

Abstract
Progressive immune-associated injury is a hallmark of severe acute respiratory syndrome (SARS). Viral evasion of innate immunity, hypercytokinemia and systemic immunopathology in the SARS coronavirus (SARS CoV) infected host have been suggested as possible mechanisms for the cause of severe pathology and morbidity in SARS patients. The molecular and cellular basis for how SARS CoV impacts the host immune system resulting in severe SARS, however, has not been elucidated. The variable clinical course of SARS may be the result of complex programs of host responses against the infectious agent. Therefore, the systematic analysis of innate and adaptive immune responses to SARS CoV is imperative in building as complete an immunological model as possible of host immunity and inflammatory responses during illness. Here we review recent advances in SARS immunopathogenesis research and present a summary of our findings regarding host responses in SARS patients. We contend that dysregulated type I and II interferon (IFN) responses during SARS may culminate in a failure of the switch from hyper-innate immunity to protective adaptive immune responses in the human host.
Keywords: SARS, Cytokine, Chemokine, Microarray, Host response, Interferon
1. Introduction
Severe acute respiratory syndrome (SARS) emerged in late 2002 from Guangdong Province, China and spread worldwide to over 30 countries by midsummer 2003 (Lau and Peiris, 2005). Before the epidemic ended, the novel coronavirus causing SARS (SARS CoV) infected over 8000 persons around the world, killed nearly 10% of those that contracted the disease and inflicted great socioeconomic strain. While the likelihood of another SARS outbreak is uncertain, the severity of SARS as compared to that of other human coronaviral infections mandates continued research into the pathogenesis of SARS CoV infection. Moreover, the emergence of new global health threats, such as avian influenza (H5N1), has refocused our attention on deriving a better understanding of how highly pathogenic viruses cause severe immunopathology in humans and our insight with SARS CoV may aid in treatment of individuals infected with emerging viruses (Mandavilli, 2006).
SARS CoV causes a spectrum of disease ranging from flu-like symptoms and pneumonia to acute respiratory distress syndrome (ARDS) (Ding et al., 2004, Farcas et al., 2005, Hwang et al., 2005, Lew et al., 2003, Salto-Tellez et al., 2005). The molecular and cellular basis for how SARS CoV impacts the host immune system resulting in severe morbidity and mortality is not well understood at this point. We believe that the variable pathogenesis and severity of SARS may be the result of complex programs of host responses against the infectious agent. Therefore, the analysis of innate and adaptive immune responses to SARS CoV on the whole host level is imperative in building as complete a model as possible of immune and inflammatory responses during the clinical evolution of SARS. Here we review recent advances in SARS immunopathogenesis research and present an overview of our findings regarding host immune responses in SARS patients.
2. Clinical evolution of SARS
The incubation period of SARS CoV infection ranges from 2 to 10 days but may last as long as 16 days (Booth et al., 2003, Lee et al., 2003). The initial symptoms of SARS are non-specific and include influenza-like symptoms such as fever, chills, rigor, headache, dizziness, malaise and myalgia, with fever being the most common symptom upon presentation. The respiratory phase of SARS begins 2–7 days after the prodrome period (Booth et al., 2003, Lee et al., 2003, Peiris et al., 2003). The early respiratory stage of SARS includes a dry, non-productive cough and mild dyspnoea. At the onset of fever, 70–80% of SARS patients have abnormal chest radiographs (Antonio et al., 2005, Wong et al., 2003a); although chest radiographs may be normal during the febrile prodrome as well as throughout the course of illness. Recent data from the Toronto cohort of SARS patients indicated that 91% of SARS cases had infiltrates by the end of the first week of illness and that very few patients never developed infiltrates (Muller et al., 2006). In other cases, radiological evidence of pneumonic changes may precede fever (Rainer et al., 2003), particularly in individuals with co-morbidities who may be impaired in their ability to mount a fever response (Fisher et al., 2003).
Early chest radiographs during SARS often show subtle peripheral pulmonary infiltrates. Radiographic involvement progresses over 1–2 days to become bilateral and generalized with interstitial or confluent infiltrates. Air-space opacities eventually develop during the course of the disease. In patients who deteriorate clinically, the air-space opacities may increase in size, extent and severity (Wong et al., 2003a, Wong et al., 2003b).
After the onset of SARS, cases may progress to a non-severe variant of the disease characterized by relatively mild respiratory symptoms with fever or a “cough variant” characterized by persistent intractable cough (Christian et al., 2004). More commonly, however, cases progress to a moderate to severe variant characterized by the development of dyspnea and hypoxia 8–12 days post onset of symptoms (Booth et al., 2003, Lee et al., 2003, Peiris et al., 2003).
The recovery phase of SARS typically begins ∼14–18 days after the onset of symptoms; however, symptoms may worsen in 10–20% of hospitalized patients to the point where mechanical ventilation is necessary (Booth et al., 2003, Fowler et al., 2003, Lee et al., 2003, Lew et al., 2003). In the latter group of SARS patients, progressive immune infiltration of the lungs and diffuse alveolar damage with unresolved viral burden may culminate in ARDS, a severe form of pulmonary failure with an immune component caused by microbial infections, transplant rejection, prolonged mechanical ventilation or trauma, etc. (Ding et al., 2004, Farcas et al., 2005, Hwang et al., 2005, Lew et al., 2003, Salto-Tellez et al., 2005). ARDS occurred in approximately 16% of all patients with SARS and was associated with a mortality rate of 50% in this context (Fowler et al., 2003, Lew et al., 2003). According to the WHO, the overall case fatality rate during the SARS outbreak in 2003 was estimated at 9.6%. Advanced age appears to be the strongest predictor of poor outcome (patients aged >60 years have a case-fatality rate of 45%). A number of other prognostic factors have been identified, including the presence of comorbidities (particularly diabetes mellitus and cardiac disease), elevations of baseline LDH and ANC, baseline hypoxemia and the extent of radiographic disease. Given the lack of a consensus therapeutic agent during the SARS outbreak, a wide range of treatment approaches were used, including ribavirin, steroids, convalescent plasma or immunoglobulin, protease inhibitors (lopinavir + ritonavir) and interferon-α (IFN-α). Stockman et al. recently reviewed these therapies and concluded that, at the present moment, it is not known if any of the potential treatments for SARS, whether antiviral or immunomodulatory, will be particularly effective in the event of a new outbreak (Stockman et al., 2006).
3. Immunopathology of SARS
SARS CoV takes hold in the airways and other organs by exploiting the renin-angiotension pathway via its main putative receptor, angiotensin-converting enzyme 2 (ACE2) expressed on many cell types including pneumocytes, enterocytes and endothelial cells (Hamming et al., 2004, Kuba et al., 2005, Li et al., 2003). SARS CoV can efficiently evade innate immune responses as exemplified by the steadily increasing viral loads found in the host during first 10 days of SARS infection (Peiris et al., 2003, Wong et al., 2003c). The consequent hyper-immune inflammation and systemic immunopathology in the SARS CoV-infected host has been well illustrated (Huang et al., 2005, Lau and Peiris, 2005, Nicholls et al., 2003, Wong et al., 2004), however the fact that the majority of SARS patients recover after a relatively moderate disease course, indicates that the general notion of deficient innate immunity against SARS CoV in concert with hyper-immune responses or a “cytokine storm” driving severe immunopathology in SARS may be oversimplified.
3.1. Cytokines and the immunopathogenesis of SARS
The induction of proinflammatory cytokine and chemokines is clearly involved in the host response to SARS CoV infection and clinical evolution of the disease. However, a great deal of controversy has arisen in the literature regarding the relationship between immune mediators and the pathophysiological events of SARS. Early work by Wong et al. demonstrated that elevation of interferon-γ (IFN-γ), IL-1, IL-6 and IL-12 occurred for at least 2 weeks after onset of SARS and that high levels of the chemokines IL-8, CCL2, and CXCL10 were associated with acute SARS infection (Wong et al., 2004). In a subsequent study, Tang et al. analyzed archival plasma samples collected during acute SARS illness and highlighted increased levels of the chemokines CXCL10, CXCL9 and IL-8 as being associated with adverse outcome (Tang et al., 2005). Focusing on elevated plasma levels of CXCL10 and their association with immunopathology, Jiang et al. showed that CXCL10 remained at high levels in SARS patients until convalescence and was highly expressed in lung and lymphoid tissues (Jiang et al., 2005). In addition to CXCL10, CXCL9 and IL-8, Huang et al. found levels of IFN-γ, transforming growth factor-β (TGF-β), IL-6 and CCL2 chemokine were pathologically relevant during acute SARS but not tumor necrosis factor-α (TNF-α), IL-2, IL-4, IL-10 or IL-13 (Huang et al., 2005). On the other hand, Chien et al. recently hypothesized that early induction of CXCL10 and IL-2 along with subsequent IL-6 induction versus a lack of IL-10 may be key during the development of SARS immunopathology (Chien et al., 2006). While some of these results conflict and the interpretation of the data has been somewhat limited by a general paucity of clinical history and status of SARS patients in these studies, it is clear that pronounced expression of a group of proinflammatory cytokines and chemokines is associated with acute and, in some cases, progressing SARS.
Our results, derived from a cohort of clinically well-defined SARS patients (n = 50) from the Toronto SARS outbreak in 2003, provide evidence that the temporal involvement of cytokines and chemokines, especially interferon (IFN) and IFN-stimulated genes (ISGs), are critical in the clinical evolution of SARS in susceptible patients as part of a larger global host response (Cameron et al., 2003). We found that SARS symptoms progress rapidly in patients during acute infection with fever peaking at median 4 days since onset of symptoms (DSO), chest radiographic involvement peaking at median 6 DSO and arterial O2 saturation (SO2) nadir occurring at median 8 DSO in surviving patients. The SO2 nadir in surviving patients was used to separate early (acute) and late phase SARS. Patients who required mechanical ventilation at any time during their clinical course were classified as having severe SARS. Severe SARS patients exhibited a higher incidence of bilateral lung infiltration over the clinical course of the disease (100% versus 40% of non-severe SARS patients). Severe SARS patients were also significantly older (median 63 years old versus median 44 years old for non-severe SARS patients, P < 0.05) and had significantly longer disease courses in those that survived versus non-severe patients (>2-fold, median 38 days in severe SARS patients versus median 15 days in non-severe patients, P < 0.05).
Using cytometric bead array technology and the manufacturer's protocol (BD Biosciences, NJ, USA), we found that non-severe SARS patients exhibited significantly increased (P < 0.05) levels of IFN-α and IFN-γ and the chemokines CXCL10 and IL-8 during the early (acute) phase of the disease in comparison to healthy controls while CXCL10 and IL-8 levels remained highly expressed with respect to healthy controls during the late phase (Table 1 ). In comparison to healthy controls, severe SARS patients showed significantly increased (P < 0.05) levels of IFN-α, IFN-γ and CXCL10 along with decreased levels of IL-12p70, IL-2 and TNF-α during acute SARS. In the late phase, severe SARS patients exhibited significantly increased (P < 0.05) levels of the chemokines IL-8, CXCL10 and CCL2 relative to the controls, but decreased levels of the cytokines IL-12p70, IL-2, TNF-α and IFN-γ. When we compared the late phases in both groups, severe SARS patients exhibited significantly higher (P < 0.05) levels of CXCL10 and CCL2 than non-severe patients. On the other hand, the severe group showed decreased levels of IL-12p70 and TNF-α with respect to the non-severe group (P < 0.05). Therefore, both SARS severity groups showed increased levels of CXCL10 throughout disease, but CXCL10 and CCL2 levels were sustained in severe SARS patients relative to non-severe late phase patients. Severe SARS patients were also associated with more diffuse pulmonary infiltrates, higher temperatures and longer periods of hospitalization (data not shown).
Table 1.
Significantly expressed cytokines and chemokines during SARS clinical evolutiona
| Expression | Early (acute) SARS | Late SARS | |
|---|---|---|---|
| Non-severe SARS | Increased | IFN-α, IFN-γ, CXCL10, IL-8 | CXCL10, IL-8 |
| Severe SARS | Increased | IFN-α, IFN-γ, CXCL10 | IL-8, CXCL10b, CCL2b |
| Decreased | IL-12p70, IL-2, TNF-α | IL-12p70b, IL-2, TNF-αb, IFN-γ |
Compared to healthy controls (P < 0.05).
Also significantly different compared to non-severe SARS patients (P < 0.05).
3.2. Modeling host responses to SARS infection
Assembling a comprehensive model of how the human immune system responds to SARS CoV infection and plays a role in the pathogenesis of SARS is a high research priority. Microarray analysis of gene expression has represented a potentially valuable tool in this regard. A paucity of SARS genomics literature exists and previous studies have generally been unable to assess human host responses to SARS CoV due to insufficient patient numbers, time points, clinical information or platform coverage of immune-related genes. Nonetheless, Yu et al. found that inflammatory genes, particularly proinflammatory cytokine genes such as IL-1, TNF-α and IL-8, were upregulated in an acute SARS patient compared with a healthy control and a convalescent SARS patient (Yu et al., 2005). Surprisingly, IFNs were not expressed during acute SARS, however this study included few patients. In comparing SARS-infected patients to healthy controls, Reghunathan et al. also found that acute SARS was associated with strong proinflammatory gene responses, albeit in the complete absence of cytokine gene expression (Reghunathan et al., 2005). Again, IFN-related genes were under-represented in this study. Lastly, Lee et al. compared SARS patients to healthy controls and developed a distinctive gene signature to accurately discriminate SARS patients from non-SARS patients (Lee et al., 2005). The primary goal of this study, however, was to develop a gene signature of SARS infection rather than model host immune responses. We have strived to analyze innate and adaptive immune responses to SARS CoV on the whole host level throughout the natural history of SARS using multiple techniques and a full clinical history to build as complete a model as possible of host immunity and inflammatory responses during the illness.
To this end, we used cDNA microarrays (∼16,000 sequence verified ESTs) to analyze host responses in peripheral blood mononuclear cells from a Toronto cohort of 50 SARS patients from onset of symptoms to discharge from hospital or death. Our longitudinal ANOVA (F-test) analysis between SARS patients and healthy controls is summarized in Fig. 1 in the form of three representative patients sampled throughout the early (acute) and late (progression/recovery) phase of SARS. We focused on 102 significantly deviated genes (P < 0.05) involved with IFN-responses (ISGs), adaptive immunity, inflammation, antigen presentation (human leukocyte antigen, HLA/major histocompatibility complex, MHC) and immunoglobulin (Ig) gene clusters. These genes are hierarchically clustered over multiple time points (at onset of symptoms and every 5–7 days) throughout the clinical course of SARS in three representative patients (Fig. 1). Patient 1 exhibited a non-severe case of SARS and recovered by day 25. Patient 2 developed severe SARS (increased O2 support) but recovered by day 50. Patient 3 also developed severe SARS and died on day 40.
Fig. 1.
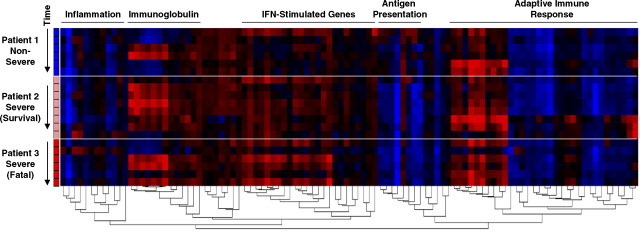
Microarray analysis of host gene expression during SARS infection. Detailed microarray procedures are posted at the UHN Microarray Facility website (http://www.microarrays.ca) and described previously (Bosinger et al., 2004). 102 significantly deviated genes (P < 0.05) involved with IFN-responses (ISGs), adaptive immune responses, inflammation, antigen presentation (HLA/MHC class I and II) and immunoglobulin (IgL and IgH) gene clusters were chosen from larger gene lists identified by ANOVA (F-test) analysis of early (n = 15, 5369 genes) and late phase (n = 13, 3860 genes) SARS patients versus healthy controls (n = 10). GO annotation (http://www.geneontology.org) and the Interferon Stimulated Gene Database (de Veer et al., 2001) were used to classify genes by related function. SARS patient datasets were then normalized gene by gene against healthy control means. Agglomerative hierarchical clustering with Pearson correlation and average linkage distance metrics was used to organize datasets corresponding to multiple time points (at onset of symptoms and every 5–7 days) throughout SARS in three representative patients described in the text. Genes shown in red are upregulated and genes shown in blue are downregulated >1.5-fold relative to healthy controls.
As shown in Fig. 1, the ISG cluster, including myxovirus (influenza) resistance 1 (MXA), peaks in expression during acute SARS in patient 1 (non-severe) and quickly resolves by the end of the first week since onset of symptoms. ISGs are similarly upregulated during acute SARS in the two severe patients; however, many of these genes, including the IFN-α/β receptor 1 (IFNAR1) for example, are strongly expressed for a longer period of time and are unresolved prior to the time of death in patient 3. These ISG profiles are echoed in the expression of the inflammatory gene cluster (includes CD14 for example). Interestingly, there is evidence of growing adaptive immunity as exemplified by Ig gene expression and increased expression of the adaptive immune response cluster (includes VAV1 and VAV2 oncogenes) in all three patients; although peak gene expression in the adaptive immune response cluster is delayed in patient 2 (severe) relative to patient 1 (non-severe) and is not evident in patient 3 who ultimately dies. Strikingly, only the non-severe SARS patient exhibits appreciable expression of antigen presentation (MHC Class I and II/HLA) genes. Collectively, these data indicate that key signatures of IFNs and ISGs are associated with developing innate and adaptive immune responses during the clinical evolution of SARS.
4. Conclusions
It appears that the current understanding of the potential role of cytokines and chemokines during SARS, i.e. hypercytokinemia or a “cytokine storm” driving SARS immunopathology, may be oversimplified. Type I IFNs are key mediators in innate immunity to viral infections and also provide a connection between the innate and adaptive arms of the immune response (Takaoka and Yanai, 2006). Likewise, the sole type II IFN, IFN-γ, is a critical contributor to adaptive immunity, especially in T helper 1 (Th1)-type responses (Malmgaard, 2004) and can potentiate type I IFN antiviral activity (Takaoka and Yanai, 2006). We contend that IFNs and IFN-stimulated chemokines, such as CXCL10, are induced during acute SARS infection in severe and non-severe patients as part of a robust innate immune response (Fig. 2 ). IFN responses may function as expected during inflammatory responses to SARS CoV infection in the majority of SARS patients who recover. Nevertheless, unregulated expression of IFNs and IFN-stimulated chemokines, e.g. CXCL10 and CCL2, in severe SARS patients as the illness progresses may lead to widespread immune dysregulation and serious pathogenic events (Fig. 2). The self-sustaining expression of proinflammatory chemokines during late SARS in susceptible patients may be a compensatory mechanism to the incapacity to mount an effective adaptive immune response to clear the virus. Those patients who ultimately succumb to SARS behave as a differentiated group; not only clinically but also from an immunological point of view. The term severe acute respiratory syndrome may therefore best describe a subgroup of particularly at-risk patients characterized by persistent induction of CXCL10 and CCL2 along with evidence of deficient adaptive immunity, more extensive thoracic involvement and a more severe compromise of respiratory indicators.
Fig. 2.

Model of immunopathological events associated with SARS clinical evolution. SARS patients mount robust IFN-mediated innate immune responses during early (acute) illness and peak symptomology. Following the median SO2 nadir in surviving patients (late SARS), most SARS patients resolve inflammation and mount effective adaptive immunity against SARS CoV. Immune dysregulation in severe SARS patients is hallmarked by continued expression of inflammatory chemokines and ISGs and deficiencies in MHC and Ig gene expression. Immune-mediated pathology worsens in the minority of susceptible SARS patients and unresolved CXCL10 expression and viral burden is associated with poor outcome.
Viral evasion during SARS infection may benefit in part by downregulation of type I IFN-directed innate immunity as has been proposed in in vitro studies (Castilletti et al., 2005, Cheung et al., 2005, Ziegler et al., 2005). Our results argue that SARS patients mount robust type I and II IFN responses and even express innate antiviral ISGs, such as MXA, during acute illness (Fig. 2). Type I and II IFN-responses may therefore act to maintain homeostasis between the development of effective versus autoinflammatory innate and adaptive immune responses. Cytokine and chemokine levels subside in the vast majority of SARS patients after acute infection and a critical switch from IFN-driven innate immunity to protective adaptive immunity and viral clearance occurs as patients recover. Conversely, the prolonged burden of SARS CoV in the lungs of at-risk patients, i.e. those with comorbidities as described above, may be the event that breaks down this homeostatic regulation. Persistent expression of distinct ISGs, unremitting induction of CXCL10 and CCL2 protein levels and deficient function of antigen presentation genes hallmark patients with progressing hypoxemia and poor disease course. Moreover, deficiencies in the expression of IL-12p70, IL-2, TNF-α and IFN-γ in severe SARS patients may indicate the incapacity to develop an effective adaptive immune response in these patients. Ultimately, unmanageable viral load, self-sustaining chemokine-mediated proinflammation and progressive pulmonary injury/ARDS are characteristic of those SARS patients at risk for fatal outcome (Fig. 2).
The host immune response to SARS may be the first to be mapped in detail in terms of an emerging infectious disease. While we do not know the exact mechanism that leads to a malfunction in the switch from innate to adaptive immunity in severe SARS patients, the gene signatures we describe in this study may help assess the immunopathology and management of severe viral infections. Using genomic technology to study host responses during severe viral illnesses, such as SARS and avian influenza (H5N1), may identify parallel gene signatures that are diagnostic and/or prognostic during clinical evolution.
Acknowledgements
This study was supported by the Canadian Institutes of Health Research, Canadian Network for Vaccines and Immunotherapeutics (CANVAC), Fondo de Investigaciones Sanitarias (J.F.B.M.) and GenomeCanada/Genome Quebec/Ontario Genomics Institute.
Special thanks to all members of the laboratory, especially Longsi Ran and Luoling Xu, for their contributions. Thanks to the Canadian SARS Research Network (http://www.canadiansarsresearchnetwork.ca) investigators and the Toronto health care community whose collaborations were critical to this study. We also express our respect and gratitude to Toronto and area clinicians, nurses, laboratory personnel and health care workers who cared for SARS patients.
References
- Antonio G.E., Ooi C.G., Wong K.T., Tsui E.L., Wong J.S., Sy A.N., Hui J.Y., Chan C.Y., Huang H.Y., Chan Y.F., Wong T.P., Leong L.L., Chan J.C., Ahuja A.T. Radiographic-clinical correlation in severe acute respiratory syndrome: study of 1373 patients in Hong Kong. Radiology. 2005;237:1081–1090. doi: 10.1148/radiol.2373041919. [DOI] [PubMed] [Google Scholar]
- Booth C.M., Matukas L.M., Tomlinson G.A., Rachlis A.R., Rose D.B., Dwosh H.A., Walmsley S.L., Mazzulli T., Avendano M., Derkach P., Ephtimios I.E., Kitai L., Mederski B.D., Shadowitz S.B., Gold W.L., Hawryluck L.A., Rea E., Chenkin J.S., Cescon D.W., Poutanen S.M., Detsky A.S. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. J. Am. Med. Assoc. 2003;289:2801–2809. doi: 10.1001/jama.289.21.JOC30885. [DOI] [PubMed] [Google Scholar]
- Bosinger S.E., Hosiawa K.A., Cameron M.J., Persad D., Ran L., Xu L., Boulassel M.R., Parenteau M., Fournier J., Rud E.W., Kelvin D.J. Gene expression profiling of host response in models of acute HIV infection. J. Immunol. 2004;173:6858–6863. doi: 10.4049/jimmunol.173.11.6858. [DOI] [PubMed] [Google Scholar]
- Cameron, M.J., Gold, W., Ran, L., Poutanen, S., Louie, M., Phillips, E., Mederski, B., Loutfy, M., Mazzulli, T., Willey, B., Muller, M., Persad, D., Xu, L., Brunton, J., Low, D., McGeer, A., Kelvin, D., 2003. Identification of gene expression profiles in patients with severe acute respiratory syndrome (SARS) that may be predictive of diagnosis, severity and clinical outcome of the illness. [Abstr. 43rd Intersci Conf Antimicrob Agents Chemother, Abstract no. K-452j].
- Castilletti C., Bordi L., Lalle E., Rozera G., Poccia F., Agrati C., Abbate I., Capobianchi M.R. Coordinate induction of IFN-alpha and -gamma by SARS-CoV also in the absence of virus replication. Virology. 2005;341:163–169. doi: 10.1016/j.virol.2005.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C.Y., Poon L.L., Ng I.H., Luk W., Sia S.F., Wu M.H., Chan K.H., Yuen K.Y., Gordon S., Guan Y., Peiris J.S. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J. Virol. 2005;79:7819–7826. doi: 10.1128/JVI.79.12.7819-7826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien J.Y., Hsueh P.R., Cheng W.C., Yu C.J., Yang P.C. Temporal changes in cytokine/chemokine profiles and pulmonary involvement in severe acute respiratory syndrome. Respirology. 2006;11:715–722. doi: 10.1111/j.1440-1843.2006.00942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M.D., Poutanen S.M., Loutfy M.R., Muller M.P., Low D.E. Severe acute respiratory syndrome. Clin. Infect. Dis. 2004;38:1420–1427. doi: 10.1086/420743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Veer M.J., Holko M., Frevel M., Walker E., Der S., Paranjape J.M., Silverman R.H., Williams B.R. Functional classification of interferon-stimulated genes identified using microarrays. J. Leuk. Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- Ding Y., He L., Zhang Q., Huang Z., Che X., Hou J., Wang H., Shen H., Qiu L., Li Z., Geng J., Cai J., Han H., Li X., Kang W., Weng D., Liang P., Jiang S. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and vims transmission pathways. J. Pathol. 2004;203:622–630. doi: 10.1002/path.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farcas G.A., Poutanen S.M., Mazzulli J., Willey B.M., Butany J., Asa S.L., Faure P., Akhavan P., Low D.E., Kain K.C. Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J. Infect. Dis. 2005;191:193–197. doi: 10.1086/426870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher D.A., Chew M.H., Lim Y.T., Tambyah P.A. Preventing local transmission of SARS: lessons from Singapore. Med. J. Aust. 2003;178:555–558. doi: 10.5694/j.1326-5377.2003.tb05358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler R.A., Lapinsky S.E., Hallett D., Detsky A.S., Sibbald W.J., Slutsky A.S., Stewart T.E. Critically ill patients with severe acute respiratory syndrome. J. Am. Med. Assoc. 2003;290:367–373. doi: 10.1001/jama.290.3.367. [DOI] [PubMed] [Google Scholar]
- Hamming I., Timens W., Bulthuis M.L., Lely A.T., Navis G.J., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.J., Su I.J., Theron M., Wu Y.C., Lai S.K., Liu C.C., Lei H.Y. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 2005;75:185–194. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D.M., Chamberlain D.W., Poutanen S.M., Low D.E., Asa S.L., Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod. Pathol. 2005;18:1–10. doi: 10.1038/modpathol.3800247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Xu J., Zhou C., Wu Z., Zhong S., Liu J., Luo W., Chen T., Qin Q., Deng P. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 2005;171:850–857. doi: 10.1164/rccm.200407-857OC. [DOI] [PubMed] [Google Scholar]
- Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W., Bao L., Zhang B., Liu G., Wang Z., Chappell M., Liu Y., Zheng D., Leibbrandt A., Wada T., Slutsky A.S., Liu D., Qin C., Jiang C., Penninger J.M. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau Y.L., Peiris J.S. Pathogenesis of severe acute respiratory syndrome. Curr. Opin. Immunol. 2005;17:404–410. doi: 10.1016/j.coi.2005.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt G.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., Lui S.F., Szeto C.C., Chung S., Sung J.J. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- Lee Y.S., Chen C.H., Chao A., Chen E.S., Wei M.L., Chen L.K., Yang K.D., Lin M.C., Wang Y.H., Liu J.W., Eng H.L., Chiang P.C., Wu T.S., Tsao K.C., Huang C.G., Tien Y.J., Wang T.H., Wang H.S., Lee Y.S. Molecular signature of clinical severity in recovering patients with severe acute respiratory syndrome coronavirus (SARS-CoV) BMC Genom. 2005;6:132. doi: 10.1186/1471-2164-6-132. 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew T.W., Kwek T.K., Tai D., Earnest A., Loo S., Singh K., Kwan K.M., Chan Y., Yim C.F., Bek S.L., Kor A.C., Yap W.S., Chelliah Y.R., Lai Y.C., Goh S.K. Acute respiratory distress syndrome in critically ill patients with severe acute respiratory syndrome. J. Am. Med. Assoc. 2003;290:374–380. doi: 10.1001/jama.290.3.374. [DOI] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmgaard L. Induction and regulation of IFNs during viral infections. J. Interferon Cytokine Res. 2004;24:439–454. doi: 10.1089/1079990041689665. [DOI] [PubMed] [Google Scholar]
- Mandavilli A. China: open season. Nature. 2006;439:382–383. doi: 10.1038/439382a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M.P., Richardson S.E., McGeer A., Dresser L., Raboud J., Mazzulli T., Loeb M., Louie M. Early diagnosis of SARS: lessons from the Toronto SARS outbreak. Eur. J. Clin. Microbiol. Infect. Dis. 2006;25:230–237. doi: 10.1007/s10096-006-0127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls J.M., Poon L.L., Lee K.C., Ng W.F., Lai S.T., Leung C.Y., Chu C.M., Hui P.K., Mak K.L., Lim W., Yan K.W., Chan K.H., Tsang N.C., Guan Y., Yuen K.Y., Peiris J.S. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361:1773–1778. doi: 10.1016/S0140-6736(03)13413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L., Law K.I., Tang B.S., Hon T.Y., Chan C.S., Chan K.H., Ng J.S., Zheng B.J., Ng W.L., Lai R.W., Guan Y., Yuen K.Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainer T.H., Cameron P.A., Smit D., Ong K.L., Hung A.N., Nin D.C., Ahuja A.T., Si L.C., Sung J.J. Evaluation of WHO criteria for identifying patients with severe acute respiratory syndrome out of hospital: prospective observational study. Br. Med. J. 2003;326:1354–1358. doi: 10.1136/bmj.326.7403.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reghunathan R., Jayapal M., Hsu L.Y., Chng H.H., Tai D., Leung B.P., Melendez A.J. Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salto-Tellez M., Tan E., Lim B. ARDS in SARS: cytokine mediators and treatment implications. Cytokine. 2005;29:92–94. doi: 10.1016/j.cyto.2004.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockman L.J., Bellamy R., Garner P. SARS: systematic review of treatment effects. PLoS Med. 2006:3. doi: 10.1371/journal.pmed.0030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A., Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- Tang N.L., Chan P.K., Wong C.K., To K.F., Wu A.I., Sung Y.M., Hui D.S., Sung J.J., Lam C.W. Early enhanced expression of interferon-inducible protein-10 (CXCL-10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin. Chem. 2005;51:2333–2340. doi: 10.1373/clinchem.2005.054460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C.K., Lam C.W., Wu A.K., Ip W.K., Lee N.L., Chan I.H., Lit L.C., Hui D.S., Chan M.H., Chung S.S., Sung J.J. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004;136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K.T., Antonio G.E., Hui D.S., Lee N., Yuen E.H., Wu A., Leung C.B., Rainer T.H., Cameron P., Chung S.S., Sung J.J., Ahuja A.T. Severe acute respiratory syndrome: radiographic appearances and pattern of progression in 138 patients. Radiology. 2003;228:401–406. doi: 10.1148/radiol.2282030593. [DOI] [PubMed] [Google Scholar]
- Wong K.T., Antonio G.E., Hui D.S., Lee N., Yuen E.H., Wu A., Leung C.B., Rainer T.H., Cameron P., Chung S.S., Sung J.J., Ahuja A.T. Thin-section CT of severe acute respiratory syndrome: evaluation of 73 patients exposed to or with the disease. Radiology. 2003;228:395–400. doi: 10.1148/radiol.2283030541. [DOI] [PubMed] [Google Scholar]
- Wong R.S., Wu A., To K.F., Lee N., Lam C.W., Wong C.K., Chan P.K., Ng M.H., Yu L.M., Hui D.S., Tam J.S., Cheng G., Sung J.J. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. Br. Med. J. 2003;326:1358–1362. doi: 10.1136/bmj.326.7403.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.Y., Hu Y.W., Liu X.Y., Xiong W., Zhou Z.T., Yuan Z.H. Gene expression profiles in peripheral blood mononuclear cells of SARS patients. World J. Gastroenterol. 2005;11:5037–5043. doi: 10.3748/wjg.v11.i32.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler T., Matikainen S., Ronkko E., Osterlund P., Sillanpaa M., Siren J., Fagerlund R., Immonen M., Melen K., Julkunen I. Severe acute respiratory syndrome coronavirus fails to activate cytokine-mediated innate immune responses in cultured human monocyte-derived dendritic cells. J. Virol. 2005;79:13800–13805. doi: 10.1128/JVI.79.21.13800-13805.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]