

Abstract
Vaccine design is progressing from empiricism towards the increasingly rational presentation of the targets of protective immunity. Nevertheless, most current vaccine antigens are essentially the native macromolecules of pathogens. These molecules are adapted to evade, not induce, immunity. High resolution structures reveal the electrostatic surfaces recognized by neutralizing antibodies and the architectures underlying these surfaces, thereby identifying which substructures must be left intact and which can be changed to optimize biochemical and immunologic performance. Armed with detailed structural information, we can engineer optimized antigens that are more stable, homogeneous, and efficiently produced, making immunization more practical and affordable. Understanding the structural basis for immunogenicity and immunodominance will allow us to improve vaccine efficacy and broaden the range of vaccine-preventable diseases.
Historical overview
The vaccines in use today evolved from two old and empirical methodologies, pioneered by Edward Jenner and Louis Pasteur. In 1796, Jenner used an attenuated infectious agent as a vaccine, inoculating with cowpox to protect against smallpox. In the late 19th century, Pasteur pioneered immunization with nonreplicating preparations of pathogens. Pasteur's technique is succinctly summarized as isolate (the causative agent), inactivate, and inject. This technique formed the basis for his effective vaccines against both bacteria (anthrax) and viruses (rabies).
Although vaccines have progressed considerably since the early complex and poorly defined preparations (pustule contents, heat inactivated bacilli, and extracts of infected spinal cords), the principles of vaccine design remain fundamentally the same. Some modern vaccines, such as those against rabies and tick-borne encephalitis, still contain entire inactivated pathogens. Others, such as vaccines against tetanus, diphtheria, human papillomavirus, and hepatitis B, contain isolated molecules known to be targets of protective immunity. The most refined modern vaccines are the glycoconjugates, in which oligosaccharide structures specific to bacterial pathogens are conjugated to carriers.
This progressive refinement has reduced nonprotective and reactogenic vaccine components and increased the safety and tolerability of immunization. Nevertheless, modern vaccines still use native antigens. With the exception of the genetically engineered modification of the polybasic cleavage site in the hemagglutinins of pre-pandemic influenza vaccines [1], the only significant antigen engineering in licensed vaccines consists of denaturation, detoxification, conjugation, and mixture with or adsorption to adjuvants. Vaccine antigens are not generally engineered using modern genetic techniques and the best available structural data. One exception is a genetically detoxified pertussis toxin [2] that was used as an acellular pertussis vaccine (alone or in combination with diphtheria and tetanus antigens) for several years in Europe. This modification was guided by the structures of the homologous toxins of Pseudomonas aeruginosa [3] and the diphtheria corynephage [4].
By contrast, small molecule therapeutic agents often result from de novo synthesis or extensive structure-based modification of naturally derived substances. Rational optimization of antigens could improve their properties as vaccine components. After all, native surface proteins of pathogens are adapted to evade, not induce, immunity. They are certainly not adapted to be stably stored, efficiently produced, or safely inoculated.
The need for structural vaccinology
The use of native antigens has proven remarkably successful against a wide range of pathogens, but not against all agents for which vaccines are needed. The pathogens for which we have successful vaccines share certain characteristics. They are, in general, agents against which humoral immunity is protective, and their surface antigens do not vary more quickly than new variants can be incorporated into updated vaccines. Examples include diphtheria, poliovirus, and hepatitis B virus, which have not undergone significant antigenic change over many decades [5].
The pathogens against which traditional techniques have not been successful also share certain characteristics. One of these characteristics is a lack of highly expressed surface antigens that are suitable for use as vaccine components. For some complex pathogens, the harnessing of genomics through reverse vaccinology is overcoming that barrier. In reverse vaccinology, whole genome sequences of bacteria, fungi, and parasites are mined to discover new protective determinants that were not previously known to be targets of natural immunity [6]. Recombinant expression of these molecules can overcome the low natural abundance that, in many cases, prevented recognition of their potential as vaccine antigens. By increasing the set of native antigens from which to choose, this technique is enabling the development of a new generation of vaccines. The technique has led to the discovery of new protective antigens for pneumococcus [7] and group B streptococcus [8]. Reverse vaccinology vaccine candidates for Staphylococcus aureus [9] and meningococcus B 10, 11 are now in clinical trials.
Vaccines against the most refractory pathogens will require different solutions. For example, most viruses do not encode as-yet undiscovered protective antigens. HIV and hepatitis C virus vary their surface antigens so rapidly and extensively that antibodies elicited by their envelope glycoproteins can only neutralize strains that are closely related to strains from which the immunogens are derived. Furthermore, the immune response against the determinants of certain viral agents, such as respiratory syncytial virus or dengue virus, can actually exacerbate disease 12, 13. Protecting against new targets faces practical challenges as well. In the impoverished settings where new vaccines are urgently needed, the cost of production and distribution of vaccines is a major barrier to use. Even in the developed world, where expense is less of an issue, the number of immunizations a child receives is becoming increasingly burdensome.
Optimizing vaccine antigens by combining the tools of genetic engineering with the insights provided by high resolution structural analysis may allow us to overcome these barriers (Figure 1 ). One important goal of structural vaccinology is to selectively present the conserved determinants of complex and variable antigens. Distinguishing structural components that elicit protective and disease-enhancing immunity could avoid vaccine-mediated exacerbation of disease. Using high resolution structures, antigens can be designed to be more efficiently produced and stably stored than native molecules, thereby lowering costs and eliminating barriers to distribution. By engineering antigens amenable for use in combination vaccines, immunization regimens can be simplified. By knowing which parts of an antigen must be retained to preserve basic characteristics and which can be altered, vaccine antigens can be modified more rapidly in response to changing epidemiology.
Figure 1.
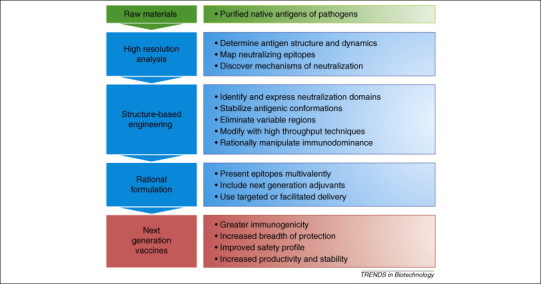
Flow chart of the structural vaccinology approach.
The basis for structural engineering of antigens
Immunization against pathogens relies on priming the immune system with the specific components of a microbe that are the targets of protective immunity. In the case of T cell mediated immunity, these epitopes are short peptides, which lack a defined three-dimensional structure [14]. Therefore, knowledge of the three dimensional structure of an antigen is not essential for designing peptide-based T cell vaccines, which are often considered for therapeutic purposes. Nevertheless, knowledge of the structural basis for peptide presentation by major histocompatibility antigens has provided insights into peptide design. In the future, increased knowledge of the structural basis for antigen processing may be incorporated into protein antigen design to allow for more efficient elicitation of T cell help [14].
By contrast, the epitopes that are recognized by neutralizing antibodies are three-dimensional, structured electrostatic landscapes. The primary mechanism of action of most vaccines in clinical use is to present these target landscapes, thereby inducing the clonal expansion of B cells that express antibodies that specifically bind these surfaces and promoting the affinity maturation of those antibodies. Most neutralizing epitopes are formed from residues that are discontinuous in the primary amino acid sequence.
Although a high affinity antibody that recognizes a pathogen's neutralization determinant may also bind an isolated, unstructured peptide, immunization with the peptide is unlikely to elicit high titer neutralizing antibodies against the pathogen reliably. This is because a neutralizing antibody may trap a flexible peptide in a conformation that matches the surface of an intact protein. However, upon immunization with the peptide, any B cell that bears an immunoglobulin that recognizes one of the many conformations of the peptide can be stimulated to expand. With peptide immunization, there is no mechanism to selectively activate only those B cells bearing immunoglobulins that bind both the flexible peptide and the complete, folded antigen. Furthermore, the flexible peptide could guide the affinity maturation of antibodies down many alternative paths, not only those that also optimize binding to the pathogen surface. Reliable affinity maturation of antibodies that efficiently recognize a specific surface requires an immunogen that stably presents that surface rather than transiently writhes through a state resembling the desired target epitope.
Identifying neutralizing epitopes
The electrostatic landscapes of neutralizing epitopes can be identified through a combination of antigenic and structural analyses (techniques listed in Table 1 ). Sequence analysis of viral variants that escape neutralization by monoclonal antibodies (mAbs) identifies those residues that, when altered, prevent antibody binding. Escape mutant analysis is particularly useful for RNA viruses and retroviruses, whose error-prone polymerases lead to high mutation rates and the efficient generation of variants that are no longer neutralized by antibodies. In general, when antibody binding sites are also delineated by physical methods, antibody neutralization escape mutations are found to be located within antibody binding sites 15, 16, 17. A key limitation of escape mutant analysis is that only those mutations that are compatible with viral replication can be identified. This limitation can be overcome by the targeted mutagenesis of recombinant viral antigens followed by directly assaying antibody binding. However, failure of antibodies to bind antigens that have undergone targeted mutagenesis must be interpreted carefully – mutations may prevent antibody binding by yielding mis-folded domains rather than by falling within the antibody binding site. Screening phage display libraries for neutralizing antibody binding [18] provides an efficient screen of many sequences. However, the utility of phage display is limited by the inability of the relatively short peptides displayed in most libraries to reproduce conformational epitopes. Deuterium exchange mass spectrometry [19] and (for bacterially expressed antigens with high solubility) nuclear magnetic resonance spectroscopy [20] can identify antibody bound residues in an intact protein.
Table 1.
A toolkit for structural vaccinology
| Property analyzed | Techniques | Utility |
|---|---|---|
| Three-dimensional structure of antigens and antigen-antibody complexes | X-ray crystallography, NMR, cryo-EM | Allow rational engineering by defining domain boundaries, epitope structure, and underlying architecture |
| Antigenic structure | ELISA, IP, escape mutant analysis, DXMS, phage display | Define the link between physical structure and the landscapes recognized by antibodies |
| Post-translational modification | SDS–PAGE, MS, glycosidic linkage analysis, X-ray crystallography, NMR | Assess the authenticity and homogeneity of modifications on recombinantly expressed proteins |
| Protein folding and stability | CD, ITC, DXMS, NMR, DSC, protease protection, native- and SDS–PAGE | Assess antigen conformation and integrity in solution over time for vaccine stability. |
| Non-covalent association and hydrodynamic radius | AUC, DLS, SEC, SPR | Assess antigen valency and aggregation |
Selection of optimized protein antigens will require a comprehensive evaluation of the structural features of the recombinantly produced protein. Ideally, these analytical tools will be applied in a high throughput manner to many candidate structures. Abbreviations: AUC, analytical ultracentrifugation; CD, circular dichroism spectroscopy; cryo-EM, electron cryomicroscopy; DLS, dynamic light scattering; DSC, differential scanning calorimetry; DXMS, deuterium exchange mass spectrometry; ELISA, enzyme-linked immunosorbent assay; IP, immunoprecipitation; ITC, isothermal titration calorimetry; mAb, monoclonal antibody; MS, mass spectrometry; NMR, nuclear magnetic resonance spectroscopy; SEC, size exclusion chromatography. SPR, surface plasmon resonance;
High resolution imaging of antigen-antibody complexes can precisely delineate epitope boundaries and provides essential information for antigen engineering. X-ray crystallography provides the most definitive data (examples include high resolution structures of antibody complexes with rhinovirus virions, influenza virus hemagglutinin, and HIV gp120) 15, 17, 21. Combining electron cryomicroscopy with molecular modeling can provide approximations when crystal structures of complexes cannot be obtained (examples include image reconstructions of antibody complexes with rhinovirus and rotavirus virions) 15, 22, 23. A basic conclusion from such studies is that an antibody epitope is a large patch on most antigens. A bound antibody typically covers ∼1250 Å2 [17]. Even when a limited number of residues make the major contacts between antigen and antibody, additional residues are usually necessary to maintain epitope structure. Other residues, if altered, could sterically hinder antibody binding.
A limitation inherent to all epitope mapping methods involving monoclonal antibodies (mAbs) is that any given immunization and screening protocol will yield only a subset of potential antibodies, so that neutralizing epitopes may be missed. For example, due to the cumbersome nature of neutralization assays, mAbs are often screened primarily for binding to recombinant proteins or for inhibition of microbial binding to receptors. Such screens are systematically biased against neutralizing antibodies that recognize epitopes only found on assembled viruses [24] or antibodies that block infection at post-binding steps [25].
Engineering stable antigen conformations
Many viral neutralization determinants function as membrane fusion or disruption devices. During viral maturation, receptor binding, and cell entry, they may undergo extensive conformational changes. Conserved epitopes may be present only in specific but transient conformations. Therefore, optimizing these molecules as immunogens requires stabilizing them in one or more advantageous conformations.
For example, because HIV gp120's binding site for its secondary cellular receptor (a chemokine receptor) is conserved, this surface provides an epitope that can be recognized by broadly neutralizing antibodies [26]. However, this conserved epitope assembles from dispersed structural elements only after gp120 has bound its primary receptor (CD4) 27, 28. Thus, the conserved surface is only stably presented when gp120 has already bound the cell, just before virus entry. In a second example, the parainfluenza virus F glycoproteins rearrange to radically different structures as the viruses fuse with cellular membranes 29, 30 (Figure 2 ). Some neutralization escape mutations of the related respiratory syncytial virus F glycoprotein map to structural elements that, by analogy, are also likely to rearrange radically during entry [31]. Different conformations of such glycoproteins are likely to elicit antibodies with different specificities and neutralizing capacities.
Figure 2.
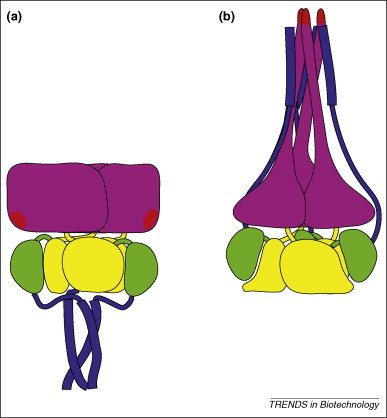
Parainfluenza virus fusion protein rearrangements. The F glycoprotein is a major target of antibodies that neutralize parainfluenza viruses. (a) Pre-fusion F. (b) Post-fusion F. During the rearrangement, a coiled-coil tipped by the fusion peptide extends from the purple domain, and the helices that make up the pre-fusion coiled-coil separate and pack between the new extension to form a six-helix bundle. This rearrangement brings the cellular and viral membranes into close apposition and extensively alters the solvent exposed surface of F. Red indicates the fusion peptide. The other colors indicate structural domains. The drawing is based on structures presented in [29] and [30].
A protein can be locked in a desired conformation by engineering cysteines that form new disulfide bonds that are only compatible with one conformation. Structural data guide the selection of the residues that are mutated to cysteine. For example, the prefusion conformation of influenza HA has been stabilized by tethering the three heads of the HA trimer together through engineered inter-subunit cross links [32]. Similarly, the prefusion state of the measles virus F glycoprotein has been stabilized by a disulfide cross-link that was introduced between the top of the coiled-coil and the base of the head of an adjacent subunit [33]. The effects of such modifications on immunogenicity are yet to be determined. Other types of mutations that favor a preferred conformation include those that reinforce or disrupt hydrophobic cores, hydrogen bond networks, or salt bridges, as has been demonstrated in studies of other paramyxovirus F glycoproteins 30, 34. In theory, engineered mutations could also stabilize other more transient protein conformations, such as those that a fusion protein passes through during the transition between the more stable pre- and post-fusion conformations. Stabilizing transient intermediates could allow the induction of antibodies against conserved epitopes that are exposed too transiently in native molecules to elicit protective antibodies efficiently [35].
Link between structure, mechanism of neutralization, and antigen design
The structure of neutralizing epitopes can be linked to the mechanism of neutralization by antibodies, as demonstrated by the rotavirus outer capsid protein, VP7. Rotavirus infection efficiently elicits neutralizing antibodies against VP7, but immunization with recombinant VP7 does not 36, 37. During cell entry, VP7 must be shed from the virion, a process termed uncoating [38]. Rotavirus virions uncoat in vitro when calcium is removed by chelation [39]. Uncoating is caused by the dissociation of calcium-dependent VP7 trimers [40]. Neutralizing antibodies inactivate rotavirus by binding specifically to VP7 trimers on the virion, stabilizing them, and preventing uncoating, even in the presence of a calcium chelator [41]. Therefore, VP7 trimers engineered for enhanced stability could more effectively elicit neutralizing antibodies than native VP7, which is in a dynamic equilibrium between monomers and trimers in solution [42]. In this case, a high resolution structural understanding of a neutralization mechanism could guide the engineering of an optimized antigen.
Optimized domains
To engineer antigens, it is not sufficient simply to know the surfaces available for antibody binding. It is also necessary to understand the underlying protein architectures that form these surfaces. For example, understanding the underlying HIV gp120 architecture guides deletion of variable loops that bias the repertoire of elicited antibodies away from more conserved parts of the molecule 43, 44. Understanding underlying architecture also allows the sensible dissection of macromolecules, ‘carving proteins at their joints’ (interdomain boundaries) into isolated, autonomously folding structural domains that are suitable for independent recombinant expression. In some cases, such as picornavirus capsids, neutralizing epitopes require an intimate co-folding and co-assembly of proteins, so that isolated domains are not apparent, and carving the protein shell into components is not feasible [45]. (An extruded picronavirus protein, VP4, is an exception [46]). By contrast, other complex neutralization determinants are collections of self-contained subunits, which can be expressed separately. Examples of such subunits include the receptor binding domain of the SARS coronavirus S glycoprotein [47] and the head and body domains of the rotavirus spike protein, VP4 48, 49.
Studies of rotavirus VP4 (Figure 3 ) illustrate some advantages of reducing a complex and flexible macromolecule to its neutralizing epitope-containing compact domains. Rotavirus VP4 misfolds when expressed in bacteria, and it is susceptible to proteolysis and aggregation during storage. However, all known neutralizing epitopes are present on two compactly folded domains 48, 49. One of these, corresponding to the ‘head’ domain of the spike, can be expressed efficiently in Escherichia coli, resists protease digestion, and is stable in solution for years [50]. Similar efficiently produced, compact, and stable neutralization domains from several pathogens are likely to have more homogeneous biochemical characteristics than the multidomain assemblages from which they are derived. Such domains could be optimized further by altering surface-exposed hydrophobic residues that cause aggregation and modifying protease cleavage sites that compromise protein stability. A collection of optimized domains could form the raw materials for combination vaccines that are tailored to particular patient populations and epidemiological circumstances.
Figure 3.
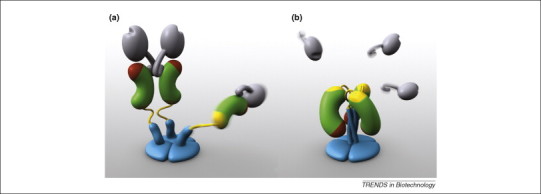
Rotavirus spike protein VP4 rearrangements. VP4 is made up of discrete, compactly folded domains that undergo rearrangements during membrane penetration. The trimeric molecule is thought to progress from an initial state in which all three subunits are flexible (not shown); to (a), a primed state in which two subunits are rigid and the third remains flexible; and then to (b), a folded back state. The grey receptor binding head and the green and red antigen domain contain all known neutralizing epitopes on VP4. The red patch depicts the hydrophobic apex of the antigen domain, which is thought to interact with host membranes to effect entry. Motions and alternative packings of flexible elements, such as the cyan foot and stalk domains, drive the rearrangements of the rigid domains. Produced by Digizyme, Inc. and originally published in [49].
Understanding and engineering immunogenicity
There appears to be a trade-off between immunogenicity and the biochemically ideality that promotes efficient antigen production and stability during storage. For example, the compact, stable, soluble, efficiently expressed, neutralizing epitope-containing domains of the rotavirus spike protein, VP4 49, 50 are less effective at eliciting neutralizing antibodies than the intact protein [51], which is protease sensitive and more prone to aggregation. The compactness, stability, solubility, and monodispersity of isolated, well-folded domains appear to compromise their effectiveness as immunogens. Indeed, protein misfolding is associated with enhanced immunogenicity [52]. Therefore, optimization of vaccine antigens requires understanding the structural basis for immunogenicity as well as biochemical ideality.
Multivalency is one manipulable structural characteristic that appears to enhance immunogenicity. Ordered arrays of antigens can elicit B cell activation (but not antibody class switching or affinity maturation) in the absence of antigen presenting cells or T cell help [53]. Studies of the immune response to immunization with the vesicular stomatitis virus G glycoprotein indicate that the more repetitively and rigidly the G glycoprotein is arrayed, the more immunogenic it becomes [53]. In another example, although the isolated body domain of rotavirus VP4 [49] is a poor immunogen, it can elicit high titer neutralizing antibodies when linked in a stably trimeric form ([23] and unpublished observations). The greater immunogenicity of multivalent antigens may reflect an adaptation of the immune system to recognize the repetitive structures that characterize many microorganisms.
These observations suggest that mounting compactly folded domains on multivalent display platforms could increase their immunogenicity. Such platforms could include self-oligomerizing proteins that form multivalent particles used for human immunization, such as human papillomavirus VP1 [54] and hepatitis B virus surface antigen [55]. Other strategies to enhance the immunogenicity of antigens include engineering linkages to strong T cell epitopes, as exemplified by glycoconjugate vaccines, in which linkage to proteins that bear T cell epitopes enhances the immunogenicity of bacterial carbohydrate antigens [56]. Linking antigens to B cell epitopes that are recognized by highly prevalent antibodies is another approach to increasing immunogenicity. For example linking domains to tetanus toxoid (against which pre-existing immunity is prevalent as a result of widespread immunization against tetanus) can increase immunogenicity, probably by promoting opsonization [57]. However, in some circumstances, this linkage can also decrease immunogenicity, possibly due to T cell-mediated suppression, masking by pre-existing antibody, or enhanced clearance of immune complexes [57].
Because humans do not produce oligosaccharides terminating with α-galactoside [58], which is a common glycan on proteins of other organisms, this sugar is recognized as a foreign antigen. Approximately 1% of the human serum antibody repertoire recognizes α-galactoside [59]. In a knockout mouse model that mimics this aspect of human immunity, addition of α-galactoside to antigens dramatically increases their immunogenicity, most likely by promoting opsonization by pre-existing antibodies that recognize this carbohydrate structure [60]. Ultimately, an increased understanding of native immunity may also allow the engineering of antigens that can directly potentiate immune recognition and presentation.
Understanding and engineering immunodominance
In addition to engineering antigens to make them more immunogenic overall, it would be desirable to manipulate the relative immunodominance of different epitopes on a given antigen. For vaccine purposes, it would be best if conserved epitopes elicited antibodies more efficiently than variable epitopes. However, on native proteins, the opposite is often the case. For example, a high proportion of mAbs that neutralize the HIV envelope glycoprotein recognize variable loops, whereas more conserved regions elicit neutralizing antibodies less efficiently 27, 61. Similarly, bactericidal antibodies elicited by immunization with a group B meningococcal vaccine produced from outer membrane vesicles primarily recognize a single variable loop of the Por A protein [62].
One explanation for immunodominance would attribute the phenomenon to the limited set of available germ line antibodies. The fit of an epitope to the antigen combining sites of germ line antibodies would dictate its relative dominance. In this case, an individual's germline antibody repertoire would determine the epitopes to which he or she would respond preferentially. In theory, primary immunization with antigens presenting epitopes that have been engineered to bind a prevalent germline antibody could efficiently promote clonal expansion. Subsequent boosting with antigens that present epitopes that have been engineered to match more precisely the conserved surfaces of a pathogen's protective determinants could guide affinity maturation of antibodies from the stimulated B cell clones.
Alternatively, more general topological or electrostatic characteristics of epitopes may predict their ability to efficiently stimulate B cell expansion. Some characteristics certainly disfavor antibody binding. For example, modifying the HIV gp120 surface by adding glycosylation sites masks epitopes on the altered surfaces [63]. More generally, glycosylation can lead to tolerance, because glycans on viruses replicating in a host are generally recognized as ‘self.’ This is the most likely explanation for the immunological ‘silence’ of the most heavily glycosylated face of HIV gp120 [27]. A role for glycosylation in evading immunity in previously exposed or immunized hosts is suggested by the evolution of H3 subtype influenza hemagglutinins since H3N2 strains emerged as human pathogens in 1968. Since then, the number of glycans has progressively increased, particularly on the membrane distal part of the HA1 fragment, where most neutralizing antibodies bind [64]. Analogously, it might be possible to abrogate the immunodominance of variable regions by adding masking oligosaccharides to these regions [63]. Conversely, determining which glycans are important for protein folding and solubility, and which glycans mask epitopes but have no essential structural role could allow elimination of masking glycans to enhance the presentation of conserved neutralizing epitopes 65, 66, 67.
High throughput protein modification
The application of modern drug discovery approaches to vaccine development has the potential to markedly expand our ability to screen and select high quality vaccine antigens. High throughput, robotic protein modification systems have been developed to optimize proteins for crystallization [68]. These systems can introduce a series of mutations into a protein-encoding gene, express and purify the mutant proteins, and carry out basic characterization in a fully automated and extremely efficient manner. This technology could also be used to optimize vaccine antigens. A major challenge is to set up high throughput in vitro screens for the characteristics that are relevant to immunogen performance in vivo. The ultimate characteristic of interest, the ability to elicit protective immunity, is not amenable to automated screening. Eventually, ex vivo systems to evaluate immune priming that are capable of predicting immunogenicity in vivo may be developed, thereby greatly facilitating the vaccine antigen discovery process. A research consortium (funded by the United States Defense Advanced Research Projects Agency) is currently working towards this goal. Until an artificial immune system that can be used to rapidly screen antigens for immunogenicity is available, automated screening will be limited to other desirable antigen characteristics, such as expression level, solubility, stability, aggregation state, protease resistance, and epitope presentation (Table 1).
If combined with sufficiently efficient assays for desired characteristics, very high throughput protein modification could provide a practical alternative to structure based design in some cases. For example, by expressing a sufficient number of empiric truncations and assaying the gene products for solubility (by centrifugation), monodispersity (by light scattering), and antigenicity (by solution immunoassays) one could identify autonomously folding antigenic domains without knowledge of the underlying protein conformation. By using a conformation-specific mAb to detect molecules with an advantageous conformation, high throughput mutagenesis, expression, and immunoassay could also yield protein variants stabilized in a desired conformation.
Concluding remarks
The biophysical, biochemical, and genetic engineering technologies needed for structural vaccinology are sophisticated, but the concept is simple: first, study the native molecular architecture of neutralization determinants; and then, use this knowledge to modify the molecules and engineer immunogens that are optimally designed for inclusion in protective vaccines. Success of structural vaccinology will require the application of high throughput methods to generate a large number of antigenic structures and assays to predict their effectiveness as immunogens. With current knowledge, we can already optimize protein antigens for efficient production, conformational homogeneity, and stable storage. However, fully realizing the potential of structure based design of vaccine antigens will require longer-term basic research into the structural determinants of immunodominance and immunogenicity. This research must take into account the diversity of the antibody repertoire generated not only in a single individual, but also in those of different genetic backgrounds and histories of antigen exposure. New techniques, such sorting IgG+ antibody secreting plasma cells to rapidly clone human monoclonal antibodies specific for a recent immunogen [69], make such analyses feasible.
The application of structural insight to vaccine design is especially important for new vaccines against viral pathogens. The greater number of protective determinants of bacteria, fungi, and parasites make them suitable for a reverse vaccinology approach to discovering protective determinants with favorable vaccine antigen characteristics [6]. The limited number of protective determinants on most viruses necessitates optimizing a relatively small set of macromolecules. Antigen optimization is not limited to subunit vaccines. Live attenuated, vectored, and virus-like-particle vaccines could also incorporate antigens that have been optimized based on structural insights. Matching the rational design of antigens to advances in the rational design of adjuvants can enable a new generation of vaccines that are more safe, broadly protective, practical, and affordable than those available today.
Acknowledgements
We thank Rafi Ahmed, Stephen Harrison, Scott Lesley, John Skehel, and Nicholas Valiante for helpful discussions; Nelle Cronen and Giorgio Corsi for illustrations; and Catherine Mallia for administrative assistance.
Glossary
- Adjuvant
a substance added to an antigen to enhance or modulate the immune response to the antigen in an immunized host.
- Affinity maturation
the process by which B cells produce antibodies with increased affinity for an antigen upon repeated or prolonged stimulation with the antigen. First, rearranged immunoglobulin genes in activated B cells undergo somatic hypermutation, which alters the complementarity-determining regions of the encoded antibodies. Second, B cells that express the mutated immunoglobulins undergo clonal selection. During clonal selection, B cells bearing immunoglobulins with greater affinity for a limited supply of antigen proliferate more than those bearing immunoglobulins with lower affinity. Affinity maturation requires T cell help.
- Analytical ultracentrifugation
a technique to measure hydrodynamic characteristics of particles. The distribution of the particles in solution is measured over time in a spinning rotor, allowing observation of the particles’ sedimentation and diffusion. The technique provides information on the size and shape of particles, including noncovalently associated complexes, and on the equilibrium between different associated states of the molecules.
- Circular dichroism spectroscopy
a technique that measures the differential absorption of left- or right-handed circularly polarized light by chiral molecules, such as proteins, in solution. A spectrum of the differential absorption (or ellipticity) of light as a function of wavelength provides information on protein secondary structure composition. Circular dichroism spectroscopy can be used to determine the thermal stability of a protein by determining the temperature at which ordered secondary structure is lost.
- Conjugation
linking a carbohydrate antigen to a protein carrier. The protein carrier elicits T cell help to generate an immune response that includes antibodies against the carbohydrate.
- Deuterium exchange mass spectrometry
a technique that allows information about protein conformation to be obtained by mass spectrometry. Solvent exposed backbone amide (and some side chain) hydrogens exchange with deuterium in solution. At lowered pH and temperature, exchange slows. The charge-to-mass ratios of protein fragments are then determined by mass spectrometry. Comparison of masses from native and deuterium exchanged samples allows determination of which regions of a protein were exposed to solvent and which were buried when deuterium exchange was taking place.
- Differential scanning calorimetry
a technique that measures the absorption of heat by a solution as a function of temperature. If the solution contains a macromolecule, the variation in the absorption of heat as the temperature is raised gives information on the unfolding of the macromolecule.
- Domain
as used by structural biologists, a part of a protein that forms a coherent folded unit. The structural elements of a domain have more intimate interactions with other parts of the domain than they do with parts of the protein outside the domain.
- Dynamic light scattering
a technique that allows measurement of diffusion coefficients based on the variation in the amount of light scattered by a small volume of suspended particles over time. Based on assumptions about the shapes of the particles, the diffusion coefficients may be used to estimate the size distribution of the particles. The technique is often used to measure polydispersity, the amount of variation in particle size.
- Electron cryomicroscopy
an electron microscopic technique in which specimens are imaged after being embedded in vitreous (glass-like, rather than crystalline) ice. Computational averaging and refinement of the resulting low contrast data can yield image reconstructions of biological specimens at considerably higher resolution than conventional electron micrographs of specimens embedded in negative stains that contain heavy atoms to enhance contrast. In the most favorable cases, near atomic resolution reconstructions can be achieved.
- Epitope
the part of an antigen that is recognized by an antibody (or group of antibodies) or a T cell receptor.
- Escape mutation
a mutation in a gene encoding a microbial antigen that allows the microbe to escape killing or neutralization, typically by an antibody. The position of the altered amino acid residue in an antigen's structure allows inferences about where the selecting neutralizing antibody binds the antigen.
- Germline antibody repertoire
an organism's collection of antibodies that have not yet been altered by somatic hypermutation and affinity maturation.
- Isothermal titration calorimetry
a technique that measures the absorption or production of heat by a solution as a second solution is mixed in. If the two solutions contain interacting molecules, the resulting thermodynamic information allows conclusions about the affinity, stoichiometry, and enthalpy of the interaction between the molecules.
- Mass spectrometry
a technique that measures the mass-to-charge ratio of particles. The technique's very high accuracy and throughput combined with protein fragmentation and computational analysis have greatly expanded the applications of mass spectrometry in protein biochemistry. The technique can be used to identify the proteins present in a mixture and to provide detailed information about a protein's primary structure and post-translational modifications.
- Nuclear magnetic resonance spectroscopy
a technique that measures the properties of the magnetic fields that are generated by the protons in some atomic nuclei. The ability of NMR to distinguish protons in a molecule that have different electronic environments; measure the response of a molecule's protons to applied magnetic pulses; and detect the interactions between the magnetic fields generated by protons that are close to each other allow conclusions about molecular structure and dynamics.
- Opsonization
the enhancement, usually by bound antibodies, of an antigen's uptake into phagocytes, such as antigen presenting cell.
- Size exclusion chromatography
a technique that separates particles in solution on the basis of their sieving through beads with pores. The rate at which particles pass through a size exclusion column is determined by a combination of their size, shape, and any affinity for the beads.
- Surface plasmon resonance
a technique that measures the binding of molecules in solution to molecules immobilized on a planar metal substrate (typically gold). Binding is detected by changes in the refractive index immediately adjacent to the metal surface as molecules in solution bind the immobilized molecules. The data allow determination of on-rates, off-rates, and binding constants (KD).
- X-ray crystallography
a technique that uses the diffraction of X-rays by molecules packed in a crystalline array to provide information about molecular structure. X-ray crystallography can produce atomic resolution structures of objects ranging in size from very simple compounds to entire viruses.
References
- 1.Subbarao K. Evaluation of a genetically modified reassortant H5N1 influenza A virus vaccine candidate generated by plasmid-based reverse genetics. Virology. 2003;305:192–200. doi: 10.1006/viro.2002.1742. [DOI] [PubMed] [Google Scholar]
- 2.Pizza M. Mutants of pertussis toxin suitable for vaccine development. Science. 1989;246:497–500. doi: 10.1126/science.2683073. [DOI] [PubMed] [Google Scholar]
- 3.Allured V.S. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc. Natl. Acad. Sci. U. S. A. 1986;83:1320–1324. doi: 10.1073/pnas.83.5.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choe S. The crystal structure of diphtheria toxin. Nature. 1992;357:216–222. doi: 10.1038/357216a0. [DOI] [PubMed] [Google Scholar]
- 5.Rappuoli R. Bridging the knowledge gaps in vaccine design. Nat. Biotechnol. 2007;25:1361–1366. doi: 10.1038/nbt1207-1361. [DOI] [PubMed] [Google Scholar]
- 6.Rappuoli R. Reverse vaccinology. Curr. Opin. Microbiol. 2000;3:445–450. doi: 10.1016/s1369-5274(00)00119-3. [DOI] [PubMed] [Google Scholar]
- 7.Giefing C. Discovery of a novel class of highly conserved vaccine antigens using genomic scale antigenic fingerprinting of pneumococcus with human antibodies. J. Exp. Med. 2008;205:117–131. doi: 10.1084/jem.20071168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maione D. Identification of a universal Group B streptococcus vaccine by multiple genome screen. Science. 2005;309:148–150. doi: 10.1126/science.1109869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weichhart T. Functional selection of vaccine candidate peptides from Staphylococcus aureus whole-genome expression libraries in vitro. Infect. Immun. 2003;71:4633–4641. doi: 10.1128/IAI.71.8.4633-4641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giuliani M.M. A universal vaccine for serogroup B meningococcus. Proc. Natl. Acad. Sci. U. S. A. 2006;103:10834–10839. doi: 10.1073/pnas.0603940103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pizza M. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science. 2000;287:1816–1820. doi: 10.1126/science.287.5459.1816. [DOI] [PubMed] [Google Scholar]
- 12.Kim H.W. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 1969;89:422–434. doi: 10.1093/oxfordjournals.aje.a120955. [DOI] [PubMed] [Google Scholar]
- 13.Halstead S.B. Pathogenesis of dengue: challenges to molecular biology. Science. 1988;239:476–481. doi: 10.1126/science.3277268. [DOI] [PubMed] [Google Scholar]
- 14.Purcell A.W. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007;6:404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 15.Hewat E.A. Structure of a neutralizing antibody bound monovalently to human rhinovirus 2. J. Virol. 1998;72:4396–4402. doi: 10.1128/jvi.72.5.4396-4402.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gulati U. Antibody epitopes on the neuraminidase of a recent H3N2 influenza virus (A/Memphis/31/98) J. Virol. 2002;76:12274–12280. doi: 10.1128/JVI.76.23.12274-12280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bizebard T. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature. 1995;376:92–94. doi: 10.1038/376092a0. [DOI] [PubMed] [Google Scholar]
- 18.Smothers J.F. Tech. Sight. Phage display. Affinity selection from biological libraries. Science. 2002;298:621–622. doi: 10.1126/science.298.5593.621. [DOI] [PubMed] [Google Scholar]
- 19.Baerga-Ortiz A. Epitope mapping of a monoclonal antibody against human thrombin by H/D-exchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein. Protein Sci. 2002;11:1300–1308. doi: 10.1110/ps.4670102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller M.D. A human monoclonal antibody neutralizes diverse HIV-1 isolates by binding a critical gp41 epitope. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14759–14764. doi: 10.1073/pnas.0506927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou T. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tihova M. Localization of membrane permeabilization and receptor binding sites on the VP4 hemagglutinin of rotavirus: implications for cell entry. J. Mol. Biol. 2001;314:985–992. doi: 10.1006/jmbi.2000.5238. [DOI] [PubMed] [Google Scholar]
- 23.Dormitzer P.R. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature. 2004;430:1053–1058. doi: 10.1038/nature02836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dormitzer P.R. Neutralizing epitopes on herpes simplex virus-1-expressed rotavirus VP7 are dependent on coexpression of other rotavirus proteins. Virology. 1992;187:18–32. doi: 10.1016/0042-6822(92)90291-v. [DOI] [PubMed] [Google Scholar]
- 25.Ruggeri F.M., Greenberg H.B. Antibodies to the trypsin cleavage peptide VP8 neutralize rotavirus by inhibiting binding of virions to target cells in culture. J. Virol. 1991;65:2211–2219. doi: 10.1128/jvi.65.5.2211-2219.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thali M. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 1993;67:3978–3988. doi: 10.1128/jvi.67.7.3978-3988.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyatt R. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 28.Kwong P.D. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin H.S. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin H.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crowe J.E. Monoclonal antibody-resistant mutants selected with a respiratory syncytial virus-neutralizing human antibody fab fragment (Fab 19) define a unique epitope on the fusion (F) glycoprotein. Virology. 1998;252:373–375. doi: 10.1006/viro.1998.9462. [DOI] [PubMed] [Google Scholar]
- 32.Godley L. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell. 1992;68:635–645. doi: 10.1016/0092-8674(92)90140-8. [DOI] [PubMed] [Google Scholar]
- 33.Lee J.K. Reversible inhibition of the fusion activity of measles virus F protein by an engineered intersubunit disulfide bridge. J. Virol. 2007;81:8821–8826. doi: 10.1128/JVI.00754-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell C.J. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J. Cell Biol. 2003;163:363–374. doi: 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamburger A.E. Steric accessibility of the HIV-1 gp41 N-trimer region. J. Biol. Chem. 2005;280:12567–12572. doi: 10.1074/jbc.M412770200. [DOI] [PubMed] [Google Scholar]
- 36.Both G.W. Relocation of antigens to the cell surface membrane can enhance immune stimulation and protection. Immunol. Cell Biol. 1992;70:73–78. doi: 10.1038/icb.1992.11. [DOI] [PubMed] [Google Scholar]
- 37.Dormitzer P.R. Presentation of neutralizing epitopes by engineered rotavirus VP7's expressed by recombinant vaccinia viruses. Virology. 1994;204:391–402. doi: 10.1006/viro.1994.1543. [DOI] [PubMed] [Google Scholar]
- 38.Ludert J.E. Penetration and uncoating of rotaviruses in cultured cells. Intervirology. 1987;27:95–101. doi: 10.1159/000149726. [DOI] [PubMed] [Google Scholar]
- 39.Cohen J. Ribonucleic acid polymerase activity associated with purified calf rotavirus. J. Gen. Virol. 1977;36:395–402. doi: 10.1099/0022-1317-36-3-395. [DOI] [PubMed] [Google Scholar]
- 40.Dormitzer P.R., Greenberg H.B. Calcium chelation induces a conformational change in recombinant herpes simplex virus-1-expressed rotavirus VP7. Virology. 1992;189:828–832. doi: 10.1016/0042-6822(92)90616-w. [DOI] [PubMed] [Google Scholar]
- 41.Ludert J.E. Antibodies to rotavirus outer capsid glycoprotein VP7 neutralize infectivity by inhibiting virion decapsidation. J. Virol. 2002;76:6643–6651. doi: 10.1128/JVI.76.13.6643-6651.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dormitzer P.R. Purified recombinant rotavirus VP7 forms soluble, calcium-dependent trimers. Virology. 2000;277:420–428. doi: 10.1006/viro.2000.0625. [DOI] [PubMed] [Google Scholar]
- 43.Srivastava I.K. Changes in the immunogenic properties of soluble gp140 human immunodeficiency virus envelope constructs upon partial deletion of the second hypervariable region. J. Virol. 2003;77:2310–2320. doi: 10.1128/JVI.77.4.2310-2320.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stamatatos L., Cheng-Mayer C. An envelope modification that renders a primary, neutralization-resistant clade B human immunodeficiency virus type 1 isolate highly susceptible to neutralization by sera from other clades. J. Virol. 1998;72:7840–7845. doi: 10.1128/jvi.72.10.7840-7845.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hogle J.M. Three-dimensional structure of poliovirus at 2.9 A resolution. Science. 1985;229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- 46.Bubeck D. The structure of the poliovirus 135S cell entry intermediate at 10-angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J. Virol. 2005;79:7745–7755. doi: 10.1128/JVI.79.12.7745-7755.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li F. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 48.Dormitzer P.R. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 2002;21:885–897. doi: 10.1093/emboj/21.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoder J.D., Dormitzer P.R. Alternative intermolecular contacts underlie the rotavirus VP5* two- to three-fold rearrangement. EMBO J. 2006;25:1559–1568. doi: 10.1038/sj.emboj.7601034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monnier N. High-resolution molecular and antigen structure of the VP8* core of a sialic acid-independent human rotavirus strain. J. Virol. 2006;80:1513–1523. doi: 10.1128/JVI.80.3.1513-1523.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mackow E.R. Immunization with baculovirus-expressed VP4 protein passively protects against simian and murine rotavirus challenge. J. Virol. 1990;64:1698–1703. doi: 10.1128/jvi.64.4.1698-1703.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maas C. A role for protein misfolding in immunogenicity of biopharmaceuticals. J. Biol. Chem. 2007;282:2229–2236. doi: 10.1074/jbc.M605984200. [DOI] [PubMed] [Google Scholar]
- 53.Bachmann M.F., Zinkernagel R.M. Neutralizing antiviral B cell responses. Annu. Rev. Immunol. 1997;15:235–270. doi: 10.1146/annurev.immunol.15.1.235. [DOI] [PubMed] [Google Scholar]
- 54.Modis Y. Atomic model of the papillomavirus capsid. EMBO J. 2002;21:4754–4762. doi: 10.1093/emboj/cdf494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gilbert R.J. Hepatitis B small surface antigen particles are octahedral. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14783–14788. doi: 10.1073/pnas.0505062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vliegenthart J.F. Carbohydrate based vaccines. FEBS Lett. 2006;580:2945–2950. doi: 10.1016/j.febslet.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 57.Peeters C.C. Effect of carrier priming on immunogenicity of saccharide-protein conjugate vaccines. Infect. Immun. 1991;59:3504–3510. doi: 10.1128/iai.59.10.3504-3510.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koike C. Functionally important glycosyltransferase gain and loss during catarrhine primate emergence. Proc. Natl. Acad. Sci. U. S. A. 2007;104:559–564. doi: 10.1073/pnas.0610012104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galili U. A unique natural human IgG antibody with anti-alpha-galactosyl specificity. J. Exp. Med. 1984;160:1519–1531. doi: 10.1084/jem.160.5.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abdel-Motal U. Increased immunogenicity of human immunodeficiency virus gp120 engineered to express Galα1-3Galβ1-4GlcNAc-R epitopes. J. Virol. 2006;80:6943–6951. doi: 10.1128/JVI.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burton D.R. HIV vaccine design and the neutralizing antibody problem. Nat. Immunol. 2004;5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- 62.Martin D.R. The VR2 epitope on the PorA P1.7-2,4 protein is the major target for the immune response elicited by the strain-specific group B meningococcal vaccine MeNZB. Clin. Vaccine Immunol. 2006;13:486–491. doi: 10.1128/CVI.13.4.486-491.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pantophlet R. Hyperglycosylated mutants of human immunodeficiency virus (HIV) type 1 monomeric gp120 as novel antigens for HIV vaccine design. J. Virol. 2003;77:5889–5901. doi: 10.1128/JVI.77.10.5889-5901.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skehel J.J., Wiley D.C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 65.Skehel J.J. A carbohydrate side chain on hemagglutinins of Hong Kong influenza viruses inhibits recognition by a monoclonal antibody. Proc. Natl. Acad. Sci. U. S. A. 1984;81:1779–1783. doi: 10.1073/pnas.81.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reitter J.N. A role for carbohydrates in immune evasion in AIDS. Nat. Med. 1998;4:679–684. doi: 10.1038/nm0698-679. [DOI] [PubMed] [Google Scholar]
- 67.Cole K.S. Removal of N-linked glycosylation sites in the V1 region of simian immunodeficiency virus gp120 results in redirection of B-cell responses to V3. J. Virol. 2004;78:1525–1539. doi: 10.1128/JVI.78.3.1525-1539.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McMullan D. High-throughput protein production for X-ray crystallography and use of size exclusion chromatography to validate or refute computational biological unit predictions. J. Struct. Funct. Genomics. 2005;6:135–141. doi: 10.1007/s10969-005-2898-1. [DOI] [PubMed] [Google Scholar]
- 69.Wrammert J. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]