

Abstract
Bovine torovirus (BToV), a member of the Coronaviridae family, is an established gastrointestinal infectious agent in cattle. No epidemiological research on BToV has been reported from Japan. In this study, we performed a survey to detect BToV in Japan in 2004 and 2005 using 231 fecal samples (167 from diarrheic cattle and 64 from asymptomatic cattle) that were analyzed by nested reverse transcription (RT) PCR using primers located in the consensus sequences of the reported BToV nucleocapsid (N), membrane (M), and spike (S) genes. BToV N, M, and S genes were detected in 6.5% (15/231), 6.1% (14/231), and 5.6% (13/231) of samples by nested-RT-PCR, respectively. In conclusion, detectability was improved compared to the results of the first round of RT-PCR. BToV was detected at a significantly higher rate in diarrheic samples than in asymptomatic samples (14/167 diarrheic samples [8.4%] and 1/64 asymptomatic samples [1.6%]), suggesting that BToV may act as a risk factor for diarrhea in Japanese cattle. The nucleotide sequence of M fragments from the BToV isolates including the newly identified Japanese isolates showed more than 97% identity. A similar degree of homology was observed in the N gene fragment among BToV isolates with the exception of BRV-1 and BRV-2. Domestic samples were classified into three clusters by phylogenetic analysis of the S gene fragment, which were considerably correlated with the geographic origin of the samples. BToV positive areas did not adjoin each other but were spread across a wide range, suggesting that BToV exists conventionally in Japan and is geographically differentiated. We also developed an RFLP method to distinguish these clusters using two restriction enzymes, HaeIII and AccI. This method should be useful for comparing newly acquired BToV-positive samples with the reported BToVs.
Abbreviations: PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism
Keywords: Bovine, Torovirus, Phylogenetic analysis
1. Introduction
Bovine torovirus (BToV), a member of the Coronaviridae family, was first detected in the USA during an outbreak of diarrhea in cattle in 1982 (Woode et al., 1982). Since then, epidemiological studies of BToV have shown that it is widespread throughout the world (Duckmanton et al., 1998a, Koopmans et al., 1991, Haschek et al., 2006, Hoet et al., 2002, Hoet et al., 2003, Liebler et al., 1992). An artificial inoculation trial also demonstrated the pathogenesis of BToV in cattle (Pohlenz et al., 1984, Woode et al., 1982). These data strongly suggest that BToV has relevance to diarrhea in cattle. However, no epidemiological research has been reported from Japan.
To detect BToV, electron microscopy (EM), enzyme-linked immunosorbent assay (ELISA), and polymerase chain reaction (PCR) are performed because this virus cannot be grown in cell culture (Koopmans et al., 1991, Liebler et al., 1992, Scott et al., 1996). PCR is more sensitive than ELISA, and its detectability is nearly identical to EM (Duckmanton et al., 1998a, Hoet et al., 2002). Consequently, the accumulation of a genetic database has progressed by analyzing gene products from different BToV isolates (Duckmanton et al., 1998b, Smits et al., 2003). The BToV genome consists of RNA polymerase, spike (S), membrane (M), hemagglutinin-estrase (HE), and nucleocapsid (N) genes (Draker et al., 2006). In particular, the N and M coding regions show high homology among the published BToV isolates, but can distinguish them from coronavirus and other toroviruses (equine isolate Berne virus, porcine torovirus, human torovirus; Koopmans and Horzinek, 1994). Therefore, detecting BToV N and M genes is thought be the most sensitive and specific method for detecting BToV in fecal samples. The S gene is known to affect antigenicity in other coronaviruses (Gallagher and Buchmeier, 2001). Therefore, analysis of the S gene in BToV positive samples is important in determining its antigenicity.
In this study, we analyzed 231 fecal samples from Japanese cattle for BToV. We used nested reverse transcription (RT) PCR with primers designed for BToV-specific N, M, and S genes. We also compared the nucleotide sequences to investigate genetic diversity among the samples.
2. Materials and methods
2.1. Specimens
A total of 231 individual fecal samples (167 from diarrheic cattle and 64 control samples from asymptomatic cattle) were collected from 32 farms in 12 prefectures in Japan (Fig. 1 ) between April 2004 and March 2005. All 231 fecal samples were used for BToV, bovine coronavirus (BCV), and rotavirus detection. The majority of cattle were between 1 week and 6 years old at the time of sample collection.
Fig. 1.
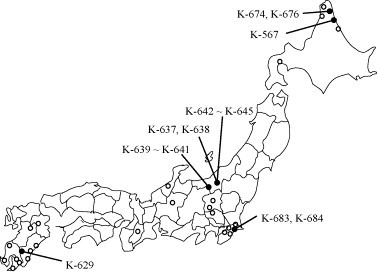
Distribution of BToV-positive samples in Japan. Open circles (○) indicate BToV-negative areas tested in this study.
2.2. RNA extraction
The fecal samples were diluted in 10× volume of phosphate-buffered saline (PBS, pH 7.5) and centrifuged at 1000 × g for 1 min at room temperature. The supernatant was transferred to a new tube and centrifuged again at 8000 × g for 5 min at room temperature. RNA was extracted from the supernatant using an RNeasy Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer's instructions. For each extraction period, ddH2O was used as a negative control.
2.3. Detection of BoTV and BCV
We used nested RT-PCR for BToV and RT-PCR for BCV detection. Primers targeting BToV N, M, and S genes were designed based on reported BToV sequences (Duckmanton et al., 1998b, Smits et al., 2003). The primers, reference sequences, and the expected sizes of the fragments obtained in the first and nested round of amplification are shown in Table 1 . The primers targeting the BCV N gene were as follows: forward: 5′-TGC CAG GAT GAT GGC GCG TG-3′ and reverse: 5′-AGA AGC ACA TCA GGG GAT TC-3′. Superscript II (Invitrogen Corp., Carlsbad, CA, USA) was used for reverse transcription using the reverse primer of each first round PCR primer. PCR was performed using TaKaRa Taq (TaKaRa, Tokyo, Japan). The mixture of reaction reagents was treated according to the manufacturer's instructions. The conditions of PCR were as follows: 30 cycles of denaturing at 94 °C for 1 min, annealing at 53 °C for 1 min, and extension at 72 °C for 1 min, followed by a final cycle of extension at 72 °C for 5 min. For each reaction, ddH2O was used as a negative control. PCR products were detected by electrophoresis on a 2.0% agarose gel.
Table 1.
Oligonucleotide primer pairs used for RT-PCR
| Target | Usage | Polarity | Positiona | Primer sequence | Expected fragment sizeb | Reference sequences and content |
|---|---|---|---|---|---|---|
| BToV N | First step PCR | Forward | 27775 bp ∼ 27795 bp | 5′-ATG AAT TCT ATG CTT AAT CCA-3′ | 471 bp | AJ575389, AJ575388, AJ575387, AJ575386, AJ575385 |
| Reverse | 28265 bp ∼ 28245 bp | 5′-AAT TCA AAG CCA CTT TTA TTG-3′ | ||||
| Nested PCRc | Forward | 27793 bp ∼ 27813 bp | 5′-CAA ATG CTA TGC CAT TTC AGC-3′ | 395 bp | ||
| Reverse | 28207 bp ∼ 28187 bp | 5′-TGG AAA CTT CAA CAG TGG CAT-3′ | ||||
| BToV M | First step PCR | Forward | 25759 bp ∼ 25779 bp | 5′-TGT TTG AGA CCA ATT ATT GGC-3′ | 682 bp | AJ575374, AJ575375, AJ575376, AY427798 |
| Reverse | 26460 bp ∼ 26440 bp | 5′-TAC TCA AAC TTA ACA CTA GAC-3′ | ||||
| Nested PCR | Forward | 25797 bp ∼ 25817 bp | 5′-CCA AAC CCA TTT ACT GCT CAA-3′ | 637 bp | ||
| Reverse | 26413 bp ∼ 26433 bp | 5′-GTA TAA TCT GCA ACA CCT TGC-3′ | ||||
| BToV S | First step PCR | Forward | 20957 bp ∼ 20977 bp | 5′-GTG TTA AGT TTG TGC AAA AAT-3′ | 722 bp | AJ575373, AY427798, AF076621 |
| Reverse | 21698 bp ∼ 21678 bp | 5′-TGC ATG AAC TCT ATA TGG TGT-3′ | ||||
| Nested PCR | Forward | 21025 bp ∼ 21045 bp | 5′-CAG AGG TGC CGT TGT TGT GTC-3′ | 616 bp | ||
| Reverse | 21660 bp ∼ 21640 bp | 5′-ACA TAG AGC GGT GTC TGT TGA-3′ | ||||
Position with respect to the AY427798 strain.
Including primer length.
First round PCR products was directly used as template in nested PCR.
2.4. Detection of rotavirus
We followed the methods of Okada and Matsumoto (2002) for detection of rotavirus using the fecal supernatant, as in the samples for RNA extraction.
2.5. Genetic analysis of BToV PCR products
The nested RT-PCR products corresponding to the BToV N, M, and S open reading frames (ORFs) were directly sequenced by Dragon GenomicsCtr (TaKaRa Bio Inc., Mie, Japan) with the ABI Prism BigDye Terminator version 3.1 cycle sequencing kit and an Applied Biosystems 3730 × l DNA analyzer (Applied Biosystems Inc., CA, USA). For the analysis of sequence relationships, BRV-1, BRV-2, and B145 BToV isolates, which have been detected in cattle from other countries, were used as reference. The accession numbers (National Center for Biotechnology Information) of the reference sequences are given in Table 3, Table 4, Table 5. RT-PCR products were purified using a DNA purification kit (QIAGEN) and digested by several restriction enzymes (TOYOBO, Tokyo, Japan) according to the manufacturer's instructions. The size of the DNA fragments was estimated using a 1-kb plus DNA ladder (Invitrogen).
Table 3.
Nucleotide (above the diagonal) and amino acid (below the diagonal) identities of the N gene sequences
| Strain | Nucleotide identity (%) |
|||||
|---|---|---|---|---|---|---|
| Group I | Group II | Group III | B145 | BRV-1 | BRV-2 | |
| Group I | 99.5 | 98.1 | 100 | 68.1 | 68.1 | |
| Group II | 100 | 97.6 | 99.5 | 68.1 | 68.1 | |
| Group III | 99.2 | 99.2 | 98.1 | 68.1 | 68.4 | |
| B145 | 100 | 100 | 99.2 | 68.1 | 68.1 | |
| BRV-1 | 68.8 | 68.8 | 68.8 | 68.8 | 97.6 | |
| BRV-2 | 69.6 | 69.6 | 69.6 | 69.6 | 96.8 | |
| Amino acid identity (%) | ||||||
Group I includes samples K-567, K-683, and K-684; Group II includes K-629 and K-637 through K645; and Group III includes K-674 and K-676. Isolates included within a group have identical sequences. The reference BToVs used for sequence comparison and accession numbers are as follows: B145 (AJ575388), BRV-1 (AY427798), and BRV-2 (AF076621).
Table 4.
Nucleotide (above the diagonal) and amino acid (below the diagonal) identities of the M gene sequences
| Strain | Nucleotide identity (%) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| K-567 | Group I | K-645 | K-674 | K-676 | Group II | B145 | BRV-1 | BRV-2 | |
| K-567 | 99.3 | 99.2 | 98.7 | 98.5 | 99.2 | 99.2 | 94.6 | 93.3 | |
| Group I | 100 | 99.8 | 99 | 98.8 | 99.2 | 99.2 | 94.4 | 93.3 | |
| K-645 | 100 | 100 | 98.8 | 98.7 | 99 | 99 | 94.4 | 93.1 | |
| K-674 | 99.5 | 99.5 | 99.5 | 99.8 | 98.5 | 98.5 | 95.1 | 93.6 | |
| K-676 | 99 | 99 | 99 | 99.5 | 98.3 | 98.3 | 94.9 | 93.4 | |
| Group II | 99.5 | 99.5 | 99.5 | 99 | 98.5 | 99 | 94.3 | 93.3 | |
| B145 | 100 | 100 | 100 | 99.5 | 99 | 99.5 | 95.3 | 94 | |
| BRV-1 | 98 | 98 | 98 | 97.4 | 96.9 | 97.4 | 98 | 97.7 | |
| BRV-2 | 96.9 | 96.9 | 96.9 | 96.4 | 95.9 | 96.4 | 96.1 | 98.3 | |
| Amino acid identity (%) | |||||||||
Table 5.
Nucleotide (above the diagonal) and amino acid (below the diagonal) identities of the S gene sequences
| Strain | Nucleotide identity (%) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| K-567 | Group I | K-639 | K-674 | K-676 | K-683 | K-684 | B145 | BRV-1 | BRV-2 | |
| K-567 | 98.3 | 98.1 | 92.6 | 92.8 | 96.8 | 96.6 | 94.3 | 93.9 | 93.3 | |
| Group I | 99 | 99.8 | 92.3 | 92.4 | 96.5 | 96.3 | 94.1 | 93.4 | 93.1 | |
| K-639 | 98.5 | 99.5 | 92.1 | 92.3 | 96.3 | 96.1 | 93.9 | 93.3 | 92.9 | |
| K-674 | 91.4 | 92.4 | 91.9 | 99.8 | 91.6 | 91.8 | 92.4 | 96 | 90.6 | |
| K-676 | 91.9 | 92.9 | 92.4 | 99.5 | 91.8 | 91.6 | 92.6 | 96.1 | 90.4 | |
| K-683 | 98 | 99 | 98.5 | 91.9 | 92.4 | 99.8 | 93.4 | 92.8 | 93.1 | |
| K-684 | 97.5 | 98.5 | 98 | 92.4 | 91.9 | 99.5 | 93.3 | 92.6 | 93.3 | |
| B145 | 94.4 | 95.4 | 94.9 | 92.4 | 92.9 | 94.4 | 93.9 | 92.9 | 91.6 | |
| BRV-1 | 93.9 | 94.9 | 94.4 | 94.4 | 94.9 | 94.4 | 93.9 | 93.9 | 92.4 | |
| BRV-2 | 93.9 | 94.9 | 94.4 | 90.4 | 89.9 | 94.9 | 95.4 | 91.4 | 92.4 | |
| Amino acid identity (%) | ||||||||||
2.6. Phylogenetic analysis of the BToV S gene
Phylogenetic analysis of the deduced amino acid sequence of the BToV S gene was preformed using the neighbor-joining method (Saitou and Nei, 1987).
3. Results
3.1. Condition of BToV-positive samples
BToV gene products corresponding to the N, M, and S ORFs were detected in 2.1% (5/231), 3.5% (8/231), and 0% (0/231) of the samples, respectively, by first round PCR and were detected in 6.5% (15/231), 6.1% (14/231), and 5.6% (13/231) of samples by subsequent nested PCR (Table 2 ). Among the 15 samples that were positive for the BToV N gene, 14 were derived from diarrheic samples (8.4%) and 1 was derived from an asymptomatic sample (1.6%). The positive samples were collected in 4 of 12 prefectures in Japan (Fig. 1). Of these 14 positive diarrheic samples, 7 were also positive for BCV, but all were negative for rotavirus (Table 2). BCV was detected in 47 samples (20.3%) and rotavirus was detected in 15 samples (6.5%). These tests were performed two or three times to confirm the initial results.
Table 2.
BToV-positive samples
| Sample name | Sampling month | Age | Fecal condition | BToV PCR result |
Bovine rotavirus | Bovine coronavirus | ||
|---|---|---|---|---|---|---|---|---|
| Nucleocapsid | Membrane | Spike | ||||||
| K-567 | 2004 April | 1 Week | Diarrhea | + | + | + | – | – |
| K-629 | 2004 April | 1 Week | Diarrhea | + | – | – | – | – |
| K-637 | 2004 September | Adulta | Diarrhea | ++ | ++ | + | – | – |
| K-638 | 2004 September | Adult | Diarrhea | ++ | ++ | + | – | – |
| K-639 | 2004 December | Adult | Diarrhea | ++ | ++ | + | – | ++ |
| K-640 | 2004 December | Adult | Diarrhea | ++ | ++ | + | – | ++ |
| K-641 | 2004 December | Adult | Diarrhea | + | ++ | + | – | ++ |
| K-642 | 2005 January | Adult | Diarrhea | + | + | + | – | ++ |
| K-643 | 2005 January | Adult | Diarrhea | ++ | + | – | – | ++ |
| K-644 | 2005 January | Adult | Diarrhea | + | ++ | + | – | ++ |
| K-645 | 2005 January | Adult | Diarrhea | + | + | + | – | ++ |
| K-674 | 2005 March | Adult | Normal | + | + | + | – | – |
| K-676 | 2005 March | Adult | Diarrhea | + | + | + | – | – |
| K-683 | 2005 March | Adult | Diarrhea | + | ++ | + | – | – |
| K-684 | 2005 March | Adult | Diarrhea | + | ++ | + | – | – |
(++) Indicates a positive result in the first step of PCR and (+) indicates a positive result in nested PCR.
Between 1 and 6 years after birth.
3.2. Genetic diversity of PCR products
The nucleotide sequences of N and M selected gene fragments showed more than 97% identity among the BToV isolates, except for the N genes of BRV-1 and BRV-2 (Table 3, Table 4 ). The nucleotide sequences of the BToV S gene fragment showed lower identity than those of N and M, even among the domestic samples (Table 5 ). Representative sequence data have been deposited in the nucleotide database (DNA Data Bank of Japan) and assigned the following accession numbers: K-567 (N: AB270904, M: AB270905, S: AB270906), K-629 (N: AB270907), K-637 (M: AB270908, S: AB270909), K-639 (S: AB270910), K-645 (M: AB270911), K-674 (N: AB270912, M: AB270913, S: AB270914), K-676 (M: AB270915, S: AB270916), K-683 (M: AB270917, S: AB270918), K-684 (S: AB270919).
We performed further analysis of the BToV S gene to characterize these samples. We generated a phylogenetic tree with the deduced amino acid sequences of the BToV S gene fragments, including previously reported BToV sequences, to investigate the phylogenetic relationship among these viruses. The BToV S gene found in Japanese isolates was divided into three clusters (Fig. 2 ): Cluster 1 (samples K-567, K-637 through K-642, K-644, and K-645), Cluster 2 (samples K-674 and K-676), and Cluster 3 (samples K-683 and K-684).
Fig. 2.
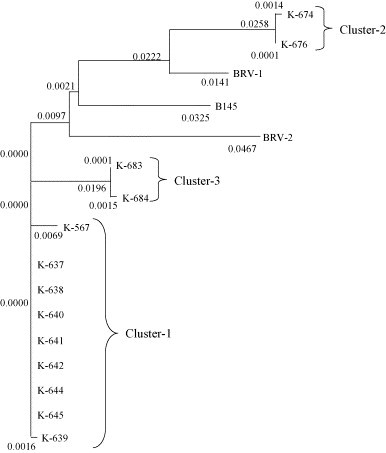
Neighbor-joining phylogenetic tree showing the relationships among the deduced S amino acid sequences of BToV from Japan and from the reference strains described in Table 5. The numbers represent the distance to the nearest node.
3.3. RFLP analysis of BToV-positive samples
We established a method to rapidly confirm the phylogenetic cluster to which a sample belonged. Based on the nucleotide sequences, we screened restriction enzymes and performed RFLP analysis of the BToV S fragment. The three clusters could be differentiated using HaeIII or AccI. Within cluster-1, K-567 could be differentiated from K-637 through K-642, K-644, and K-645 by detection of a distinctive 142-bp fragment using AccI (Fig. 3 ).
Fig. 3.
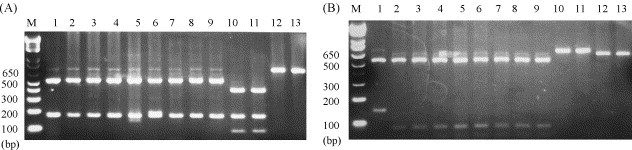
RFLP patterns of PCR-amplified BToV S gene products. (A) Digestion using HaeIII. (B) Digestion using AccI. Lane 1: K-567, Lane 2: K-637, Lane 3: K-638, Lane 4: K-639, Lane 5: K-640, Lane 6: K-641, Lane 7: K-642, Lane 8: K-644, Lane 9: K-645, Lane 10: K-674, Lane 11: K-676, Lane 12: K-683, Lane 13: K-684.
4. Discussion
We performed an epidemiological analysis of BToV in Japan by looking for BToV-specific genes in 231 fecal samples collected in 2004 and 2005. We performed nested RT-PCR to improve the detectability of BToV by using primers located in the consensus sequence of the reported BToVs. Consequently, we obtained 15 BToV-positive samples. Improvements in the detectability of BToV N, M, and S PCR products were observed in all samples by nested PCR as compared to first round PCR, suggesting the usefulness of this method in detecting BToV. The M gene could not be amplified from sample K-629, and this sample and sample K-643 were both negative for the S gene. However, since the N gene could be amplified from both samples, it is likely that nucleotide differences may exist in the primer annealing region used for M and S amplification and the corresponding sequences of these isolates.
Of the 14 BToV-positive diarrheic samples, 7 were negative for BCV and rotavirus, well known diarrheic agents of cattle. However, since infections with BCV, rotavirus, or other enteropathogens could have occurred before the samples were taken, it is impossible to determine whether BToV was the primary cause of diarrhea in these samples. However, there have been several reports indicating the relationship of BToV and diarrhea in cattle. Further, we detected BToV at a significantly higher rate in diarrheic samples than in asymptomatic samples. These results suggest that BToV may act as a risk factor for diarrhea in Japanese cattle.
The nucleotide sequences of the BToV S fragments had lower identity compared to the sequence identity of N and M fragments. In coronaviruses, which possess a similar structure to torovirus, a hypervariable region exists in the S gene (Wang et al., 1992). One amino acid substitution causes a change in virus neutralization activity in BCV (Yoo and Deregt, 2001). Thus, the S gene has been used to genetically characterize each strain of coronaviruses (Jackwood et al., 2005, Phillips et al., 2001, Yoo and Deregt, 2001). The BToV S molecule is thought to be located on the surface of the virus membrane and to have a similar function to the coronavirus spike (Horzinek et al., 1987).
Two BToV serotypes exist (BRV-1 and BRV-2; Duckmanton et al., 1998b), but further research on their antigenic properties is required to characterize them. There are few reports studying BToV, probably due to the fact that BToV cannot be yet propagated in cell culture. For the same reason, there are few reports characterizing BToV. In this study, we genetically characterized BToV samples by phylogenetic analysis of the deduced amino acid sequence of the BToV S gene, including the previously reported BRV-1, BRV-2, and B145 sequences. The detected BToVs were separated into three clusters. Cluster 2, which included samples K-674 and K-676, was located relatively closer to BRV-1. Cluster 1, the most frequently detected BToV cluster, was located distal to BRV-1, BRV-2, and B145. Cluster 3, which included samples K-683 and K-684, also seems to be separeted from these reference strains but relatively closer to cluster 1. In addition, high divergence was observed in the N sequence between the domestic samples and BRV-1 and BRV-2. These results raise the possibility that the predominant BToV in Japan are closer to the European strain B145 than to BRV-1 and BRV-2 regarding N gene properties. Therefore, it is important to identify the antigenic properties and to clarify their correlation with genetic properties.
The phylogenetic clusters were correlated with the geographic sources of the samples. The BToV-positive areas did not adjoin each other but were spread throughout Japan, spanning the BToV-negative areas. These results suggest that BToV is geographically differentiated in Japan.
Additionally, intertypic recombination occurs in torovirus (Smits et al., 2003). Therefore, it is important to continue the investigation of the genetic divergence of BToV. To support this, we developed an RFLP method to distinguish the clusters indicated by the phylogenetic classification. Although both HaeIII and AccI discriminate among the clusters, the cutting patterns differ. From the predicted cutting pattern, it is also possible to distinguish among BRV-1, BRV-2, and B145. The combination of these two enzymes will further help to determine the polymorphism of novel BToV isolates in the future.
In addition to the analysis of the S gene performed in this study, analysis of the HE gene would further aid to establish the phylogenetic and evolutionary relationship among all known BToV isolates. And it is also important to identify the retention of the BToV antibody to predict the prevalence and epidemiplogy of BToV in Japan.
Acknowledgment
We thank Emi Shirahase of Kyoto Biken Laboratories, Inc. in Kyoto, Japan, for collecting the data.
References
- Draker R., Roper R.L., Petric M., Tellier R. The complete sequence of the bovine torovirus genome. Virus Res. 2006;115(1):56–68. doi: 10.1016/j.virusres.2005.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckmanton L., Carman S., Nagy E., Petric M. Detection of bovine torovirus in fecal specimens of calves with diarrhea from Ontario farms. J. Clin. Microbiol. 1998;36(5):1266–1270. doi: 10.1128/jcm.36.5.1266-1270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckmanton L.M., Tellier R., Liu P., Petric M. Bovine torovirus: sequencing of the structural genes and expression of the nucleocapsid protein of Breda virus. Virus Res. 1998;58(1–2):83–96. doi: 10.1016/S0168-1702(98)00104-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.M., Buchmeier M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279(2):371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haschek B., Klein D., Benetka V., Herrera C., Sommerfeld-Stur I., Vilcek S., Moestl K., Baumgartner W. Detection of bovine torovirus in neonatal calf diarrhoea in Lower Austria and Styria (Austria) J. Vet. Med. B. Infect. Dis. Vet. Public Health. 2006;53(4):160–165. doi: 10.1111/j.1439-0450.2006.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoet A.E., Cho K.O., Chang K.O., Loerch S.C., Wittum T.E., Saif L.J. Enteric and nasal shedding of bovine torovirus (Breda virus) in feedlot cattle. Am. J. Vet. Res. 2002;63(3):342–348. doi: 10.2460/ajvr.2002.63.342. [DOI] [PubMed] [Google Scholar]
- Hoet A.E., Smiley J., Thomas C., Nielsen P.R., Wittum T.E., Saif L.J. Association of enteric shedding of bovine torovirus (Breda virus) and other enteropathogens with diarrhea in veal calves. Am. J. Vet. Res. 2003;64(4):485–490. doi: 10.2460/ajvr.2003.64.485. [DOI] [PubMed] [Google Scholar]
- Horzinek M.C., Flewett T.H., Saif L.J., Spaan W.J., Weiss M., Woode G.N. A new family of vertebrate viruses: Toroviridae. Intervirology. 1987;27(1):17–24. doi: 10.1159/000149710. [DOI] [PubMed] [Google Scholar]
- Jackwood M.W., Hilt D.A., Lee C.W., Kwon H.M., Callison S.A., Moore K.M., Moscoso H., Sellers H., Thayer S. Data from 11 years of molecular typing infectious bronchitis virus field isolates. Avian Dis. 2005;49(4):614–618. doi: 10.1637/7389-052905R.1. [DOI] [PubMed] [Google Scholar]
- Koopmans M., Horzinek M.C. Toroviruses of animals and humans: a review. Adv. Virus Res. 1994;43:233–273. doi: 10.1016/S0065-3527(08)60050-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans M., van Wuijckhuise-Sjouke L., Schukken Y.H., Cremers H., Horzinek M.C. Association of diarrhea in cattle with torovirus infections on farms. Am. J. Vet. Res. 1991;52(11):1769–1773. [PubMed] [Google Scholar]
- Liebler E.M., Kluver S., Pohlenz J., Koopmans M. The significance of bredavirus as a diarrhea agent in calf herds in Lower Saxony. Dtsch. Tierärztl. Wochenschr. 1992;99(5):195–200. [PubMed] [Google Scholar]
- Okada N., Matsumoto Y. Bovine rotavirus G and P types and sequence analysis of the VP7 gene of two G8 bovine rotaviruses from Japan. Vet. Microbiol. 2002;84(4):297–305. doi: 10.1016/s0378-1135(01)00445-x. [DOI] [PubMed] [Google Scholar]
- Phillips J.J., Chua M., Seo S.H., Weiss S.R. Multiple regions of the murine coronavirus spike glycoprotein influence neurovirulence. J. Neurovirol. 2001;7(5):421–431. doi: 10.1080/135502801753170273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlenz J.F., Cheville N.F., Woode G.N., Mokresh A.H. Cellular lesions in intestinal mucosa of gnotobiotic calves experimentally infected with a new unclassified bovine virus (Breda virus) Vet. Pathol. 1984;21(4):407–417. doi: 10.1177/030098588402100407. [DOI] [PubMed] [Google Scholar]
- Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Scott F.M., Holliman A., Jones G.W., Gray E.W., Fitton J. Evidence of torovirus infection in diarrhoeic cattle. Vet. Rec. 1996;138(12):284–285. doi: 10.1136/vr.138.12.284. [DOI] [PubMed] [Google Scholar]
- Smits S.L., Lavazza A., Matiz K., Horzinek M.C., Koopmans M.P., de Groot R.J. Phylogenetic and evolutionary relationships among torovirus field variants: evidence for multiple intertypic recombination events. J. Virol. 2003;77(17):9567–9577. doi: 10.1128/JVI.77.17.9567-9577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.I., Fleming J.O., Lai M.M. Sequence analysis of the spike protein gene of murine coronavirus variants: study of genetic sites affecting neuropathogenicity. Virology. 1992;186(2):742–749. doi: 10.1016/0042-6822(92)90041-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woode G.N., Reed D.E., Runnels P.L., Herrig M.A., Hill H.T. Studies with an unclassified virus isolated from diarrheic calves. Vet. Microbiol. 1982;7(3):221–240. doi: 10.1016/0378-1135(82)90036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo D., Deregt D. A single amino acid change within antigenic domain II of the spike protein of bovine coronavirus confers resistance to virus neutralization. Clin. Diagn. Lab. Immunol. 2001;8(2):297–302. doi: 10.1128/CDLI.8.2.297-302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]