

Abstract
Emerging infectious diseases such as SARS and the bioterror attacks with anthrax spores that occurred after September 11th, 2001 have highlighted the need to be better prepared for the detection and management of infectious pathogens that threaten public health. Negative staining electron microscopy is one method used to screen environmental and clinical samples for relevant infectious pathogens. Unfortunately, bacterial endospores, like those of Bacillus anthracis, are difficult to identify using this method because of their density that prevents imaging of structural details. Thin section electron microscopy would be an alternative method but this usually requires a few days for preparation and diagnosis. In the present paper we describe the development of a rapid thin section protocol, using mainly Bacillus subtilis spores as a model, which allows an unequivocal diagnosis of endospores within 2 h. The protocol involves chemical fixation assisted by heat or microwaves, rapid dehydration, embedding in the low-viscosity resin LR White and chemically enhanced polymerization. Structural preservation of spores is comparable to preservation after standard Epon embedding. Immunolabeling experiments using B. atrophaeus spores and a specific antibody suggest that the protocol preserves significant antigenicity for on-section immunocytochemistry and therefore offers the possibility for the strain typing of spores using specific antibodies. Further experiments with vegetative bacteria, viruses and cell cultures indicate that the rapid thin section protocol not only preserves spores but also other biological structures. Because of its universality and speed the described protocol complements negative staining electron microscopy as a front line method for the morphology-based diagnosis of pathogens in environmental and clinical samples.
Keywords: Anthrax, Bacterial endospore, Diagnostic electron microscopy, Immunogold labeling, Rapid embedding, Ultrastructure
1. Introduction
Diagnostic electron microscopy is a beneficial method for the diagnosis of infectious diseases (Hazelton and Gelderblom, 2003, Curry et al., 2006). In particular, negative staining electron microscopy is used to visualize the causative agent of infectious diseases in certain clinical and environmental samples. The method is quick and involves the adsorption of particles in a suspension onto a particular support followed by staining with a heavy metal salt and drying. Transmission electron microscopy of such preparations allows a diagnosis to the group or family level based on morphological criteria without the need for any specific primers or antibodies that limit the diagnostic spectrum. Even viruses and bacteria that are not on the list for routine diagnostics or are genetically modified to avoid detection can therefore be identified within a few tens of minutes. The value of the method was recently demonstrated during the SARS epidemic where diagnostic electron microscopy gave the first indications that the causative virus was a member of the coronavirus family (WHO, 2003).
Since the events of 11th September, 2001 and the receipt of letters containing weapon grade spores of Bacillus anthracis (Anthrax) in the USA shortly after, diagnostic electron microscopy became a front line method for the inspection of suspect material in many public health institutions (Miller, 2003, Hazelton and Gelderblom, 2003). However, negative staining electron microscopy is limited to small particles, depending on their density and the acceleration voltage of the electron microscope. Bacterial endospores, such as those of B. anthracis and related strains, are too dense to allow visualization of structural details other than size and form with negative staining electron microscopy (Fig. 1 ), which prevents their reliable diagnosis. The safe detection or exclusion of relevant infectious agents is particularly of paramount importance in a bioterroristic scenario where environmental samples are collected.
Fig. 1.
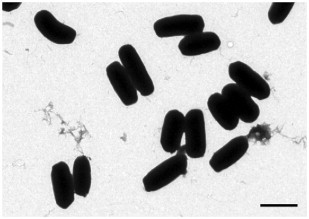
Negative staining transmission electron microscopy of Bacillus subtilis spores. Spores appear as black, brick-like particles revealing no structural detail apart from their form and size. Bar = 1 μm.
The necessary structural information about samples of high density can be obtained by thin section electron microscopy. This method produces thin sections of a sample that are transparent enough for the electron beam, and visualizes the internal ultrastructure in the transmission electron microscope (see for an overview Bozzola and Russell, 1998). In the case of bacterial endospores, the particular structural signature, i.e. the complex coat structures that surround the dense membrane-bound spore core (Setlow, 2005), can provide an unequivocal diagnosis. However, a major drawback of this method is the comparatively long preparation time using standard protocols, which usually requires several days. Much effort has therefore been invested in reducing the processing time. For instance, increasing the curing temperature of resin polymerization reduces the processing time significantly (Doane et al., 1974) and the use of microwaves allows better heat distribution in the sample and could have non-thermal effects, which increases the rate of diffusion and/or the speed of chemical reactions (Kok and Boon, 1990, Leong and Sormunen, 1998). In all, the total processing time from native sample to a microscopic diagnosis can be reliably reduced to about 3–4 h (Giberson et al., 2003, Schröder et al., 2006) or even less (Gove et al., 1990) using optimized methods. Most protocols use epoxy resin as an embedding medium, often in conjunction with osmium tetroxide post-fixation, which provides a good representation of the ultrastructure but usually does not preserve antigenicity sufficiently for on-section immunocytochemistry.
The major goal of the study presented in this paper was to develop a simple and rapid thin section protocol for bacterial endospores that also allows on-section immunolabeling for additional molecular typing using antibodies. Our strategy involved reducing the sample size and using the low-viscosity acrylic resin LR White (Newman et al., 1982), which efficiently reduced both the diffusion time during dehydration and infiltration with the resin. In addition, the polymerization speed of LR White was drastically increased by the use of a chemical accelerator (Newman and Hobot, 1987). These changes, together with an improved method for chemical fixation, reduced the total processing time for spores that have significant diffusion barriers (Driks, 1999) and are therefore difficult to process to about 2 h including diagnosis. The paper describes and evaluates the rapid LR White embedding protocol using bacterial endospores and other samples, including virus and cell cultures.
2. Materials and methods
2.1. Bacterial, viral and cellular strains
Vegetative cells of Bacillus subtilis (ATCC, strain 6633) were plated on semolina agar (50% semolina, 2% agar) to induce sporulation. Spores were harvested after 3 weeks of culture and stored in 0.05 M Hepes buffer at 4 °C until use.
Bacillus atrophaeus spores (strains DSM 2277 and DSM 675) were prepared according to the protocol of a European standard test procedure (EN 14347, 2004).
Escherichia coli (E. coli wild type strain RL 462-05; kindly provided by Dr. Lothar Beutin, Federal Institute for Risk Assessment) was cultivated on blood agar. Colonies were harvested with a loop and resuspended in distilled water for 1 h before fixation.
Vaccinia virus (strain VR-1536) was propagated in Hep G2 cells. The culture supernatant was harvested and fixed as indicated below.
Jurkat cells (clone E-6; ATCC TIB 152) originally derived from an acute T cell leukemia patient and have the morphology of lymphocytes. These were grown in suspension in complete medium to a cell density of approximately 106 cells/ml.
Mouse L20B cells are fibroblast cells expressing the human poliovirus receptor (Pipkin et al., 1993) and were cultivated in complete medium on a plastic substrate to near confluency.
2.2. Chemical fixation
All samples were chemically fixed in Hepes-buffered (0.05 M, pH 7.2) aldehyde (formaldehyde and/or glutaraldehyde). Fixatives were prepared immediately before use. Formaldehyde was freshly depolymerized from paraformaldehyde or from a frozen stock of concentrated formaldehyde solution by heating to 60–70 °C for at least 30 min. Virus suspensions were mixed with concentrated buffered formaldehyde solution at a final concentration of 2% formaldehyde and 0.05 M Hepes (pH 7.2). Bacteria and spores were spun down and resuspended with 2.5% glutaraldehyde in 0.05 M Hepes (pH 7.2) or 10% formaldehyde, 0.05% glutaraldehyde in 0.05 M Hepes (pH 7.2) (standard fixative for spores). L20B cells were fixed in the culture flask by removing the medium and adding 2.5% glutaraldehyde in 0.05 M Hepes (pH 7.2). Jurkat cells were spun down and resuspended in 2.5% glutaraldehyde in Hepes buffer (pH 7.2). Fixation was carried out at room temperature for at least 1 h (2 h in case of spores) unless otherwise stated.
2.3. Microwave-assisted chemical fixation
Spore suspensions were heated in a microwave oven (Rapid Electron Microscopy Tissue Processing System; Milestone, Italy) that uses and regulates a defined microwave energy output to achieve and maintain the desired sample temperature. All experiments were performed using the Histomodule F/H, a cylinder of 2.5 cm diameter, filled with distilled water. For microwave-assisted chemical fixation, spore suspensions were mixed with concentrated fixative (to give a final concentration of 10% formaldehyde, 0.05% glutaraldehyde in 0.05 M Hepes buffer) in a reaction tube (1.5 ml total volume, containing 1.2 ml of the suspension), which was then placed into the Histomodule. The adjustable variables for microwaving were temperature, time and microwave power (relative units in percentage; 15% of 800 W is recommended by the manufacturer). Various time–temperature profiles were applied (compare Section 3.2 and Table 1 ) and actual sample temperatures in the microwave oven were determined beforehand using a thermocouple and a reaction tube filled with distilled water.
Table 1.
Structural preservation of Bacillus subtilis spores after microwave-assisted chemical fixation
| No. | Fixation parameters | Microwave parameters | Round [%] | Condensed [%] | n |
|---|---|---|---|---|---|
| 1.1 | 24 h, RTa | – | 74 | 26 | 54 |
| 1.2 | 10′, RT | – | 0 | 100 | 51 |
| 1.3 | 5′, RT/5′, 37°, MW | 2′, RT→37°/3′, 37° | 35 | 65 | 54 |
| 1.4 | 10′, RT/5′, 37°, MW | 2′, RT→37°/3′, 37° | 8 | 92 | 53 |
| 2.1 | 24 h, RTa | – | 59 | 41 | 56 |
| 2.2 | 10′, 37°, MW | 2′, RT→37°/8′, 37° | 44 | 56 | 66 |
| 2.3 | 5′, RT/10′, 37°, MW | 2′, RT→37°/8′, 37° | 40 | 60 | 63 |
| 2.4 | 5′, RT/5′, 37°, MW | 2′, RT→37°/3′, 37°, 100% | 19 | 81 | 59 |
| 3.1 | > 12 h, RTa | – | 83 | 17 | 60 |
| 3.2 | 15′, 37°, MW | 2′, RT→37°/13′, 37° | 42 | 58 | 59 |
| 3.3 | 5′, RT/10′, 37°, MW/5′, RT/5′, 37°, MW | 2′, RT→37°/8′, or 3′, 37° | 16 | 84 | 61 |
| 3.4 | 5′, RT/5′, 37–50°, MW | 2′, RT→37°/3′, 37→50° | 79 | 21 | 62 |
| 4.1 | 24 h, RTa | – | 34 | 66 | 58 |
| 4.2 | 5′, RT/10′, 37–50°, MW | 2′, RT→37°/2′, 37°→50°/6′, 50° | 81 | 19 | 59 |
| 4.3 | 5′, RT/10′, 50–60°, MW | 2′, RT→37°/2′, 37°→50°/4′, 50°/2′, 50°→60° | 85b | 15 | 60 |
| 4.4 | 5′, RT/10′, 37–50°, MW | 2′, RT→37°/5′, 37°/3′ 37°→50° | 63 | 37 | 62 |
| 5.1 | 24 h, RTa | – | 47 | 53 | 62 |
| 5.2 | 24 h, RTa | Depolymerization of PFA/2′ RT→70°, 100%/8′, 70° | 51 | 49 | 61 |
| 5.3 | 5′, RT/10′, 37–50°, MW | 2′, RT→37°/2′, 37°→50°/6′, 50° | 93 | 7 | 59 |
| 5.4 | 5′, RT/5′, 37–50°, MW | 2′, RT→37°/3′, 37°→50° | 31 | 69 | 64 |
In five experiments (1–5) spore core preservation was evaluated. Round spore cores indicate a sufficient ultrastructural preservation.
MW = microwave.
RT = room temperature.
Microwave parameters: x′, RT→37° = increase from room temperature to 37 °C within x min; x′, 37° = holding at 37 °C for x min.
Microwave power was set to 15% except of No. 2.4 and 5.2.
Fixation was done at room temperature (RT) for 1 h and the remaining time at 4 °C.
Membrane preservation was insufficient.
2.4. Negative staining
For the direct visualization of B. subtilis spores in the transmission electron microscope a drop (5 μl) of a spore suspension was added to a filmed grid surface. The grid surface had been pre-treated with Alcian blue for better adsorption of particles (Biel and Gelderblom, 1999). After 10 min incubation at room temperature excess suspension was removed by blotting with a filter paper and grids were washed on four drops of distilled water. Finally the grid was placed briefly on a drop of 1% aqueous uranyl acetate and blotted dry.
2.5. Airfuge concentration of virus suspension
Prior to centrifugation, antibodies coupled to 10 nm gold particles were mixed with the virus suspension (10 μl with 160 μl suspension per tube). The gold colloids stain the pellet a dark red color allowing it to be localized in the centrifugation tube. Virus suspensions were spun down with the Airfuge desktop ultracentrifuge (Beckman Coulter, USA) at 20 psi (approx. 100.000 g) in an A-100 rotor using polyallomer tubes (5 × 20 mm; 240 μl) for 15 min. The supernatant was then removed and the pellet covered with a thin layer of low-melting point agarose (3% in distilled water) and embedded in situ.
2.6. Embedding of suspensions in thin agarose gels
To allow better handling during processing and to guarantee a reproducible sample thickness, suspensions were embedded in agarose gels according to De Camilli et al. (1983). Briefly, spore or cell suspensions were sedimented by centrifugation. Pellets were resuspended with low-melting point agarose (3% in distilled water; mixed 1:1 with the pellet) at 40 °C and filled between two microscope slides separated by 0.3 mm spacers. After solidification at 4 °C, the gel was cut into small strips and immersed briefly in Alcian blue (0.1% in 0.1% acetic acid) and excess Alcian blue was washed away by immersion in 0.05 M Hepes buffer (pH 7.2). This staining increases the visibility of the strips during processing, particularly during trimming and sectioning.
2.7. Embedding in LR White
The general principles for embedding in LR White at low temperature (i.e. dehydration, chemically accelerated polymerization) have already been described by Newman and Hobot (1987). Incubation times in the following basic protocol were determined empirically. In some experiments samples were additionally fixed in 1% osmium tetroxide buffered with 0.05 M Hepes for 30 min prior to dehydration.
Dehydration, infiltration with LR White (hard grade; The London Resin Ltd.) and final polymerization were carried out on ice to reduce extraction. For dehydration, samples were immersed in ethanol (70%, 100%, 100%; 5 min each). Sample infiltration with the resin was performed with a mixture of ethanol and LR White (1:1) and pure LR White (twice) for 5 min each. For embedding, LR White monomer was thoroughly mixed with LR White accelerator (2.5 μl/ml monomer) and added to reaction tubes (0.5 ml, Eppendorf Safe lock). Agarose gel strips were then transferred into the tubes for polymerization. Virus pellets were processed in their centrifuge tubes, filled with the monomer (including accelerator) and sealed for polymerization with a tight fitting gelatin capsule cap (size “3”). Samples were then left for 40 min on ice and 5 min at 60 °C for final polymerization. A thin layer of unpolymerized monomer resin usually remains on top of the block and was removed by soaking with a tissue. Blocks were finally cut out of the reaction vials or centrifuge tubes.
2.8. Embedding in epoxy resin (Epon)
Embedding in epoxy resin (Epon, i.e. glycidyl ether; mixture according to Luft, 1961) was done without any prior post-fixation or block contrasting. Samples were dehydrated in a series of ethanol (30%, 50%, 70%, 95%, 100%) and infiltrated with the resin using mixtures of propylene oxide and resin followed by pure resin. Polymerization was carried out at 60 °C for 48 h.
2.9. Ultramicrotomy and electron microscopy
Stained gels or pellets were easily identified within the resin block using a stereo microscope and trimmed with a razor blade or a trimming device equipped with a diamond blade (TM-30; Reichert-Jung, Austria). Ultrathin sections (60–80 nm) were cut with an ultramicrotome (Ultracut S or UCT; Leica, Germany) and picked up on slot grids covered with a pioloform supporting film. To add contrast, sections were stained with uranyl acetate (2% in distilled water) and lead citrate (0.1% in distilled water) or with uranyl acetate mixed with methyl cellulose (1.8%/0.2% in distilled water) according to Roth et al. (1990). The latter method is very simple and quick (1 min incubation time; no washing steps) and gives a mixture of negative and positive contrast.
Sections were inspected with a Tecnai Spirit transmission electron microscope (FEI Co., USA) at 120 kV. Either a 1 k (Megaview III; Olympus Soft Imaging Solutions GmbH, Germany) or a 2 k (TVIPS 214; Tietz, Germany) slow-scan camera was used for image acquisition. Negatively stained spores were visualized with an EM902 (ZEISS, Germany) at 80 kV and a 1 k slow-scan camera (Proscan, Germany).
2.10. Post-embedding immunolabeling
On-section immunolabeling was done by incubating the grids, sections down, on drops (30 μl) of the following solutions: glycine (50 mM) in phosphate-buffered saline (PBS, pH 7.2); blocking solution of fish gelatin (0.5%), bovine serum albumin (0.5%) and Tween 20 (0.01%) in PBS; primary antibody (diluted in blocking solution; incubated overnight at 4 °C); blocking solution; secondary antibody (coupled to 10 nm gold; 1.5 h at room temperature); blocking solution; PBS; distilled water. To stabilize the binding between antigen and antibody complexes, sections were treated with 0.25% glutaraldehyde in PBS for 30 min at room temperature. The primary antibody was a purified rabbit antibody (IgG fraction) raised against formaldehyde inactivated spores of B. atrophaeus. A mixture of sera raised against green fluorescent protein (BD Biosciences, USA) was used at the same concentration as a control. Finally, sections were contrasted with a mixture of uranyl acetate and methyl cellulose (compare Section 2.9).
3. Results
3.1. Comparison between rapid LR White and standard Epon embedding
To check the quality of the rapid thin section protocol, B. subtilis spores were chemically fixed (10% formaldehyde, 0.05% glutaraldehyde in 0.05 M Hepes) and either embedded in LR White or in Epon. Treatment with osmium tetroxide or uranyl acetate was omitted with both protocols. Sections were stained with uranyl acetate followed by lead citrate. Both embedding protocols, LR White and Epon, allowed ultrastructural details of the spores to be visualized with comparable quality (Fig. 2A, B). The spore core appeared round and the delimiting membrane was preserved. Outer and inner spore coats could be distinguished. However, the overall contrast was slightly lower in the LR White embedded samples than in Epon samples making differentiation of coat substructures difficult. To improve the visibility of structures and to reduce the staining time from 10–20 min to 1 min, LR White samples were stained with a mixture of uranyl acetate and methyl cellulose (compare Section 2.9 for details). This staining procedure resulted in a mixture of positive and negative contrast where membranes in particular appear negative (i.e. white) because of the surrounding stain (Fig. 2C, D). Using this very simple and quick on-section staining procedure, substructures of the spore coats became visible and the core membrane appeared to be better defined (Fig. 2C).
Fig. 2.
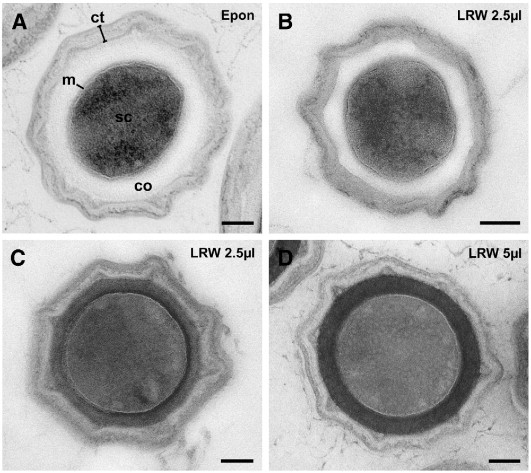
Cross sections through Bacillus subtilis spores embedded using different protocols. Apart from the contrast, no significant differences between the spores are visible. A. Epon embedding (without block contrasting and osmium tetroxide post-fixation). On-section staining with uranyl acetate and lead citrate. B. LR White rapid embedding using 2.5 μl accelerator per ml monomer resin (LRW 2.5 μl). On-section staining as in A. C. LR White rapid embedding as in B but on-section staining with uranyl acetate/methyl cellulose. D. LR White rapid embedding using 5 μl accelerator per ml monomer resin (LRW 5 μl) and on-section staining as in C. (ct) outer and inner spore coats; (co) cortex; (sc) spore core; (m) core membrane. Bars = 100 nm.
3.2. Testing and improving different parameters of the rapid LR White embedding protocol
The quality of the rapid LR White embedding protocol depends on various parameters. Three of these parameters, sample size, polymerization time and fixation strength, were tested for their influence on the structural preservation using a B. subtilis spore suspension as a test sample.
Sample sizes of 0.18, 0.3, 0.85 and 1.1 mm thickness were made by increasing the distance between the slides forming the gel chamber. No significant differences were detectable between the different sample sizes, with infiltration and polymerization always being sufficient and structural details being equally preserved (not shown).
The polymerization time (i.e. time until the resin had completely solidified) depends on the concentration of the accelerator. Polymerization experiments with the pure resin employing different concentrations of accelerator at 0 °C showed that the polymerization time decreased with increasing concentration of the accelerator: 2.5 μl ≈ 40 min; 3 μl ≈ 30 min; 4 μl ≈ 20 min; 5 μl ≈ 15 min. To check whether rapid polymerization affects ultrastructural preservation, 0.3 mm thick agarose gels of B. subtilis spore suspensions were embedded in LR White using differing amounts of accelerator. Even the highest concentration of accelerator gave ultrastructural preservation that could not be distinguished from that found using lower concentrations of accelerator (Fig. 2 C, D). Concentrations above 5 μl accelerator per ml monomer were not useful for sample embedding because the resin gelled very quickly while the sample remained soft (not shown). Using 5 μl accelerator per ml resin, it was possible to complete dehydration, infiltration with resin monomer, embedding and polymerization within an hour. Together with sample preparation before embedding (not including fixation), ultramicrotomy, staining and microscopy an overall processing time of well below 2 h was achieved.
In all of the experiments described above, samples were stabilized using the standard fixative consisting of 10% formaldehyde and 0.05% glutaraldehyde in Hepes buffer for at least 2 h at room temperature. To cut down the overall processing time for the procedure (including fixation), different fixation times and fixatives were tested. Reducing the fixation time for the standard fixative from 2 h to 30 min or 10 min also reduced the degree of structural preservation: the shorter the fixation time, the worse the degree of structural preservation. The cores of almost all spores fixed for 10 min at room temperature were condensed (Table 1, No. 1.2) and unfixed material appeared to leak out of the spores during sectioning to cover other structures (Fig. 3A). Fixation with 2.5% glutaraldehyde for 30 min up to 4 h also did not result in acceptable structural preservation (Fig. 3B), with the spore cores especially again being condensed.
Fig. 3.
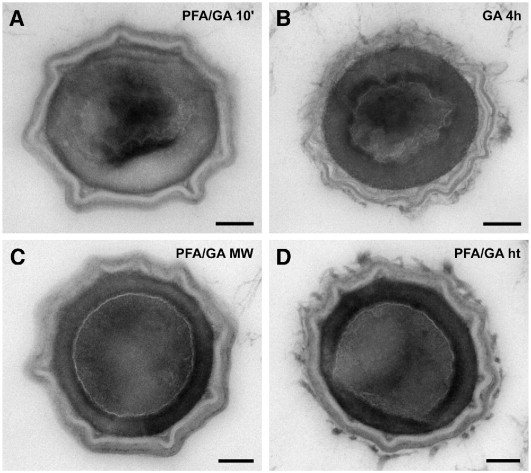
Cross sections through Bacillus subtilis spores after rapid embedding in LR White (2.5 μl accelerator per ml monomer) and using different fixation protocols. Fixation was done with a final concentration of: A. 10% formaldehyde, 0.05% glutaraldehyde in 0.05 M Hepes buffer (pH 7.2) for 10 min at room temperature (PFA/GA 10′); B. 2.5% glutaraldehyde for 4 h at room temperature (GA 4 h); C. 10% formaldehyde, 0.05% glutaraldehyde in 0.05 M Hepes buffer (pH 7.2) for 5 min at room temperature followed by 2 min heating to 37 °C, 2 min heating from 37 °C to 50 °C and 6 min holding at 50 °C in a microwave oven (PFA/GA MW); D. 10% formaldehyde, 0.05% glutaraldehyde in 0.05 M Hepes buffer (pH 7.2) for 5 min at room temperature followed by 10 min at 50 °C in a water bath (PFA/GA ht). Bars = 100 nm.
To reduce the rather long fixation times, a microwave-assisted procedure was tested in a series of experiments using the standard fixative (Table 1). Fixation for at least 12 h (1 h at room temperature and the remainder at 4 °C) served as a control. Remarkably, variation was seen in the preservation of control samples between experiments (Table 1, No. 1.1, 2.1, 3.1, 4.1, 5.1). In order to rule out the possibility that depolymerization of the formaldehyde by heat was incomplete, depolymerization by microwaves was compared with depolymerization by heat in one experiment. One beneficial effect of the microwave fixation might be the efficient depolymerization of formaldehyde polymers (Giberson and Elliott, 2001) that usually form in formaldehyde solutions (Griffiths, 1993). However, no differences in structural preservation could be detected using the two depolymerization procedures (Table 1, No. 5.1, 5.2).
A sufficient and reliable preservation of spore ultrastructure was achieved using 5 min of pre-fixation at room temperature followed by a step-wise increase of temperature to 50 °C in a microwave oven (Table 1, No. 4.2 and 5.3; Fig. 3C). Microwave-assisted fixation without pre-fixation at room temperature (Table 1, No. 2.2, 3.2) or shorter fixation times (Table 1, No. 3.4, 5.4) did not give acceptable structural preservation (Note that the sufficient core preservation by the shorter microwave-assisted fixation at 50 °C [Table 1, No. 3.4] could not be reproduced [Table 1, No. 5.4]). Heating to 50 °C appeared to be essential because lower (Table 1, No. 2.3, 3.3) and higher (Table 1, No. 4.3) microwave-assisted fixation temperatures were also unable to effectively preserve spore ultrastructure. The spores that were heated to 60 °C, although having round non-compressed cores, had no visible membranes or even in many cases showed blebs. However, heating the spore suspension to 50 °C in a water bath resulted in a degree of structural preservation acceptable for diagnostic purposes. Spore coats remained intact and the core membrane was visible in most of the spores (96%, n = 108). However, the cores of many spores (≈ 70%) showed some degree of deformation (Fig. 3D).
In summary the experiments allowed an improved protocol to be developed that involved a modified chemical fixation using microwaves or heat and polymerization of the LR White using 5 μl accelerator per ml of monomer. As a consequence the total processing time for spore suspensions, including chemical fixation and diagnosis under the electron microscope, could be reduced to about 2 h (Table 2 ).
Table 2.
Rapid 2 h protocol for thin section electron microscopy of bacterial endospores
| Operation | Time [min] |
|---|---|
| Fixation | 15 |
| Centrifugation or agarose gel preparation | 10 |
| Dehydration with ethanol (70, 100, 100%) | 15 |
| Infiltration with LR White (1:1 with ethanol, pure, pure) | 15 |
| Polymerization (5 μl accelerator/ml monomer) | 20 |
| Sectioning | 15 |
| Staining | 1 |
| Microscopy | 10 |
| Time between steps | 15 |
| Total | 116 |
Processing time is given for 1–2 samples.
3.3. Preservation of antigenicity
Samples embedded in LR White resin usually retain at least a degree of antigenicity that can be used for post-embedding immunocytochemistry. However, it was unclear if antigenicity would still be preserved using the rapid dehydration and embedding protocol. To evaluate the retention of antigenicity, spores of B. atrophaeus were embedded with 2.5 μl of accelerator per ml resin. Sections were incubated with an antibody that had been raised against inactivated B. atrophaeus spores. A control serum raised against peptides of the green fluorescence protein was used at the same concentration as the specific antiserum. The results clearly demonstrate labeling of the spore coat by the relevant antibodies and an absence of labeling with the controls (Fig. 4 ).
Fig. 4.
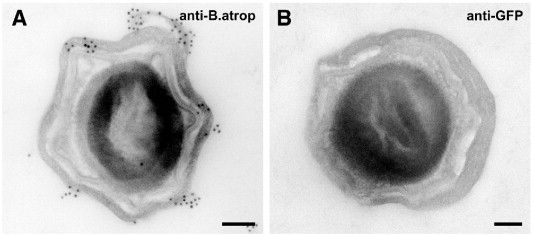
Post-embedding immunogold labeling of cross sections through Bacillus atrophaeus spores. Sections were labeled with an antibody raised against formaldehyde inactivated B. atrophaeus spores (A, anti-B. atrophaeus) or an irrelevant antibody raised against the green fluorescent protein (B, anti-GFP) followed by a secondary antibody coupled to 10 nm gold colloid. Spores treated with the anti-B. atrophaeus antibody are decorated with gold colloids (dark round particles) mainly at their outer coats (A) while spores treated with the anti-GFP antibody showed no such labeling (B). Bars = 100 nm.
3.4. Rapid embedding of viruses, bacteria and eukaryotic cells
The versatility of the rapid thin section protocol was evaluated in experiments using a variety of specimens and 2.5 μl accelerator per ml of resin.
E. coli bacteria were embedded into agarose gels (0.3 mm thick) and processed for LR White embedding. The bacteria were found to be well preserved (Fig. 5A, B), with both membranes and the periplasmatic space distinguishable (Fig. 5 B). The cytoplasm appeared homogenous with regions of higher and lower density but without significant condensation (Fig. 5A). The addition of 1% osmium tetroxide as a post-fixation step increased the contrast but did not change the overall structural appearance (not shown).
Fig. 5.
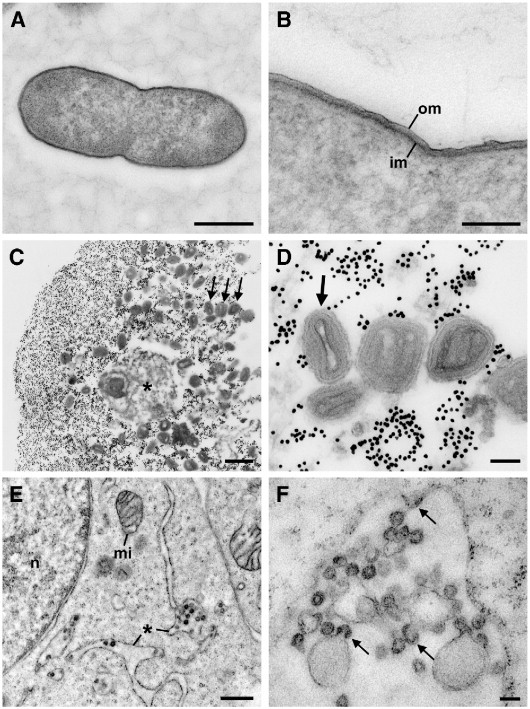
Ultrathin sections of bacteria, virus and cell cultures after rapid embedding in LR White (2.5 μl accelerator per ml monomer). A, B. Longitudinal sections through a dividing bacterial cell of Escherichia coli. The cytoplasm appears granular and dense except for the centre where it shows fine strands, probably indicating localization of the DNA (A). The cell wall shows two membranes, the outer (om) and the inner membrane (im), typical for gram-negative bacteria (B). Note that the sample was neither post-fixed with osmium tetroxide nor treated by en bloc staining with uranyl acetate. C, D. Sections through a pellet of vaccina virus (cell culture supernatant mixed with gold colloids). C. Overview of the pellet in cross section. Virus particles (arrows) and cellular debris (⁎) are surrounded by gold particles (black dots of about 10 nm size). D. Sections through virus particles in different orientations. The side view (arrow) shows the typical dumbbell shape of the core. The different membranes of the virus particles are well preserved. E, F. Retrovirus-infected L20B cells (post-fixed in 1% osmium tetroxide). Virus particles are localized in membrane-bound compartments (⁎) of the cytoplasm that are continuous with the endoplasmic reticulum (E). Virus particles are formed by budding at the compartment membrane into the lumen (arrows in F). Particles consist of a membrane and an inner ring-like structure giving in a double ring-appearance typical for type-A retrovirus particles (F). (mi) mitochondrion; (n) nucleus. Bar in A, C, E = 0.5 μm and in B, D, F = 100 nm.
Vaccinia virus suspensions (fixed in 2% formaldehyde) were mixed with gold colloids and spun down using a desktop ultracentrifuge. The pellet was easily detected by the dark red color of the concentrated gold colloid. Virus particles and debris from the host cells were embedded with numerous gold particles at the pellet rim (Fig. 5 C, D) while in central regions of the pellet, the gold particle concentration was greatly reduced and viral particles plus cellular debris formed a thick layer (not shown). Viral particles showed the typical structure of orthopoxviruses, e.g. with a dumbbell shaped core when viewed from the side (Fig. 5D).
Fixed L20B cells were scraped from the bottom of their culture flasks, resuspended and finally spun down using a desktop microfuge. Pellets were mixed with low-melting point agarose and made into 0.3 mm thick gels. Samples were either post-fixed with 1% osmium tetroxide (30 min) before dehydration and embedding or directly dehydrated and embedded in LR White. Cellular ultrastructure was sufficiently preserved to allow identification of relevant organelles and retrovirus particles (Fig. 5E). The virus particles were localized in membrane-bound compartments that were most probably continuous with the endoplasmic reticulum. Membrane bound particles were fixed in the act of budding from the cytoplasm into the lumen of the compartment (Fig. 5F). The virus particles had the morphology characteristics of type A particles, i.e. a double-ring structure of membrane and protein lacking a typical core (Fig. 5F). All of these structural hallmarks, albeit with a reduced contrast, could also be seen in samples that had not been post-fixed with osmium tetroxide (not shown).
Jurkat cells embedded without prior osmium tetroxide post-fixation showed reasonable contrast at low power if the sections were treated with uranyl acetate/methyl cellulose on-section staining (Fig. 6A). Even at higher power, structural details important for diagnostic purposes, such as membranes and cytoskeletal elements, could be identified, although membrane contrast was relatively low (Fig. 6B, C, D).
Fig. 6.
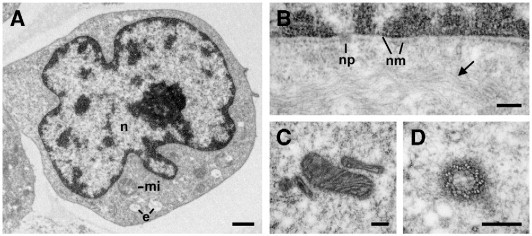
Structural preservation of Jurkat cells processed with the rapid thin section embedding protocol using LR White (2.5 μl accelerator per ml monomer). A. View of a typical cell with the morphology of a lymphocyte. The central nucleus (n) is surrounded by a thin layer of cytoplasm with only few organelles, such as mitochondria (mi) and endosomes (e). B. Nuclear membrane (nm) with nuclear pores (np). The cytoplasm contains bundles of microfilaments (arrow). C. Mitochondria displaying their inner membrane system. D. A centriole with the typical triplet organization of microtubules (9 × 3). Bar in A = 1 μm and in B, C, D = 0.2 μm.
4. Discussion
The diagnosis of pathogens in clinical or environmental samples that are suspected to pose a risk to the public must be performed as quickly as possible and therefore needs a rapid method for sample preparation. Because of their inherent speed, microwave-assisted preparation protocols in particular have been developed and widely applied to diagnostic thin section electron microscopy (Kok and Boon, 1990, Leong and Sormunen, 1998, Giberson et al., 2003). In this paper we described a rapid and rather simple protocol using LR White as a resin that allows the diagnosis of bacterial endospores within 2 h. All the relevant details of spore ultrastructure (spore core and coats) could be distinguished, although it should be noted that the appearance of these structures was somewhat unconventional, as the particular on-section staining method used (Roth et al., 1990) gives a mixture of positive and negative contrast. However, providing the characteristic signature of bacterial endospores is preserved, unequivocal diagnosis is straightforward. This has been impressively demonstrated by the diagnosis of spores in mummies at least 1000 years old (Hino et al., 1982, Pabst et al., 2005).
Recently, Schröder et al. (2006) described a rapid Epon-embedding protocol for spores that uses microwaves in many of the incubation steps and that results in virtually the same structural appearance of spores as that after conventional processing. Nevertheless, already the embedding protocol takes 3 h 15 min, resulting in total processing times well over 4 h — too long for a first diagnosis needed to assess the risk posed by a sample. In some cases even the 2 h protocol presented in this paper may be too long. However, there is little room for further improving the processing time for thin section electron microscopy because the individual preparation steps have now been optimized to the point that handling times (changing solutions, transferring samples, ultramicrotomy) now contribute significantly to the time needed. Reducing dehydration, infiltration and microwave times may gain a few minutes and the polymerization times for the acrylic resins could also be reduced using microwaves alone (Giammara, 1993) or in combination with a chemical accelerator (Hillmer et al., 1991). In all, a further reduction of 15–20 min might be possible and this will be evaluated in the future.
Spores are extremely resistant to environmental stress (i.e. heat, chemicals, radiation; Setlow, 2005) and are particularly difficult to prepare for thin section electron microscopy because their spore coats, being efficient barriers to diffusion (Driks, 1999), prevent the rapid entry of fixatives and other media into the core. We initially used a high concentration of formaldehyde in conjunction with a low concentration of glutaraldehyde for at least 2 h at room temperature because this procedure not only results in acceptable structural preservation but also inactivates the spores (Gelderblom et al., 2007). In an attempt to reduce this time we tried microwave-assisted chemical fixation using the same fixative, resulting, after several trials, in a protocol involving 5 min pre-fixation at room temperature followed by heating to 50 °C for 10 min in the microwave oven. This protocol is very similar to that used by Schröder et al. (2006) to process the various samples in their study although they used a modified Karnovsky fixative (1% paraformaldehyde, 2.5% glutaraldehyde in cacodylate buffer, pH 7.3) for 15 or 20 min at 50 °C in the same microwave oven. In contrast, studies with other samples have used microwave-assisted fixation times of a few minutes or even seconds (Gove et al., 1990, Giberson and Demaree, 1995, Login et al., 1995). In our study, however, shorter times for microwave-assisted fixation, with or without higher microwave power, failed to preserve spore ultrastructure, probably due to the unusual characteristics of spores (i.e coats and low water content) or even to the microwave oven used.
An interesting result of the fixation experiments was that acceptable preservation of spore ultrastructure was achieved by incubating for 10 min at 50 °C in a water bath after an initial 5 min at room temperature. Structural preservation was very similar to that seen with microwave-assisted fixation, with only the cores being slightly deformed. This procedure may be useful for the fixation of highly infectious species in a biosafety level 3 or 4 lab where the operation of particular microwave machines may be difficult because of space and safety restrictions. However, further studies should determine whether chemical fixation of bacterial endospores can be further optimized or not, for instance by modifying the temperature–time course of the fixation or the composition of the fixative. Furthermore, sample inactivation is important for laboratory safety (Gelderblom et al., 2007) and must be assessed for any modified fixation protocol individually.
The fact that the rapid LR White embedding protocol was able to preserve antigenicity was demonstrated by the specific labeling of the B. atrophaeus spores with an antibody raised against formaldehyde-inactivated spores. Therefore, the use of a comparatively high concentration of LR White accelerator (i.e. 2.5 μl/ml monomer), which produces more heat per time unit during polymerization than polymerization without accelerator, did not prevent specific immunolabeling. This is in line with other studies which have showed that low concentration of the accelerator (i.e. 1 μl/ml of monomer) has no significant influence on the specific immunolabeling density but can result in an increase of unspecific labeling (Goping et al., 1992). However, it is unclear whether antigenicity will be preserved if even higher accelerator concentrations (e.g. 5 μl/ml monomer) are used. This parameter, together with others that may also affect sample antigenicity, such as fixation conditions (e.g. temperature, fixative concentration, time), will be addressed in future studies.
The diagnosis of environmental or clinical samples requires that the preparation procedure maintains adequate structural preservation of most if not all possible pathogens, including those that are unexpected. We therefore prepared samples other than bacterial endospores, including vegetative bacteria, mammalian cells, and both RNA and DNA viruses using the rapid LR White protocol. In every case, structural preservation was adequate for diagnosis of the pathogen group. Although these tests were done using the slower protocol (2.5 μl accelerator), the rapid protocol (5 μl accelerator) is probably universally useable because no significant differences in ultrastructural preservation between the rapid and slower methods were observed with both spores (this paper) and cells (unpublished data).
Speed, universality and retention of antigenicity are key features of the rapid LR White preparation protocol presented in this paper. The protocol will serve as the basis for further studies addressing the strain typing of endospores from environmental samples using morphological criteria alone or in conjunction with strain-specific antibodies. Strain-typing using antibodies is particularly attractive because sectioning exposes both external and internal antigens, increasing the number of potential targets for strain-specific antibodies. In conclusion, the new protocol described here should provide a much-needed improvement for the diagnosis of environmental samples in the practical setting.
Acknowledgements
The authors would like to thank the following colleagues of the Robert Koch Institute for their support: Gabi Schlier, Marianne Hohoff, Janett Piesker and Karoline Trescher for technical assistance, Dr. Daniela Jacob for a donation of B. atrophaeus spores, Dr. Kazimiriez Madela for the preparation of B. subtilis spores, Dr. Ingeborg Schwebke for the cultivation of the L20B cells, Bakhtiyor Asomuddinov for culturing Jurkat cells, Gudrun Holland for critical reading of the manuscript and Dr. Steve Norley for “polishing” the wording of the manuscript.
References
- Biel S.S., Gelderblom H.R. Electron microscopy of viruses. In: Cann A., editor. Virus Cell Culture. Oxford University Press; 1999. pp. 111–147. [Google Scholar]
- Bozzola J.J., Russell L.D. Jones and Bartlett Publishers; Boston: 1998. Electron Microscopy: Principles and Techniques for Biologists. [Google Scholar]
- Curry A., Appleton H., Dowsett B. Application of transmission electron microscopy to the clinical study of viral and bacterial infections: present and future. Micron. 2006;37:91–106. doi: 10.1016/j.micron.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P., Harris S.M., Huttner W.B., Greengard P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J. Cell Biol. 1983;96:1355–1373. doi: 10.1083/jcb.96.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doane F.W., Anderson N., Chao J., Noonan A. Two-hour embedding procedure for intracellular detection of viruses by electron microscopy. Appl. Microbiol. 1974;27:407–410. doi: 10.1128/am.27.2.407-410.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driks A. Bacillus subtilis spore coat. Microbiol. Mol. Biol. Rev. 1999;63:1–20. doi: 10.1128/mmbr.63.1.1-20.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EN 14347, 2004. Chemische Desinfektionsmittel und Antiseptika–Sporizide Wirkung Basistest–Prüfverfahren und Anforderungen Phase 1, Stufe 1, Deutsche Fassung.
- Gelderblom H., Bannert N., Pauli G. Arguments pro disinfection in diagnostic electron microscopy: a response to Madeley and Biel. J. Infect. 2007;54:307–308. doi: 10.1016/j.jinf.2006.03.032. [DOI] [PubMed] [Google Scholar]
- Giammara B.L. Microwave embedment for light and electron microscopy using epoxy resins, LR White, and other polymers. Scanning. 1993;15:82–87. [Google Scholar]
- Giberson R.T., Demaree R.S., Jr. Microwave fixation: understanding the variables to achieve rapid reproducible results. Microsc. Res. Tech. 1995;32:246–254. doi: 10.1002/jemt.1070320307. [DOI] [PubMed] [Google Scholar]
- Giberson R.T., Elliott D.E. Microwave-assisted formalin fixation of fresh tissue. In: Giberson R.T., Demaree R.S. Jr., editors. Microwave Techniques and Protocols. Human Press; Totowa: 2001. pp. 191–208. [Google Scholar]
- Giberson R.T., Austin R.L., Charlesworth J., Adamson G., Herrera G.A. Microwave and digital imaging technology reduce turnaround times for diagnostic electron microscopy. Ultrastruct. Pathol. 2003;27:187–196. doi: 10.1080/01913120309937. [DOI] [PubMed] [Google Scholar]
- Goping G., Yedgar S., Pollard H.B., Kuijpers G.A.J. Flat embedding and immunolabeling of SW 1116 colon carcinoma cells in LR White: an improved technique in light and electron microscopy. Microsc. Res. Tech. 1992;21:1–9. doi: 10.1002/jemt.1070210102. [DOI] [PubMed] [Google Scholar]
- Gove D.W., Lang C.A., Waterhouse L.K., Leong A.S.-Y. Rapid microwave-stimulated fixation of fine-needle aspiration biopsies for transmission electron microscopy. Diagn. Cytopathol. 1990;6:68–71. doi: 10.1002/dc.2840060115. [DOI] [PubMed] [Google Scholar]
- Griffiths G. Springer; Heidelberg, Berlin: 1993. Fine Structure Immunocytochemistry. [Google Scholar]
- Hazelton P.R., Gelderblom H.R. Electron microscopy for rapid diagnosis of infectious agents in emergent situations. Emerg. Infect. Dis. 2003;9:294–303. doi: 10.3201/eid0903.020327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmer S., Joachim S., Robinson D.G. Rapid polymerization of LR-White for immunocytochemistry. Histochemistry. 1991;95:315–318. doi: 10.1007/BF00266782. [DOI] [PubMed] [Google Scholar]
- Hino H., Ammitzboll T., Möller R., Asboe-Hansen G. The ultrastructure of bacterial spores in skin of an Egyptian mummy. Acta Pathol. Microbiol. Immunol. Scand. [B] 1982;90:21–24. doi: 10.1111/j.1699-0463.1982.tb00077.x. [DOI] [PubMed] [Google Scholar]
- Kok L.P., Boon M.E. Microwaves for microscopy. J. Microsc. 1990;158:291–322. doi: 10.1111/j.1365-2818.1990.tb03003.x. [DOI] [PubMed] [Google Scholar]
- Leong A.D.-Y., Sormunen R.T. Microwave procedures for electron microscopy and resin-embedded sections. Micron. 1998;29:397–409. doi: 10.1016/s0968-4328(98)00018-3. [DOI] [PubMed] [Google Scholar]
- Login G.R., Ku T.-C., Dvorak A.M. Rapid primary microwave-aldehyde and microwave-osmium fixation: improved detection of rat parotid acinar cell secretory granule alpha-amylase using a post-embedding immunogold ultrastructural morphometric analysis. J. Histochem. Cytochem. 1995;43:515–523. doi: 10.1177/43.5.7730590. [DOI] [PubMed] [Google Scholar]
- Luft J.H. Improvements in epoxy resin embedding methods. J. Biophys. Biochem. Cytol. 1961;9:409–416. doi: 10.1083/jcb.9.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.E. Bioterrorism and electron microscopy differentiation of poxviruses from herpesviruses: does and don'ts. Ultrastruct. Pathol. 2003;27:133–140. doi: 10.1080/01913120309932. [DOI] [PubMed] [Google Scholar]
- Newman G.R., Hobot J.A. Modern acrylics for post-embedding immunostaining techniques. J. Histochem. Cytochem. 1987;35:971–981. doi: 10.1177/35.9.3302021. [DOI] [PubMed] [Google Scholar]
- Newman G.R., Jasani B., Williams E.D. The preservation of ultrastructure and antigenicity. J. Microsc. 1982;127:5–6. [Google Scholar]
- Pabst M.A., Letofsky-Pabst I., Bock E., Spindler K., Guillén S., Hofer F. Proceedings of the Microscopy Conference 2005, Davos. 2005. Bacterial spores in the skin of a 1000-year-old Peruvian mummy; p. 179. [Google Scholar]
- Pipkin P.A., Wood D.J., Racaniello V.R., Minor P.D. Characterisation of L cells expressing the human poliovirus receptor for the specific detection of polioviruses in vitro. J. Virol. Methods. 1993;41:333–340. doi: 10.1016/0166-0934(93)90022-j. [DOI] [PubMed] [Google Scholar]
- Roth J., Taatjes D.J., Tokuyasu K.T. Contrasting of Lowicryl K4M thin sections. Histochemistry. 1990;95:123–136. doi: 10.1007/BF00266584. [DOI] [PubMed] [Google Scholar]
- Setlow P. Spores of Bacillus subtilis: their resistance to and killing by radiation, heat and chemicals. J. Appl. Microbiol. 2005;101:514–525. doi: 10.1111/j.1365-2672.2005.02736.x. [DOI] [PubMed] [Google Scholar]
- Schröder J.A., Gelderblom H.R., Hauröder B., Schmetz C., Milios J., Hofstädter F. Microwave-assisted tissue processing for same-day EM-diagnosis of potential bioterrorism and clinical samples. Micron. 2006;37:577–590. doi: 10.1016/j.micron.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organisation A multicentre collaboration to investigate the cause of severe acute respiratory syndrome. Lancet. 2003;361:1730–1733. doi: 10.1016/S0140-6736(03)13376-4. [DOI] [PMC free article] [PubMed] [Google Scholar]