

Abstract
There are virtually no antiviral drugs available for the treatment of infections with RNA viruses. This is particularly worrisome since most of the highly pathogenic and emerging viruses are, and will likely continue to be, RNA viruses. These viruses can cause acute, severe illness, including severe respiratory disease, hemorrhagic fever and encephalitis, with a high case fatality rate. It is important to have potent and safe drugs at hand that can be used for the treatment or prophylaxis of such infections. Drugs approved for the treatment of RNA virus infections (other than HIV) are the influenza M2 channel inhibitors, amantadine and rimantadine; the influenza neuraminidase inhibitors, oseltamivir and zanamivir, and ribavirin for the treatment of infections with respiratory syncytial virus and hepatitis C virus. The molecular mechanism(s) by which ribavirin inhibits viral replication, such as depletion of intracellular GTP pools and induction of error catastrophe, may not readily allow the design of analogues that are more potent/selective than the parent drug. Highly pathogenic RNA viruses belong to a variety of virus families, each having a particular replication strategy, thus offering a wealth of potential targets to selectively inhibit viral replication. We here provide a non-exhaustive review of potential experimental strategies, using small molecules, to inhibit the replication of several RNA viruses. Other approaches, such as the use of interferon or other host-response modifiers, immune serum or neutralizing antibodies, are not addressed in this review.
Keywords: RNA viruses, Emerging viruses, Arenaviruses, Bunyaviruses, Flaviviruses, Filoviruses, Orthomyxoviruses, Ribavirin, Influenza, Avian influenza, Antiviral therapy
1. Introduction: need for antiviral drugs against highly pathogenic RNA viruses
This paper, one of three that introduce a special issue of Antiviral Research, describes potential antiviral drug strategies that may be employed to treat or prevent infections with highly pathogenic RNA viruses. A second article reviews the epidemiology and pathogenesis of representative diseases, and a third discusses experimental sequence-based approaches to therapy (Bray, 2008, Spurgers et al., 2008). Two other papers in this issue cover animal models of hemorrhagic fever (Gowen and Holbrook, 2008) or encephalitis (Holbrook and Gowen, 2008) caused by highly pathogenic RNA viruses, while others describe opportunities for research support and collaboration or focus on the treatment of specific diseases (see below).
Highly pathogenic RNA viruses belong to various virus families. These include the Arenaviridae (Lassa and Argentine hemorrhagic fever (AHF) viruses), the Bunyaviridae (Crimean-Congo hemorrhagic fever (CCHF) virus and hantaviruses), the Coronaviridae (severe acute respiratory syndrome (SARS) coronavirus), the Filoviridae (Marburg and Ebola virus), the Flaviviridae (yellow fever virus, Japanese encephalitis virus and other flaviviruses that cause encephalitis), the Paramyxoviridae (Nipah and Hendra virus), the Orthomyxoviridae (influenza) and the Togaviridae (chikungunya, and western, eastern and Venezuelan equine encephalitis viruses). In addition to pathogens that are a serious threat to human health, viruses such as the foot-and-mouth disease virus (Picornaviridae) and classical swine fever virus (Flaviviridae) are a threat to livestock, and can have an enormous impact on the economy (reviewed by Goris et al., 2008).
At present, some 40 antiviral drugs have been formally licensed for use in humans, mostly for the treatment of infections with the human immunodeficiency virus (HIV), hepatitis B virus (HBV) and herpesviruses. The number of licensed antiviral drugs that can be used for the treatment of highly pathogenic RNA virus infections is very limited. These are for influenza, the M2 channel inhibitors, amantadine and rimantadine, and the neuraminidase inhibitors, oseltamivir and zanamivir. Ribavirin is licensed for the treatment of respiratory syncytial virus (RSV) and hepatitis C virus (HCV) infections and is also being used for the treatment of Lassa fever. There is thus an unmet need for potent inhibitors of RNA virus replication, which should have as broad a spectrum as possible. Here we will first discuss the use of licensed antiviral drugs for the treatment of infections caused by highly pathogenic RNA viruses. Next, we provide a (non-exhaustive) review of potential targets for inhibiting the replication of RNA viruses.
2. Licensed drugs against highly pathogenic RNA viruses
Because the market for drugs for the treatment or prophylaxis of infections with highly pathogenic RNA viruses is rather limited, very few such drugs have been brought to the market. Because influenza causes worldwide epidemics with significant morbidity and mortality, it is an exception to this rule (see paper by Beigel and Bray, 2008).
2.1. Influenza drugs
For infections with the influenza virus, the M2 channel inhibitors (amantadine and rimantadine) and the neuraminidase inhibitors (oseltamivir and zanamivir) have been formally licensed.
2.1.1. M2 channel blockers
The first synthetic compound shown to inhibit influenza replication was amantadine (Fig. 1A). This compound blocks the migration of H+ ions via the viral M2 ion channel into the interior of the virus particles residing in the endosomes (Davies et al., 1964). Acidification of the interior of the viral particle results in destabilization of the capsid and results in uncoating; amantadine prevents this acidification process and thus uncoating (Horimoto and Kawaoka, 2005, De Clercq, 2006). M2 channel inhibitors are, however, only effective against influenza A strains and resistance against the drugs develops readily (Monto, 2003). In recent years, there has been a dramatic increase in the prevalence of amantadine-resistant influenza virus strains (both seasonal influenza and H5N1 avian influenza). The extensive use of amantadine in poultry farms in Asia may be one of reasons of the high amantadine-resistance incidence (Bright et al., 2005, He et al., 2008).
Fig. 1.
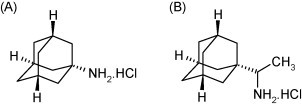
Structure of amantadine (A) and rimantadine (B).
2.1.2. Neuraminidase inhibitors
The mechanism by which influenza virus is released from the host cell by the viral neuraminidase has been extensively documented. After budding from the host cell, the viral hemagglutinin interacts with the host cell receptor (a glycoprotein) bearing N-acetylneuraminic acid (NANA). The neuraminidase cleaves off NANA from the cell-surface glycoprotein at a specific bond, the sialic acid linked to galactose by an alfa 2,3 or alfa 2,6 linkage. This enables the progeny virus to leave the infected cells and to spread to other host cells. Inhibition of the neuraminidase prevents virus release and thus spread of the virus (Moscona, 2005a, Moscona, 2005b).
The neuraminidase inhibitors were designed following a similar “transition-state” principle (Abdel-Magid et al., 2001) as applied for the peptidomimetic HIV protease inhibitors. This provided the influenza inhibitors DANA (2-deoxy-2,3-didehydro-N-acetylneuraminic acid, Fig. 2A) and FANA (2-deoxy-2,3-didehydro-N-trifluoroacetylneuraminic acid, Fig. 2B) and further rational design led to the development of the neuraminidase inhibitors zanamivir (Fig. 2C) and oseltamivir (Fig. 2D) (von Itzstein et al., 1993, Kim et al., 1997). Both compounds are highly potent inhibitors of the influenza neuraminidase and influenza A and B virus replication in vitro and in vivo (mice, ferrets). They are both prophylactically and therapeutically effective against influenza in humans (reviewed by De Clercq, 2006). The H274Y mutation may engender resistance of both influenza A H1N1 and influenza A H5N1 to oseltamivir but not zanamivir. In both H5N1 and H1N1 neuraminidases, tyrosine at position 252 is involved in a network of hydrogen bonds, with histidine at position 274 (Russell et al., 2006).
Fig. 2.
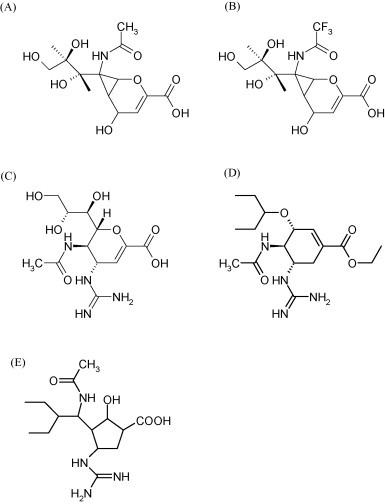
Structure of DANA (A), FANA (B), zanamivir (C), oseltamivir (D) and peramivir (E).
Further insight into the structure of the type 1 neuraminidase suggests new opportunities for the future design of new neuraminidase inhibitors (Russell et al., 2006). Peramivir (Fig. 2E) is a cyclopentane neuramidinase inhibitor that, like oseltamivir and zanamivir, acts as a sialic acid analogue transition-state intermediate in the neuraminidase active site. The development of peramivir is supported by the US Department of Health and Human Services (http://www.hhs.gov/news/press/2007pres/20070104.html). An injectable version of peramivir is being developed, as is also the case for zanamivir (see also Beigel and Bray, 2008). The availability of an injectable neuraminidase inhibitor may be an important asset in treating patients hospitalized with severe and potentially life-threatening influenza; an injectable formulation may ensure appropriate dosing which may be a concern with oral or inhaled anti-influenza agents.
2.2. Ribavirin
Ribavirin (1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide, Fig. 3A) is a broad-spectrum inhibitor of RNA virus replication, that has been formally approved to treat HCV infections in combination with pegylated interferon and in aerosol form for the treatment of pediatric RSV infections. The drug has also been used experimentally against a number of other conditions, including Lassa fever (see Khan et al., 2008), CCHF virus (Ergonul, 2008) and hantaviruses (Jonsson et al., 2008). The synthesis and antiviral activity of ribavirin was first reported in the early 1970s. The compound was found to inhibit the replication of a number of viruses (almost all of which were RNA viruses) in vitro and was shown to exert a protective effect in some experimental animal models (Sidwell et al., 1973, Huffman et al., 1973). Ribavirin has since been shown to inhibit the in vitro replication of virtually any RNA virus studied, although the potency may vary considerably, depending on the nature of the virus (Graci and Cameron, 2006).
Fig. 3.
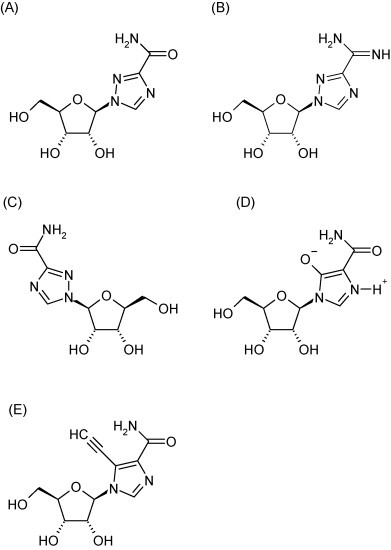
Structure of ribavirin (A), viramidine (B), levovirin (C), mizoribine (D) and EICAR (E).
2.2.1. Spectrum of activity and molecular mechanism of action
Although almost all RNA viruses are sensitive to the in vitro antiviral activity of ribavirin, some viruses are more susceptible than others. Overall, ribavirin is not very potent as an antiviral drug, and 50% effective concentrations (EC50) values are mostly (well above) 1 μM. In particular the replication of RSV is relatively efficiently inhibited by ribavirin; other viruses such as several picorna- flavi-, and filoviridae are less susceptible to the drug. Ribavirin was reported to inhibit the replication of several arenaviruses, including Lassa virus in cell culture (Huggins et al., 1984, McKee et al., 1988, Andrei and De Clercq, 1990, Gunther et al., 2004) and has beneficial effects in experimental animal models of Lassa fever (Stephen and Jahrling, 1979, Jahrling et al., 1980, Dvoretskaia et al., 1991). In vitro antiviral activity of ribavirin has also been demonstrated against bunyaviruses including CCHF virus, Rift Valley fever virus and hantaviruses (Watts et al., 1989, Huggins, 1989, Severson et al., 2003). Ribavirin inhibits the replication of coronavirus, including the SARS-coronavirus (Saijo et al., 2005, Barnard et al., 2006) and flaviviruses (Neyts et al., 1996) but has not shown efficacy in experimental infections in animals with these viruses. Likewise, ribavirin is not effective in animal models of filovirus infections (Huggins, 1989). Like other paramyxoviruses such as RSV, Nipah virus is relatively sensitive to ribavirin in cell culture and limited efficacy of the compound has been demonstrated in animal models (Snell, 2004, Georges-Courbot et al., 2006). Ribavirin has also been shown to inhibit the in vitro replication of alphaviruses such as Chikungunya and equine encephalitis viruses (Briolant et al., 2004, Markland et al., 2000).
Despite the fact that the antiviral activity of ribavirin was reported 35 years ago, the molecular mechanism by which the compound exerts its antiviral activity still remains a matter of debate. Inosine 5′-monophosphate (IMP) dehydrogenase, a cellular enzyme that converts IMP to xanthosine 5′-monophosphate in the de novo synthesis pathway of guanosine 5′-monophosphate (GMP), is inhibited by ribavirin 5′-monophosphate (Streeter et al., 1973) (Fig. 4 ). The subsequent depletion of intracellular GTP-pools compromises the supply of this nucleotide for the synthesis of progeny viral RNA. This mechanism provided a reasonable explanation for the broad-spectrum anti-RNA virus activity of ribavirin (Streeter et al., 1973). Ribavirin-resistant Sindbis virus was shown to carry mutations in the viral guanylyltransferase gene. This is in line with the observation that drug-resistant Sindbis virus replicates relatively efficiently in cells with low intracellular GTP concentrations, either induced by ribavirin or by other IMP dehydrogenase inhibitors (Malinoski and Stollar, 1981, Scheidel and Stollar, 1991).
Fig. 4.
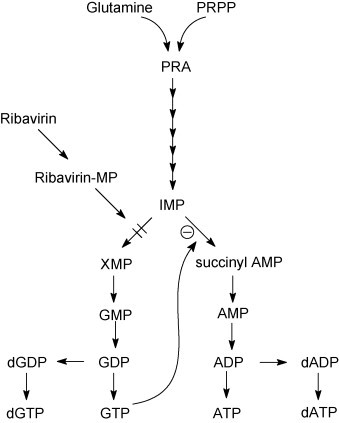
Mechanism of action of ribavirin. Target enzyme: IMP dehydrogenase. Ribavirin 5′-monophosphate inhibits the conversion of IMP to XMP resulting in a reduced supply of GTP, and indirectly a reduced supply of ATP.
Later, several other mechanisms were suggested that may contribute to the antiviral activity of ribavirin, including (i) inhibition of viral capping as mediated by an effect on the viral guanylyltransferase or inhibition of viral methyltransferases of viruses that encode such enzymes (Goswami et al., 1979, Bougie and Bisaillon, 2003, Bougie and Bisaillon, 2004, Benarroch et al., 2004), (ii) inhibition of RNA-dependent RNA polymerase activity by ribavirin 5′-triphosphate as shown for influenza and HCV (Eriksson et al., 1977, Maag et al., 2001, Bougie and Bisaillon, 2003) and (iii) inhibition of the viral helicase activity (as suggested for reoviruses) (Rankin et al., 1989). The poliovirus RNA-dependent RNA polymerase was shown to incorporate ribavirin opposite to cytidine and uridine in the growing RNA chain (Crotty et al., 2000) and poliovirus, that was cultured in the presence of high concentrations of ribavirin (1 mM), accumulated 10-fold more mutations in its genome than virus grown in untreated cultures. The accumulation of mutations in the viral genome results in a process called error catastrophe (Crotty et al., 2001). A single mutation (G64S) in the poliovirus RNA-dependent RNA polymerase gene was found to increase the enzyme's fidelity and to render the virus less susceptible to the mutagenic effect of ribavirin (Pfeiffer and Kirkegaard, 2003). Error catastrophe has also been proposed to be involved in the antiviral activity of ribavirin against Hantaan virus replication (Severson et al., 2003) as well as the replication of foot-and-mouth disease virus (Airaksinen et al., 2003) and GB virus B virus (Lanford et al., 2001).
It remains, however, unclear to what extent error catastrophe, depletion of intracellular GTP-pools or one or more of the other proposed mechanisms contribute to the actual antiviral effect of ribavirin, and whether or not these effects also play a part in the antiviral activity of the compound in vivo. For flaviviruses and paramyxoviruses, we demonstrated that GTP pool depletion is the predominant mechanism by which ribavirin exerts its antiviral activity (Leyssen et al., 2005); for flaviviruses, error catastrophe does not appear to contribute to the in vitro antiviral activity of the drug (Leyssen et al., 2006).
2.2.2. Clinical use of ribavirin
Ribavirin was first approved in 1985 for the aerosol treatment of severe respiratory tract infections in children caused by the RSV. Until today, the actual clinical benefit of this treatment has been debated. Review of reported cases and clinical trials indicate that ribavirin may reduce the duration of mechanical ventilation and may reduce days of hospitalization. In addition, use of ribavirin may be associated with a decrease in the long-term incidence of recurrent wheezing following RSV disease (Ventre and Randolph, 2007).
In patients chronically infected with HCV, ribavirin alone induces either a transient early decline or no decrease in HCV viral load, but, in combination with interferon, it significantly improves long-term response rates. The precise mechanism by which the compound improves the efficacy of interferon (Dixit et al., 2004) remains a matter of debate. Conflicting evidence has been reported regarding the potential of ribavirin to induce error catastrophe. On the one hand, no increase in mutation rate was detected in HCV genomes isolated from patients treated with ribavirin (Lutchman et al., 2004, Pawlotsky et al., 2004a, Pawlotsky et al., 2004b) or ribavirin/interferon combination therapy (Schinkel et al., 2003, Pawlotsky et al., 2004a, Pawlotsky et al., 2004b, Chevaliez et al., 2007a, Chevaliez and Pawlotsky, 2007b), on the other hand, in a subset of ribavirin-treated, HCV-infected patients, an increased number of mutations was observed (Asahina et al., 2005).
Ribavirin is used off-label for the treatment of Lassa fever; this use, however, is based on a single published study using a historical control group (McCormick et al., 1986). Intravenously administrated ribavirin appears to be more efficacious than when the drug is given orally, which is likely related to the fact that plasma concentrations reached following oral dosing are lower. For the treatment of Lassa fever, ribavirin is effective when given in the early stages of the course of the disease, but reduced or no efficacy was noted if ribavirin was given in the late stages of the disease (McCormick et al., 1986, Schmitz et al., 2002). The drug has also been used as post-exposure prophylaxis, but there are no published data to support this use. For further information on the use of ribavirin in the treatment of Lassa fever or another arenavirus infection, Argentine hemorrhagic fever, the reader is referred to other articles by Khan et al. (2008) and Enria et al. (2008).
Ribavirin showed a beneficial effect in a placebo-controlled study in patients infected with Hantaan virus (hemorrhagic fever with renal syndrome, HFRS) when treatment was initiated early in the course of the disease (Huggins et al., 1991). However, in two double-blind placebo-controlled studies in patients infected with a New World hantavirus (hantavirus (cardio)pulmonary syndrome, HPS) the drug had no, or virtually no, clinical efficacy (Chapman et al., 1999, Collaborative Antiviral Study Group, 2004). The lack of activity may be related to the phase of infection and severity of the disease; which may call for early intervention (see Jonsson et al., 2008). HPS is to a large extent an immunopathological syndrome; thus even the most effective antiviral may be of little utility without controlling the deleterious host response that is in full-force at the time patients seek medical attention.
Ribavirin has also been used, administered either orally or intravenously, to treat CCHF (Fisher-Hoch et al., 1995, Mardani et al., 2003, Bossi et al., 2004). As described by Ergonul (2008), evidence for efficacy is limited to observational studies. Placebo-controlled studies, may be for ethical reasons impossible to perform in patients with CCHF. Despite this shortcoming, post-exposure prophylaxis and treatment with ribavirin in every stage of the disease is recommended Ribavirin has also been used without much success as a first-line-of-defence against emerging viruses such as Nipah virus and the SARS-coronavirus (Chong et al., 2001, Stockman et al., 2006). Finally, ribavirin is not effective in the treatment of filovirus infections (reviewed by Bausch et al., 2008) or for the treatment of Japanese encephalitis, yellow fever or other flaviviral infections (see articles by Gould et al., 2008, Monath, 2008).
2.2.3. Ribavirin analogues and prodrugs
To increase the oral bioavailability of ribavirin and to decrease side-effects such as a dose-limiting hemolytic anemia, various prodrugs of ribavirin have been developed. Viramidine (Fig. 3B) is an aminated prodrug of ribavirin that is deaminated in vivo by adenosine deaminase (Wu et al., 2006). The drug, following absorption, is rapidly converted to ribavirin (Lin et al., 2006). The activity of viramidine on influenza virus replication was comparable to that of ribavirin (Sidwell et al., 2005). Viramidine is being developed for the treatment of HCV infection.
Several other analogues of ribavirin, such as levovirine and mizoribine have been studied. Levovirin (ICN-17261, Fig. 3C) is the l-analogue of ribavirin; it does not undergo phosphorylation, and has no antiviral activity in vitro. It has been proposed that levovirin possesses, akin to ribavirin, immunomodulatory activity (Tam et al., 1999, Tam et al., 2000). Clinical studies with levovirin in HCV-infected patients were unsuccessful, which suggest that the immunomodulatory activity of levovirin is not sufficient to result in clinical efficacy. Levovirin was not further developed (Fang et al., 2003).
Mizoribine (Fig. 3D) is a hydroxylated analogue of ribavirin and inhibits, akin to ribavirin, cellular IMP dehydrogenase; this molecule has been developed as an immunosuppressive agent (Gan et al., 2003) rather than an antiviral agent, although some antiviral activity in combination with other agents has been reported (Saijo et al., 2005, Naka et al., 2005, Yanagida et al., 2004, Stuyver et al., 2002). A potent reversible uncompetitive IMP dehydrogenase inhibitor, VX-497 (imepodib, Fig. 5 ), structurally unrelated to other inhibitors of this enzyme, was developed and shown to exhibit in vitro antiviral activity that was superior to that of ribavirin. However, the compound was not further developed (Markland et al., 2000).
Fig. 5.
Structure of VX-497.
2.2.4. Ribavirin as a lead for improved analogues?
Although ribavirin exerts broad-spectrum activity in vitro, its clinical use is, as discussed, limited to only a few viral infections. The mechanism by which ribavirin inhibits viral replication varies with the virus studied and can be considered as not very selective (GTP depletion, error catastrophe, etc.). The design of safe and more potent analogues of ribavirin with broad-spectrum activity that can be used in the treatment of a variety of RNA virus infections will therefore likely be very difficult to achieve. EICAR, the 5-ethynyl analogue of ribavirin (Fig. 3E) was shown to be in vitro roughly 10- to 30-fold more potent in inhibiting the replication of various RNA viruses (Leyssen et al., 2005). However, this improved activity came at the price of a concomitant increase in toxicity; which is explained by the fact that the 5′-monophosphate metabolite of EICAR is also more potent than ribavirin monophosphate in inhibiting the IMP dehydrogenase (Balzarini et al., 1993).
3. Viral targets for therapy
3.1. Entry and uptake
The initial interaction of the virus with the host cell represents an attractive target for therapeutic intervention. We will discuss briefly possible approaches to inhibit these early events in the replication cycle of some RNA viruses, including picorna-, corona- arena- and paramyxoviruses, for which strategies that inhibit entry or uptake have been reported.
3.1.1. Picornaviruses
The attachment of picornaviruses occurs through binding of the host cell receptor into a canyon situated around each 5-fold vertex of the capsid; binding of the receptor is believed to trigger also the uncoating process. Binding of specific molecules in a pocket immediately underneath the floor of the canyon, results in an increase in protein rigidity and stabilizes the particle in such a manner that uncoating cannot proceed (reviewed by De Palma et al., submitted for publication). Numerous compounds have been identified that inhibit picornavirus replication by specific interaction with this pocket. Two compounds that bind into this pocket, pleconaril (Fig. 6A) and pirodavir (Fig. 6B) have been evaluated in the clinical setting, but further development has been halted because of lack of activity or adverse side-effects (De Palma et al., submitted for publication).
Fig. 6.
Structure of pleconaril (A) and pirodavir (B).
3.1.2. Coronaviruses
Many enveloped viruses carry carbohydrate-containing proteins on their surface. These glycoproteins are key to the infection process as they are mediators in binding and/or uptake and are thus attractive targets for the development of novel antiviral therapies. Recently, carbohydrate-binding agents were shown to exert in vitro antiviral activity towards a number of viruses, including coronaviruses (van der Meer et al., 2007, Balzarini et al., 2007, Balzarini, 2007a, Balzarini, 2007b).
3.1.3. Paramyxoviruses
A human monoclonal antibody, palivizumab (Synagis®) that targets the RSV fusion protein (Johnson et al., 1997) has been approved as a prophylactic therapy for RSV infection, but it is only administered in high-risk pediatric patients. Under preclinical development are the fusion inhibitors VP-14637 (Fig. 7A) (Douglas et al., 2003, Douglas et al., 2005) and JNJ-2408068 (Fig. 7B) (Douglas et al., 2005), which both interact with the RSV F polyprotein, albeit at different sites, as indicated by the resistance mutations selected in the presence of these inhibitors. Peptides derived from the F protein (the HRN and HRC regions) that interfere with fusion intermediates of F are candidate molecules for impeding virus entry. Fusion peptides derived from human parainfluenza or Hendra virus were shown to inhibit Hendra virus replication in vitro (Porotto et al., 2006). A similar strategy would also be amenable for coronaviruses (Yan et al., 2006). The ability of peptides that interfere with the class I fusion process has led to the creation of a clinically effective peptide inhibitor of HIV-1 fusion (enfuvirtide, T-20, Fig. 8 ) and related molecules (Dwyer et al., 2007). The efficacy of T-20 as a therapy for HIV-1 infection supports the concept of designing effective therapies based on understanding the mechanism of viral fusion and the use of peptides as antivirals.
Fig. 7.
Structure of VP-14637 (A) and JNJ-2408068 (B).
Fig. 8.

Structure of enfuvirtide.
Recently, a recombinant fusion protein composed of the catalytic domain of the neuraminidase (sialidase) derived from Actinomyces viscosus, fused with a cell-surface-anchoring sequence was reported as a novel broad-spectrum inhibitor of influenza virus replication. The fusion protein is to be applied by aerosol to remove sialic acids (the influenza receptor) from the airway epithelium. A sialidase fusion protein, DAS181 (fludase), has been reported that, at subnanomolar concentrations, inhibits both human and avian influenza virus replication. Significant in vivo efficacy of the DAS181 was noted in both prophylactic and therapeutic approaches in experimental influenza virus infections in animals (Malakhov et al., 2006). The compound is in advanced preclinical development at NexBio.
3.1.4. Arenaviruses
A high-throughput screening effort resulted in the identification of a potent and broad-spectrum inhibitor of arenavirus entry (Bolken et al., 2006).
3.2. Uncoating
As discussed for picornaviruses, inhibition of viral uncoating results in inhibition of viral replication. For picornaviruses, the uncoating process is “linked” to receptor binding. For the enveloped viruses, the uncoating process takes place after the virus adsorption and virus–cell fusion processes. Perhaps the best known antiviral uncoating inhibitors are the adamantan(amin)e derivatives, which selectively inhibit the uncoating process of influenza A viruses (see above).
3.3. Translation and processing
3.3.1. Capping and IRES inhibitors
The 5′-capping of viral RNA and Internal Ribosomal Entry Site (IRES)-mediated transcription of viral RNA may also seem to be attractive and largely unexplored targets for antiviral therapy. The following enzymes play a crucial role in the capping machinery: 2′-O-methyltransferase, guanylyltransferase and RNA-triphosphatase (Egloff et al., 2002, Egloff et al., 2007, Benarroch et al., 2004). The potential of the methyl transferase as drug target for flaviviruses has recently been demonstrated (Luzhkov et al., 2007). Inhibition of RNA virus replication by targeting the host SAH-hydrolase is discussed in Section 3.3.2. Various inhibitors of the HCV IRES have been synthesized, but none of these have so far proven to be sufficiently potent to warrant further development (Jubin, 2003).
3.3.2. Protease inhibitors
Many RNA viruses encode one or more proteases. Perhaps the best characterized are those of the Flaviviridae (HCV and flaviviruses) and of the Picornaviridae. Although HCV is not discussed in this review, it is important to mention the successful development of highly potent and selective inhibitors of the HCV NS3 serine protease. Lamarre and his colleages were the first to develop a potent HCV NS3 inhibitor, BILN-2061 (Fig. 9A) and to demonstrate efficacy of the drug in patients chronically infected with HCV (Lamarre et al., 2003, Pause et al., 2003). Because of cardiotoxicity in experimental models, the development of this compound was discontinued. However, other HCV NS3 protease inhibitors were developed and excellent efficacy has been demonstrated in the clinical setting, the most advanced is VX-950 (Fig. 9B, De Francesco and Migliaccio, 2005, Forestier et al., 2007). The crystal structure of dengue virus NS3 proteases has been solved (Murthy et al., 2000) and the first inhibitors of these enzymes have been reported (Ganesh et al., 2005).
Fig. 9.
Structure of BILN-2061 (A) and VX-950 (B).
For the prevention and/or treatment of rhinovirus infections, inhibitors of the human rhinovirus (HRV) 3C protease have been extensively investigated. Ruprintrivir (Fig. 10 ) is an irreversible 3C protease inhibitor, which upon intranasal administration in human volunteers, appeared to be safe and well tolerated. In experimentally induced rhinovirus colds in healthy volunteers, prophylaxis with the drug reduced the proportion of subjects with positive viral cultures but did not reduce the frequency of colds (Binford et al., 2005, Binford et al., 2007). An analogue of ruprintrivir with excellent oral bioavailability was developed; the compound appeared safe in phase I studies, but has not further progressed toward clinical development (Patick et al., 2005, Patick, 2006). The crystal structures for the main protease (Mpro) of human coronavirus was determined; this revealed a remarkable degree of conservation with the substrate-binding sites of the picornavirus 3C protease. Molecular modeling suggests that ruprintrivir and analogues may be modified to make them efficient inhibitors of coronavirus (such as SARS-coronavirus) replication (Anand et al., 2003, Yang et al., 2003, Yang et al., 2005).
Fig. 10.
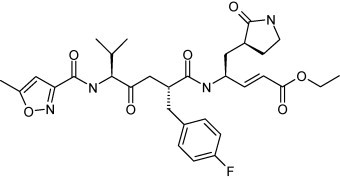
Structure of ruprintrivir.
3.4. Replication machinery
3.4.1. The viral RNA-dependent RNA polymerase
Numerous selective inhibitors of HCV replication have been identified that target the viral RNA-dependent RNA polymerase (RdRp). As in the case for the HIV reverse transcriptase, these compounds can largely been classified as nucleoside and non-nucleoside inhibitors. Nucleoside analogues need to be phosphorylated to their 5′-triphosphate metabolite to inhibit the function of the enzyme at the active site. Non-nucleoside analogues act as allosteric site inhibitors. Several nucleoside and non-nucleoside inhibitors are, or have been, in clinical development for the treatment of infection with HCV (De Francesco and Migliaccio, 2005). The nucleoside HCV inhibitors 2′-C-methylcytidine (Fig. 11A), 2′-C-methyl-7-deazaguanosine (Fig. 11B) and related 2′-C-methyl nucleosides exert broad-spectrum activity against single plus-stranded RNA viruses, including activity against the picornavirus foot-and-mouth disease virus (Goris et al., 2007). Other nucleoside inhibitors of HCV replication, such as 4′-azidocytidine (Fig. 11C), a drug that is being developed by Roche, is solely active against HCV (Klumpp et al., 2006 and our unpublished findings). As for HCV, it is also possible to develop highly selective non-nucleoside inhibitors of the polymerase of other RNA viruses, as demonstrated for example with pestiviruses (Baginski et al., 2000, Paeshuyse et al., 2006b, Paeshuyse et al., 2007). Thus, developing potent non-nucleoside inhibitors of related viruses such as other members of the Flaviviridae or the highly pathogenic agents covered in this issue, may well be feasible.
Fig. 11.
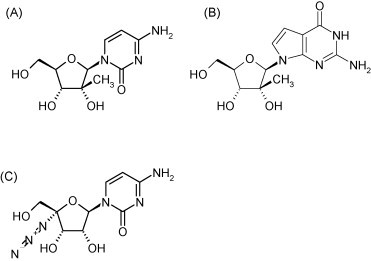
Structure of 2′-C-methylcydtidine (A), 2′-C-methyl-7-deazaguanosine (B) and 4′-azidocytidine (C).
The influenza virus RNA polymerase consists of a complex of three virus-encoded enzymes (PB1, PB2 and PA), which in addition to RNA replicative activity, also contains an endonuclease activity so as to ensure “cap snatching” to initiate the transcription and subsequent translation process (Deng et al., 2005). Only few compounds have been reported that target the influenza polymerase (replicase). Like the inhibitors that have been shown to inhibit the reverse transcriptase (RNA-dependent DNA polymerase) of HIV or the RNA replicase of HCV, influenza replicase inhibitors can (should) in principle also be divided into two classes: nucleosides and non-nucleosides. More than 10 years ago, 2′-deoxy-2′-fluoroguanosine (Fig. 12A) was described as an inhibitor of influenza virus transcription, although it has not been further studied in this capacity (Tisdale et al., 1995).
Fig. 12.
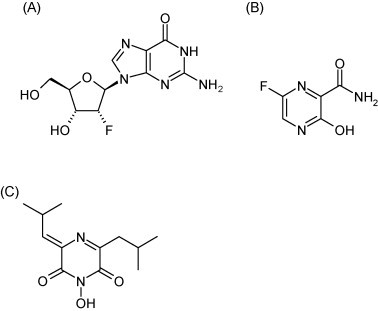
Structure of 2′-deoxy-2′-fluoroguanosine (A), T-705 (B) and flutimide (C).
More recently, a substituted pyrazine, T-705 (Fig. 12B) was reported that has potent in vitro activity against influenza A, B and C (Furuta et al., 2002, Furuta et al., 2005). A polymerase inhibitor can be expected to have a resistance profile that does not overlap with those of the neuraminidase or M2 inhibitors. According to a comparative study, T-705 would even be more potent than oseltamivir when increasing the multiplicity of infection (in vitro) or using a higher virus challenge dose (in vivo) (Takahashi et al., 2003). Against avian influenza A (H5N1), T-705 was found to be less effective than oseltamivir in cell culture, but more effective than ribavirin (Sidwell et al., 2007). T-705 proved also to be effective, when administered orally, against a lethal avian influenza A virus infection in mice (Sidwell et al., 2007). It has been postulated that T-705 is converted intracellularly to the ribonucleotide T-705-ribofuranosyl-5′-monophosphate (T-705 RMP) by a phosphoribosyl transferase, and, upon phosphorylation to its 5′-triphosphate, T-705 RTP would inhibit influenza virus RNA polymerase in a GTP-competitive manner (Furuta et al., 2005, Fig. 13 ). Unlike ribavirin 5′-monophosphate, T-705 RMP does not significantly inhibit IMP dehydrogenase, indicating that it may owe its anti-influenza virus activity mainly, if not exclusively, to inhibition of the influenza virus RNA polymerase.
Fig. 13.
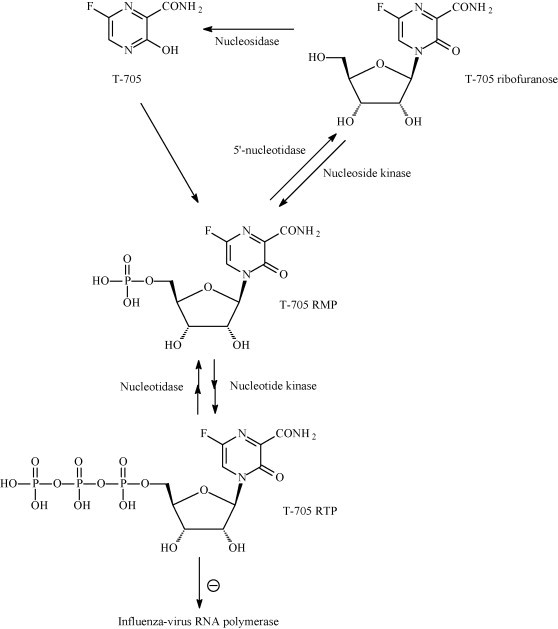
Intracellular metabolism and mechanism of action of T-705 against influenza A virus. T-705 would first be converted to its ribonucleotide (T-705 RMP), which would subsequently be converted to its 5′-triphosphate (T-705 RTP) that would then inhibit influenza virus RNA polymerase through competition with GTP (Furuta et al., 2005).
The “cap snatching” or “cap scavenging” endonuclease activity associated with the influenza polymerase complex may be an attractive target for drug therapy. A number of molecules have been shown to inhibit the influenza endonuclease activity. Flutimide (Fig. 12C), a 2,6-diketopiperazine isolated from extracts of the fungal species Delitschia confertaspora, inhibits the cap-dependent endonuclease activity and influenza virus (A and B strains) replication in cell culture (Tomassini et al., 1996).
Recently, the anti-influenza inhibitor T-705 has also been accredited with both in vitro and in vivo activities against a panel of arenaviruses (Pichinde) and bunyaviruses (Punta Toro) (Gowen et al., 2007). In general, T-705 was more active than ribavirin, as reflected by substantially greater therapeutic indexes and it was suggested that T-705 may be a viable alternative for the treatment of life-threatening infections with bunya- or arenaviruses (Gowen et al., 2007). It may be assumed that the mechanism by which T-705 inhibits the replication of bunya- and arenaviruses, is similar to the mechanism by which it presumably inhibits influenza replication, that is inhibition of the viral polymerases by T-705-ribofuranosyl-5′-triphosphate.
3.4.2. The viral helicase and the herewith associated NTPase
Almost all RNA viruses encode a helicase/NTPase that is responsible for the unwinding of the double-stranded RNA formed during replication. The helicase may represent an attractive target for therapy. There have been substantial efforts to identify inhibitors of the HCV NS3 helicase/NTPase. Inhibitors of both functions have been described, but have so far not been further developed (Frick, 2007, Borowski et al., 2007, Ujjinamatada et al., 2007). We recently identified a class of thiazolobenzimidazoles (Fig. 14 ) as potent inhibitors of the replication of various enteroviruses (De Palma et al., 2007). Mechanistic studies indicate that the compounds target the picornavirus 2C gene product, which has been predicted to be a helicase/NTPase. Coxsackie virus 2C was expressed and shown to have functional NTPase activity, that, however, was not inhibited by this class of compounds (De Palma et al., accepted for publication). Nevertheless, our unpublished data suggest that the thiazolibenzimidazoles might interfere with the assembly/functioning of the viral helicase. The helicase can be considered as a valuable but yet largely unexplored target for antiviral therapy.
Fig. 14.
General structure of thiazolobenzimidazoles.
3.5. Assembly
Recently a novel inhibitor of RSV replication has been identified, the benzodiazepine RSV604 (Fig. 15 ), the first of a new class of RSV inhibitors (currently in phase II clinical trials) that appear to be targeted at the viral nucleocapsid (N) protein (Chapman et al., 2007). This compound may possibly prevent the assembly of the virus particles. RSV604 represents one of the most promising candidate drugs to date for the treatment of RSV infections in humans.
Fig. 15.
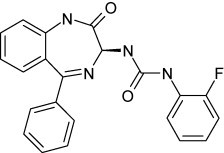
Structure of RSV604.
3.6. Release
An essential step in the viral life cycle is the release of the new progeny virions from the infected cell. As discussed in Section 2.1 this process has been extensively studied for influenza virus, resulting in the identification of the neuraminidase inhibitors. For many other RNA viruses, there is very little information about the precise molecular events leading to release of the progeny virions.
4. Cellular targets for therapy
Quite a number of antiviral agents have been reported to be targeted at cellular factors. Some of them may prevent the interaction of a cellular factor with the replication machinery of the virus, while others may influence host cell metabolism in such a way that efficient viral replication is prevented.
4.1. Essential virus/host interactions
Various host cell factors play a key role in the efficient replication of RNA viruses. In 2004, it was discovered that the immunosuppressive agent cyclosporin A inhibits HCV replication in vitro (Nakagawa et al., 2004). It was proposed that this effect is mediated by blockade of cyclophilins (Nakagawa et al., 2005) and that cyclosporin A (Fig. 16A) would suppress HCV replication at the level of the HCV RNA polymerase (Watashi et al., 2005). More specifically, cyclosporin A would block the ability of peptidyl-prolyl cis–trans isomerase (PPIase), cyclophilin B (CyPB), to bind to the RNA replicase (NS5B), thus inhibiting the function of the RNA replicase (Rice and You, 2005).
Fig. 16.
Structure of cyclosporine A (A) and Debio-025 (B).
We reported that Debio-025 (Fig. 16B), a non-immunosuppressive analogue of CsA, is a much more potent inhibitor of HCV replication than CsA (Paeshuyse et al., 2006a). Our recent data indicate that the mechanism of action of Debio-025 and cyclosporin A may be different from the proposed interaction with NS5B (Coelmont et al., 2007). Debio-025 is currently in phase II clinical studies for the treatment of HCV infections. The clinical efficacy of Debio-025 in the treatment of HCV infections supports the concept that molecules that prevent crucial interactions between viral and cellular factors may be interesting antiviral agents. The advantage of such approach (as we have also shown for Debio-025) is that it may make antiviral resistance development more difficult.
4.2. Glycosylation inhibitors
Endoplasmic reticulum (ER) alpha-glucosidase inhibitors, which block the trimming step of N-linked glycosylation, have been shown to impede the production of several ER-budding viruses, including flavi- and pestiviruses as well as HCV (Dwek et al., 2002, Wu et al., 2002). In a lethal mouse model of Japanese encephalitis, oral delivery of N-nonyl-deoxynojirimycin (Fig. 17 ) reduced the mortality rate (Wu et al., 2002).
Fig. 17.
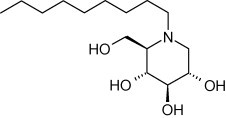
Structure of N-nonyl-deoxynojirimycin.
4.3. Compounds that interfere with nucleoside metabolism
Since viral replication predominantly relies on building blocks present in the host cell, it is apparent that interfering with this supply will slow down or halt the production of progeny viruses. However, antiviral drugs that target the nucleoside metabolism of the host cell will not only result in inhibition of viral replication, but will also be endowed with a cytotoxic and/or cytostatic effect. Thus, although these compounds may prove to be antivirally active, they lack selectivity.
4.3.1. IMP dehydrogenase inhibitors
As discussed earlier, inhibition of IMP dehydrogenase results in a depletion of the intracellular GTP-pools, which contributes to the antiviral activity of ribavirin and structurally related molecules such as viramidine, EICAR and structurally unrelated compounds such as mycophenolic acid (Fig. 18 ) and VX-497, which are putatively targeted at the coenzyme (NAD+) rather than the apoenzyme (at which the ribavirin congeners are targeted).
Fig. 18.
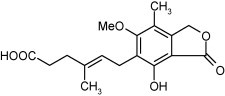
Structure of mycophenolic acid.
4.3.2. SAH hydrolase inhibitors
Ever since S-adenosylhomocysteine hydrolase (SAH) was recognized as a pharmacological target for antiviral agents (Montgomery et al., 1982), an increasing number of adenosine, acyclic adenosine and carbocyclic adenosine analogues have been described as potent SAH hydrolase inhibitors endowed with broad-spectrum antiviral activity (De Clercq, 2005). The antiviral activity spectrum of SAH hydrolase inhibitors against RNA viruses includes rhabdo-, filo-, arena-, paramyxo-, reo-, and retroviruses. Among the most potent SAH hydrolase inhibitors and antiviral agents rank carbocyclic 3-deazaadenosine (C-c3Ado, Fig. 19A), neplanocin A (Fig. 19B), 3-deazaneplanocin A (Fig. 19C), the 5′-nor derivatives of carbocyclic adenosine (C-Ado, aristeromycin, Fig. 19D), and the 2-halo (2-fluoro) and 6′-R-alkyl (6′-methyl) derivatives of neplanocin A.
Fig. 19.
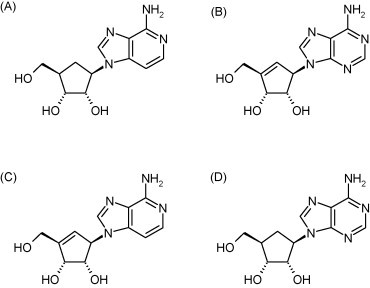
Structure of carbocyclic 3-deaza-adenosine (A), neplanocin A (B), 3-deazaneplanocin A (C) and aristeromycin (D).
These compounds are particularly active against rhabdoviruses, and have proven to be effective against filoviruses such as Ebola virus (Huggins et al., 1999). In fact, when administered as a single dose of 1 mg/kg on the first or second day after an Ebola Zaire virus infection in mice, 3-deazaneplanocin A reduced peak viremia by more than 1000-fold compared with mock-treated controls, and most or all of the animals survived (Bray et al., 2000). This protective effect was accompanied, and probably mediated, by the production of high concentration of IFN-α in the Ebola virus-infected mice (Bray et al., 2002). It could be postulated that by blocking the 5′-capping of the nascent viral (+)RNA strands, of which the maturation is dependent on methylation (Fig. 20 ), 3-deazaneplanocin A prevented the dissociation of these strands from the viral (−)RNA genome, thereby leading to an accumulation of replicative intermediates. These replicative intermediates (containing double-stranded RNA stretches) could, according to the concept proposed many years ago (Carter and De Clercq, 1974), be implicated in the high-level production of interferon.
Fig. 20.
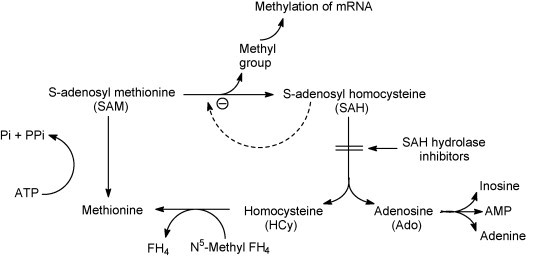
Mechanism of action of S-adenosylhomocysteine hydrolase (SAH) inhibitors. SAH is a product-inhibitor of methylation reactions including those that are required for the maturation (5′-capping) of viral mRNAs. Through the hydrolysis of SAH, SAH hydrolase prevents the accumulation of SAH that would otherwise lead to inhibition of the SAM-dependent methylation reaction.
4.3.3. OMP decarboxylase inhibitors
Pyrazofurin [3-(β-d-ribofuranosyl)-4-hydroxypyrazole-5-carboxamide, Fig. 21A], as prototype of the OMP decarboxylase inhibitors, is active against both (+)RNA viruses [picornaviruses (poliovirus, Coxsackie B4), togaviruses (Sindbis)] and (−)RNA viruses [paramyxoviruses (measles, RSV), orthomyxoviruses (influenza A, B, C), rhabdoviruses (vesicular stomatitis virus) and arenaviruses (Junin, Tacaribe)] (Andrei and De Clercq, 1990, Andrei and De Clercq, 1993, Shigeta et al., 1988). Also paramyxoviruses, RSV (Kawana et al., 1987) and measles virus (Hosoya et al., 1989) have been reported to be sensitive to inhibition by pyrazofurin. Recently, this also has been proven to be the case for Nipah virus, another paramyxovirus (Georges-Courbot et al., 2006).
Fig. 21.
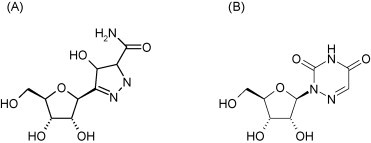
Structure of pyrazofurin (A) and 6-azauridine (B).
To act as an OMP decarboxylase inhibitor, pyrazofurin must first be converted by cellular kinase(s) to its 5′-monophosphate (Fig. 22 ). Another compound known to interfere with the conversion of OMP to UMP is 6-azauridine (Fig. 21B), and this again requires intracellular phosphorylation of the compound to its 5′-monophosphate. Not much is known about the antiviral activity spectrum of 6-azauridine (Rada and Dragun, 1977). However, this should be similar to that of pyrazofurin. Like pyrazofurin, 6-azauridine has been found to be effective against Nipah virus (Georges-Courbot et al., 2006).
Fig. 22.
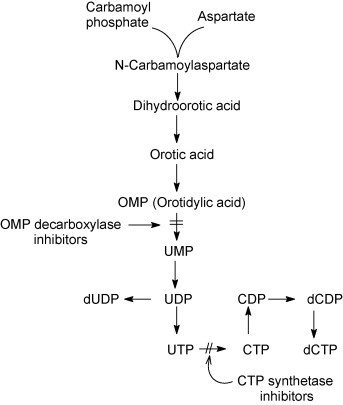
Mechanisms of action of OMP decarboxylase inhibitors and CTP synthetase inhibitors. OMP decarboxylase inhibitors (pyrazofurin, 6-azauridine) need to be phosphorylated to their 5′-monophosphate before they are able to interact with OMP decarboxylase. CTP synthetase inhibitors (C-Cyd, Ce-Cyd) need to be phosphorylated to their 5′-triphosphate before they are able to interact with CTP synthetase.
4.3.4. CTP synthetase inhibitors
The carbocyclic analogues of the normal nucleoside cytidine, such as cyclopentylcytosine (C-Cyd, carbodine) (Fig. 23A) and cyclopentenylcytosine (Ce-Cyd) (Fig. 23B) have been considered as potential antiviral and antitumor agents. C-Cyd was first described as an anti-influenza virus agent (Shannon et al., 1981), but it has also shown activity against DNA viruses [poxviruses (vaccinia)], (+)RNA viruses [togaviruses (Sindbis, Semliki forest)], (−)RNA viruses [orthomyxoviruses (influenza), paramyxoviruses (parainfluenza, measles), rhabdoviruses (vesicular stomatitis)] and dsRNA viruses (reovirus) (De Clercq et al., 1990). Ce-Cyd is a more potent antiviral agent than its counterpart, C-Cyd, and, in addition, it has also proved active against picornaviruses (poliovirus, Coxsackie B, rhinovirus) and herpesviruses (De Clercq et al., 1991). The antiviral activity spectrum of Ce-Cyd extends to bunyaviruses (Punta Toro), flaviviruses (Japanese encephalitis) and arenaviruses (Junin, Tacaribe) (Marquez et al., 1988, Andrei and De Clercq, 1990). For both the antitumor and antiviral effects of C-Cyd and Ce-Cyd, the putative target is assumed to be CTP synthetase, the enzyme that catalyses the conversion of UTP to CTP, which is the final step in the de novo pyrimidine biosynthetic pathway. The fact that both the antiviral and cytotoxic effect of C-Cyd and Ce-Cyd are readily reversed by cytidine and, to a lesser extent, by uridine, but not at all by other nucleosides, such as 2′-deoxythymidine or 2′-deoxycytidine, supports this hypothesis.
Fig. 23.
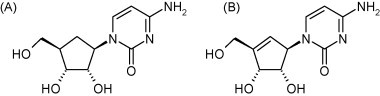
Structure of cyclopentylcytosine (A) and cyclopentylcytosine (B).
5. Conclusion
Many targets exist for selective inhibition of the replication of RNA viruses, some of which have been discussed in this paper. Many other (potential) interesting targets have remained unexplored. Concerted efforts of dedicated laboratories acting in international networks, such as the VIZIER network, described by Coutard et al. (this issue), that study the structural genomics of enzymes involved in RNA virus replication may be instrumental in identifying novel antiviral strategies to inhibit RNA virus resplication. For example, it may be assumed that the strictly regulated nuclear import of the eight influenza RNA segments or the encapsidation of one single copy of each of these RNA segment into the progeny viral particles, may represent potential targets for therapeutic intervention (Greber and Fornerod, 2005). Although it is technically feasible to develop drugs for the treatment of various families or genera of RNA viruses (for example flaviviruses, orthomyxoviruses, paramyxoviruses, picornaviruses, etc.), the pharmaceutical industry is unlikely to develop antivirals for diseases for which the market is too small or uncertain. Ideally, antiviral drugs should be developed that exert broad-spectrum anti-RNA virus activity. The fact that the influenza polymerase inhibitor T-705 also inhibits the replication of other RNA viruses (Gowen et al., 2007) and that 2′-C-methyl nucleoside analogue inhibitors of the HCV RdRp inhibit also the replication of various other (+)ssRNA viruses, suggest that nucleoside polymerase inhibitors may likely to be broad-spectrum antiviral drug candidates. If such drugs were to be licensed for the treatment of such frequent infections as influenza or HCV, they could eventually be used “off-label”, or on a compassionate basis, for the treatment of other important, but less common infections with highly pathogenic RNA viruses.
Acknowledgements
The authors are supported by RiviGene (Risk Virus Genome Database) “Genomic inventory, forensic markers, and assessment of potential therapeutic and vaccine targets for viruses relevant in biological crime and terrorism” from the EU (FP6 Contract no.: 022639). We thank Mrs. Dominique Brabants for editorial help.
References
- Abdel-Magid A.F., Maryanoff C.A., Mehrman S.J. Synthesis of influenza neuraminidase inhibitors. Curr. Opin. Drug Discov. Dev. 2001;4:776–791. [PubMed] [Google Scholar]
- Airaksinen A., Pariente N., Menéndez-Arias L., Domingo E. Curing of foot-and-mouth disease virus from persistently infected cells by ribavirin involves enhanced mutagenesis. Virology. 2003;311:339–349. doi: 10.1016/s0042-6822(03)00144-2. [DOI] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Andrei G., De Clercq E. Inhibitory effect of selected antiviral compounds on arenavirus replication in vitro. Antivir. Res. 1990;14:287–300. doi: 10.1016/0166-3542(90)90009-v. [DOI] [PubMed] [Google Scholar]
- Andrei G., De Clercq E. Molcular approaches for the treatment of hemorrhagic fever virus infections. Antivir. Res. 1993;22:45–75. doi: 10.1016/0166-3542(93)90085-w. [DOI] [PubMed] [Google Scholar]
- Asahina Y., Izumi N., Enomoto N., Uchihara M., Kurosaki M., Onuki Y., Nishimura Y., Ueda K., Tsuchiya K., Nakanishi H., Kitamura T., Miyake S. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J. Hepatol. 2005;43:623–629. doi: 10.1016/j.jhep.2005.05.032. [DOI] [PubMed] [Google Scholar]
- Baginski S.G., Pevear D.C., Seipel M., Sun S.C., Benetatos C.A., Chunduru S.K., Rice C.M., Collett M.S. Mechanism of action of a pestivirus antiviral compound. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7981–7986. doi: 10.1073/pnas.140220397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J., Karlsson A., Wang L., Bohman C., Horská K., Votruba I., Fridland A., Van Aerschot A., Herdewijn P., De Clercq E. Eicar (5-ethynyl-1-beta-d-ribofuranosylimidazole-4-carboxamide). A novel potent inhibitor of inosinate dehydrogenase activity and guanylate biosynthesis. J. Biol. Chem. 1993;268:24591–24598. [PubMed] [Google Scholar]
- Balzarini J. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat. Rev. Microbiol. 2007;5:583–597. doi: 10.1038/nrmicro1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J. Carbohydrate-binding agents: a potential future cornerstone for the chemotherapy of enveloped viruses? Antivir. Chem. Chemother. 2007;18:1–11. doi: 10.1177/095632020701800101. (review) [DOI] [PubMed] [Google Scholar]
- Balzarini J., Van Laethem K., Daelemans D., Hatse S., Bugatti A., Rusnati M., Igarashi Y., Oki T., Schols D. Pradimicin A, a carbohydrate-binding nonpeptidic lead compound for treatment of infections with viruses with highly glycosylated envelopes, such as human immunodeficiency virus. J. Virol. 2007;81:362–373. doi: 10.1128/JVI.01404-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard D.L., Day C.W., Bailey K., Heiner M., Montgomery R., Lauridsen L., Winslow S., Hoopes J., Li J.K., Lee J., Carson D.A., Cottam H.B., Sidwell R.W. Enhancement of the infectivity of SARS-CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antivir. Res. 2006;71:53–63. doi: 10.1016/j.antiviral.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch D., Sprecher A.G., Jeffs B., Boumandouki P. Treatment of Marburg and Ebola hemorrhagic fevers: a strategy for testing new drugs and vaccines under outbreak conditions. Antivir. Res. 2008;78:150–161. doi: 10.1016/j.antiviral.2008.01.152. [DOI] [PubMed] [Google Scholar]
- Beigel J., Bray M. Current and future antiviral therapy of severe seasonal and avian influenza. Antivir. Res. 2008;78:91–102. doi: 10.1016/j.antiviral.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch D., Egloff M.P., Mulard L., Guerreiro C., Romette J.L., Canard B. A structural basis for the inhibition of the NS5 dengue virus mRNA 2′-O-methyltransferase domain by ribavirin 5′-triphosphate. J. Biol. Chem. 2004;279:35638–35643. doi: 10.1074/jbc.M400460200. [DOI] [PubMed] [Google Scholar]
- Binford S.L., Maldonado F., Brothers M.A., Weady P.T., Zalman L.S., Meador J.W., 3rd, Matthews D.A., Patick A.K. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 2005;49:619–626. doi: 10.1128/AAC.49.2.619-626.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binford S.L., Weady P.T., Maldonado F., Brothers M.A., Matthews D.A., Patick A.K. In vitro resistance study of rupintrivir, a novel inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 2007;51(12):4366–4373. doi: 10.1128/AAC.00905-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolken T.C., Laquerre S., Zhang Y., Bailey T.R., Pevear D.C., Kickner S.S., Sperzel L.E., Jones K.F., Warren T.K., Amanda Lund S., Kirkwood-Watts D.L., King D.S., Shurtleff A.C., Guttieri M.C., Deng Y., Bleam M., Hruby D.E. Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antivir. Res. 2006;69:86–97. doi: 10.1016/j.antiviral.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowski P., Lang M., Haag A., Baier A. Tropolone and its derivatives as inhibitors of the helicase activity of hepatitis C virus nucleotide triphosphatase/helicase. Antivir. Chem. Chemother. 2007;18:103–109. doi: 10.1177/095632020701800206. [DOI] [PubMed] [Google Scholar]
- Bossi P., Tegnell A., Baka A., Van Loock F., Hendriks J., Werner A., Maidhof H., Gouvras G. Task force on biological and chemical agent threats, Public Health Directorate, European Commission, Luxembourg. Bichat guidelines for the clinical management of haemorrhagic fever viruses and bioterrorism-related haemorrhagic fever viruses. Eur. Surveill. 2004;9:E11–E12. [PubMed] [Google Scholar]
- Bougie I., Bisaillon M. Initial binding of the broad spectrum antiviral nucleoside ribavirin to the hepatitis C virus RNA polymerase. J. Biol. Chem. 2003;278:52471–52478. doi: 10.1074/jbc.M308917200. [DOI] [PubMed] [Google Scholar]
- Bougie I., Bisaillon M. The broad spectrum antiviral nucleoside ribavirin as a substrate for a viral RNA capping enzyme. J. Biol. Chem. 2004;279:22124–22130. doi: 10.1074/jbc.M400908200. [DOI] [PubMed] [Google Scholar]
- Bray M. Highly pathogenic RNA viral infections: challenges for antiviral research. Antivir. Res. 2008;78:1–8. doi: 10.1016/j.antiviral.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Bray M., Driscoll J., Huggins J.W. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-l-homocysteine hydrolase inhibitor. Antivir. Res. 2000;45:135–147. doi: 10.1016/s0166-3542(00)00066-8. [DOI] [PubMed] [Google Scholar]
- Bray M., Raymond J.L., Geisbert T., Baker R.O. 3-Deazaneplanocin A induces massively increased interferon-alpha production in Ebola virus-infected mice. Antivir. Res. 2002;55:151–159. doi: 10.1016/s0166-3542(02)00018-9. [DOI] [PubMed] [Google Scholar]
- Bright R.A., Medina M.J., Xu X., Perez-Oronoz G., Wallis T.R., Davis X.M., Povinelli L., Cox N.J., Klimov A.I. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366(9492):1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- Briolant S., Garin D., Scaramozzino N., Jouan A., Crance J.M. In vitro inhibition of Chikungunya and Semliki Forest viruses replication by antiviral compounds: synergistic effect of interferon-alpha and ribavirin combination. Antivir. Res. 2004;61:111–117. doi: 10.1016/j.antiviral.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Carter W.A., De Clercq E. Viral infection and host defense. Science. 1974;186:1172–1178. doi: 10.1126/science.186.4170.1172. [DOI] [PubMed] [Google Scholar]
- Chapman L.E. Intravenous ribavirin for hantavirus pulmonary syndrome: safety and tolerance during 1 year of open-label experience. Antivir. Ther. 1999;4:211–219. doi: 10.1177/135965359900400404. [DOI] [PubMed] [Google Scholar]
- Chapman J., Abbott E., Alber D.G., Baxter R.C., Bithell S.K., Henderson E.A., Carter M.C., Chambers P., Chubb A., Cockerill G.S., Collins P.L., Dowdell V.C., Keegan S.J., Kelsey R.D., Lockyer M.J., Luongo C., Najarro P., Pickles R.J., Simmonds M., Taylor D., Tyms S., Wilson L.J., Powell K.L. RSV604, a novel inhibitor of respiratory syncytial virus replication. Antimicrob. Agents Chemother. 2007;51:3346–3353. doi: 10.1128/AAC.00211-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaliez S., Brillet R., Lazaro E., Hezode C., Pawlotsky J.M. Analysis of ribavirin mutagenicity in human hepatitis C virus infection. J. Virol. 2007;81(14):7732–7741. doi: 10.1128/JVI.00382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaliez S., Pawlotsky J.M. Hepatitis C virus: virology, diagnosis and management of antiviral therapy. World J. Gastroenterol. 2007;13:2461–2466. doi: 10.3748/wjg.v13.i17.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H.T., Kamarulzaman A., Tan C.T., Goh K.J., Thayaparan T., Kunjapan S.R., Chew N.K., Chua K.B., Lam S.K. Treatment of acute Nipah encephalitis with ribavirin. Ann. Neurol. 2001;49:810–813. doi: 10.1002/ana.1062. [DOI] [PubMed] [Google Scholar]
- Coelmont L., Paeshuyse J., Kaptein S., Vliegen I., Kaul A., De Clercq E., Rosenwirth B., Scalfaro P., Crabbe R., Bartenschlager R., Dumont J.M., Neyts J. The cyclophilin inhibitor Debio-025 is a potent inhibitor of hepatitis C virus replication in vitro with a unique resistance profile. Antivir. Res. 2007;74:A39. (abstract no. 29) [Google Scholar]
- Collaborative Antiviral Study Group Placebo-controlled, double-blind trial of intravenous ribavirin for the treatment of hantavirus cardiopulmonary syndrome in North America. Clin. Infect. Dis. 2004;39(9):1307–1313. doi: 10.1086/425007. [DOI] [PubMed] [Google Scholar]
- Crotty S., Maag D., Arnold J.J., Zhong W., Lau J.Y., Hong Z., Andino R., Cameron C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Crotty S., Cameron C.E., Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies W.L., Grunert R.R., Haff R.F., MCGahen J.W., Neumayer E.M., Paulschock M., Watts J.C., Wood T.R., Hermann E.C., Hoffmann C.E. Antiviral activity of 1-adamantanamine (amantadine) Science. 1964;144:862–863. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- De Clercq E., Bernaerts R., Shealy Y.F., Montgomery J.A. Broad-spectrum antiviral activity of carbodine, the carbocyclic analogue of cytidine. Biochem. Pharmacol. 1990;39:319–325. doi: 10.1016/0006-2952(90)90031-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E., Murase J., Marquez V.E. Broad-spectrum antiviral and cytocidal activity of cyclopentenylcytosine, a carbocyclic nucleoside targeted at CTP synthetase. Biochem. Pharmacol. 1991;41:1821–1829. doi: 10.1016/0006-2952(91)90120-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Antiviral drug discovery and development: where chemistry meets with biomedicine. Antivir. Res. 2005;67:56–75. doi: 10.1016/j.antiviral.2005.05.001. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco R., Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature. 2005;436:953–960. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- De Palma, A.M., Vliegen, I., De Clercq, E., Neyts, J. Selective inhibitors of picornaviruses, submitted for publication.
- De Palma, A.M., Heggermont, W., Lanke, K., Coutard, C., Canard, B., De Clercq, E., Chimirri, A., Rohayem, J., Norder, H., Van Kuppeveld, F., Neyts, J. TBZ inhibits replication of enterovirus species B and D by targeting a short conserved domain in the non-structural protein 2C. J. Virol., accepted for publication.
- De Palma A.M., Heggermont W., Leyssen P., Pürstinger G., Wimmer E., De Clercq E., Rao A., Monforte A.M., Chimirri A., Neyts J. Anti-enterovirus activity and structure-activity relationship of a series of 2,6-dihalophenyl-substituted 1H, 3H-thiazolo[3,4-a]benzimidazoles. Biochem. Biophys. Res. Commun. 2007;353:628–632. doi: 10.1016/j.bbrc.2006.12.063. [DOI] [PubMed] [Google Scholar]
- Deng T., Sharps J., Fodor E., Brownlee G.G. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J. Virol. 2005;79:8669–8674. doi: 10.1128/JVI.79.13.8669-8674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit N.M., Layden-Almer J.E., Layden T.J., Perelson A.S. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature. 2004;432:922–924. doi: 10.1038/nature03153. [DOI] [PubMed] [Google Scholar]
- Dwek R.A., Butters T.D., Platt F.M., Zitzmann N. Targeting glycosylation as a therapeutic approach. Nat. Rev. Drug Discov. 2002;1:65–75. doi: 10.1038/nrd708. [DOI] [PubMed] [Google Scholar]
- Douglas J.L., Panis M.L., Ho E., Lin K.Y., Krawczyk S.H., Grant D.M., Cai R., Swaminathan S., Cihlar T. Inhibition of respiratory syncytial virus fusion by the small molecule VP-14637 via specific interactions with F protein. J. Virol. 2003;77:5054–5064. doi: 10.1128/JVI.77.9.5054-5064.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J.L., Panis M.L., Ho E., Lin K.Y., Krawczyk S.H., Grant D.M., Cai R., Swaminathan S., Chen X., Cihlar T. Small molecules VP-14637 and JNJ-2408068 inhibit respiratory syncytial virus fusion by similar mechanisms. Antimicrob. Agents Chemother. 2005;49:2460–2466. doi: 10.1128/AAC.49.6.2460-2466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoretskaia I., Bogatikov G.V., Pshenichnov V.A., Evseev A.A. The therapeutic efficacy of ribamidil and virazole in experimental Lassa fever in monkeys. Vopr. Virusol. 1991;36:55–57. [PubMed] [Google Scholar]
- Dwyer J.J., Wilson K.L., Davison D.K., Freel S.A., Seedorff J.E., Wring S.A., Tvermoes N.A., Matthews T.J., Greenberg M.L., Delmedico M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M.P., Benarroch D., Selisko B., Romette J.L., Canard B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M.P., Decroly E., Malet H., Selisko B., Benarroch D., Ferron F., Canard B. Structural and functional analysis of methylation and 5′-RNA sequence requirements of short capped RNAs by the methyltransferase domain of dengue virus NS5. J. Mol. Biol. 2007;372:723–736. doi: 10.1016/j.jmb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Enria D.A., Briggliler A.M., Sanchez Z. Treatment of argentine hemorrhagic fever. Antivir. Res. 2008;78:132–139. doi: 10.1016/j.antiviral.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergonul O. Treatment of Crimean-Congo hemorrhagic fever. Antivir. Res. 2008;78:125–131. doi: 10.1016/j.antiviral.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Eriksson B., Helgstrand E., Johansson N.G., Larsson A., Misiorny A., Norén J.O., Philipson L., Stenberg K., Stening G., Stridh S., Oberg B. Inhibition of influenza virus ribonucleic acid polymerase by ribavirintriphosphate. Antimicrob. Agents Chemother. 1977;11:946–951. doi: 10.1128/aac.11.6.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C., Srivastava P., Lin C.C. Effect of ribavirin, levovirin and viramidine on liver toxicological gene expression in rats. J. Appl. Toxicol. 2003;23:453–459. doi: 10.1002/jat.938. [DOI] [PubMed] [Google Scholar]
- Fisher-Hoch S.P., Khan J.A., Rehman S., Mirza S., Khurshid M., McCormick J.B. Crimean Congo-haemorrhagic fever treated with oral ribavirin. Lancet. 1995;346:472–475. doi: 10.1016/s0140-6736(95)91323-8. [DOI] [PubMed] [Google Scholar]
- Forestier N., Reesink H.W., Weegink C.J., McNair L., Kieffer T.L., Chu H.M., Purdy S., Jansen P.L., Zeuzem S. Antiviral activity of telaprevir (VX-950) and peginterferon alfa-2a in patients with hepatitis C. Hepatology. 2007;46:640–648. doi: 10.1002/hep.21774. [DOI] [PubMed] [Google Scholar]
- Frick D.N. The hepatitis C virus NS3 protein: a model RNA helicase and potential drug target. Curr. Issues Mol. Biol. 2007;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- Furuta Y., Takahashi K., Kuno-Maekawa M., Sangawa H., Uehara S., Kozaki K., Nomura N., Egawa H., Shiraki K. Mechanism of action of T-705 against influenza virus. Antimicrob. Agents Chemother. 2005;49:981–986. doi: 10.1128/AAC.49.3.981-986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y., Takahashi K., Fukuda Y., Kuno M., Kamiyama T., Kozaki K., Nomura N., Egawa H., Minami S., Watanabe Y., Narita H., Shiraki K. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 2002;46:977–981. doi: 10.1128/AAC.46.4.977-981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L., Seyedsayamdost M.R., Shuto S., Matsuda A., Petsko G.A., Hedstrom L. The immunosuppressive agent mizoribine monophosphate forms a transition state analogue complex with inosine monophosphate dehydrogenase. Biochemistry. 2003;42:857–863. doi: 10.1021/bi0271401. [DOI] [PubMed] [Google Scholar]
- Ganesh V.K., Muller N., Judge K., Luan C.H., Padmanabhan R., Murthy K.H. Identification and characterization of nonsubstrate based inhibitors of the essential dengue and West Nile virus proteases. Bioorg. Med. Chem. 2005;13:257–264. doi: 10.1016/j.bmc.2004.09.036. [DOI] [PubMed] [Google Scholar]
- Georges-Courbot M.C., Contamin H., Faure C., Loth P., Baize S., Leyssen P., Neyts J., Deubel V. Poly-(I)-poly(C12U) but not ribavirin prevents death in a hamster model of Nipah virus infection. Antimicrob. Agents Chemother. 2006;50:1768–1772. doi: 10.1128/AAC.50.5.1768-1772.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris N., De Palma A., Toussaint J.F., Musch I., Neyts J., De Clercq K. 2′-C-methylcytidine as a potent and selective inhibitor of the replication of foot-and-mouth disease virus. Antivir. Res. 2007;73:161–168. doi: 10.1016/j.antiviral.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Goris N., Vandenbussche F., De Clercq K. Potential of antiviral prophylaxis and therapy for controlling RNA viral infections of livestock. Antivir. Res. 2008;78:170–178. doi: 10.1016/j.antiviral.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Goswami B.B., Borek E., Sharma O.K., Fujitaki J., Smith R.A. The broad spectrum antiviral agent ribavirin inhibits capping of mRNA. Biochem. Biophys. Res. Commun. 1979;89:830–836. doi: 10.1016/0006-291x(79)91853-9. [DOI] [PubMed] [Google Scholar]
- Gould E., Solomon T., Mackenzie J. Does antiviral therapy have a role in the control of Japanese encephalitis? Antivir. Res. 2008;78:140–149. doi: 10.1016/j.antiviral.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Gowen B., Holbrook M. Animal models of highly pathogenic RNA viral infections: hemorrhagic fever viruses. Antivir. Res. 2008;78:79–90. doi: 10.1016/j.antiviral.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Gowen B.B., Wong M.H., Jung K.H., Sanders A.B., Mendenhall M., Bailey K.W., Furuta Y., Sidwell R.W. In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob. Agents Chemother. 2007;51:3168–3176. doi: 10.1128/AAC.00356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graci J.D., Cameron C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006;16:37–48. doi: 10.1002/rmv.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber U.F., Fornerod M. Nuclear import in viral infections. Curr. Top. Microbiol. Immunol. 2005;285:109–138. doi: 10.1007/3-540-26764-6_4. [DOI] [PubMed] [Google Scholar]
- Gunther S., Asper M., Roser C., Luna L.K., Drosten C., Becker-Ziaja B., Borowski P., Chen H.M., Hosmane R.S. Application of real-time PCR for testing antiviral compounds against Lassa virus, SARS coronavirus and Ebola virus in vitro. Antivir. Res. 2004;63:209–215. doi: 10.1016/j.antiviral.2004.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G., Qiao J., Dong C., He C., Zhao L., Tian Y. Amantadine-resistance among H5N1 avian influenza viruses isolated in Northern China. Antivir. Res. 2008;77(1):72–76. doi: 10.1016/j.antiviral.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Holbrook M., Gowen B. Animal models of highly pathogenic RNA viral infections: encephalitis viruses. Antivir. Res. 2008;78:69–78. doi: 10.1016/j.antiviral.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Horimoto T., Kawaoka Y. Influenza: lessons from past pandemics, warnings from current incidents. Nat. Rev. Microbiol. 2005;3:591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- Hosoya M., Shigeta S., Nakamura K., De Clercq E. Inhibitory effect of selected antiviral compounds on measeles (SSPE) virus replication in vitro. Antivir. Res. 1989;12:87–98. doi: 10.1016/0166-3542(89)90072-7. [DOI] [PubMed] [Google Scholar]
- Huggins J.W., Robins R.K., Canonico P.G. Synergistic antiviral effects of ribavirin and the C-nucleoside analogs tiazofurin and selenazofurin against togaviruses, bunyaviruses, and arenaviruses. Antimicrob. Agents Chemother. 1984;26:476–480. doi: 10.1128/aac.26.4.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins J.W. Prospects for treatment of viral hemorrhagic fevers with ribavirin, a broad-spectrum antiviral drug. Rev. Infect. Dis. 1989;11(Suppl. 4):S750–S761. doi: 10.1093/clinids/11.supplement_4.s750. [DOI] [PubMed] [Google Scholar]
- Huggins J.W., Hsiang C.M., Cosgriff T.M., Guang M.Y., Smith J.I., Wu Z.O., LeDuc J.W., Zheng Z.M., Meegan J.M., Wang Q.N. Prospective, double-blind, concurrent, placebo-controlled clinical trial of intravenous ribavirin therapy of hemorrhagic fever with renal syndrome. J. Infect. Dis. 1991;164:1119–1127. doi: 10.1093/infdis/164.6.1119. [DOI] [PubMed] [Google Scholar]
- Huggins J., Zhang Z.X., Bray M. Antiviral drug therapy of filovirus infections: S-adenosylhomocysteine hydrolase inhibitors inhibit Ebola virus in vitro and in a lethal mouse model. J. Infect. Dis. 1999;179(Suppl. 1):S240–S247. doi: 10.1086/514316. [DOI] [PubMed] [Google Scholar]
- Huffman J.H., Sidwell R.W., Khare G.P., Witkowski J.T., Allen L.B., Robins R.K. In vitro effect of 1-beta-d-ribofuranosyl-1,2,4-triazole-3-carboxamide (virazole, ICN 1229) on deoxyribonucleic acid and ribonucleic acid viruses. Antimicrob. Agents Chemother. 1973;3:235–241. doi: 10.1128/aac.3.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahrling P.B., Hesse R.A., Eddy G.A., Johnson K.M., Callis R.T., Stephen E.L. Lassa virus infection of rhesus monkeys: pathogenesis and treatment with ribavirin. J. Infect. Dis. 1980;141:580–589. doi: 10.1093/infdis/141.5.580. [DOI] [PubMed] [Google Scholar]
- Johnson S., Oliver C., Prince G.A., Hemming V.G., Pfarr D.S., Wang S.C., Dormitzer M., O’Grady J., Koenig S., Tamura J.K., Woods R., Bansal G., Couchenour D., Tsao E., Hall W.C., Young J.F. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J. Infect. Dis. 1997;176:1215–1224. doi: 10.1086/514115. [DOI] [PubMed] [Google Scholar]
- Jonsson C.B., Hooper J., Mertz G. Treatment of hantavirus pulmonary syndrome. Antivir. Res. 2008;78:162–169. doi: 10.1016/j.antiviral.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubin R. Targeting hepatitis C virus translation: stopping HCV where it starts. Curr. Opin. Invest. Drugs. 2003;4:162–167. [PubMed] [Google Scholar]
- Kawana F., Shigeta S., Hosoya M., Suzuki H., De Clercq E. Inhibitory effects of antiviral compounds on respiratory syncytial virus replication in vitro. Antimicrob. Agents Chemother. 1987;31:1225–1230. doi: 10.1128/aac.31.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S.H., Goba A., Chu M., Roth C., Healing T., Marx A., Fair J., Guttieri M.C., Ferro P., Imes T., Monagin C., Garry R.F., Bausch D.G. New opportunities for field research on the pathogenesis and treatment of Lassa fever. Antivir. Res. 2008;78:103–115. doi: 10.1016/j.antiviral.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Kim C.U., Lew W., Williams M.A., Liu H., Zhang L., Swaminathan S., Bischofberger N., Chen M.S., Mendel D.B., Tai C.Y., Laver W.G., Stevens R.C. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- Klumpp K., Leveque V., Le Pogam S., Ma H., Jiang W.R., Kang H., Granycome C., Singer M., Laxton C., Hang J.Q., Sarma K., Smith D.B., Heindl D., Hobbs C.J., Merrett J.H., Symons J., Cammack N., Martin J.A., Devos R., Najera I. The novel nucleoside analog R1479 (4′-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J. Biol. Chem. 2006;281:3793–3799. doi: 10.1074/jbc.M510195200. [DOI] [PubMed] [Google Scholar]
- Lamarre D., Anderson P.C., Bailey M., Beaulieu P., Bolger G., Bonneau P., Bos M., Cameron D.R., Cartier M., Cordingley M.G., Faucher A.M., Goudreau N., Kawai S.H., Kukolj G., Lagace L., LaPlante S.R., Narjes H., Poupart M.A., Rancourt J., Sentjens R.E., St George R., Simoneau B., Steinmann G., Thibeault D., Tsantrizos Y.S., Weldon S.M., Yong C.L., Llinas-Brunet M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- Lanford R.E., Chavez D., Guerra B., Lau J.Y., Hong Z., Brasky K.M., Beames B. Ribavirin induces error-prone replication of GB virus B in primary tamarin hepatocytes. J. Virol. 2001;75:8074–8081. doi: 10.1128/JVI.75.17.8074-8081.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyssen P., Balzarini J., De Clercq E., Neyts J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 2005;79:1943–1947. doi: 10.1128/JVI.79.3.1943-1947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyssen P., De Clercq E., Neyts J. The anti-yellow fever virus activity of ribavirin is independent of error-prone replication. Mol. Pharmacol. 2006;69:1461–1467. doi: 10.1124/mol.105.020057. [DOI] [PubMed] [Google Scholar]
- Lin C.C., Xu C., Zhu N., Yeh L.T. Absorption, metabolism, and excretion of [14C]viramidine in humans. Antimicrob. Agents Chemother. 2006;50:2368–2373. doi: 10.1128/AAC.00118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutchman G.A., Danehower S., Park Y., Ward C., Liang J.T., Hoofnagle J.H. Mutation rate of hepatitis C virus in patients during ribavirin monotherapy. Hepatology. 2004;40(Suppl. 1):385A. [Google Scholar]
- Luzhkov V.B., Selisko B., Nordqvist A., Peyrane F., Decroly E., Alvarez K., Karlen A., Canard B., Qvist J. Virtual screening and bioassay study of novel inhibitors for dengue virus mRNA cap (nucleoside-2′O)-methyltransferase. Bioorg. Med. Chem. 2007;15(24):7795–7802. doi: 10.1016/j.bmc.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Maag D., Castro C., Hong Z., Cameron C.E. Hepatitis C virus RNA-dependent RNA polymerase (NS5B) as a mediator of the antiviral activity of ribavirin. J. Biol. Chem. 2001;276(49):46094–46098. doi: 10.1074/jbc.C100349200. [DOI] [PubMed] [Google Scholar]
- Malakhov M.P., Aschenbrenner L.M., Smee D.F., Wandersee M.K., Sidwell R.W., Gubareva L.V., Mishin V.P., Hayden F.G., Kim D.H., Ing A., Campbell E.R., Yu M., Fang F. Sialidase fusion protein as a novel broadspectrum inhibitor of influenza virus infection. Antimicrob. Agents Chemother. 2006;50:1470–1479. doi: 10.1128/AAC.50.4.1470-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinoski F., Stollar V. Inhibitors of IMP dehydrogenase prevent sindbis virus replication and reduce GTP levels in Aedes albopictus cells. Virology. 1981;30:281–289. doi: 10.1016/0042-6822(81)90060-x. [DOI] [PubMed] [Google Scholar]
- Mardani M., Jahromi M.K., Naieni K.H., Zeinali M. The efficacy of oral ribavirin in the treatment of Crimean-Congo hemorrhagic fever in Iran. Clin. Infect. Dis. 2003;36:1613–1618. doi: 10.1086/375058. [DOI] [PubMed] [Google Scholar]
- Markland W., McQuaid T.J., Jain J., Kwong A.D. Broad-spectrum antiviral activity of the IMP dehydrogenase inhibitor VX-497: a comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob. Agents Chemother. 2000;44:859–866. doi: 10.1128/aac.44.4.859-866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez V.E., Lim M.I., Treanor S.P., Plowman J., Priest M.A., Markovac A., Khan M.S., Kaskar B., Driscoll J.S. Cyclopentenylcytosine. A carbocyclic nucleoside with antitumor and antiviral properties. J. Med. Chem. 1988;31:1687–1694. doi: 10.1021/jm00117a004. [DOI] [PubMed] [Google Scholar]
- McCormick J.B., King I.J., Webb P.A., Scribner C.L., Craven R.B., Johnson K.M., Elliott L.H., Belmont-Williams R. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986;314:20–26. doi: 10.1056/NEJM198601023140104. [DOI] [PubMed] [Google Scholar]
- McKee K.T., Jr., Huggins J.W., Trahan C.J., Mahlandt B.G. Ribavirin prophylaxis and therapy for experimental argentine hemorrhagic fever. Antimicrob. Agents Chemother. 1988;32:1304–1309. doi: 10.1128/aac.32.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monath T.P. Treatment of yellow fever. Antivir. Res. 2008;78:116–124. doi: 10.1016/j.antiviral.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Monto A.S. The role of antivirals in the control of influenza. Vaccine. 2003;21(16):1796–1800. doi: 10.1016/s0264-410x(03)00075-6. [DOI] [PubMed] [Google Scholar]
- Montgomery J.A., Clayton S.J., Thomas H.J., Shannon W.M., Arnett G., Bodner A.J., Kion I.K., Cantoni G.L., Chiang P.K. Carbocyclic analogue of 3-deazaadenosine: a novel antiviral agent using S-adenosylhomocysteine hydrolase as a pharmacological target. J. Med. Chem. 1982;25:626–629. doi: 10.1021/jm00348a004. [DOI] [PubMed] [Google Scholar]
- Moscona A. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J. Clin. Invest. 2005;115:1688–1698. doi: 10.1172/JCI25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005;29:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- Murthy H.M., Judge K., DeLucas L., Padmanabhan R. Crystal structure of Dengue virus NS3 protease in complex with a Bowman-Birk inhibitor: implications for flaviviral polyprotein processing and drug design. J. Mol. Biol. 2000;301:759–767. doi: 10.1006/jmbi.2000.3924. [DOI] [PubMed] [Google Scholar]
- Naka K., Ikeda M., Abe K., Dansako H., Kato N. Mizoribine inhibits hepatitis C virus RNA replication: effect of combination with interferon-alpha. Biochem. Biophys. Res. Commun. 2005;13:871–879. doi: 10.1016/j.bbrc.2005.03.062. [DOI] [PubMed] [Google Scholar]
- Nakagawa M., Sakamoto N., Enomoto N., Tanabe Y., Kanazawa N., Koyama T., Kurosaki M., Maekawa S., Yamashiro T., Chen C.H., Itsui Y., Kakinuma S., Watanabe M. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 2004;313:42–47. doi: 10.1016/j.bbrc.2003.11.080. [DOI] [PubMed] [Google Scholar]
- Nakagawa M., Sakamoto N., Tanabe Y., Koyama T., Itsui Y., Takeda Y., Chen C.H., Kakinuma S., Oooka S., Maekawa S., Enomoto N., Watanabe M. Suppression of hepatitis C virus replication by cyclosporin a is mediated by blockade of cyclophilins. Gastroenterology. 2005;129:1031–1041. doi: 10.1053/j.gastro.2005.06.031. [DOI] [PubMed] [Google Scholar]
- Neyts J., Meerbach A., McKenna P., De Clercq E. Use of the yellow fever virus vaccine strain 17D for the study of strategies for the treatment of yellow fever virus infections. Antivir. Res. 1996;30(2–3):125–132. doi: 10.1016/0166-3542(96)89697-5. [DOI] [PubMed] [Google Scholar]
- Paeshuyse J., Kaul A., De Clercq E., Rosenwirth B., Dumont J.M., Scalfaro P., Bartenschlager R., Neyts J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43:761–770. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- Paeshuyse J., Leyssen P., Mabery E., Boddeker N., Vrancken R., Froeyen M., Ansari I.H., Dutartre H., Rozenski J., Gil L.H., Letellier C., Lanford R., Canard B., Koenen F., Kerkhofs P., Donis R.O., Herdewijn P., Watson J., De Clercq E., Pürstinger G., Neyts J. A novel, highly selective inhibitor of pestivirus replication that targets the viral RNA-dependent RNA polymerase. J. Virol. 2006;80:149–160. doi: 10.1128/JVI.80.1.149-160.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeshuyse J., Chezal J.M., Froeyen M., Leyssen P., Dutartre H., Vrancken R., Canard B., Letellier C., Li T., Mittendorfer H., Koenen F., Kerkhofs P., De Clercq E., Herdewijn P., Pürstinger G., Gueiffier A., Chavignon O., Teulade J.C., Neyts J. The imidazopyrrolopyridine analogue AG110 is a novel, highly selective inhibitor of pestiviruses that targets the viral RNA-dependent RNA polymerase at a hot spot for inhibition of viral replication. J. Virol. 2007;81:11046–11053. doi: 10.1128/JVI.00388-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patick A.K., Brothers M.A., Maldonado F., Binford S., Maldonado O., Fuhrman S., Petersen A., Smith G.J., 3rd, Zalman L.S., Burns-Naas L.A., Tran J.Q. In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 2005;49:2267–2275. doi: 10.1128/AAC.49.6.2267-2275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patick A.K. Rhinovirus chemotherapy. Antivir. Res. 2006;71:391–396. doi: 10.1016/j.antiviral.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A., Kukolj G., Bailey M., Brault M., Do F., Halmos T., Lagace L., Maurice R., Marquis M., McKercher G., Pellerin C., Pilote L., Thibeault D., Lamarre D. An NS3 serine protease inhibitor abrogates replication of subgenomic hepatitis C virus RNA. J. Biol. Chem. 2003;278:20374–20380. doi: 10.1074/jbc.M210785200. [DOI] [PubMed] [Google Scholar]
- Pawlotsky J.M., Brillet R., Hezode C., Dhumeaux D. Is ribavirin antiviral effect in humans mediated b mutagenic properties resulting in error catastrophe? J. Hepatol. 2004;40(Suppl. 1):23. [Google Scholar]
- Pawlotsky J.M., Dahari H., Neumann A.U., Hezode C., Germanidis G., Lonjon I., Castera L., Dhumeaux D. Antiviral action of ribavirin in chronic hepatitis C. Gastroenterology. 2004;126:703–714. doi: 10.1053/j.gastro.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Porotto M., Doctor L., Carta P., Fornabaio M., Greengard O., Kellogg G.E., Moscona A. Inhibition of hendra virus fusion. J. Virol. 2006;80:9837–9849. doi: 10.1128/JVI.00736-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer J.K., Kirkegaard K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada B., Dragun M. Antiviral action and selectivity of 6-azauridine. Ann. N. Y. Acad. Sci. 1977;284:410–417. doi: 10.1111/j.1749-6632.1977.tb21977.x. [DOI] [PubMed] [Google Scholar]
- Rankin J.T., Jr., Eppes S.B., Antczak J.B., Joklik W.K. Studies on the mechanism of the antiviral activity of ribavirin against reovirus. Virology. 1989;168:147–158. doi: 10.1016/0042-6822(89)90413-3. [DOI] [PubMed] [Google Scholar]
- Rice C.M., You S. Treating hepatitis C: can you teach old dogs new tricks? Hepatology. 2005;42:1455–1458. doi: 10.1002/hep.20975. [DOI] [PubMed] [Google Scholar]
- Russell R.J., Haire L.F., Stevens D.J., Collins P.J., Lin Y.P., Blackburn G.M., Hay A.J., Gamblin S.J., Skehel J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- Saijo M., Morikawa S., Fukushi S., Mizutani T., Hasegawa H., Nagata N., Iwata N., Kurane I. Inhibitory effect of mizoribine and ribavirin on the replication of severe acute respiratory syndrome (SARS)-associated coronavirus. Antivir. Res. 2005;66:159–163. doi: 10.1016/j.antiviral.2005.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidel L.M., Stollar V. Mutations that confer resistance to mycophenolic acid and ribavirin on Sindbis virus map to the nonstructural protein nsP1. Virology. 1991;181:490–499. doi: 10.1016/0042-6822(91)90881-b. [DOI] [PubMed] [Google Scholar]
- Schinkel J., de Jong M.D., Bruning B., van Hoek B., Spaan W.J., Kroes A.C. The potentiating effect of ribavirin on interferon in the treatment of hepatitis C: lack of evidence for ribavirin-induced viral mutagenesis. Antivir. Ther. 2003;8:535–540. [PubMed] [Google Scholar]
- Schmitz H., Köhler B., Laue T., Drosten C., Veldkamp P.J., Günther S., Emmerich P., Geisen H.P., Fleischer K., Beersma M.F., Hoerauf A. Monitoring of clinical and laboratory data in two cases of imported Lassa fever. Microb. Infect. 2002;4:43–50. doi: 10.1016/s1286-4579(01)01508-8. [DOI] [PubMed] [Google Scholar]
- Severson W.E., Schmaljohn C.S., Javadian A., Jonsson C.B. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 2003;77:481–488. doi: 10.1128/JVI.77.1.481-488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon W.M., Arnett G., Westbrook L., Shealy Y.F., O’Dell C.A., Brockman R.W. Evaluation of carbodine, the carbocyclic analog of cytidine, and related carbocyclic analogs of pyrimidine nucleosides for antiviral activity against human influenza type A viruses. Antimicrob. Agents Chemother. 1981;20:769–776. doi: 10.1128/aac.20.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeta S., Konno K., Yokota T., Nakamura K., De Clercq E. Comparative activities of several nucleoside analogs against influenza A, B, and C viruses in vitro. Antimicrob. Agents Chemother. 1988;32:906–911. doi: 10.1128/aac.32.6.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidwell R.W., Allen L.B., Khare G.P., Huffman J.H., Witkowski J.T., Simon L.N., Robins R.K. Effect of 1-beta-d-ribofuranosyl1-1,2,4-triazole-3-carboxamide (virazole, ICN 1229) on herpes and vaccinia keratitis and encephalitis in laboratory animals. Antimicrob. Agents Chemother. 1973;3:242–246. doi: 10.1128/aac.3.2.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidwell R.W., Bailey K.W., Wong M.H., Barnard D.L., Smee D.F. In vitro and in vivo influenza virus-inhibitory effects of viramidine. Antivir. Res. 2005;68:10–17. doi: 10.1016/j.antiviral.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Sidwell R.W., Barnard D.L., Day C.W., Smee D.F., Bailey K.W., Wong M.H., Morrey J.D., Furuta Y. Efficacy of orally administered T-705 on lethal avian influenza A (H5N1) virus infections in mice. Antimicrob. Agents Chemother. 2007;51:845–851. doi: 10.1128/AAC.01051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell N.J. Ribavirin therapy for Nipah virus infection. J. Virol. 2004;78:10211. doi: 10.1128/JVI.78.18.10211.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurgers K., Sharkey C.M., Warfield K., Bavari S. Oligonucleotide antiviral therapeutics: antisense and RNA interference for highly pathogenic RNA viral infections. Antivir. Res. 2008;78:26–36. doi: 10.1016/j.antiviral.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen E.L., Jahrling P.B. Experimental Lassa fever virus infection successfully treated with ribavirin. Lancet. 1979;1:268–269. doi: 10.1016/s0140-6736(79)90790-6. [DOI] [PubMed] [Google Scholar]
- Stockman L.J., Bellamy R., Garner P. SARS: systematic review of treatment effects. PLoS Med. 2006;3:e343. doi: 10.1371/journal.pmed.0030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeter D.G., Witkowski J.T., Khare G.P., Sidwell R.W., Bauer R.J., Robins R.K., Simon L.N. Mechanism of action of 1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc. Natl. Acad. Sci. U.S.A. 1973;70:1174–1178. doi: 10.1073/pnas.70.4.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuyver L.J., Lostia S., Patterson S.E., Clark J.L., Watanabe K.A., Otto M.J., Pankiewicz K.W. Inhibitors of the IMPDH enzyme as potential anti-bovine viral diarrhoea virus agents. Antivir. Chem. Chemother. 2002;13:345–352. doi: 10.1177/095632020201300602. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Furuta Y., Fukuda Y., Kuno M., Kamiyama T., Kozaki K., Nomura N., Egawa H., Minami S., Shiraki K. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antivir. Chem. Chemother. 2003;14(5):235–241. doi: 10.1177/095632020301400502. [DOI] [PubMed] [Google Scholar]
- Tam R.C., Pai B., Bard J., Lim C., Averett D.R., Phan U.T., Milovanovic T. Ribavirin polarizes human T cell responses towards a Type 1 cytokine profile. J. Hepatol. 1999;30:376–382. doi: 10.1016/s0168-8278(99)80093-2. [DOI] [PubMed] [Google Scholar]
- Tam R.C., Ramasamy K., Bard J., Pai B., Lim C., Averett D.R. The ribavirin analog ICN 17261 demonstrates reduced toxicity and antiviral effects with retention of both immunomodulatory activity and reduction of hepatitis-induced serum alanine aminotransferase levels. Antimicrob. Agents Chemother. 2000;44:1276–1283. doi: 10.1128/aac.44.5.1276-1283.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale M., Ellis M., Klumpp K., Court S., Ford M. Inhibition of influenza virus transcription by 2′-deoxy-2′-fluoroguanosine. Antimicrob. Agents Chemother. 1995;39:2454–2458. doi: 10.1128/aac.39.11.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassini J.E., Davies M.E., Hastings J.C., Lingham R., Mojena M., Raghoobar S.L., Singh S.B., Tkacz J.S., Goetz M.A. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 1996;40:1189–1193. doi: 10.1128/aac.40.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujjinamatada R.K., Baier A., Borowski P., Hosmane R.S. An analogue of AICAR with dual inhibitory activity against WNV and HCV NTPase/helicase: synthesis and in vitro screening of 4-carbamoyl-5-(4,6-diamino-2,5-dihydro-1,3,5-triazin-2-yl)imidazole-1-beta-d-ribofuranoside. Bioorg. Med. Chem. Lett. 2007;17:2285–2288. doi: 10.1016/j.bmcl.2007.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer F.J., de Haan C.A., Schuurman N.M., Haijema B.J., Verheije M.H., Bosch B.J., Balzarini J., Egberink H.F. The carbohydrate-binding plant lectins and the non-peptidic antibiotic pradimicin A target the glycans of the coronavirus envelope glycoproteins. J. Antimicrob. Chemother. 2007;60:741–749. doi: 10.1093/jac/dkm301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventre K., Randolph A.G. Ribavirin for respiratory syncytial virus infection of the lower respiratory tract in infants and young children. Cochrane Database Syst. Rev. 2007;24 doi: 10.1002/14651858.CD000181.pub3. CD000181. [DOI] [PubMed] [Google Scholar]
- von Itzstein M., Wu W.Y., Kok G.B., Pegg M.S., Dyason J.C., Jin B., Van Phan T., Smythe M.L., White H.F., Oliver S.W., Colman P.M., Vargehse J.N., Ryan D.M., Woods J.M., Bethell R.C., Hotham V.J., Cameron J.M., Penn C.R. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- Watashi K., Ishii N., Hijikata M., Inoue D., Murata T., Miyanari Y., Shimotohno K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell. 2005;19:111–122. doi: 10.1016/j.molcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Watts D.M., Ussery M.A., Nash D., Peters C.J. Inhibition of Crimean-Congo hemorrhagic fever viral infectivity yields in vitro by ribavirin. Am. J. Trop. Med. Hyg. 1989;41:581–585. doi: 10.4269/ajtmh.1989.41.581. [DOI] [PubMed] [Google Scholar]
- Wu S.F., Lee C.J., Liao C.L., Dwek R.A., Zitzmann N., Lin Y.L. Antiviral effects of an iminosugar derivative on flavivirus infections. J. Virol. 2002;76:3596–3604. doi: 10.1128/JVI.76.8.3596-3604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.Z., Yeh L.T., Lin C.C., Hong Z. Conversion of viramidine to ribavirin in vivo by adenosine deaminase and its inhibition by 2′-deoxycoformycin. Antivir. Chem. Chemother. 2006;17:33–39. doi: 10.1177/095632020601700105. [DOI] [PubMed] [Google Scholar]
- Yan Z., Tripet B., Hodges R.S. Biophysical characterization of HRC peptide analogs interaction with heptad repeat regions of the SARS-Coronavirus Spike fusion protein core. J. Struct. Biol. 2006;155:162–175. doi: 10.1016/j.jsb.2006.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagida K., Baba C., Baba M. Inhibition of bovine viral diarrhea virus (BVDV) by mizoribine: synergistic effect of combination with interferon-alpha. Antivir. Res. 2004;64:195–201. doi: 10.1016/j.antiviral.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main proteaseand its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]