

Abstract
Category A arenaviruses as defined by the National Institute of Allergy and Infectious Diseases (NIAID) are human pathogens that could be weaponized by bioterrorists. Many of these deadly viruses require biosafety level-4 (BSL-4) containment for all laboratory work, which limits traditional laboratory high-throughput screening (HTS) for identification of small molecule inhibitors. For those reasons, a related BSL-2 New World arenavirus, Tacaribe virus, 67–78% identical to Junín virus at the amino acid level, was used in a HTS campaign where approximately 400,000 small molecule compounds were screened in a Tacaribe virus-induced cytopathic effect (CPE) assay. Compounds identified in this screen showed antiviral activity and specificity against not only Tacaribe virus, but also the Category A New World arenaviruses (Junín, Machupo, and Guanarito). Drug resistant variants were isolated, suggesting that these compounds act through inhibition of a viral protein, the viral glycoprotein (GP2), and not through cellular toxicity mechanisms. A lead compound, ST-294, has been chosen for drug development. This potent and selective compound, with good bioavailability, demonstrated protective anti-viral efficacy in a Tacaribe mouse challenge model. This series of compounds represent a new class of inhibitors that may warrant further development for potential inclusion in a strategic stockpile.
Keywords: Arenavirus, Antiviral, Tacaribe, Junín, Hemorrhagic fever
1. Introduction
The National Institute of Allergy and Infectious Diseases (NIAID) and the Centers for Disease Control and Prevention (CDC) have classified a number of viruses as potential agents of bioterrorism (http://www.bt.cdc.gov/agent/agentlist-category.asp). The highest threat agents, the Category A pathogens, have the greatest potential for adverse public health impact and mass casualties if used in ill-intentioned ways. Within the Category A pathogens, there are a number of viruses that can cause viral hemorrhagic fevers with high case fatality rates. The Category A hemorrhagic fever viruses pose serious threats as potential biological weapons because they can: (1) be disseminated through aerosols, (2) cause disease at a low dose (1–10 plaque forming unit (pfu)), (3) cause severe morbidity and mortality (case fatality rates of 15–30%), (4) cause fear and panic in the general public, and (5) can be readily produced in large quantities (Charrel and de Lamballerie, 2003). The availability of antiviral drugs directed at these viruses would provide treatment and a strong deterrent against their use as biowarfare agents. Since antiviral drugs can be easily administered (oral pill or liquid) and exert their antiviral effect within hours of administration, they will serve to effectively treat diseased patients, protect those suspected of being exposed to the pathogen (post-exposure prophylaxis), and assist in the timely containment of an outbreak.
Arenaviruses are enveloped viruses with a genome that consists of two single-stranded RNA segments designated small (S, 3.5 kb) and large (L, 7.5 kb), both with an ambisense coding arrangement (Southern, 2001). The S RNA segment encodes the major structural proteins, nucleocapsid protein (NP) and a precursor envelope protein (GPC) encoding two envelope glycoproteins (external GP1 and transmembrane GP2) (Beyer et al., 2003, Froeschke et al., 2003, Kunz et al., 2003, Lenz et al., 2001), and the L RNA segment encodes the RNA polymerase protein L and an 11 kDa protein, Z protein, with putative regulatory function (Bowen et al., 1996). GP1 and GP2, which form the tetrameric surface glycoprotein spike, are responsible for virus entry into targeted host cells. The Arenaviridae family has been divided into two groups according to sequence-based phylogeny. The ‘Old World’ group, endemic in Africa, includes the human pathogens lymphocytic choriomeningitis (LCM) virus and Lassa virus. The ‘New World’ group, endemic in Latin America, is divided into three clades. Clade B includes in addition to Tacaribe and Amapari viruses, the Category A human pathogenic viruses Junín (Argentine hemorrhagic fever), Machupo (Bolivian hemorrhagic fever), Guanarito (Venezuelan hemorrhagic fever), and Sabiá (Brazilian hemorrhagic fever). These Category A viruses are capable of causing severe and often fatal hemorrhagic fever disease in humans.
Rodents are the natural host of arenaviruses, although Tacaribe virus is found in bats. The arenaviruses characteristically produce chronic viremic infections in their natural host (Rawls et al., 1976), which in turn shed virus in their urine and feces, ultimately infecting humans in close contact with these infected materials either by aerosol or direct contact with skin abrasions or cuts. The natural history of the human disease is determined by the pathogenicity of the virus, its geographical distribution, the habitat and the habits of the rodent reservoir host, and the nature of the human–rodent interaction (Childs and Peters, 1993).
Currently, there are no virus-specific treatments approved for use against arenavirus hemorrhagic fevers. A number of in vitro inhibitors of arenavirus replication have been reported in the literature (Candurra et al., 1996, Glushakova et al., 1990, Petkevich et al., 1981, Wachsman et al., 2000, Rawls et al., 1976) with modest activity and significant toxicity. Ribavirin is the only compound that has shown partial efficacy against some arenavirus infections, but with a high level of undesirable secondary reactions (Enria and Maiztegui, 1994). Ribavirin is a nonimmunosuppressive nucleoside analogue with broad antiviral properties (Jahrling, 1997). Ribavirin has been shown to reduce mortality from Lassa fever in high-risk patients (Jahrling, 1997). Treatment of viral hemorrhagic fever currently consists primarily of supportive care (Enria and Maiztegui, 1994, Garcia et al., 2000, Leifer et al., 1970). The only arenavirus hemorrhagic fever for which studies have been undertaken toward development of a vaccine has been Argentine hemorrhagic fever (AHF) caused by Junín virus. A live-attenuated vaccine, called Candid 1, has been evaluated in controlled trials among agricultural workers in AHF-endemic areas, where it appeared to reduce the number of reported AHF cases with no serious side effects (Enria et al., 1999). It is not known if the Candid 1 vaccine would be useful against other arenavirus hemorrhagic fevers.
Tacaribe virus is a biosafety level-2 (BSL-2) New World arenavirus (NWA) that is found in clade B and phylogenetically related to the Category A NWA (Junín, Machupo, Guanarito, and Sabiá). Tacaribe virus is 67–78% identical to Junín virus at the amino acid level for all four viral proteins. In order to screen for inhibitors of NWA a high-throughput screening (HTS) assay for virus replication was developed using Tacaribe virus as a surrogate for Category A NWA. A 400,000 small molecule library was screened using this HTS assay. A lead series was chosen based on drug properties and this series was optimized through iterative chemistry resulting in the identity of a highly active and specific small molecule inhibitor of Tacaribe virus with selective activity against human pathogenic NWA (Junín, Machupo, Guanarito, and Sabiá). This molecule demonstrates favorable pharmacodynamic properties which permitted the demonstration of in vivo anti-arenavirus activity in a newborn mouse model.
2. Materials and methods
2.1. Compound library
The chemical library screened represents a broad and well-balanced collection of 400,000 compounds accumulated over a number of years from a variety of distinct sources. The library achieves broad coverage across property space involving the following chemical descriptors: calculated logarithm of n-octanol/water partition coefficient (ClogP), polar water-accessible surface area (PSA), globularity (three dimensional structure) and molecular weight (average: 394.5 Da).
2.2. Cells and viruses
Vero (African green monkey kidney epithelial, ATCC #CCL-81) cells were grown in Eagle's minimum essential medium (MEM, Gibco) supplemented with 2 mM l-glutamine, 25 μg/ml gentamicin, and 10% heat-inactivated fetal bovine serum (FBS). For infection medium (IM), the serum concentration was reduced to 2%. HEp-2 cells (human carcinoma of the larynx epithelial; ATCC #CCL-23) were cultured in MEM containing 10% heat-inactivated FBS and 1% penicillin/streptomycin. MRC-5 cells (human normal lung fibroblast; ATCC #CCL-171) were cultured in MEM containing 10% heat-inactivated FBS, 1% penicillin/streptomycin, 1% l-glutamine (Invitrogen 25030-081), 1% non-essential amino acids (Invitrogen #11140-050), 1% sodium pyruvate (Invitrogen #11360-070), and 2% sodium bicarbonate. MA104 cells (epithelial African green monkey kidney, ATCC CRL-2378.1) were cultured in MEM with 1% penicillin/streptomycin, 1% l-glutamine, 1% non-essential amino acids, 1% sodium pyruvate, and 2% sodium bicarbonate and 62.5 μg/ml trypsin and no serum during virus infection. All cell lines were incubated at 37 °C and 5% CO2. Respiratory syncytial virus (RSV; A isolate), lymphocytic choriomeningitis virus (LCMV; Armstrong E350 isolate), cytomegalovirus (CMV; AD-169 isolate), herpes simplex virus 1 (HSV-1; KOS isolate), Vaccinia virus (Strain WR), Tacaribe virus (strain TRVL 11573) and rotavirus (strain WA) were obtained from ATCC (#VR-1422, #VR-1540, #VR-134, #VR-538, #VR-1493, #VR-1354, #VR-114, and # VR-2018, respectively). Candid 1 and Amapari BeAn 70563 were obtained from Dr. Robert Tesh at the University of Texas Medical Branch (Galveston, TX). Work done with BSL-4 viruses (Lassa Josiah strain, Machupo Carvallo strain, Guanarito INH-95551 strain, and Junín Romero strain) as well as severe acute respiratory syndrome-associated coronavirus (SARS-CoV) was conducted by collaborators at USAMRIID (Fort Detrick, MD).
2.3. Antiviral assays for specificity screening: cytopathic effect (CPE) assay, virus plaque reduction assay, and ELISA
A viral CPE assay was used to evaluate the antiviral effect of compounds against Tacaribe virus (Vero cells), Candid-1 vaccine virus (Vero cells), Amapari virus (Vero cells), SARS-CoV (Vero cells), HSV-1 (Vero cells), RSV (HEp-2 cells), vaccinia virus (Vero cells), and Rotavirus (MA104). An enzyme-linked immunosorbent assay (ELISA) was used to evaluate the antiviral effect of compounds against CMV (MRC-5 cells) and LCMV (Vero cells). All of these assays were carried out in the appropriate media containing 2% heat-inactivated FBS. Ninety-six-well cell culture plates were seeded 24 h before use with 1.5 × 104 cells per well (Vero), 2.2 × 104 cells per well (HEp-2 and MA104), and 4.5 × 104 cells per well (MRC-5). For compound susceptibility testing, compounds (solubilized with 100% DMSO) were added to duplicate wells at final concentrations of 50, 15.8, 5, 1.6, 0.5, 0.16, 0.05, 0.016, and 0 μM. The final concentration of DMSO in the assays was 0.5%. Virus stocks were titrated in a separate experiment to determine the concentration that resulted in 90% destruction of the cell monolayer (CPE assay) after 3 days (HSV-1, Rotavirus and vaccinia) or 4 days (SARS-CoV, RSV, Tacaribe virus, Candid 1 vaccine virus and Amapari virus) or the concentration that generated an ELISA signal of 2.5 at an optical density of 650 nm (OD650) after 3 days (LCMV) or 4 days (CMV). These pre-established dilutions of virus (250 pfu for Tacaribe virus) were added to wells containing serial dilutions of compound. Uninfected cells and cells receiving virus without compound were included on each assay plate. In addition, reference agents, when available, were included on each assay plate (gancyclovir for HSV-1 and CMV, Sigma #G2536; ribavirin for LCMV and RSV, Sigma #R9644; rifampicin for vaccinia virus, Sigma #R3501). Plates were incubated at 37 °C and 5% CO2 for either 3 days (HSV-1, Rotavirus, LCMV, Vaccinia virus) or 4 days (Tacaribe virus, Amapari virus, Candid 1 virus, SARS-CoV, RSV, and CMV). HSV-1, SARS-CoV, Rotavirus, Vaccinia virus, RSV, Tacaribe virus, Amapari virus, Candid 1 vaccine virus infected plates were processed for crystal violet staining while plates infected with CMV and LCMV were processed for ELISA analysis. For crystal violet staining, the plates were fixed with 5% glutaraldehyde and stained with 0.1% crystal violet. After rinsing and drying, the optical density at 570 nm (OD570) was measured using a Microplate Reader. For ELISA analysis, the medium from the LCMV and CMV-infected plates was removed and the cells were fixed with 100% methanol (Fisher, CAS #67-56-1, HPLC grade) for 20 min at room temperature. The methanol solution was removed and the plates were washed three times with PBS. Non-specific binding sites were blocked by the addition of 130 μl of Superblock Blocking Buffer (Pierce #37515) for 1 h at 37 °C. The blocking agent was removed and the wells were washed three times with PBS. Thirty microliters of a 1:20 dilution of LCMV nuclear protein (NP) specific monoclonal antibody (generous gift of Juan Carlos de la Torre, The Scripps Research Institute, La Jolla, CA) or 30 μl of a 1:200 dilution of CMV (protein 52 and unique long gene 44 product) specific cocktail monoclonal antibodies (Dako, #M0854) in Superblock Blocking Buffer containing 0.1% Tween-20 was added. Following 1 h incubation at 37 °C, the primary antibody solution was removed and the wells were washed three times with PBS containing 0.1% Tween-20. Forty microliters of goat anti-mouse horseradish peroxidase conjugated monoclonal antibody (Bio-Rad #172-1011) diluted 1:4000 (LCMV) or 1:400 (CMV) in Superblock Blocking Buffer containing 0.1% Tween-20 was added to the wells and the plates were incubated for 1 h at 37 °C. The secondary antibody solution was removed and the wells were washed five times with PBS. The assay was developed for 15 min by the addition of 130 μl of 3,3′,5,5-tetramethylbenzidine substrate (Sigma #T0440) to quantify peroxidase activity. The OD650 of the resulting reaction product was measured using a Molecular Devices Kinetic Microplate Reader with a 650 nm filter.
Antiviral activity against Tacaribe virus was evaluated by three methods: CPE assay, plaque reduction, and virus yield inhibition assay. For the HTS CPE assay, Vero cells were plated at 80% confluency on 96-well plates. Test compounds (80 per plate) from the library were added to wells at a final concentration of 5 μM. Tacaribe virus was then added at a virus dilution that would result in 90% CPE after 5 days (multiplicity of infection [MOI] approximately 0.001) from a seed stock of 1 × 1010 pfu/ml (10% of final volume). Plates were incubated at 37 °C and 5% CO2 for 5 days, then fixed with 5% glutaraldehyde and stained with 0.1% crystal violet. The extent of virus CPE was quantified spectrometrically at OD570 using an Envision Microplate Reader. The inhibitory activity of each compound was calculated by subtracting from the OD570 of test compound well from the average OD570 of virus-infected cell wells, then dividing by the average OD570 of mock-infected cell wells. The result represents the percent protection against Tacaribe virus CPE activity conferred by each compound. “Hits” in this assay were defined as compound that inhibited virus-induced CPE by greater than 50% at the test concentration (5 μM). Hits that possessed chemical tractability were further evaluated for their inhibitory potency. The inhibitory concentration 50% (EC50) values were determined from a plot of the compound inhibitory activity following the CPE assay across eight compound concentrations (50, 15, 5, 1.5, 0.5, 0.15, 0.05, and 0.015 μM). All determinations were performed in duplicate.
In the plaque reduction assay, Vero cell monolayers grown in six-well plates were infected with about 50 pfu/well in the absence or presence of various concentrations of the compounds. After 1 h of virus adsorption at 37 °C, residual inoculum was replaced by a 50:50 mix of 1% seaplaque agarose (in de-ionized water) and 2× MEM. Plaques were counted after 5–7 days of incubation at 37 °C. The EC50 was calculated as the compound concentration required to reduce virus plaque numbers by 50%. Under BSL-4 conditions at USAMRIID the plaque reduction assays (with Lassa, Machupo, Guanarito, and Junín viruses) were performed as follows: 200 pfu of each virus was used to infect Vero cells. After virus adsorption, cell monolayers were rinsed and overlaid with complete medium containing 1% agarose and either lacking test compound or with different concentrations ranging from 15 to 0.05 μM. After 5 days incubation at 37 °C, the monolayers were stained with neutral red and the numbers of plaques were counted.
In virus yield reduction assays, Vero cells grown in 24-well plates were infected with Tacaribe virus at a MOI of 0.1 in the presence of different concentrations of the compounds, two wells per concentration. After 48 h of incubation at 37 °C virus was harvested and the virus yields were determined by plaque formation in Vero cells. The EC50 values were calculated as indicated above and similar calculations were performed to determine EC90 and EC99.
2.4. Cytotoxicity assay
Cell viability was measured by a cell proliferation assay to determine a compound's effect on cellular functions so that a 50% cytotoxicity concentration (CC50) could be calculated; the ratio of this value to the EC50 is referred to as the selective index (SI = CC50/EC50). Two types of assays were used to determine cytotoxicity. One was a colorimetric method that measures the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), and the other uses fluorimetry to measure the reduction of resazurin (Alamar Blue). Both methods produced similar data. Confluent as well as rapidly dividing cell cultures in 96-well plates were exposed to different concentrations of the compounds, with two wells for each concentration, using incubation conditions equivalent to those used in the antiviral assays.
2.5. Medicinal chemistry
Several potent compounds were identified by the Tacaribe HTS and were grouped into several clusters of structure type. One cluster of compounds, with ST-336 (FW = 407.3) representing the prototype based on antiviral activity and chemical tractability, was chosen for further development. Through retrosynthetic analysis of ST-336, it was determined that a library of analogues could be prepared convergently in a single synthetic step by combining an isocyanate with an acyl hydrazide. Using this chemistry, 165 analogues were prepared and the most potent examined for in vitro metabolic stability in S9 liver extracts (described below).
2.6. Time of addition experiment
This experiment was designed to characterize the mechanism of action of the anti-viral compounds. Vero cells were grown in 24 well culture plates. The medium was removed when the cells reached 70–80% confluency and replaced with infection medium. Cells were infected with Tacaribe virus at MOI = 0.1. After 1 h adsorption, the viral inoculum was removed and replaced with fresh infection medium. Duplicate wells were treated with 3 μM ST-336 1 h prior to infection, at the time of infection or at specific times post-infection (from 1 to 20 h p.i.). Control infected cell cultures were treated with drug vehicle (DMSO) only. ST-336 was removed 1 h post-absorption and the monolayer was washed twice with cold PBS-M and replaced with fresh infection medium. The cells were harvested at 24 h p.i. and were titrated as described above.
In a separated experiment, Vero cells plated in a 6 well dish were infected with Tacaribe virus at MOI = 4. Absorption was carried out for 1 h. A 3 μM of ST-336 was added for 1 h at 1 h before infection, during infection, and 1 h following infection. Following drug addition and virus infection, monolayers were washed three times with complete media. Four hours following last drug addition, monolayers were overlaid with 1% agarose without compound until plaques developed. At 5–7 days post-infection, monolayers were fixed, crystal violet stained and plaque numbers counted.
2.7. Assay for compound binding to intact virus
This experiment was designed to test the binding/fusion inhibitory properties of ST-336 towards Tacaribe virus. Vero cells were grown in MEM with 2% fetal calf serum. For this experiment, cells were grown to 70–80% confluency in 24-well culture plates. In one set of tubes Tacaribe virus (4000 pfu) was treated with 1% DMSO, serially diluted tenfold in infection medium and treated with the specific concentrations of ST-336 (400 pfu + 0.5 μM ST-336, 40 pfu + 0.05 μM ST-336) or DMSO only (400 or 40 pfu + DMSO). Final DMSO concentration is each well is 0.5%. In another set of tubes Tacaribe virus (4000 pfu) was treated with 5 μM ST-336 then serially diluted tenfold in infection medium. The suspensions were plated in wells and after adsorption for one hour inocula were removed and overlaid with 0.5% Seaplaque agarose in MEM. The plate was incubated at 37 °C until cytopathic effect was observed in the DMSO control well. The cells were fixed with 5% gluteraldehyde and stained with 0.1% crystal violet for plaque visualization.
Another assay employed to test the binding properties of ST-336 to pre-fusion F-proteins on virions was a dialysis experiment. Purified Tacaribe virus (1000 pfu) was incubated with 5 μM of ST-336 or 0.5% DMSO. The suspensions were dialyzed overnight at 4 °C in a dialysis chamber. Twenty-four hours post-dialysis viral suspensions were titrated on Vero cells. One hour post-adsorption, inocula were removed and a 0.5% Seaplaque agarose in MEM overlay was applied. The plate was incubated at 37 °C until cytopathic effect was observed. The cells were fixed with 5% gluteraldehyde and stained with 0.1% crystal violet. To confirm absence of free drug in dialysed virus–drug sample, virus was spiked in dialysed mixture at time of infection and plaques developed as described above.
2.8. Isolation of drug resistant variant viruses
Initially, single plaques of WT Tacaribe virus were isolated. For this plaque-purification Vero cells in a six-well plate were infected with 50 pfu/well of WT Tacaribe virus for 1 h at 37 °C. Following virus adsorption the inoculum was removed and each well was overlaid with 0.5% Seaplaque agarose in MEM and incubated at 37 °C until plaques were visible (5–7 days). Four plaques were picked and further amplified in Vero cells in a 24-well plate until CPE developed (5–7 days). Virus-infected cell extracts were harvested by scraping cells into the media and then collected in 1.5 ml microcentrifuge tubes. Each plaque-purified isolate was further amplified in 150 mm plates, and then each virus stock that originated from one virus plaque was titrated.
For the isolation of compound-resistant Tacaribe virus variants, each wild type plaque-purified isolate was titrated in the presence of 3 μM ST-336 as described. Vero cells in a six-well plate were infected with 104–106 pfu/well in media containing 3 μM ST-336 for 1 h, then the cells were overlaid with 0.5% seaplaque agarose in MEM containing 3 μM ST-336 and incubated until plaques formed. Plaques were picked and used to infect Vero cells in a 24-well plate without compound. When CPE developed the infected wells were harvested. Each drug-resistant isolate was then titrated on a 96-well plate in 0.5 log dilutions, starting with 25 μl of pure virus stock, without compound and with 1 and 3 μM ST-336. Each mutant went through several rounds of plaque purification before final virus stocks were made.
2.9. Sequencing
RNA was extracted from each of the Tacaribe WT isolates (1–4) and four of the drug resistant isolates (DR#1–4) and used for reverse transcription PCR. Primers specific to the GPC (Tac-forward: 5′ GCCTAACTGAACCAGGTGAATC and Tac-reverse: 5′ TAAGACTTCCGCACCACAGG) from Tacaribe were used for amplification and sequencing.
2.10. Solubility
Two tests were used to assess compound solubility: solubility in cell culture medium with and without various concentrations of serum and solubility in aqueous buffer at pH 7.4. The solutions were stirred overnight and then filtered through an Amicon Centrifree YM-30 column with a 30,000 MW cut off to remove potentially precipitated compound and compound bound to protein. The compound was quantified by LC/MS or UV spectrometry.
2.11. Stability
In vitro metabolic stability was determined by Absorption Systems (Exton, PA) using the 9000 × g supernatant (S9) of homogenized liver from various species as a source of oxidative conjugation enzymes (e.g., cytochromes P450, UdP-glucuronosyl transferase) that are known to be the primary pathways of biotransformation for most drugs. The metabolic stability was measured as the persistence of parent compound over incubation time in the S9 fractions by mass spectrometry. Briefly, human, rat, mouse and guinea pig S9 fractions were obtained from Xenotech (Lenexa, KS). The reaction mixture, minus cofactor cocktails, was prepared (1 mg/ml liver S9 fractions, 1 mM NADPH, 1 mM UDPGA, 1 mM PAPS, 1 mM GSH, 100 mM potassium phosphate pH 7.4, 10 mM magnesium chloride, 10 μM test article) and equilibrated at 37 °C for 3 min. An aliquot of reaction mixture was taken as a negative control. The reaction was initiated by the addition of cofactor cocktails to the reaction mixture only, and then the reaction mixture and negative control were incubated in a shaking water bath at 37 °C. Aliquots (100 μl) were withdrawn in triplicate at 0, 15, 30, and 60 min and combined with 900 μl of ice-cold 50/50 acetonitrile/dH2O to terminate the reaction. Each sample was analyzed via LC/MS/MS. The natural log of the percent remaining was plotted versus time. A linear fit was used to determine the rate constant. The fit was truncated when percent remaining of test article was less than 10%. The elimination half-lives associated with the disappearance of test and control articles were determined to compare their relative metabolic stability.
2.12. Genotoxicity
An exploratory bacterial mutagenicity assay (Ames test) was used to assess the potential of the compound genotoxicity. This assay utilized S. typhimurium tester strains TA7007 and TA7006 (single base pair mutations) and TA98 (frame shift mutation) with and without metabolic activation (Arochlor-induced rat liver S9) as described previously (Maron and Ames, 1983).
2.13. Pharmacokinetic (PK) assessments in rats and newborn mice
Analysis of the oral pharmacokinetics of selected compounds was performed in Sprague–Dawley rats in a single dose study with serum samples taken over a 24 h period. For the newborn mice PK evaluation, 4 day old BALB/c mice were dosed intraperitoneally (IP) and serum samples were taken over a 24 h period (5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, and 24 h). A 50 μl aliquot of plasma was combined with 150 μl of 100% acetonitrile containing an internal standard (100 ng/ml tolbutamide) in a 1.5 ml centrifuge tube. Samples were vortexed and centrifuged at 13,000 rpm for 10 min. An 80 μl aliquot of the resulting supernatant was then transferred to an HPLC for vial analysis. Plasma levels of each compound were determined by LC/MS/MS, and pharmacokinetic parameters were determined using WinNolin software.
2.14. Efficacy in newborn mouse model
To determine tolerability of ST-294, newborn (4 days old) BALB/c mice were given IP dosages of 0 (vehicle), 10, 25, or 100 mg/kg/day of ST-294 for 5 days with assessment of clinical status daily.
To test the efficacy of ST-294 in the Tacaribe newborn mouse model, four day old BALB/c mice (eight per dose group) were challenged with 3 × 103 pfu (30XLD50) of Tacaribe virus per mouse by IP injection with death as the end point. Mice were either treated with placebo (vehicle), ribavirin (MP Biomedical) administered IP at 25 mg/kg once a day for 10 days, or ST-294 administered IP at 100 mg/kg once a day or at 50 mg/kg twice a day for 10 days. Mice were monitored daily and weighed every other day throughout the study. Any mice showing signs of morbidity were euthanized by CO2 asphyxiation. All animal studies conformed to the Institute for Laboratory Animal Research and were approved through appropriate IACUC review.
3. Homology
3.1. Homology between Tacaribe and other BSL-4 NWA
There are currently 23 recognized viral species of the Arenaviridae family (Clegg and Bowen, 2000). These viruses have been classified into two groups: the Old World (Lassa/LCM) arenaviruses and the New World (Tacaribe complex) group. The New World Tacaribe complex comprises three phylogenetic lineages, designated clades A–C. Clade B includes the prototypic Tacaribe virus, Amapari virus and the four South American Category A pathogens (Junín, Machupo, Guanarito, and Sabiá). Tacaribe virus is 67–78% identical to Junín virus at the amino acid level for all four viral proteins (Clegg, 2002). Working with authentic Category A arenaviruses requires maximum laboratory containment (BSL-4), and therefore presents significant logistical and safety issues. Since Tacaribe virus is closely related to the Category A pathogens it was chosen as a surrogate BSL-2 NWA for the development of a HTS assay to screen for inhibitors of virus replication.
4. Results
4.1. Tacaribe HTS assay
Since Tacaribe virus grows well in cell culture and causes clear virus-induced cytopathic effect (CPE) a robust HTS CPE assay was developed in a 96-well plate. The CPE assay is a whole cell assay which allows for calculation of the selective index of the compounds and identification of inhibitors of any essential steps in the virus life cycle. Of the 400,000 compounds screened in the Tacaribe virus HTS assay, 2347 hits were identified (0.58% hit rate). All of these hits had EC50 values ≤5 μM. The 2347 hits were then qualified based on four criteria: (i) chemical tractability, (ii) inhibitory potency, (iii) inhibitory selectivity, and (iv) antiviral specificity. A chemically tractable compound is defined as an entity that is synthetically accessible using reasonable chemical methodology, and which possesses chemically stable functionalities and potential drug-like qualities. Hits that passed this medicinal chemistry filter were evaluated for their inhibitory potency. EC50, CC50, and selective index (SI) values were determined to assess whether the hit was a selective inhibitor. Hits with SI values of at least 10 were considered further. Of the 2347 hits identified, 36 compounds exhibited all the characteristics of quality hits. These compounds were chemically tractable, had EC50 values ≤5 μM and SI values ≥10. Among the 36 quality hits, there were several clusters of structure type. One structure type was chosen for further development and ST-336 is the representative prototype for this series. ST-336 is a 407.33 Da compound and its structure is shown in Fig. 1 .
Fig. 1.
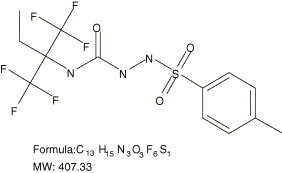
Chemical structure, formula, and molecular weight of ST-336.
4.2. Characterization of ST-336
As seen in Table 1 , ST-336 has submicromolar potency, good selectivity and antiviral specificity against Tacaribe virus as well as the Category A NWA. Evaluation of ST-336 in a virus yield reduction assay against Tacaribe virus produced EC90 and EC99 values of 0.068 and 0.085 μM, respectively. The CC50 value for ST-336 on Vero cells is >20 μM, which represents the solubility limit of this compound in cell culture media, giving it a selective index of >363. The activity of ST-336 against Tacaribe virus was tested on multiple cell lines and all the EC50 values were similar to those achieved on Vero cells (data not shown). When tested against several arenaviruses, ST-336 showed no inhibitory activity against OWA, either LCM virus or authentic Lassa virus (Table 1). This drug also lacked activity against the NWA Amapari virus. This was a surprising result given the close phylogenetic relationship between Amapari and Tacaribe viruses (Clegg, 2002, Bowen et al., 1996). This discrepancy is later discussed following sequencing of GP2 of all NWA. However, importantly ST-336 showed potent antiviral activity against the vaccine strain of Junín virus (Candid 1) as well as Machupo, Guanarito, and Junín viruses (Table 1).
Table 1.
Specificity of ST-336
| Assay | ST-336 (μM) | |
|---|---|---|
| NWA viruses | ||
| Tacaribe | CPE EC50 | 0.055 |
| CPE EC90 | 0.125 | |
| Virus yield EC90 | 0.068 | |
| Virus yield EC99 | 0.085 | |
| Plaque reduction EC50 | 0.100 | |
| Candid 1 | CPE EC50 | 0.062 |
| Amapari | CPE EC50 | >20a |
| Machupo | Plaque reduction EC50 | 0.150 |
| Guanarito | Plaque reduction EC50 | 0.300 |
| Juinín | Plaque reduction EC50 | 0.150 |
| OWA viruses | ||
| Lassa | Plaque reduction EC50 | >20 |
| LCMV | ELISA EC50 | >20 |
Results represent the average of at least two independent determinations.
20 μM represents limit of compound solubility.
The specificity of the antiviral activity exhibited by ST-336 was determined by testing against a number of related and unrelated viruses. ST-336 showed no activity against a variety of unrelated DNA (HSV, CMV, vaccinia virus) and RNA (RSV, Rotavirus and SARS-CoV) viruses.
4.3. Mechanism of action of ST-336
A single cycle (24 h) time of addition experiment was done to determine when during the virus replication cycle ST-336 exerts its antiviral activity. The effect of ST-336 on Tacaribe virus yield was determined following addition of compound to Vero cell cultures at various times before or after infection. ST-336 was added at 1 h before infection (−1 h), during virus adsorption (0 h), and at several times post-infection. Drug was kept, following sequential addition, on infected cell cultures for the entire time of the experiment. Control infected cultures were treated with drug vehicle (DMSO) only. At 24 h post-infection, samples were collected, and virus yields were determined by plaque assay. As shown in Fig. 2 A ST-336 exerted its inhibitory effect only at the very early stage in the virus life cycle. Addition of ST-336 at any time points post-infection had no effect on virus yield. These data suggest that ST-336 is an early stage inhibitor of virus replication.
Fig. 2.
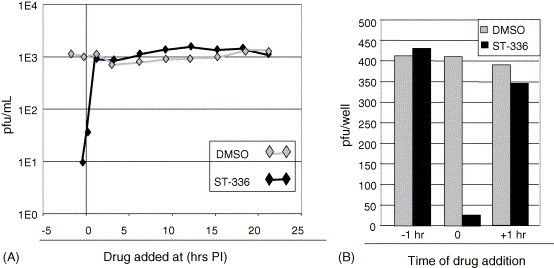
Effect of the time of addition of ST-336 on Tacaribe virus yield and plaque formation. (A) Vero cells were infected with Tacaribe virus at a MOI = 0.01. ST-336 was added prior to or during Tacaribe infection (−1, 3, 6, 9, 12, 15, 18 or 21 h p.i.). At 24 h p.i. virus yields were determined by plaque assay. (B) Vero cells were infected with 400 pfu Tacaribe virus. ST-336 was added for 1 h before the infection (−1), for 1 h during adsorption (0), and for 1 h after the infection (+1). Infected monolayers were washed with PBS and overlaid with medium containing agarose. Five days post-infection, cells were glutaraldehyde fixed and crystal violet stained prior to plaque counting.
These results were confirmed in a second type of time addition experiment. In this experiment, compound was spiked in the culture medium for only 1 h, at 1 h before infection (−1 h), during infection (0) and at 1 h post-infection (+1 h), and then removed. The cultures were washed to remove any residual compound and overlaid with agarose. Virus plaque numbers were then determined at 5 days post-infection. Data in Fig. 2B showed that while compound added before and after virus adsorption for 1 h had no effect on plaque formation, compound added during the 1 h adsorption/entry process dramatically reduced Tacaribe plaque formation. These data are consistent with ST-336 being an adsorption/entry inhibitor.
Two approaches were taken to determine if ST-336 is binding to intact virions. In the first experiment, 1000 pfu of purified Tacaribe virus was incubated with ST-336 or DMSO and dialyzed overnight at 4 °C and titrated. While no virus was titrated from the dialyzed bag originally incubated with drug, more than 300 pfu of virus was titrated from the DMSO vehicle dialyzed bag (data not shown). No drug was biologically detected in the dialysis bag originally containing 5 μM of drug as measured by the incapability of the virus plus drug dialyzed mixture to inhibit freshly added Tacaribe virus (300 pfu). These data suggested that ST-336 binds intact virions with a very slow dissociation constant. In the second experiment (Fig. 3 ), Tacaribe virus was incubated in a test tube with 5 μM of ST-336 or DMSO. Serial 1:10 dilutions were performed and for some samples ST-336 was added as a specified dilution representing the concentration of drug expected following sample dilution. As virus and compound are diluted with media, the compound concentration will reach a concentration without an inhibitory effect, unless the compound was capable of binding to virus. Test virus without compound in the initial tube was also diluted in media and compound concentrations corresponding to that found in the tubes where virus and compound were diluted together was added to each virus dilution. Titration on Vero cells showed that ST-336 present in excess in the initial tube was carried over for two additional 1:10 dilutions through specific virus binding and inhibits virus infection. Whereas when drug was added at a specified dilution virus was not inhibited to the same degree as virus diluted with drug (data not shown). These data suggest that ST-336 binds with at least a slow K off to intact protein present on Tacaribe virus.
Fig. 3.
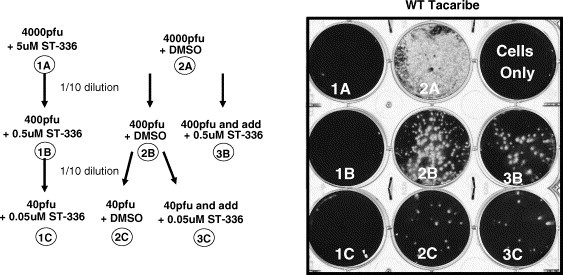
ST-336 binds with slow Koff to intact Tacaribe virion in absence of cells. (A) Diagram of virus dilution scheme prior to plating. Virus mixed with ST-336 and diluted (left side) or virus diluted and ST-336 added after dilution (right side). (B) Picture of plaques that resulted after plating each dilution shown in A on Vero cells.
4.4. Isolation of drug resistant variants
The expected mutation rate of RNA viruses is very high (∼1 resistant variant in 10,000 virions) and a common approach to determining the target of an antiviral is to isolate virus resistance to the antiviral and then map the site of resistance. Virus variants with reduced susceptibility to ST-336 were isolated from wild type Tacaribe virus stocks plated in the presence of ST-336. The observed frequency of ST-336 drug resistant (ST-336DR) variants was as expected for RNA viruses. Sixteen ST-336DR isolates from four independent wild type Tacaribe virus stocks were isolated and plaque purified three times. All ST-336DR isolates were tested for their ability to grow in the presence of ST-336. The growth of ST-336DR isolates was unaffected by the presence of ST-336 at concentrations that completely inhibited wild type Tacaribe virus replication (data not shown). The isolation and confirmation of drug resistant virus variants strongly suggest that ST-336 acts as a direct antiviral inhibitor.
To determine the genetic basis for resistance and the molecular target of ST-336, RNA was isolated from the wild type and ST-336DR isolates. Based on the time of addition experiments, it was suspected that the viral glycoproteins might be the target of ST-336. The entire glycoprotein precursor GPC region of the S segment was sequenced. Sequence analysis was performed on four wild type isolates (WT#1–4) and four ST-336DR isolates derived from drug selection applied to each corresponding parental wild type isolate (DR#1.1 from WT#1, DR#2.1 from WT#2, DR#3.1 from WT#3 and DR#4.1 from WT#4). The sequence analysis showed that the GPC gene from the four parental wild type isolates had identical sequences. When compared to the GPC sequences of four drug resistant variants, each possessed a single nucleotide change that in all cases resulted in an amino acid change. Fig. 4 A shows the location of each of the mutations which are located in or around the transmembrane domain of GP2. The sequence alignments of the region of the GP2 containing the changes is presented in Fig. 4B. The single change in DR#1.1 was at amino acid position 418 (I418T), in DR#2.1 at amino acid 416 (T416N), in DR#3.1 at amino acid 433 (S433I) and in DR#4.1 at amino acid 436 (F436I). I418 is similarly conserved (I or L, but never a T) in all clade B New World arenavirus, while T416 is conserved among all clade B NWA. F436 is similarly conserved with one exception; Amapari virus encodes a leucine at position 436. This change in Amapari virus may explain its lack of susceptibility to ST-336. I418, T416, S433 and F436 lie near the N- and C-terminal limits of the putative transmembrane domain of GP2, a region known to play a vital role in enveloped virus fusion (Bagai and Lamb, 1996, Harman et al., 2002, Jeetendra et al., 2003, West et al., 2001, Yao and Compans, 1995). Taken together, these data suggest that amino acid changes in arenavirus GP2 at either position 416, 418, 433 or 436 are sufficient to confer reduced susceptibility to ST-336 and are consistent with the proposed fusion inhibition mechanism suggested by virological experiments.
Fig. 4.
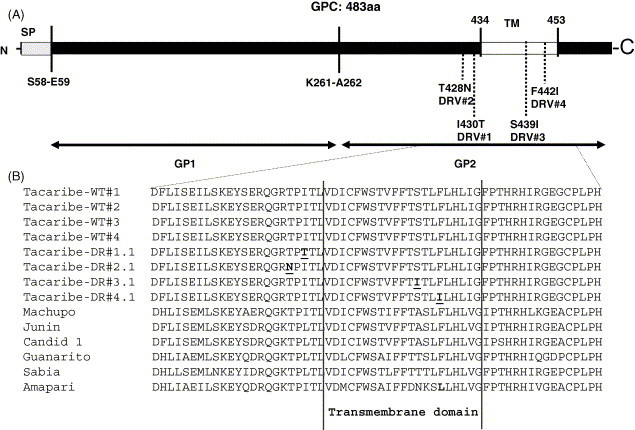
Mapping of ST-336 drug resistant variants (DRV's). (A) Linear map of the glycoprotein precursor (GPC) showing the location of the signal peptide (SP), transmembrane domain (TM), the cleavage site between GP1 and GP2 (K261-A262) and the location of the four ST-336 resistant mutants (DRV#1–4) and the amino acid change for each. (B) Amino acid sequence alignment of GP2 from wild type NWA and ST-336 DRV's. Shown is the amino acid sequence of the C-terminal portion of GP2 (aa397–457) containing the transmembrane domain (marked by vertical lines), the location of the mutations for DR#1–4 (underlined) and the amino acid difference in Amapari (bold).
4.5. Hit-to-lead optimization
Preliminary data showed that ST-336, while demonstrating interesting antiviral activity and specificity, had poor pharmacokinetic (PK) properties in rodents (mouse and rats, data not shown). In order to improve the PK properties of ST-336, a lead optimization chemistry campaign was initiated. The objective of the optimization program was to develop compounds that possess attributes consistent with the ultimate drug product profile. Lead optimization activities comprised a series of iterations involving design and chemical synthesis of analogs of the lead structure, followed by a series of biological, physiochemical, and pharmacological evaluations of the new compounds. Chemical analogs flowed through a compound evaluation paradigm that involved first in vitro virological and cytotoxicity assessments, followed by a series of evaluations as listed: in vitro metabolic stability (S9), solubility, exploratory bacterial mutagenesis and pharmacokinetic assessments. One hundred and sixty-five analogues were prepared and the most potent were examined for in vitro metabolism in S9 liver extracts. The most stable were dosed in rats, and ST-294 emerged as a potent, orally bioavailable representative of the compounds.
4.6. Characterization of ST-294
The structure of ST-294 (N-2-(1,1,1,3,3,3-hexafluoro-1-methylpropyl)-2-[(4-difluoromethoxyphenyl)sulfonyl]hydrazine-1-carboxamide) is show in Fig. 5 . ST-294 was tested against the drug resistant Tacaribe mutants generated with ST-336 (DR#1–4) and all of the mutants elicited cross-resistance to ST-294 suggesting that this compound is targeting the same area of GP2 as ST-336 (data not shown). The activity of ST-294 against Tacaribe, Machupo, Guanarito, and Junín viruses was similar to that seen with ST-336 (Table 1). The CC50 of ST-294 on Vero cells is >50 μM yielding a selective index of >416. Further characterization of ST-294 showed that this compound is soluble up to 23 μM in media containing 10% fetal calf serum and up to 480 μM in buffer at pH 7.4 (Table 2 ). The metabolic stability of ST-294 was tested in S9 liver extracts from rat, mouse, human, and guinea pigs and was found to be most stable in human S9 followed by mouse, rat and guinea pig respectively (Table 2). Analysis of the oral pharmacokinetics of ST-294 was initially performed in the rat as this species is well characterized for this type of study. The rats were dosed with ST-294 by oral gavage and samples were taken over a 24 h period. Serum levels were very high (C max = 6670 ng/ml) and ST-294 has good oral bioavailability (68.2%) (Table 2).
Fig. 5.
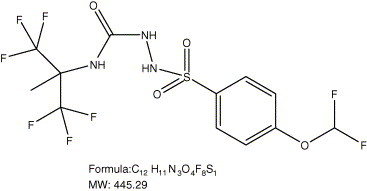
Chemical structure, formula, and molecular weight of ST-294.
Table 2.
Characterization of ST-294
| ST-294 | |
|---|---|
| Virus (assay) | |
| Tacaribe (CPE EC50) | 0.120 μM |
| Tacaribe (plaque reduction EC50) | 0.100 μM |
| Machupo (plaque reduction EC50) | 0.300 μM |
| Guanarito (plaque reduction EC50) | 1.0 μM |
| Junín (plaque reduction EC50) | 0.300 μM |
| Properties | |
| Solubility (0%, 2%, 10% FBS) | 18, 21 and 23 μM |
| Solubility (pIon, pH 7.4) | 480 μM |
| Stability (S9) rat/mouse/human/guinea pig | 26/74/100/23 min |
| Genotoxicity (Ames test) | Negative |
| PK | |
|---|---|
| Rat/oral | |
| Half-life | 2 h |
| Bioavailability (F) | 68% |
| Newborn mouse/IP | |
| Half-life | 3 h |
| Cmax | 2910 ng/ml |
4.7. Efficacy study with ST-294 in newborn mouse model
ST-294 has potent antiviral activity against NWA and good drug-like properties, so the next step was to test the ability of ST-294 to inhibit NWA-induced disease in an animal model. For the Category A agents, the experiments require BSL-4 containment. However, in an effort to obtain an initial readout, a Tacaribe virus challenge model in newborn mice was established. In preparation for this study, PK and tolerability experiments were performed with ST-294 in newborn mice prior to conducting an efficacy trial. Newborn (4-day old) BALB/c mice were dosed IP with 10 mg/kg of ST-294 and blood samples were collected for analysis. Relative to in vitro antiviral concentrations required to inhibit Tacaribe virus CPE (EC50 = 66 ng/ml), mean plasma concentrations in newborn mice were well above this level for prolonged periods of time (>15× through 8 h and 6× at 24 h after dosing, data not shown). In this model the drug is delivered via the IP route due to the difficulty of performing multiple oral gavages on newborn mice. To test tolerability, newborn mice were given IP dosages ranging from 0 to 100 mg/kg/day of ST-294 for 5 days. Dosages of 100 mg/kg/day for 5 days were well tolerated by the newborn mice as there were no clinical signs of toxicity and the mice gained weight at the same rate as the control mice (data not shown). This highest tested concentration of ST-294 of 100 mg/kg/day was used in a Tacaribe animal efficacy study.
The drug levels and half-life shown in the PK study in the newborn mice was not equivalent to that seen in the rats, but the serum levels seemed sufficient to perform a proof-of-concept animal study in the Tacaribe animal model. Four day old mice were challenged with 30XLD50 of Tacaribe virus and treated with placebo, ribavirin as a control or ST-294. As the results in Fig. 6 demonstrate, ST-294 showed efficacy in the Tacaribe infected newborn mice with both survival and a delay in death similar to the drug control (ribavirin). Taken together these data suggest that ST-294 is a promising and appropriate drug candidate to advance into definitive animal studies where guinea pigs and primates will be challenged with authentic NWA (Junín and Guanarito viruses) and treated at various times post-infection and prophylatically with ST-294.
Fig. 6.
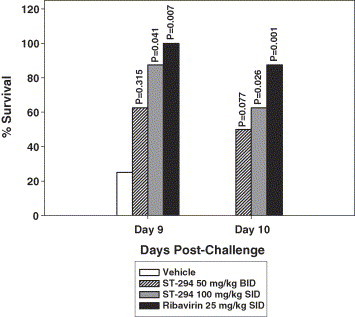
Effect of ST-294 in newborn mice challenged with Tacaribe virus. Four day old BALB/c mice were infected IP with 30XLD50 Tacaribe virus and treated daily for 10 days with vehicle (control), ribavirin at 25 mg/kg, ST-294 twice a day (BID) at 50 mg/kg or once a day (SID) at 100 mg/kg. Shown are the percent survivors in each treatment group on days 9 and 10 after infection. Significance (P values) was determined using the Fisher exact test.
5. Discussion
Through a successful HTS and medicinal chemistry program, a NWA antiviral drug candidate, ST-294, has been identified. This drug potently and selectively inhibits NWA viruses in vitro including the three NIAID/CDC Category A viruses (Junín, Machupo, and Guanarito viruses). This compound was also evaluated for stability in S9 liver extracts and for it is pharmacokinetic properties and was found to be metabolically stable and orally bioavailable. In a preliminary animal efficacy study, ST-294 showed significant protection against Tacaribe virus induced disease in newborn mice. Through mechanism of action studies it is apparent that this series of compounds targets GP2 and are viral entry inhibitors. Further studies will be undertaken to elucidate the experimental affinity of ST-294 to GP2 and the precise binding site. Also the fitness of drug resistant variants will be analyzed both in vitro and in vivo.
From the dialysis and dilution experiments (Fig. 3) it is apparent that the drug binds to virus and is carried over during dilutions. This phenomenon could potentially have an effect when titrating virus samples during other experiments. However, in the time of addition experiment, there was not enough drug carry over due to high dilution to affect the titers when added 1 h or more after infection (Fig. 2).
Since ST-294 has better S9 stability than ST-336 does, it is thought that metabolism occurs at the methyl group on the aromatic ring (Fig. 1). The benzylic position is susceptible to oxidation. When there is no benzylic hydrogen present as in ST-294 (Fig. 2), the oxidation is blocked and thus eliminates the fastest metabolism pathway. The addition of the difluoromethoxy group in ST-294 gave this compound increased S9 stability, but did not reduce antiviral activity.
In the Tacaribe newborn mouse model the mice appear to die of a neurological disease (indicated by hind quarter paralysis) and it is not known whether ST-294 can cross the blood brain barrier (BBB). Ribavirin does not cross the BBB in adult species; however the extent that ribavirin can cross the barrier in newborn mice is unknown. Also the drug levels and half-life of this drug candidate given IP in newborn mice is not as good as oral dosing in rats so serum levels and compound getting to the brain may have compromised the ability to obtain complete protection in this model. The more appropriate animal models for hemorrhagic fever caused by arenaviruses are in guinea pigs and non-human primates where the virus replicates predominantly in the spleen, lymph nodes and bone marrow causing hemorrhagic diathesis. Guinea pig models are well established for Junín, Machupo, and Guanarito virus diseases, and represent the best small animal model for evaluation during preclinical studies (Hall et al., 1996, Peters et al., 1987). Guinea pigs infected with pathogenic strains of Junín virus develop a fatal disease akin to human AHF (Weissenbacher et al., 1982).
There are many reports of the role of transmembrane in the function of viral fusion proteins. In the case of influenza virus hemagglutinin, it is clear that a transmembrane anchor is required for full fusion activity (Harman et al., 2002). In contrast, specific sequence requirements within the transmembrane domain have been identified, for example, in human immunodeficiency virus (HIV) type 1, murine leukemia virus, foamy viruses, coronavirus, Newcastle disease virus and measles virus (Harman et al., 2002). Based on the drug resistant variants generated during these studies, the ST-336 class of compounds targets the GP2 envelope protein, with mutations eliciting reduced susceptibility to the drug arising in or around the transmembrane region (Fig. 4).
Drugs that target the interactions between the virus envelope and the cellular receptor represent a new class of antiviral drugs. For HIV therapy, entry inhibitors have recently raised great interest because of their activity against multi-drug resistant viruses. A new antiviral against HIV was recently approved by the FDA called enfuvirtide. Enfuvirtide (Fuzeon) is a potent fusion inhibitor that blocks formation of the six-helix bundle and thus prevents membrane fusion (Kinomoto et al., 2005). Enfuvirtide has been successful in improving the virological and immunological response in treatment-experienced HIV-infected patients (Oldfield et al., 2005). There are several other compounds that counter HIV entry that are in different developmental stages, among them: (1) the attachment inhibitor dextrin-2-sulfate; (2) the inhibitors of the glycoprotein (gp) 120/CD4 interaction PRO 542, TNX 355 and BMS 488043; (3) the co-receptor inhibitors subdivided in those targeting CCR5 or CXCR4 (Castagna et al., 2005). The success of enfuvirtide and others in the development pathway are proof that virus entry inhibitors can be used to treat viral diseases in humans.
ST-294 also has the potential for prophylactic use since this drug appears to bind to the virus (Fig. 3) and would prevent infection. Other virus entry inhibitors have demonstrated protection when given prophylactically (Cianci et al., 2004). This is an indication that can be pursued to determine its feasibility.
The results presented here show that ST-294 is a potent specific inhibitor of New World arenaviruses including the Category A hemorrhagic fever viruses (Junín, Machupo, and Guanarito). More importantly the target of ST-294 (virus entry into the cell) serves as a viable target for antiviral development. Since virus infection can be completely inhibited at concentrations in the nanomolar range, the target for ST-294 would seem to be both accessible and extremely sensitive to reagents that disrupt its role in the infection process. Therefore, it will be important to further define the mechanism involved in ST-294 mediated inhibition.
Acknowledgements
This research was supported by grants from the National Institutes of Health (5 R44 A1056525-04 and 7 R43 AI056525-02). The authors would like to thank Juan Carlos de la Torre for personal communications on the Tacaribe virus newborn mouse model and for monoclonal antibody to the nuclear protein of LCMV. The authors would also like to thank Robert Jordan and Theodore Nitz for scientific discussions and for critical input on the publication; Mark Collett for initiation of this program.
References
- Bagai S., Lamb R.A. J. Cell Biol. 1996;135:73–84. doi: 10.1083/jcb.135.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer W.R., Popplau D., Garten W., von Laer D., Lenz O. J. Virol. 2003;77:2866–2872. doi: 10.1128/JVI.77.5.2866-2872.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen M.D., Peters C.J., Nichol S.T. Virology. 1996;219:285–290. doi: 10.1006/viro.1996.0248. [DOI] [PubMed] [Google Scholar]
- Candurra N.A., Maskin L., Damonte E.B. Antiviral Res. 1996;31:149–158. doi: 10.1016/0166-3542(96)06956-2. [DOI] [PubMed] [Google Scholar]
- Castagna A., Biswas P., Beretta A., Lazzarin A. Drugs. 2005;65:879–904. doi: 10.2165/00003495-200565070-00001. [DOI] [PubMed] [Google Scholar]
- Charrel R.N., de Lamballerie X. Antiviral Res. 2003;57:89–100. doi: 10.1016/s0166-3542(02)00202-4. [DOI] [PubMed] [Google Scholar]
- Childs J.E., Peters C.J. In: The Arenaviridae. Salvato M.S., editor. Plenum Press; New York: 1993. pp. 331–384. [Google Scholar]
- Cianci C., Genovesi E.V., Lamb L., Medina I., Yang Z., Zadjura L., Yang H., D’Arienzo C., Sin N., Yu K.L., Combrink K., Li Z., Colonno R., Meanwell N., Clark J., Krystal M. Antimicrob. Agents Chemother. 2004;48:2448–2454. doi: 10.1128/AAC.48.7.2448-2454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg J.C. Curr. Top. Microbiol. Immunol. 2002;262:1–24. doi: 10.1007/978-3-642-56029-3_1. [DOI] [PubMed] [Google Scholar]
- Clegg J.C., Bowen M.D. In: Virus Taxonomy. Seven Report on the International Committee for the Taxonomy of Viruses. Van Regenmortel M.H.V., Fauquet C.M., Bishop D.H.L., Carsten E., Estes M.K., Lemon S.M., Maniloff J., Mayo M., McGeoch D.J., Pringle C.R., Wickner R.B., editors. Academic Press; New York: 2000. pp. 633–640. [Google Scholar]
- Enria D.A., Feuillade M.R., Levis S., Briggiler A.M., Ambrosio A.M., Saavedra M.C., Becker J.L., Aviles G., Garcia J., Sabattini M. In: Factors in the Emergence and Control for Rodent-Borne Viral Diseases. Saluzzo J.F., Dodet B., editors. Elsevier; Paris: 1999. pp. 273–279. [Google Scholar]
- Enria D.A., Maiztegui J.I. Antiviral Res. 1994;23:23–31. doi: 10.1016/0166-3542(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Froeschke M., Basler M., Groettrup M., Dobberstein B. J. Biol. Chem. 2003;278:41914–41920. doi: 10.1074/jbc.M302343200. [DOI] [PubMed] [Google Scholar]
- Garcia C.C., Candurra N.A., Damonte E.B. Antiviral Chem. Chemother. 2000;11:231–237. doi: 10.1177/095632020001100306. [DOI] [PubMed] [Google Scholar]
- Glushakova S.E., Iakuba A.I., Vasiuchkov A.D., Mar’iankova R.F., Kukareko T.M., Stel’makh T.A., Kurash T.P., Lukashevich I.S. Vop. Virusol. 1990;35:146–150. [PubMed] [Google Scholar]
- Hall W.C., Geisbert T.W., Huggins J.W., Jahrling P.B. Am. J. Trop. Med. Hyg. 1996;55:81–88. doi: 10.4269/ajtmh.1996.55.81. [DOI] [PubMed] [Google Scholar]
- Harman A., Browne H., Minson T. J. Virol. 2002;76:10708–10716. doi: 10.1128/JVI.76.21.10708-10716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahrling P.B. In: Medical Aspects of Chemical and Biological Warfare. Zajtchuk R.M.C., editor. Office of The Surgeon General; Bethesda: 1997. pp. 591–602. [Google Scholar]
- Jeetendra E., Ghosh K., Odell D., Li J., Ghosh H.P., Whitt M.A. J. Virol. 2003;77:12807–12818. doi: 10.1128/JVI.77.23.12807-12818.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinomoto M., Yokoyama M., Sato H., Kojima A., Kurata T., Ikuta K., Sata T., Tokunaga K. J. Virol. 2005;79:5996–6004. doi: 10.1128/JVI.79.10.5996-6004.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz S., Edelmann K.H., de la Torre J.C., Gorney R., Oldstone M.B. Virology. 2003;314:168–178. doi: 10.1016/s0042-6822(03)00421-5. [DOI] [PubMed] [Google Scholar]
- Leifer E., Gocke D.J., Bourne H. Am. J. Trop. Med. Hyg. 1970;19:677–679. doi: 10.4269/ajtmh.1970.19.677. [DOI] [PubMed] [Google Scholar]
- Lenz O., ter Meulen J., Klenk H.D., Seidah N.G., Garten W. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12701–12705. doi: 10.1073/pnas.221447598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron D.M., Ames B.N. Mutat. Res. 1983;113:173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- Oldfield V., Keating G.M., Plosker G. Drugs. 2005;65:1139–1160. doi: 10.2165/00003495-200565080-00007. [DOI] [PubMed] [Google Scholar]
- Peters C.J., Jahrling P.B., Liu C.T., Kenyon R.H., McKee K.T., Jr., Barrera Oro J.G. Curr. Top. Microbiol. Immunol. 1987;134:5–68. doi: 10.1007/978-3-642-71726-0_2. [DOI] [PubMed] [Google Scholar]
- Petkevich A.S., Sabynin V.M., Lukashevich I.S., Galegov G.A., Votiakov V.I. Vop. Virusol. 1981:244–245. [PubMed] [Google Scholar]
- Rawls W.E., Banerjee S.N., McMillan C.A., Buchmeier M.J. J. Gen. Virol. 1976;33:421–434. doi: 10.1099/0022-1317-33-3-421. [DOI] [PubMed] [Google Scholar]
- Southern P.J. In: Knipe D.M., Howley P.M., editors. vol. 2. Lippincott & Wilkins; Pennsylvania: 2001. pp. 1505–1551. (Virology). [Google Scholar]
- Wachsman M.B., Lopez E.M., Ramirez J.A., Galagovsky L.R., Coto C.E. Antiviral Chem. Chemother. 2000;11:71–77. doi: 10.1177/095632020001100107. [DOI] [PubMed] [Google Scholar]
- Weissenbacher M.C., Coto C.E., Calello M.A., Rondinone S.N., Damonte E.B., Frigerio M.J. Infect. Immun. 1982;35:425–430. doi: 10.1128/iai.35.2.425-430.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J.T., Johnston P.B., Dubay S.R., Hunter E. J. Virol. 2001;75:9601–9612. doi: 10.1128/JVI.75.20.9601-9612.2001. http://www.bt.cdc.gov/agent/agentlist-category.asp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q., Compans R.W. J. Virol. 1995;69:7045–7053. doi: 10.1128/jvi.69.11.7045-7053.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]