this lysosomal degradation pathway in the progression of the disease (Anglade et al., 1997; Chu, Dodiya, Aebischer, Olanow, & Kordower, 2009). Given the association of LRRK2 with autophagy pathways, several groups assessed whether iPSC‐derived mesDA neuronal cultures show alterations in autophagic clearance. Sanchez‐Danes and colleagues found increased levels of LC3‐II (the activated form of autophagy marker microtubule‐associated protein light chain 3/LC3) and inhibition of autophagosome–lysosome fusion in LRRK2G2019S and idiopathic PD‐derived mesDA neurons compared to healthy controls, suggesting a general impairment of autophagic flux in PD‐derived mesDA neurons (Sanchez‐Danes et al., 2012). In addition, electron microscopy of these cells showed massive accumulation of autophagosomes indicative for a defect in autophagic flux through inhibited fusion of autophagosomes with lysosomes. Reinhardt and colleagues (Reinhardt, Schmid et al., 2013) also found evidence for a decreased autophagic flux in G2019S versus isogenic control mesDA neurons under starvation conditions. Interestingly, another study interpreted similar findings as indication for excessive autophagy induction in G2019S iPSC‐derived neurons (Su & Qi, 2013). Along this line, Borgs and colleagues showed an upregulation of autophagy genes (e.g. LC3A, LC3B, autophagy‐related (ATG)5, ATG7) in immature LRRK2G2019S neurons (Borgs et al., 2016). These data might collectively be interpreted as a compensatory effect to counteract impairment of the autophagic system. Strikingly, Ohta et al. (2015) found that the LRRK2 variant I2020T, too, is associated with increased LC3‐II levels. In a recent study, Ho and colleagues used several cell models including iPSC‐derived mesDA neurons from G2019S carriers to demonstrate that the mTOR regulator leucyl‐tRNA synthetase (LRS) is phosphorylated by LRRK2 in a genotype‐dependent way, and that G2019S‐associated hyperphosphorylation was linked to impairment of the autophagic system (Ho et al., 2018). These data suggest that LRRK2 may affect macroautophagy at several levels, including phosphorylation of LRS and Beclin‐1 (Manzoni et al., 2016).
As outlined above, LRRK2 may be degraded via both proteasomal and lysosomal pathways, in particular via CMA, under normal conditions. The Cuervo laboratory was able to demonstrate that CMA is impaired in LRRK2G2019S mesDA neurons (Orenstein et al., 2013). Investigation of the underlying mechanisms revealed a dramatic increase in co‐localization of LAMP‐2A with α‐syn (a known substrate of CMA) (Vogiatzi, Xilouri, Vekrellis, & Stefanis, 2008). As LRRK2 itself also represents a prominent CMA substrate, these data point toward an impairment of CMA by competition between α‐syn and LRRK2 for binding to and import via the CMA receptor LAMP‐2A. It is tempting to speculate that this inhibitory effect on CMA is pivotally involved in PD‐associated accumulation of α‐syn. Along the same line, it has been discussed that accumulation of (hyperphosphorylated) tau results from dysregulated autophagic activity caused by PD‐associated LRRK2 variants (Guerreiro et al., 2016).
Importantly, impairment of the autophagy‐lysosomal degradation system and its enzymatic activities (e.g. glucocerebrosidase/GCase activity) is not restricted to LRRK2G2019S and LRRK2R1441C/G variants (Nguyen & Krainc, 2018) and has been convincingly demonstrated for several PD risk factors: GBA (Fernandes et al., 2016; Schöndorf et al., 2014; Woodard et al., 2014), SNCAtrp (Mazzulli, Zunke, Isacson et al., 2016), OPA1 (Iannielli et al., 2018), DJ‐1 (Burbulla et al., 2017), for iPD (Burbulla et al., 2017; Mazzulli, Zunke, Isacson et al., 2016; Sanchez‐Danes et al., 2012) and for GBA mutant cells from GD patients (Aflaki et al., 2016; Mazzulli et al., 2011; Schöndorf et al., 2014).
Taken together, the observations summarized above demonstrate significant functional crosstalk between LRRK2 and autophagy pathways in the context of PD etiopathology which could be linked to at least two possible underlying mechanisms: Direct regulation of macroautophagy by LRRK2 at the level of Beclin‐1 and LRS, and competition between CMA substrates, ultimately resulting in a breakdown of the proteostatic system.
3.3. Disrupted mitochondrial dynamics and mitophagy
Mitochondria are highly dynamic cell organelles that are essential for a variety of cellular functions, including energy metabolism, regulation of Ca2+ homeostasis as well as cell death pathways, stress responses and immune signaling. Since the 1980s, mitochondria dysfunction is known to be strongly linked to the progression of PD, based on the demonstration that the pro‐drug 1‐methyl‐4‐phenyl‐1‐2‐3‐6‐tetrahydropyridine (MPTP) can induces PD‐like features in rodents (Heikkila, Hess, & Duvoisin, 1985). MPTP is capable to cross the blood–brain barrier and is then metabolized into its active form MPP+ (1‐methyl‐4‐phenyl‐4‐dihydropyridine). MPP+ is imported into mesDA neurons where it induces mitochondrial adenosine triphosphate (ATP) depletion and oxidative stress, ultimately resulting in neuronal cell death (reviewed in Cannon & Greenamyre, 2010). Further evidence for mitochondrial dysfunction in PD were the identification of defects in the mitochondrial respiratory chain in post‐mortem samples of patients with sporadic PD (reviewed by Larsen et al., 2018), and the fact that several autosomal recessive genetic risk factors for PD (PRKN, PINK1, DJ‐1) are associated with mitochondrial function (see the chapter by Anindita Bose and Flint Beal in this Special Issue). In consequence, many studies focused on various aspects of mitochondrial biology in the context of PD pathogenesis. Basal oxygen consumption rate (OCR) was decreased in LRRK2G2019S, LRRK2R1441C, SNCAA53T, DJ‐1mut and OPA1mut compared to controls, in mesDA neurons or neural progenitor cells (NPCs) (Burbulla et al., 2017; Cooper et al., 2012; Iannielli et al., 2018; Ryan et al., 2013). Schwab and colleagues confirmed these findings in cortical and mesDA neurons of LRRK2G2019S patients (Schwab et al., 2017). The authors found increased expression of sirtuins, a family of nicotinamide adenine dinucleotide (NAD)+‐dependent protein deacetylases involved in mitochondrial metabolism, but a decrease in deacetylase activity and complex III levels. LRRK2 kinase inhibitor treatment was not sufficient to normalize levels of sirtuins in LRRK2G2019S mesDA neurons (Schwab et al., 2017). Interestingly, the authors could not observe these phenotypes in iPSC‐derived peripheral neurons, indicating neuronal subtype‐specific susceptibility. Moreover, LRRK2G2019S neurons were shown to display decreased ATP content (Schwab et al., 2017; Su & Qi, 2013), accompanied by a decrease in mitochondrial membrane potential (Hsieh et al., 2016; Su & Qi, 2013). Both findings were also observed in other PD models in SNCAtrp and mutant OPA1 neural progenitor cells (NPCs) (Flierl et al., 2014; Iannielli et al., 2018). Decreased mitochondrial membrane potential was also reported for SNCAtrp and SNCAA53T mesDA neurons (Little et al., 2018). As these observations hint towards a profound disturbance of mitochondrial function in familial forms of PD, several groups consequently investigated effects of PD risk factors on mitochondrial redox homeostasis and found increased levels of mitochondrial reactive oxygen species (ROS) in mesDA neurons carrying polymorphisms in LRRK2, DJ‐1, PINK1, PRKN and SNCA (Burbulla et al., 2017; Chung et al., 2016; Ryan et al., 2013; Su & Qi, 2013; Suzuki et al., 2017), as well as in NPCs with SNCAtrp and OPA1 variants (Flierl et al., 2014; Iannielli et al., 2018). Intriguingly, damage to mitochondrial DNA, presumably as a result of increased ROS production, was detectable already at the NPC stage of LRRK2G2019S and LRRK2R1441C carriers (Sanders et al., 2014).
Several groups have analyzed mitochondrial length in iPSC models of familiar PD. The results are ambiguous, underscoring the complexity of this cellular trait. Su and Qi reported shortened mitochondrial length in mesDA neurons carrying the LRRK2G2019S variant, and Schwab and colleagues revealed a reduced mitochondrial distribution in neurites (Schwab et al., 2017; Su & Qi, 2013). In contrast, Hsieh et al. showed evidence that mitochondria were significantly longer in LRRK2G2019S neurons as compared to controls (Hsieh et al., 2016). Cooper and colleagues reported no significant difference in mitochondrial length compared to controls for PINK1 and LRRK2G2019S neural cultures, but decreased mitochondrial length in LRRK2R1441C cultures (Cooper et al., 2012). These discrepancies may reflect differences in the methodology used for morphological analyses or different cell types analyzed. Schwab and colleagues revealed decreased mitochondrial content in mesDA neurons carrying LRRK2G2019S or PRKN variants (Schwab et al., 2017; Shaltouki et al., 2015), while this was not observed in cortical or peripheral neurons (Schwab et al., 2017). Focusing on movement of mitochondria within neuronal cells along the cytoskeleton, two independent groups showed that the G2019S and R1441C variants in LRRK2 induced a significant increase in mitochondrial mobility in iPSC‐derived neurons compared to controls (Cooper et al., 2012; Schwab et al., 2017).
In summary, alterations in mitochondrial morphology were repeatedly shown not only for LRRK2 variants (Su & Qi, 2013) but also for mutants of SNCAtrp/A53T PINK1, PRKN and OPA1 (Chung et al., 2016; Iannielli et al., 2018; Imaizumi et al., 2012; Little et al., 2018; Shaltouki et al., 2015; Zanon et al., 2017).
Our own data (Figure 3) confirm that LRRK2G2019S mesDA progenitor cells show alterations in mitochondrial morphology as compared to isogenic controls. Figure 3c+d depicts representative images of cells displaying mitochondria that were manually classified according to their morphology. At day 14 of differentiation (d14), the number of cells with filamentous mitochondria was significantly decreased in LRRK2G2019S, while intermediate and punctate morphologies were increased (Figure 3c+d). Our data are in line with the results of other studies showing a prominent effect of LRRK2 and its variants on regulation of mitochondrial dynamics and indicate that alterations in mitochondrial morphology can already be detected at early time points of mesDA neuronal differentiation.
Several lines of evidence are furthermore hinting toward PD‐associated impairment in the turnover of damaged mitochondria from the cytosol, referred to as ‘mitophagy’ (Zhang, Duan, & Yang, 2015). This process by which aged or dysfunctional mitochondria are trapped within an autophagosome and transported toward the lysosomal compartment is tightly regulated and crucial to maintain the balance between mitochondrial biogenesis and degradation, as well as to prevent the accumulation of ROS (Von Stockum, Nardin, Schrepfer, & Ziviani, 2016). Hsieh and colleagues demonstrated that LRRK2 mediates mitophagy by removing the adaptor protein MIRO from the MIRO/MILTON/KINESIN motor complex that facilitates mitochondrial movement along the cytoskeleton (Hsieh et al., 2016). LRRK2‐dependent removal of MIRO results in retarded movement of mitochondria, thus enabling engulfment by the LC3‐coated autophagosomes and subsequent mitophagic degradation. In addition, the authors were able to demonstrate that the LRRK2G2019S polymorphism results in delayed MIRO degradation in mesDA neurons upon induction of mitochondrial damage by complex III inhibition, thus leading to impaired recruitment of LC3 and the mitophagy receptor optineurin. Importantly, the authors observed this also in mesDA neuronal cultures from idiopathic PD patients, which points to a general role of compromised mitophagy in the pathology of PD. Evidence for a connection between altered mitochondrial morphology and mitophagy was further provided by the finding that LRRK2G2019S fibroblasts display increased numbers of fragmented mitochondria, and that LRRK2 is involved in regulation of mitochondrial fission by regulating the phosphorylation and activation status of DRP1 (Su & Qi, 2013).
Using a well‐described tandem‐fluorochrome‐based read‐out for mitophagy, we could confirm the impact of the LRRK2G2019S variant on mitophagy. This system is based on a lentivirally expressed mito‐RFP‐EGFP fusion protein, which is imported into the mitochondrial matrix and indicates mitophagy by a color shift from yellow to red upon lysosomal delivery (Figure 3b) (Kim et al., 2013; Klionsky et al., 2016; Till et al., 2015). We found that impaired mitophagy in LRRK2G2019S cells is already apparent at an early stage of in vitro differentiation. Specifically, we assessed mitophagy in isogenic LRRK2G2019S and control cells at day 14 of the floor plate‐based differentiation protocol. Figure 3e shows a representative image displaying active mitophagy as evident from appearance of ‘red only’ structures. LRRK2G2019S cultures exhibited significantly decreased levels of active mitophagy compared to the control (Figure 3e). Together with the results of previously published studies, these data point to a vital and hitherto underappreciated detrimental effect of LRRK2 dysfunction on mitochondrial homeostasis already at early stages of differentiation.
As evident from the studies outlined above, impairment of mitophagy is not restricted to LRRK2‐associated PD but emerges as a central pathomechanism of the disease. While LRRK2 appears to modulate mitophagy by affecting mitochondrial movement, other PD risk factors are more directly involved in mitochondrial turnover. Upon mitochondrial damage, the kinase PINK1 is stabilized on the cytosolic face of the outer mitochondrial membrane and recruits parkin via a series of phosphorylation steps. Parkin (encoded by PD risk gene PRKN) represents a E3 ubiquitin ligase that subsequently mediates polyubiquitination of mitochondrial substrates, ultimately forming the molecular basis for attraction of autophagy receptors and initiation of mitophagy (Menzies, Fleming, & Rubinsztein, 2015). Consequently, several independent studies have focused on the pivotal role of PRKN and PINK1 mutants in iPSC‐based and other models of PD (Chung et al., 2016; Imaizumi et al., 2012; Seibler et al., 2011; Shaltouki et al., 2015; Suzuki et al., 2017).
Taken together, these data underscore the vital role of mitochondrial function and quality control in mesDA neurons and point to the functional implications of LRRK2 variants (and, although by different mechanisms, variants of PINK1, PRKN and SNCA) that result in impaired mitophagy, accumulation of damaged mitochondria and disturbed cellular redox homeostasis.
3.4. Increased susceptibility toward cellular stress
Differential vulnerability of distinct neuronal subpopulations is a hallmark of numerous neurodegenerative diseases. Stunningly, in many of these disorders it is a specific neuronal population or even subpopulation of neurons that is susceptible to neurodegeneration while other, even closely related, neuronal subsets remain completely or largely unaffected. Identification of the factors responsible for this differential vulnerability is expected to improve our understanding of disease mechanisms and to support the development of novel therapeutic approaches for the treatment of neurodegenerative diseases. In the context of PD, selective vulnerability of mesDA neurons has already been described almost 100 years ago (Foix & Nicolesco, 1925). MesDA neurons are commonly distinguished by their anatomical position, projection field, function and marker expression. The largest population of mesDA neurons is localized in the SNpc and the ventral tegmental area (VTA) from where they project to motor and corticolimbic structures, respectively (A9 and A10 dopamine pathways). The initial cell loss observed in PD is almost entirely restricted to mesDA neurons in the ventral tier of the SNpc. These neurons project almost exclusively to the dorsal striatum. MesDA cells from the neighboring dorsal tier of the SNpc and the VTA display a significantly lower degree and a later onset of degeneration (Alberico, Cassell, & Narayanan, 2015; Damier, Hirsch, Agid, & Graybiel, 1999; Dauer & Przedborski, 2003; Hassler, 1938). Loss or functional impairment of SNpc mesDA neurons results in a severe reduction in dopamine release in the dorsal striatum, ultimately causing the main motor symptoms of PD. While several genetic risk factors for PD are known (see above and chapter by Marie‐Thérèse Fuzzati‐Armentero in this Special Issue) the majority of them is ubiquitously expressed. Thus, their expression profile cannot account for the cell type‐selective degeneration of mesDA neurons in the SNpc. The current view is that mesDA neuron degeneration is based on genetically determined intrinsic susceptibility to cell death but is ultimately triggered by cellular stress conditions (such as proteotoxic or oxidative stress). Thus, both cell‐intrinsic factors (during the first steps of disease manifestation) and environmental/extrinsic factors (during disease progression) are involved in the etiopathology (Surmeier, Obeso, & Halliday, 2017). With respect to intrinsic factors, the emerging concept is that SNpc‐mesDA neurons display a set of cellular and molecular features that renders them more vulnerable toward external stress triggers than neighboring VTA‐mesDA neurons (for a comprehensive review see Brichta & Greengard, 2014). The following cellular characteristics of SNpc‐mDA neurons are discussed in this context: (a) high expression levels of certain subtypes of dopamine receptors and glycosylated (and thus matured) dopamine transporter (DAT) may mediate the uptake of neurotoxic substances (Reyes, Cottam, Kirik, Double, & Halliday, 2013). (b) The differential expression and/or selective activity of ATP‐sensitive potassium (K‐ATP) channels or their subunits (i.e. Kir6.2 and SUR1). K‐ATP channels display selective sensitivity toward external stimuli that may alter cell viability, potentially through mitochondria‐triggered ROS generation (Liss et al., 2005; Schiemann et al., 2012). (c) The specific electrophysiological properties of SNpc mesDA neurons, in particular, their endogenous pacemaking activity (Lammel et al., 2008) driven by CaV1‐type calcium channels may render them increasingly dependent on well‐orchestrated calcium homeostasis (Bishop et al., 2010; Mercuri et al., 1994; Nedergaard, Flatman, & Engberg, 1993). This might be particularly detrimental in SNpc mesDA neurons as they have a low capacity for intrinsic calcium buffering and display particular sensitivity to excitatory amino acid‐induced damage (Bywood & Johnson, 2000; Foehring, Zhang, Lee, & Callaway, 2009). (d) SNpc mesDA neurons are described to form much larger axon terminal arbors and display a higher number of synapses than VTA mesDA neurons (Aransay, Rodriguez‐Lopez, Garcia‐Amado, Clasca, & Prensa, 2015; Bolam & Pissadaki, 2012; Matsuda et al., 2009; Surmeier, Guzman, Sanchez‐Padilla, & Goldberg, 2011). Thus, these cells are supposed to have increased energy demands, which necessitate high motility and plasticity of mitochondria (Pacelli et al., 2015; Pissadaki & Bolam, 2013). Impairment of mitochondrial function and/or homeostasis would thus be particularly detrimental in SNpc‐mesDA neurons (Liang, Wang, Luby‐Phelps, & German, 2007). (e) Metabolism of the neurotransmitter dopamine requires detoxification systems to prevent neurotoxic effects due to metabolite accumulation. Differential expression of metabolite clearing enzymes such as aldehydedehydrogenases (ALDHs) may contribute to increased vulnerability of subsets of mesDA neurons at various stages of disease progression (Fitzmaurice et al., 2013; Liu et al., 2014). In how far these molecular features of specific mesDA subpopulations, or a specific combination thereof, contribute to the observed higher vulnerability during PD progression, and whether these may at some point offer entry points for prevention or therapy remains to be elucidated in future studies, for example by direct comparison of specific neuronal subpopulations.
Among the various types of external stress conditions, increased sensitivity toward oxidative stress is thought to be one of the most common underlying mechanisms for PD leading to dysfunction of the cells and finally neuronal cell loss. Major evidence for this idea is based on a large number of studies in postmortem samples of PD patients showing that these samples display evidence for oxidative stress, for example increased levels of oxidized proteins and deregulation of antioxidant proteins (Toulorge, Schapira, & Hajj, 2016). Surprisingly, in vitro model systems have contributed to the notion that patient neurons display increased vulnerability even under normal cultivation conditions: Several studies provide evidence for increased neuronal degeneration (indicated by detection of cleaved‐caspase 3 [c‐casp3]) in cultured mesDA neurons from LRRK2G2019S, LRRK2I2020T, iPD background, PRKNmutant as well as SNCA triplication carriers compared to healthy controls (Lin et al., 2016; Ohta et al., 2015; Sanchez‐Danes et al., 2012; Suzuki et al., 2017). In the context of apoptosis regulation, it is important to mention that Lopez and colleagues recently described an impaired NF‐κB response in mesDA neuronal cultures of both G2019S and R1441G variants, which might point to fundamental defects in antiapoptotic signaling (Lopez de Maturana et al., 2016). A recent study linked LRRK2 to neuronal cell death regulation via the direct phosphorlyation and activation of apoptosis signal‐regulating kinase 1 (ASK1). The authors demonstrated that MG132‐induced susceptibility toward apoptosis, nuclear aberration and impairment of colony formation were rescued by either LRRK2 kinase inhibitor‐1 (IN‐1) or ASK1 inhibitor treatment in isogenic LRRK2G2019S NPCs (Yoon et al., 2017). In addition, Sandor and colleagues performed transcriptomic profiling of purified LRRK2G2019S and control mesDA neuronal cultures, thereby revealing significant functional convergence among differentially expressed genes, with ‘oxidative stress’ representing one of the top four affected gene ontology terms (Sandor et al., 2017). The relevance of oxidative stress in PD pathogenesis is supported by the observation that exposure to pro‐oxidative industrial chemicals or cellular stressors like rotenone, hydrogen peroxide (H2O2) or 6‐hydroxydopamine (6‐OHDA) can suffice to induce PD‐related neurodegeneration in various model systems (Goldstein, Kopin, & Sharabi, 2014). In consequence, several groups assessed whether patient‐derived mesDA neuronal cultures exhibit increased susceptibility toward these stressors. Cellular models including LRRK2G2019S, LRRK2I2020T and SNCAtrp variants revealed increased c‐casp3 levels upon treatment with H2O2 compared to control cultures (Byers et al., 2011; Nguyen et al., 2011; Ohta et al., 2015). Intriguingly, decreased survival after exposure to H2O2 has been shown previously in mouse primary cortical neurons transfected with either wild‐type or mutant LRRK2 (G2019S/I2020T) (West et al., 2007).
In a similar approach, 6‐OHDA treatment decreased cell survival in LRRK2G2019S neurons as well as in SNCAtrp cultures (Lin et al., 2016; Nguyen et al., 2011; Reinhardt, Glatza et al., 2013; Reinhardt, Schmid et al., 2013). Further underscoring these findings, LRRK2 variant mesDA neuronal cultures showed decreased cell survival after exposure to rotenone (Reinhardt, Glatza et al., 2013; Reinhardt, Schmid et al., 2013) and increased sensitivity toward the mitochondrial stressors valinomycin and concanamycin A (Cooper et al., 2012).
Increased vulnerability of patient‐derived mesDA neurons is not restricted to oxidative stress: This is exemplified by the finding that LRRK2G2019S mesDA neurons and NPCs (Liu et al., 2012; Nguyen et al., 2011; Yoon et al., 2017) display significantly increased sensitivity toward treatment with the proteasomal inhibitor MG132, pointing to a higher susceptibility of mutant carriers toward proteotoxic stress. In this context, it is important to note that, aside from LRRK2, several models for familial PD show evidence for increased sensitivity toward various forms of cellular stress, e.g. PRKN (Chang et al., 2016; Chung et al., 2016; Imaizumi et al., 2012; Suzuki et al., 2017), SNCAtrp (Flierl et al., 2014), A53Tmut (Kouroupi et al., 2017), PINK1 (Chung et al., 2016; Cooper et al., 2012) and GBA (Schöndorf et al., 2014).
In summary, LRRK2 variant carriers display increased vulnerability under normal growth conditions and under oxidative as well as proteotoxic stress conditions. This increased stress sensitivity may derive from the discussed role of LRRK2 in regulation of mitochondrial function and proteostasis pathways.
3.5. Alterations in subcellular organization
LRRK2 has been described to be involved in regulation of subcellular architecture and organelle organization (reviewed by Roosen & Cookson, 2016). In 2012, the Belmonte lab focused on alterations of nuclear morphology in isogenic NPCs carrying the G2019S mutations (Liu et al., 2012). Using lamin B staining, the authors revealed the appearance of deformed nuclei in late passages of G2019S carriers, accompanied by impaired clonal expansion and defective neuronal differentiation, which could be rescued either by LRRK2‐kinase inhibitor treatment or targeted correction of the LRRK2 gene. Interestingly, disorganized nuclear envelope architecture manifested already at the neural progenitor stage at late passages and was not conserved upon differentiation of the NPCs into neurons, suggesting that this phenotype manifests early during development and may be harder to detect at later stages. Besides affecting nuclear organization, LRRK2 has also been linked to regulation of GA morphology: LRRK2 interacts with cyclin‐G‐associated kinase (GAK) and RAB29 (also known as RAB7L1), both of which represent risk factors for sporadic PD that are controlling function and morphology of the GA (Beilina et al., 2014). Moreover, RAB7L1 is a direct LRRK2 kinase substrate, and the G2019S mutation of LRRK2 significantly increases RAB7L1 phosphorylation and recruitment of LRRK2 to the GA cisternae (Liu et al., 2018; MacLeod et al., 2013). Joseph Mazzulli and colleagues showed in a different genetic model for PD that SNCAtrp iPSC‐derived dopaminergic cultures contained increased numbers of neurons with a highly fragmented GA (Mazzulli, Zunke, Isacson et al., 2016). These findings support the notion of overlapping GA‐associated phenotypes in different genetic models of PD. In addition to modulation of the architecture of the nucleus and GA, LRRK2 has been linked to formation of the primary cilium through interaction with Rab10 and RILPLs in a recent publication (Dhekne et al., 2018). The authors have shown that pathogenic LRRK2 has a profound inhibitory effect on cilia formation in human cancer cells, in human G2019S iPSCs as well as R1441C mouse primary neurons. They conclude that this impairment of ciliogenesis may inhibit cilia‐mediated signaling via the SHH signaling pathway. It is important to note that SHH signaling is not only pivotally involved in neurogenesis but also has been shown to protect dopaminergic neurons against cytotoxic effects of the neurotoxin MPP+ (Miao et al., 1997). Further studies are warranted to assess the details underlying LRRK2's role in ciliogenesis and SHH signaling. Taken together, LRRK2 and its variants are tightly linked to the organization and function of various subcellular structures.
3.6. Changes in cell morphology
One key function of LRRK2 with respect to cell homeostasis is the regulation of cytoskeleton balance (as outlined above). The vital importance of this process in the context of neurodegenerative diseases is underscored by the essential role of the cytoskeleton in maintaining structural polarity of neurons and thus their physiological function. In particular, the extensive axon length of DA neurons, their high level of arborization (see below) and the requirement for orchestrated dopamine vesicle transport points to the necessity of tight cooperation between microtubule dynamics, motor protein activity and maintenance of cell morphology through actin fibers and membrane anchoring components. Moreover, it is intriguing to realize that Lewy bodies have been shown to contain tubulins, microtubule‐associated proteins and neurofilament components. Finally, several PD‐associated proteins, including α‐syn, PINK1 and parkin have been linked to actin remodeling (Kim & Son, 2010; Lim et al., 2007). These data emphasize the central role of LRRK2 as key regulator of cytoskeletal dynamics and point to potential connections between this homeostatic role and PD etiopathology. Consequently, several studies focused on the analysis of neurite length and complexity of mesDA neuronal cultures. It was shown by independent studies that neurite length and complexity are significantly reduced in LRRK2G2019S mesDA neurons (Borgs et al., 2016; Lin et al., 2016; Sanchez‐Danes et al., 2012; Schwab & Ebert, 2015; Su & Qi, 2013). Reinhardt and colleagues demonstrated a reduced neurite outgrowth velocity of LRRK2G2019S mesDA neuronal cultures which could be rescued by continuous treatment with the LRRK2 kinase inhibitor‐1 (IN‐1) or ERK inhibitor (Reinhardt, Schmid et al., 2013). Surprisingly, in a recent study, the same group demonstrated that a decrease in neurite outgrowth in G2019S cultures was not corrected by CZC‐251146, another well characterized inhibitor of LRRK2 kinase activity (Marrone et al., 2018). As shown by Schwab and Ebert, peripheral neurons carrying the LRRK2G2019S variant exhibit no significant reduction in neuronal length, but display significantly larger and more neurite aggregates compared to controls, a phenotype which could be partially rescued by the LRRK2 kinase inhibitor IN‐1 (Schwab & Ebert, 2015). Recently, the results of another study corroborated the phenotype of reduced neurite length and showed that treatment of LRRK2G2019S neurons with IN‐1 could rescue the shortened neurite length (Qing et al., 2017).
From our own data, we would conclude that treatment with IN‐1 can rescue the reduced neurite lengths in LRRK2G2019S mesDA neuronal cultures. For these experiments, immature dopaminergic cultures generated according to the floor plate protocol were dissociated at day 24 of differentiation and replated for 24 hr (Figure 4a). Staining for tyrosine hydroxylase (TH) was used to identify mesDA neurons, and neurite length was determined using quantitative image analysis. LRRK2G2019S mesDA neurons displayed significantly reduced neurite lengths when compared to controls. This decrease in neurite length was rescued by treatment with IN‐1 (Figure 4b,c).
Figure 4.
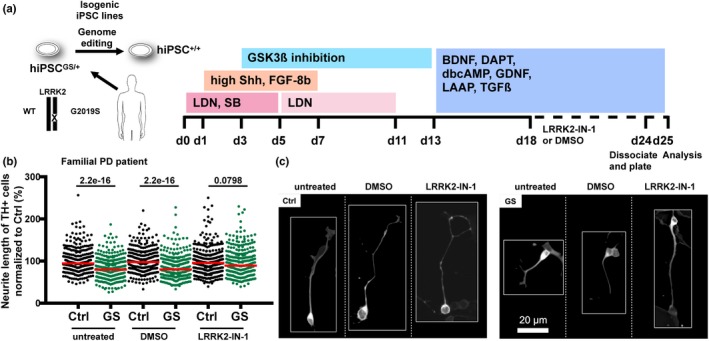
LRRK2G2019S mesDA neuronal cultures exhibit decreased neurite length which can be rescued by LRRK2 kinase inhibitor treatment. (a) Schematic overview of the experimental set up. Control (Ctrl) and isogenic G2019S (GS) mesDA neuronal cultures were either untreated (left), treated with DMSO or treated with 1 μM LRRK2‐IN1 (IN‐1) and subjected to a neurite outgrowth assay. LDN, LDN193189; SB, SB431542. (b) Quantification of neurite length demonstrates that decreased neurite lengths in LRRK2G2019S neurons can be rescued by treatment with IN‐1. For each condition, 2–3 wells were analyzed in three independent experimental sets (n = 3). A total of 4982 neurites were quantified. Each dot represents the neurite length of one individual neurite (expressed as percentage from the mean of the respective healthy control). Red lines indicate the median. (c) Exemplary images of isogenic control and LRRK2G2019S TH + neurons under different experimental conditions. For details on the methodology, see Supporting Information
Interestingly, reduced neurite length appears to be specific for only a subset of PD risk genes. While this phenotype was repeatedly demonstrated for LRRK2 and also for SNCAtrp (Lin et al., 2016), A53Tmut (Kouroupi et al., 2017) and iPD (Sanchez‐Danes et al., 2012), conflicting or inconclusive data have been presented for PRKN and PINK1 (Lin et al., 2016; Miller et al., 2013; Ren et al., 2015). In addition, a model for GBA1N370S did not show clear evidence for reduced neurite length (Woodard et al., 2014). Neurite aggregates as described by Schwab and Ebert for LRRK2G2019S (Schwab & Ebert, 2015) were also observed in A53Tmut cultures (Kouroupi et al., 2017). Given the functional involvement of LRRK2 in regulation of cytoskeleton dynamics and directed vesicle transport, these findings may point to LRRK2‐specific defects in the establishment of neuronal morphology.
3.7. Cell–cell communication
As outlined above, LRRK2 function is linked to regulation of Ca2+ homeostasis, cytoskeleton dynamics, cell morphology and vesicle trafficking as well as several other pathways. Considering the contribution of all these cellular mechanisms to neuronal functionality and the role of LRRK2 in synapse formation and function (discussed above), it is tempting to hypothesize that LRRK2 variants may alter neuronal activity and network function in mesDA neurons. Consequently, electrophysiological properties of LRRK2G2019S and control mesDA neuronal cultures were analyzed in detail: Using multi‐electrode array (MEA) measurements Lin and colleagues found that neuronal networks composed of LRRK2G2019S cells showed significantly reduced firing rates and synchrony as compared to control cultures (Lin et al., 2016), which points to a LRRK2G2019S‐based impairment of neuronal network activity. Focusing on the key neurotransmitter dopamine, several groups assessed dopamine metabolism of mesDA neuronal cultures of control and PD patients. Nguyen and colleagues observed reduced dopamine levels in G2019S cultures compared to controls (Nguyen et al., 2011). Along the same line, LRRK2I2020T mesDA neuronal cultures were found to exhibit a decreased Ca2+‐dependent dopamine release compared to controls (Ohta et al., 2015). While reliable detection of altered dopamine release remains challenging in the context of LRRK2‐focused iPSC models (as demonstrated by two studies that could not confirm alterations in dopamine release of LRRK2G2019S mesDA neurons (Reinhardt, Schmid et al., 2013; Lin et al., 2016), several PD risk factors aside from LRRK2 have been shown to elicit alterations in either release or uptake of DA, including GBA (Woodard et al., 2014) and PRKN (Jiang et al., 2012). A negative effect of LRRK2 mutations on dopamine release, likely caused by altered burst firing in SNpc‐mesDA neurons, has been confirmed in an in vivo rat model in which LRRK2G2019S and LRRK2R1441C are overexpressed. In addition, experimental data from overexpression studies and animal models suggest a central role for LRRK2 in release of dopamine‐containing vesicles as well as in regulation of dopamine receptor abundance and internalization (Cirnaru et al., 2014; Migheli et al., 2013; Rassu et al., 2017; Sloan et al., 2016). A recent study demonstrated that the LRRK2 variants R1441C/G and G2019S increase phosphorylation of auxilin (a protein that mediates uncoating of clathrin‐coated vesicles) and thus impair synaptic vesicle endocytosis (SVE) (Nguyen & Krainc, 2018). Consequences of impaired SVE were a lower synaptic vesicle density and increased oxidized DA levels found in LRRK2G2019S and LRRK2R1441C/G variants (Nguyen & Krainc, 2018), similar to findings in DJ‐1mut mesDA neurons in a previous publication (Burbulla et al., 2017).
While the majority of studies on LRRK2 variants in PD pathogenesis focus on the role of the variants in dopaminergic neurons as the main site of manifestation of PD (see also the chapter by Emma Lane in this Special Issue), recent evidence suggests a role for LRRK2 in the maturation of striatal projection neurons (SPNs), one of the neuronal populations that receives dopaminergic input. LRRK2 is highly expressed in SPNs (Parisiadou et al., 2014). It also interacts with the PKA regulatory subunit II beta to regulate activity of protein kinase A (PKA) during synapse formation and in response to dopamine. Loss of LRRK2 results in increased, PKA‐mediated phosphorylation of the actin‐cytoskeletal regulator cofilin and the glutamate receptor Glut1, and deficient synaptogenesis (Parisiadou et al., 2014). Importantly, disease‐associated mutations in LRRK2 mutation interfere with synapse formation and synaptic function, both in SPNs and in hippocampal neurons (Matikainen‐Ankney et al., 2016, 2018; Sweet, Saunier‐Rebori, Yue, & Blitzer, 2015).
As the detailed molecular analysis of cell–cell communication represents a key challenge in the investigation of various disorders of the CNS (including neurodegeneration), establishment of iPSC‐centered technologies for quantitative assessment of associated parameters warrants further investigation.
3.8. Neuroinflammation
Inflammation in the CNS (and to a certain degree also in the periphery) is a major pathological hallmark of PD (Doorn et al., 2014; Imamura et al., 2003; Vawter, Dillon‐Carter, Tourtellotte, Carvey, & Freed, 1996), accompanied by microglia activation and the presence of reactive astrocytes (Miklossy, Doudet et al., 2006) (see also the chapter by Malú Tansey and Marina Romero‐Ramos in this Special Issue). Currently, it still needs to be elucidated if mesDA neuronal loss is causative for changes in microglia activation status and behavior or a consequence of neuron demise (as reviewed by Le, Wu, & Tang, 2016). Interestingly, microglia and macrophages express high levels of LRRK2 and further upregulate LRRK2 expression in response to pro‐inflammatory cytokines. In addition, the LRRK2 protein is involved in regulation of phagocytosis, cytokine release as well as migration, suggesting a pivotal role for LRRK2 in immune cells (Lee, James, & Cowley, 2017). LRRK2 is also expressed in astrocytes (Miklossy, Arai et al., 2006), and several studies implicate a role for the protein in the autophagy–lysosomal pathway in those cells (reviewed by Booth, Hirst, & Wade‐Martins, 2017). Given the fact that astrocytes and microglia have been linked to the progression of PD and both express LRRK2, future studies involving iPSC‐derived astro‐ and microglia alone or in co‐culture with mesDA neurons could provide further insight into potential contribution of these cell types to the progression of the disease. So far, only few iPSC studies have focused on non‐neuronal cells. Haenseler and co‐workers derived macrophages (which have a similar ontogeny as microglia) from iPSCs carrying different variants in the SNCA gene (Haenseler et al., 2017). The authors reported that SNCAtrp but not SNCAA53T variant macrophages exhibited significantly elevated intracellular α‐syn levels and increased release of α‐synuclein into the medium. Furthermore, phagocytosis was reduced in SNCAtrp macrophages compared to A53T variant and control macrophages (Haenseler et al., 2017). These data point to the relevance of the macrophage lineage in the pathogenesis of PD and warrant a detailed analysis of other PD risk factors such as LRRK2 in the context of immune cell activity.
In summary, the plethora of iPSC‐based studies presented here, including our own exemplary data, underscore the multifaceted role of LRRK2 and its variants in PD etiopathology. While some pathologically relevant mechanisms (including proteostasis, mitochondrial function, stress response) emerge as common disease pathways that are also affected by polymorphisms in other PD‐related candidate genes (SNCA, PINK1, PRKN), other traits (such as effects on cell morphology) appear to be more specific for LRRK2. Unraveling the functional implications of disease‐linked variants (e.g. LRRK2G2019S) in combination with detailed knowledge about the affected pathways will be vital for gaining a comprehensive understanding of PD etiopathology for a given genotype. As outlined here, iPSC technology (and especially an isogenic approach) is a powerful experimental tool to identify and study specific phenotypes in vitro and to establish suitable experimental routines and cell‐based assays for disease modeling and drug discovery.
4. CURRENT LIMITATIONS AND FUTURE DIRECTIONS OF IN VITRO DISEASE MODELING
The examples outlined above illustrate how iPSC technology can be used to explore potential disease mechanisms and to assess candidate drugs in the context of a neurodegenerative disease (i.e. PD). This methodology offers a number of advantages over more conventional model systems such as animal models, human cancer cell lines or immortalized cells: (a) easy access to human neurons which are otherwise not accessible; (b) derivation of desired cell type(s) (e.g. mesDA neurons) presumably directly affected by the disease; (c) application of non‐immortal cells that may react authentically to internal and external stress conditions (in contrast to e.g. cancer cell lines); and (d) usability of patient‐specific material for individualized analysis and/or drug development approaches. In addition, the advances in genome‐editing technologies have enabled the establishment of target cells with desired genomic make‐up, expanding the possibilities of using genotype‐specific cells in combination with highly selective reporter systems and/or genetic interference technology. Intriguingly, iPSC‐based in vitro models appear to exhibit disease‐related phenotypes (such as protein aggregation, altered cell morphology or stress‐induced cell death) much earlier than in the patient in vivo. While it takes years to decades for a certain defect to appear in patient tissue, in vitro models display pathophenotypes sometimes within weeks or months of continued cultivation. Reasons for this accelerated manifestation may be stress‐inducing cell cultivation conditions (high oxygen levels, minimal media composition, non‐physiological oxidant/antioxidant levels), accelerated proliferation and forced differentiation due to high levels of growth and patterning factors, and the lack of adaptation and compensatory mechanisms present in a multicellular organismic context (e.g. contact inhibition, extracellular matrix, glial and endothelial cells). Supporting this notion, Chen and colleagues (Chen et al., 2014) reported neurofilament aggregation and neurite degeneration in an iPSC model for SOD1‐associated amyotrophic lateral sclerosis within weeks in the absence of glial cells. In this context, it is also important to note that the respective in vitro conditions may favor appearance of phenotypes that are genotype‐ and cell type‐specific but not directly linked to the diseased condition in vivo. While these features may prove helpful for screening purposes and for unraveling of underlying general principles linked to disease manifestation and progression, accelerated phenotype manifestation in vitro might further contribute to phenotypic variability. In order to address this issue and to optimize experimental efforts, a careful statistical power analysis for the observed data sets and their intrinsic variations is highly desirable. While proper power analysis and sample size estimation is routinely applied in the context of animal studies, systematic use of these tools in the field of in vitro disease modeling is still to be improved. One reason for this might be that to estimate how many replicates are necessary to detect a given phenomenon, it is crucial to not only define the analysis’ alpha‐level and power but also to appreciate the expected effect size. This is not trivial, especially in cases where the effect under investigation is not well studied and potentially not even described for the cell type under investigation. It becomes even more challenging once several techniques are implemented to analyze one effect or in cases where technical variation is comparably high (e.g. protein quantification by western blot). Moreover, iPSC‐based studies are typically very laborious, thus limiting the experimental sample sizes and number of repeats feasible in the context of a single study. However, choosing appropriate experimental designs a priori and suitable analysis methods reduces the risk of identifying effects unspecific to the investigated condition, for instance effects caused by genetic or epigenetic variability instead of being truly disease‐associated. In 2017, Germain and Testa published a study demonstrating the importance of these aspects by analyzing the effect of sample sizes in iPSC‐based models on detecting differentially expressed genes by transcriptomic profiling (Germain & Testa, 2017). In this study, the authors addressed the high degree of genetic variation between different donors and critically reflected on the common habit in the iPSC field to make use of various clones of one donor to address this biological variability. The results of their study clearly argue for an increase in the number of individuals analyzed, although using different clones of one individual can be additionally implemented to increase the sensitivity of the performed analyses. However, in case of using more than a single clone per donor, it is mandatory to carefully consider the choice of statistics performed during analysis in order to prevent a drastic increase of the false discovery rate (Germain & Testa, 2017). In conclusion, careful experiment planning, such as to implement isogenic systems whenever applicable to mitigate genetic variability from the beginning, is mandatory to ensure the validity of data provided by iPSC in vitro systems.
In addition to interindividual genetic heterogeneity between different iPSC donors, genetic heterogeneity in neurons of the same individual is increasingly emerging as a further confounder. As revealed by single cell whole genome sequencing, neurons accumulate somatic single‐nucleotide variants (SNVs) as a result of active transcription or retrotransposon activity (Coufal et al., 2009; Kurnosov et al., 2015; Lodato et al., 2015; Muotri et al., 2005). Recent evidence suggests that this process could also be instrumental in the development of neurodegenerative diseases such as Alzheimer's disease (Lee et al., 2018). It has yet to be explored whether and to what extent this phenomenon can be recapitulated using in vitro systems such as iPSC models.
Despite the obvious advantages offered by iPSC‐based approaches , it is important to realize that iPSC models come with a number of limitations intrinsic to the current technology. In vitro models naturally represent reductionist systems and can only serve as proxy for the physiological situation. In a typical setting, one would focus on one gene or a small number of variants at a time and analyze a limited number of phenotypes. A global assessment of the plethora of the disease‐related mechanisms usually exceeds the scope of a single study. Also, while some of the phenotypes observed in a cell culture scenario may not directly be related to in vivo pathogenesis, they can still provide valuable information on molecular pathways affected by distinct genetic variants in a defined and disease‐relevant human cell type.
A further challenge in iPSC‐based disease modeling is that in vitro differentiation protocols may yield variable results with respect to quality, quantity and purity of the target cell population. An illustrative example in the context of PD research is the derivation of different populations of mesDA neurons from iPSCs by using distinct differentiation paradigms: While the ‘floor plate protocol’ (Kirkeby et al., 2012; Kriks et al., 2011; Nolbrant, Heuer, Parmar, & Kirkeby, 2017; Xi et al., 2012) is based on early patterning of the differentiating cells toward a floor plate stage and thus recapitulates the developmental origin of primary mesDA neurons, other protocols include a rosette‐like stage for expansion of neural progenitor‐like cells (e.g. Perrier et al., 2004). A number of elegant studies indicate that floor plate‐derived mesDA neurons are superior to dopamine‐like neurons generated from expanded NPC populations with respect to both functionality (Kirkeby et al., 2012; Kriks et al., 2011; Steinbeck et al., 2015) and the induction of disease‐associated phenotypes in vitro (Chung et al., 2016). Addressing the topic of cell type authenticity, an array of mesDA‐specific markers is typically used to characterize in vitro differentiated dopamine neurons, including enzymes involved in the synthesis of dopamine (TH, AADC), dopamine and monoamine transporter (DAT and VMAT, respectively) as well as transcription factors that indicate ventral midbrain specification (e.g. LMX1A, FOXA2, OTX2) and specificity (NURR1, PITX3, GIRK2, ALDH1A1, CALB) (Arenas, Denham, & Villaescusa, 2015). It is important to note that none of these factors is specifically expressed in mesDA neurons and that only the combined expression of these markers validates that the generated cell types are bona fide mesDA neurons.
Today, developmental specification of human pluripotent stem cells (hPSCs) into mesDA neurons is relatively well understood. Recent protocols for generating mesDA neurons from hPSCs offer the possibility of generating an unlimited number of subtype‐specific mesDA neurons in vitro that mature and are functional after transplantation in vivo (Chen et al., 2016; Doi et al., 2014; Kirkeby et al., 2012, 2017; Kriks et al., 2011). Importantly, the iPSC‐derived mesDA neurons even display characteristics of the PD‐susceptible SNpc (A9‐subtype specific) mesDA neurons (Kee et al., 2017; Kirkeby et al., 2012, 2017; Kriks et al., 2011; Mendez et al., 2005). In animal models of PD, transplanted human PSC‐derived mesDA neurons have been shown to innervate distant target structures and to be functionally similar to bona fide mesDA neurons derived from fetal ventral midbrain with regard to morphology, marker expression and axonal projection patterns (Grealish et al., 2014; Hallett et al., 2015). Grafted iPSC‐derived mesDA neurons were found to release dopamine and reverse motor deficits in animal models of PD (Kirkeby et al., 2012; Kriks et al., 2011). Moreover, there is evidence that they modulate the glutamatergic input into striatal projection neurons (Steinbeck et al., 2015). Thus, it is fair to say that human PSC‐derived mesDA neurons represent a suitable in vitro model that recapitulates phenotypical and physiological hallmarks as well as functional properties of its in vivo counterparts.
In addition to authenticity, maturity of the in vitro generated neurons is of critical importance. This is particularly relevant as neurons differentiated from PSCs typically exhibit maturation stages reflecting an early fetal age. While automated cell culture systems can facilitate prolonged cultivation across several months under strictly defined conditions (Terstegge et al., 2006), this strategy severely limits feasibility and throughput of disease modeling applications. One way to expedite the generation of mature functional neurons from PSCs is direct cell fate conversion by forced expression of lineage‐specific transcription factors (‘forward programming’). For instance, doxycyclin‐induced expression of neurogenin 2 (NEUROG2) in hESCs is sufficient to derive highly enriched neuronal cultures which, after maturation on mouse glial cells, exhibit mature properties of excitatory cortical neurons and even form functional synapses with mouse host neurons upon transplantation (Zhang et al., 2013). Consequently, forward programmed neurons are increasingly employed for modeling neurological diseases. For example, the Südhof group utilized genome editing in hESCs and subsequent NEUROG2 expression to derive neurons carrying knock‐out mutations in the autism‐associated SH and multiple ankyrin repeat domains 3 (SHANK3) gene (Yi et al., 2016) or in the autism‐ as well as schizophrenia‐associated neurexin 1 (NRXN1) gene (Pak et al., 2015). The functional characterization of these in vitro models revealed phenotypes that are characterized by changes in electrophysiological properties and/or defects in synaptic function and plasticity (Pak et al., 2015; Yi et al., 2016).
As an alternative to forward reprogramming, somatic cells such as fibroblasts may be directly converted into ‘induced’ neurons (iNs). This approach has been pioneered by the Wernig laboratory which initially screened a pool of nineteen transcription factors involved in neural tissue development or epigenetic reprogramming for their ability to transdifferentiate mouse fibroblasts into neurons (Vierbuchen et al., 2010). This set was finally reduced to three factors, which proved to be crucial for an efficient fibroblast‐to‐iN conversion: achaete‐scute homolog 1 (ASCL1), POU class 3 homeobox 2 (POU3F2, aka BRN2) and myelin transcription factor 1 like (MYT1L), a combination that is now commonly known as ‘BAM’ (Vierbuchen et al., 2010). Although overexpression of BAM alone was shown to be insufficient to obtain iNs from human fibroblasts, adding neurogenic differentiation 1 (NEUROD1) to the transcription factor cocktail resulted in the derivation of mostly excitatory glutamatergic neurons, which expressed mature neuronal and synaptic markers after five to six weeks of differentiation and were capable of functionally integrating into pre‐existing mouse cortical neuronal networks after co‐culture in vitro (Pang et al., 2011). Meanwhile, the iN approach has been refined to induce specific neuronal subtypes such as induced dopamine neurons (iDAs): For generating human iDA, the BAM cocktail or the neurogenic transcription factor ASCL1 were combined with overexpression of genes associated with the establishment of the dopaminergic fate such as nuclear receptor related‐1 protein (NURR1), LIM homeobox transcription factor 1 A and B (LMX1A/B), forkhead box protein A2 (FOXA2) and orthodenticle homeobox 2 (OTX2) (Caiazzo et al., 2011; Pfisterer et al., 2011; Torper et al., 2013). However, these approaches still suffer from a rather low yield of mesDA neurons and are limited with regard to the degree of iDA maturation (Caiazzo et al., 2011; Pereira et al., 2014; Pfisterer et al., 2011).
A major limitation of the iPSC technology is posed by the fact that reprogramming of somatic cells into the pluripotent state resets not only the cell's identity but also erases its age‐associated epigenetic memory (Horvath, 2013). Consequently, iPSCs exhibit embryonic‐like epigenetic profiles with only very weak residual aging signatures (Lo Sardo et al., 2017), thus posing a severe limitation for modeling age‐related pathologies. As a potential work‐around, aging hallmarks (such as shortened telomeres, nuclear morphology abnormalities, global loss of the heterochromatin markers, increased mitochondrial ROS production, protein oxidation, DNA damage and senescence) may be promoted in iPSC‐derived fibroblasts and partially also iPSC‐derived mesDA‐like neurons (which otherwise lack these cellular aging signatures) by overexpression of progerin (Miller et al., 2013). This protein represents a truncated variant of the nuclear envelope complex component lamin A known to be involved in the pathology of the premature aging disease Hutchinson–Gilford progeria syndrome (Miller et al., 2013). Demonstrating the applicability of this approach for modeling late‐onset age‐related disease traits, the Studer laboratory showed that ectopic expression of progerin induces various aging‐related markers, including dopamine‐specific phenotypes (e.g. neuromelanin accumulation), in iPSC‐derived mesDA neurons (Miller et al., 2013). An alternative to the artificial induction of cellular aging in iPSC‐derived neurons might be the iN approach (described above), which as such bypasses the pluripotent stage and thereby might omit cellular rejuvenation. For example, Mertens and colleagues found that directly converted iNs preserve donor age‐dependent transcriptomic signatures after comparing the transcriptomes of human fibroblasts, fibroblast‐derived iNs, iPSCs, iPSC‐derived neurons and brain biopsies from young and aged donors (Mertens et al., 2015). Besides transcriptomic signatures, iN preserve the donor cell's age‐associated epigenetic memory and other aging hallmarks such as shortened telomere lengths, elevated ROS levels and accumulated DNA damage (Huh et al., 2016). Thus, it is not surprising that iNs have also entered the arena of disease modeling (reviewed by Drouin‐Ouellet, Pircs, Barker, Jakobsson, & Parmar, 2017). As a recent example, Huntington's disease (HD) patient fibroblasts converted into induced medium spiny neurons (iMSNs) were found to exhibit multifaceted mitochondrial dysfunctions, leading to oxidative DNA damage and spontaneous cell death (Victor et al., 2018). More importantly, HD‐iMSNs but not iPSC‐derived HD‐MSNs were shown to develop cytoplasmic and intranuclear mutant Huntingtin aggregates, a key hallmark of the HD pathology. Noteworthy, some phenotypes (e.g. mitochondrial dysfunction, proteostasis collapse and cell death) are depending on the patient's age and not present in iMSNs of young, pre‐symptomatic HD patients, thus indicating that iNs constitute a promising tool for modeling age‐dependent neurological disorders.
The conventional way of culturing cells as two‐dimensional (2D) cell monolayers is increasingly regarded as limited when it comes to modeling pathological changes in CNS tissue. Here, three‐dimensional (3D) cultivation techniques giving rise to self‐organizing entities (‘organoid cultures’) appear to be a promising strategy to depict the complexity of the brain. Pioneered in the context of cerebral organoids exhibiting cortical layer formation (Lancaster et al., 2013; Sasai, 2013), this field now moves toward the generation of other brain region‐specific organoids as exemplified by midbrain‐specific 3D cultures. Junghyun Jo and colleagues have recently reported on an experimental approach for establishing a 3D culture model of the human midbrain from hESCs (Jo et al., 2016). They demonstrate that hESCs can be differentiated into mesencephalic progenitors, which form midbrain floor plate‐like aggregates that differentiate in an organized manner (ventricular zone, intermediate zone, mantle layer) into mesDA neurons that display physiological properties of mature mesDA neurons and form functional synapses and neuronal networks. Intriguingly, midbrain‐like organoids contained mesDA neurons that produce neuromelanin, a pigment emerging in the context of SNpc‐mesDA neuron degeneration (Zecca, Zucca, Wilms, & Sulzer, 2003). So far, neuromelanin granule formation has only been shown for iPSC‐derived mesDA neurons in 2D when progerin was overexpressed to mimic cellular aging (Miller et al., 2013). Thus, the novel 3D midbrain‐like organoid system may provide a valuable platform to study the role of neuromelanin production in authentic and functional mesDA neurons, especially in the context of PD etiopathogenesis. Another study further underscores the utility of 3D systems for recapitulating cellular brain architecture: Jens Schwamborn's group did not use PSCs, but already pre‐patterned NPCs as starting population for the derivation of 3D midbrain floor plate‐like aggregates. Using this experimental shortcut, they demonstrated emergence of mesDA neurons, astrocytes and oligodendrocytes in an organized structure, accompanied by formation of synaptic connections and the appearance of myelinated axons (Monzel et al., 2017).
Although the current 3D models still suffer from significant limitations (e.g. the absence of microglia and vasculature in brain organoids and the lack of non‐dopaminergic midbrain progenitor domains), they clearly illustrate that neural aggregates and organoids are emerging as an exciting intermediate between 2D cultures and an organismic tissue context.
While today's in vitro differentiation protocols can deliver iPSC‐derived neurons closely resembling their physiological counterparts with respect to marker expression, ion channel abundance, neurotransmitter metabolism and cell‐intrinsic electrophysiological properties, it is by far more difficult to determine their functionality in a complex tissue context. In particular, the establishment of the input–output circuitry of a specific neuron is impossible to model in vitro as of yet. One promising experimental approach toward comprehensive analysis of circuit formation and synaptic connectivity may be rabies virus‐based retrograde transsynaptic tracing. Employing tissue clearing and subsequent light sheet microscopy, this approach has recently been used to delineate the innervation of grafted human neurons within a host mouse brain in a whole‐mount 3D (Doerr et al., 2017). Intriguingly, rabies‐based synaptic tracing has revealed that ventral midbrain‐patterned human ESC‐derived progenitor cells after grafting to midbrain extend axonal projections toward respective target structures in the forebrain and integrate into the host circuitry, resulting even in functional recovery in an OHDA‐based PD model (Cardoso et al., 2018). Future models might aim at translating circuitry back to an in vitro scenario, e.g. via the generation of defined neuronal networks in cell culture, which could serve as reductionist models for pharmaceutical circuit modulation.
Thus, despite a number of challenges, the current convergence of major advances in cell programming technology, genome editing and translation of cell culture paradigms from 2D toward organoids and defined networks provides exciting opportunities for the refinement of cell‐based disease modeling. It is to be expected that progress along this line will have a major impact on increasing our understanding of disease pathogenesis, the establishment of human cell‐based drug discovery platforms and personalized medicine.
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
BW performed experiments, analyzed data, assembled the figures, performed the literature searches and wrote the manuscript. SH, JJ, MH, MP and AT designed experiments, analyzed data and contributed text parts. LJF analyzed data, performed statistical analyses and wrote parts of the manuscript. GL, KS and JCIB critically revised and edited the manuscript. SB critically reviewed the manuscript and contributed individual sections. SH, MP, AT and OB supervised the experimental work and wrote sections of the manuscript. AT and OB supervised the entire project and handled the submission. All authors approved the final version of the manuscript.
Supporting information
ACKNOWLEDGEMENTS
We thank Michaela Segschneider, Cornelia Thiele, Melanie Bloschies and Iris Laufenberg for their outstanding technical assistance. This work was supported by grants from the German Ministry for Education and Research (BMBF, grant 01EK1603A‐Neuro2D3, to O.B., M.P. and A.T), the German Research Foundation (BL 767/3‐1 and BL 767/4‐1 to S.B.), the European Regional Development Fund (Stem Cell Factory III; grants EFRE‐0800977; EFRE‐0800978), and the European Union (grant FP7‐HEALTH‐F4‐2013‐602278‐Neurostemcellrepair to O.B.). Work in the laboratory of J.C.I.B. was supported by Universidad Catolica San Antonio de Murcia (UCAM) and G. Harold and Leila Y. Mathers Charitable Foundation.
Weykopf B, Haupt S, Jungverdorben J, et al. Induced pluripotent stem cell‐based modeling of mutant LRRK2‐associated Parkinson's disease. Eur J Neurosci. 2019;49:561–589. 10.1111/ejn.14345
Edited by Yoland Smith. Reviewed by Matt Farrer and Sabine Hilfiker
All peer review communications can be found with the online version of the article.
Contributor Information
Andreas Till, Email: a.till@uni-bonn.de.
Oliver Brüstle, Email: brustle@uni-bonn.de.
REFERENCES
- Aflaki, E. , Borger, D. K. , Moaven, N. , Stubblefield, B. K. , Rogers, S. A. , Patnaik, S. , … Sidransky, E. (2016). A new glucocerebrosidase chaperone reduces alpha‐synuclein and glycolipid levels in iPSC‐derived dopaminergic neurons from patients with Gaucher Disease and Parkinsonism. Journal of Neuroscience, 36, 7441–7452. 10.1523/JNEUROSCI.0636-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberico, S. L. , Cassell, M. D. , & Narayanan, N. S. (2015). The vulnerable ventral tegmental area in Parkinson's Disease. Basal Ganglia, 5, 51–55. 10.1016/j.baga.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglade, P. , Vyas, S. , Javoy‐Agid, F. , Herrero, M. T. , Michel, P. P. , Marquez, J. , … Agid, Y. (1997). Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histology and Histopathology, 12, 25–31. [PubMed] [Google Scholar]
- Aransay, A. , Rodriguez‐Lopez, C. , Garcia‐Amado, M. , Clasca, F. , & Prensa, L. (2015). Long‐range projection neurons of the mouse ventral tegmental area: A single‐cell axon tracing analysis. Frontiers in Neuroanatomy, 9, 59 10.3389/fnana.2015.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas, E. , Denham, M. , & Villaescusa, J. C. (2015). How to make a midbrain dopaminergic neuron. Development, 142, 1918–1936. 10.1242/dev.097394 [DOI] [PubMed] [Google Scholar]
- Avior, Y. , Sagi, I. , & Benvenisty, N. (2016). Pluripotent stem cells in disease modelling and drug discovery. Nature Reviews Molecular Cell Biology, 17, 170–182. 10.1038/nrm.2015.27 [DOI] [PubMed] [Google Scholar]
- Bardien, S. , Lesage, S. , Brice, A. , & Carr, J. (2011). Genetic characteristics of leucine‐rich repeat kinase 2 (LRRK2) associated Parkinson's disease. Parkinsonism & Related Disorders, 17, 501–508. [DOI] [PubMed] [Google Scholar]
- Barker, R. A. , Parmar, M. , Studer, L. , & Takahashi, J. (2017). Human trials of stem cell‐derived dopamine neurons for Parkinson's Disease: Dawn of a new era. Cell Stem Cell, 21, 569–573. 10.1016/j.stem.2017.09.014 [DOI] [PubMed] [Google Scholar]
- Bedford, C. , Sears, C. , Perez‐Carrion, M. , Piccoli, G. , & Condliffe, S. B. (2016). LRRK2 regulates voltage‐gated calcium channel function. Frontiers in Molecular Neuroscience, 9, 35 10.3389/fnmol.2016.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beevers, J. E. , Lai, M. C. , Collins, E. , Booth, H. D. E. , Zambon, F. , Parkkinen, L. , … Caffrey, T. M. (2017). MAPT genetic variation and neuronal maturity alter isoform expression affecting axonal transport in iPSC‐derived dopamine neurons. Stem Cell Reports, 9, 587–599. 10.1016/j.stemcr.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina, A. , Rudenko, I.N. , Kaganovich, A. , Civiero, L. , Chau, H. , Kalia, S.K. , … Cookson, M.R. (2014). Unbiased screen for interactors of leucine‐rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America, 111, 2626–2631. 10.1073/pnas.1318306111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, D. , Schweitzer, K. J. , Leitner, P. , Zimprich, A. , Lichtner, P. , Belcredi, P. , … Gasser, T. (2005). Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease*. Brain, 128, 3000–3011. 10.1093/brain/awh666 [DOI] [PubMed] [Google Scholar]
- Berger, Z. , Smith, K. , & LaVoie, M. (2010). Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry, 49, 5511–5523. 10.1021/bi100157u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, M. W. , Chakraborty, S. , Matthews, G. A. , Dougalis, A. , Wood, N. W. , Festenstein, R. , & Ungless, M. A. (2010). Hyperexcitable substantia nigra dopamine neurons in PINK1‐ and HtrA2/Omi‐deficient mice. Journal of Neurophysiology, 104, 3009–3020. 10.1152/jn.00466.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaess, S. , Bodea, G. O. , Kabanova, A. , Chanet, S. , Mugniery, E. , Derouiche, A. , … Joyner, A. L. (2011). Temporal‐spatial changes in Sonic Hedgehog expression and signaling reveal different potentials of ventral mesencephalic progenitors to populate distinct ventral midbrain nuclei. Neural Development, 6, 29 10.1186/1749-8104-6-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam, J. P. , & Pissadaki, E. K. (2012). Living on the edge with too many mouths to feed: Why dopamine neurons die. Movement Disorders, 27, 1478–1483. 10.1002/mds.25135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati, V. , Dekker, M. C. J. , Vanacore, N. , Fabbrini, G. , Squitieri, F. , Marconi, R. , … Italian Parkinson Genetics Network . (2002). Autosomal recessive early onset parkinsonism is linked to three loci: PARK2, PARK6, and PARK7. Neurological Sciences, 23, s59–s60. 10.1007/s100720200069 [DOI] [PubMed] [Google Scholar]
- Booth, H. D. E. , Hirst, W. D. , & Wade‐Martins, R. (2017). The role of astrocyte dysfunction in Parkinson's Disease pathogenesis. Trends in Neurosciences, 40, 358–370. 10.1016/j.tins.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgs, L. , Peyre, E. , Alix, P. , Hanon, K. , Grobarczyk, B. , Godin, J. D. , … Nguyen, L. (2016). Dopaminergic neurons differentiating from LRRK2 G2019S induced pluripotent stem cells show early neuritic branching defects. Scientific Reports, 6, 33377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosgraaf, L. , Waijer, A. , Engel, R. , Visser, A. J. , Wessels, D. , Soll, D. , & van Haastert, P. J. (2005). RasGEF‐containing proteins GbpC and GbpD have differential effects on cell polarity and chemotaxis in Dictyostelium. Journal of Cell Science, 118, 1899–1910. 10.1242/jcs.02317 [DOI] [PubMed] [Google Scholar]
- Brichta, L. , & Greengard, P. (2014). Molecular determinants of selective dopaminergic vulnerability in Parkinson's disease: an update. Frontiers in Neuroanatomy, 8, 152 10.3389/fnana.2014.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla, L. F. , Song, P. , Mazzulli, J. R. , Zampese, E. , Wong, Y. C. , Jeon, S. , … Krainc, D. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science, 357, 1255–1261. 10.1126/science.aam9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers, B. , Cord, B. , Nguyen, H. N. , Schüle, B. , Fenno, L. , Lee, P. C. , … Palmer, T. D. (2011). SNCA triplication Parkinson's patient's iPSC‐derived DA neurons accumulate α‐synuclein and are susceptible to oxidative stress. PLoS ONE, 6, e26159 10.1371/journal.pone.0026159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywood, P. T. , & Johnson, S. M. (2000). Differential vulnerabilities of substantia nigra catecholamine neurons to excitatory amino acid‐induced degeneration in rat midbrain slices. Experimental Neurology, 162, 180–188. 10.1006/exnr.2000.7310 [DOI] [PubMed] [Google Scholar]
- Caiazzo, M. , Dell'Anno, M. T. , Dvoretskova, E. , Lazarevic, D. , Taverna, S. , Leo, D. , … Broccoli, V. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature, 476, 224–227. 10.1038/nature10284 [DOI] [PubMed] [Google Scholar]
- Cannon, J. R. , & Greenamyre, J. T . (2010). Chapter 2 – Neurotoxic in vivo models of Parkinson's disease: recent advances In Björklund A. & Cenci M. A. (Eds.), Progress in Brain Research (pp. 17–33). Amsterdam, the Netherlands: Elsevier; 10.1016/S0079-6123(10)84002-6 [DOI] [PubMed] [Google Scholar]
- Cardoso, T. , Adler, A. F. , Mattsson, B. , Hoban, D. B. , Nolbrant, S. , Wahlestedt, J. N. , … Parmar, M. (2018). Target‐specific forebrain projections and appropriate synaptic inputs of hESC‐derived dopamine neurons grafted to the midbrain of Parkinsonian rats. Journal of Comparative Neurology, 526, 2133–2146. 10.1002/cne.24500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, K.‐H. , Lee‐Chen, G.‐J. , Wu, Y.‐R. , Chen, Y.‐J. , Lin, J.‐L. , Li, M. , … Chen, C.‐M. (2016). Impairment of proteasome and anti‐oxidative pathways in the induced pluripotent stem cell model for sporadic Parkinson's disease. Parkinsonism & Related Disorders, 24, 81–88. 10.1016/j.parkreldis.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Chen, H. , Qian, K. , Du, Z. , Cao, J. , Petersen, A. , Liu, H. , … Zhang, S.C. (2014). Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell, 14, 796–809. 10.1016/j.stem.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Xiong, M. , Dong, Y. , Haberman, A. , Cao, J. , Liu, H. , … Zhang, S. C. (2016). Chemical control of grafted human PSC‐derived neurons in a mouse model of Parkinson's Disease. Cell Stem Cell, 18, 817–826. 10.1016/j.stem.2016.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, H. J. , Yu, J. , Xie, C. , Rudrabhatla, P. , Chen, X. , Wu, J. , … Cai, H. (2014). Leucine‐rich repeat kinase 2 regulates Sec16A at ER exit sites to allow ER–Golgi export. The EMBO Journal, 33, 2314 10.15252/embj.201487807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, I. , Kim, B. , Byun, J.‐W. , Baik, S. H. , Huh, Y. H. , Kim, J.‐H. , … Joe, E.‐H. (2015). LRRK2 G2019S mutation attenuates microglial motility by inhibiting focal adhesion kinase. Nature Communications, 6, 8255 10.1038/ncomms9255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, Y. , Dodiya, H. , Aebischer, P. , Olanow, C. W. , & Kordower, J. H. (2009). Alterations in lysosomal and proteasomal markers in Parkinson's disease: Relationship to alpha‐synuclein inclusions. Neurobiology of Disease, 35, 385–398. 10.1016/j.nbd.2009.05.023 [DOI] [PubMed] [Google Scholar]
- Chung, S. Y. , Kishinevsky, S. , Mazzulli, J. R. , Graziotto, J. , Mrejeru, A. , Mosharov, E. V. , … Shim, J. W. (2016). Parkin and PINK1 patient iPSC‐derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha‐synuclein accumulation. Stem Cell Reports, 7, 664–677. 10.1016/j.stemcr.2016.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirnaru, M. D. , Marte, A. , Belluzzi, E. , Russo, I. , Gabrielli, M. , Longo, F. , … Piccoli, G. (2014). LRRK2 kinase activity regulates synaptic vesicle trafficking and neurotransmitter release through modulation of LRRK2 macro‐molecular complex. Frontiers in Molecular Neuroscience, 7, 49 10.3389/fnmol.2014.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiero, L. , Cirnaru, M. D. , Beilina, A. , Rodella, U. , Russo, I. , Belluzzi, E. , … Greggio, E. (2015). Leucine‐rich repeat kinase 2 interacts with p21‐activated kinase 6 to control neurite complexity in mammalian brain. Journal of Neurochemistry, 135, 1242–1256. 10.1111/jnc.13369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiero, L. , Cogo, S. , Kiekens, A. , Morganti, C. , Tessari, I. , Lobbestael, E. , … Greggio, E. (2017) PAK6 phosphorylates 14‐3‐3gamma to regulate steady state phosphorylation of LRRK2. Frontiers in Molecular Neuroscience, 10, 417 10.3389/fnmol.2017.00417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiero, L. , Dihanich, S. , Lewis, P. A. , & Greggio, E. (2014). Genetic, structural, and molecular insights into the function of Ras of complex proteins domains. Chemistry & Biology, 21, 809–818. 10.1016/j.chembiol.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L. , Ran, F. A. , Cox, D. , Lin, S. , Barretto, R. , Habib, N. , … Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson, M. R. (2010). The role of leucine‐rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nature Reviews Neuroscience, 11, 791–797. 10.1038/nrn2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, O. , Seo, H. , Andrabi, S. , Guardia‐Laguarta, C. , Graziotto, J. , Sundberg, M. , … Isacson, O. (2012) Pharmacological rescue of mitochondrial deficits in iPSC‐derived neural cells from patients with familial Parkinson's disease. Science Translational Medicine, 4, 141ra190. 10.1126/scitranslmed.3003985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal, N. G. , Garcia‐Perez, J. L. , Peng, G. E. , Yeo, G. W. , Mu, Y. , Lovci, M. T. , … Gage, F. H. (2009). L1 retrotransposition in human neural progenitor cells. Nature, 460, 1127–1131. 10.1038/nature08248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damier, P. , Hirsch, E. C. , Agid, Y. , & Graybiel, A. M. (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine‐containing neurons in Parkinson's disease. Brain, 122 (Pt 8), 1437–1448. [DOI] [PubMed] [Google Scholar]
- Dauer, W. , & Przedborski, S. (2003). Parkinson's disease: mechanisms and models. Neuron, 39, 889–909. [DOI] [PubMed] [Google Scholar]
- Dhekne, H. S. , Yanatori, I. , Gomez, R.C. , Tonelli, F. , Diez, F. , Schule, B. , … Pfeffer, S.R. (2018) A pathway for Parkinson's Disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. Elife, 7, e40202. https://doi.org/10.7554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio, R. , Hoffman, E.K. , Rocha, E.M. , Keeney, M.T. , Sanders, L.H. , De Miranda, B.R. , … Greenamyre, J.T . (2018). LRRK2 activation in idiopathic Parkinson's disease. Science Translational Medicine, 10, eaar5429. https://doi.org/10.1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr, J. , Schwarz, M. K. , Wiedermann, D. , Leinhaas, A. , Jakobs, A. , Schloen, F. , … Brustle, O. (2017). Whole‐brain 3D mapping of human neural transplant innervation. Nature Communications, 8, 14162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi, D. , Samata, B. , Katsukawa, M. , Kikuchi, T. , Morizane, A. , Ono, Y. , … Takahashi, J. (2014). Isolation of human induced pluripotent stem cell‐derived dopaminergic progenitors by cell sorting for successful transplantation. Stem Cell Reports, 2, 337–350. 10.1016/j.stemcr.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorn, K. J. , Moors, T. , Drukarch, B. , van de Berg, W. , Lucassen, P. J. , & van Dam, A. M. (2014). Microglial phenotypes and toll‐like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathologica Communications, 2, 90 10.1186/s40478-014-0090-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin‐Ouellet, J. , Pircs, K. , Barker, R.A. , Jakobsson, J. , & Parmar, M . (2017). Direct neuronal reprogramming for disease modeling studies using patient‐derived neurons: What have we learned? Frontiers in Neuroscience, 11, 530 10.3389/fnins.2017.00530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko, N. , Inesta‐Vaquera, F. , Zhang, J. , Xie, C. , Cai, H. , Arthur, S. , … Alessi, D. R. (2012). The IkappaB kinase family phosphorylates the Parkinson's Disease kinase LRRK2 at Ser935 and Ser910 during Toll‐like receptor signaling. PLoS ONE, 7, e39132 10.1371/journal.pone.0039132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava, V. M. , Manry, J. , Cobat, A. , Orlova, M. , Van Thuc, N. , Ba, N. N. , … Canadian Lrrk2 in Inflammation Team . (2016). A missense LRRK2 variant is a risk factor for excessive inflammatory responses in leprosy. PLoS Neglected Tropical Diseases, 10, e0004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes, H. J. , Hartfield, E. M. , Christian, H. C. , Emmanoulidou, E. , Zheng, Y. , Booth, H. , … Wade‐Martins, R. (2016). ER stress and autophagic perturbations lead to elevated extracellular alpha‐synuclein in GBA‐N370S Parkinson's iPSC‐derived dopamine neurons. Stem Cell Reports, 6, 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzmaurice, A. G. , Rhodes, S. L. , Lulla, A. , Murphy, N. P. , Lam, H. A. , O'Donnell, K. C. , … Bronstein, J. M. (2013). Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America, 110, 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flierl, A. , Oliveira, L. M. A. , Falomir‐Lockhart, L. J. , Mak, S. K. , Hesley, J. , Soldner, F. , … Schüle, B. (2014). Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha‐synuclein gene triplication. PLoS ONE, 9, e112413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehring, R. C. , Zhang, X. F. , Lee, J. C. , & Callaway, J. C. (2009). Endogenous calcium buffering capacity of substantia nigral dopamine neurons. Journal of Neurophysiology, 102, 2326–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foix, C. , & Nicolesco, N. J . (1925). Anatomie cérébrale. Les noyaux gris centraux et la región Mésencéphalo‐sous‐optique., Suivi d'un apéndice sur l'anatomie pathologique de la maladie de Parkinson.
- Foo, J. N. , Tan, L. C. , Irwan, I. D. , Au, W.‐L. , Low, H. Q. , Prakash, K.‐M. , … Tan, E.‐K. (2017). Genome‐wide association study of Parkinson's disease in East Asians. Human Molecular Genetics, 26, 226–232. [DOI] [PubMed] [Google Scholar]
- Franke, A. , McGovern, D. P. , Barrett, J. C. , Wang, K. , Radford‐Smith, G. L. , Ahmad, T. , … Parkes, M. (2010). Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nature Genetics, 42, 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard, A. , Pavese, N. , Hotton, G. , Turkheimer, F. , Es, M. , Hammers, A. , … Brooks, D. J. (2006). In vivo imaging of microglial activation with [11C](R)‐PK11195 PET in idiopathic Parkinson's disease. Neurobiology of Diseases, 21, 404–412. [DOI] [PubMed] [Google Scholar]
- Germain, P. L. , & Testa, G. (2017). Taming human genetic variability: Transcriptomic meta‐analysis guides the experimental design and interpretation of iPSC‐based disease modeling. Stem Cell Reports, 8, 1784–1796. https://doi.org/10.1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner, C. J. , Kinkl, N. , Schumacher, A. , Braun, R. J. , O'Neill, E. , Meitinger, T. , … Ueffing, M. (2006). The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Human Molecular Genetics, 15, 223–232. https://doi.org/10.1073 [DOI] [PubMed] [Google Scholar]
- Goetz, S. C. , & Anderson, K. V. (2010). The primary cilium: a signalling centre during vertebrate development. Nature Reviews Genetics, 11, 331–344. 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, D. S. , Kopin, I. J. , & Sharabi, Y. (2014). Catecholamine autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacology & Therapeutics, 144, 268–282. 10.1152/jn.00038.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Suaga, P. , Fdez, E. , Blanca Ramirez, M. , & Hilfiker, S. (2012). A link between autophagy and the pathophysiology of LRRK2 in Parkinson's Disease. Parkinson's Disease, 2012, 324521 10.1093/hmg/ddw379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Suaga, P. , Luzon‐Toro, B. , Churamani, D. , Zhang, L. , Bloor‐Young, D. , Patel, S. , … Hilfiker, S. (2012). Leucine‐rich repeat kinase 2 regulates autophagy through a calcium‐dependent pathway involving NAADP. Human Molecular Genetics, 21, 511–525. 10.1038/ng.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grealish, S. , Diguet, E. , Kirkeby, A. , Mattsson, B. , Heuer, A. , Bramoulle, Y. , … Parmar, M. (2014). Human ESC‐derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson's disease. Cell Stem Cell, 15, 653–665. https://doi.org/10.1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro, P. S. , Gerhardt, E. , Lopes da Fonseca, T. , Bähr, M. , Outeiro, T. F. , & Eckermann, K. (2016). LRRK2 promotes tau accumulation, aggregation and release. Molecular Neurobiology, 53, 3124–3135. 10.1007/s12035-015-9209-z [DOI] [PubMed] [Google Scholar]
- Haenseler, W. , Zambon, F. , Lee, H. , Vowles, J. , Rinaldi, F. , Duggal, G. , … Cowley, S. A. (2017). Excess alpha‐synuclein compromises phagocytosis in iPSC‐derived macrophages. Scientific Reports, 7, 9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett, P. J. , Deleidi, M. , Astradsson, A. , Smith, G. A. , Cooper, O. , Osborn, T. M. , … Isacson, O. (2015). Successful function of autologous iPSC‐derived dopamine neurons following transplantation in a non‐human primate model of Parkinson's disease. Cell Stem Cell, 16, 269–274. 10.1016/j.stem.2015.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassler, R . (1938). Zur Pathologie der paralysis agitans und des postencephalitschen Parkinsonismus. Journal of Psychology and Neurology, 48, 387–476. [Google Scholar]
- Heikkila, R. E. , Hess, A. , & Duvoisin, R. C. (1985). Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6- tetrahydropyridine in mice. Science, 224, 1451–1453. [DOI] [PubMed] [Google Scholar]
- Hirsch, E. C. , Hunot, S. , Damier, P. , & Faucheux, B. (1998). Glial cells and inflammation in Parkinson's disease: a role in neurodegeneration? Annals of Neurology, 44, S115–120. [DOI] [PubMed] [Google Scholar]
- Ho, D. H. , Kim, H. , Nam, D. , Sim, H. , Kim, J. , Kim, H. G. , … Seol, W. (2018). LRRK2 impairs autophagy by mediating phosphorylation of leucyl‐tRNA synthetase. Cell Biochemistry and Function, 36, 431–442. [DOI] [PubMed] [Google Scholar]
- Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biology, 14, 3156 10.1002/cbf.3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, C. H. , Shaltouki, A. , Gonzalez, A. E. , Bettencourt da Cruz, A. , Burbulla, L. F. , St Lawrence, E. , … Wang, X. (2016). Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson's Disease. Cell Stem Cell, 19, 709–724. 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh, C. J. , Zhang, B. , Victor, M. B. , Dahiya, S. , Batista, L. F. , Horvath, S. , & Yoo, A. S. (2016). Maintenance of age in human neurons generated by microRNA‐based neuronal conversion of fibroblasts. ELife, 5, e18648. 10.1016/j.stem.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui, K.Y. , Fernandez‐Hernandez, H. , Hu, J. , Schaffner, A. , Pankratz, N. , Hsu, N.Y. , … Peter, I . (2018) Functional variants in the LRRK2 gene confer shared effects on risk for Crohn's disease and Parkinson's disease. Science Translational Medicine, 10, eaar5429 10.1126/scitranslmed.aai7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannielli, A. , Bido, S. , Folladori, L. , Segnali, A. , Cancellieri, C. , Maresca, A. , … Broccoli, V. (2018). Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson's Disease models. Cell Reports, 22, 2066–2079. 10.1016/j.celrep.2018.01.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi, Y. , Okada, Y. , Akamatsu, W. , Koike, M. , Kuzumaki, N. , Hayakawa, H. , … Okano, H. (2012). Mitochondrial dysfunction associated with increased oxidative stress and alpha‐synuclein accumulation in PARK2 iPSC‐derived neurons and postmortem brain tissue. Molecular Brain, 5, 35 10.1186/1756-6606-5-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura, K. , Hishikawa, N. , Sawada, M. , Nagatsu, T. , Yoshida, M. , & Hashizume, Y. (2003). Distribution of major histocompatibility complex class II‐positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathologica, 106, 518–526. 10.1007/s00401-003-0766-2 [DOI] [PubMed] [Google Scholar]
- Jaleel, M. , Nichols, R. J. , Deak, M. , Campbell, D. G. , Gillardon, F. , Knebel, A. , & Alessi, D. R. (2007). LRRK2 phosphorylates moesin at threonine‐558: characterization of how Parkinson's disease mutants affect kinase activity. Biochemical Journal, 405, 307–317. 10.1042/BJ20070209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, H. , Ren, Y. , Yuen, E. Y. , Zhong, P. , Ghaedi, M. , Hu, Z. , … Feng, J. (2012). Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nature Communications, 3, 668 10.1038/ncomms1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J. A. , & Charpentier, E. (2012). A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo, J. , Xiao, Y. , Sun, Alfred X. , Cukuroglu, E. , Tran, H.‐D. , Göke, J. , … Ng, H.‐H. (2016). Midbrain‐like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin‐producing neurons. Cell Stem Cell, 19, 248–257. 10.1016/j.stem.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksimovic, M. , Anderegg, A. , Roy, A. , Campochiaro, L. , Yun, B. , Kittappa, R. , … Awatramani, R. (2009). Spatiotemporally separable Shh domains in the midbrain define distinct dopaminergic progenitor pools. Proceedings of the National Academy of Sciences of the United States of America, 106, 19185–19190. 10.1073/pnas.0904285106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins, L. , Ripke, S. , Weersma, R. K. , Duerr, R. H. , McGovern, D. P. , Hui, K. Y. , … Cho, J. H. (2012). Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 491, 119 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachergus, J. , Mata, I. F. , Hulihan, M. , Taylor, J. P. , Lincoln, S. , Aasly, J. , … Toft, M. (2005). Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. American Journal of Human Genetics, 76, 672–680. 10.1086/429256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik, S. , & Cuervo, A. M. (2009). Chapter 19: Methods to monitor chaperone‐mediated autophagy. Methods in Enzymology, 452, 297–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami, F. , Shimada, N. , Ohta, E. , Kagiya, G. , Kawashima, R. , Maekawa, T. , … Ichikawa, T. (2014). Leucine‐rich repeat kinase 2 regulates tau phosphorylation through direct activation of glycogen synthase kinase‐3β. The FEBS Journal, 281, 3–13. 10.1111/febs.12579 [DOI] [PubMed] [Google Scholar]
- Kee, N. , Volakakis, N. , Kirkeby, A. , Dahl, L. , Storvall, H. , Nolbrant, S. , … Perlmann, T. (2017). Single‐cell analysis reveals a close relationship between differentiating dopamine and subthalamic nucleus neuronal lineages. Cell Stem Cell, 20, 29–40. 10.1016/j.stem.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Kikuchi, T. , Morizane, A. , Doi, D. , Magotani, H. , Onoe, H. , Hayashi, T. , … Takahashi, J. (2017). Human iPS cell‐derived dopaminergic neurons function in a primate Parkinson's disease model. Nature, 548, 592–596. [DOI] [PubMed] [Google Scholar]
- Kim, S.‐J. , Khan, M. , Quan, J. , Till, A. , Subramani, S. , & Siddiqui, A. (2013). Hepatitis B Virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLOS Pathogens, 9, e1003722 10.1371/journal.ppat.1003722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. H. , & Son, J. H. (2010). PINK1 gene knockdown leads to increased binding of parkin with actin filament. Neuroscience Letters, 468, 272–276. 10.1016/j.neulet.2009.11.011 [DOI] [PubMed] [Google Scholar]
- Kirkeby, A. , Grealish, S. , Wolf, Daniel A. , Nelander, J. , Wood, J. , Lundblad, M. , … Parmar, M. (2012). Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Reports, 1, 703–714. 10.1016/j.celrep.2012.04.009 [DOI] [PubMed] [Google Scholar]
- Kirkeby, A. , Nolbrant, S. , Tiklova, K. , Heuer, A. , Kee, N. , Cardoso, T. , … Parmar, M. (2017). Predictive markers guide differentiation to improve graft outcome in clinical translation of hESC‐based therapy for Parkinson's Disease. Cell Stem Cell, 20, 135–148. 10.1016/j.stem.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, R. L. , Dayton, R. D. , Henderson, K. M. , & Petrucelli, L. (2006). Parkin is protective for substantia nigra dopamine neurons in a tau gene transfer neurodegeneration model. Neuroscience Letters, 401, 130–135. 10.1016/j.neulet.2006.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, R. L. , Dayton, R. D. , Lin, W. L. , & Dickson, D. W. (2005). Tau gene transfer, but not alpha‐synuclein, induces both progressive dopamine neuron degeneration and rotational behavior in the rat. Neurobiology of Diseases, 20, 64–73. 10.1016/j.nbd.2005.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J. , Abdelmohsen, K. , Abe, A. , Abedin, M.J. , Abeliovich, H. , Zois, C.E. , … Zughaier, S.M . (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy, 12, 1–222. 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouroupi, G. , Taoufik, E. , Vlachos, I. S. , Tsioras, K. , Antoniou, N. , Papastefanaki, F. , … Matsas, R. (2017). Defective synaptic connectivity and axonal neuropathology in a human iPSC‐based model of familial Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America, 114, E3679–E3688. 10.1073/pnas.1617259114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks, S. , Shim, J. W. , Piao, J. , Ganat, Y. M. , Wakeman, D. R. , Xie, Z. , … Studer, L. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature, 480, 547–551. 10.1038/nature10648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurnosov, A. A. , Ustyugova, S. V. , Nazarov, V. I. , Minervina, A. A. , Komkov, A. Y. , Shugay, M. , … Lebedev, Y. B. (2015). The evidence for increased L1 activity in the site of human adult brain neurogenesis. PLoS ONE, 10, e0117854 10.1371/journal.pone.0117854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuss, M. , Adamopoulou, E. , & Kahle, P. J. (2014). Interferon‐γ induces leucine‐rich repeat kinase LRRK2 via extracellular signal‐regulated kinase ERK5 in macrophages. Journal of Neurochemistry, 129, 980–987. 10.1111/jnc.12668 [DOI] [PubMed] [Google Scholar]
- Kyttala, A. , Moraghebi, R. , Valensisi, C. , Kettunen, J. , Andrus, C. , Pasumarthy, K. K. , … Trokovic, R. (2016). Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Reports, 6, 200–212. 10.1016/j.stemcr.2015.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel, S. , Hetzel, A. , Hackel, O. , Jones, I. , Liss, B. , & Roeper, J. (2008). Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron, 57, 760–773. 10.1016/j.neuron.2008.01.022 [DOI] [PubMed] [Google Scholar]
- Lancaster, M. A. , Renner, M. , Martin, C. A. , Wenzel, D. , Bicknell, L. S. , Hurles, M. E. , … Knoblich, J. A. (2013). Cerebral organoids model human brain development and microcephaly. Nature, 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, S. B. , Hanss, Z. , & Krüger, R. (2018). The genetic architecture of mitochondrial dysfunction in Parkinson's disease. Cell and Tissue Research, 373, 21–37. 10.1007/s00441-017-2768-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, W. , Wu, J. , & Tang, Y. (2016). Protective microglia and their regulation in Parkinson's Disease. Frontiers in Molecular Neuroscience, 9, 89 10.3389/fnmol.2016.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. , James, W. S. , & Cowley, S. A. (2017). LRRK2 in peripheral and central nervous system innate immunity: its link to Parkinson's disease. Biochemical Society Transactions, 45, 131–139. 10.1042/BST20160262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. H. , Siddoway, B. , Kaeser, G. E. , Segota, I. , Rivera, R. , Romanow, W. J. , … Chun, J. (2018). Somatic APP gene recombination in Alzheimer's disease and normal neurons. Nature, 563, 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, P. A. (2009). The function of ROCO proteins in health and disease. Biology of the Cell, 101, 183–191. 10.1042/BC20080053 [DOI] [PubMed] [Google Scholar]
- Lewis, P. A. , Greggio, E. , Beilina, A. , Jain, S. , Baker, A. , & Cookson, M. R. (2007). The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochemical and Biophysical Research Communications, 357, 668–671. 10.1016/j.bbrc.2007.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. Q. , Tan, L. , & Yu, J. T. (2014). The role of the LRRK2 gene in Parkinsonism. Molecular Neurodegeneration, 9, 47 10.1186/1750-1326-9-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, C. L. , Wang, T. T. , Luby‐Phelps, K. , & German, D. C. (2007). Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson's disease. Experimental Neurology, 203, 370–380. 10.1016/j.expneurol.2006.08.015 [DOI] [PubMed] [Google Scholar]
- Lim, M. K. , Kawamura, T. , Ohsawa, Y. , Ohtsubo, M. , Asakawa, S. , Takayanagi, A. , & Shimizu, N. (2007). Parkin interacts with LIM Kinase 1 and reduces its cofilin‐phosphorylation activity via ubiquitination. Experimental Cell Research, 313, 2858–2874. 10.1016/j.yexcr.2007.04.016 [DOI] [PubMed] [Google Scholar]
- Lin, L. , Goke, J. , Cukuroglu, E. , Dranias, M. R. , VanDongen, A. M. , & Stanton, L. W. (2016). Molecular features underlying neurodegeneration identified through in vitro modeling of genetically diverse Parkinson's Disease patients. Cell Reports, 15, 2411–2426. 10.1016/j.celrep.2016.05.022 [DOI] [PubMed] [Google Scholar]
- Liss, B. , Haeckel, O. , Wildmann, J. , Miki, T. , Seino, S. , & Roeper, J. (2005). K‐ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nature Neuroscience, 8, 1742–1751. 10.1038/nn1570 [DOI] [PubMed] [Google Scholar]
- Little, D. , Luft, C. , Mosaku, O. , Lorvellec, M. , Yao, Z. , Paillusson, S. , … Gissen, P. (2018). A single cell high content assay detects mitochondrial dysfunction in iPSC‐derived neurons with mutations in SNCA. Scientific Reports, 8, 9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Bryant, N. , Kumaran, R. , Beilina, A. , Abeliovich, A. , Cookson, M. R. , & West, A. B. (2018). LRRK2 phosphorylates membrane‐bound Rabs and is activated by GTP‐bound Rab7L1 to promote recruitment to the trans‐Golgi network. Human Molecular Genetics, 27, 385–395. 10.1093/hmg/ddx410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Lee, J. , Krummey, S. , Lu, W. , Cai, H. , & Lenardo, M. J. (2011). The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nature Immunology, 12, 1063 10.1038/ni.2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G. H. , Qu, J. , Suzuki, K. , Nivet, E. , Li, M. , Montserrat, N. , … Izpisua Belmonte, J. C. (2012). Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature, 491, 603–607. 10.1038/nature11557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G. , Yu, J. , Ding, J. , Xie, C. , Sun, L. , Rudenko, I. , … Cai, H. (2014). Aldehyde dehydrogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. Journal of Clinical Investigation, 124, 3032–3046. 10.1172/JCI72176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Sardo, V. , Ferguson, W. , Erikson, G. A. , Topol, E. J. , Baldwin, K. K. , & Torkamani, A. (2017). Influence of donor age on induced pluripotent stem cells. Nature Biotechnology, 35, 69–74. 10.1038/nbt.3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato, M. A. , Woodworth, M. B. , Lee, S. , Evrony, G. D. , Mehta, B. K. , Karger, A. , … Walsh, C. A. (2015). Somatic mutation in single human neurons tracks developmental and transcriptional history. Science, 350, 94–98. 10.1126/science.aab1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Maturana, R. , Lang, V. , Zubiarrain, A. , Sousa, A. , Vazquez, N. , Gorostidi, A. , … Sanchez‐Pernaute, R. (2016). Mutations in LRRK2 impair NF‐kappaB pathway in iPSC‐derived neurons. Journal of Neuroinflammation, 13, 295 10.1186/s12974-016-0761-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, B. , Xu, L. , Pan, X. , Sun, L. , Ding, J. , Xie, C. , … Cai, H. (2016). LRRK2 modulates microglial activity through regulation of chemokine (C‐X3‐C) receptor 1 ‐mediated signalling pathways. Human Molecular Genetics, 25, 3515–3523. 10.1093/hmg/ddw194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod, David A. , Rhinn, H. , Kuwahara, T. , Zolin, A. , Di Paolo, G. , McCabe, Brian D. , … Abeliovich, A. (2013). RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson Disease risk. Neuron, 77, 425–439. 10.1016/j.neuron.2012.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P. , Yang, L. , Esvelt, K. M. , Aach, J. , Guell, M. , DiCarlo, J. E. , … Church, G. M. (2013). RNA‐guided human genome engineering via Cas9. Science, 339, 823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni, C. , Mamais, A. , Roosen, D. A. , Dihanich, S. , Soutar, M. P. M. , Plun‐Favreau, H. , … Lewis, P. A. (2016). mTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin‐1. Scientific Reports, 6, 35106 10.1038/srep35106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinek, P. , Jha, A. N. , Shinde, V. , Sundaramoorthy, A. , Rajkumar, R. , Suryadevara, N. C. , … Velavan, T. P. (2013). LRRK2 and RIPK2 variants in the NOD 2‐mediated signaling pathway are associated with susceptibility to Mycobacterium leprae in Indian populations. PLoS ONE, 8, e73103 10.1371/journal.pone.0073103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone, L. , Bus, C. , Schöndorf, D. , Fitzgerald, J. C. , Kübler, M. , Schmid, B. , … Sterneckert, J. (2018). Generation of iPSCs carrying a common LRRK2 risk allele for in vitro modeling of idiopathic Parkinson's disease. PLoS ONE, 13, e0192497 10.1371/journal.pone.0192497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata, I. F. , Wedemeyer, W. J. , Farrer, M. J. , Taylor, J. P. , & Gallo, K. A. (2006). LRRK2 in Parkinson's disease: protein domains and functional insights. Trends in Neurosciences, 29, 286–293. 10.1016/j.tins.2006.03.006 [DOI] [PubMed] [Google Scholar]
- Matikainen‐Ankney, B. A. , Kezunovic, N. , Menard, C. , Flanigan, M. E. , Zhong, Y. , Russo, S. J. , … Huntley, G. W. (2018). Parkinson's Disease‐linked LRRK2‐G2019S mutation alters synaptic plasticity and promotes resilience to chronic social stress in young adulthood. Journal of Neuroscience, 38, 9700–9711. 10.1523/JNEUROSCI.1457-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen‐Ankney, B. A. , Kezunovic, N. , Mesias, R. E. , Tian, Y. , Williams, F. M. , Huntley, G. W. , & Benson, D. L. (2016). Altered development of synapse structure and function in striatum caused by Parkinson's Disease‐linked LRRK2‐G2019S mutation. Journal of Neuroscience, 36, 7128–7141. 10.1523/JNEUROSCI.3314-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda, W. , Furuta, T. , Nakamura, K. C. , Hioki, H. , Fujiyama, F. , Arai, R. , & Kaneko, T. (2009). Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. Journal of Neuroscience, 29, 444–453. 10.1523/JNEUROSCI.4029-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli, J. R. , Xu, Y. H. , Sun, Y. , Knight, A. L. , McLean, P. J. , Caldwell, G. A. , … Krainc, D. (2011). Gaucher disease glucocerebrosidase and alpha‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell, 146, 37–52. 10.1016/j.cell.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli, J. R. , Zunke, F. , Isacson, O. , Studer, L. , & Krainc, D. (2016). α‐Synuclein–induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proceedings of the National Academy of Sciences of the United States of America, 113, 1931 10.1073/pnas.1520335113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli, J. R. , Zunke, F. , Tsunemi, T. , Toker, N. J. , Jeon, S. , Burbulla, L. F. , … Krainc, D. (2016). Activation of beta‐glucocerebrosidase reduces pathological alpha‐synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. Journal of Neuroscience, 36, 7693–7706. 10.1523/JNEUROSCI.0628-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer, P. L. , Itagaki, S. , Boyes, B. E. , & McGeer, E. G. (1988). Reactive microglia are positive for HLA‐DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology, 38, 1285–1291. [DOI] [PubMed] [Google Scholar]
- Mendez, I. , Sanchez‐Pernaute, R. , Cooper, O. , Vinuela, A. , Ferrari, D. , Bjorklund, L. , … Isacson, O. (2005). Cell type analysis of functional fetal dopamine cell suspension transplants in the striatum and substantia nigra of patients with Parkinson's disease. Brain, 128, 1498–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies, F. M. , Fleming, A. , & Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nature Reviews Neuroscience, 16, 345–357. 10.1093/brain/awh510 [DOI] [PubMed] [Google Scholar]
- Mercuri, N. B. , Bonci, A. , Calabresi, P. , Stratta, F. , Stefani, A. , & Bernardi, G. (1994). Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. British Journal of Pharmacology, 113, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens, J. , Paquola, A. C. M. , Ku, M. , Hatch, E. , Böhnke, L. , Ladjevardi, S. , … Gage, F. H. (2015). Directly reprogrammed human neurons retain aging‐associated transcriptomic signatures and reveal age‐related nucleocytoplasmic defects. Cell Stem Cell, 17, 705–718. 10.1016/j.stem.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, N. , Wang, M. , Ott, J. A. , D'Alessandro, J. S. , Woolf, T. M. , Bumcrot, D. A. , … Pang, K. (1997). Sonic hedgehog promotes the survival of specific CNS neuron populations and protects these cells from toxic insult in vitro. Journal of Neuroscience, 17, 5891–5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migheli, R. , Del Giudice, M. G. , Spissu, Y. , Sanna, G. , Xiong, Y. , Dawson, T. M. , … Iaccarino, C. (2013). LRRK2 affects vesicle trafficking, neurotransmitter extracellular level and membrane receptor localization. PLoS ONE, 8, e77198 10.1371/journal.pone.0077198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy, J. , Arai, T. , Guo, J. P. , Klegeris, A. , Yu, S. , McGeer, E. G. , & McGeer, P. L. (2006). LRRK2 expression in normal and pathologic human brain and in human cell lines. Journal of Neuropathology and Experimental Neurology, 65, 953–963. 10.1097/01.jnen.0000235121.98052.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy, J. , Doudet, D. D. , Schwab, C. , Yu, S. , McGeer, E. G. , & McGeer, P. L. (2006). Role of ICAM‐1 in persisting inflammation in Parkinson disease and MPTP monkeys. Experimental Neurology, 197, 275–283. 10.1016/j.expneurol.2005.10.034 [DOI] [PubMed] [Google Scholar]
- Miller, Justine D. , Ganat, Yosif M. , Kishinevsky, S. , Bowman, Robert L. , Liu, B. , Tu, Edmund Y. , … Studer, L. (2013). Human iPSC‐based modeling of late‐onset disease via progerin‐induced aging. Cell Stem Cell, 13, 691–705. 10.1016/j.stem.2013.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzel, A. S. , Smits, L. M. , Hemmer, K. , Hachi, S. , Moreno, E. L. , van Wuellen, T. , … Schwamborn, J. C. (2017). Derivation of human midbrain‐specific organoids from neuroepithelial stem cells. Stem Cell Reports, 8, 1144–1154. 10.1016/j.stemcr.2017.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri, A. R. , Chu, V. T. , Marchetto, M. C. , Deng, W. , Moran, J. V. , & Gage, F. H. (2005). Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature, 435, 903–910. 10.1038/nature03663 [DOI] [PubMed] [Google Scholar]
- Nedergaard, S. , Flatman, J. A. , & Engberg, I. (1993). Nifedipine‐ and omega‐conotoxin‐sensitive Ca2 + conductances in guinea‐pig substantia nigra pars compacta neurones. Journal of Physiology, 466, 727–747. [PMC free article] [PubMed] [Google Scholar]
- Nguyen, Ha N. , Byers, B. , Cord, B. , Shcheglovitov, A. , Byrne, J. , Gujar, P. , … Pera, Renee R. (2011). LRRK2 mutant iPSC‐derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell, 8, 267–280. 10.1016/j.stem.2011.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, M. , & Krainc, D. (2018). LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America, 115, 5576–5581. 10.1073/pnas.1717590115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolbrant, S. , Heuer, A. , Parmar, M. , & Kirkeby, A. (2017). Generation of high‐purity human ventral midbrain dopaminergic progenitors for in vitro maturation and intracerebral transplantation. Nature Protocols, 12, 1962–1979. [DOI] [PubMed] [Google Scholar]
- Ohta, E. , Nihira, T. , Uchino, A. , Imaizumi, Y. , Okada, Y. , Akamatsu, W. , … Okano, H. (2015). I2020T mutant LRRK2 iPSC‐derived neurons in the Sagamihara family exhibit increased Tau phosphorylation through the AKT/GSK‐3beta signaling pathway. Human Molecular Genetics, 24, 4879–4900. 10.1093/hmg/ddv212 [DOI] [PubMed] [Google Scholar]
- Ono, Y. , Nakatani, T. , Sakamoto, Y. , Mizuhara, E. , Minaki, Y. , Kumai, M. , … Imai, T. (2007). Differences in neurogenic potential in floor plate cells along an anteroposterior location: midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development, 134, 3213–3225. 10.1242/dev.02879 [DOI] [PubMed] [Google Scholar]
- Orenstein, S. J. , Kuo, S. H. , Tasset, I. , Arias, E. , Koga, H. , Fernandez‐Carasa, I. , … Cuervo, A. M. (2013). Interplay of LRRK2 with chaperone‐mediated autophagy. Nature Neuroscience, 16, 394–406. 10.1038/nn.3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacelli, C. , Giguere, N. , Bourque, M. J. , Levesque, M. , Slack, R. S. , & Trudeau, L. E. (2015). Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Current Biology, 25, 2349–2360. 10.1016/j.cub.2015.07.050 [DOI] [PubMed] [Google Scholar]
- Pak, C. , Danko, T. , Zhang, Y. , Aoto, J. , Anderson, G. , Maxeiner, S. , … Sudhof, T. C. (2015). Human neuropsychiatric disease modeling using conditional deletion reveals synaptic transmission defects caused by heterozygous mutations in NRXN1. Cell Stem Cell, 17, 316–328. 10.1016/j.stem.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, Z. P. , Yang, N. , Vierbuchen, T. , Ostermeier, A. , Fuentes, D. R. , Yang, T. Q. , … Wernig, M. (2011). Induction of human neuronal cells by defined transcription factors. Nature, 476, 220–223. 10.1038/nature10202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou, L. , & Cai, H. (2010). LRRK2 function on actin and microtubule dynamics in Parkinson's disease. Communicative & Integrative Biology, 3, 396–400. 10.4161/cib.3.5.12286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou, L. , Yu, J. , Sgobio, C. , Xie, C. , Liu, G. , Sun, L. , … Cai, H. (2014). LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nature Neuroscience, 17, 367–376. 10.1038/nn.3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, M. , Pfisterer, U. , Rylander, D. , Torper, O. , Lau, S. , Lundblad, M. , … Parmar, M. (2014). Highly efficient generation of induced neurons from human fibroblasts that survive transplantation into the adult rat brain. Scientific Reports, 4, 6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier, A. L. , Tabar, V. , Barberi, T. , Rubio, M. E. , Bruses, J. , Topf, N. , … Studer, L. (2004). Derivation of midbrain dopamine neurons from human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America, 101, 12543–12548. https://doi.org/10.1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfisterer, U. , Kirkeby, A. , Torper, O. , Wood, J. , Nelander, J. , Dufour, A. , … Parmar, M. (2011). Direct conversion of human fibroblasts to dopaminergic neurons. Proceedings of the National Academy of Sciences of the United States of America, 108, 10343–10348. https://doi.org/10.1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissadaki, E. K. , & Bolam, J. P. (2013). The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Frontiers in Computational Neuroscience, 7, 13 10.3389/fncom.2013.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A. , Manzoni, C. , Cookson, M. R. , & Lewis, P. A. (2018). The LRRK2 signalling system. Cell and Tissue Research, 373, 39–50. 10.1007/s00441-017-2759-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purlyte, E. , Dhekne, H. S. , Sarhan, A. R. , Gomez, R. , Lis, P. , Wightman, M. , … Alessi, D. R. (2018). Rab29 activation of the Parkinson's disease‐associated LRRK2 kinase. EMBO Journal, 37, 1–18. 10.15252/embj.201798099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing, X. , Walter, J. , Jarazo, J. , Arias‐Fuenzalida, J. , Hillje, A.‐L. , & Schwamborn, J. C. (2017). CRISPR/Cas9 and piggyBac‐mediated footprint‐free LRRK2‐G2019S knock‐in reveals neuronal complexity phenotypes and α‐Synuclein modulation in dopaminergic neurons. Stem Cell Research, 24, 44–50. 10.1016/j.scr.2017.08.013 [DOI] [PubMed] [Google Scholar]
- Rajput, A. , Dickson, D. W. , Robinson, C. A. , Ross, O. A. , Dachsel, J. C. , Lincoln, S. J. , … Farrer, M. J. (2006). Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology, 67, 1506–1508. 10.1212/01.wnl.0000240220.33950.0c [DOI] [PubMed] [Google Scholar]
- Rassu, M. , Del Giudice, M. G. , Sanna, S. , Taymans, J. M. , Morari, M. , Brugnoli, A. , … Iaccarino, C. (2017). Role of LRRK2 in the regulation of dopamine receptor trafficking. PLoS ONE, 12, e0179082 10.1371/journal.pone.0179082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt, P. , Glatza, M. , Hemmer, K. , Tsytsyura, Y. , Thiel, C. S. , Höing, S. , … Sterneckert, J. (2013). Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE, 8, e59252 10.1371/journal.pone.0059252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt, P. , Schmid, B. , Burbulla, Lena F. , Schöndorf, David C. , Wagner, L. , Glatza, M. , … Sterneckert, J. (2013). Genetic correction of a LRRK2 mutation in human iPSCs links Parkinsonian neurodegeneration to ERK‐dependent changes in gene expression. Cell Stem Cell, 12, 354–367. 10.1016/j.stem.2013.01.008 [DOI] [PubMed] [Google Scholar]
- Ren, Y. , Jiang, H. , Hu, Z. , Fan, K. , Wang, J. , Janoschka, S. , … Feng, J. (2015). Parkin mutations reduce the complexity of neuronal processes in iPSC‐derived human neurons. Stem Cells, 33, 68–78. 10.1002/stem.1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes, S. , Cottam, V. , Kirik, D. , Double, K. L. , & Halliday, G. M. (2013). Variability in neuronal expression of dopamine receptors and transporters in the substantia nigra. Movement Disorders, 28, 1351–1359. 10.1002/mds.25493 [DOI] [PubMed] [Google Scholar]
- Roosen, D. A. , & Cookson, M. R. (2016). LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Molecular Neurodegeneration, 11, 73 10.1186/s13024-016-0140-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbusch, K. E. , & Kortholt, A. (2016). Activation mechanism of LRRK2 and its cellular functions in Parkinson’s disease. Parkinson’s Disease, 2016, 7351985 10.1155/2016/7351985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, O. A. , Soto‐Ortolaza, A. I. , Heckman, M. G. , Aasly, J. O. , Abahuni, N. , Annesi, G. , … Genetic Epidemiology of Parkinson's Disease (GEO-PD) Consortium . (2011). Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case‐control study. The Lancet Neurology, 10, 898–908. 10.1016/S1474-4422(11)70175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhani, F. , Kumasaka, N. , de Brito, M. C. , Bradley, A. , Vallier, L. , & Gaffney, D. (2014). Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genetics, 10, e1004432 10.1371/journal.pgen.1004432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, I. , Berti, G. , Plotegher, N. , Bernardo, G. , Filograna, R. , Bubacco, L. , & Greggio, E. (2015). Leucine‐rich repeat kinase 2 positively regulates inflammation and down‐regulates NF‐kappaB p50 signaling in cultured microglia cells. Journal of Neuroinflammation, 12, 230 10.1186/s12974-015-0449-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, I. , Bubacco, L. , & Greggio, E. (2014). LRRK2 and neuroinflammation: partners in crime in Parkinson's disease? Journal of Neuroinflammation, 11, 52 10.1186/1742-2094-11-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, S. D. , Dolatabadi, N. , Chan, S. F. , Zhang, X. , Akhtar, M. W. , Parker, J. , … Lipton, S. A. (2013). Isogenic human iPSC Parkinson's model shows nitrosative stress‐induced dysfunction in MEF2‐PGC1alpha transcription. Cell, 155, 1351–1364. 10.1016/j.cell.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, B. J. , Hoek, S. , Fon, E. A. , & Wade‐Martins, R. (2015). Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends in Biochemical Sciences, 40, 200–210. 10.1016/j.tibs.2015.02.003 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Danes, A. , Richaud‐Patin, Y. , Carballo‐Carbajal, I. , Jimenez‐Delgado, S. , Caig, C. , Mora, S. , … Raya, A. (2012). Disease‐specific phenotypes in dopamine neurons from human iPS‐based models of genetic and sporadic Parkinson's disease. EMBO Molecular Medicine, 4, 380–395. 10.1002/emmm.201200215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, L. H. , Laganière, J. , Cooper, O. , Mak, S. K. , Vu, B. J. , Huang, Y. A. , … Schüle, B. (2014). LRRK2 mutations cause mitochondrial DNA damage in iPSC‐derived neural cells from Parkinson's disease patients: Reversal by gene correction. Neurobiology of Disease, 62, 381–386. 10.1016/j.nbd.2013.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandor, C. , Robertson, P. , Lang, C. , Heger, A. , Booth, H. , Vowles, J. , … Webber, C. (2017). Transcriptomic profiling of purified patient‐derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson's disease. Human Molecular Genetics, 26, 552–566. 10.1093/hmg/ddw412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai, Y. (2013). Cytosystems dynamics in self‐organization of tissue architecture. Nature, 493, 318–326. [DOI] [PubMed] [Google Scholar]
- Schiemann, J. , Schlaudraff, F. , Klose, V. , Bingmer, M. , Seino, S. , Magill, P. J. , … Roeper, J. (2012). K‐ATP channels in dopamine substantia nigra neurons control bursting and novelty‐induced exploration. Nature Neuroscience, 15, 1272–1280. 10.1038/nn.3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöndorf, D. C. , Aureli, M. , McAllister, F. E. , Hindley, C. J. , Mayer, F. , Schmid, B. , … Deleidi, M. (2014). iPSC‐derived neurons from GBA1‐associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nature Communications, 5, 4028 10.1038/ncomms5028 [DOI] [PubMed] [Google Scholar]
- Schwab, A. J. , & Ebert, A. D. (2015). Neurite aggregation and calcium dysfunction in iPSC‐derived sensory neurons with Parkinson's Disease‐related LRRK2 G2019S mutation. Stem Cell Reports, 5, 1039–1052. 10.1016/j.stemcr.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, A. J. , Sison, S. L. , Meade, M. R. , Broniowska, K. A. , Corbett, J. A. , & Ebert, A. D. (2017). Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC‐derived dopaminergic neurons. Stem Cell Reports, 9, 1839–1852. 10.1016/j.stemcr.2017.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibler, P. , Graziotto, J. , Jeong, H. , Simunovic, F. , Klein, C. , & Krainc, D. (2011). Mitochondrial parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. The Journal of Neuroscience, 31, 5970 10.1523/JNEUROSCI.4441-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltouki, A. , Sivapatham, R. , Pei, Y. , Gerencser, A. A. , Momcilovic, O. , Rao, M. S. , & Zeng, X. (2015). Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient‐specific and PARK2 knockout isogenic iPSC lines. Stem Cell Reports, 4, 847–859. 10.1016/j.stemcr.2015.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, Z. , Zhang, S. , Bustos, D. , Kleinheinz, T. , Le Pichon, C.E. , Dominguez, S.L. , … Zhu, H . (2012). Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Science Translational Medicine, 4, 164ra161. 10.1126/scitranslmed.3004485 [DOI] [PubMed] [Google Scholar]
- Skibinski, G. , Hwang, V. , Ando, D. M. , Daub, A. , Lee, A. K. , Ravisankar, A. , … Finkbeiner, S. (2017). Nrf2 mitigates LRRK2‐ and alpha‐synuclein‐induced neurodegeneration by modulating proteostasis. Proceedings of the National Academy of Sciences of the United States of America, 114, 1165–1170. 10.1073/pnas.1522872114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski, G. , Nakamura, K. , Cookson, M. R. , & Finkbeiner, S. (2014). Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. The Journal of Neuroscience, 34, 418 10.1523/JNEUROSCI.2712-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan, M. , Alegre‐Abarrategui, J. , Potgieter, D. , Kaufmann, A. K. , Exley, R. , Deltheil, T. , … Wade‐Martins, R. (2016). LRRK2 BAC transgenic rats develop progressive, L‐DOPA‐responsive motor impairment, and deficits in dopamine circuit function. Human Molecular Genetics, 25, 951–963. 10.1093/hmg/ddv628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafa, K. , Tsika, E. , Moser, R. , Musso, A. , Glauser, L. , Jones, A. , … Moore, D. J. (2014). Functional interaction of Parkinson's disease‐associated LRRK2 with members of the dynamin GTPase superfamily. Human Molecular Genetics, 23, 2055–2077. 10.1093/hmg/ddt600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger, M. , Tonelli, F. , Ito, G. , Davies, P. , Trost, M. , Vetter, M. , … Mann, M. (2016). Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife, 5, e12813 10.7554/eLife.12813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck, J. A. , Choi, S. J. , Mrejeru, A. , Ganat, Y. , Deisseroth, K. , Sulzer, D. , … Studer, L. (2015). Optogenetics enables functional analysis of human embryonic stem cell‐derived grafts in a Parkinson's disease model. Nature Biotechnology, 33, 204–209. 10.1038/nbt.3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner, J. A. , Quansah, E. , & Brundin, P. (2018). The concept of alpha‐synuclein as a prion‐like protein: ten years after. Cell and Tissue Research, 373, 161–173. 10.1007/s00441-018-2814-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, Y. C. , & Qi, X. (2013). Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Human Molecular Genetics, 22, 4545–4561. 10.1093/hmg/ddt301 [DOI] [PubMed] [Google Scholar]
- Surmeier, D. J. , Guzman, J. N. , Sanchez‐Padilla, J. , & Goldberg, J. A. (2011). The origins of oxidant stress in Parkinson's disease and therapeutic strategies. Antioxidants & Redox Signaling, 14, 1289–1301. 10.1089/ars.2010.3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier, D. J. , Obeso, J. A. , & Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nature Reviews Neuroscience, 18, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, S. , Akamatsu, W. , Kisa, F. , Sone, T. , Ishikawa, K.‐I. , Kuzumaki, N. , … Okano, H. (2017). Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochemical and Biophysical Research Communications, 483, 88–93. 10.1016/j.bbrc.2016.12.188 [DOI] [PubMed] [Google Scholar]
- Sweet, E. S. , Saunier‐Rebori, B. , Yue, Z. , & Blitzer, R. D. (2015). The Parkinson's Disease‐associated mutation LRRK2‐G2019S impairs synaptic plasticity in mouse hippocampus. Journal of Neuroscience, 35, 11190–11195. 10.1523/JNEUROSCI.0040-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabar, V. , & Studer, L. (2014). Pluripotent stem cells in regenerative medicine: Challenges and recent progress. Nature Reviews Genetics, 15, 82–92. 10.1038/nrg3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K. , Tanabe, K. , Ohnuki, M. , Narita, M. , Ichisaka, T. , Tomoda, K. , & Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–872. 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- Takahashi, K. , & Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126, 663–676. 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- Takahashi, K. , & Yamanaka, S. (2016). A decade of transcription factor‐mediated reprogramming to pluripotency. Nature Reviews Molecular Cell Biology, 17, 183–193. 10.1038/nrm.2016.8 [DOI] [PubMed] [Google Scholar]
- Terstegge, S. , Laufenberg, I. , Pochert, J. , Schenk, S. , Itskovitz‐Eldor, J. , Endl, E. , & Brüstle, O. (2006). Automated maintenance of embryonic stem cell cultures. Biotechnology and Bioengineering, 96, 195–201. 10.1002/bit.21061 [DOI] [PubMed] [Google Scholar]
- Till, A. , Saito, R. , Merkurjev, D. , Liu, J.‐J. , Syed, G. H. , Kolnik, M. , … Subramani, S. (2015). Evolutionary trends and functional anatomy of the human expanded autophagy network. Autophagy, 11, 1652–1667. 10.1080/15548627.2015.1059558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torper, O. , Pfisterer, U. , Wolf, D. A. , Pereira, M. , Lau, S. , Jakobsson, J. , … Parmar, M. (2013). Generation of induced neurons via direct conversion in vivo. Proceedings of the National Academy of Sciences of the United States of America, 110, 7038–7043. 10.1073/pnas.1303829110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulorge, D. , Schapira, A. H. , & Hajj, R. (2016). Molecular changes in the postmortem parkinsonian brain. Journal of Neurochemistry, 139(Suppl. 1), 27–58. 10.1111/jnc.13696 [DOI] [PubMed] [Google Scholar]
- Vawter, M. P. , Dillon‐Carter, O. , Tourtellotte, W. W. , Carvey, P. , & Freed, W. J. (1996). TGFbeta1 and TGFbeta2 concentrations are elevated in Parkinson's disease in ventricular cerebrospinal fluid. Experimental Neurology, 142, 313–322. 10.1006/exnr.1996.0200 [DOI] [PubMed] [Google Scholar]
- Verma, M. , Callio, J. , Otero, P. A. , Sekler, I. , Wills, Z. P. , & Chu, C. T. (2017). Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD‐associated LRRK2 mutants. The Journal of Neuroscience, 37, 11151–11165. 10.1523/JNEUROSCI.3791-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor, M. B. , Richner, M. , Olsen, H. E. , Lee, S. W. , Monteys, A. M. , Ma, C. , … Yoo, A. S. (2018). Striatal neurons directly converted from Huntington's disease patient fibroblasts recapitulate age‐associated disease phenotypes. Nature Neuroscience, 21, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen, T. , Ostermeier, A. , Pang, Z. P. , Kokubu, Y. , Sudhof, T. C. , & Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature, 463, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi, T. , Xilouri, M. , Vekrellis, K. , & Stefanis, L. (2008). Wild type alpha‐synuclein is degraded by chaperone‐mediated autophagy and macroautophagy in neuronal cells. The Journal of Biological Chemistry, 283, 23542–23556. 10.1074/jbc.M801992200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Stockum, S. , Nardin, A. , Schrepfer, E. , & Ziviani, E. (2016). Mitochondrial dynamics and mitophagy in Parkinson's disease: A fly point of view. Neurobiology of Disease, 90, 58–67. 10.1016/j.nbd.2015.11.002 [DOI] [PubMed] [Google Scholar]
- Wallings, R. , Manzoni, C. , & Bandopadhyay, R. (2015). Cellular processes associated with LRRK2 function and dysfunction. The FEBS Journal, 282, 2806–2826. 10.1111/febs.13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Yan, M. H. , Fujioka, H. , Liu, J. , Wilson‐Delfosse, A. , Chen, S. G. , … Zhu, X. (2012). LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Human Molecular Genetics, 21, 1931–1944. 10.1093/hmg/dds003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, A. B. , Moore, D. J. , Choi, C. , Andrabi, S. A. , Li, X. , Dikeman, D. , … Dawson, T. M. (2007). Parkinson's disease‐associated mutations in LRRK2 link enhanced GTP‐binding and kinase activities to neuronal toxicity. Human Molecular Genetics, 16, 223–232. 10.1093/hmg/ddl471 [DOI] [PubMed] [Google Scholar]
- Witoelar, A. , Jansen, I.E. , Wang, Y. , Desikan, R.S. , Gibbs, J.R. , Blauwendraat, C. , … United Kingdom Brain Expression Consortium (UKBEC) Investigators . (2017). Genome‐wide pleiotropy between Parkinson Disease and autoimmune diseases. JAMA Neurology, 74, 780–792. 10.1001/jamaneurol.2017.0469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard, C. M. , Campos, B. A. , Kuo, S. H. , Nirenberg, M. J. , Nestor, M. W. , Zimmer, M. , … Noggle, S. A. (2014). iPSC‐derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson's disease. Cell Reports, 9, 1173–1182. 10.1016/j.celrep.2014.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, T. H. , Lu, Y. N. , Chuang, C. L. , Wu, C. L. , Chiang, A. S. , Krantz, D. E. , & Chang, H. Y. (2013). Loss of vesicular dopamine release precedes tauopathy in degenerative dopaminergic neurons in a Drosophila model expressing human tau. Acta Neuropathologica, 125, 711–725. 10.1007/s00401-013-1105-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi, J. , Liu, Y. , Liu, H. , Chen, H. , Emborg, M. E. , & Zhang, S. C. (2012). Specification of midbrain dopamine neurons from primate pluripotent stem cells. Stem Cells, 30, 1655–1663. 10.1002/stem.1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S. , Xia, C. , Li, S. , Du, L. , Zhang, L. , & Hu, Y . (2014) Mitochondrial dysfunction driven by the LRRK2‐mediated pathway is associated with loss of Purkinje cells and motor coordination deficits in diabetic rat model. Cell Death & Disease, 5, e1217. 10.1038/cddis.2014.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, F. , Danko, T. , Botelho, S.C. , Patzke, C. , Pak, C. , Wernig, M. , & Sudhof, T.C . (2016). Autism‐associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science, 352, aaf2669. 10.1126/science.aaf2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, J.‐H. , Mo, J.‐S. , Kim, M.‐Y. , Ann, E.‐J. , Ahn, J.‐S. , Jo, E.‐H. , … Park, H.‐S . (2017). LRRK2 functions as a scaffolding kinase of ASK1‐mediated neuronal cell death. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, 1864, 2356–2368. 10.1016/j.bbamcr.2017.09.001 [DOI] [PubMed] [Google Scholar]
- Zanon, A. , Kalvakuri, S. , Rakovic, A. , Foco, L. , Guida, M. , Schwienbacher, C. , … Seibler, P. (2017). SLP‐2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin‐deficient human iPSC‐derived neurons and Drosophila. Human Molecular Genetics, 26, 2412–2425. 10.1093/hmg/ddx132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca, L. , Zucca, F. A. , Wilms, H. , & Sulzer, D. (2003). Neuromelanin of the substantia nigra: a neuronal black hole with protective and toxic characteristics. Trends in Neurosciences, 26, 578–580. 10.1016/j.tins.2003.08.009 [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Duan, C. , & Yang, H. (2015). Defective autophagy in Parkinson's disease: Lessons from genetics. Molecular Neurobiology, 51, 89–104. 10.1007/s12035-014-8787-5 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Pak, C. , Han, Y. , Ahlenius, H. , Zhang, Z. , Chanda, S. , … Südhof, T. C. (2013). Rapid single‐step induction of functional neurons from human pluripotent stem cells. Neuron, 78, 785–798. 10.1016/j.neuron.2013.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich, A. , Biskup, S. , Leitner, P. , Lichtner, P. , Farrer, M. , Lincoln, S. , … Gasser, T. (2004). Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601–607. 10.1016/j.neuron.2004.11.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials