

Abstract
The pharmacological inhibitor SP600125 [anthra(1,9-cd)pyrazol-6(2H)-one 1,9-pyrazoloanthrone] has been largely employed as a c-JUN N-terminal kinase (JNK1/2) inhibitor. In this study, we evaluated whether pretreatment with SP600125 was able to prevent Orthopoxviruses Vaccinia virus (VACV), Cowpox virus (CPXV) and modified Vaccinia virus Ankara (MVA) replication. We found that incubation with SP600125 not only blocked virus-stimulated JNK phosphorylation, but also, significantly reduced virus production. We observed 1–3 log decline in viral yield depending on the cell line infected (A31, BSC-40 or BHK-21). The reduction in viral yield correlated with a dramatic impact on virus morphogenesis progress, intracellular mature viruses (IMV) were barely detected. Despite the fact that SP600125 can act as an efficient anti-orthopoxviral compound, we also provide evidence that this antiviral effect is not specifically exerted through JNK1/2 inhibition. This conclusion is supported by the fact that viral titers measured after infections of JNK1/2 knockout cells were not altered as compared to those of wild-type cells. In contrast, a decline in viral titers was verified when the infection of KO cells was carried out in the presence of the pharmacological inhibitor. SP600125 has been the focus of recent studies that have evaluated its action on diverse viral infections including DNA viruses. Our data support the notion that SP600125 can be regarded as a potential antipoxviral compound.
Keywords: Poxvirus, SP600125, Antipoxviral, Antiviral, JNK, Morphogenesis
1. Introduction
The Orthopoxviruses encompass a family of large, double-stranded DNA viruses, approximately 200 kbp in size, whose replication is entirely carried out in the cytoplasm of infected cells (Condit et al., 2006, Moss, 2007). In 1980, the World Health Organization (WHO) declared that smallpox (Variola) – a devastating human disease caused by Variola virus (VARV) – was eradicated (Fenner et al., 1988, Barquet and Domingo, 1997, Smith and McFadden, 2002). With its eradication, vaccination was discontinued. As a consequence, much of the world’s population has either never been immunized or has not been immunized for more than 30 years. Either scenario results in a population that is extremely susceptible to variola or other poxviruses.
Our laboratory is interested in dissecting poxvirus-host cell interactions. We have observed that pharmacological inhibition of the MEK/ERK pathway with UO126 or PD98059 decreased virus yield by at least one order of magnitude (de Magalhães et al., 2001, Andrade et al., 2004). Moreover, pretreatment of cells with LY294002, a pharmacological inhibitor of the PI3K/Akt pathway, decreased Vaccinia virus (VACV) or Cowpox virus (CPXV) replication by 99% (Soares et al., 2009). Here we show that SP600125, an anthrapyrazolone inhibitor of the c-JUN N-terminal kinases 1/2 (JNK1/2) (Bennett et al., 2001), caused a significant decrease in viral yield of VACV, CPXV and modified Vaccinia virus Ankara (MVA). Although SP600125 is regarded as a specific JNK inhibitor (Bennett et al. 2001), our findings demonstrate that its antipoxviral effect is mediated through the target of a yet undefined kinase(s) other than JNK1/2. Since SP600125 has proved to be efficient in vitro against diverse viral infections such as influenza (Mehrotra et al., 2007), rotavirus (Holloway et al., 2006) and herpesvirus (Zapata et al., 2007, Hamza et al., 2004, Perkins et al., 2003, Chen et al., 2002), we propose a potential use of this compound to treat poxviruses infection or complications associated with vaccination.
2. Materials and methods
2.1. Cell culture, antibodies and chemicals
A31 cells (a clone derived from mouse Balb/c 3T3), BSC-40, BHK-21 and mouse embryonic fibroblasts (MEFs) from WT and double knockout (KO) JNK1/2−/− cells (Tournier et al., 2000), were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with heat-inactivated fetal bovine serum (FBS), (% v/v), as follows: BSC-40 (6%); BHK-21 (10%) and JNK (5%), and antibiotics in 5% CO2 at 37 °C. FBS was purchased from Cultilab, Campinas, SP, Brazil. A31 cells were kindly provided by Sogayar (Department of Biochemistry, University of São Paulo, Brazil). Davis (Howard Hughes Medical Institute, University of Massachusetts Medical School, Worcester, MA) gently provided us with WT and JNK1/2 KO cells. The following rabbit polyclonal antibodies were purchased from Sigma–Aldrich (São Paulo, Brazil): anti β-Tubulin or Cell Signaling Technology (Beverly, MA): anti-phospho JNK1/2 (Thr183/Tyr185), anti-c-JUN (Ser73), anti-total ERK1/2, as were the horse radish peroxidase (HRP) conjugated anti-rabbit and anti-mouse secondary antibodies. Both SP600125 [anthra(1,9-cd)pyrazol-6(2H)-one 1,9-pyrazoloanthrone] (structural formula below) and the JNK Inhibitor VIII (JNKi VIII) - (N-(4-amino-5-cyano-6-ethoxypyridin-2-yl)-2-(2,5-dimethoxyphenylacetamide), were purchased from Calbiochem (São Paulo, Brazil); inhibitors were diluted in DMSO to a final concentration of 25 mM (SP600125) and 4 mM (JNKi VIII) and stored at −20 °C.
2.2. Viruses
(A) Viral stocks: Wild-type VACV (strain WR) and CPXV (strain BR) were propagated in Vero or BSC-40 cells. MVA was propagated in BHK-21 cells. Viruses were then highly purified by sucrose gradient sedimentation as described (Joklik, 1962). The experiments presented in this study were carried out using the intracellular mature virus (IMV) form of the virus. (B) Viral infection: Cells were allowed to reach 80–90% confluence and starved by changing the media to 1% FBS for 12 h. Cells were infected at the indicated multiplicity of infection (MOI) for the times shown. When needed, cells were treated with the indicated compound for 30 min prior to viral infection and incubated in the continued presence of the drug.
2.3. Multi-step viral growth curves
Thirty five millimeter dishes of A31, BSC-40, BHK-21 and JNK1/2 KO cells (density 5 × 105 cells/dish) were starved and infected at an MOI of 10 for the indicated times 3, 6, 12, 24, 36 and 48 h either in the absence or in the presence of SP600125 (40 μM) or JNKi VIII (4 μM). At each time point, cultures were washed with cold PBS, and cells were disrupted by freeze/thawing. Supernatant were collected and the viral yield was quantified by viral plaque assay as described (da Silva et. al., 2006). Data were confirmed by at least three independent experiments with similar results.
2.4. Electron microscopy
BSC-40 cells were infected with VACV (MOI of 2) either in the absence or in the presence of SP600125 (40 μM) and incubated at 37 °C for 18 h. Cells were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 1 h at room temperature, scraped gently, and collected by centrifugation. The cells were washed with cacodylate buffer, postfixed with 1% osmium tetroxide, dehydrated in acetone and processed for conventional transmission electron microscopy. Thin sections were examined with a Morgagni transmission electron microscope operating at 80 kV.
2.5. Cytotoxicity assays
Confluent 35 mm dishes of A31 or BSC-40 cells were treated with increasing concentrations (10, 20, 40 and 50 μM) of SP600125. At 48 h, an equal volume of Trypan Blue stain was added to each well. Cells were stained for 10 min at room temperature after which time the stain was removed and cells were observed for any evidence of stain absorption (an indication of cellular membrane permeability and death). We found that ⩾90% of the cells pretreated with SP600125 at 40 μM were not stained. This concentration was used throughout the experiments. A dose response including 0.4, 4 and 40 μM of JNKi VIII was also performed for cytotoxicity assays and 4 μM was employed in our experiments.
2.6. Western blotting
(A) Lysate preparation – A31 and BSC-40 cells were starved and infected with VACV or CPXV (MOI = 10) in the presence or absence of SP600125. At the indicated times, cells were washed with cold PBS and disrupted on ice with lysis buffer [100 mM Tris–HCl (pH 8,0), 1% Triton X-100, 0.2 mM EDTA, 20% glycerol (v/v), 200 mM NaCl, 1 mM NaVO3 (sodium orthovanadate), 1 mM PMSF (phenylmethanesulfonyl fluoride), 5 μg/mL aprotinin, 2.5 μg/mL leupeptin, 1 mM DTT]. Whole cell lysates were collected by centrifugation at 13,500 rpm for 15 min at 4 °C. Protein concentration was determined by the Bio-Rad assay. (B) Electrophoresis and immunoblotting – Forty microgram of protein per sample were separated by electrophoresis on a 10% SDS polyacrylamide gel and transferred to nitrocellulose membranes (de Magalhães et al., 2001). Briefly, membranes were blocked at room temperature for 1 h with PBS containing 0.1% Tween-20 and 5% (w/v) non-fat milk. The membranes were washed three times with PBS containing 0.1% Tween-20, incubated with specific polyclonal or monoclonal antibody (1:1000–1:3000) in PBS containing 0.1% Tween-20 and 5% (w/v) BSA, followed by incubation with the HRP-conjugated secondary anti-rabbit Ab (1:3000) or anti-mouse Ab (1:1000). Immunoreactive bands were visualized by the ECL detection system as described in the Manufacturer’s instructions (GE Healthcare, UK).
3. Results
3.1. VACV and CPXV infection stimulate JNK1/2 phosphorylation
In order to investigate whether the cellular stress associated with orthopoxvirus infection led to the activation of the stress-associated protein kinases (SAPKs)/c-Jun N-terminal kinase (JNKs), BSC-40 cells were infected with VACV or CPXV. At 3, 6, 12, 24 and 36 h post-infection (h.p.i) whole cell lysates were collected and subjected to western blot to evaluate the phosphorylation status of JNK1/2. Our data (Fig. 1 A and B) demonstrated that both CPXV and VACV infections were able to stimulate JNK1/2 phosphorylation (JNK1/2-P) as early as 3 h.p.i., and to a maximal level reached at late times in the infective cycle, 36 h.p.i. As a control, and as expected, we observed no difference in the levels of ERK1/2 during infection. Additionally, viral stimulation of JNK1/2-P was blocked in a dose-dependent manner [10, 20, 40 and 50 μM (Fig. 1C, lanes 4–7)] when VACV infection was performed in the continued presence of SP600125. Similar results were obtained with CPXV infection (data not shown).
Fig. 1.
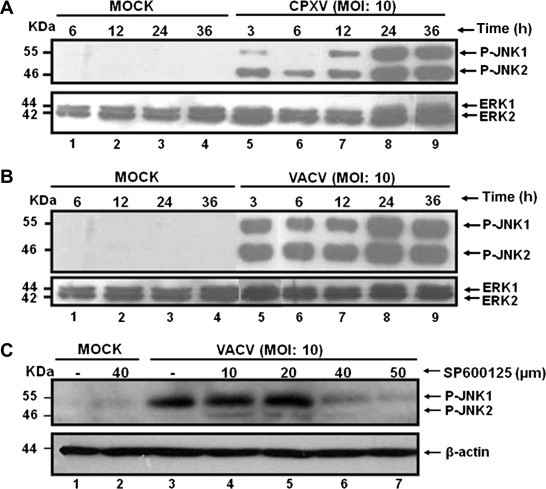
VACV and CPXV infection stimulate JNK1/2 phosphorylation. BSC-40 cells were left uninfected (MOCK) or were infected with CPXV (A) or VACV (B) for the times shown. (C) Cells were left uninfected (MOCK) or infected (VACV) and treated with SP600125 (10, 20, 40 or 50 μM) prior to and during viral infection (lanes 4–7). Cell lysates (40 μg) were prepared, subjected to western blot and probed with anti-phospho JNK1/2 (Thr183/Tyr185) – upper panels, or probed with anti-ERK1/2 or anti-β actin antibody as a loading control – lower panels. Data are representative of three distinct experiments with similar results.
3.2. SP600125 inhibits VACV, CPXV and MVA growth
In order to investigate whether the Orthopoxvirus-stimulated JNK1/2-P was biological relevant to the virus, we performed multi-step viral growth curves (MOI = 10) in the presence or absence of SP600125. Cellular extracts were collected at 3, 6, 12, 24, 36 and 48 h.p.i and assayed for viral yield. We observed that the SP600125-mediated inhibition played a relevant role in both VACV and CPXV biology. A significant reduction in the viral titers (⩾1 log reduction) was observed when VACV (Fig. 2 A) or CPXV (Fig. 2B) infections were carried out in the continued presence of SP600125. To verify that the inhibitory effect associated with SP600125 was not restricted to the A31 cells, BSC-40 were infected with VACV or CPXV as described above. As shown in Fig. 2C and D, treatment with SP600125 resulted in a severe decrease in viral production (2–3 log reduction) thereby demonstrating that viral growth inhibition is not cell-type specific. Additionally, we investigated whether SP600125 was able to affect MVA replication. To that end, BHK-21 cells were infected with MVA as described above. Again, our results showed (Fig 2E) that the inhibitor caused a significant decline in virus yield (nearly 3 log reduction); while a more mild decrease (1 log) in infectivity was noted with VACV and CPXV (2F and 2G). The variation in the levels of inhibition caused by SP600125 might be due to the viruses’ tropism within different species such as murine (A31 cells), monkey (BSC-40 cells) and hamster (BHK-21 cells).
Fig. 2.
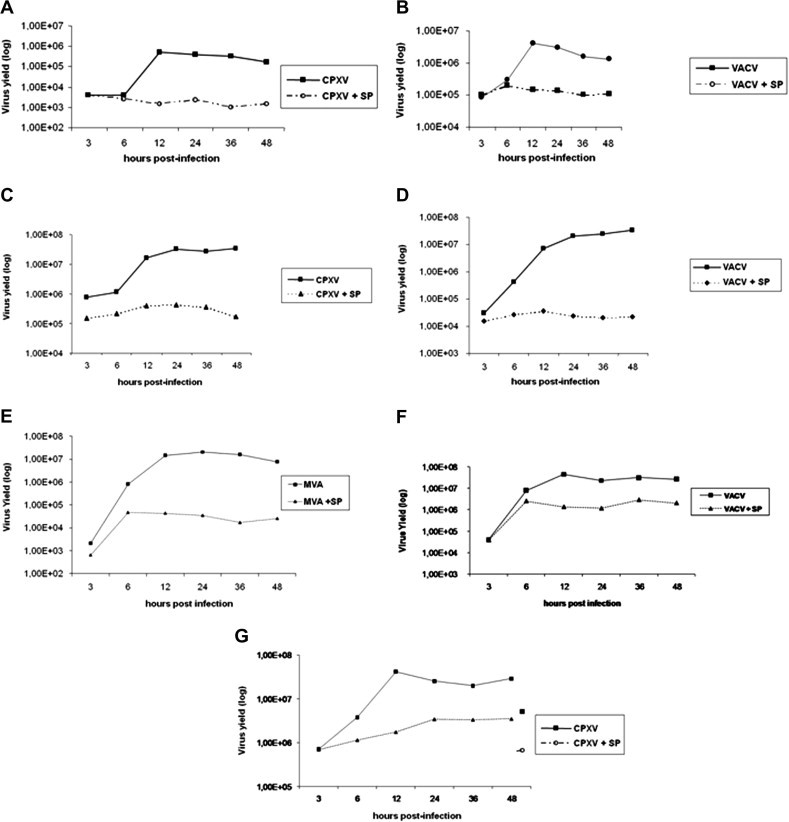
SP600125 inhibits VACV, CPXV and MVA growth. Multi-step growth curves - A31 cells (A, B), BSC-40 (C, D) and BHK-21(E–G) were CPXV- (A, C, G), VACV- (B, D, F) and MVA- (E) infected at an MOI of 10, for the indicated times, either in the absence or presence of SP600125 (40 μM). Cell lysates were collected and viral yields were quantitated. Data are representative of three distinct experiments with similar results.
3.3. SP600125 severely compromise progress of Orthopoxviruses morphogenesis
In order to investigate at what stage the progression of the viral cycle was affected by SP600125, BSC-40 cells were left untreated (Fig 3 A, B and C) or were pretreated with the inhibitor (Fig 3D, E, F and G) and infected with VACV at an MOI of 2. At 18 h.p.i, infected cells were harvested and examined by electron microscopy. While infected cells in the absence of inhibitor (panels A, B and C) contained the full spectrum of virion morphogenesis forms characterized by the identification of crescent, spherical, immature virions (IV), immature virions with nucleoids (IVN) and brick-shaped mature virions (IMV), cells pre-incubated with SP600125 (panels D, E, F and G) showed a severe impairment of morphogenesis progression. Large virosomes surrounded by crescents were repeatedly detected. IVs could be also observed, however IVNs or IMVs were rarely seen. Identical phenotype was also observed when cells were infected with CPXV in the presence of SP600125 (data not shown).
Fig. 3.
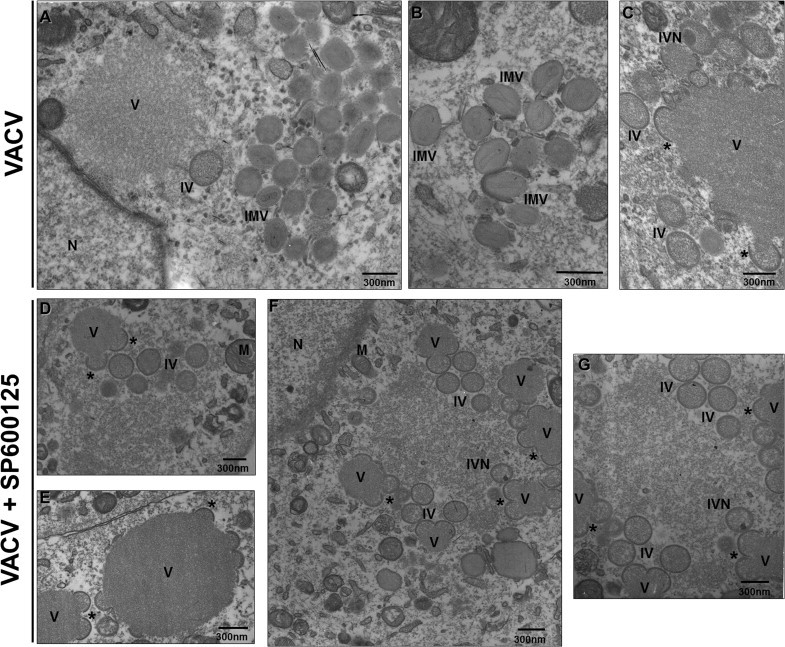
SP600125 blocks virion maturation during orthopoxvirus morphogenesis. Electron microscopy analysis – A31 cells were either left untreated (A–C) or treated with SP600125 prior to VACV (D–G) infection at MOI of 2 for 18 h. Cells were then fixed and prepared for transmission electron microscopy. Electron micrographs are shown with their scale indicated by the bars. Abbreviations:∗ – Crescents, IV – immature virus, IVN – immature virions with nucleoids, IMV – intracellular mature virus, N – nucleus, M – mitochondria, V - virosome. Panel G is a close up image of panel F. Data is representative of two independent experiments with similar results.
3.4. Inhibition of VACV and CPXV growth by SP600125 is independent of JNK1/2
Although SP600125 has been characterized as a specific JNK pharmacological inhibitor (Bennett et al., 2001), a growing body of evidence suggests that SP600125 may be an inhibitor of other kinases as well (Bain et al., 2003, Bain et al., 2007, Bogoyevitch and Arthur, 2008). Thus, to further define whether the reduction in viral yields associated with SP600125 treatment was a direct consequence of JNK1/2 inhibition, WT (Fig. 4 A) or JNK1/2 KO MEF cells (Fig. 4B) were infected with VACV or with CPXV. Infections were carried out either in the absence or presence of SP600125 (40 μM) or the pharmacological inhibitor of JNK (JNKi VIII - 4 μM). After 24 h, infected cells were collected and assayed for viral production. As shown in Fig. 4A and B, in the absence of any inhibitor, the viral titers were comparable when produced in either cell line (WT or JNK KO cells lines). This observation strongly suggests that neither VACV nor CPXV require JNK for productive infection. Furthermore, both the WT and JNK KO cells were equally susceptible to SP600125, while being refractory to JNKi VIII treatment.
Fig. 4.
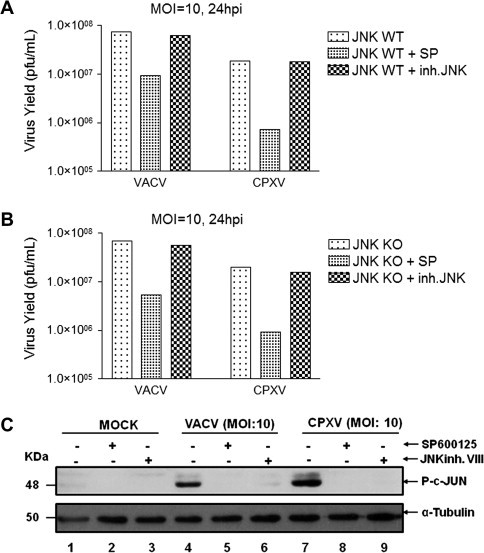
Inhibition of VACV and CPXV growth by SP600125 is independent of JNK1/2. WT (A) and JNK1/2 KO (B) cells were either left untreated or treated with SP600125 (40 μM), or with JNK Inhibitor VIII (4 μM) prior to VACV or CPXV infection at an MOI of 10. At 24 h.p.i, total cell lysates were assayed for viral production. (C) Cells were left uninfected (MOCK) or were treated with SP600125 or JNKi at (4 μM) (lanes 5, 6 and 8, 9), as indicated. Total cell lysates (40 μg) were prepared, subjected to western blot and probed with anti-phospho c-JUN (Ser 73) – upper panels, or probed with anti-β tubulin antibody (1:3000) as a loading control – lower panels. Data are representative of three distinct experiments with similar results.
In order to confirm that JNK does not contribute to the viral replication, we evaluated the phosphorylation levels of its substrate, c-Jun, during viral infection in the presence or absence of either SP600125 or JNKi VIII. Both compounds are known as reversible ATP-competitive JNK inhibitors that ultimately block phosphorylation of JNK substrates such as c-Jun (Bennett et al., 2001, Vivanco et al., 2007). Fig. 4C shows that both SP600125 and JNKi VIII affected VACV- and CPXV-stimulated c-Jun phosphorylation (c-Jun-P). Taken together these findings demonstrated that even though both pharmacological inhibitors targeted the same downstream substrate of JNK (c-Jun), viral replication was only affected in the presence of SP600125. Thus, our data strongly suggest that SP600125 is targeting kinase(s) other than JNK1/2 and, therefore, provide evidence of its JNK-independent inhibitory action.
4. Discussion
Smallpox was announced eradicated by WHO in 1980 and since then, vaccination has been discontinued. As a consequence, much of the world’s population is vulnerable and, therefore, under continuous threat. Moreover, even though the smallpox vaccine (VACV) was successfully used in the WHO’s eradication program, the vaccine has an imperfect safety record and cannot be used with those having immunological deficiency or eczema (Fenner et al., 1988, Barquet and Domingo, 1997, Smith and McFadden, 2002). Furthermore, the re-emergence of CPXV in Europe (Vorou et al., 2008), Monkeypox virus (MPXV) outbreaks in Africa and the United States (Sejvar et al., 2004, Reynolds et al., 2004, Formenty et al., 2010), and the emergence of VACV in Brazil (Fonseca et al., 1998, Damaso et al., 2000, Trindade et al., 2007), emphasizes the need for searching for new antipoxviral compounds with potential use in clinical trials.
Currently, the only antiviral agent currently approved by FDA (Food and Drug Administration) for use against Orthopoxviruses is cidofovir (CDV), an acyclic nucleoside phosphonate analogue, which is known to inhibit not only poxvirus replication but also the replication of a variety of other DNA viruses such as herpesvirus, adenovirus, papillomavirus, and polyomavirus (De Clercq, 2003). In 2009, it was shown that cidofovir impairs Vaccinia DNA encapsidation and, consequently, affects viral morphogenesis (Jesus et al., 2009). In humans, cidofovir has been used successfully against Molluscum contagiosum virus and ORF virus, however renal toxicity is a known side effect caused by this drug (De Clercq, 2002). Importantly, cidofovir-resistant strains of camelpox, cowpox, monkeypox and vaccinia viruses have also been isolated (Smee et al., 2002). To overcome nephrotoxicity, a derivative form of CDV has been generated and tested. CMX001 is a lipid conjugate of the acyclic nucleotide phosphonate and is currently in Phase II clinical trials for the prophylaxis of human cytomegalovirus infection and under development using the Animal Rule for smallpox infection. CMX001 has demonstrated in vitro and in vivo efficacy against orthopoxvirus infections, and no evidence of nephrotoxicity in either animals or humans was found. Both drugs target the viral DNA polymerase, and VACV strains have been shown to be cross resistant to CMX001 as well.
A new class of anti-poxvirus drugs, which affects both viral spread and dissemination, has also emerged. One of them, ST-246, has been intensely tested against a number of Orthopoxvirus species in animal studies (Yang et al., 2005a, Yang et al., 2005b, Sbrana et al., 2007, Quenelle et al., 2007). ST-246 specifically inhibits the viral protein F13, which is required for the formation of enveloped virus forms. Similar to CDV in which viral resistance is conferred by point mutations in the DNA polymerase gene (Becker et al., 2008), it has also been described that a single point mutation in F13 conferred resistance to ST-246 (Yang et al., 2005a, Yang et al., 2005b). ST-246 was recently tested in a Phase I clinical trial and found to be well tolerated and safe in healthy humans (Jordan et al., 2008, Jordan et al., 2010).
An additional approach to inhibit viral multiplication is targeting cellular signaling pathways stimulated and required for successful replication and dissemination. In the past years, we and others have shown the ability of the Orthopoxviruses VACV and CPXV to induce protein kinases pathways to provide an adequate environment to favor their viral replication cycles (de Magalhães et al., 2001, Andrade et al., 2004, da Silva et al., 2006, Mercer and Helenius, 2008, Soares et al., 2009, McNulty et al., 2010). It is also known that poxviruses use the Src and Abl family kinase activities to modulate intracellular spread and release (Frischknecht et al., 1999, Reeves et al., 2005, Reeves et al., 2011) but only the Abl family of kinases mediate release of CEV to form EEV (Reeves et al., 2005). Therefore, tyrosine kinase inhibitors originally developed for treating cancers have also been tested against many poxviruses. One of these drugs is imatinib mesylate (STI-571; Gleevec), which is approved for treating human cancers (Tolomeo et al., 2009, Wolf and Rumpold, 2009). Gleevec specifically inhibits the Abl family of kinases, reducing VACV dissemination in vivo (Reeves et al., 2005). It has been suggested that cardiotoxicity can be a side-effect caused by this drug; but even targeting cellular kinases may bring attention about unwanted side effects (Kerkelä et al., 2006), it seems that drug resistance cannot readily develop, which is a benefit for antiviral chemotherapy.
The anthrapyrazolone inhibitor of c-JUN N-terminal kinases 1/2 (JNK1/2), SP600125 (Bennett et al., 2001, Bogoyevitch et al., 2004), the focus of this manuscript, has been largely utilized as a potential therapeutic for the treatment of cancer and diseases caused by inflammation and neurodegeneration (Sharma et al., 2010, Holm et al., 2011, Hu and Liu, 2009, de Borst et al., 2009, Wang et al., 2009, Song et al., 2008). Some derivatives of SP600125 are being tested in diverse clinical trials (Manning and Davis, 2003, Bogoyevitch et al., 2004, Bennett, 2006, Bogoyevitch and Arthur, 2008). In addition, the antiviral effects of SP600125 have been investigated in diverse viral models suggesting that JNK inhibitors may provide new therapeutic interventions (Manning and Davis, 2003, Bogoyevitch and Arthur, 2008). For instance, it has been shown that the viral kinase ORF36 of the Kaposi’s sarcoma-associated herpesvirus activates JNK1/2 and its inhibition by SP600125 blocks viral gene expression at late stages of infection (Hamza et al., 2004). Varicella-zoster virus (VZV) replication was also decreased in a dose-dependent manner by treatment with SP600125 (Zapata et al., 2007). Another report showed that SP600125 inhibited the activation of JNK by the hepatitis C virus protein NS3, which contributes to hepatitis C related hepatocarcinogenesis (Hassan et al., 2005). Furthermore, the use of signal transduction pathways modulators, either singly (Yang et al., 2005a, Yang et al., 2005b, Reeves et al., 2005) or in combination, could be the most appropriate therapeutic strategy. In fact, it has been shown that SP600125 used together with inhibitors of phosphatidylinositol 3-kinase/Akt prevented the establishment of persistent SARS-CoV infection (Mizutani et al., 2005).
While studying the Orthopoxviruses VACV, CPXV, and MVA-cell host- interaction, we found that SP600125 exerted an antiviral effect. Our results showed a dramatic reduction in virus yield when infections were performed in the presence of this inhibitor. Electron microscope images demonstrated that in the presence of SP600125, Orthopoxviruses replication is compromised; normal-looking IVs are frequently seen but IVN are very rare and no IMVs could be detected (Fig 3, Bottom panel). SP600125 is considered as a specific pharmacological inhibitor of JNK1/2, not only in response to cytokine stimulation (Dong et al., 2000), but also with viral infections (Bogoyevitch and Arthur, 2008). Our results show that upon VACV or CPXV infection JNK1/2 is activated during the entire viral cycle and SP600125, indeed, inhibits JNK1/2 phosphorylation in a dose-dependent manner (Fig 1C). However, the block identified in the viral cycle caused by SP600125 is an event that occurs independently of JNK1/2 since no effect on viral yield was observed when infections were performed in JNK1/2 KO MEF cells. Similar results were found with the use of JNKi VIII inhibitor.
Previous reports have shown that SP600125 inhibits cellular kinases in vitro other than JNK1/2 (Bain et al., 2003, Bain et al., 2007), but even in the face of the concerns raised on the specificity of this inhibitor, several studies still rely on this drug for a possible therapeutic application regarding treatment of human diseases. Furthermore, since its discovery in 2001, SP600125 has been extensively studied for treatment of numerous non-viral diseases in murine model (Ikezumi et al., 2004, Gao et al., 2005, Han et al., 2005, Gunawan et al., 2006, Guan et al., 2006, Takamura et al., 2007, Syrkina et al., 2007, Hu and Liu, 2009). However, up to the publication of this work, a search in the literature did not show a single report demonstrating that SP600125 is effective against viral infection in animal studies to support the results observed in cell culture system. Furthermore, studies have shown that viral infection can lead to JNK activation and the inhibition of these cellular kinases by SP600125 affects viral multiplication (Hamza et al., 2004, Hassan et al., 2005, Zapata et al., 2007, Gupta et al., 2011).
Most of these studies make a strict connection between the inhibition of JNK by SP600125 and its impact on viral infection. Because JNK is only one of the kinases targeted by this drug, additional analyses with other inhibitors of JNK1/2 or cell lines knockouts for those kinases or even RNAi approach should be taken into consideration to confirm this direct relationship. Therefore, since animal studies are a cost, time and energy-dependent system, it is possible that researchers are more careful about taking a step further and testing SP600125 in mice, for instance, and do not succeed in correlating the data observed in tissue culture. Additional disadvantages of SP600125 may be considerable off-target activity, or perhaps its poor solubility in aqueous solution or/and possible undesirable side-effects (Bennett et al., 2001, Bain et al., 2003, Begleiter et al., 2006). In effort to get around these complications, a derivative of SP600125 (CC-401) was developed by Celgene has successfully completed a Phase I trial in healthy volunteers as stated by the pharmaceutical company. CC-401 has also been reported in Phase II evaluation for the treatment of acute myelogenous leukemia, and has also been considered for the treatment of respiratory diseases (Roberts and Der, 2007, Bogoyevitch and Arthur, 2008). Nevertheless, a shortest path to evaluate SP600125 in vivo against an orthopoxvirus infection would be a viral challenge in a murine model.
Taken together, questions still remain regarding the potential protein kinase(s) targeted by SP600125 during Orthopoxvirus infection causing the impairment of viral morphogenesis. Poxviruses encode two essential serine/threonine kinases, B1 (Traktman et al., 1989, Lin et al., 1992, Rempel and Traktman, 1992) and F10 (Lin and Broyles, 1994). While B1 plays a function during viral DNA replication (Traktman et al., 1989, Rempel et al., 1990, Domi and Beaud, 2000), F10 plays a role in the very early stages of virion morphogenesis (Wang and Shuman, 1995, Traktman et al., 1995). When B1 or F10 proteins are repressed or inactive, none of the hallmarks of morphogenesis are identified. Therefore, it is doubtful that SP600125 would target one or both viral kinases. In addition, some viral proteins that play a role in morphogenesis are proposed to be also phosphorylated by cellular kinases (Resch et al., 2005, Trindade et al., 2007, Wickramasekera and Traktman, 2010). By comparison with electron microscopy images of VACV mutants, under nonpermissive conditions, we observed that some of them phenotypically copy our results when infections are performed in the presence of SP600125. The repression of the phosphoprotein A13L arrests morphogenesis at the stage of IV formation. Essentially, no IMVs are seen and IVNs are rare; DNA crystalloids accumulate in the cytoplasm (Unger and Traktman, 2004). A similar phenotype is also seen when H3L, a major immunodominant protein, is repressed or deleted (da Fonseca et al., 2000). When the myristoylated L1R protein is repressed, the transition from IV to IMV is blocked (Ravanello et al., 1994). Thus far, it is hard to predict a putative cellular target for SP600125 that would affect viral morphogenesis. Steps that prior and subsequently lead to the formation and maturation of IMVs are very complex and not fully understood. Protein phosphorylation, protein–protein interactions and proteolytic processing are some of the events involved. Since cellular kinases are likely thought to contribute to phosphorylation of viral proteins, it is plausible that their inhibition by SP600125 could affect those events blocking morphogenesis progress.
In conclusion, our results demonstrate the use of SP600125 inhibits Orthopoxviruses replication in a JNK independent-manner. This suggests that other cellular and/or viral substrates are affected by the action of SP600125. While significant progress has been made in the discovery of novel compounds against Orthopoxviruses, the need for a range of antiviral drugs is imperative since the occurrence of resistance to antiviral drugs is not a rare event. Our data support the notion that SP600125 can be regarded as a potential antipoxviral compound; the combined use of SP600125 with other antipoxviral drugs may enhance their antiviral activity and, perhaps, minimize major side effects with the advantage of reducing drug resistance.
Acknowledgements
The authors are grateful to Angela S. Lopes, Ilda M. V. Gama, João R. dos Santos and Andreza A. Carvalho for their secretarial/technical assistance. We also thank Dr. M. C. Sogayar (Department of Biochemistry, University of São Paulo, Brazil), who kindly provided us with the A31 cell line and Dr. R. Davis (Howard Hughes Medical Institute, University of Massachusetts Medical School, Worcester, MS) for the WT and JNK1/2 KO cells. VACV WR and CPXV BR were from Dr. C. Jungwirth (Universität Würzburg, Germany). MVA was from Dr. B. Moss (NIAID, Bethesda, MD)/Dr. Flávio G. da Fonseca (Universidade Federal de Minas Gerais). Dr. Kathleen A. Boyle, Department of Microbiology and Molecular Genetics, Medical College of Wisconsin, Milwaukee, WI, for critically reading the manuscript. This work was supported by grants from Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG), Coordenadoria de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazilian Ministry of Culture, Science and Technology and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). Drs. ACTCP, JAPSM AND FGGL were recipients of pre-doctoral fellowships from CNPq. AFPC and AAT were recipients of undergraduate students from CNPq (PIBIC) and CAB, EGK, TSP, and PCPF are recipients of research fellowships from CNPq.
References
- Andrade A.A., Silva P.N., Pereira A.C., De Sousa L.P., Ferreira P.C., Gazzinelli R.T., Kroon E.G., Ropert C., Bonjardim C.A. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 2004;381:437–446. doi: 10.1042/BJ20031375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J., McLauchlan H., Elliott M., Cohen P. The specificities of protein kinase inhibitors: an update. Biochem. J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J., Plater L., Elliott M., Shapiro N., Hastie C.J., McLauchlan H., Klevernic I., Arthur J.S., Alessi D.R., Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barquet N., Domingo P. Smallpox: the triumph over the most terrible of the ministers of death. Ann. Intern. Med. 1997;127:635–642. doi: 10.7326/0003-4819-127-8_part_1-199710150-00010. [DOI] [PubMed] [Google Scholar]
- Begleiter A., Lin D., Larson K.K., Lang J., Wu X., Cabral T., Taylor H., Guziec L.J., Kerr P.D., Hasinoff B.B., Guziec F.S., Jr Structure-activity studies with cytotoxic anthrapyrazoles. Oncol. Rep. 2006;15:1575–1580. [PubMed] [Google Scholar]
- Bennett B.L., Sasaki D.T., Murray B.W., O’Leary E.C., Sakata S.T., Xu W., Leisten J.C., Motiwala A., Pierce S., Satoh Y., Bhagwat S.S., Manning A.M., Anderson D.W. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B.L. C-Jun N-terminal kinase-dependent mechanisms in respiratory disease. Eur. Respir. J. 2006;28:651–661. doi: 10.1183/09031936.06.00012106. [DOI] [PubMed] [Google Scholar]
- Becker M.N., Obraztsova M., Kern E.R., Quenelle D.C., Keith K.A., Prichard M.N., Luo M., Moyer R.W. Isolation and characterization of cidofovir resistant vaccinia viruses. Virol. J. 2008;5:58. doi: 10.1186/1743-422X-5-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch M.A., Boehm I., Oakley A., Ketterman A.J., Barr R.K. Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential. Biochim. Biophys. Acta. 2004;1697:89–101. doi: 10.1016/j.bbapap.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch M.A., Arthur P.G. Inhibitors of c-Jun N-terminal kinases: JuNK no more? Biochim. Biophys. Acta. 2008;1784:76–93. doi: 10.1016/j.bbapap.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.Y., Lu J., Shih Y.C., Tsai C.H. Epstein-Barr virus latent membrane protein 2A regulates c-Jun protein through extracellular signal-regulated kinase. J. Virol. 2002;76:9556–9561. doi: 10.1128/JVI.76.18.9556-9561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condit R.C., Moussatche N., Traktman P. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 2006;66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- da Fonseca F.G., Wolffe E.J., Weisberg A., Moss B. Effects of deletion or stringent repression of the H3L envelope gene on vaccinia virus replication. J. Virol. 2000;74:7518–7528. doi: 10.1128/jvi.74.16.7518-7528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva P.N., Soares J.A., Brasil B.S., Nogueira S.V., Andrade A.A., de Magalhaes J.C., Bonjardim M.B., Ferreira P.C., Kroon E.G., Bruna-Romero O., Bonjardim C.A. Differential role played by the MEK/ERK/EGR-1 pathway in orthopoxviruses vaccinia and cowpox biology. Biochem. J. 2006;398:83–95. doi: 10.1042/BJ20060509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaso C.R., Esposito J.J., Condit R.C., Moussatche N. An emergent poxvirus from humans and cattle in Rio de Janeiro State: Cantagalo virus may derive from Brazilian smallpox vaccine. Virology. 2000;277:439–449. doi: 10.1006/viro.2000.0603. [DOI] [PubMed] [Google Scholar]
- de Borst M.H., Prakash J., Sandovici M., Klok P.A., Hamming I., Kok R.J., Navis G., van Goor H. C-Jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. J. Pharmacol. Exp. Ther. 2009;331:896–905. doi: 10.1124/jpet.109.154179. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Cidofovir in the therapy and short-term prophylaxis of poxvirus infections. Trends Pharmacol. Sci. 2002;23:456–458. doi: 10.1016/S0165-6147(02)02091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin. Microbiol. Rev. 2003;16:569–596. doi: 10.1128/CMR.16.4.569-596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhães J.C., Andrade A.A., Silva P.N., Sousa L.P., Ropert C., Ferreira P.C., Kroon E.G., Gazzinelli R.T., Bonjardim C.A. A mitogenic signal triggered at an early stage of vaccinia virus infection: implication of MEK/ERK and protein kinase A in virus multiplication. J. Biol. Chem. 2001;276:38353–38360. doi: 10.1074/jbc.M100183200. [DOI] [PubMed] [Google Scholar]
- Domi A., Beaud G. The punctate sites of accumulation of vaccinia virus early proteins are precursors of sites of viral DNA synthesis. J. Gen. Virol. 2000;81:1231–1235. doi: 10.1099/0022-1317-81-5-1231. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang D.D., Tournier C., Whitmarsh A.J., Xu J., Davis R.J., Flavell R.A. JNK is required for effector T-cell function but not for T-cell activation. Nature. 2000;405:91–94. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- Fenner, F., Anderson, D.A., Arita, I., Jezek, Z., Ladnyi, I.D., 1988. Smallpox and its eradication. World Health Organization.
- Fonseca F.G., Lanna M.C., Campos M.A., Kitajima E.W., Peres J.N., Golgher R.R., Ferreira P.C., Kroon E.G. Morphological and molecular characterization of the poxvirus BeAn 58,058. Arch. Virol. 1998;143:1171–1186. doi: 10.1007/s007050050365. [DOI] [PubMed] [Google Scholar]
- Formenty P., Muntasir M.O., Damon I., Chowdhary V., Opoka M.L., Monimart C., Mutasim E.M., Manuguerra J.C., Davidson W.B., Karem K.L., Cabeza J., Wang S., Malik M.R., Durand T., Khalid A., Rioton T., Kuong-Ruay A., Babiker A.A., Karsani M.E., Abdalla M.S. Human monkeypox outbreak caused by novel virus belonging to Congo Basin clade, Sudan, 2005. Emerg. Infect. Dis. 2010;16:1539–1545. doi: 10.3201/eid1610.100713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht F., Cudmore S., Moreau V., Reckmann I., Rottger S., Way M. Tyrosine phosphorylation is required for actin-based motility of vaccinia but not Listeria or Shigella. Curr. Biol. 1999;9:89–92. doi: 10.1016/s0960-9822(99)80020-7. [DOI] [PubMed] [Google Scholar]
- Gao Y., Signore A.P., Yin W., Cao G., Yin X.M., Sun F., Luo Y., Graham S.H., Chen J. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J. Cereb. Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- Guan Q.H., Pei D.S., Liu X.M., Wang X.T., Xu T.L., Zhang G.Y. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res. 2006;1092:36–46. doi: 10.1016/j.brainres.2006.03.086. [DOI] [PubMed] [Google Scholar]
- Gunawan B.K., Liu Z.X., Han D., Hanawa N., Gaarde W.A., Kaplowitz N. C-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- Gupta N., Bhaskar A.S., Lakshmana Rao P.V. Transcriptional regulation and activation of the mitogen-activated protein kinase pathway after Japanese encephalitis virus infection in neuroblastoma cells. FEMS Immunol. Med. Microbiol. 2011;62:110–121. doi: 10.1111/j.1574-695X.2011.00792.x. [DOI] [PubMed] [Google Scholar]
- Hamza M.S., Reyes R.A., Izumiya Y., Wisdom R., Kung H.J., Luciw P.A. ORF36 protein kinase of Kaposi’s sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 2004;279:38325–38330. doi: 10.1074/jbc.M400964200. [DOI] [PubMed] [Google Scholar]
- Han Y.L., Qi Y.M., Kang J., Liang M., Chen X.H. Role of MAPK in the migration of human coronary artery smooth muscle cell into three-dimensional fibrin gel. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2005;21:388–392. [PubMed] [Google Scholar]
- Hassan M., Ghozlan H., Abdel-Kader O. Activation of c-Jun NH2-terminal kinase (JNK) signaling pathway is essential for the stimulation of hepatitis C virus (HCV) non-structural protein 3 (NS3)-mediated cell growth. Virology. 2005;333:324–336. doi: 10.1016/j.virol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Holloway G., Coulson B.S. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J. Virol. 2006;80:10624–10633. doi: 10.1128/JVI.00390-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm T.M., Habashi J.P., Doyle J.J., Bedja D., Chen Y., van Erp C., Lindsay M.E., Kim D., Schoenhoff F., Cohn R.D., Loeys B.L., Thomas C.J., Patnaik S., Marugan J.J., Judge D.P., Dietz H.C. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.B., Liu X.Y. Protective effects of SP600125 in a diet-induced rat model of non-alcoholic steatohepatitis. Scand. J. Gastroenterol. 2009;44:1356–1362. doi: 10.3109/00365520903312441. [DOI] [PubMed] [Google Scholar]
- Ikezumi Y., Hurst L., Atkins R.C., Nikolic-Paterson D.J. Macrophage-mediated renal injury is dependent on signaling via the JNK pathway. J. Am. Soc. Nephrol. 2004;15:1775–1784. doi: 10.1097/01.asn.0000131272.06958.de. [DOI] [PubMed] [Google Scholar]
- Jordan R., Tien D., Bolken T.C., Jones K.F., Tyavanagimatt S.R., Strasser J., Frimm A., Corrado M.L., Strome P.G., Hruby D.E. Single-dose safety and pharmacokinetics of ST-246, a novel orthopoxvirus egress inhibitor. Antimicrob. Agents Chemother. 2008;52:1721–1727. doi: 10.1128/AAC.01303-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R., Chinsangaram J., Bolken T.C., Tyavanagimatt S.R., Tien D., Jones K.F., Frimm A., Corrado M.L., Pickens M., Landis P., Clarke J., Marbury T.C., Hruby D.E. Safety and pharmacokinetics of the antiorthopoxvirus compound ST-246 following repeat oral dosing in healthy adult subjects. Antimicrob. Agents Chemother. 2010;54:2560–2566. doi: 10.1128/AAC.01689-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesus D.M., Costa L.T., Goncalves D.L., Achete C.A., Attias M., Moussatche N., Damaso C.R. Cidofovir inhibits genome encapsidation and affects morphogenesis during the replication of vaccinia virus. J. Virol. 2009;83:11477–11490. doi: 10.1128/JVI.01061-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joklik W.K. The purification for four strains of poxvirus. Virology. 1962;18:9–18. doi: 10.1016/0042-6822(62)90172-1. [DOI] [PubMed] [Google Scholar]
- Kerkelä R., Grazette L., Yacobi R., Iliescu C., Patten R., Beahm C., Walters B., Shevtsov S., Pesant S., Clubb F.J., Rosenzweig A., Salomon R.N., Van Etten R.A., Alroy J., Durand J.B., Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- Lin S., Chen W., Broyles S.S. The vaccinia virus B1R gene product is a serine/threonine protein kinase. J. Virol. 1992;66:2717–2723. doi: 10.1128/jvi.66.5.2717-2723.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Broyles S.S. Vaccinia protein kinase 2: a second essential serine/threonine protein kinase encoded by vaccinia virus. Proc. Natl. Acad. Sci. USA. 1994;91:7653–7657. doi: 10.1073/pnas.91.16.7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning A.M., Davis R.J. Targeting JNK for therapeutic benefit: from junk to gold? Nat. Rev. Drug. Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- McNulty S., Bornmann W., Schriewer J., Werner C., Smith S.K., Olson V.A., Damon I.K., Buller R.M., Heuser J., Kalman D. Multiple phosphatidylinositol 3-kinases regulate vaccinia virus morphogenesis. PLoS One. 2010;5:e10884. doi: 10.1371/journal.pone.0010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra S., Chhabra A., Hegde U., Chakraborty N.G., Mukherji B. Inhibition of c-Jun N-terminal kinase rescues influenza epitope-specific human cytolytic T lymphocytes from activation-induced cell death. J. Leukoc. Biol. 2007;81:539–547. doi: 10.1189/jlb.0706479. [DOI] [PubMed] [Google Scholar]
- Mercer J., Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochim. Biophys. Acta. 2005;1741:4–10. doi: 10.1016/j.bbadis.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, B. 2007. Poxviridae, In: Fields, B.N., Knipe, D.M., Howley, P.M. (Ed.), Virology, fifth ed, vol. 2. Lippincott-Raven, Philadelphia. pp. 2905–2946.
- Perkins D., Gyure K.A., Pereira E.F., Aurelian L. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J. Neurovirol. 2003;9:101–111. doi: 10.1080/13550280390173427. [DOI] [PubMed] [Google Scholar]
- Quenelle D.C., Buller R.M., Parker S., Keith K.A., Hruby D.E., Jordan R., Kern E.R. Efficacy of delayed treatment with ST-246 given orally against systemic orthopoxvirus infections in mice. Antimicrob. Agents Chemother. 2007;51:689–695. doi: 10.1128/AAC.00879-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravanello M.P., Hruby D.E. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virion assembly. J. Virol. 1994;68:6401–6410. doi: 10.1128/jvi.68.10.6401-6410.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves P.M., Bommarius B., Lebeis S., McNulty S., Christensen J., Swimm A., Chahroudi A., Chavan R., Feinberg M.B., Veach D., Bornmann W., Sherman M., Kalman D. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat. Med. 2005;11:731–739. doi: 10.1038/nm1265. [DOI] [PubMed] [Google Scholar]
- Reeves P.M., Smith S.K., Olson V.A., Thorne S.H., Bornmann W., Damon I.K., Kalman D. Variola and monkeypox viruses utilize conserved mechanisms of virion motility and release that depend on abl and SRC family tyrosine kinases. J. Virol. 2011;85:21–31. doi: 10.1128/JVI.01814-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel R.E., Anderson M.K., Evans E., Traktman P. Temperature-sensitive vaccinia virus mutants identify a gene with an essential role in viral replication. J. Virol. 1990;64:574–583. doi: 10.1128/jvi.64.2.574-583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel R.E., Traktman P. Vaccinia virus B1 kinase: phenotypic analysis of temperature-sensitive mutants and enzymatic characterization of recombinant proteins. J. Virol. 1992;66:4413–4426. doi: 10.1128/jvi.66.7.4413-4426.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resch W., Weisberg A.S., Moss B. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 2005;79:6598–6609. doi: 10.1128/JVI.79.11.6598-6609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M.G., Cono J., Curns A., Holman R.C., Likos A., Regnery R., Treadwell T., Damon I. Human monkeypox. Lancet Infect. Dis. 2004;4:604–605. doi: 10.1016/S1473-3099(04)01139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts P.J., Der C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Sbrana E., Xiao S.Y., Newman P.C., Tesh R.B. Comparative pathology of North American and central African strains of monkeypox virus in a ground squirrel model of the disease. Am. J. Trop. Med. Hyg. 2007;76:155–164. [PubMed] [Google Scholar]
- Sejvar J.J., Chowdary Y., Schomogyi M., Stevens J., Patel J., Karem K., Fischer M., Kuehnert M.J., Zaki S.R., Paddock C.D., Guarner J., Shieh W.J., Patton J.L., Bernard N., Li Y., Olson V.A., Kline R.L., Loparev V.N., Schmid D.S., Beard B., Regnery R.R., Damon I.K. Human monkeypox infection: a family cluster in the midwestern United States. J. Infect. Dis. 2004;190:1833–1840. doi: 10.1086/425039. [DOI] [PubMed] [Google Scholar]
- Sharma N., Deshmukh R., Bedi K.L. SP600125, a competitive inhibitor of JNK attenuates streptozotocin induced neurocognitive deficit and oxidative stress in rats. Pharmacol. Biochem. Behav. 2010;96:386–394. doi: 10.1016/j.pbb.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Smee D.F., Sidwell R.W., Kefauver D., Bray M., Huggins J.W. Characterization of wild-type and cidofovir-resistant strains of camelpox, cowpox, monkeypox, and vaccinia viruses. Antimicrob. Agents Chemother. 2002;46:1329–1335. doi: 10.1128/AAC.46.5.1329-1335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G.L., McFadden G. Smallpox: anything to declare? Nat. Rev. Immunol. 2002;2:521–527. doi: 10.1038/nri845. [DOI] [PubMed] [Google Scholar]
- Soares J.A., Leite F.G., Andrade L.G., Torres A.A., De Sousa L.P., Barcelos L.S., Teixeira M.M., Ferreira P.C., Kroon E.G., Souto-Padron T., Bonjardim C.A. Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J. Virol. 2009;83:6883–6899. doi: 10.1128/JVI.00245-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z.F., Ji X.P., Li X.X., Wang S.J., Wang S.H., Zhang Y. Inhibition of the activity of poly (ADP-ribose) polymerase reduces heart ischaemia/reperfusion injury via suppressing JNK-mediated AIF translocation. J. Cell. Mol. Med. 2008;12:1220–1228. doi: 10.1111/j.1582-4934.2008.00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrkina O.L., Quinn D.A., Jung W., Ouyang B., Hales C.A. Inhibition of JNK activation prolongs survival after smoke inhalation from fires. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:984–991. doi: 10.1152/ajplung.00248.2006. [DOI] [PubMed] [Google Scholar]
- Takamura M., Matsuda Y., Yamagiwa S., Tamura Y., Honda Y., Suzuki K., Ichida T., Aoyagi Y. An inhibitor of c-Jun NH2-terminal kinase, SP600125, protects mice from d-galactosamine/lipopolysaccharide-induced hepatic failure by modulating BH3-only proteins. Life Sci. 2007;80:1335–1344. doi: 10.1016/j.lfs.2006.12.034. [DOI] [PubMed] [Google Scholar]
- Tolomeo M., Dieli F., Gebbia N., Simoni D. Tyrosine kinase inhibitors for the treatment of chronic myeloid leukemia. Anticancer Agents Med. Chem. 2009;9:853–863. doi: 10.2174/187152009789124637. [DOI] [PubMed] [Google Scholar]
- Tournier C., Hess P., Yang D.D., Xu J., Turner T.K., Nimnual A., Bar-Sagi D., Jones S.N., Flavell R.A., Davis R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- Traktman P., Anderson M.K., Rempel R.E. Vaccinia virus encodes an essential gene with strong homology to protein kinases. J. Biol. Chem. 1989;264:21458–21461. [PubMed] [Google Scholar]
- Traktman P., Caligiuri A., Jesty S.A., Liu K., Sankar U. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of virion morphogenesis. J. Virol. 1995;69:6581–6587. doi: 10.1128/jvi.69.10.6581-6587.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trindade G.S., Emerson G.L., Carroll D.S., Kroon E.G., Damon I.K. Brazilian vaccinia viruses and their origins. Emerg. Infect. Dis. 2007;13:965–972. doi: 10.3201/eid1307.061404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger B., Traktman P. Vaccinia virus morphogenesis: a13 phosphoprotein is required for assembly of mature virions. J. Virol. 2004;78:8885–8901. doi: 10.1128/JVI.78.16.8885-8901.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I., Palaskas N., Tran C., Finn S.P., Getz G., Kennedy N.J., Jiao J., Rose J., Xie W., Loda M., Golub T., Mellinghoff I.K., Davis R.J., Wu H., Sawyers C.L. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell. 2007;11:555–569. doi: 10.1016/j.ccr.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Vorou R.M., Papavassiliou V.G., Pierroutsakos I.N. Cowpox virus infection: an emerging health threat. Curr. Opin. Infect. Dis. 2008;21:153–156. doi: 10.1097/QCO.0b013e3282f44c74. [DOI] [PubMed] [Google Scholar]
- Wang S., Shuman S. Vaccinia virus morphogenesis is blocked by temperature-sensitive mutations in the F10 gene, which encodes protein kinase 2. J. Virol. 1995;69:6376–6388. doi: 10.1128/jvi.69.10.6376-6388.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhang Y., Wei Z., Li H., Zhou H., Zhang Z. JNK inhibitor protects dopaminergic neurons by reducing COX-2 expression in the MPTP mouse model of subacute Parkinson’s disease. J. Neurol. Sci. 2009;285:172–177. doi: 10.1016/j.jns.2009.06.034. [DOI] [PubMed] [Google Scholar]
- Wickramasekera N.T., Traktman P. Structure/function analysis of the vaccinia virus F18 phosphoprotein, an abundant core component required for virion maturation and infectivity. J. Virol. 2010;84:6846–6860. doi: 10.1128/JVI.00399-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D., Rumpold H. A benefit-risk assessment of imatinib in chronic myeloid leukaemia and gastrointestinal stromal tumours. Drug Safety. 2009;32:1001–1015. doi: 10.2165/11314600-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Yang G., Pevear D.C., Davies M.H., Collett M.S., Bailey T., Rippen S., Barone L., Burns C., Rhodes G., Tohan S., Huggins J.W., Baker R.O., Buller R.L., Touchette E., Waller K., Schriewer J., Neyts J., De Clercq E., Jones K., Hruby D., Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus Challenge. J. Virol. 2005;79:13139–13149. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Kim S.K., Kim M., Reche P.A., Morehead T.J., Damon I.K., Welsh R.M., Reinherz E.L. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J. Clin. Invest. 2005;115:379–387. doi: 10.1172/JCI23220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata H.J., Nakatsugawa M., Moffat J.F. Varicella-zoster virus infection of human fibroblast cells activates the c-Jun N-terminal kinase pathway. J. Virol. 2007;81:977–990. doi: 10.1128/JVI.01470-06. [DOI] [PMC free article] [PubMed] [Google Scholar]