

Highlights
► PMOs can be rapidly designed and produced in response to emergency medical needs. ► PMOs exhibit antiviral activity against a variety of viruses in in vivo models. ► Novel design strategies may improve the broad-spectrum antiviral activity of PMOs.
Keywords: Antisense, Morpholino, Phosphorodiamidate, Biodefense, Ebola virus, Marburg virus
Abstract
Phosphorodiamidate morpholino oligomers (PMOs) are synthetic antisense oligonucleotide analogs that are designed to interfere with translational processes by forming base-pair duplexes with specific RNA sequences. Positively charged PMOs (PMOplus™) are effective for the postexposure protection of two fulminant viral diseases, Ebola and Marburg hemorrhagic fever in nonhuman primates, and this class of antisense agent may also have possibilities for treatment of other viral diseases. PMOs are highly stable, are effective by a variety of routes of administration, can be readily formulated in common isotonic delivery vehicles, and can be rapidly designed and synthesized. These are properties which may make PMOs good candidates for use during responses to emerging or reemerging viruses that may be insensitive to available therapies or for use during outbreaks, especially in regions that lack a modern medical infrastructure. While the efficacy of sequence-specific therapies can be limited by target-site sequence variations that occur between variants or by the emergence of resistant mutants during infections, various PMO design strategies can minimize these impacts. These strategies include the use of promiscuous bases such as inosine to compensate for predicted base-pair mismatches, the use of sequences that target conserved sites between viral strains, and the use of sequences that target host products that viruses utilize for infection.
1. Introduction
Antisense oligonucleotides (ASOs) are designed to bind to complementary RNA by Watson–Crick hybridization. ASOs are typically designed to arrest translational processes either by inducing cleavage mechanisms involving endogenous cellular nucleases such as RNAse H (Giles et al., 1993) or by sterically blocking enzymes involved in target gene translation (Fig. 1 ) (Stein et al., 1997). Alternatively, ASOs can be designed to target viral genomic regions or RNA secondary structures involved in transcription or genomic-replication processes (Deas et al., 2005, Stone et al., 2008). ASO-mediated interference with these virological processes inhibits production of infectious virus particles in infected cells, potentially reducing cytopathic effects. As illustrated in Fig. 1, one unproven hypothesis about the mechanism of action of ASOs postulates that production of defective interfering particles following ASO treatment may facilitate the generation of productive immunological responses that clear virus following infection.
Fig. 1.
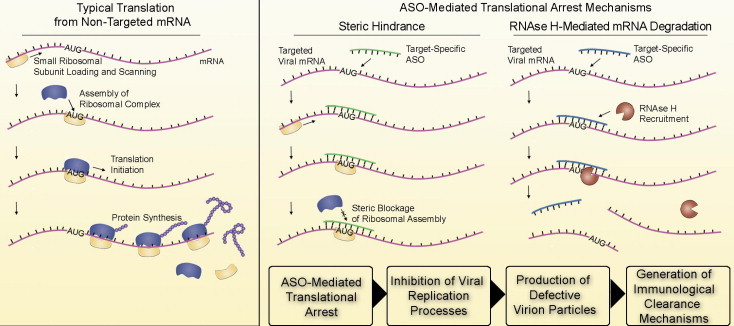
Comparison of translational arrest mechanisms mediated by antisense molecules. During typical translational processes, assembly of the ribosomal complex at the AUG start codon facilitates de novo protein synthesis from template RNA. Complementary antisense molecules can arrest translation either by sterically blocking ribosomal assembly or by supporting RNAse H cleavage activity. ASO mediated translational arrest may interfere with critical processes required for virion production and assembly and could promote the formation of defective interfering viral particles. The presence of these altered proteins or defective particles may serve to stimulate productive immunological clearance mechanisms.
Since the discovery of antisense molecules, there has been keen interest in optimizing the molecular scaffolds of antisense molecules to improve the pharmaceutical properties and in vivo activity for therapeutic applications. Improvements to increase solubility, reduce toxicity and increase broad spectrum activity have been engineered into phosphorodiamidate morpholino oligomers (PMOs) over the last few years of research. This article focuses on PMO-based antiviral therapeutic agents. Overviews of efforts to develop other antisense-based therapeutics for treatment of both viral and bacterial pathogens have been presented in several recent reviews (Haasnoot and Berkhout, 2009, Spurgers et al., 2008, Stein, 2008, Warfield et al., 2006a).
2. A brief history of antisense technology
In 1978, Zamecnik and Stephensen provided the first report of the use of a synthetic oligonucleotide to inhibit pathogen replication through antisense mechanisms. In these experiments, a tridecamer oligodeoxyribonucleotide (Fig. 2 ) complementary to Rous sarcoma virus RNA introduced to chick embryo fibroblast cells inhibited production of virus (Zamecnik and Stephenson, 1978). Further investigations revealed that hybridization of this oligodeoxyribonucleotide likely inhibited replication by blocking viral protein translation (Campbell et al., 1990, Stephenson and Zamecnik, 1978, Wickstrom, 1986). While the findings of Zamecnik and Stephensen suggested that administration of complementary antisense oligonucleotides (ASOs) might prove to be a useful therapeutic approach to achieve gene-specific knockdown, limitations to this approach were quickly realized.
Fig. 2.
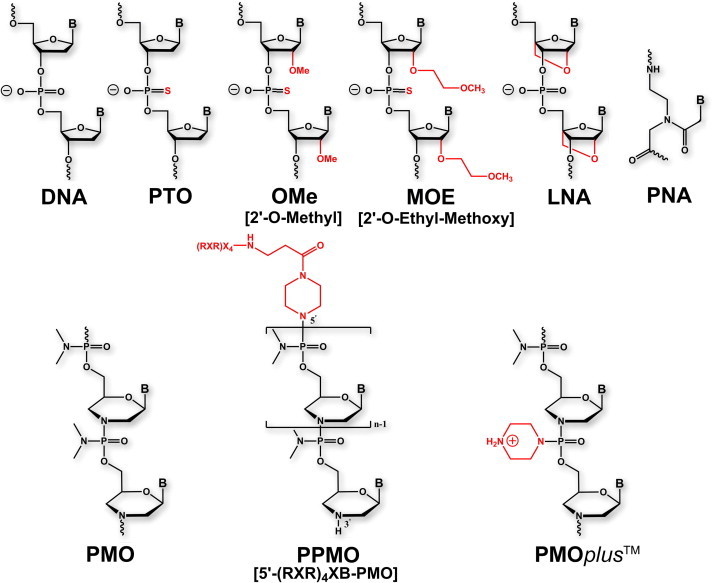
Nucleic acids and ASO analogs discussed in this review.
Unmodified oligodeoxynucleotides are susceptible to degradation by endogenous nucleases, which cleave phosphodiester linkages, present in bodily fluids (Wickstrom, 1986). In one of the first oligonucleotide analog derivatives designed to resist nuclease degradation, linkages of natural oligonucleotides were replaced with phosphorothioate groups (Fig. 2) (Campbell et al., 1990). Findings from wheat-germ and reticulocyte cell-free translation assays showed that these phosphorothioate oligonucleotides (PTOs) inhibited translation through RNAse H-dependent cleavage of mRNA (Minshull and Hunt, 1986). Matsukura et al. first exploited the antisense properties of PTOs, to inhibit replication of HIV (Matsukura et al., 1987). The benefits of resistance to nuclease degradation conferred by the PTO chemical modifications are diminished by a number of off-target effects, including cleavage of non-target sequences, interaction with a variety of proteins, activation of B lymphocytes, and activation of complement (Henry et al., 1997, Krieg and Stein, 1995, Krieg et al., 1995, Stein et al., 1997, Summerton, 2007). Despite these limitations, the FDA has approved a PTO antisense drug – fomiversen, for intravitreal treatment of cytomegalovirus retinitis in patients with AIDS – which to date is the only antisense agent to receive approval for therapeutic application (Geary et al., 2002, Perry and Balfour, 1999).
Various other strategies have been adopted in attempts to improve the resistance of ASOs to degradation by nucleases, to limit off-target effects, and to enhance binding affinity with target RNA. Incorporating O-methyl or O-methoxyethyl groups at the 2′ position of the PTO ribose ring increases the potency and nuclease resistance compared to unmodified PTOs (Fig. 2) (Dean and Bennett, 2003, Turner et al., 2006). These modifications do not support RNAse H-activity, which has led to development of chimeric strategies, in which gaps of contiguous unmodified phosphorothioate nucleotide analogs are incorporated to maintain RNAse H activity while retaining the favorable properties of modified PTOs (Turner et al., 2006, Zheng, 1999). The locked nucleic acid (LNA) ASO design incorporates a O2′-C4′-methylene linkage of the ribose rings, a design which supports RNAse H recruitment (Fig. 2) (Kurreck et al., 2002, Veedu and Wengel, 2009). Peptide nucleic acids (PNAs) possess no ribose rings and rely instead on a peptide-based structure for the molecular backbone (Fig. 2). As a result, PNAs do not support RNAse activity but effect translational arrest through steric-block mechanisms. PNAs investigated to date have exhibited poor cellular penetration and in vivo pharmacokinetic properties (Larsen et al., 1999, McMahon et al., 2002).
3. Morpholino ASOs
3.1. Phosphorodiamidate morpholino oligomers (PMOs)
Phosphorodiamidate morpholino oligomers are antisense molecules in which the ribose rings of DNA are replaced with 6-membered morpholine rings and phosphodiester linkages are replaced with phosphorodiamidate linkages (Fig. 2). PMOs are nonionic, a charge status that contrasts with the negative charges associated with native nucleotides and many other ASO design approaches. PMOs do not support RNAse H activity and effect translational arrest or mRNA processing, depending on the target site, through steric block mechanisms. The 5′ untranslated region (5′ UTR) and transcript sites at or near the AUG translational start codons are most often targeted to achieve translation arrest. Although it has been suggested that the nonionic characteristics of PMOs hinder cellular uptake, PMOs readily enter primary cells (Arora et al., 2004, Iversen, 2008). The in vivo activity of PMO antisense agents against a variety of viral pathogens has been demonstrated and is discussed in greater detail herein. While a number of neutral-charge PMOs have entered human clinical trials (Iversen, 2008, Iversen et al., 2003), none has yet been licensed for treatment of viral infection or other indications.
3.2. Peptide-conjugated PMOs (PPMOs)
Attempts to improve the in vivo efficacy of PMOs have included conjugation with peptides designed to enhance transport across cellular membranes. Peptide conjugated PMOs (PPMOs; Fig. 2), which include positively charged amino acid residues such as arginine, have been viewed as particularly promising transporters and have shown efficacy in multiple in vivo models of viral infection (Enterlein et al., 2006, Paessler et al., 2008, Stein, 2008, Swenson et al., 2009). The enhanced efficacy of PPMOs has been hypothesized to result from the enhanced affinity resulting from ionic interactions between the net positive charge of the peptides and the negative charge of the complementary RNA and through enhanced pharmacokinetic properties and cellular uptake (Iversen, 2008). However, MALDI-TOF mass spectrometry analyses have shown that while the PMO portion of PPMO conjugates are stable in cells, serum or tissue homogenate, the peptide portion is susceptible to degradation (Amantana et al., 2007, Youngblood et al., 2007). This degradation impacts the effectiveness of PPMOs by preventing the efficient escape of the conjugated molecule from endosomal or lysosomal vesicles (Youngblood et al., 2007).
While PPMOs are efficacious in multiple in vivo models of viral infections, PPMOs are more poorly tolerated in vivo compared to neutral-charge PMOs. In mice, dose-dependent reductions in weight, behavioral alterations, and mild liver histopathology were observed following repeat administration of 200–300 μg doses of a PMO conjugated to an arginine-rich peptide (Deas et al., 2007). In rats, administration of 30 or 150 mg/kg of an anti-c-myc PPMO (containing an arginine-rich peptide) produced reduction in body weight and elevation of serum blood urea nitrogen and creatinine concentrations, changes not observed following dose-matched administration of an unconjugated version of this PMO (Amantana et al., 2007). Various attempts to introduce non-natural amino acids or other chemical changes in the conjugated peptide to offset toxicity and improve cellular uptake and intracellular distribution have met with varying degrees of success (Abes et al., 2008, Wu et al., 2007, Youngblood et al., 2007).
3.3. Positively-charged PMOs
The recognition that positively-charged peptides can enhance the potency of neutrally charged PMOs led to the design of a new class of positively charged PMOs (PMOplus TM), which contain positively-charged piperazine groups along the molecular backbone (Fig. 2). Two recent reports (Swenson et al., 2009, Warren et al., 2010), showing that these therapeutics are well tolerated and provide improved efficacy in numerous in vivo viral infection models, have broad implications for therapeutic development against viruses and other highly pathogenic microbes. In 2009, Swenson et al. provided preliminary findings showing that intraperitoneal administration of PMOplus TM molecules targeting Ebola virus (EBOV) VP35 mRNA protected mice against lethal infection (Swenson et al., 2009). These results were expanded upon in a report by Warren et al. that showed that treatment conferred postexposure protection against EBOV and, separately, Marburg virus (MARV) in nonhuman primates (NHPs) (Warren et al., 2010). To achieve postexposure efficacy against EBOV in NHPs, a combination therapeutic (AVI-6002) containing PMOplus TM molecules targeting EBOV VP35 and VP24 mRNA was administered either intravenously or at intraperitoneal and subcutaneous sites (Warren et al., 2010). Reducing production of VP24 and VP35 may facilitate viral clearance by host innate and adaptive immunological mechanisms by limiting the interference of these viral products on type I interferon induction and signaling processes with which they are involved (Hartman et al., 2008, Kash et al., 2006, Reid et al., 2006).
In studies showing protection against MARV, AVI-6003, a combination of PMOplus TM molecules targeting MARV VP24 and nucleoprotein (NP), was delivered either intravenously, subcutaneously, or both intraperitoneally and subcutaneously. Inhibiting translation of MARV NP and VP24 may interfere with the release of virus from infected cells (Bamberg et al., 2005) and with MARV genome synthesis by affecting formation and activity of nucleoprotein complexes (DiCarlo et al., 2007).
The inclusion of cross-over, negative control treatments in NHP trials confirmed that the efficacy of both AVI-6002 and AVI-6003 was sequence-specific and was not mediated by nonspecific mechanisms. In all in vivo studies, treatment was well tolerated in all species tested (rat, mouse, guinea pig, rhesus monkey, and cynomolgous macaque). Open Investigational New Drug applications are on file with the US Food and Drug Administration (FDA) and placebo-controlled, double-blinded Phase I clinical trials of 30 patients for each of AVI-6002 and AVI-6003 are in progress to evaluate the safety of up to 9 mg/kg dose levels. An advanced development plan for these candidates is being actively pursued and relies on licensure under the FDA’s Animal Efficacy Rule, a regulatory mechanism designed to facilitate the development of new drug products for indications in which clinical efficacy studies are neither feasible nor ethical (Shurtleff et al., 2011, Snoy, 2010).
There currently are no licensed vaccine or therapeutic products for treatment of EBOV or MARV infection in humans, and reports of therapeutic agents besides PMOplus TM showing substantive post-exposure protection against these highly virulent pathogens in nonhuman primate models are few (Geisbert et al., 2010a, Geisbert et al., 2010b). Findings showing these therapeutics protect against MARV and EBOV – viruses associated with high case fatality rates, up to 90% for certain outbreaks – provide encouragement that this class of therapeutics may be efficacious against other, more prevalent, human viruses.
4. Potential of PMOplusTM to counter pathogenic viruses
4.1. Chemical properties
Antisense therapeutics possess multiple drug properties that are particularly favorable for use as therapeutics during outbreak scenarios. Because the timing of viral outbreaks can be highly unpredictable, an ideal therapeutic will need to be stable and amenable to large-scale manufacturing procedures to allow the drug to be prepared in advance of an outbreak. PMOplus TM agents have been amenable to the mid-scale, quality-controlled manufacturing needs required for Phase I clinical trials and nonhuman primate efficacy trials conditions reported herein, and these molecules are highly stable in lyophilized form following purification or in solution. Further, they are highly soluble in aqueous solutions or other isotonic solutions in which they may be administered in clinical settings. Initial investigations in nonhuman primates demonstrate that the agents are well tolerated by intravenous administration (Warren et al., 2010), suggesting that their use in emergency-response personnel are not likely to impact the ability of these individuals to perform their critical duties during public health or medical crises.
4.2. Pharmacokinetic properties
The pharmacokinetic properties of the agents comprising AVI-6002 and AVI-6003 have been examined in rats following administration of a single IV or IP dose (Warren et al., 2010). The plasma elimination half-life following IV administration of 32.8–35 mg/kg ranges from 0.78 to 2.98 h (Warren et al., 2010). These half-lives are comparable to that reported from a rat pharmacokinetic analysis involving a neutral-charge c-myc PMO but somewhat shorter than that of an arginine-rich peptide conjugated version of the c-myc PMO (Amantana et al., 2007). The plasma elimination half-life for neutral-charge PMOs is sequence-dependent and can range from 1.8 to 15 h (Amantana et al., 2007, Arora et al., 2004, Iversen, 2001). Further studies are required to determine whether the pharmacokinetic behavior of these agents is sequence-specific. For the two PMOplus TM components of AVI-6002, the primary route of excretion is renal with 23–34 percent recovery of the total IV dose from urine within 24 h of administration. The agents of AVI-6003 exhibited reduced renal clearance relative to AVI-6002, with 3.0–8.0 percent of the total dose excreted in urine within 24 h. Administered via the IV route, renal half-life for the components of AVI-6002 was 4.9–5.2 h and for AVI-6003 was 3.9–4.4 h. Following IP administration, the agents are generally rapidly aborbed with a time to maximal plasma concentration of 0.8–1.3 h. Plasma elimination half-life following IP administration ranges from 2.9 to 5.0 h.
The plasma elimination half-life may underestimate tissue residence time, which may have important implications for efficacy. For neutral-charge PMOs, tissue residence time in kidney and liver has been reported to range from 7 to 14 days (Amantana and Iversen, 2005, Iversen, 2001). Studies in rats have shown that conjugation of a c-myc PMO with an arginine-rich peptide increases the tissue uptake and increases tissue retention, especially in liver, kidney, and spleen (Amantana et al., 2007). Tissue distribution studies and interspecies pharmacokinetic comparisons involving AVI-6002, AVI-6003 or other agents have not yet been reported.
4.3. Routes of administration
While investigations are ongoing to further explore the routes by which PMOplus TM agents can be administered to confer in vivo efficacy against viral pathogens, results from studies involving neutrally charged PMOs or PPMOs suggest that efficacy can be achieved using administration via any of a number of routes. Recent mouse studies (Eide et al., 2010, Moerdyk-Schauwecker et al., 2009) have shown that PPMOs directed against immediate early genes ICP(0) and ICP27 of HSV-1 and HSV-2, when administered topically to the eye or genital tract epithelium, for these viruses, respectively, significantly reduced the viral loads in tissues and reduced the number of deaths from these infections. PPMOs can be successfully administered intranasally for treatment of respiratory diseases such as influenza in experimental models. Lupfer et al. showed that fluorescently labeled PPMOs given by intranasal instillation to mice could be detected in lung tissue by fluorescence visualization (Lupfer et al., 2008). Grossly, they observed the presence of fluorescent PMOs in a concentration gradient, which decreased towards the lower lobes of the lung (Lupfer et al., 2008). Microscopically, PPMOs were at highest concentrations in and around bronchioles (Lupfer et al., 2008). Using this intranasal instillation method for PPMO delivery, mice were protected from challenge with influenza virus A subtype H3N8, demonstrating that PPMOs were present in situ at concentrations that were sufficient for antiviral activity (Lupfer et al., 2008).
Mouse models of other systemic viral infections, such as alphaviruses, flaviviruses and filoviruses, have been used to test intraperitoneal, subcutaneous, intramuscular or intravenous routes, or even some combination thereof, of neutral-charge PMOs, PPMOs, and PMOplus therapeutic agents (Paessler et al., 2008, Stein et al., 2008, Warfield et al., 2006b). A limited number of published studies describe administration of PPMOs, neutrally charged PMOs, or PMOplus TM to larger animals such as guinea pigs, cats, or nonhuman primates for treatment of infectious diseases. Cats were treated for calicivirus infection with 2 mg doses of neutrally charged PMOs delivered subcutaneously, resulting in up to 89% protection from disease (Smith et al., 2008). Intraperitoneal treatment of guinea pigs with 10 mg doses of neutrally charged PMOs directed against EBOV proteins greatly enhanced survival in this lethal infection model (Warfield et al., 2006b). A prophylactic dosing regimen consisting of subcutaneous, intraperitoneal, and intramuscular doses of up to 100 mg of EBOV-specific PMOs protected rhesus macaques against lethal EBOV infection (Warfield et al., 2006b).
There exists a keen interest in developing antiviral therapeutics that can be administered rapidly to a large number of individuals with minimal involvement by medical professionals. Accordingly, therapeutics that can be delivered by oral, intramuscular, or transdermal routes are particularly appealing for use in emergency response scenarios and for use in remote regions that lack a modern medical infrastructure. Moreover, a host of promising new technologies are currently under development for drug administration, including gene-gun methods (Tavernier et al., 2010), aerosol and nasal deliveries (Wong et al., 2010), and transdermal dissolving microneedle technologies (Escobar-Chavez et al., 2010). Exploration of the efficacy of antisense therapeutics delivered by methods such as these may further expand the treatment options.
4.4. Use of morpholino ASOs during viral infections
Through use of animal models for investigations of experimental efficacy, PMOs and PPMOs have shown activity against human pathogenic viruses from a number of families of DNA and RNA viruses. Published efficacy data against these viruses range from early in vitro antiviral activity studies to in vivo advanced preclinical development studies in animal models of viral infection, with most of the published work concentrating on RNA viruses (Moerdyk-Schauwecker et al., 2009, Neuman et al., 2004, Smith et al., 2008, Smith et al., 2002, van den Born et al., 2005, Warren et al., 2010). Because viruses that belong to the same taxonomic class or order generally have similar genomic structure and gene functions, a gene target that can be inhibited effectively by ASOs for one virus, is likely to implicate promising targets in related viruses (Neuman et al., 2004, van den Born et al., 2005). In fact, some viruses possess conserved sequence regions across different virus isolates or strains, and these areas sometimes offer targets against which PMOs can be designed (Gabriel et al., 2008, Holden et al., 2006).
PPMOs have inhibited replication of herpes simplex virus-1 (HSV-1) in cell culture and in vivo when directed against HSV-1 ICP0 or ICP27 (Moerdyk-Schauwecker et al., 2009). Gene expression for another large DNA virus, the iridovirus, has been further characterized using morpholinos to knock down viral gene expression to assess the roles of viral regulatory genes and proteins (Sample et al., 2007).
The replication of a calicivirus, an RNA virus, has been reduced up to 70% in cell culture by neutrally charged PMOs at a concentration of 20 μM (Smith et al., 2002). PMOs used in this study were designed to target the 5′ feline calicivirus (FCV) ORF-1. FCV can cause a severe and contagious febrile respiratory disease in cats and kittens, and human caliciviruses, categorized as Category B Priority Pathogens by NIAID (NIAID, 2003), cause serious infections. When given at the time of disease onset and visible signs, subcutaneous administration of 2.0 mg/kg of this 5′ FCV ORF-1 PMO resulted in protection of up to 89% of cats tested in a veterinary field trial (Smith et al., 2008). Prophylactic efficacy was not investigated in this veterinary clinical setting.
Viruses of the order Nidovirales include viruses from the Coronaviridae family, e.g. severe acute respiratory syndrome coronavirus (SARS-CoV), and human coronaviruses which cause respiratory and gastrointestinal diseases. Coronaviruses and arteriviruses are RNA viruses that share similar genomic structures. PMOs and arginine-rich peptide-conjugated PPMOs directed against the 5′ UTR of equine arteritis virus (EAV) and against the 3′ UTR of porcine reproductive and respiratory syndrome virus (PRRSV), respectively, have exhibited antiviral activity in in vitro translation assays and in cell culture (Han et al., 2009, Patel et al., 2008, van den Born et al., 2005, Zhang et al., 2006). Regions of the arterivirus genome which can be inhibited by PMOs have also shown to be target regions for inhibiting CoV. These viruses possess a region within the 5′ UTR of the genome termed the transcription regulatory sequence (TRS). Arginine-rich peptide-conjugated PPMOs directed against this sequence have been shown to reduce SARS CoV yield in vitro by 104 fold (Neuman et al., 2005). Prophylactic administration of a PPMO targeting the 5′ UTR genomic region reduced liver viral load and diminished hepatic pathophysiology in mice infected with murine hepatitis virus (MHV), another betacoronavirus related to SARS CoV (Burrer et al., 2007, Neuman et al., 2004). However, viral challenge with delayed PPMO treatment given as a therapeutic regimen did not protect the animals (Burrer et al., 2007).
A variety of studies of other human viruses have been published demonstrating the antiviral efficacy of PMOs or PPMOs, either in cell culture or in mouse infection models. Subtypes of influenza A viruses, such as H1N1 and H3N2, represent common seasonal respiratory pathogens for humans, yet highly pathogenic H5N1 viruses can emerge and cause natural outbreaks. A collection of in vitro and in vivo studies have shown that PPMOs targeting the translation start site for PB1 and the 3′ terminal region of NP RNA have high efficacy against several subtypes of influenza A: H1N1, H3N2, H3N8, H5N1 and H7N7 (Gabriel et al., 2008, Ge et al., 2006, Lupfer et al., 2008). The same PPMOs, which were tested in vitro against strains of H7N7 and H3N8, protected up to 50% of mice from lethal influenza infection when administered intranasally before and after virus challenge (Gabriel et al., 2008, Lupfer et al., 2008).
Venezuelan equine encephalitis virus (VEEV) is a fast-replicating Category B alphavirus that causes a rapid onset of debilitating illness in humans which is usually nonfatal. Arginine-rich peptide-conjugated PPMOs directed against the 5′ UTR and the AUG region of the genomic RNA of VEEV were administered as a combination therapeutic to mice before and/or after challenge with a lethal dose of virulent VEEV strain ZPC738 (Paessler et al., 2008). The combination PPMO therapy was administered intranasally and intraperitoneally, with 40 μg and 160 μg given at each location, respectively. The prechallenge doses given both at 1 day and 4 h before infection imparted superior protection, compared to simple daily administration of the combination therapeutic at both dose sites after virus challenge. Mice receiving PPMOs before and after challenge were 100% protected from mortality due to VEEV infection, while only 63% of mice survived challenge when given the post-infection dose regimen of PPMOs within a few hours after infection, as a post-exposure prophylactic (Paessler et al., 2008).
Flaviviruses, many of which are categorized as NIAID Category A or B Priority Pathogens, rely on conserved structures within the 5′ and 3′ UTRs of their viral genomes to form secondary stem-loop structures to direct and regulate viral transcription and translation [reviewed in (Shurtleff et al., 2001)]. West Nile virus, dengue virus types 1–4, Japanese encephalitis virus (JEV) and St. Louis encephalitis virus can be blocked by arginine-rich peptide-conjugated PPMOs directed against areas in the 5′ UTR and the 3′ conserved region number 1 (Deas et al., 2007, Deas et al., 2005, Holden et al., 2006, Kinney et al., 2005). One study described the requirement for the presence of the arginine-rich peptide conjugated to the PMO for protection of mice from WNV infection, since subsequent experimental trials using unconjugated PMOs did not protect (Deas et al., 2007). Increased survival was observed in dengue-2-infected AG129 mice treated using a prophylactic regimen, but not a post-infection regimen, with arginine-rich PPMOs directed towards viral cyclization sequences in the 3′ and 5′ UTR, in comparison to treatment with unconjugated versions of these PMOs (Stein et al., 2008). As with other flaviviruses, arginine-rich PPMOs targeting the 3′ viral cyclization sequence were able to inhibit JEV infection of 3 different cell types in vitro and prophylactically in a mouse model of intracerebral infection (Anantpadma et al., 2010).
Arenaviruses are another virus family that causes significant human disease and for which there are limited treatment options. Peptide-linked PMOs, termed PPMO TERM-S and PPMO TERM-L, were designed against conserved sequences in the 5′ termini of the arenavirus short and long RNA segments, and evaluated in vitro against 4 related arenaviruses: Lymphocytic choriomeningitis virus (LCMV), Tacaribe virus, Pichinde virus and Junin virus vaccine strain Candid #1 (Neuman et al., 2011). In vitro, PPMOs directed against the L terminus were able to reduce viral NP and GP expression and reduce LCMV growth by 1000 fold (Neuman et al., 2011). When the PPMOs were administered prophylactically to mice at 6 or 9 mg/kg, just prior to infection with LCMV and daily for 3 days thereafter, significant reduction of viral growth in spleen and liver tissues was observed upon sampling at day 4 (Neuman et al., 2011). These PPMOs were only administered using a prophylactic regimen, and were not evaluated as strictly therapeutics.
Collectively, these studies suggest that modifying PMOs to include positive charges improves their in vivo efficacy. Additional investigations are currently in progress to evaluate the in vivo efficacy of PMOplus TM agents against flaviviruses, influenza viruses, hemorrhagic fever viruses, and other clinically important viral pathogens.
4.5. Accommodation of sequence mismatches
While the complementary design of antisense molecules for target RNA confers a high degree of selectivity to these agents, this aspect of antisense therapeutics poses limitations on the use of antisense molecules against virus variants that possess mismatches at the target site. Two design strategies were implemented in AVI-6003 to enhance the binding of PMOplus TM agents with sequence variations known to occur in MARV variants (Fig. 3 ). First, positive piperazine moieties were selectively positioned at sites where mismatches were predicted, in an effort to stablilize non-complementary bases by strengthening the ionic interactions of the PMOplus TM molecules with the targeted viral RNA. Second, inosine bases, which indiscriminately pair with adenine, uracil, or cytosine, were incorporated at two positions in the agent targeting MARV VP24 mRNA and at one position in the agent targeting MARV NP mRNA. Inosine residues were positioned to produce complementarity at locations where bases were known to vary between MARV variants, based on previous sequence analyses. Efficacy results obtained in mouse, guinea-pig, and NHP infection models suggest that the PMOplus TM design strategy used in AVI-6003 successfully accommodated the sequences predicted in the mouse-adapted Ravn virus isolate and in the MARV-Musoke isolates used in guinea-pig and NHP models of MARV infection (Warren et al., 2010). A similar strategy may prove useful in designing agents against other viruses, to accommodate limited mispairing that would be predicted among virus variants or closely related species.
Fig. 3.

Accommodation of sequence mismatches by PMOplusTM therapeutics. Sequences represent an alignment of a generalized MARV consensus genome with the PMOplusTM molecules comprising AVI-6003. Positively charged piperazine groups (+) and inosine bases (I) are positioned in the molecules at sites where mismatches occur. Genomic sequences from representative MARV variants/isolates are shown as cDNA complementary to genomic RNA. The MARV isolates shown include those against which AVI-6003 has exhibited antiviral activity in in vivo models (mouse-adapted Ravn virus, guinea-pig adapted MARV-Musoke, and non-adapted MARV-Musoke), or which were isolated during outbreaks (Ang0998, 07DRC, 09DRC), or which may have been involved in weaponization programs (Poppinga).
4.6. Rapid response to emerging pathogens
The ability to rapidly develop and produce therapeutics is integral to efficiently responding to emergency situations involving pathogens that may be resistant or otherwise insensitive to available drugs. Highly specific PMOs can be rapidly synthesized in response to such a need. In 2004, anti-EBOV morpholino-based oligomers were designed, synthesized, and shipped to a biocontainment medical facility as a contingency therapeutic within 7 days of a suspected accidental laboratory exposure event (Kortepeter et al., 2008). PMOplus TM-based therapeutics have also been designed and prepared as part of formal rapid-response exercises in only 11 days (Patrick Iversen, personal communication). For these events, the viral agent was identified and the complete viral genomic RNA target sequence was available at the time PMOs were requested for immediate synthesis. While some of these efforts have formed part of biodefense preparations, they will also serve to refine the rapid responses required for scenarios involving emerging viruses and other pathogens.
A rapid response to a veterinary medical crisis was generated in 2002 when a lethal outbreak of WNV in Humboldt penguins occurred at the Milwaukee County Zoo. PMOs were designed, synthesized and delivered for treatment within 7 days (Iversen, 2001). All 3 PMO-treated penguins survived, while 7 of 8 untreated penguins died. The small number of animals involved in this event limits any conclusions that can be drawn regarding the efficacy of the PMO treatment, but the event illustrates that PMOs can be quickly designed and delivered in response to a medical crisis. This situation required only a relatively small-scale synthesis of PMOs; obstacles associated with large-scale production technologies will need to be overcome to produce the large quantities of therapeutics needed for emergency mass treatment during outbreaks involving large numbers of patients or potentially infected individuals.
4.7. Genetic resistance to PMOs
It is well established that viruses can escape the effects of antiviral therapeutics through genomic alterations. This is a concern for ASO-based therapeutics, for which antiviral resistance could conceivably occur through the propagation of minority variants containing nucleotide polymorphisms that prevent the efficient binding of the antisense agents to the target nucleotides.
Several reports document the rapid selection of RNA viruses containing compensatory mutations – specifically SARS CoV, WNV, poliovirus type 1 (PV1), and foot-and-mouth disease virus (FMDV) – enabling these viruses to overcome PMO antiviral activity following serial passage in cultured cells (Deas et al., 2007, Neuman et al., 2005, Stone et al., 2008, Vagnozzi et al., 2007). Nucleotide variations that alter the complementarity of the target RNA with the antiviral antisense agents were responsible for conferring resistance in SARS CoV, FMDV, and PV1; however, in WNV, resistance determinants were identified outside of a PPMO target site when virus was serially passaged in the presence of a PPMO targeting the untranslated 3′ conserved sequence I (3′ CSI) (Deas et al., 2007). It has been hypothesized that these mutations compensate for 3′ CSI-mediated disruptions of WNV genome cyclization (Stein, 2008, Stein and Shi, 2008). WNV isolated from brains of infected mice treated with 3′ CSI PPMO showed no mutations compared to original virus challenge stock (Deas et al., 2007). Likewise, sequence obtained from virus isolated from the muscle of PV1 PPMO-treated mice did not show mutations (Stone et al., 2008).
Whether escape mutants appear during the replication of EBOV and MARV in infected nonhuman primates treated with antiviral PMOplus TM agents is an active area of research in our laboratory. Rigorous deep-sequence-based investigations are in progress to characterize the occurrence and prevalence of potentially resistant minority quasispecies in virus challenge stocks and in viruses isolated from infected nonhuman primates.
5. Conclusions
PMOs possess multiple chemical properties favorable for use as therapeutic agents: they are highly soluble, stable during storage and in biological fluids, and can be designed with either a fine degree of antiviral specificity or with broad-spectrum efficacy against multiple viruses. Investigators striving to develop antisense-based therapeutics have been challenged to identify ASO chemical modifications that enhance the in vivo efficacy of candidate therapeutics while minimizing the potential for toxicity. Recent studies have shown that PMOplus TM therapeutic agents confer a high degree of protection against filoviruses. Earlier-generation morpholino oligomers, i.e. neutrally charged PMOs and PPMOs, have been shown to be efficacious by a variety of routes of administration and against a wide range of pathogens. The stability and multiple-route efficacy of PMOplus TM agents make them well suited for use in a variety of settings, including regions lacking a modern medical infrastructure.
Two therapeutic agents, AVI-6002 and AVI-6003, have entered Phase I clinical safety studies, and preclinical efficacy evaluations are in progress for additional therapeutics in efforts to enhance the antiviral activity already achieved by other morpholino ASOs against a number of viral pathogens. Because the molecular backbone of all PMOplus TM agents is identical, establishment of clinical safety profiles for one therapeutic may serve to expedite the regulatory review and approval of additional agents.
While the PMOplus TM design can accommodate limited sequence mispairing, development of broad-spectrum antisense-based therapeutic agents will likely benefit from a parallel approach targeting both pathogen-specific products and host-based processes that viruses exploit during infection. As new discoveries are made of host pathways that divergent viruses rely on for entry, replication, or exit from infected cells, new potentially targetable genes will be identified to assist in efforts to develop broad-spectrum antisense-based therapeutics.
Disclosures
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the US Army.
Acknowledgements
The authors thank Dr. James C. Burnett for providing the structural figures presented in Fig. 2. Support for this work has been provided by the US Defense Threat Reduction Agency and by Transformational Medical Technologies (W9113 M-10-C-0056).
References
- Abes R., Moulton H.M., Clair P., Yang S.T., Abes S., Melikov K., Prevot P., Youngblood D.S., Iversen P.L., Chernomordik L.V., Lebleu B. Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: structure–activity studies. Nucleic Acids Res. 2008;36:6343–6354. doi: 10.1093/nar/gkn541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amantana A., Iversen P.L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr Opin Pharmacol. 2005;5:550–555. doi: 10.1016/j.coph.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Amantana A., Moulton H.M., Cate M.L., Reddy M.T., Whitehead T., Hassinger J.N., Youngblood D.S., Iversen P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem. 2007;18:1325–1331. doi: 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- Anantpadma M., Stein D.A., Vrati S. Inhibition of Japanese encephalitis virus replication in cultured cells and mice by a peptide-conjugated morpholino oligomer. J Antimicrob Chemother. 2010;65:953–961. doi: 10.1093/jac/dkq074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora V., Devi G.R., Iversen P.L. Neutrally charged phosphorodiamidate morpholino antisense oligomers: uptake, efficacy and pharmacokinetics. Curr Pharm Biotechnol. 2004;5:431–439. doi: 10.2174/1389201043376706. [DOI] [PubMed] [Google Scholar]
- Bamberg S., Kolesnikova L., Moller P., Klenk H.D., Becker S. VP24 of Marburg virus influences formation of infectious particles. J Virol. 2005;79:13421–13433. doi: 10.1128/JVI.79.21.13421-13433.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrer R., Neuman B.W., Ting J.P., Stein D.A., Moulton H.M., Iversen P.L., Kuhn P., Buchmeier M.J. Antiviral effects of antisense morpholino oligomers in murine coronavirus infection models. J Virol. 2007;81:5637–5648. doi: 10.1128/JVI.02360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J.M., Bacon T.A., Wickstrom E. Oligodeoxynucleoside phosphorothioate stability in subcellular extracts, culture media, sera and cerebrospinal fluid. J Biochem Biophys Methods. 1990;20:259–267. doi: 10.1016/0165-022x(90)90084-p. [DOI] [PubMed] [Google Scholar]
- Dean N.M., Bennett C.F. Antisense oligonucleotide-based therapeutics for cancer. Oncogene. 2003;22:9087–9096. doi: 10.1038/sj.onc.1207231. [DOI] [PubMed] [Google Scholar]
- Deas T.S., Bennett C.J., Jones S.A., Tilgner M., Ren P., Behr M.J., Stein D.A., Iversen P.L., Kramer L.D., Bernard K.A., Shi P.Y. In vitro resistance selection and in vivo efficacy of morpholino oligomers against West Nile virus. Antimicrob Agents Chemother. 2007;51:2470–2482. doi: 10.1128/AAC.00069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas T.S., Binduga-Gajewska I., Tilgner M., Ren P., Stein D.A., Moulton H.M., Iversen P.L., Kauffman E.B., Kramer L.D., Shi P.Y. Inhibition of flavivirus infections by antisense oligomers specifically suppressing viral translation and RNA replication. J Virol. 2005;79:4599–4609. doi: 10.1128/JVI.79.8.4599-4609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo A., Moller P., Lander A., Kolesnikova L., Becker S. Nucleocapsid formation and RNA synthesis of Marburg virus is dependent on two coiled coil motifs in the nucleoprotein. Virol J. 2007;4:105. doi: 10.1186/1743-422X-4-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide K., Moerdyk-Schauwecker M., Stein D.A., Bildfell R., Koelle D.M., Jin L. Reduction of herpes simplex virus type-2 replication in cell cultures and in rodent models with peptide-conjugated morpholino oligomers. Antivir Ther. 2010;15:1141–1149. doi: 10.3851/IMP1694. [DOI] [PubMed] [Google Scholar]
- Enterlein S., Warfield K.L., Swenson D.L., Stein D.A., Smith J.L., Gamble C.S., Kroeker A.D., Iversen P.L., Bavari S., Muhlberger E. VP35 knockdown inhibits Ebola virus amplification and protects against lethal infection in mice. Antimicrob Agents Chemother. 2006;50:984–993. doi: 10.1128/AAC.50.3.984-993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Chavez J.J., Bonilla-Martinez D., Villegas-Gonzalez M.A., Molina-Trinidad E., Casas-Alancaster N., Revilla-Vazquez A.L. Microneedles: A Valuable Physical Enhancer to Increase Transdermal Drug Delivery. J Clin Pharmacol. 2010:964–977. doi: 10.1177/0091270010378859. [DOI] [PubMed] [Google Scholar]
- Gabriel G., Nordmann A., Stein D.A., Iversen P.L., Klenk H.D. Morpholino oligomers targeting the PB1 and NP genes enhance the survival of mice infected with highly pathogenic influenza A H7N7 virus. J Gen Virol. 2008;89:939–948. doi: 10.1099/vir.0.83449-0. [DOI] [PubMed] [Google Scholar]
- Ge Q., Pastey M., Kobasa D., Puthavathana P., Lupfer C., Bestwick R.K., Iversen P.L., Chen J., Stein D.A. Inhibition of multiple subtypes of influenza A virus in cell cultures with morpholino oligomers. Antimicrob Agents Chemother. 2006;50:3724–3733. doi: 10.1128/AAC.00644-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary R.S., Henry S.P., Grillone L.R. Fomivirsen: clinical pharmacology and potential drug interactions. Clin Pharmacokinet. 2002;41:255–260. doi: 10.2165/00003088-200241040-00002. [DOI] [PubMed] [Google Scholar]
- Geisbert T.W., Hensley L.E., Geisbert J.B., Leung A., Johnson J.C., Grolla A., Feldmann H. Postexposure treatment of Marburg virus infection. Emerg Infect Dis. 2010;16:1119–1122. doi: 10.3201/eid1607.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert T.W., Lee A.C., Robbins M., Geisbert J.B., Honko A.N., Sood V., Johnson J.C., de Jong S., Tavakoli I., Judge A., Hensley L.E., Maclachlan I. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference. a proof-of-concept study. Lancet. 2010;375:1896–1905. doi: 10.1016/S0140-6736(10)60357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles R.V., Spiller D.G., Tidd D.M. Chimeric oligodeoxynucleotide analogues: enhanced cell uptake of structures which direct ribonuclease H with high specificity. Anticancer Drug Des. 1993;8:33–51. [PubMed] [Google Scholar]
- Haasnoot J., Berkhout B. Nucleic acids-based therapeutics in the battle against pathogenic viruses. Handb Exp Pharmacol. 2009:243–263. doi: 10.1007/978-3-540-79086-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Fan S., Patel D., Zhang Y.J. Enhanced inhibition of porcine reproductive and respiratory syndrome virus replication by combination of morpholino oligomers. Antiviral Res. 2009;82:59–66. doi: 10.1016/j.antiviral.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A.L., Bird B.H., Towner J.S., Antoniadou Z.A., Zaki S.R., Nichol S.T. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J Virol. 2008;82:2699–2704. doi: 10.1128/JVI.02344-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry S.P., Giclas P.C., Leeds J., Pangburn M., Auletta C., Levin A.A., Kornbrust D.J. Activation of the alternative pathway of complement by a phosphorothioate oligonucleotide: potential mechanism of action. J Pharmacol Exp Ther. 1997;281:810–816. [PubMed] [Google Scholar]
- Holden K.L., Stein D.A., Pierson T.C., Ahmed A.A., Clyde K., Iversen P.L., Harris E. Inhibition of dengue virus translation and RNA synthesis by a morpholino oligomer targeted to the top of the terminal 3′ stem-loop structure. Virology. 2006;344:439–452. doi: 10.1016/j.virol.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Iversen P. Antisense Drug Technology. In: Crooke S.T., editor. Antisense Drug Technology. Marcel Dekker, Inc.; New York: 2001. pp. 375–389. [Google Scholar]
- Iversen P. Morpholinos. In: Crooke S.T., editor. Antisense drug technology: principles, strategies, and applications. CRC Press; Boca Raton FL: 2008. [Google Scholar]
- Iversen P.L., Arora V., Acker A.J., Mason D.H., Devi G.R. Efficacy of antisense morpholino oligomer targeted to c-myc in prostate cancer xenograft murine model and a Phase I safety study in humans. Clin Cancer Res. 2003;9:2510–2519. [PubMed] [Google Scholar]
- Kash J.C., Muhlberger E., Carter V., Grosch M., Perwitasari O., Proll S.C., Thomas M.J., Weber F., Klenk H.D., Katze M.G. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol. 2006;80:3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney R.M., Huang C.Y., Rose B.C., Kroeker A.D., Dreher T.W., Iversen P.L., Stein D.A. Inhibition of dengue virus serotypes 1–4 in vero cell cultures with morpholino oligomers. J Virol. 2005;79:5116–5128. doi: 10.1128/JVI.79.8.5116-5128.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortepeter M.G., Martin J.W., Rusnak J.M., Cieslak T.J., Warfield K.L., Anderson E.L., Ranadive M.V. Managing potential laboratory exposure to ebola virus by using a patient biocontainment care unit. Emerg Infect Dis. 2008;14:881–887. doi: 10.3201/eid1406.071489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg A.M., Stein C.A. Phosphorothioate oligodeoxynucleotides: antisense or anti-protein? Antisense Res Dev. 1995;5:241. doi: 10.1089/ard.1995.5.241. [DOI] [PubMed] [Google Scholar]
- Krieg A.M., Yi A.K., Matson S., Waldschmidt T.J., Bishop G.A., Teasdale R., Koretzky G.A., Klinman D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- Kurreck J., Wyszko E., Gillen C., Erdmann V.A. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002;30:1911–1918. doi: 10.1093/nar/30.9.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen H.J., Bentin T., Nielsen P.E. Antisense properties of peptide nucleic acid. Biochim Biophys Acta. 1999;1489:159–166. doi: 10.1016/s0167-4781(99)00145-1. [DOI] [PubMed] [Google Scholar]
- Lupfer C., Stein D.A., Mourich D.V., Tepper S.E., Iversen P.L., Pastey M. Inhibition of influenza A H3N8 virus infections in mice by morpholino oligomers. Arch Virol. 2008;153:929–937. doi: 10.1007/s00705-008-0067-0. [DOI] [PubMed] [Google Scholar]
- Matsukura M., Shinozuka K., Zon G., Mitsuya H., Reitz M., Cohen J.S., Broder S. Phosphorothioate analogs of oligodeoxynucleotides: inhibitors of replication and cytopathic effects of human immunodeficiency virus. Proc Natl Acad Sci U S A. 1987;84:7706–7710. doi: 10.1073/pnas.84.21.7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon B.M., Mays D., Lipsky J., Stewart J.A., Fauq A., Richelson E. Pharmacokinetics and tissue distribution of a peptide nucleic acid after intravenous administration. Antisense Nucleic Acid Drug Dev. 2002;12:65–70. doi: 10.1089/108729002760070803. [DOI] [PubMed] [Google Scholar]
- Minshull J., Hunt T. The use of single-stranded DNA and RNase H to promote quantitative ‘hybrid arrest of translation’ of mRNA/DNA hybrids in reticulocyte lysate cell-free translations. Nucleic Acids Res. 1986;14:6433–6451. doi: 10.1093/nar/14.16.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerdyk-Schauwecker M., Stein D.A., Eide K., Blouch R.E., Bildfell R., Iversen P., Jin L. Inhibition of HSV-1 ocular infection with morpholino oligomers targeting ICP0 and ICP27. Antiviral Res. 2009;84:131–141. doi: 10.1016/j.antiviral.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Neuman B.W., Bederka L.H., Stein D.A., Ting J.P., Moulton H.M., Buchmeier M.J. Development of peptide-conjugated morpholino oligomers as pan-arenavirus inhibitors. Antimicrob Agents Chemother. 2011;55:4631–4638. doi: 10.1128/AAC.00650-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman B.W., Stein D.A., Kroeker A.D., Churchill M.J., Kim A.M., Kuhn P., Dawson P., Moulton H.M., Bestwick R.K., Iversen P.L., Buchmeier M.J. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J Virol. 2005;79:9665–9676. doi: 10.1128/JVI.79.15.9665-9676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman B.W., Stein D.A., Kroeker A.D., Paulino A.D., Moulton H.M., Iversen P.L., Buchmeier M.J. Antisense morpholino-oligomers directed against the 5′ end of the genome inhibit coronavirus proliferation and growth. J Virol. 2004;78:5891–5899. doi: 10.1128/JVI.78.11.5891-5899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIAID. 2003. NIAID Biodefense Research Agenda for Category B and C Agents. In: U.D.o.H.a.H. Services (Ed).
- Paessler S., Rijnbrand R., Stein D.A., Ni H., Yun N.E., Dziuba N., Borisevich V., Seregin A., Ma Y., Blouch R., Iversen P.L., Zacks M.A. Inhibition of alphavirus infection in cell culture and in mice with antisense morpholino oligomers. Virology. 2008;376:357–370. doi: 10.1016/j.virol.2008.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D., Opriessnig T., Stein D.A., Halbur P.G., Meng X.J., Iversen P.L., Zhang Y.J. Peptide-conjugated morpholino oligomers inhibit porcine reproductive and respiratory syndrome virus replication. Antiviral Res. 2008;77:95–107. doi: 10.1016/j.antiviral.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry C.M., Balfour J.A. Fomivirsen. Drugs. 1999;57:375–380. doi: 10.2165/00003495-199957030-00010. discussion 381. [DOI] [PubMed] [Google Scholar]
- Reid S.P., Leung L.W., Hartman A.L., Martinez O., Shaw M.L., Carbonnelle C., Volchkov V.E., Nichol S.T., Basler C.F. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol. 2006;80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample R., Bryan L., Long S., Majji S., Hoskins G., Sinning A., Olivier J., Chinchar V.G. Inhibition of iridovirus protein synthesis and virus replication by antisense morpholino oligonucleotides targeted to the major capsid protein, the 18 kDa immediate-early protein, and a viral homolog of RNA polymerase II. Virology. 2007;358:311–320. doi: 10.1016/j.virol.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Shurtleff A.C., Beasley D.W., Chen J.J., Ni H., Suderman M.T., Wang H., Xu R., Wang E., Weaver S.C., Watts D.M., Russell K.L., Barrett A.D. Genetic variation in the 3′ non-coding region of dengue viruses. Virology. 2001;281:75–87. doi: 10.1006/viro.2000.0748. [DOI] [PubMed] [Google Scholar]
- Shurtleff A.C., Warren T., Bavari S. Nonhuman primates as models for the discovery and development of ebolavirus therapeutics. Expert Opinions on Drug Discovery. 2011;6:1–18. doi: 10.1517/17460441.2011.554815. [DOI] [PubMed] [Google Scholar]
- Smith A.W., Iversen P.L., O’Hanley P.D., Skilling D.E., Christensen J.R., Weaver S.S., Longley K., Stone M.A., Poet S.E., Matson D.O. Virus-specific antiviral treatment for controlling severe and fatal outbreaks of feline calicivirus infection. Am J Vet Res. 2008;69:23–32. doi: 10.2460/ajvr.69.1.23. [DOI] [PubMed] [Google Scholar]
- Smith A.W., Matson D.O., Stein D.A., Skilling D.E., Kroeker A.D., Berke T., Iversen P.L. Antisense treatment of caliciviridae: an emerging disease agent of animals and humans. Curr Opin Mol Ther. 2002;4:177–184. [PubMed] [Google Scholar]
- Snoy P.J. Establishing efficacy of human products using animals: the US food and drug administration’s “animal rule”. Vet Pathol. 2010;47:774–778. doi: 10.1177/0300985810372506. [DOI] [PubMed] [Google Scholar]
- Spurgers K.B., Sharkey C.M., Warfield K.L., Bavari S. Oligonucleotide antiviral therapeutics: antisense and RNA interference for highly pathogenic RNA viruses. Antiviral Res. 2008;78:26–36. doi: 10.1016/j.antiviral.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein D., Foster E., Huang S.B., Weller D., Summerton J. A specificity comparison of four antisense types: morpholino, 2′-O-methyl RNA, DNA, and phosphorothioate DNA. Antisense Nucleic Acid Drug Dev. 1997;7:151–157. doi: 10.1089/oli.1.1997.7.151. [DOI] [PubMed] [Google Scholar]
- Stein D.A. Inhibition of RNA virus infections with peptide-conjugated morpholino oligomers. Curr Pharm Des. 2008;14:2619–2634. doi: 10.2174/138161208786071290. [DOI] [PubMed] [Google Scholar]
- Stein D.A., Huang C.Y., Silengo S., Amantana A., Crumley S., Blouch R.E., Iversen P.L., Kinney R.M. Treatment of AG129 mice with antisense morpholino oligomers increases survival time following challenge with dengue 2 virus. J Antimicrob Chemother. 2008;62:555–565. doi: 10.1093/jac/dkn221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein D.A., Shi P.Y. Nucleic acid-based inhibition of flavivirus infections. Front Biosci. 2008;13:1385–1395. doi: 10.2741/2769. [DOI] [PubMed] [Google Scholar]
- Stephenson M.L., Zamecnik P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J.K., Rijnbrand R., Stein D.A., Ma Y., Yang Y., Iversen P.L., Andino R. A morpholino oligomer targeting highly conserved internal ribosome entry site sequence is able to inhibit multiple species of picornavirus. Antimicrob Agents Chemother. 2008;52:1970–1981. doi: 10.1128/AAC.00011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerton J.E. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr Top Med Chem. 2007;7:651–660. doi: 10.2174/156802607780487740. [DOI] [PubMed] [Google Scholar]
- Swenson D.L., Warfield K.L., Warren T.K., Lovejoy C., Hassinger J.N., Ruthel G., Blouch R.E., Moulton H.M., Weller D.D., Iversen P.L., Bavari S. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob Agents Chemother. 2009;53:2089–2099. doi: 10.1128/AAC.00936-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernier G., Andries O., Demeester J., Sanders N.N., De Smedt S.C., Rejman J. MRNA as gene therapeutic: How to control protein expression. J Control Release. 2010;150:238–247. doi: 10.1016/j.jconrel.2010.10.020. [DOI] [PubMed] [Google Scholar]
- Turner J.J., Fabani M., Arzumanov A.A., Ivanova G., Gait M.J. Targeting the HIV-1 RNA leader sequence with synthetic oligonucleotides and siRNA: chemistry and cell delivery. Biochim Biophys Acta. 2006;1758:290–300. doi: 10.1016/j.bbamem.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Vagnozzi A., Stein D.A., Iversen P.L., Rieder E. Inhibition of foot-and-mouth disease virus infections in cell cultures with antisense morpholino oligomers. J Virol. 2007;81:11669–11680. doi: 10.1128/JVI.00557-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Born E., Stein D.A., Iversen P.L., Snijder E.J. Antiviral activity of morpholino oligomers designed to block various aspects of Equine arteritis virus amplification in cell culture. J Gen Virol. 2005;86:3081–3090. doi: 10.1099/vir.0.81158-0. [DOI] [PubMed] [Google Scholar]
- Veedu R.N., Wengel J. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2009;6:321–323. doi: 10.4161/rna.6.3.8807. [DOI] [PubMed] [Google Scholar]
- Warfield K.L., Panchal R.G., Aman M.J., Bavari S. Antisense treatments for biothreat agents. Curr Opin Mol Ther. 2006;8:93–103. [PubMed] [Google Scholar]
- Warfield K.L., Swenson D.L., Olinger G.G., Nichols D.K., Pratt W.D., Blouch R., Stein D.A., Aman M.J., Iversen P.L., Bavari S. Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog. 2006;2:e1. doi: 10.1371/journal.ppat.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren T.K., Warfield K.L., Wells J., Swenson D.L., Donner K.S., Van Tongeren S.A., Garza N.L., Dong L., Mourich D.V., Crumley S., Nichols D.K., Iversen P.L., Bavari S. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat Med. 2010;16:991–994. doi: 10.1038/nm.2202. [DOI] [PubMed] [Google Scholar]
- Wickstrom E. Oligodeoxynucleotide stability in subcellular extracts and culture media. J Biochem Biophys Methods. 1986;13:97–102. doi: 10.1016/0165-022x(86)90021-7. [DOI] [PubMed] [Google Scholar]
- Wong J.P., Christopher M.E., Viswanathan S., Schnell G., Dai X., Van Loon D., Stephen E.R. Aerosol and nasal delivery of vaccines and antiviral drugs against seasonal and pandemic influenza. Expert Rev Respir Med. 2010;4:171–177. doi: 10.1586/ers.10.15. [DOI] [PubMed] [Google Scholar]
- Wu R.P., Youngblood D.S., Hassinger J.N., Lovejoy C.E., Nelson M.H., Iversen P.L., Moulton H.M. Cell-penetrating peptides as transporters for morpholino oligomers: effects of amino acid composition on intracellular delivery and cytotoxicity. Nucleic Acids Res. 2007;35:5182–5191. doi: 10.1093/nar/gkm478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood D.S., Hatlevig S.A., Hassinger J.N., Iversen P.L., Moulton H.M. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjug Chem. 2007;18:50–60. doi: 10.1021/bc060138s. [DOI] [PubMed] [Google Scholar]
- Zamecnik P.C., Stephenson M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.J., Stein D.A., Fan S.M., Wang K.Y., Kroeker A.D., Meng X.J., Iversen P.L., Matson D.O. Suppression of porcine reproductive and respiratory syndrome virus replication by morpholino antisense oligomers. Vet Microbiol. 2006;117:117–129. doi: 10.1016/j.vetmic.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R. Technology evaluation: GEM-92, Hybridon Inc. Curr Opin Mol Ther. 1999;1:521–523. [PubMed] [Google Scholar]