

Abstract
The feline gastrointestinal microbiota have direct influence on feline health and also human health as a reservoir for potential zoonotic pathogens and antibiotic resistant bacterial strains. In order to describe the feline gastrointestinal microbial diversity, fecal samples from cats have been characterized using both culture-dependent and culture-independent methods. However, data correlating total microbial composition and their functions are lacking. Present descriptive study evaluated both phylogenetic and metabolic diversity of the feline intestinal microbiota using GS Junior titanium shotgun pyrosequencing. A total of 152,494 pyrosequencing reads (5405 assembled contigs) were generated and classified into both phylogenetic and metabolic profiles of the feline intestinal microbiota. The Bacteroides/Chlorobi group was the most predominant bacterial phylum comprising ~ 68% of total classified diversity, followed by Firmicutes (~ 13%) and Proteobacteria (~ 6%) respectively. Archaea, fungi and viruses made up the minor communities in the overall microbial diversity. Interestingly, this study also identified a range of potential enteric zoonotic pathogens (0.02–1.25%) and genes involved in antimicrobial resistance (0.02–0.7%) in feline fecal materials. Based on clustering among nine gastrointestinal metagenomes from five different monogastric hosts (dog, human, mice, cat and chicken), the cat metagenome clustered closely together with chicken in both phylogenetic and metabolic level (> 80%). Future studies are required to provide deeper understandings on both intrinsic and extrinsic effects such as impact of age, genetics and dietary interventions on the composition of the feline gastrointestinal microbiome.
Keywords: Feline intestinal microbiome, Metagenomics, Zoonotic pathogens, Pyrosequencing
Highlights
► We described the gene-centric metagenomics analysis of feline intestinal microbiota. ► Comparisons were made with available metagenomes from other species. ► Potential pathogens and antimicrobial resistance genes were identified.
1. Introduction
The gastrointestinal microbiota influences the health of the host, supports the immune system and provides competitive exclusion, which acts to enhance protection against intestinal pathogens (Falk et al., 1998, Hooper et al., 2001). Moreover, microorganisms and their ecology are important for most of the biochemical transformations in the environment. Dynamic interactions and overall composition of the microbial community in the gastrointestinal tract of animals and humans ultimately reflect the co-evolution or selection of microorganism with their host and the environments, and diets adopted by the host (Qu et al., 2008). Domestic cats are “obligate carnivores,” their diet is composed of primarily animal-based protein and supplemented by plant-based fibrous material diet. Unlike the case for other monogastric animals (e.g., rats, mice, pigs, and chickens), knowledge regarding the composition of the feline gastrointestinal microbiota is still very limited. Domestic cats are one of the most common companion animals and the close interactions between human and their cats are well known. Attention to the feline intestinal microbial community is important not only to animal health (Inness et al., 2007, Jergens, 2002, Johnston et al., 2001, Simpson, 1998) but also as a reservoir for potential zoonotic pathogens and antibiotic resistant bacterial strains which could be opportunistic risks to human health (Leener et al., 2005, Moyaert et al., 2006). In addition, the distribution of certain bacteria such as Bifidobacterium and Lactobacillus spp. is of particular interest due to the usage of these bacteria as probiotics in veterinary medicine (Ritchie et al., 2010).
Previous conventional culture techniques showed that facultative and obligate anaerobic bacteria were predominant in the feline intestine (Johnston et al., 1993, Johnston et al., 2001, Papasouliotis et al., 1998). From culture-independent 16S rRNA and cpn60 sequence analysis of pooled feline fecal samples, Firmicutes have been shown to be the most predominant bacterial group, followed by Proteobacteria, Bacteroidetes, Fusobacteria, and Actinobacteria respectively (Desai et al., 2009, Ritchie et al., 2010, Ritchie et al., 2008). Bacteroidetes and Firmicutes were the predominant microbial phyla in human gut as well (Eckburg et al., 2005, Gill et al., 2006), and a recent study also indicated a similar trend in dog fecal samples (Swanson et al., 2011). Similarly, Bacteroidetes were also highly represented in the metagenomes of chicken and turkey poult ceca (Qu et al., 2008, Scupham, 2007).
Besides the microbial diversity, association with beneficial microorganisms, enteric zoonotic agents in domestic cats are also a concern. Domestic cats in general are considered to pose only a small risk for carrying and transmitting enteric zoonotic agents, but cat owners are still at a higher risk for contracting zoonoses from their pets than individuals who do not reside with a companion animal (Angulo et al., 1994, Glaser et al., 1994). Due to the absence of more definitive risk estimations, some physicians recommend that immunosuppressive patients should not reside together with pets (Spencer, 1992). On the other hand, animal companionship offers psychological benefits particularly those who may be socially isolated because of illness e.g. people with acquired immunodeficiency syndrome (AIDS) (Carmack, 1991, Siegel, 1990). Salmonella, Campylobacter, and Cryptosporidium are potential enteric zoonosis pathogens in kittens because the consequences of human infection can be severe especially in immunocompromised individuals (Angulo and Swerdlow, 1995, Glaser et al., 1994). In addition, Toxoplasma gondii, a protozoan parasite causes clinician manifestations in cats and can also be transmitted to humans by different transmission routes causing serious effects on both human fetuses and immunocompromised patients (Dabritz and Conrad, 2008). The fecal shedding of aforementioned enteric zoonosis pathogens has been reported in kittens without showing any clinical signs (Spain et al., 2001). For these reasons, arguments on animal companionship for immunosuppressive patients continue. Therefore, enhancing our knowledge of the feline intestinal microbiome is important for both animal and human health. Although there were reports that the microbiota vary along the gastrointestinal tract (Ritchie et al., 2008, Suchodolski et al., 2005), fecal samples are readily available for clinical studies, and continued efforts to characterize the domestic feline fecal microbiota is ecologically important. To our knowledge, this study is the primary endeavor to provide a more comprehensive overview of the feline gastrointestinal metagenome using pyrosequencing.
2. Materials and methods
2.1. Fecal sample collection
Fresh fecal samples were collected from five house-hold cats (n = 5) including four domestic shorthaired (DSH) breed and one domestic longhaired (DLH) breed. All cats were from Soares Avenue Paws and Claws Veterinary Clinic's client population in Hong Kong SAR. Sampled cats live in indoor home environments in different locations across Hong Kong and were fed with commercially available cat diets (Royal Canine Inc., St. Charles, MO, USA) for at least a year before sampling. The cats were clinically healthy with no history of antibiotic therapy 4 months prior to the sample collection. The age of cats ranged from 3 to 16 years (median of 8 years), while body condition scores ranged from 4 to 7 (median 5) on a 9-point scale. All were vaccinated against feline infectious diseases (feline rhinotracheitis, calicivirus, and panleukopenia). Fresh fecal samples (within ~ 20 min of defecation) were collected aseptically and transported to the laboratory on dry ice, then stored at − 80 °C until DNA extraction.
2.2. DNA extraction and shotgun pyrosequencing
Genomic DNA was extracted from fecal samples using the method of Yu and Morrison (2004) with slight modification. Briefly, 0.4 g of sterile glass beads (425–600 microns; SIGMA-ALDRICH CHEMIE Gmbh, Steinheim, Germany) were added to each fecal sample (~ 250 mg) and vortexed (full speed, 3 min) to facilitate the breaking of the fecal mass. The aggressive beads beater steps were skipped to reduce DNA shearing. Subsequently, 1 ml of lysis buffer containing 500 mM Tris–HCl, pH 8.0, 50 mM EDTA and 4% sodium dodecyl sulfate (SDS) was applied to each sample. 2 μl of RNase and 15 μl of proteinase K were also applied together with QIAamp DNA stool mini kit (Qiagen GmBH, Hilden, Germany) for removal of RNA, protein, and purification. After extraction, quality and quantity of the DNAs were determined using a nanodrop (ND-2000) spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). DNA from each of all five samples was pooled on an equimolar basis. Pooled fecal DNA samples were subjected to shotgun pyrosequencing by 454 GS Junior using titanium chemistry (454 Life Sciences-a Roche Company, Branford, CT, USA).
2.3. Bioinformatics and statistical analysis
Sequence reads derived from 454 GS Junior pyrosequencer were trimmed and a de novo assembly was performed by Newbler Assembler software (Margulies et al., 2005). Both unassembled reads and assembled contigs were analyzed separately by available metagenomics analysis software. For unassembled analyses, both sequence data and quality data were submitted to Galaxy metagenomics analysis pipeline to generate taxonomic phylogeny (Kosakovsky Pond et al., 2009). In addition, data were also submitted to MG-RAST (Glass et al., 2010) and WebCARMA (Gerlach et al., 2009). Assembled contigs were analyzed in MG-RAST, Galaxy and NBC (the naïve Bayes classification tool webserver) (Rosen et al., 2011). For statistical analysis, both phylogenetic and metabolic clustering were performed with eight public-assembled data sets of monogastric animals from MG-RAST including chicken cecum A contigs (CCA 4440285) and B contigs (CCB), two dog metagenome data sets (K9C 4444164 and K9BP 4444165), lean mouse cecum (LMC 4440463.3), obese mouse cecum (OMC 4440464), human stool metagenome (HSM 4444130) and human F1-S feces metagenomes (F1S 4440939). For parameter settings in analysis, a maximum e-value of 0.01, minimum percent identity of 50, minimum alignment length of 50 and a raw score maximum of 0.3 were applied. Clustering was performed by Ward's minimum variance using unscaled Manhattan distances (Kaufman and Rousseeuw, 1990). Heat maps were drawn by hierarchal clustering performed in NCSS/GESS 2007. Non-metric multidimensional scaling analysis based on Bray–Curtis' similarity index were performed using PRIMER 6 statistical software (PRIMER E Ltd., Plymouth Marine Laboratory, Plymouth, U.K.).
3. Results and discussion
Next generation sequencing technologies have recently been used to characterize the microbial diversity and functional capacity of a range of microbial communities in the gastrointestinal tracts of several mammalian species (Eckburg et al., 2005, Gill et al., 2006, Guan et al., 2007, Qu et al., 2008, Swanson et al., 2010). Recently, the newly introduced bench-top next generation sequencing (NGS) machine, 454 GS Junior system allows individual research laboratory access to high through-put sequencing technology. Similar to the parental 454 GS systems, the 454 GS Junior system enables a comprehensive view into the diversity and metabolic profile of microbial communities of complex samples. Analysis of the reads can yield a high percentage of species identification in complex metagenomes and even higher in less complex samples. Long sequence reads from 454 GS Junior pyrosequencing can also provide the enormous specificity needed to compare sequenced reads against DNA or protein databases and give unambiguous assignment of even closely related species. 454 GS junior pyrosequencing generated a total of 152,494 sequences with average read length of 405.24 bp. De novo assembly of our feline metagenome sequences gave 5405 contigs with 83 contigs > 2 kb and 2659 contigs > 500 bp. The average contig length was 656.19 bp while the longest contig was 5231 bp and the shortest was 100 bp. The sequences have been submitted to NCBI Gene Bank with genome project ID 62073 under the cat fecal metagenome (CFM) project, and also available within a short read archive under the accession number SRA029158.2.
3.1. Phylogenetic profiles of microbial diversity
A total of 78.3% of contigs were matched to the SEED database using an e-value of 1e-5. There were 4232 hits against the non-redundant protein database. The overview of phylogenetic computations (based on assembled data) provided 97.78% bacteria, 1.23% Eukaryota, 0.9% Archaea and 0.09% Viruses. In this feline intestinal metagenome, the Bacteroides/Chlorobi group was the most predominant phylum ~ 68%, followed by Firmicutes ~ 13% and Proteobacteria ~ 6% (Table 1 ), and a similar trend was demonstrated by WebCARMA analysis (Supplementary Fig. 1). Galaxy's taxonomy also provided the same pattern of phyla distribution (Supplementary Fig. 2). Our findings differ from those in recent studies involving 16S rRNA pyrosequencing (Handl et al., 2011) and gene clone library analysis of healthy feline fecal samples by universal bacterial primers. In these studies, Firmicutes were reported as the most abundant phylum (87.3%), followed by Proteobacteria (7.9%), Bacteroidetes (2.4%), Actinobacteria (2.3%), and Fusobacteria (0.2%) (Ritchie et al., 2010). Within Bacteroidetes/Chlorobi group, Bacteroidales were the most predominant including Bacteroides vulgatus, Bacteroides thetaiotaomicron, Bacteroides fraglis, Porphyromonas gingivalis and Bacteroides ruminocola. Firmicutes were the second predominant phylum in feline gastrointestinal tract (Supplementary Fig. 1) with Clostridia being the primary contributor to the Firmicutes populations followed by Bacilli, Mollicutes and Lactobacillales. In comparison with previous 16S rRNA gene-based data (Ritchie et al., 2010), there were significantly lower percentages of Firmicutes in this feline intestinal metagenome. Reasons for variation are difficult to identify and not obvious, biases involved in selection of clones, sequencing depth and primer biases, etc., may contribute to this divergence. It also has been shown that different regions of 16S rRNA gene reveal different diversity, and therefore a certain region may serve well for profiling a certain spectrum of bacteria but not all. Comparison between 16S rRNA profiling and metagenomics profiling did not make sense, and ideally we could only do the comparison using only the 16S rRNA gene that cover the same region. Although microbial diversity analysis using 16S rRNA gene-based pyrosequencing analysis is a more cost-effective method for microbiome characterization 16S rRNA gene based data, a shotgun metagenomics approach as used here is a more ideal approach to characterize the unbiased bacterial phyla diversity in feline gastrointestinal tract.
Table 1.
Bacterial phylum profiles in feline intestinal metagenome.
| Bacterial phylum | % Composition in CFM |
|---|---|
| (No. of contigs in parenthesis) | |
| Bacteroidetes/Chlorobi group | 67.54% (2795) |
| Firmicutes | 12.98% (537) |
| Bacteroidetes | 8.68% (359) |
| Proteobacteria | 5.85% (242) |
| Actinobacteria | 1.16% (48) |
| Fusobacteria | 0.68% (28) |
| Synergistetes | 0.58% (24) |
| Cyanobacteria | 0.51% (21) |
| Thermotogae | 0.48% (20) |
| Spirochaetes | 0.41% (17) |
| Chlamydiae/Verrucomicrobia group | 0.34% (14) |
| Fibrobacteres/Acidobacteria group | 0.27% (11) |
| Deinococcus-Thermus | 0.27% (11) |
| Chloroflexi | 0.14% (6) |
| Planctomycetes | 0.05% (2) |
| Aquificae | 0.02% (1) |
| Chlorobi | 0.02% (1) |
| Unclassified bacteria | 0.02% (1) |
However, both approaches have their limitations in which 16S rRNA sequencing may be biased due to unequal amplification among 16S rRNA genes of different bacterial spices, whereas shotgun metagenomic sequencing may not be deep enough to detect the rare species and only the most abundant species are usually detected. The shotgun approach may able to detect the rare species when deeper metagenomic sequencing becomes available. On the other hand, shotgun metagenomic sequencing gives more precise information in species level of microbial community, when 16S rRNA sequencing often provides information on genus level. Moreover, the shotgun metagenomic approach could provide us additional information of other microbes (archaea, fungi, etc.) as well as the functional roles of microbiome in their ecological niches.
As both dogs and cats are common companion animals, it is noteworthy to provide comparison between these species. Although both domestic cats and dogs are members of Carnivora, the intestinal microbial diversity in cats showed only minor differences from those reported for dogs. The predominant phylum of dogs, humans and mice are Fusobacteria and that dogs have a relatively greater percentage of Firmicutes (Kurokawa et al., 2007, Swanson et al., 2010, Turnbaugh et al., 2006) than we observed in cats.
Recent studies have demonstrated that dietary components affect intestinal microbiota of cats (Lubbs et al., 2009), and also reported by the experiment done in mice, rats and human (Lubbs et al., 2009, Turnbaugh et al., 2009). In this study, we only demonstrate a comprehensive microbial diversity in a small population of normal feline gastrointestinal tracts and future efforts focused on characterizing the changes of microbiome in respect to dietary components are warranted.
In our feline metagenome, Eukaryota was a minor constituent (1.23%) after the Bacteria. Very low abundant fungi sequences (0.02%) were identified in this study. Dikarya was the classification for all the fungi sequences in which Gibberella zeae PH-1 was only distinct fungi species. Interestingly, there were more diverse phyla of fungi species in dog metagenomes (K9C and K9BP) (Swanson et al., 2011) as well as in mouse (OMC) in which two more species such as Neurospora crassa and Saccharomyces cerevisiae were identified (Turnbaugh et al., 2006). The biases were noted in the study of human distal gut fungi using both culture-dependent and culture-independent methods. In that study, culture-independent methods based upon molecular techniques demonstrated more diverse fungi species (Scanlan and Marchesi, 2008). Candida, Cladosporium, Penicillum and Saccharomyces have also been identified in stool samples from patients with human inflammatory bowel disease and from healthy controls (Ott et al., 2008). Our study demonstrated low percentage of fungi with least diversity among overall fecal microflora. The fungal diversity in the feline gastrointestinal tract has not yet been studied, and no doubt that the next generation sequencing should be used in future studies.
Archaea also represented as a minor component of the feline metagenome, comprising ~ 0.09% of total sequences in which Euryarchaeota, Crenarchaeota and Korarcheota diverged into nine orders (Supplementary Table 1). Among these groups of archaea, the methanogenic archaea were the most predominant and diverse in the feline metagenome. Methanogenic archaea were also the most abundant group in dogs, human, mice and chicken (Kurokawa et al., 2007, Qu et al., 2008, Swanson et al., 2010, Turnbaugh et al., 2006). In the cat fecal Metagenome (CFM), Crenarchaeota were the lowest proportion of archaea, whereas they were more abundant in other metagenomes (Fig. 1 ). In the gastrointestinal tract of ruminants, archaea are commensal organisms and have also been described in the intestine of monogastric hosts including human in which Methanobacteriales are the most common group (Eckburg et al., 2005). Although the diversity of archaea have been described in dogs previously (Swanson et al., 2010), the diversity of this domain in cats have not yet been elucidated. Despite archaea playing a commensal role in the intestine, the interaction with other microorganisms may also contribute to pathogenicity in humans. Methanogens have been associated with various pathologies in human such as periodontitis, endodontitis, colon cancer and diverticulosis (Conway de Macario and Macario, 2009). Therefore, it is also important for future efforts to further characterize the actual prevalence and clinical importance of archaea in cats.
Fig. 1.
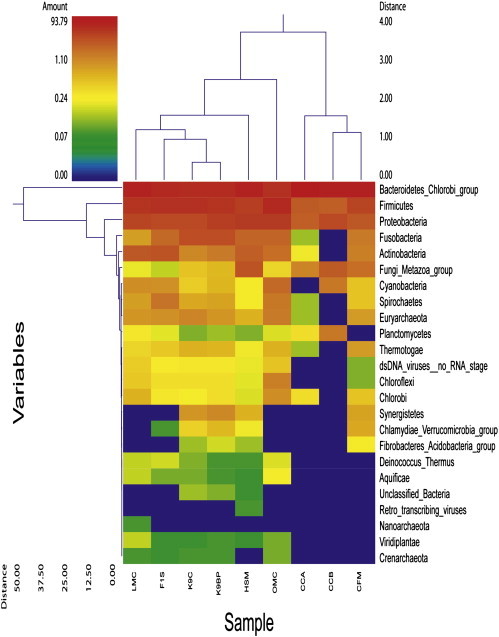
Phylogenetic clustering among feline, canine, human, mouse and chicken gastrointestinal metagenomes. A double hierarchical dendogram was performed using weight-pair group clustering method based on non-scaling Manhattan distance. It shows phylogenetic prevalence of microorganisms among nine metagenomes from five different hosts including feline (CFM), canine (K9C and K9BP), human (F1S and HSM), murine (LMC and OMC) and chicken (CCA and CCB). The linkages of the dendogram are not showing phylogenetic relationship of the bacterial classes, but it based on relative abundance of taxonomic profiles. The heat map color represents the relative percentage of the microbial descriptions within each sample, with the legend indicated at the upper left corner. Branch length indicate Manhattan distances of the samples along the x axis (scale at the upper right corner) and of the microbial classes along the y axis (scale at the lower left corner).
Only ~ 0.09% of total contigs were identified as viral, with only the order Caudovirales being present, represented by Staphylococcus phage Twort. Cats can be infected by several viruses such as rotavirus, coronavirus, parvovirus, influenza virus and feline immunodeficiency virus (Kapil and Lamm, 2008). Recent viral metagenomics studies also showed the diversity of viruses in human gastrointestinal tract with several phylotypes (Breitbart et al., 2003). Domestic cats and dogs are the most common pets and live in close proximity to humans in daily life. Therefore, studies on the viral community in companion animals and the potential for cross-transmission between human and pets are needed. Due to the methodology (shotgun DNA pyrosequencing approach) the current study only allowed the evaluation of dsDNA viruses. In the future, surveillance of RNA and ssDNA viruses in cats should be performed in order to improve knowledge on diversity and ecology of intestinal viruses.
3.2. Metabolic profiles of feline metagenome
Metagenomics based metabolic profiling in feline has not been studied until present study provides the characterization of the metabolic profiles of gastrointestinal microbiota in healthy cats. About half (46.51%) of total contigs were characterized as functional categories in normal healthy cats and summarized data (based on assembled analysis) are shown in Table 2 (MG-RAST) and Supplementary Fig. 3 (WebCARMA). As in canine metagenome, carbohydrates; protein metabolism; cell wall and capsule; cofactors, vitamins, prosthetic groups and pigments; DNA metabolism; RNA metabolism; amino acids and derivatives; and virulence are the most represented functional categories.
Table 2.
Metabolic profiles of feline intestinal microbiome.
| Functional metabolic category | % Composition in CFM |
|---|---|
| (no. of contigs in parenthesis) | |
| Cofactors, vitamins, prosthetic groups and pigments | 5.73% (144) |
| Cell wall and capsule | 8.95% (225) |
| Potassium metabolism | 0.44% (11) |
| Miscellaneous | 0.56% (14) |
| Membrane transport | 1.79% (45) |
| RNA metabolism | 5.77% (145) |
| Protein metabolism | 8.99% (226) |
| Nucleosides and nucleotides | 3.82% (96) |
| Cell division and cell cycle | 1.95% (49) |
| Motility and chemotaxis | 0.56% (14) |
| Regulation and cell signaling | 1.47% (37) |
| DNA metabolism | 7.84% (197) |
| Prophage | 0.04% (1) |
| Unclassified | 3.54% (89) |
| Virulence | 6.88% (173) |
| Nitrogen metabolism | 0.32% (8) |
| Clustering-based subsystems | 14.36% (361) |
| Respiration | 2.47% (62) |
| Stress response | 1.59% (40) |
| Metabolism of aromatic compounds | 0.24% (6) |
| Amino Acids and derivatives | 6.13% (154) |
| Fatty acids and lipids | 0.76% (19) |
| Sulfur Metabolism | 0.88% (22) |
| Phosphorus metabolism | 1.63% (41) |
| Carbohydrates | 13.33% (335) |
According to the percentage of sequences in metabolic profiling of feline metagenome, microbial carbohydrate and protein metabolism (13.33% and 8.99% respectively) were similar to the previous canine metagenome (Swanson et al., 2011). Gene involved with biosynthesis of folate (0.99%), coenzyme B12 (0.44%), biotin (0.08%), vitamin B6 (0.16%), thiamin (0.48%), riboflavin (0.44%) and menaquinones and phylloquinones (0.48%) were predominant in the cofactors, vitamins, prosthetic groups and pigments subsystem in this study. The data set also includes the sequences associated with the biosynthesis of nicotinamide adenine dinucleotide (NAD) and NADP (0.44%), both of which function as hydride acceptors and are important in biochemical redox reactions.
Genes involved in DNA metabolism of feline metagenome is similar with canine metagenome, however, genes associated with RNA metabolism of feline (5.77%) is apparently higher than those in canine. When analyzing solely at the feline gastrointestinal metagenome and the SEED Virulence Subsystem, there were a variety of genes associated with resistance to antibiotics and toxic compounds. Details on the presence of antibiotic resistance genes will be discussed in the next section. Other resistance-related genes having the greatest sequence number were those associated with acriflavine (0.52%), cobalt-zinc-cadmium (0.36%) and zinc (0.24%). Acriflavine is a common antiseptic agent and its resistance is thought to be due to an overexpression of efflux pump genes. In the cell wall and capsule subsystem, genes associated with biosynthesis of peptidoglycan (1.35%), KDO2-Lipid A (0.72%) and LOS core oligosaccharides (0.32%), rhamnose containing glycans (0.56%) and sialic acid metabolism (0.76%) also had a high prevalence.
Three functional categories (dormancy and sporulation, macromolecular synthesis and secondary metabolism) were absent in our feline metagenome by assembled data analysis. However, we can identify least abundance of those functional genes (0.01%, 0.03% and 0.04% respectively) in unassembled data analysis. Microbial secondary metabolites contain antibiotics, pigments, toxins, effectors of ecological competition and symbiosis, pheromones, enzyme inhibitors, immunomodulating agents, receptor antagonists and agonists, pesticides, antitumor agents and growth promoters of animals and plants. Such metabolites have a major effect on the health, nutrition and economics of our society (Demain, 1992). The factors contributing to these profiles are still uncertain but their formation is certainly affected by factors including nutrients, growth rate, feedback control, enzyme inactivation, and enzyme induction (Demain, 1998). The other two functional categories (dormancy and sporulation, and macromolecular synthesis) are also important in microbial growth and microbe–microbe interactions (Dworkin and Shah, 2010). Our present results least abundant of dormancy and sporulation in feline gastrointestinal microbiota indicating that either most of the bacteria in fresh fecal sample are vegetative cells or the lack of a bead beating step in our DNA processing may have reduced the contribution of spore forming bacteria. Also the assembly of the sequencing reads and sequencing depth are other possibilities to exclude those least abundant genes. The least abundant of these functional genes in our feline metagenome is interesting and deserves future experimentation.
3.3. Opportunistic zoonotic agents
In addition to the DNA profiling, the findings of the enteric zoonotic agents in domestic cats are also important. Domestic cats are generally believed to pose a small risk for carrying and transmitting enteric zoonotic agents, however, litter boxes may be a possible source of zoonotic infections due to everyday human contacts. As noted previously it has been recommended by some physicians that immunosuppressive patients should not live with pets (Spencer, 1992), yet animal companionship also provides psychological benefits particularly for socially isolated patients (Carmack, 1991). The current gene-centric metagenomics study adds further understanding on the potential risks of domestic cats to human health. From our analysis, we identified 12 genera of enteric zoonotic bacteria composed of 41 species in the feline fecal microbiome (Table 3 ). Most of these organisms are primarily considered opportunistic foodborne pathogens which were in relatively low abundance (< 0.1%) in relation to the rest of the bacterial community. The identification of zoonotic pathogens showed that the abundance of enteric zoonotic bacteria in domestic cats certainly highlight the potential for transmission of opportunistic infections to human. Further study is needed to determine the prevalence and importance of such pathogens in more diverse cat populations.
Table 3.
Phylogenetic classification of opportunistic enteric zoonotic pathogens in the feline metagenome sample.
| Genus | Species | ~% in CFM |
|---|---|---|
| Streptococcus | Streptococcus thermophilus | 0.05% |
| Streptococcus agalactiae | 0.07% | |
| Streptococcus pneumonia | 0.09% | |
| Streptococcus pyogens | 0.4% | |
| Streptococcus uberis | 0.02% | |
| Streptococcus equi subsp. zooepidemicus | 0.07% | |
| Streptococcus mitis | 0.02% | |
| Streptococcus sanguinis | 0.05% | |
| Streptococcus suis | 0.07% | |
| Staphylococcus | Staphylococcus aureus | 0.07% |
| Staphylococcus warneri | 0.02% | |
| Staphylococcus sciuri | 0.05% | |
| Staphylococcus epidemidis | 0.05% | |
| Staphylococcus haemolyticus | 0.02% | |
| Bacillus | Bacillus anthracis | 0.05% |
| Bacillus cereus | 0.21% | |
| Listeria | Listeria monocytogenes | 0.09% |
| Clostridium | Clostridium tetani | 0.24% |
| Clostridium botulinum | 0.5% | |
| Clostridium perfringens | 0.35% | |
| Clostridium difficile | 1.25% | |
| Salmonella | Salmonella enteric subsp. Enterica serovar Thyphi | 0.02% |
| Salmonella enteric subsp. Enterica serovar Gallinarum | 0.07% | |
| Salmonella enteric subsp. Enterica serovar Choleraesuis | 0.05% | |
| Escherichia coli | Escherichia coli O157.H7 | 0.02% |
| Escherichia coli E110019 | 0.02% | |
| Yersinia | Yersinia enterocolitica | 0.02% |
| Yersinia pseudotuberculosis | 0.02% | |
| Shigella | Shigella flexneri | 0.07% |
| Shigella sonnei | 0.02% | |
| Shigella boydi | 0.02% | |
| Vibrio | Vibrio vulnificus | 0.02% |
| Vibrio cholera | 0.19% | |
| Vibrio spp. | 0.05% | |
| Campylobacter | Campylobacter coli | 0.05% |
| Campylobacter jejuni subsp. jejuni | 0.02% | |
| Campylobacter upsaliensis | 0.02% | |
| Campylobacter fetus subsp. fetus | 0.02% | |
| Helicobacter | Helicobacter hepaticus | 0.02% |
| Helicobacter pylori | 0.02% | |
| Helicobacter acinonychis | 0.02% |
All over recent years, there has been a concern about the increasing prevalence of antimicrobial resistance in human as well as in animal medicine. It is generally accepted that the increased use of antimicrobial agents led to the emergence and spread of resistant bacteria and/or their resistant genes. In addition, several studies suggested a possible exchange of resistant organisms and/or their resistance genes between pet animals and humans (Guardabassi et al., 2004, Simjee et al., 2002). The current functional gene profiling also documents the existing antimicrobial resistance genes in feline gastrointestinal tract (Table 4 ). Multidrug resistance efflux pumps, fluroquinolones resistance and beta-lactamase are major predominant antimicrobial resistance genes. Previous studies also provided data on antimicrobial resistance in the intestinal microbiota of cats (Moyaert et al., 2006). However, these cultural based studies relied on the resistance level of indicator bacteria. Our study provides further the molecular data on the antibiotic resistant genes present in feline gastrointestinal microbiota. Due to the presence of enteric zoonotic bacteria and antibiotic resistance genes in cat fecal materials, litter boxes certainly represent potential sources of infection between cats and humans. Although healthy individuals are at low risks of getting zoonotic infections from domestic cats, immunocompromised people may be of particular concern.
Table 4.
Identification of antimicrobial resistant genes in the feline metagenome sample.
| Antibiotic-resistant genes | ~% in CFM |
|---|---|
| Methicillin resistance in Staphylococci | 0.02% |
| Multidrug resistance tripartite systems found in gram negative bacteria | 0.05% |
| Multidrug resistance 2-protein version found in gram positive bacteria | 0.02% |
| Multidrug efflux pump in Camphylobacter jejuni | 0.02% |
| Intergrons | 0.02% |
| Tetracycline resistance ribosome protection type | 0.14% |
| Fluroquinolones resistance | 0.24% |
| Beta-lactamase | 0.24% |
| Multidrug resistance efflux pumps | 0.64% |
3.4. Comparative metagenomics analysis
The results of this study were compared with eight publicly available metagenome data sets from four different species within MG-RAST. Among the eight data sets, two chicken intestinal metagenomes including chicken cecum contigs A (CCA) and contigs B (CCB) (Qu et al., 2008), two mouse intestinal metagenomes which included lean (LMC) and obese (OMC) mouse cecum metagenomes (Turnbaugh et al., 2006), two dog fecal metagenomes (K9C and K9BP) (Swanson et al., 2010) and two human fecal metagenomes (F1S and HSM) (Kurokawa et al., 2007) were included to compare with the current feline fecal metagenome (CFM). HSM was defined as a human fecal sample from a malnourished person, while CCB was the cecum sample from Camphylobacter jejuni NCTC11168 challenged chicken. The comprehensive overviews of all nine data sets are shown in Supplementary Table 2. Clustering among those metagenomes was performed using unscaled Manhattan variance distances and presented by a double hierarchical dendogram. The clustering based comparisons were demonstrated at the phylogenetic level (Fig. 1) as well as the metabolic level (Fig. 2 ). Hierarchical clustering demonstrated that our cat sample clustered together with two chicken cecum metagenomes (CCA and CCB) and separated from other mammalian samples including dogs, humans and mice. The Bacteroidetes/Chlorobi group, Firmicutes and Proteobacteria were the most abundant phyla in all metagenomic samples. The heat map also demonstrated that Fusobacteria and Actinobacteria are more predominant in CFM rather than that of both CCA and CCB. Fungi and metazoa groups are also found to be predominated in CFM, CCA, CCB and HSM. Non-metric MDS plot illustrated that the similarity among CFM, CCA and CCB as well as among F1S, K9C, K9BP and LMC are more than 80%, whereas all metagenomics data have maximum of 60–80% similarity at the phylogenetic level (Fig. 3A). The clustering of cat metagenome with that of chicken may be due to similar bacterial diversity influenced by similar dietary components. However, the distances among all metagenomes were not distinct.
Fig. 2.
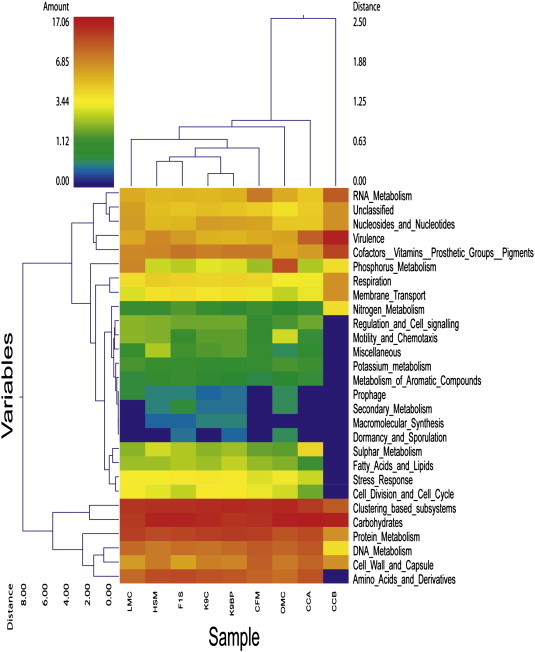
Metabolic clustering among feline, canine, human, mouse and chicken gastrointestinal metagenomes. A double hierarchical dendogram was performed using weight-pair group clustering method based on non-scaling Manhattan distance. It shows distribution of functional categories among nine metagenomes from five different hosts including feline (CFM), canine (K9C and K9BP), human (F1S and HSM), murine (LMC and OMC) and chicken (CCA and CCB). The linkages of the dendogram are based on relative abundance of metabolic profiles. The heat map color represent the relative percentage of functional categories within each sample, with the legend indicated at the upper left corner. Branch length indicate Manhattan distances of the samples along the x axis (scale at the upper right corner) and of the functional categories along the y axis (scale at the lower left corner).
Fig. 3.
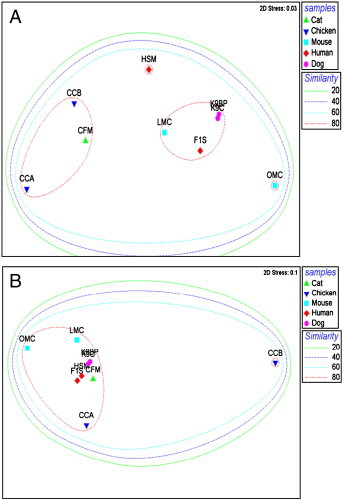
Non-metric multidimensional analysis based on relative abundance of (A) taxonomic profiles and (B) metabolic profiles among feline, canine, human, mouse and chicken gastrointestinal metagenomes. Samples of the same host species are indicated by the same symbol. Superimposed circles represent clusters of samples at different similarity values of 20%, 40%, 60% and 80% (Bray–Curtis similarity).
Metabolic based hierarchical clustering demonstrated that cat, dog and human samples were clustered together due to their similar host system whereas chicken cecum and mice samples were least similar to cats (Fig. 2). These findings are also comparable to the previous dog metagenome study. Non-metric MDS plot also illustrated that all metagenomics data have more than 80% similarity except CCB which was from infected chicken (Fig. 3B). The extent of functional capacity changes in infected animals should be of interest and warrant further experimentation.
In summary, this study is the first metagenomics approach using shotgun pyrosequencing to characterize a more comprehensive overview of phylogenetic and functional capacity of the feline intestinal microbiome. Metagenomics sequencing of the community DNA extracted from cat feces revealed the most dominant microbes of feline intestinal microbiome are bacteria (especially Bacteroides/ Chlorobi group) while archaea, fungi and viruses are minor constituents. According to the similarity and distances based clustering among nine metagenomics data sets from five different hosts, feline metagenome clustered closely together with chicken at the phylogenetic contribution (> 80% relative similarity), and at the metabolic level, all metagenomes were clustered together (> 80%) except CCB which presented a notable out group. Our feline fecal metagenomics study also demonstrated the diverse species of enteric zoonotic pathogens as well as antimicrobial resistant genes in cat feces. Together, these data will serve to improve our understanding of potential public health risks from companion cats. Further studies involving deeper sequencing of the metagenomes are required to characterize the gastrointestinal microbiome of populations of individual cats in healthy and disease states, of ages and breeds variations, and dietary interventions.
Conflict of interests
The authors declare that they have no competing interests.
The following are the supplementary materials related to this article.
Bacterial phylum counts in cat fecal metagenome (CFM) determined using WebCARMA (http://webcarma.cebitec.uni-bielefeld.de/cgi-bin/webcarma.cgi).
A class-level phylogenetic profile of feline fecal metagenome derived from Galaxy metagenomic pipeline (http://main.g2.bx.psu.edu/screencast).
Functional profile of feline metagenome determined using WebCARMA (http://webcarma.cebitec.uni-bielefeld.de/cgi-bin/webcarma.cgi).
Phylogenetic classification of Archaea in Feline Metagenome.
Overview of the MG-RAST metagenomes chosen for comparison.
Acknowledgements
We would like to thank volunteer pet owners, veterinary assistants and nurses from Soares Avenue Paws and Claws Veterinary Clinic for sampling. We also thank Dr. Kelly S Swanson for providing canine metagenomics data.
References
- Angulo F.J., Swerdlow D.L. Bacterial enteric infections in persons infected with human immunodeficiency virus. Clin. Infect. Dis. 1995;21(Suppl. 1):S84–S93. doi: 10.1093/clinids/21.supplement_1.s84. [DOI] [PubMed] [Google Scholar]
- Angulo F.J., Glaser C.A., Juranek D.D., Lappin M.R., Regnery R.L. Caring for pets of immunocompromised persons. J. Am. Vet. Med. Assoc. 1994;205:1711–1718. [PubMed] [Google Scholar]
- Breitbart M., Hewson I., Felts B., Mahaffy J.M., Nulton J., Salamon P., Rohwer F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003;185:6220–6223. doi: 10.1128/JB.185.20.6220-6223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmack B.J. The role of companion animals for persons with AIDS/HIV. Holist. Nurs. Pract. 1991;5:24–31. doi: 10.1097/00004650-199101000-00007. [DOI] [PubMed] [Google Scholar]
- Conway de Macario E., Macario A.J. Methanogenic archaea in health and disease: a novel paradigm of microbial pathogenesis. Int. J. Med. Microbiol. 2009;299:99–108. doi: 10.1016/j.ijmm.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Dabritz H.A., Conrad P.A. Cats and Toxoplasma: implications for public health. Zoonoses Public Health. 2008;57:34–52. doi: 10.1111/j.1863-2378.2009.01273.x. [DOI] [PubMed] [Google Scholar]
- Demain A.L. Microbial secondary metabolism: a new theoretical frontier for academia, a new opportunity for industry. Ciba Found. Symp. 1992;171:3–16. doi: 10.1002/9780470514344.ch2. (discussion 16-23) [DOI] [PubMed] [Google Scholar]
- Demain A.L. Induction of microbial secondary metabolism. Int. Microbiol. 1998;1:259–264. [PubMed] [Google Scholar]
- Desai A.R., Musil K.M., Carr A.P., Hill J.E. Characterization and quantification of feline fecal microbiota using cpn60 sequence-based methods and investigation of animal-to-animal variation in microbial population structure. Vet. Microbiol. 2009;137:120–128. doi: 10.1016/j.vetmic.2008.12.019. [DOI] [PubMed] [Google Scholar]
- Dworkin J., Shah I.M. Exit from dormancy in microbial organisms. Nat. Rev. Microbiol. 2010;8:890–896. doi: 10.1038/nrmicro2453. [DOI] [PubMed] [Google Scholar]
- Eckburg P.B., Bik E.M., Bernstein C.N., Purdom E., Dethlefsen L., Sargent M., Gill S.R., Nelson K.E., Relman D.A. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk P.G., Hooper L.V., Midtvedt T., Gordon J.I. Creating and maintaining the gastrointestinal ecosystem: what we know and need to know from gnotobiology. Microbiol. Mol. Biol. Rev. 1998;62:1157–1170. doi: 10.1128/mmbr.62.4.1157-1170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach W., Junemann S., Tille F., Goesmann A., Stoye J. WebCARMA: a web application for the functional and taxonomic classification of unassembled metagenomic reads. BMC Bioinformatics. 2009;10:430. doi: 10.1186/1471-2105-10-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S.R., Pop M., Deboy R.T., Eckburg P.B., Turnbaugh P.J., Samuel B.S., Gordon J.I., Relman D.A., Fraser-Liggett C.M., Nelson K.E. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser C.A., Angulo F.J., Rooney J.A. Animal-associated opportunistic infections among persons infected with the human immunodeficiency virus. Clin. Infect. Dis. 1994;18:14–24. doi: 10.1093/clinids/18.1.14. [DOI] [PubMed] [Google Scholar]
- Glass E.M., Wilkening J., Wilke A., Antonopoulos D., Meyer F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb. Protoc. 2010 doi: 10.1101/pdb.prot5368. pdb prot5368. [DOI] [PubMed] [Google Scholar]
- Guan C., Ju J., Borlee B.R., Williamson L.L., Shen B., Raffa K.F., Handelsman J. Signal mimics derived from a metagenomic analysis of the gypsy moth gut microbiota. Appl. Environ. Microbiol. 2007;73:3669–3676. doi: 10.1128/AEM.02617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardabassi L., Schwarz S., Lloyd D.H. Pet animals as reservoirs of antimicrobial-resistant bacteria. J. Antimicrob. Chemother. 2004;54:321–332. doi: 10.1093/jac/dkh332. [DOI] [PubMed] [Google Scholar]
- Handl S., Dowd S.E., Garcia-Mazcorro J.F., Steiner J.M., Suchodolski J.S. Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiol. Ecol. 2011;76:301–310. doi: 10.1111/j.1574-6941.2011.01058.x. [DOI] [PubMed] [Google Scholar]
- Hooper L.V., Wong M.H., Thelin A., Hansson L., Falk P.G., Gordon J.I. Molecular analysis of commensal host–microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- Inness V.L., McCartney A.L., Khoo C., Gross K.L., Gibson G.R. Molecular characterisation of the gut microflora of healthy and inflammatory bowel disease cats using fluorescence in situ hybridisation with special reference to Desulfovibrio spp. J. Anim. Physiol. Anim. Nutr. (Berl) 2007;91:48–53. doi: 10.1111/j.1439-0396.2006.00640.x. [DOI] [PubMed] [Google Scholar]
- Jergens A.E. Feline inflammatory bowel disease—current perspectives on etiopathogenesis and therapy. J. Feline Med. Surg. 2002;4:175–178. doi: 10.1053/jfms.2002.0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston K., Lamport A., Batt R.M. An unexpected bacterial flora in the proximal small intestine of normal cats. Vet. Rec. 1993;132:362–363. doi: 10.1136/vr.132.14.362. [DOI] [PubMed] [Google Scholar]
- Johnston K.L., Swift N.C., Forster-van Hijfte M., Rutgers H.C., Lamport A., Ballevre O., Batt R.M. Comparison of the bacterial flora of the duodenum in healthy cats and cats with signs of gastrointestinal tract disease. J. Am. Vet. Med. Assoc. 2001;218:48–51. doi: 10.2460/javma.2001.218.48. [DOI] [PubMed] [Google Scholar]
- Kapil S., Lamm C.G. Emerging and reemerging viruses of dogs and cats. Preface. Vet. Clin. North Am. Small Anim. Pract. 2008;38:xiii-xiv. doi: 10.1016/j.cvsm.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Kaufman L., Rousseeuw P.J. John Wiley & Sons, Inc.; New York, NY, USA: 1990. Finding Groups in Data: An Introduction to Cluster Analysis. [Google Scholar]
- Kosakovsky Pond S., Wadhawan S., Chiaromonte F., Ananda G., Chung W.Y., Taylor J., Nekrutenko A. Windshield splatter analysis with the Galaxy metagenomic pipeline. Genome Res. 2009;19:2144–2153. doi: 10.1101/gr.094508.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K., Itoh T., Kuwahara T., Oshima K., Toh H., Toyoda A., Takami H., Morita H., Sharma V.K., Srivastava T.P., Taylor T.D., Noguchi H., Mori H., Ogura Y., Ehrlich D.S., Itoh K., Takagi T., Sakaki Y., Hayashi T., Hattori M. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leener E.D., Decostere A., De Graef E.M., Moyaert H., Haesebrouck F. Presence and mechanism of antimicrobial resistance among enterococci from cats and dogs. Microb. Drug Resist. 2005;11:395–403. doi: 10.1089/mdr.2005.11.395. [DOI] [PubMed] [Google Scholar]
- Lubbs D.C., Vester B.M., Fastinger N.D., Swanson K.S. Dietary protein concentration affects intestinal microbiota of adult cats: a study using DGGE and qPCR to evaluate differences in microbial populations in the feline gastrointestinal tract. J. Anim. Physiol. Anim. Nutr. (Berl) 2009;93:113–121. doi: 10.1111/j.1439-0396.2007.00788.x. [DOI] [PubMed] [Google Scholar]
- Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Dewell S.B., Du L., Fierro J.M., Gomes X.V., Godwin B.C., He W., Helgesen S., Ho C.H., Irzyk G.P., Jando S.C., Alenquer M.L., Jarvie T.P., Jirage K.B., Kim J.B., Knight J.R., Lanza J.R., Leamon J.H., Lefkowitz S.M., Lei M., Li J., Lohman K.L., Lu H., Makhijani V.B., McDade K.E., McKenna M.P., Myers E.W., Nickerson E., Nobile J.R., Plant R., Puc B.P., Ronan M.T., Roth G.T., Sarkis G.J., Simons J.F., Simpson J.W., Srinivasan M., Tartaro K.R., Tomasz A., Vogt K.A., Volkmer G.A., Wang S.H., Wang Y., Weiner M.P., Yu P., Begley R.F., Rothberg J.M. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyaert H., De Graef E.M., Haesebrouck F., Decostere A. Acquired antimicrobial resistance in the intestinal microbiota of diverse cat populations. Res. Vet. Sci. 2006;81:1–7. doi: 10.1016/j.rvsc.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Ott S.J., Kuhbacher T., Musfeldt M., Rosenstiel P., Hellmig S., Rehman A., Drews O., Weichert W., Timmis K.N., Schreiber S. Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand. J. Gastroenterol. 2008;43:831–841. doi: 10.1080/00365520801935434. [DOI] [PubMed] [Google Scholar]
- Papasouliotis K., Sparkes A.H., Werrett G., Egan K., Gruffydd-Jones E.A., Gruffydd-Jones T.J. Assessment of the bacterial flora of the proximal part of the small intestine in healthy cats, and the effect of sample collection method. Am. J. Vet. Res. 1998;59:48–51. [PubMed] [Google Scholar]
- Qu A., Brulc J.M., Wilson M.K., Law B.F., Theoret J.R., Joens L.A., Konkel M.E., Angly F., Dinsdale E.A., Edwards R.A., Nelson K.E., White B.A. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS One. 2008;3:e2945. doi: 10.1371/journal.pone.0002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie L.E., Steiner J.M., Suchodolski J.S. Assessment of microbial diversity along the feline intestinal tract using 16S rRNA gene analysis. FEMS Microbiol. Ecol. 2008;66:590–598. doi: 10.1111/j.1574-6941.2008.00609.x. [DOI] [PubMed] [Google Scholar]
- Ritchie L.E., Burke K.F., Garcia-Mazcorro J.F., Steiner J.M., Suchodolski J.S. Characterization of fecal microbiota in cats using universal 16S rRNA gene and group-specific primers for Lactobacillus and Bifidobacterium spp. Vet. Microbiol. 2010;144:140–146. doi: 10.1016/j.vetmic.2009.12.045. [DOI] [PubMed] [Google Scholar]
- Rosen G.L., Reichenberger E.R., Rosenfeld A.M. NBC: the Naive Bayes Classification tool webserver for taxonomic classification of metagenomic reads. Bioinformatics. 2011;27:127–129. doi: 10.1093/bioinformatics/btq619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan P.D., Marchesi J.R. Micro-eukaryotic diversity of the human distal gut microbiota: qualitative assessment using culture-dependent and -independent analysis of faeces. ISME J. 2008;2:1183–1193. doi: 10.1038/ismej.2008.76. [DOI] [PubMed] [Google Scholar]
- Scupham A.J. Succession in the intestinal microbiota of preadolescent turkeys. FEMS Microbiol. Ecol. 2007;60:136–147. doi: 10.1111/j.1574-6941.2006.00245.x. [DOI] [PubMed] [Google Scholar]
- Siegel J.M. Stressful life events and use of physician services among the elderly: the moderating role of pet ownership. J. Pers. Soc. Psychol. 1990;58:1081–1086. doi: 10.1037//0022-3514.58.6.1081. [DOI] [PubMed] [Google Scholar]
- Simjee S., White D.G., McDermott P.F., Wagner D.D., Zervos M.J., Donabedian S.M., English L.L., Hayes J.R., Walker R.D. Characterization of Tn1546 in vancomycin-resistant Enterococcus faecium isolated from canine urinary tract infections: evidence of gene exchange between human and animal enterococci. J. Clin. Microbiol. 2002;40:4659–4665. doi: 10.1128/JCM.40.12.4659-4665.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.W. Diet and large intestinal disease in dogs and cats. J. Nutr. 1998;128:2717S–2722S. doi: 10.1093/jn/128.12.2717S. [DOI] [PubMed] [Google Scholar]
- Spain C.V., Scarlett J.M., Wade S.E., McDonough P. Prevalence of enteric zoonotic agents in cats less than 1 year old in central New York State. J. Vet. Intern. Med. 2001;15:33–38. doi: 10.1892/0891-6640(2001)015<0033:poezai>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- Spencer L. Study explores health risks and the human/animal bond. J. Am. Vet. Med. Assoc. 1992;201:1669. [PubMed] [Google Scholar]
- Suchodolski J.S., Ruaux C.G., Steiner J.M., Fetz K., Williams D.A. Assessment of the qualitative variation in bacterial microflora among compartments of the intestinal tract of dogs by use of a molecular fingerprinting technique. Am. J. Vet. Res. 2005;66:1556–1562. doi: 10.2460/ajvr.2005.66.1556. [DOI] [PubMed] [Google Scholar]
- Swanson K.S., Dowd S.E., Suchodolski J.S., Middelbos I.S., Vester B.M., Barry K.A., Nelson K.E., Torralba M., Henrissat B., Coutinho P.M., Cann I.K., White B.A., Fahey G.C., Jr. Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. Isme J. 2011;5:639–649. doi: 10.1038/ismej.2010.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J., Ley R.E., Mahowald M.A., Magrini V., Mardis E.R., Gordon J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Turnbaugh P.J., Ridaura V.K., Faith J.J., Rey F.E., Knight R., Gordon J.I. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z., Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;36:808–812. doi: 10.2144/04365ST04. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial phylum counts in cat fecal metagenome (CFM) determined using WebCARMA (http://webcarma.cebitec.uni-bielefeld.de/cgi-bin/webcarma.cgi).
A class-level phylogenetic profile of feline fecal metagenome derived from Galaxy metagenomic pipeline (http://main.g2.bx.psu.edu/screencast).
Functional profile of feline metagenome determined using WebCARMA (http://webcarma.cebitec.uni-bielefeld.de/cgi-bin/webcarma.cgi).
Phylogenetic classification of Archaea in Feline Metagenome.
Overview of the MG-RAST metagenomes chosen for comparison.