

Abstract
The discovery and development of new, highly potent anti-coronavirus agents and effective approaches for controlling the potential emergence of epidemic coronaviruses still remains an important mission. Here, we identified tylophorine compounds, including naturally occurring and synthetic phenanthroindolizidines and phenanthroquinolizidines, as potent in vitro inhibitors of enteropathogenic coronavirus transmissible gastroenteritis virus (TGEV). The potent compounds showed 50% maximal effective concentration (EC50) values ranging from 8 to 1468 nM as determined by immunofluorescent assay of the expression of TGEV N and S proteins and by real time-quantitative PCR analysis of viral yields. Furthermore, the potent tylophorine compounds exerted profound anti-TGEV replication activity and thereby blocked the TGEV-induced apoptosis and subsequent cytopathic effect in ST cells. Analysis of the structure–activity relations indicated that the most active tylophorine analogues were compounds with a hydroxyl group at the C14 position of the indolizidine moiety or at the C3 position of the phenanthrene moiety and that the quinolizidine counterparts were more potent than indolizidines. In addition, tylophorine compounds strongly reduced cytopathic effect in Vero 76 cells induced by human severe acute respiratory syndrome coronavirus (SARS CoV), with EC50 values ranging from less than 5 to 340 nM. Moreover, a pharmacokinetic study demonstrated high and comparable oral bioavailabilities of 7-methoxycryptopleurine (52.7%) and the naturally occurring tylophorine (65.7%) in rats. Thus, our results suggest that tylophorine compounds are novel and potent anti-coronavirus agents that may be developed into therapeutic agents for treating TGEV or SARS CoV infection.
Keywords: Coronavirus, Human severe acute respiratory syndrome coronavirus, Porcine transmissible gastroenteritis virus, Phenanthroindolizine, Phenanthroquinolizidine, Tylophorine
1. Introduction
Coronaviruses are enveloped viruses with a positive-sense single-stranded RNA genome and a helical symmetry. As animal viruses, they belong to the family Coronaviridae and are divided into three groups based on serological cross-reactivity and confirmed by genome sequence analysis. Group I coronaviruses include animal pathogens, such as porcine transmissible gastroenteritis coronavirus (TGEV) and human coronaviruses HCoV-229E. Group II also includes pathogens of veterinary relevance, such as murine coronavirus mouse hepatitis virus (MHV)-JHM strain and human severe acute respiratory syndrome coronavirus (SARS CoV) as well as Group III includes only avian coronaviruses (Weiss and Navas-Martin, 2005). They primarily infect the upper respiratory and gastrointestinal tract of birds and mammals (Brian and Baric, 2005).
Vaccines are currently available for some animal coronaviruses. Some vaccines are efficacious, and others cause adverse side effects. For instance, some vaccines against feline coronaviruses exacerbated disease in vaccinated animals exposed to the wild-type virus (Olsen, 1993). Moreover, antibody enhancement of disease is a potential risk of SARS CoV vaccines in humans (Weiss and Navas-Martin, 2005). Although no cases of SARS have been reported since April 2004, the nature of the unpredictable outbreak of SARS CoV is still a potential threat to the global economy and public health (Weiss and Navas-Martin, 2005). No effective therapy exists for SARS CoV infection, despite tremendous effort invested in finding anti-SARS CoV drugs, including targeting SARS CoV-specific main protease or viral attachment, entry, and fusion for intervention. Thus, SARS CoV or some variants thereof could easily remerge to cause disease. The discovery and development of new, highly potent anti-coronavirus agents and effective approaches for controlling the potential emergence of epidemic coronaviruses remains an important mission.
The phenanthroindolizidine alkaloids, such as tylophorine, tylocrebrine, and tylophorinine, are characteristic constituents of Cynanchum, Pergularia, Tylophora, and other genera of the Asclepiadaceae. These phenanthroindolizidines have activities against leukemia, asthma, anaphylaxis, other inflammations and bacterial infection (Bhutani et al., 1987, Chemler, 2009, Gopalakrishnan et al., 1980), as well as against cancer (Gao et al., 2004, Li et al., 2001b, Staerk et al., 2002, Yang et al., 2006, Yang et al., 2007a). The herbal plant Tylophora indica has been traditionally used in India as a folk remedy to treat bronchial asthma, bronchitis, rheumatism, and dermatitis (Li et al., 2001a). In a clinical trial, six Tylophora leaves (one per day) were efficacious as an herbal medicine for asthma (Shivpuri et al., 1968). Tylophora ovata has been used as traditional herbal medicine for rheumatism, asthma, and even traumatic injury even though it possesses mildly toxic properties (Lin, 2003). Phenanthroquinolizidines, typified by cryptopleurine, isolated from Boehmeria siamensis or through chemical synthesis, have anti-cancer and anti-inflammatory activities (Chuang et al., 2006, Li et al., 2001b, Luo et al., 2003, Yang et al., 2006). Tylophora plants used as alternative medicine and their derived alkaloids have been under extensive investigation for drug development (Chemler, 2009).
Here, we report on the discovery, synthesis, isolation and identification of tylophorine compounds as novel, potent inhibitors of TGEV and SARS CoV. Structure–activity relations were also analyzed. This inhibition of TGEV and SARS CoV by tylophorine compounds with high potency in vitro at low nanomolar range and high oral availability in rats suggests that they may be potential therapeutic agents for coronavirus infections such as TGEV and SARS CoV.
2. Materials and methods
2.1.1. Cells, viruses, immunofluorescent assay (IFA), and cytopathic effect (CPE)
Swine testicular (ST) epithelial cells and the Taiwan field-isolated virulent strain of TGEV were grown and propagated as described (Yang et al., 2007b). IFA and CPE and cytotoxicity assays were also as described (Yang et al., 2007b). Briefly, ST cells in 96-well plates, with or without a 2 h pretreatment with test compounds, were infected with TGEV at a multiplicity of infection (MOI) of 10 for IFA. The IFA was performed at 6 h post-infection (hpi) with antibodies against the spike (S) and nucleocapsid (N) proteins of TGEV. The cells were treated with 10 different concentrations of test compounds. The results of these assays were used to obtain the dose–response curves from which 50% maximal effective concentration (EC50) values were determined. For the CPE assay, TGEV was inoculated into a monolayer of ST cells in 12-well plates at 5 MOI, and the CPE was determined at 24 hpi for detection of anti-TGEV activity. TGEV-induced CPE is characterized by a rounding and enlargement of cells, formation of syncytia, and detachment of cells into the medium. Results presented are representative of at least 3 independent experiments. For cytotoxicity assay, ST cells cultured in minimal essential medium (Invitrogen, Inc.) and 10% fetal bovine serum (HyClone Co.) in 96-well plates were treated with 10 different concentrations of test compounds for 24 h. The resulting dose–response curves were used to determine 50% maximal cytotoxic concentration (CC50) values.
Vero 76 cells and SARS CoV Urbani strain were grown and propagated as described (Barnard et al., 2006). SARS CoV-related CPE inhibition and neutral red uptake assays for determination of antiviral efficacy (EC50) and compound cytotoxicity (CC50) were performed as described (Barnard et al., 2006).
2.2. Chemicals, western blot analysis, viral RNA isolation and relative quantification by real-time RT-PCR
These assays were performed as described (Yang et al., 2007b); antibody against human GAPDH was purchased from Cell Signaling Technology Inc. (MA, USA). DMSO, PEG400 and DMA were purchased from Sigma Aldrich Inc. (MO, USA). Infergen™ was interferon alfacon-1 lot 002586 kindly provided by Intermune, Inc. (Brisbane, CA).
2.3. Pharmacokinetic analysis
The Sprague–Dawley rats for the pharmacokinetic study were obtained from BioLASCO Taiwan Co. (Ilan, Taiwan) and housed in the animal facility at the National Health Research Institutes, Taiwan. The animal studies were performed according to committee-approved procedures. Male rats (330–380 g, 9–10 weeks old) were quarantined for 1 week before use. The animals were surgically implanted with a jugular-vein cannula 1 day before treatment and were fasted before treatment. Compounds 1a and 1c were given to rats (n = 3) at 3 mg/kg by intravenous or oral administration. 1a was prepared in a mixture of DMSO/PEG400 (90/10, v/v) for intravenous and oral administration and 1c in 100% DMA for oral administration and in a mixture of DMSO/PEG400 (80/20, v/v) for intravenous administration. The volume of the dosing solution given was adjusted according to the body weight recorded before the drug was administered. At 0 (immediately before dosing), 2, 5 (intravenous only), 15 and 30 min and 1, 2, 4, 6, 8, 12 and 24 h after compound administration, a blood sample (150 μl) was taken from each animal via the jugular-vein cannula and stored in ice (0–4 °C). The processing of the plasma and subsequent analysis by high-performance liquid chromatography–tandem mass spectrometry (HPLC–MS) were as described (Yao et al., 2007). The plasma concentration data were analyzed by a standard non-compartmental method with the Kinetica software (InnaPhase, Philadelphia, PA, USA).
2.4. Chemical synthesis
Tylophorine (1a) and analogues 1c, 2a, 2c, 3a, 3b and 3c were prepared as described (Chuang et al., 2006, Yang et al., 2006). NMR data for compounds 2a, 2c, 3a, and 3c were previously reported (Chuang et al., 2006) and identification for 3b were previously reported (Wu et al., 2002, Yang et al., 2006). A novel and concise route with high yield was developed to synthesize a novel intermediate, 2,3,5,6,7-pentamethoxy-phenanthrene-9-carbaldehyde. An intermediate, 2,3,6-trimethoxy-phenanthrene-9-carbaldehyde, was synthesized by the same scheme. This high yield-synthesis scheme will be published elsewhere. These two intermediates were further used for the synthesis of 1b, 1d, 1i, 1j, 2b, and 2d. Consequently, two naturally occurring compounds, 4-methoxytylophorine (Rao, 1970) and Boehmeriasin A (Luo et al., 2003), 1b and 1j, were chemically synthesized for the first time. Compounds 4, 7-dimethoxycryptoplerine (1i) (a novel compound) and deoxypergularinine (1d) were similarly synthesized. Collectively, compounds 1b, 1d, 1i, 1j, 2b, and 2d were synthesized from phenanthrene-9-carbaldehyde intermediates and further completely synthesized with slight modifications of a reported method (Chuang et al., 2006). Synthesized compounds were verified on HPLC–MS (data not shown), 1H NMR, and 13C NMR. Compounds 4a (tylophorine N-oxide, a naturally occurring compound) and 4b (7-methoxycryptopleurine N-oxide, a novel compound) were synthesized by use of 1a and 1c, respectively, and reacted with H2O2, as follows. Compounds 1a or 1c (10 mg, 0.025 mmol) were dissolved in acetone (15 ml) before the drop-wise addition of 30% H2O2 (16.8 ml, 0.15 mmol). The resulting solutions were stirred at room temperature for 24 h and then extracted with CHCl3 (4 × 30 ml). The organic phases were combined, dried, and evaporated. Finally, the residue was purified by column chromatography on silica gel (CH2Cl2:MeOH = 15:1) to give the compound 4a (yield 80.1%) and the novel compound 4b (yield 60.5%). All compounds derived from chemical synthesis are racemic except 3a–3c compounds. See Fig. 1 for chemical structures and the supplementary data for the physical properties of the synthesized tylophorine compounds.
Fig. 1.
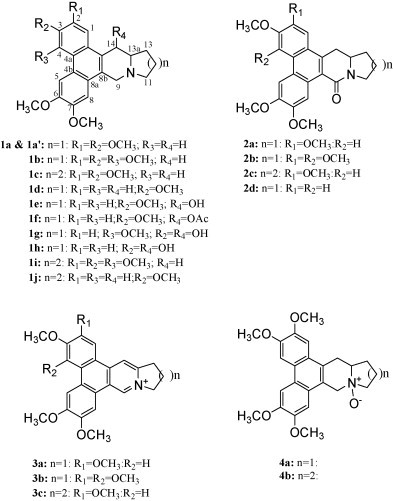
Chemical structures of phenanthroindolizidine and phenanthroquinolizidine compounds. Phenanthroindolizidines, 1a (1a′), 1b, 1d–1h, 2a, 2b, 2d, 3a, 3b, and 4a, consist of the moieties of phenanthrene and indolizidine and phenanthroquinolizidines, 1c, 1i, 1j, 2c, 3c, and 4b of phenanthrene and quinolizidine. These phenanthroindolizidines and phenanthroquinolizidines are tri-, tetra-, or penta-methoxylated at C2, C3, C4, C6, or C7 in the moiety of phenanthrene and with or without hydroxylated or OAc- at C14, formation of C9-one, or N-oxide in the indolizidine/quinolizidine moiety as shown.
2.5. Isolation and purification of tylophorine compounds from T. indica and T. ovata
The tylophorine compounds 1a′, 1e, 1g, and 1h were obtained from the methanol extracts of T. indica and T. ovata, which were further purified by a chromatography series including silica-gel open-column chromatography and HPLC. The details of isolation and purification will be published elsewhere. LC–MS, HREIMS, 1H NMR, 13C NMR, DEPT, COSY, NOESY, HSQC, and HMBC were used to identify and verify the structure of the obtained tylophorine analogues (data not shown except 1H NMR and 13C NMR).
Compounds 1a′ and 1e were validated as tylophorine and tylophorinine with 1H NMR and 13C NMR data previously reported (Abe et al., 1995, Chuang et al., 2006, Nordlander and Njoroge, 1987, Zeng and Chemler, 2008, Zhen et al., 2002). Tylophorine analogue 1f was semi-synthesized by mixing acetic anhydride (Ac2O), pyridine, and 1e with overnight stirring at room temperature (Govindachari et al., 1973) and validated with 1H NMR and 13C NMR data previously reported (Govindachari et al., 1973, Zhen et al., 2002). Detailed information about compounds 1g and 1h will be published elsewhere; the compounds were identified as 3,14a-dihydroxy-4,6,7-trimethoxyphenanthroindolizidine and 3,14a-dihydroxy-6,7-dimethoxyphenanthroindolizidine (Komatsu et al., 2001). See Fig. 1 for chemical structures and the supplementary data for the physical properties of the isolated tylophorine compounds.
3. Results
3.1. Discovery of tylophorine and 7-methoxycryptopleurine as potent anti-TGEV agents
Because of the laboratory constraints of biosafety levels 3 and 4, ST cells infected with TGEV and MRC5 or Vero E6 cells infected with human CoV 229E were used as surrogate systems for screening agents that inhibit the activity of SARS CoV. Our laboratory has established cell-based and 3CLpro enzymatic assays for TGEV to search for anti-viral agents for TGEV and SARS CoV. Some benzothiazolium compounds, e.g. A38120, were found to inhibit 3CLpro enzymatic activities of TGEV and SARS CoV through hit compounds obtained from IFA screening of agents against TGEV, which may reflect the highly conserved substrate specificity and structure of CoV 3CLpros. These active benzothiazolium compounds inhibit TGEV with EC50 values or 50% maximal inhibition concentration (IC50) values ranging from ∼2 to ∼36 μM (Yang et al., 2007b).
Tylophorine compounds were not included in the compound library for our primary IFA screening for agents against TGEV or SARS CoV because of a shortage of compound resources at that time, although we carried out studies of the anti-inflammatory and anti-cancerous mechanisms of tylophorine (Wu et al., 2009, Yang et al., 2006). Accidentally, our laboratory found that tylophorine (1a) and 7-methoxycryptopleurine (1c) potently inhibited TGEV replication, as assessed by IFA with antibodies against the N and S proteins of TGEV. EC50 values for the compounds were 58 ± 4 and 20 ± 1 nM, respectively (Fig. 2A and Table 1 ). However, the compounds at up to 20 μM did not significantly inhibit TGEV or SARS CoV 3CLpro enzymatic activity (data not shown). Moreover, the two compounds exerted long-lasting, for at least 64 hpi, inhibition of virus CPE in TGEV-infected ST cells, in contrast to TGEV-infected ST cells without compound treatment which exhibited significant CPE for about 20–24 hpi (Fig. 2B). We examined the other apoptosis indicator for TGEV-infected ST cells, activation of caspase 3, and found good agreement with the anti-CPE effects of tylophorine (1a) and 7-methoxycryptopleurine (1c) (Fig. 2C).
Fig. 2.
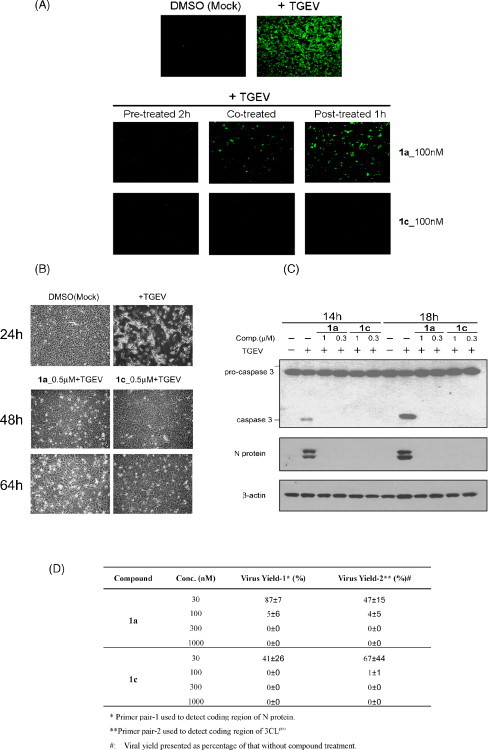
Anti-transmissible gastroenteritis virus (anti-TGEV) activities of tylophorine (1a) and 7-methoxycryptopleurine (1c). (A) Immunofluorescent assay against S and N protein of TGEV in ST cells infected with TGEV (10 multiplicities of infection [MOI]) at 6 hpi treated with vehicle (1% DMSO) and 100 nM 1a and 1c. The compounds were co-administered or administered 2 h before or 1 h after viral infection for prophylactic and therapeutic effects on anti-viral replication. (B) Anti-cytopathic effect (CPE) in ST cells infected with TGEV at 5 MOI. Shown are the CPE of ST cells with TGEV infection and mock infection at 24 hpi, as well as compound treatments (500 nM) at 48 and 64 hpi in TGEV-infected ST cells. (C) Western blot analysis for effect of compounds 1a and 1c at the indicated concentrations on caspase 3 activation, N protein production, and β-actin (as an internal loading control) in TGEV (5 MOI)-infected ST cells at 14 and 18 hpi. Shown for A, B, and C are representative of 3 independent experiments. (D) Effect of the compounds 1a and 1c on TGEV yield. Real time quantification of RT-PCR involved viral RNA extracted from TGEV (10 MOI)-infected ST cell lysates and was performed at 6 hpi with or without compound treatment as described (Yang et al., 2007b). Shown are means ± S.D. from 3 independent experiments.
Table 1.
In vitro anti-transmissible gastroenteritis virus (anti-TGEV) activity of phenanthroindolizidines and phenanthroquinolizidines in ST cells.
| Compound | Source | EC50 (nM) | CC50 (nM) | SI |
|---|---|---|---|---|
| 1a | Synthesis | 58 ± 4 | >100,000 | >1715 |
| 1a′ | T. indica | 95 ± 17 | >100,000 | >1053 |
| 1b | Synthesis | 207 ± 25 | 83,826 ± 3288 | 406 |
| 1c | Synthesis | 20 ± 1 | 43,522 ± 7404 | 2232 |
| 1d | Synthesis | 83 ± 9 | 71,541 ± 2148 | 859 |
| 1e | T. indica | 82 ± 8 | >100,000 | >1220 |
| 1f | T. indica/semi-synthesis | 403 ± 22 | >100,000 | >248 |
| 1g | T. ovata | 8 ± 2 | 59,943 ± 2786 | 7685 |
| 1h | T. ovata | 18 ± 1 | 31,632 ± 1192 | 1719 |
| 1i | Synthesis | 170 ± 21 | 48,306 ± 3071 | 284 |
| 1j | Synthesis | 313 ± 46 | 41,876 ± 6917 | 134 |
| 2a | Synthesis | >100,000 | >100,000 | >1 |
| 2b | Synthesis | 12,798 ± 1567 | >100,000 | >8 |
| 2c | Synthesis | 74,743 ± 5377 | >100,000 | >1 |
| 2d | Synthesis | 19,949 ± 1501 | 71,541 ± 2148 | 4 |
| 3a | Synthesis | >100,000 | >100,000 | >1 |
| 3b | Ficus septicaa | 14,906 ± 2468 | >100,000 | >7 |
| 3c | Synthesis | 22,595 ± 2825 | >100,000 | >4 |
| 4a | Synthesis | 1468 ± 110 | >50,000 | >34 |
| 4b | Synthesis | 363 ± 45 | >50,000 | >137 |
| A38120b | ChemDivb | 6.2 ± 6 (μM)b | >50 (μM)b | >8b |
The compounds and their sources from chemical synthesis or isolation from Tylophora indica and T. ovata plants are shown. The 50% maximal effective concentration (EC50) (nM) values for TGEV replication were determined by immunofluorescent assay at 6 h post-infection (hpi). The 50% maximal cytotoxic concentration (CC50) (nM) values for cytotoxicity of each compound in ST cells were obtained at 24 hpi, and the selectivity index (SI) values, CC50/EC50, were calculated. Shown are means ± S.D. from 3 to 5 independent experiments, each performed in duplicate.
The effect of these compounds on inhibiting virus yields was also examined by real-time quantitative PCR with two primer pairs corresponding to 3CLpro (ORF1a/ORF1ab) and the N protein regions at the 5′-end and 3′-end of the TGEV genome, respectively (Brian and Baric, 2005, Sawicki and Sawicki, 2005). TGEV virus yields were profoundly reduced by compounds 1a and 1c, with EC50 values comparable to those measured from IFA (Fig. 2D to A). Moreover, compounds 1a and 1c prophylactically and therapeutically inhibited TGEV replication because pre-, co-, or post-treatments of 1a and 1c in TGEV-infected ST cells potently reduced fluorescent staining (Fig. 2A). Thus, these compounds strongly protected ST cells against TGEV infection.
3.2. Anti-TGEV activities of synthesized and natural tylophorine analogues
In addition to tylophorine (1a) and 7-methoxycryptopleurine (1c), another 18 compounds were obtained and analyzed for anti-TGEV activity, EC50 values, and cytotoxicity to ST cells with CC50 values (Table 1). Selective index (SI) values were calculated (CC50/EC50). The dehydro-tylophorine analogues, 3a–3c, were the least inhibitory of the compounds tested, with EC50 values ranging from 14906 ± 2468 to >100,000 nM (Table 1). The 9-one-tylophorine analogues 2a–2d were moderate inhibitors of TGEV replication, with EC50 values ranging from 12798 ± 1567 to >100,000 nM (Table 1). The tylophorine analogues 1b, 1d–1j, 4a, and 4b were the most potent and selective inhibitors of virus replication, with EC50 values from 8 to 1468 nM and high SI values from >34 to 7685 (Table 1). Moreover, the anti-TGEV activities of these compounds derived from the CPE assay were consistent with the EC50 values determined by IFA (Fig. 3A and B and Table 1). As with 1a and 1c, these potent analogues (1b, 1d–1j) exerted prolonged inhibition of CPE, inhibiting the appearance of CPE at least up to 48 h (Fig. 2, Fig. 3). Thus, they blocked TGEV replication and induced apoptosis, as indicated by the reduction of TGEV N protein expression and caspase 3 activation in the infected ST cells (Fig. 3C).
Fig. 3.
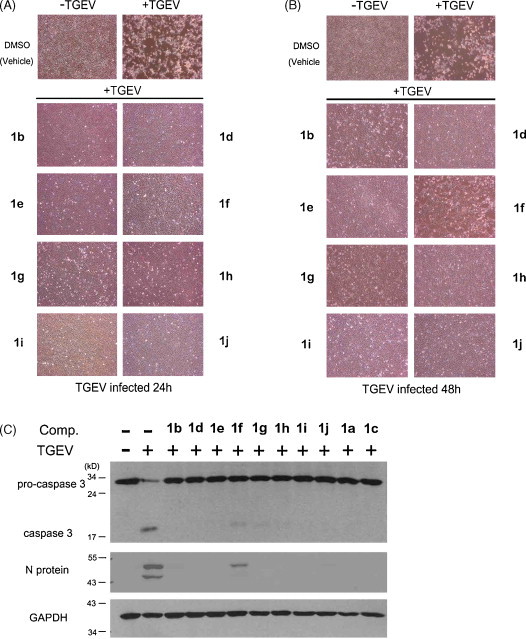
Anti-CPE and anti-apoptosis activities of phenanthroindolizidines and phenanthroquinolizidines. Shown are the CPE of ST cells with mock infection and TGEV infection (5 MOI) in the absence and in the presence of 500 nM compound treatments at 24 (A) and 48 (B) hpi and western blot analysis for protein expression of caspase 3, TGEV N protein and GAPDH (as an internal loading control) at 18 hpi (C) in the presence of 500 nM compound treatments. Shown are representative of 3 independent experiments.
3.3. Tylophorine compounds show potent anti-SARS CoV activity in vitro
Efforts have been initiated to identify small-molecule agents for anti-SARS CoV infection. Thus far, inhibitors targeting the SARS CoV protease have been reported to have IC50 values ranging from 0.5 to 7 μM and the SARS CoV papain-like protease in the 100 nM range. However, the anti-enzymatic activity of these inhibitors may not exhibit corresponding anti-SARS CoV activity in cells (Ghosh et al., 2008). Because we have used porcine TGEV as a surrogate system to search for potential anti-SARS CoV agents (Yang et al., 2007a), we tested the tylophorine compounds 1a, 1c, 1e, 4a, and 4b for anti-CPE activity induced by SARS CoV (Urbani strain) in Vero 76 cells. These compounds were found to have profound activity with EC50 values ranging from 0.34 μM to <0.005 μM and selective index (SI) values from 10 to >100 determined by CPE assay under microscopic observation whereas their EC50 values ranging from 0.62 μM to <0.005 μM and SI values from 6.8 to >15 determined by Neutral red uptake assay, respectively (Table 2 ). The CPE assay in Vero 76 cells induced by SARS CoV takes 3–4 days (Barnard et al., 2006) and tylophorine compounds have been reported to inhibit cell growth (Gao et al., 2004, Lee et al., 2003, Wu et al., 2009), so that on the day for assay the final cell numbers would have 2- to 16-fold difference and the cell volume usually larger for those treated with tylophorine compounds (our unpublished data). This also was manifested by comparing the cell numbers and cell sizes of SARS CoV infected Vero 76 cells treated by compound 1a to those without SARS infected control in the equal area under microscope (Fig. 4 ). Therefore, it is conceivable that lower SI values were obtained from Neutral red uptake assay determining relative cell numbers than those from CPE assay by observation under microscope. These compounds also decreased the formation of large syncytia and/or multinucleated giant cells resulting from virus-induced fusion of cell membranes in a dose-dependent manner (Fig. 4). Thus, these tylophorine compounds are potent small molecules for anti-SARS CoV protection in cell systems.
Table 2.
Anti-SARS CoV activity of phenanthroindolizidines and phenanthroquinolizidines in Vero 76 cells.
| Compound | Visual assay |
Neutral red uptake assay |
||||
|---|---|---|---|---|---|---|
| EC50 (μM)a | CC50 (μM)b | SIc | EC50 (μM) | CC50 (μM) | SI | |
| 1a | 0.018 | 1.6 | 88 | 0.066 | 1.1 | 17 |
| 1c | <0.005 | 0.5 | >100 | <0.005 | 0.0084 | >1.7 |
| 1e | <0.005 | 0.39 | >78 | <0.005 | >0.077 | >15 |
| 4a | 0.34 | 3.4 | 10 | 0.62 | 4.2 | 6.8 |
| 4b | 0.039 | 0.73 | 19 | 0.056 | 0.56 | 10 |
| Infergen™ (μg/ml) | <0.32 | >100 | >320 | 2.4 | >130 | >320 |
The EC50 and CC50 (μM) values were determined visually by microscopy and neutral red uptake assay on the same test plates for anti-SARS CoV-induced cytopathic effect and cytotoxicity, respectively, for the indicated compounds in Vero 76 cells as described (Barnard et al., 2006). The SI values and CC50/EC50 were also calculated.
EC50: the 50% maximal effective concentration.
CC50: the 50% maximal cytotoxic concentration.
SI: selectivity index, CC50/EC50.
Fig. 4.
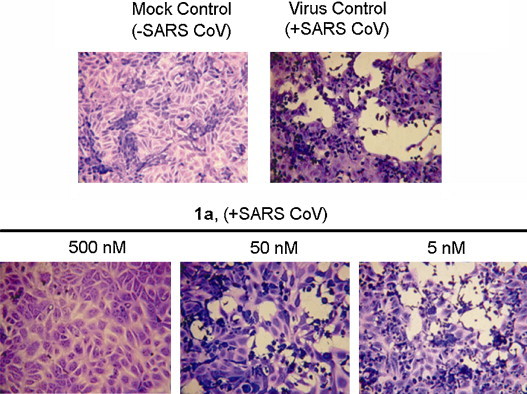
Anti-SARS CoV activity of tylophorine. SARS CoV and the indicated concentrations of tylophorine (1a) were added in equal volumes to near-confluent cell monolayers of Vero 76 cells in culture plates. The MOI used was ranged from 0.01 to 0.025 to produce virus cytopathic effects in 100% of the cells in the virus control wells within 3–4 days along with a mock infection control. The plates were incubated at 37 °C until the cells in the virus control wells showed complete viral CPE as observed by light microscopy and stained with neutral red as described (Barnard et al., 2006). Equal field areas of each treatment were shown from microscopic observation at 100× magnification.
3.4. Analysis of structure–activity relations
The increased planarity and rigidity in structure and more dehydrogenated in indolizidine/quinolizidine moiety of compounds 2a–2d and 3a–3c are suggested to be responsible for their moderate and least activities of anti-TGEV compared to the other potent compounds, 1a–1j and 4a–4b and these structure–activity relations were also found stand for their anti-inflammatory and anti-cancerous activities (Yang et al., 2006, Yang et al., 2007a). Among the most potent tylophorine analogues inhibiting TGEV were compounds 1e, 1g, and 1h with a hydroxyl group at C14 or at the phenanthrene moiety C3. EC50 values for these compounds were 82 ± 8, 8 ± 2 and 18 ± 1 nM, respectively. Thus, these hydroxyl groups contributed significantly to the inhibitory activity exhibited by the tylophorine analogues. However, introduction of an OAc-group into the C14 position decreased the activity by ∼5-fold as compared with compounds with a hydrogen or a hydroxyl group-replacement (1f to 1d and 1e, respectively). Although 1d and 1e had similar potency, they differed in cytotoxicity and thus in selectivity (Table 1), which is an important concern for future drug development. Moreover, introduction of a methoxyl group to position C4 of tylophorine decreased the inhibitory activity of the parent compound ∼4- to 8-fold for both the indolizidine and the quinolizidine counterparts (1a to 1b and 1c to 1i). Similarly, replacing the methoxyl with a hydrogen group at position C2 of tylophorine decreased the inhibitory activity of both the indolizidine and the quinolizidine counterparts but to a lesser extent (1a to 1d and 1c to 1j). In addition, compounds 4a and 4b with an oxygen atom introduced to the reactive nitrogen to form an N-oxide as compared with 1a and 1c, respectively, exerted less potency, by ∼8- to 25-fold. Introduction of the N-oxide to the indolizidine/quinolizidine moiety changed the nitrogen reactivity and might affect the position projection of hydrogen atoms for potency because dehydrogenation in this moiety profoundly decreased activity, e.g. 1a to 2a/3a and 1c to 2c/3c. Similarly, the phenanthroquinolizidines 1c and 4b were more potent than their phenanthroindolizidine counterparts 1a and 4a for anti-SARS CoV activity, and the hydroxyl group at C14 contributed significantly to the inhibitory activity exhibited by compound 1e.
3.5. Pharmacokinetic studies of the tylophorine compounds
Because tylophorine is the main active constituent of T. indica, which has been used as traditional herbal medicine in India taken as dry leaves for anti-asthma, for example, we performed a pharmacokinetic study to determine the oral bioavailability and other parameters of tylophorine (1a) and its quinolizidine counterpart 7-methoxycryptopleurine (1c) (Table 3 ). 7-Methoxycryptopleurine is a synthetic compound that is not naturally occurring (Yang et al., 2007a). After the intravenous administration of 1a and 1c at 3 mg/kg, the total body clearances were 66.8 ± 15.3 and 101.0 ± 8.8 ml/min/kg for 1a and 1c, respectively. The volumes of distribution at steady state (V ss) were 16.6 ± 2.3 and 31.6 ± 4.1 L/kg for 1a and 1c, respectively, which suggests a wide distribution of these compounds to the extravascular tissues. The apparent elimination half-life (t 1/2) with an intravenous dose was long, at ∼4 h for both compounds. After oral administration, the compounds were rapidly absorbed; the C max values of 31.9 ± 17.5 and 16.5 ± 10.4 ng/ml were reached at 1.8 and 1.0 h for 1a and 1c, respectively, after administration. The oral bioavailability of 1c was estimated to be 52.7%, which was comparable to that of 1a, 65.7%, and suggests conserved oral bioavailability for these compounds.
Table 3.
Pharmacokinetic properties of tylophorine (1a) and 7-methoxycryptopleurine (1c) in rats.
|
1a |
1c |
||||
|---|---|---|---|---|---|
| Parametera | Unit | Intravenous dose | Oral dose | Intravenous dose | Oral dose |
| N | 3 | 3 | 3 | 3 | |
| Dose | mg/kg | 3.0 | 3.0 | 3.0 | 3.0 |
| t1/2 | h | 3.9b | 30.7 | 4.2 | 15.1 |
| Clearance | ml/min/kg | 66.8 | 101.0 | ||
| Vss | L/kg | 16.6 | 31.6 | ||
| Cmax | ng/ml | 31.9 | 16.5 | ||
| Ctrough | ng/ml | 1.1 | 3.0 | 1.4 | 5.3 |
| Tmax | h | 1.8 | 1.0 | ||
| AUC(0–inf.) | ng/ml*h | 772 | 507 | 503 | 265 |
| Oral bioavailability | % | 65.7 | 52.7 | ||
t1/2: apparent elimination half-life; Vss: volume of distribution at steady state; Cmax: maximal concentration; AUC(0–inf.): area under concentration curve from time 0 to infinity.
Data are mean values.
4. Discussion
Plant materials are great resources for human therapeutic medicine, for use as alternative medicines and as great reservoirs for the discovery of naturally occurring compounds for developing therapeutic agents. The content of phenanthroindolizidines differs in each Tylophora plant and could slightly vary by harvest season and location. For instance, tylophorine is the main active constituent of T. indica (Gopalakrishnan et al., 1980), tylophorinine in T. atrofolliculata (Huang et al., 2004), and 14-hydroxytylophorine in T. ovata (our unpublished data). In addition, the potency and toxicity of each phenanthroindolizidine may vary depending on the plant source (Table 1 and our unpublished data). Thus, the doses to achieve efficacy and selectivity must be investigated as part of the drug development process, regardless of the formulation, of pure compounds or herbal medicine. Our study is the first to report and compare the pharmacokinetics of naturally occurring tylophorine and synthesized 7-methoxycryptopleurine. We demonstrated their good oral bioavailability. Because of the efficacy, selectivity and oral bioavailability of the tylophorine compounds we report, efforts to develop and optimize more drug-like derivatives of these compounds are warranted.
The SI values for 1a, 1c, and 1e are 88, >100, and >78 from microscopic observation results (Table 2) are different when comparing the values obtained for the TGEV assays and to the values obtained for the SARS CoV assay with neutral red uptake due entirely different cells from different species origins were used for each assays, the length of time the compound was exposed to cells was very different for each assay, and the type of cytotoxicity tests used were different. The compounds were exposed to the Vero 76 cells in the SARS CoV tests for 72 h, which gives the compounds longer to become toxic to cells, the neutral red uptake assays used are more sensitive evaluation of cytotoxicity or growth inhibition (Smee et al., 2002), and there always tissue to tissue differences when assessing cytotoxicity (Smee et al., 2002). Cells derived from swine testicular tissue apparently are less sensitive to toxicity than cells derived from monkey kidneys.
The main effect of the tylophorine compounds is to inhibit TGEV replication in ST cells thereby blocking TGEV-induced apoptosis signaling in ST cells (Fig. 2, Fig. 3). The tylophorine compounds may be administered prophylactically and therapeutically because both administration regimens greatly inhibited TGEV replication, as measured by IFA (Fig. 2A). Therefore, tylophorine compounds are likely to inhibit TGEV replication event(s) after the viruses enter the cells. Moreover, the ability of these compounds to inhibit other coronaviruses such as SARS CoV in Vero 76 cells (Table 2 and Fig. 4) and increase the survival rate of mice infected with MHV-JHM (data not shown) suggests that these compounds inhibit a common or highly conserved target among coronaviruses involved in coronaviral transcriptional complex, e.g. RdRp, helicase, N protein etc., or common cellular factors those are crucial for coronaviral replication. Further detailed mechanism studies of the effect of tylophorine compounds at molecular levels on TGEV replication in infected ST cells are under investigation.
For development and optimization of phenanthroindolizidines into therapeutic drugs, the variety of tylophorine derivatives must be expanded to provide fundamental data and to better understand the relationship between structure and activity. Here we have synthesized and isolated from natural sources to obtain a variety of tylophorine and cryptopleurine compounds and described the previously unreported anti-coronavirus activity of tylophorine compounds and provided more insight into structure–activity relations. Moreover, because of the compounds’ clinical relevance, with potent activity in the low nanomolar range (Table 1, Table 2) and high oral bioavailability (Table 3), the most active tylophorine compounds should be able to safely achieve clinically acceptable doses. Collectively, tylophorine compounds and the traditional herb plants from which the compounds were derived should be considered useful sources of anti-coronavirus inhibitors for coronavirus epidemics or pandemics.
Acknowledgements
This work was supported by the National Science Council of Taiwan, ROC [Grants 99-2320-B-400-009-MY3 and 95-2320-B-400-009-MY3] and the National Health Research Institutes, Taiwan, ROC [Grants BP-098-PP-05 and BP-099-PP-04]. Cheng-Wei Yang is a Ph.D. student in the Graduate Program of Biotechnology in Medicine sponsored by the National Tsing Hua University and the National Health Research Institutes. The authors also acknowledge Dr. Yu-Cheng Chou for some of the 1H NMR and LC/MS measurements and Dr. Chi-Min Chen for providing antibodies for TGEV. The work contributed by Dr. Barnard was supported in part by Contract N01-AI-30048 from the US National Institutes of Health, NIAID, Division of Virology.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.antiviral.2010.08.009.
Appendix A. Supplementary data
References
- Abe I., Yukiko, Yamauchi T., Honda K., Hayashi N. Phenanthroindolizidine alkaloids from Tylophora tanakae. Phytochemistry. 1995;39:695–699. [Google Scholar]
- Barnard D.L., Day C.W., Bailey K., Heiner M., Montgomery R., Lauridsen L., Winslow S., Hoopes J., Li J.K., Lee J., Carson D.A., Cottam H.B., Sidwell R.W. Enhancement of the infectivity of SARS-CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antiviral. Res. 2006;71:53–63. doi: 10.1016/j.antiviral.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani K.K., Sharma G.L., Ali M. Plant based antiamoebic drugs. Part I. Antiamoebic activity of phenanthroindolizidine alkaloids; common structural determinants of activity with emetine. Planta Med. 1987;53:532–536. doi: 10.1055/s-2006-962803. [DOI] [PubMed] [Google Scholar]
- Brian D.A., Baric R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005;287:1–30. doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemler S.R. Phenanthroindolizidines and phenanthroquinolizidines: promising alkaloids for anti-cancer therapy. Current Bioactive Compounds. 2009;5:2–19. doi: 10.2174/157340709787580928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang T.H., Lee S.J., Yang C.W., Wu P.L. Expedient synthesis and structure–activity relationships of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Org. Biomol. Chem. 2006;4:860–867. doi: 10.1039/b516152e. [DOI] [PubMed] [Google Scholar]
- Gao W., Lam W., Zhong S., Kaczmarek C., Baker D.C., Cheng Y.C. Novel mode of action of tylophorine analogs as antitumor compounds. Cancer Res. 2004;64:678–688. doi: 10.1158/0008-5472.can-03-1904. [DOI] [PubMed] [Google Scholar]
- Ghosh A.K., Gong G., Grum-Tokars V., Mulhearn D.C., Baker S.C., Coughlin M., Prabhakar B.S., Sleeman K., Johnson M.E., Mesecar A.D. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2008;18:5684–5688. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan C., Shankaranarayanan D., Nazimudeen S.K., Kameswaran L. Effect of tylophorine, a major alkaloid of Tylophora indica, on immunopathological and inflammatory reactions. Indian J. Med. Res. 1980;71:940–948. [PubMed] [Google Scholar]
- Govindachari T.R., Viswanthan N., Radhakrishnan J. Minor alkaloids of Tylophora asthmatica revised structure of tylophorinidine. Tetradedron. 1973;29:891–897. [Google Scholar]
- Huang X., Gao S., Fan L., Yu S., Liang X. Cytotoxic alkaloids from the roots of Tylophora atrofolliculata. Planta Med. 2004;70:441–445. doi: 10.1055/s-2004-818973. [DOI] [PubMed] [Google Scholar]
- Komatsu H., Watanabe M., Ohyama M., Enya T., Koyama K., Kanazawa T., Kawahara N., Sugimura T., Wakabayashi K. Phenanthroindolizidine alkaloids as cytotoxic substances in a Danaid butterfly, Ideopsis similis, against human cancer cells. J. Med. Chem. 2001;44:1833–1836. doi: 10.1021/jm0004042. [DOI] [PubMed] [Google Scholar]
- Lee S.K., Nam K.A., Heo Y.H. Cytotoxic activity and G2/M cell cycle arrest mediated by antofine, a phenanthroindolizidine alkaloid isolated from Cynanchum paniculatum. Planta Med. 2003;69:21–25. doi: 10.1055/s-2003-37021. [DOI] [PubMed] [Google Scholar]
- Li M., Ansari S.H., Grever M.R. Cytotoxic alkaloids form Tylophora indica. Die Pharm. 2001;56:188–190. [PubMed] [Google Scholar]
- Li Z., Jin Z., Huang R. Isolation, total synthesis and biological activity of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Synthesis. 2001;16:2365–2378. [Google Scholar]
- Lin, I.H., 2003. Compendium of Medicinal Plants Used by the Indigenous People of Taiwan, English Edition; Committee on Chinese Medicine and Pharmacy, Department of Health, Executive Yuan, ROC, Taipei, Taiwan ROC, pp. 385–386.
- Luo Y., Liu Y., Luo D., Gao X., Li B., Zhang G. Cytotoxic alkaloids from Boehmeria siamensis. Planta Med. 2003;69:842–845. doi: 10.1055/s-2003-43215. [DOI] [PubMed] [Google Scholar]
- Nordlander J.E., Njoroge F.G. A short synthesis of (S)-(+)-tylophorine. J. Org. Chem. 1987;52:1627–1630. [Google Scholar]
- Olsen C.W. A review of feline infectious peritonitis virus: molecular biology, immunopathogenesis, clinical aspects, and vaccination. Vet. Microbiol. 1993;36:1–37. doi: 10.1016/0378-1135(93)90126-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao K.V. Alkaloids of tylophora. II. Structural studies. J. Pharm. Sci. 1970;59:1608–1611. doi: 10.1002/jps.2600591113. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. Coronavirus transcription: a perspective. Curr. Top. Microbiol. Immunol. 2005;287:31–55. doi: 10.1007/3-540-26765-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivpuri D., Menon M., Parkash D. Preliminary studies in Tylophora indica in the treatment of asthma and allergic rhinitis. J. Assoc. Phys. India. 1968;16:9–15. [PubMed] [Google Scholar]
- Smee D.F., Morrison A.C., Barnard D.L., Sidwell R.W. Comparison of colorimetric, fluorometric, and visual methods for determining anti-influenza (H1N1 and H3N2) virus activities and toxicities of compounds. J. Virol. Methods. 2002;106:71–79. doi: 10.1016/s0166-0934(02)00137-4. [DOI] [PubMed] [Google Scholar]
- Staerk D, Lykkeberg A.K., Christensen J., Budnik B.A., Abe F., Jaroszewski J.W. In vitro cytotoxic activity of phenanthroindolizidine alkaloids from Cynanchum vincetoxicum and Tylophora tanakae against drug-sensitive and multidrug-resistant cancer cells. J. Nat. Prod. 2002;65:1299–1302. doi: 10.1021/np0106384. [DOI] [PubMed] [Google Scholar]
- Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005;69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.M., Yang C.W., Lee Y.Z., Chuang T.H., Wu P.L., Chao Y.S., Lee S.J. Tylophorine arrests carcinoma cells at G1 phase by downregulating cyclin A2 expression. Biochem. Biophys. Res. Commun. 2009;386:140–145. doi: 10.1016/j.bbrc.2009.05.138. [DOI] [PubMed] [Google Scholar]
- Wu P.L., Rao K.V., Su C.H., Kuoh C.S., Wu T.S. Phenanthroindolizidine alkaloids and their cytotoxicity from the leaves of Ficus Septica. Heterocycles. 2002;57:2401–2408. [Google Scholar]
- Yang C.W., Chen W.L., Wu P.L., Tseng H.Y., Lee S.J. Anti-inflammatory mechanisms of phenanthroindolizidine alkaloids. Mol. Pharmacol. 2006;69:749–758. doi: 10.1124/mol.105.017764. [DOI] [PubMed] [Google Scholar]
- Yang C.W., Chuang T.H., Wu P.L., Huang W.H., Lee S.J. Anti-inflammatory effects of 7-methoxycryptopleurine and structure–activity relations of phenanthroindolizidines and phenanthroquinolizidines. Biochem. Biophys. Res. Commun. 2007;354:942–948. doi: 10.1016/j.bbrc.2007.01.065. [DOI] [PubMed] [Google Scholar]
- Yang C.W., Yang Y.N., Liang P.H., Chen C.M., Chen W.L., Chang H.Y., Chao Y.S., Lee S.J. Novel small-molecule inhibitors of transmissible gastroenteritis virus. Antimicrob. Agents Chemother. 2007;51:3924–3931. doi: 10.1128/AAC.00408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H.T., Wu Y.S., Chang Y.W., Hsieh H.P., Chen W.C., Lan S.J., Chen C.T., Chao Y.S., Chang L., Sun H.Y., Yeh T.K. Biotransformation of 6-methoxy-3-(3′,4′,5′-trimethoxy-benzoyl)-1H-indole (BPR0L075), a novel antimicrotubule agent, by mouse, rat, dog, and human liver microsomes. Drug Metab. Dispos. 2007;35:1042–1049. doi: 10.1124/dmd.106.014597. [DOI] [PubMed] [Google Scholar]
- Zeng W., Chemler S.R. Total synthesis of (S)-(+)-tylophorine via enantioselective intramolecular alkene carboamination. J. Org. Chem. 2008;73:6045–6047. doi: 10.1021/jo801024h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen Y.Y., Huang X.S., Yu D.Q., Yu S.S. Anti-tumor alkaloids isolated from Tylophora ovata. Acta Bot. Sin. 2002;44:349–353. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.