

Highlights
► We developed an efficient pseudotype virus system for HCoV-NL63. ► We identified the domains and critical residues involved in the NL63 S-mediated viral entry. ► This viral system is useful for investigation of the effect of NL63 S on ACE2 expression and shedding as well as the pathogenicity. ► Our study may advance our understanding of the molecular mechanism of NL63 S–ACE2 interaction. ► Our data may provide important information for the development of vaccines, small peptides, compounds, or neutralizing antibodies against NL63 infections.
Keywords: HCoV-NL63, Spike protein, Receptor binding, Pseudotype virus entry
Abstract
The spike (S) protein of human coronavirus NL63 (HCoV-NL63) mediates both cell attachment by binding to its receptor hACE2 and membrane fusion during virus entry. We have previously identified the receptor-binding domain (RBD) and residues important for RBD–hACE2 association. Here, we further characterized the S protein by investigating the roles of the cytoplasmic tail and 19 residues located in the RBD in protein accumulation, receptor binding, and pseudotype virus entry. For these purposes, we first identified an entry-efficient S gene template from a pool of gene variants and used it as a backbone to generate a series of cytoplasmic tail deletion and single residue substitution mutants. Our results showed that: (i) deletion of 18 aa from the C-terminus enhanced the S protein accumulation and virus entry, which might be due to the deletion of intracellular retention signals; (ii) further deletion to residue 29 also enhanced the amount of S protein on the cell surface and in virion, but reduced virus entry by 25%, suggesting that residues 19–29 contributes to membrane fusion; (iii) a 29 aa-deletion mutant had a defect in anchoring on the plasma membrane, which led to a dramatic decrease of S protein in virion and virus entry; (iv) a total of 15 residues (Y498, V499, V531, G534, G537, D538, S540, G575, S576, E582, W585, Y590, T591, V593 and G594) within RBD were important for receptor binding and virus entry. They probably form three receptor binding motifs, and the third motif is conserved between NL63 and SARS-CoV.
1. Introduction
Human coronavirus NL63 (HCoV-NL63, NL63) was first described in 2004 in the Netherlands (van der Hoek et al., 2004). However, it was already isolated in 1988 (Fouchier et al., 2004), or even earlier (van der Hoek et al., 2006). NL63 has since been reported in more than 15 countries across the world and accounts for up to 9.3% of acute respiratory infections leading to hospitalization (Albuquerque et al., 2009, Arden et al., 2005, Bastien et al., 2005, Chiu et al., 2005, Dare et al., 2007, Ebihara et al., 2005, Esper et al., 2005, Gerna et al., 2006, Han et al., 2007, Koetz et al., 2006, Moes et al., 2005, Oosterhof et al., 2009, Smuts et al., 2008, Vabret et al., 2005). These findings suggest that NL63 has been widely circulating in the human population for decades. NL63 infection generally causes mild upper respiratory tract diseases, but may also cause more severe lower respiratory tract diseases, e.g. croup, bronchiolitis, and pneumonia in young children, the elderly, and immunocompromised people (van der Hoek et al., 2006). No vaccine or antiviral drug is currently available for NL63.
Coronaviruses can be divided into three groups based on antigenic and phylogenetic relationships. NL63 belongs to group I (van der Hoek et al., 2004). Like other coronaviruses, the 5′ proximal two-third of NL63 genome is occupied by two large replicase genes 1a and 1b, while the 3′ proximal one-third encodes four structural protein genes: spike (S), envelope (E), membrane (M), and nucleocapsid (N). These genes are arranged in a conserved order 5′-1a-1b-S-E-M-N-3′ (Fig. 1 ).
Fig. 1.
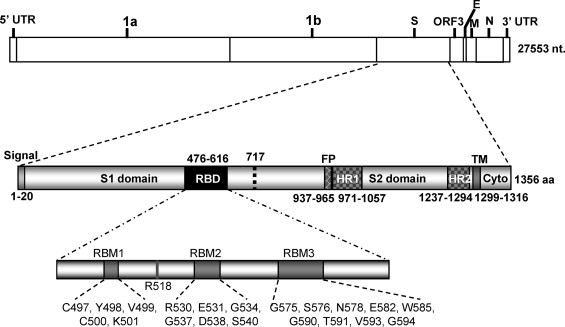
Schematic diagram of genome organization of NL63 and domain architecture of spike (S) protein. Signal: signal peptide; RBD: receptor-binding domain; FP: fusion peptide; HR: heptad repeat; TM: transmembrane domain; Cyto: cytoplasmic tail. Twenty-one residues that have been previously shown to be important for RBD–hACE2 interaction are indicated within three receptor binding motifs (RBMs) (Li et al., 2007, Lin et al., 2008).
The S protein of coronaviruses forms a layer of long petal-shaped surface spikes that give the virions a crown-like appearance when visualized by electron microscopy (Lai and Holmes, 2001). S protein is anchored on the viral envelope, a lipid bilayer derived from the intracellular membrane during virus budding. S protein is a type I transmembrane protein containing a signal peptide, long N-terminal ectodomain, transmembrane (TM) domain, and a short C-terminal cytoplasmic tail (Fig. 1). The signal peptide directs the nascent S polypeptide to the endoplasmic reticulum (ER), where the signal peptide is cleaved and monomeric precursors are synthesized. The precursors are heavily glycosylated to yield 150–200 kDa mature monomers, which are further oligomerized into trimers within the ER (Masters, 2006). S trimers interact with M protein through their cytoplasmic tails and are transported to the ER–Golgi intermediate compartment (ERGIC) where assembly and budding occur (Hogue and Machamer, 2008). This is in contrast to the assembly and budding of many enveloped viruses, e.g. HIV, that occur on the plasma membrane (Chen and Lamb, 2008).
In groups 2a, group 3 and some group 1 coronaviruses, the S proteins are cleaved in the Golgi complex by a furin-like protease into noncovalently associated S1 and S2 subunits prior to assembly into the virion (de Haan et al., 2008, de Haan and Rottier, 2005). The S protein of NL63 does not have furin-recognition site and is thus not cleaved during biogenesis. Nevertheless, the S1 (21–717 aa) and S2 (718–1356 aa) domains can still be deduced based on sequence similarity.
The S1 domain of coronavirus mediates cell attachment through interaction with a cellular receptor. As such, the particular S1–receptor interaction largely determines the host range, tissue and cell tropisms of coronaviruses. In addition, S1 is the major determinant for eliciting neutralizing antibody, which in turn blocks virion–receptor binding and protects animals from infection (Gallagher and Buchmeier, 2001, Wentworth and Holmes, 2007). Many coronaviruses have variations in S1 that evade immune response. Amazingly, variations as small as one amino acid have been shown to change receptor usage, tissue tropism and pathogenesis (Ballesteros et al., 1997, Krempl et al., 2000). The S2 domain mediates membrane fusion during viral entry into host cells. It contains several conserved regions characteristic of class I viral fusion proteins: fusion peptide (FP), two heptad repeats (HRs), TM domain and several conserved cysteine residues in the cytoplasmic tail (Fig. 1). Each of these structural features has been shown to contribute to membrane fusion (Wentworth and Holmes, 2007).
The primary entry receptor for NL63 was identified as human angiotensin-converting enzyme 2 (hACE2), which is also the primary receptor for group 2b SARS-CoV. This is particularly interesting since the S proteins of these viruses share only 21% amino acid identity (Hofmann et al., 2005, Li et al., 2003). Although they use the same receptor for entry, these two viruses cause different clinical outcomes. In contrast to NL63 that generally causes mild respiratory diseases, such as croup in children (van der Hoek et al., 2005, Wu et al., 2008, Sung et al., 2010), SARS-CoV is highly pathogenic and is characterized by acute lung failure with high mortality (Drosten et al., 2003, Ksiazek et al., 2003, Peiris et al., 2003, WHO, 2004).
This contrast has generated wide interest in comparing the roles of S proteins of these two viruses in hACE2 binding and entry. The receptor-binding domain (RBD) has been mapped to amino acid residues 318–510 for SARS-CoV (Wong et al., 2004), and 476–616 for NL63 (Li et al., 2007, Lin et al., 2008). The RBDs of these two viruses bind to a largely overlapping region of hACE2 (Li et al., 2007), although the receptor-binding affinity of NL63 S is much less compared with SARS-CoV S (Lin et al., 2008, Mathewson et al., 2008). In addition, SARS-CoV S and NL63 S may cause differential downregulation of hACE2 level by induced shedding of its ectodomain from the cell surface (Haga et al., 2008, Glowacka et al., 2010). Downregulation of hACE2 is considered to be a leading cause of acute lung failure (Imai et al., 2005, Kuba et al., 2005). Finally, SARS-CoV S, but not NL63 S, is cleaved by endosomal cathepsin L, and such proteolytic cleavage is important for SARS-CoV entry (Huang et al., 2006). It should be mentioned that these studies were greatly facilitated by the utilization of lentivirus- or VSV-based pseudotype virus bearing coronaviral S proteins. These S-mediated pseudotype viruses have identical cell tropisms to the authentic coronaviruses, and their entry is also solely dependent on cellular receptors (Broer et al., 2006, Fukushi et al., 2005, Giroglou et al., 2004, Hofmann et al., 2004, Li et al., 2003, Li et al., 2005b, Moore et al., 2004, Nie et al., 2004, Schwegmann-Wessels et al., 2009, Simmons et al., 2004). The use of pseudotype viruses not only provides a safe tool to analyze the entry process of highly pathogenic viruses under conventional biosafety condition, but also provides a unique system where the role of S protein in entry is not interfered by other viral proteins.
Nevertheless, compared to the tremendous efforts focused on SARS-CoV S, little attention has been given to the S protein of NL63. Here, we established an efficient pseudotype virus system for NL63 and used it to characterize the contributions of various regions of NL63 S protein in mediating plasma membrane translocation, incorporation into pseudotype virions, receptor association, and pseudotype virus entry.
2. Materials and methods
2.1. Virus and cells
The NL63 virus was kindly provided by Dr. L. van der Hoek at the University of Amsterdam, the Netherlands, and was propagated in a rhesus monkey kidney cell line, LLC-MK2. Human 293T cells were used for protein expression, pseudotype virus production, and viral entry assay. Huh-7 (a human hepatoma cell line) was used as target cells for pseudotype virus infection. These cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 100 IU of penicillin–streptomycin at 37 °C with 5% CO2.
2.2. Construction of expression plasmids
In this study, the domain architecture of NL63 S protein was defined by using different computer programs (Fig. 1). The border of S1 and S2 domain was deduced from Pfam (http://www.sanger.ac.uk/Software/Pfam/). The signal peptide was predicted by web-based SOSUIsignal (http://bp.nuap.nagoya-u.ac.jp/sosui/sosuisignal/sosuisignal_submit.html). The boundaries of FP and HR were defined as described previously (Zheng et al., 2006), whereas the boundaries of the TM domain were predicted by TMpred (http://www.ch.embnet.org/software/TMPRED_form.html). The full-length wild type S gene of NL63 was obtained from NL63-infected LLC-MK2 cells by RT-PCR. cDNA was synthesized using total RNAs extracted from the infected cells using RNeasy Mini kit (QIAGEN), oligo (dT)12–18 and Superscript II reverse transcriptase (Invitrogen) according to the manufacturer's instructions. The PCR products were amplified with Pfx DNA polymerase (Invitrogen) and cloned into mammalian cell expression vector pCAGGS (kindly provided by Dr. J. Miyazaki from the Kumamoto University Medical School of Japan). Based on the entry efficiency of 23 randomly selected S gene variants, No. 5 variant was chosen for further study. S gene mutants with C-terminal truncations or single amino acid substitution were generated using an overlapping PCR strategy. Two plasmids that express soluble NL63 S1/His and hACE2/Ig fusion protein, respectively, have been described previously (Lin et al., 2008). All these plasmid constructs used for this study were confirmed by DNA sequencing.
2.3. Production of pseudotype virus
HIV-based pseudotype viruses were produced as described elsewhere (Gorry et al., 2002).
Briefly, each well of 293T cells of a 6-well plate were co-transfected with 5 μg of pHIV-luc, 0.2 μg of pTat, 0.2 μg of pRev and different amounts of NL63 S gene plasmids (or the plasmid encoding SARS S or VSVG) following the standard calcium phosphate transfection method. At 48 h post-transfection, 1.5 ml of medium was collected and centrifuged at 5000 rpm for 5 min to remove large cell debris. The supernatant was further purified by passing through a 0.45 μm pore size filter. The viral titer was measured using a reverse transcriptase (RT) assay described previously (Rho et al., 1981). Briefly, 300 μl of pseudotyped virus was concentrated by centrifugation at 13,200 rpm for 2 h at 4 °C. The virus pellet was resuspended with 10 μl of RT buffer (50 mM Tris–HCl (pH 7.5), 1 mM DTT, 20% glycerol, 0.25 M KCl and 0.25% Triton X-100) and mixed with 40 μl of RT cocktail consisting of 50 mM Tris–HCl (pH 7.5), 7.5 mM MgCl2, 0.05% Triton X-100, 4.8 mM DTT, 1 μl Poly(A)×dT15 (Roche Diagnostics, Indianapolis, IN) and 1 μl TTP [methyl-3H] (PE Life and Analytical Sciences, Boston, MA). The mixture was incubated at 37 °C for 1 h and spotted on Whatman® 2.3 cm filter paper for air drying at room temperature for 15 min. After 3 successive 5 min washes with SSC buffer and a brief rinse with 95% ethanol, the filter paper was allowed to air dry on aluminum foil for 20 min. 5 ml of scintillation solution (National Diagnostics) was added into a LSC vial with the filter paper for radioactivity measurement. The titer was measured by scintillation counting and expressed as the mean count per minute (cpm)/ml.
2.4. Virus entry assay
For 96-well plate format, 5000 cpm of pseudotype virus was added into each well containing Huh-7 cells and incubated at 37 °C for 2 h, whereas 20,000 cpm of pseudotype viruses were added to each well when infection of Huh-7 or 293T cells transiently transfected with an expression plasmid encoding human ACE2 receptor (293T/hACE2 cells) was performed in a 24-well plate. The cells were washed twice with phosphate buffered saline (PBS) to remove the unbound viruses. 100 μl of new medium was added into each well followed by two days of further incubation. The cells were then lysed with 50 μl of lysis buffer (Promega), incubated at room temperature for 15 min and centrifuged at 13,000 rpm for 2 min. For measurement of luciferase (Luc) activity, 10 μl of cell lysate was mixed with 50 μl luciferase substrate (Promega). Luc activity was measured using a luminometer (Tuner Biosystem 20/20n).
2.5. Protein expression, purification and preparation of anti-NL63 S1 antibody
For production of recombinant protein, 293T cells were transfected with expression plasmid DNA using the standard calcium phosphate transfection method. Soluble NL63 S1 protein was purified as described previously (Lin et al., 2008) and used to immunize rabbits for preparation of anti-NL63 S polyclonal antibody.
2.6. Western blot and immunoprecipitation assay (IP)
NL63 S protein expressed in cells or in pseudotype virus particles was detected by anti-NL63 S1 antibody following a method described previously (Lin et al., 2008). To detect the expression level of S protein in cells, 5 μg of total protein lysate was adjusted to a total volume of 32 μl with PBS. The cell lysate was mixed with 8 μl 5× denaturing buffer, boiled for 15 min and loaded onto an 8% denaturing SDS-polyacrylamide gel. To detect the amount of S protein incorporated into pseudotype virus, 300,000 cpm of virus was centrifuged at 13,000 rpm at 4 °C for 2 h. The pellet was resuspended in 32 μl of PBS and was used for SDS-PAGE analysis. The electrophoresis running condition was 80 V for 30 min, followed by 120 V for 150 min. As a loading control, the HIV p24 protein was also detected. For this purpose, 100,000 cpm of pseudotype virus was used for western blotting with anti-p24 antibody. The density of protein bands was quantified using Quantity One software (BioRad). The association between NL63 S protein in cell lysate and soluble hACE2/Ig was measured by IP according to a previously described method (Lin et al., 2008).
2.7. Flow cytometry
To detect the S protein expression level on the cell surface, flow cytometry was performed as described previously (Lin et al., 2008). In brief, 293T cells were transfected with 1 μg/each well of the S-expressing plasmid DNA in a 24-well plate. After two days post-transfection, the cells were detached, and the two duplicated wells of cells were pooled together. The cells were incubated with anti-NL63 S1 antibody on ice for 1 h, followed by three washes with IF buffer. They were further incubated with FITC-conjugated anti-rabbit IgG on ice for 30 min. The cells were washed three times with IF buffer, fixed with 1% formaldehyde, and analyzed with a Beckman Coulter FC5000 Cytofluorometer. The X-mean fluorescent values were used for plotting.
2.8. Structural modeling
Homologous domains in the RBDs of SARS-CoV and NL63 were predicted by four proteomic analysis programs, i.e. NOMAD, MAFFT, T-COFFEE and Dialign. The structure model of SARS-CoV RBD–hACE2 complex was retrieved from protein data bank (PDB). Modeling was performed using Wincoot (Emsley and Cowtan, 2004) and PyMol Viewer (http://www.pymol.org).
3. Results
3.1. Generation and characterization of the pseudotype viruses bearing NL63 S variants
Previous studies suggested that NL63 S mediated viral entry activity was substantially lower than that of SARS S. However, the underlying mechanisms remain largely unknown. To establish an efficient NL63 pseudotype virus entry system, we first needed to choose an entry-efficient S gene template. RNA viruses are well known to exist as quasispecies, a heterogeneous population that is composed of closely related sequence variants with minor nucleotide variation (Domingo et al., 2005). The ability to mediate virus entry may vary significantly among different variants. To obtain an S variant that is highly efficient in mediating virus entry, we cloned the RT-PCR products of the S gene into a pCAGGS vector and randomly picked 23 colonies. The plasmid DNA from each variant was co-transfected with a non-replicative HIV genome harboring a luciferase gene into 293T cells. The resulting pseudotype viruses were quantified and used to infect Huh-7 cells. Luciferase activity was measured as an index of entry level or infectivity, and was normalized to the highest observed level. As illustrated in Fig. 2A, variant No. 5 consistently showed the highest efficiency in virus entry. Twelve variants had an entry level less than 20% of No. 5 and 6 variants had <40% of entry level. Variant No. 5 was thus selected for further study and is referred to as wild type S (wt S) hereafter. Sequence analysis of this variant showed that it contained three amino acid substitutions (A685 → V, L853 → F and Q998 → K) compared to the reference standard sequence in GenBank (accession No. NC_005831).
Fig. 2.
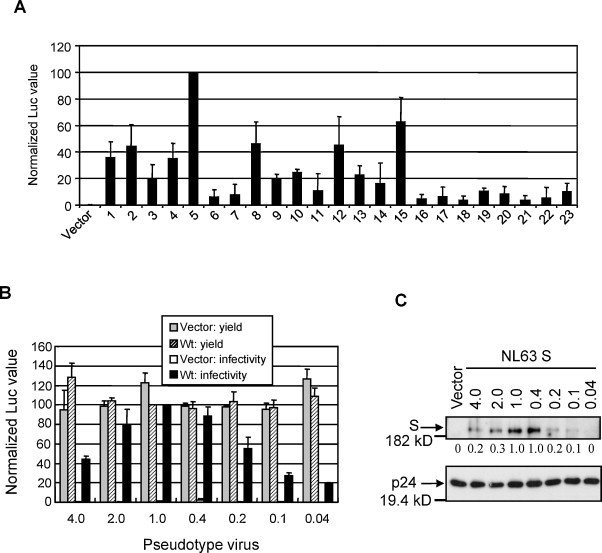
Development of NL63 pseudotype virus system. (A) Pseudotype virus entry of 23 randomly selected NL63 S gene variants. The Luc activity was measured and normalized to that of variant No. 5 that consistently showed the highest virus entry level. (B) Yield and infectivity of pseudotype virus made by transfection of different amounts of pCAGGS vector and wt S plasmid DNA (0.04–4.0 μg in each well of 6-well plate). (C) Amount of S protein incorporated into virions. The density of protein bands was quantified by Quantity One, and the relative values were shown at the bottom of the gel. HIV-1 p24 protein was detected as a loading control.
We further optimized the pseudotype virus system by testing different amounts of wt S plasmid DNA. As shown in Fig. 2B, the amount of S DNA had little effect on the virus yield, but had a profound effect on the infectivity of resulting virus. The optimal amount of DNA for virus infectivity was 1.0 μg in each well of a 6-well plate. To understand the molecular basis of this dose-dependent effect, we measured the amount of S protein incorporated into pseudotype virus particles. Since the S protein with a C-myc epitope fused either at the N- or C-terminus failed to produce infectious pseudotye virus (not shown data), we purified soluble S1 protein and prepared a rabbit polyclonal antibody. This antibody was used to detect NL63 S protein hereafter. As a loading control, HIV-1 p24 protein was probed with a p24-specific antibody to confirm that similar amounts of pseudotype virus were used in each assay. The result revealed a positive correlation between the infectivity and amount of S protein incorporated into pseudotype virus particles (Fig. 2B and C).
3.2. The cytoplasmic tail of NL63 S could affect the production and cellular entry of the pseudotyped viruses
The C-terminal cytoplasmic tail of coronaviral S proteins contains signals required for intracellular trafficking, assembly, and membrane fusion (Hogue and Machamer, 2008). A 19 amino acid (aa) deletion from the C-terminus of SARS-CoV S protein increased the pseudotype virus titer by 100-fold (Giroglou et al., 2004). To see if similar enhancement occurs in NL63 pseudotype virus, we generated four truncated S mutants, ΔC8, ΔC18, ΔC29 and ΔC38, corresponding to 8, 18, 29 and 38 aa deletion from the C-terminus, respectively (Fig. 3A). These mutants were then tested for virus production and cell entry. As shown in Fig. 3B, mutants ΔC8 and ΔC18 mediated higher levels of virus entry than wt S. However, even the best deletion mutant, ΔC18, was only about 1.5 times higher than wt S. Further deletions were deleterious to the infectivity as the entry levels of ΔC29 and ΔC38 were 75% and 3% of wt S, respectively. There is a positive correlation between the viral entry levels and the amount of S protein incorporated into virions, with the exception ΔC29 (Fig. 3C).
Fig. 3.
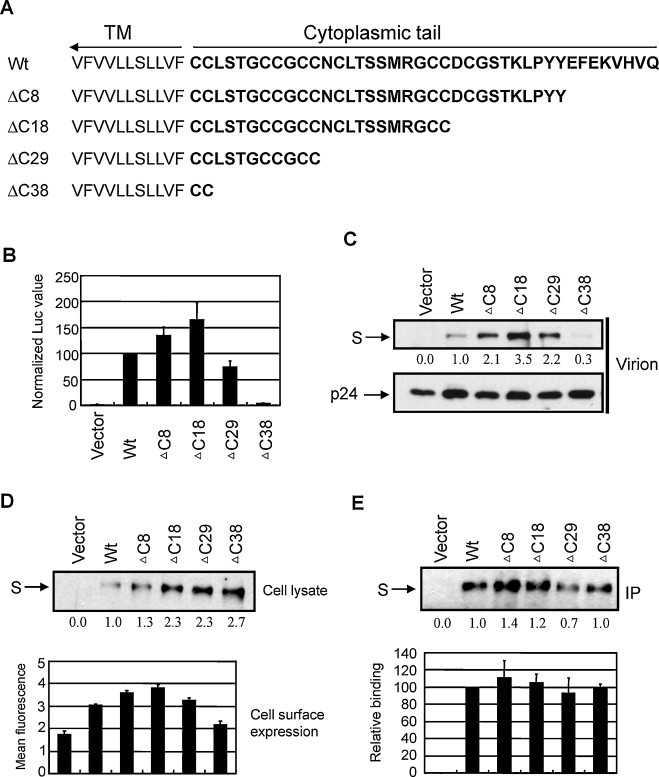
Analysis of the cytoplasmic tail of S protein in protein accumulation, receptor binding and pseudotype virus entry. (A) Schematic diagram of the truncated S mutants. (B) Pseudotype virus entry. (C) Amount of S protein incorporated into virions. (D) S protein accumulation in cell lysate and on cell surface. (E) Receptor binding ability. The density of protein bands was quantified by Quantity One, and the relative values were shown at the bottom of each gel.
To further investigate the role of cytoplasmic tail in pseudotype virus entry, we measured the S protein expression level and receptor binding ability of these mutants. Interestingly, the protein expression level of these mutants was higher than wt in cell lysate (upper panel in Fig. 3D). This was especially obvious for ΔC18, ΔC29 and ΔC38, implying that the C-terminal 18 residues of S protein harbor inhibitory signal(s) for protein accumulation. This result also ruled out the possibility that the small amount of ΔC38 S protein detected in virions was due to a protein expression defect. Since it is the S protein bound to the plasma membrane that is assembled into pseudotype virus, we checked the level of S protein on the cell surface using flow cytometry. The results showed that the expression level on the cell surface was positively correlated with the amount of S protein incorporated into virions (lower panel in Fig. 3D). Therefore, the entry defect of ΔC38 is attributed to its defect in cell surface accumulation. This data also suggests that C-terminal 38 aa region of S plays an important role in anchoring to the plasma membrane. We also did an IP assay to test the receptor binding ability of these mutants. Based on the relative expression level (upper panel in Fig. 3D), we adjusted the volume of cell lysate in order to use similar amounts of S protein for the binding assay. The analysis revealed similar receptor binding ability among these mutants in three repeated experiments (Fig. 3E). This is expected since only the S1 domain of coronaviral S protein is involved in receptor binding while the S2 domain mediates membrane fusion during virus entry.
3.3. Cellular entry mediated by NL63 S, SARS S, and VSVG
While our data clearly demonstrated that the NL63 S variant #5-ΔC18 could significantly enhance the viral infectivity, we did not know its potency compared with SARS S and VSVG. It is well-known that VSVG-pseudotyped viruses could enter a variety of mammalian cells very efficiently. Indeed, when we infected Huh-7 cells with same amount of these pseudotyped viruses (20,000 cpm/well in a 24-well plate), we consistently observed that VSVG mediated viral entry activity was about 100 times higher than that of SARS S and NL63 S (data not shown). Therefore, in order to compare the differences between NL63 S and SARS S and adjust the scales of VSVG mediated entry activity, we used 10 times less VSVG viruses (i.e. 2000 cpm/well) while the amount of NL63 and SARS viruses remained the same (20,000 cpm/well). Interestingly, we found that the viral entry of NL63-ΔC18 was 2–3 folds higher than that of SARS S in Huh-7 cells (Fig. 4A). However, when 293T/hACE2 cells were infected by these viruses, we observed that the SARS S mediated viral entry were 2–3 folds higher than that of NL63 S (Fig. 4B). In contrast, there was no significant difference in the entry activity of VSVG-pseudotyped virus between Huh-7 and 293T/hACE2 cells.
Fig. 4.
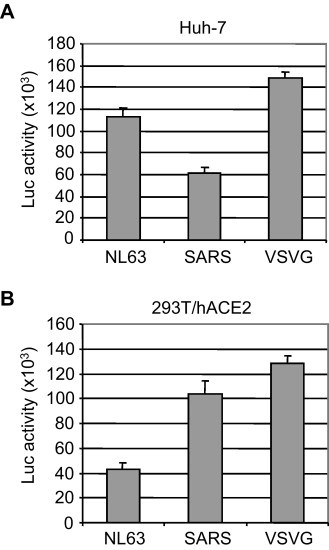
Viral entry mediated by NL63 S, SARS S and VSVG. Huh-7 (A) and 293T/hACE2 cells (B) were infected with same amount of pseudotyped NL63 and SARS viruses (20,000 cpm/well in a 24-well plate) and less amount of VSVG viruses (2000 cpm/well), respectively, and luciferase activity was measured at 48 h post-infection.
3.4. Characterization of critical residues in the NL63 RBD for viral entry
Previously, we identified 15 residues (C497, Y498, V499, C500, K501, R518, R530, V531, G534, G537, D538, S540, E582, W585 and T591) in a minimal RBD (aa 476–616) that are important for hACE2 binding by IP assay. These residues fall into three clusters that might represent three receptor binding motifs (RBMs) (Lin et al., 2008). Using the same method, Li et al. (2007) found that substitutions at 7 residues (G575, S576, N578, Y590, T591, V593, and G594) in a larger RBD (aa 301–749) affected the receptor binding ability. Interestingly, these residues fall into the proposed RBM3. However, six of them were tested via a double-substitution strategy, i.e. they were combined in three mutants, G575/S576, G590/T591 and V593/G594. Thus, the contribution of individual residues to receptor binding remained unclear. Also, all of these residues were only tested in the context of RBD–receptor interaction. To further define their roles in receptor binding and virus entry, we generated a total of 19 single substitution mutants mostly by replacing these residues with alanine in ΔC18, a more physiologically relevant context (Fig. 1). Residues C497 and C500 were not tested since it was hard to uncouple their roles in receptor binding and maintaining proper structure by forming disulphide bonds. These S mutants were then tested for their S protein accumulation and abilities to mediate virus entry and receptor association.
As shown in Fig. 5 , most of these mutants were expressed at a similar level in cell lysate compared to ΔC18. Based on the extent of entry level and receptor binding ability, mutants were placed into four categories. First, R530A behaved almost the same as ΔC18 in every aspect we examined (Fig. 5A). Second, K501A and R518A showed a slight reduction in entry level (92–96%) and receptor binding ability (60–70%) (Fig. 5A). Third, N578 → Y enhanced the entry level (144%) and receptor binding ability (220%) (Fig. 5B). Fourth, the remaining 15 mutants experienced a more pronounced drop of entry level (<60%) and receptor binding ability (≤40%). Therefore, in the context of a full-length S, 15 residues within RBD (Y498, V499, V531, G534, G537, D538, S540, G575, S576, E582, W585, Y590, T591, V593 and G594) were shown to be important for receptor binding and virus entry. They still form three RBMs, as found in the context of a minimal RBD (Lin et al., 2008). In addition, 8 mutants, W585R, G575A, V593A, G594A, V499L, D538A, E582A and T591A, also showed pronounced reduction of S protein in virions (≤40%). In the former four mutants, the reduced S protein in virions may be due to the decreased cell surface expression; however, in the latter four mutants, their cell surface expression was not reduced, therefore the reduction may reflect a defect in virion assembly. These observations suggest that substitutions at these 8 sites may change the structural conformation of S protein and adversely affect its cell surface transportation/accumulation and/or stability.
Fig. 5.
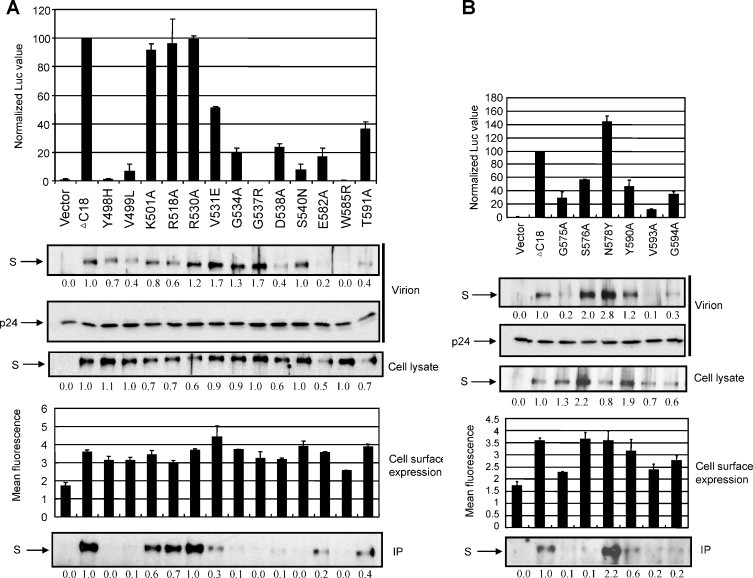
The roles of 19 residues in protein accumulation, receptor binding and pseudotype virus entry. (A) Results for 13 residues that were previously identified in a minimum RBD context (aa 476–616) (Lin et al., 2008). (B) Results for 6 residues that were previously tested in double-substitution mutants in a larger RBD context (aa 301–749) (Li et al., 2007).
4. Discussion
In this report, we established an efficient NL63 pseudotype virus entry system. An S gene variant efficient in virus entry was chosen according to the quasispecies nature of RNA viruses (Fig. 2A). Amazingly, 18 out of 23 randomly selected variants had <40% of the entry level of the best variant. This reinforces the necessity to select an efficient S gene template for research. The infectivity of pseudotype virus was further promoted by truncation of the cytoplasmic 18 aa tail of NL63 S protein (Fig. 3). A similar phenomenon was observed in the pseudotype virus systems of SARS-CoV and TGEV (Fukushi et al., 2005, Giroglou et al., 2004, Schwegmann-Wessels et al., 2009). However, compared to the 50–100-fold enhancement of infectivity by C-terminal deletion in SARS-CoV (18–19 aa deletion) and TGEV (14 aa deletion), the maximum enhancement by 18 aa deletion in NL63 S was only 2 folds (Fig. 3B). This difference presumably reflects the nature of different S proteins. Virus infectivity was positively correlated with the amount of S protein in virions. Similar to the truncated S proteins of SARS-CoV and TGEV, NL63 mutants ΔC8 and ΔC18 were more efficient than wt full-length S in assembly into pseudotype virions (Fig. 3C), which should be due to higher expression levels on plasma membrane, the site of assembly and budding of lentivirus (Fig. 3D). But why did the removal of 18 aa from the C-terminus of S protein promote cell surface expression and overall protein accumulation in cell lysate? This may be related to the intracellular retention signals contained within the cytoplasmic tail region. Bioinformatics analysis reveals that the cytoplasmic tail of NL63 S contains a putative dibasic ER-targeting signal (KVHVQ) and a putative tyrosine-based endocytic signal (YYEF) at the very C-terminus (Lontok et al., 2004) (Fig. 3A). Such tyrosine-based endocytic signal allows membrane protein to be sorted from cell surface to endosomes and lysosomes for degradation (Bonifacino and Jackson, 2003). It has been demonstrated in infectious bronchitis virus (IBV) that the dibasic ER-targeting signal allows the S protein to be retained near the assembly site when expressed alone at low levels, but a high level of expression saturates the machinery and reaches to the plasma membrane (Lontok et al., 2004). Also in IBV, the endocytic signal was able to rapidly internalize the S protein from plasma membrane (Youn et al., 2005). SARS-CoV S protein does not have such an endocytic signal at its cytoplasmic tail, and is accordingly expressed on the plasma membrane. Introduction of this signal into SARS-CoV S protein caused it to be retained intracellularly (Schwegmann-Wessels et al., 2004). The roles of these two signals have not been demonstrated for NL63, but they most likely function in a similar fashion to those in IBV and SARS-CoV mutant. This can explain why the absence of an ER-targeting signal and part of an endocytic signal (the critical tyrosine is still maintained) in ΔC8 caused more protein accumulation on the plasma membrane and increased protein in cell lysate, while complete removal of these two signals in ΔC18 further facilitated its plasma membrane translocation and protection from endocytosis and lysosomal degradation (Fig. 3A and C).
ΔC29 also showed enhanced protein accumulation in cell lysate, on plasma membrane and more efficient incorporation into virion. Furthermore, the deletion had little effect on receptor binding. However, the virus infectivity was reduced by 25% (Fig. 3). Compared to ΔC18, ΔC29 lacks 11 residues, NCLTSSMRGCC. This small region contains two cysteine residues conserved among all the coronaviruses. The homologous regions in SARS-CoV (LKGACSCGSCC) and MHV (FKKCGNCC) have been shown to play a role in cell-to-cell membrane fusion (Chang et al., 2000, Peti et al., 2005). Taken together, we suggest that these 11 residues contribute to membrane fusion during NL63 entry.
The overall protein accumulation in cell lysate of ΔC38 was similar to ΔC29; however, this mutant had a defect in accumulation on the plasma membrane, which might directly result in the drastic reduction of S protein in virions and virus infectivity (Fig. 3). Therefore, those residues (LSTGCCGCC) that are additionally deleted in ΔC38 may participate in membrane anchoring. The exact boundaries of transmembrane (TM) domains of coronaviruses have not yet been experimentally specified. Different predictions include or exclude part of the cysteine-rich domains as part of the TM domain (Bosch et al., 2005, Broer et al., 2006, Chang et al., 2000, Godeke et al., 2000, Petit et al., 2007, Thorp et al., 2006, Ye et al., 2004, Zheng et al., 2006). According to TMpred, the strongly preferred TM model of NL63 S protein predicts a TM to be located between residues 1299 and 1316 (VWLIISVVFVVLLSLLVF), which does not include any cysteine-rich domains. This prediction was adopted for the TM boundaries used in this paper. However, based on SOSUI, another web-based prediction algorithm, the TM domain is located between residues 1303 and 1325 (ISVVFVVLLSLLVFCCLSTGCCG), which includes 7 residues that were deleted in ΔC38. Our data seemingly supports the SOSUI prediction on TM domain boundary.
We also compared the cellular entry mediated by NL63 S-ΔC18, SARS S, and VSVG. Our data demonstrated that both NL63 S and SARS S could efficiently mediate viral entry into Huh-7 and 293T/hACE2 cells (Fig. 4). Interestingly, NL63 S and SARS S exhibited significant differences in the viral entry into these host cells, indicating that additional host factors and/or co-receptors may be involved in this process. Our findings may represent an important advance in the development of an efficient NL63 pseudotype viral system for studies of the viral entry.
A ΔC18-based assay system was further used to delineate the roles of 19 residues that were chosen based on our and others’ previous characterization in the context of truncated RBDs (Li et al., 2007, Lin et al., 2008). These single residue mutants functioned differently (Fig. 5). Overall, their pseudotype virus infectivity was positively correlated with their receptor binding ability, although the infectivity of some mutants was interfered by their defects in plasma membrane translocation (e.g. W585R) or incorporation into virions (e.g. E582A). In terms of receptor binding, most of the mutants tested here functioned similarly between the context of a near full-length S and a small RBD. However, some residues that were important in the RBD (aa 476–616) were not or of limited importance in ΔC18. For instance, R530 → A abolished the RBD–hACE2 association (Lin et al., 2008), but the same substitution in ΔC18 had almost no effect on hACE2 binding and virus entry (Fig. 5). Likewise, K501 → A and R518 → A reduced the receptor binding ability by 100% and 90% in RBD, respectively (Lin et al., 2008), but same substitutions reduced the receptor binding by only 40% and 30%, respectively, and had very minor effect on virus entry (Fig. 5). Therefore, these three residues may be exposed on the surface of RBD but are partially or completely buried inside the S structure. This data is consistent with our previous finding that the minimum RBD had much higher receptor binding efficiency than S1 (Lin et al., 2008).
The crystal structure information of SARS-CoV RBD–hACE2 complex and NL63-RBD–hACE2 complex has significantly advanced our understanding of the molecular interaction between the S proteins and hACE2 receptor (Li et al., 2005a, Wu et al., 2009, Wu et al., 2011). The data obtained from the NL63 RBD–hACE2 complex indicated that there are three receptor-binding motifs in the NL63 RBD (RBM1, 493–513; RBM2, 531–541; and RBM3, 585–590) (Wu et al., 2009). These results are consistent with our previously published data suggesting that NL63 RBD may contain three receptor-binding regions (RI, RII, and RIII) (Lin et al., 2008 and Fig. 1). In addition, a panel of critical residues (such as Y498, G534, G537, S540, and W585) in the NL63 RBD that appears important for receptor binding may have direct contact with hACE2 receptor (Lin et al., 2008, Wu et al., 2009). Moreover, our current study has provided functional data demonstrating that some of the residues in RBM1 (Y498, V499), RBM2 (G534, G537, D538, S540), and RBM3 (G575, S576, E582, Y590, V593, and G594) may indeed play important roles in the viral entry (Fig. 1, Fig. 5; Wu et al., 2009). To further compare the molecular interactions between the SARS-RBA/hACE2 and NL63-RBD/hACE2, we had performed the structural modeling using several bioinformatics tools as described in Section 2 and identified a short homology domain between SARS-RBD and NL63-RBD (Fig. 6A). Interestingly, in SARS, this domain is within the receptor-binding motif (RBM) that is in close contact with hACE2. Among the 14 receptor-binding residues revealed by the structure model of SARS-CoV RBD–hACE2 complex, 9 are located in this conserved region (underlined residues in Fig. 6A) (Li et al., 2005a). Among these residues, the importance of N479 and T487 has been demonstrated (Li et al., 2005b, Qu et al., 2005). More interestingly, this conserved domain in NL63 also contains 9 residues (underlined residues in Fig. 6A) that were experimentally shown to be critical for receptor binding and virus entry (Fig. 5). We therefore refer to the region as conserved binding motif (CBM). It has been shown that the RBDs of NL63 and SARS-CoV bind to a largely overlapping region in hACE2 (Li et al., 2007). Thus, it is possible that NL63 CBM may interact with hACE2 in a conformation similar to SARS-CoV CBM. To support this scenario, we used the structure model of SARS-CoV RBD–hACE2 complex to explain the opposite activities of NL63 S mutants N578Y and E582A. The residue corresponding to N578 of NL63 in SARS-CoV is Y475 (Fig. 5A). Y475 is able to interact with three residues in hACE2, T27, K31 and Y83, among which, the interaction with Y83 is the strongest since they form a hydrogen bond (left panel in Fig. 6B) (Li et al., 2005a). After we mutated Y475 to N475, mimicking the site in NL63 (N578), it was no longer able to form a hydrogen bond with Y83 although it could still weakly interact with T27 and K31 through Van der Waals contacts (right panel in Fig. 6B). Conversely, if N578 of NL63 is replaced with tyrosine Y, a hydrogen bond is acquired. This model explains why N578Y showed enhanced receptor binding ability (Fig. 5B). It can also explain the drastically reduced receptor binding ability of E582A (Fig. 5A). The residue corresponding to E582 of NL63 in SARS-CoV is N479 (Fig. 6A). This residue is able to form a hydrogen bond with H34 in hACE2 (left panel in Fig. 6C) (Li et al., 2005a). However, if N479 is substituted with a glutamic acid residue, mimicking the site in NL63 (E582), a stronger RBD–hACE2 complex is formed through a salt bridge (negative–positive charge interaction) between E479 and H34 (middle panel in Fig. 6C). When this residue is changed to A, this interaction is lost (right panel in Fig. 6C).
Fig. 6.
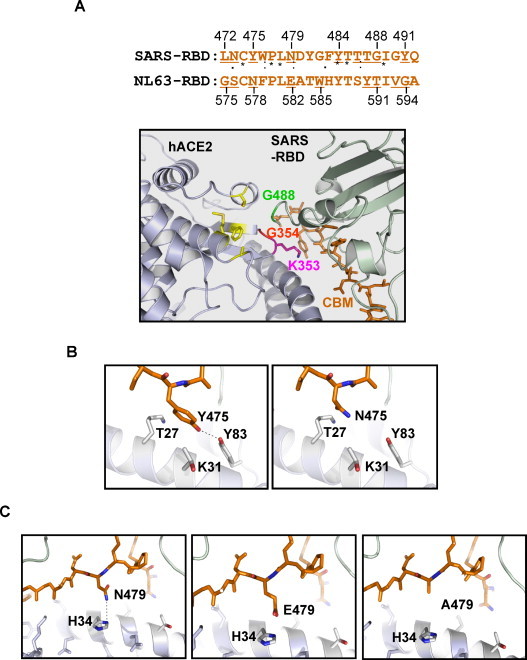
Structure models of CBM–hACE2 interaction. (A) Conserved binding motif (CBM) of NL63 and SARS-CoV S protein (upper panel) and the structure model of SARS-CoV CBM–hACE2 interaction (lower panel). The underlined residues in SARS-CoV CBM represent those residues in contact with hACE2 revealed by the RBD–hACE2 complex crystal structure of RBD–hACE2 complex (Li et al., 2005a). The underlined residues in the NL63 CMB are those that were experimentally shown to be important for hACE2 binding (Fig. 4). (B) Structural basis for the enhanced receptor binding of N578Y. The left panel illustrates that residue Y475 of SARS-CoV forms a hydrogen bond with Y83 of hACE2 and has two weak Van der Waals interactions with T27 and K31 of hACE2. The right panel shows the loss of hydrogen bond when Y475 is changed to N475, mimicking the corresponding site in NL63 (N578), while the weak interactions with T27 and K31 are still retained. (C) Structural basis for the drastically reduced receptor binding of E582A. The left panel shows the hydrogen bond formed between N479 of SARS-CoV and H34 of hACE2. The middle panel illustrates that when N479 is changed to E479, mimicking the corresponding site in NL63 (E582), the contact between virus and receptor is strengthened by formation of a salt bridge that is stronger than a single hydrogen bond. The right panel depicts the loss of contact when E479 is changed to A479.
Taken together, we have established and optimized an efficient pseudotype viral system for NL63 and used it to characterize the role of the cytoplasmic tail of the S1 domain and further analyzed the involvement of 19 residues in cell surface accumulation, receptor binding and pseudotype virus entry. This viral system would also be useful for investigation of the effect of NL63-S on hACE2 expression and shedding, which may help determine whether the difference in pathogenicity is related to the effect on the hACE2 expression/shedding. Lastly, information obtained here not only provides insights into the studies of molecular mechanism of S–receptor interaction and the function of NL63 S protein, but also provides important new information useful in development of effective vaccines, small peptides, and compounds or neutralizing antibodies to block NL63 infections.
Acknowledgements
We are grateful to Dr. L. van der Hoek from the University of Amsterdam and Dr. J. Miyazaki from Kumamoto University Medical School of Japan for providing HCoV-NL63 virus stock and pCAGGS cloning vector, respectively. This work was supported by the operating grants MOP 68955 (C. Zhang) and MOP 53209 (M. Junop) from the Canadian Institute of Health Research.
References
- Albuquerque M.C., Pena G.P., Varella R.B., Gallucci G., Erdman D., Santos N. Novel respiratory virus infections in children, Brazil. Emerg. Infect. Dis. 2009;15:806–808. doi: 10.3201/eid1505.081603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden K.E., Nissen M.D., Sloots T.P., Mackay I.M. New human coronavirus, HCoV-NL63, associated with severe lower respiratory tract disease in Australia. J. Med. Virol. 2005;75:455–462. doi: 10.1002/jmv.20288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros M.L., Sánchez C.M., Enjuanes L. Two amino acid changes at the N-terminus of transmissible gastroenteritis coronavirus spike protein result in the loss of enteric tropism. Virology. 1997;227:378–388. doi: 10.1006/viro.1996.8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien N., Anderson K., Hart L., Van Caeseele P., Brandt K., Milley D., Hatchette T., Weiss E.C., Li Y. Human coronavirus NL63 infection in Canada. J. Infect. Dis. 2005;191:503–506. doi: 10.1086/426869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino J.S., Jackson C.L. Endosome-specific localization and function of the ARF activator GNOM. Cell. 2003;112:141–142. doi: 10.1016/s0092-8674(03)00038-2. [DOI] [PubMed] [Google Scholar]
- Bosch B.J., de Haan C.A., Smits S.L., Rottier P.J. Spike protein assembly into the coronavirion: exploring the limits of its sequence requirements. Virology. 2005;334:306–318. doi: 10.1016/j.virol.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broer R., Boson B., Spaan W., Cosset F.L., Corver J. Important role for the transmembrane domain of severe acute respiratory syndrome coronavirus spike protein during entry. J. Virol. 2006;80:1302–1310. doi: 10.1128/JVI.80.3.1302-1310.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K.W., Sheng Y., Gombold J.L. Coronavirus-induced membrane fusion requires the cysteine-rich domain in the spike protein. Virology. 2000;269:212–224. doi: 10.1006/viro.2000.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.J., Lamb R.A. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology. 2008;372:221–232. doi: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S.S., Chan K.H., Chu K.W., Kwan S.W., Guan Y., Poon L.L., Peiris J.S. Human coronavirus NL63 infection and other coronavirus infections in children hospitalized with acute respiratory disease in Hong Kong, China. Clin. Infect. Dis. 2005;40:1721–1729. doi: 10.1086/430301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dare R.K., Fry A.M., Chittaganpitch M., Sawanpanyalert P., Olsen S.J., Erdman D.D. Human coronavirus infections in rural Thailand: a comprehensive study using real-time reverse-transcription polymerase chain reaction assays. J. Infect. Dis. 2007;196:1321–1328. doi: 10.1086/521308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., Haijema B.J., Schellen P., Schreur P.W., te Lintelo E., Vennema H., Rottier P.J. Cleavage of group 1 coronavirus spike proteins: how furin cleavage is traded off against heparan sulfate binding upon cell culture adaptation. J. Virol. 2008;82:6078–6083. doi: 10.1128/JVI.00074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., Rottier P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005;64:165–230. doi: 10.1016/S0065-3527(05)64006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E., Escarmís C., Lázaro E., Manrubia S.C. Quasispecies dynamics and RNA virus extinction. Virus Res. 2005;107:129–139. doi: 10.1016/j.virusres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Ebihara T., Endo R., Ma X., Ishiguro N., Kikuta H. Detection of human coronavirus NL63 in young children with bronchiolitis. J. Med. Virol. 2005;75:463–465. doi: 10.1002/jmv.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Esper F., Weibel C., Ferguson D., Landry M.L., Kahn J.S. Evidence of a novel human coronavirus that is associated with respiratory tract disease in infants and young children. J. Infect. Dis. 2005;191:492–498. doi: 10.1086/428138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier R.A., Hartwig N.G., Bestebroer T.M., Niemeyer B., de Jong J.C., Simon J.H., Osterhaus A.D. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. U.S.A. 2004;101:6212–6216. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushi S., Mizutani T., Saijo M., Matsuyama S., Miyajima N., Taguchi F., Itamura S., Kurane I., Morikawa S. Vesicular stomatitis virus pseudotyped with severe acute respiratory syndrome coronavirus spike protein. J. Gen. Virol. 2005;86:2269–2274. doi: 10.1099/vir.0.80955-0. [DOI] [PubMed] [Google Scholar]
- Gallagher T.M., Buchmeier M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerna G., Campanini G., Rovida F., Percivalle E., Sarasini A., Marchi A., Baldanti F. Genetic variability of human coronavirus OC43-, 229E-, and NL63-like strains and their association with lower respiratory tract infections of hospitalized infants and immunocompromised patients. J. Med. Virol. 2006;78:938–949. doi: 10.1002/jmv.20645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroglou T., Cinatl J., Jr., Rabenau H., Drosten C., Schwalbe H., Doerr H.W., von Laer D. Retroviral vectors pseudotyped with severe acute respiratory syndrome coronavirus S protein. J. Virol. 2004;78:9007–9015. doi: 10.1128/JVI.78.17.9007-9015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowacka I., Bertram S., Herzog P., Pfefferle S., Steffen I., Muench M.O., Simmons G., Hofmann H., Kuri T., Weber F., Eichler J., Drosten C., Pöhlmann S. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010;84:1198–1205. doi: 10.1128/JVI.01248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godeke G.J., de Haan C.A., Rossen J.W., Vennema H., Rottier P.J. Assembly of spikes into coronavirus particles is mediated by the carboxy-terminal domain of the spike protein. J. Virol. 2000;74:1566–1571. doi: 10.1128/jvi.74.3.1566-1571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorry P.R., Zhang C., Wu S., Kunstman K., Trachtenberg E., Phair J., Wolinsky S., Gabuzda D. Presistence of dual-tropic HIV-1 in an individual homozygous for the CCR5 Delta 32 allele. Lancet. 2002;359:1832–1834. doi: 10.1016/S0140-6736(02)08681-6. [DOI] [PubMed] [Google Scholar]
- Haga S., Yamamoto N., Nakai-Murakami C., Osawa Y., Tokunaga K., Sata T., Sasazuki T., Ishizaka Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7809–7814. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T.H., Chung J.Y., Kim S.W., Hwang E.S. Human Coronavirus-NL63 infections in Korean children, 2004–2006. J. Clin. Virol. 2007;38:27–31. doi: 10.1016/j.jcv.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H., Hattermann K., Marzi A., Gramberg T., Geier M., Krumbiegel M., Kuate S., Uberla K., Niedrig M., Pohlmann S. S protein of severe acute respiratory syndrome-associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J. Virol. 2004;78:6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H., Pyrc K., van der Hoek L., Geier M., Berkhout B., Pohlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogue B.G., Machamer C.E. Coronavirus structural proteins and virus assembly. In: Perlman S., Gallagher T., Snijder E.J., editors. Nidoviruses. ASM Press; Washington, DC, USA: 2008. pp. 179–200. [Google Scholar]
- Huang I.C., Bosch B.J., Li F., Li W., Lee K.H., Ghiran S., Vasilieva N., Dermody T.S., Harrison S.C., Dormitzer P.R., Farzan M., Rottier P.J., Choe H. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 2006;281:3198–3203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong-Poi H., Crackower M.A., Fukamizu A., Hui C.C., Hein L., Uhlig S., Slutsky A.S., Jiang C., Penninger J.M. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koetz A., Nilsson P., Linden M., van der Hoek L., Ripa T. Detection of human coronavirus NL63, human metapneumovirus and respiratory syncytial virus in children with respiratory tract infections in south-west Sweden. Clin. Microbiol. Infect. 2006;12:1089–1096. doi: 10.1111/j.1469-0691.2006.01506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krempl C., Ballesteros M.L., Zimmer G., Enjuanes L., Klenk H.D., Herrler G. Characterization of the sialic acid binding activity of transmissible gastroenteritis coronavirus by analysis of haemagglutination-deficient mutants. J. Gen. Virol. 2000;81:489–496. doi: 10.1099/0022-1317-81-2-489. [DOI] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W., Bao L., Zhang B., Liu G., Wang Z., Chappell M., Liu Y., Zheng D., Leibbrandt A., Wada T., Slutsky A.S., Liu D., Qin C., Jiang C., Penninger J.M. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M., Holmes K.V. Coronaviruses. In: Knipe D.M., Griffin D.E., Lamb R.A., Matin M.A., Roizman B., Strauss S.E., editors. Field's Virology. 4th ed. Lippincott Williams&Wikins; Philadelphia, PA: 2001. pp. 1163–1185. [Google Scholar]
- Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Sui J., Huang I.C., Kuhn J.H., Radoshitzky S.R., Marasco W.A., Choe H., Farzan M. The S proteins of human coronavirus NL63 and severe acute respiratory syndrome coronavirus bind overlapping regions of ACE2. Virology. 2007;367:367–374. doi: 10.1016/j.virol.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., Wong S.K., Huang I.C., Xu K., Vasilieva N., Murakami A., He Y., Marasco W.A., Guan Y., Choe H., Farzan M. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.X., Feng Y., Wong G., Wang L., Li B., Zhao X., Li Y., Smaill F., Zhang C. Identification of residues in the receptor-binding domain (RBD) of the spike protein of human coronavirus NL63 that are critical for the RBD-ACE2 receptor interaction. J. Gen. Virol. 2008;89:1015–1024. doi: 10.1099/vir.0.83331-0. [DOI] [PubMed] [Google Scholar]
- Lontok E., Corse E., Machamer C.E. Intracellular targeting signals contribute to localization of coronavirus spike proteins near the virus assembly site. J. Virol. 2004;78:5913–5922. doi: 10.1128/JVI.78.11.5913-5922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathewson A.C., Bishop A., Yao Y., Kemp F., Ren J., Chen H., Xu X., Berkhout B., van der Hoek L., Jones I.M. Interaction of severe acute respiratory syndrome-coronavirus and NL63 coronavirus spike proteins with angiotensin converting enzyme-2. J. Gen. Virol. 2008;89:2741–2745. doi: 10.1099/vir.0.2008/003962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moes E., Vijgen L., Keyaerts E., Zlateva K., Li S., Maes P., Pyrc K., Berkhout B., van der Hoek L., Van Ranst M. A novel pancoronavirus RT-PCR assay: frequent detection of human coronavirus NL63 in children hospitalized with respiratory tract infections in Belgium. BMC Infect. Dis. 2005;5:6. doi: 10.1186/1471-2334-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.J., Dorfman T., Li W., Wong S.K., Li Y., Kuhn J.H., Coderre J., Vasilieva N., Han Z., Greenough T.C., Farzan M., Choe H. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2. J. Virol. 2004;78:10628–10635. doi: 10.1128/JVI.78.19.10628-10635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y., Wang P., Shi X., Wang G., Chen J., Zheng A., Wang W., Wang Z., Qu X., Luo M., Tan L., Song X., Yin X., Ding M., Deng H. Highly infectious SARS-CoV pseudotyped virus reveals the cell tropism and its correlation with receptor expression. Biochem. Biophys. Res. Commun. 2004;321:994–1000. doi: 10.1016/j.bbrc.2004.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterhof L., Christensen C.B., Sengelov H. Fatal lower respiratory tract disease with human corona virus NL63 in an adult haematopoietic cell transplant recipient. Bone Marrow Transplant. 2009 doi: 10.1038/bmt.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti W., Johnson M.A., Herrmann T., Neuman B.W., Buchmeier M.J., Nelson M., Joseph J., Page R., Stevens R.C., Kuhn P., Wuthrich K. Structural genomics of the severe acute respiratory syndrome coronavirus: nuclear magnetic resonance structure of the protein nsP7. J. Virol. 2005;79:12905–12913. doi: 10.1128/JVI.79.20.12905-12913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit C.M., Chouljenko V.N., Iyer A., Colgrove R., Farzan M., Knipe D.M., Kousoulas K.G. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology. 2007;360:264–274. doi: 10.1016/j.virol.2006.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X.X., Hao P., Song X.J., Jiang S.M., Liu Y.X., Wang P.G., Rao X., Song H.D., Wang S.Y., Zuo Y., Zheng A.H., Luo M., Wang H.L., Deng F., Wang H.Z., Hu Z.H., Ding M.X., Zhao G.P., Deng H.K. Identification of two critical amino acid residues of the severe acute respiratory syndrome coronavirus spike protein for its variation in zoonotic tropism transition via a double substitution strategy. J. Biol. Chem. 2005;280:29588–29595. doi: 10.1074/jbc.M500662200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho H.M., Poiesz B., Ruscetti F.W., Gallo R.C. Characterization of the reverse transcriptase from a new retrovirus (HTLV) produced by a human cutaneous T-cell lymphoma cell line. Virology. 1981;112:355–360. doi: 10.1016/0042-6822(81)90642-5. [DOI] [PubMed] [Google Scholar]
- Schwegmann-Wessels C., Al-Falah M., Escors D., Wang Z., Zimmer G., Deng H., Enjuanes L., Naim H.Y., Herrler G. A novel sorting signal for intracellular localization is present in the S protein of a porcine coronavirus but absent from severe acute respiratory syndrome-associated coronavirus. J. Biol. Chem. 2004;279:43661–43666. doi: 10.1074/jbc.M407233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwegmann-Wessels C., Glende J., Ren X., Qu X., Deng H., Enjuanes L., Herrler G. Comparison of vesicular stomatitis virus pseudotyped with the S proteins from a porcine and a human coronavirus. J. Gen. Virol. 2009;90:1724–1729. doi: 10.1099/vir.0.009704-0. [DOI] [PubMed] [Google Scholar]
- Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smuts H., Workman L., Zar H.J. Role of human metapneumovirus, human coronavirus NL63 and human bocavirus in infants and young children with acute wheezing. J. Med. Virol. 2008;80:906–912. doi: 10.1002/jmv.21135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung J.Y., Lee H.J., Eun B.W., Kim S.H., Lee S.Y., Lee J.Y., Park K.U., Choi E.H. Role of human coronavirus NL63 in hospitalized children with croup. Pediatr. Infect. Dis. J. 2010;29:822–826. doi: 10.1097/INF.0b013e3181e7c18d. [DOI] [PubMed] [Google Scholar]
- Thorp E.B., Boscarino J.A., Logan H.L., Goletz J.T., Gallagher T.M. Palmitoylations on murine coronavirus spike proteins are essential for virion assembly and infectivity. J. Virol. 2006;80:1280–1289. doi: 10.1128/JVI.80.3.1280-1289.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret A., Mourez T., Dina J., van der Hoek L., Gouarin S., Petitjean J., Brouard J., Freymuth F. Human coronavirus NL63, France. Emerg. Infect. Dis. 2005;11:1225–1229. doi: 10.3201/eid1108.050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Berkhout B. Human coronavirus NL63, a new respiratory virus. FEMS Microbiol. Rev. 2006;30:760–773. doi: 10.1111/j.1574-6976.2006.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Sure K., Ihorst G., Stang A., Pyrc K., Jebbink M.F., Petersen G., Forster J., Berkhout B., Uberla K. Croup is associated with the novel coronavirus NL63. PLoS Med. 2005;2:e240. doi: 10.1371/journal.pmed.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentworth D.E., Holmes K.V. Coronavirus binding and entry. In: Thiel V., editor. Coronaviruses: Molecular and Cellular Biology. Caister Academic Press; Norfolk, UK: 2007. pp. 3–30. [Google Scholar]
- WHO, 2004. Summary of probable SARS cases with onset of illness from November 1, 2002 to July 31, 2003. http://www.who.int/csr/sars/country/table2004_04_21/en/index.html (accessed 21.04.04).
- Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Li W., Peng G., Li F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. U.S.A. 2009;106:19970–19974. doi: 10.1073/pnas.0908837106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Chen L., Peng G., Zhou W., Pennell C.A., Mansky L.M., Geraghty R.J., Li F. A virus-binding hot spot on human angiotensin-converting enzyme 2 is critical for binding of two different coronaviruses. J. Virol. 2011;85:5331–5337. doi: 10.1128/JVI.02274-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.S., Chang L.Y., Berkhout B., van der Hoek L., Lu C.Y., Kao C.L., Lee P.I., Shao P.L., Lee C.Y., Huang F.Y., Huang L.M. Clinical manifestations of human coronavirus NL63 infection in children in Taiwan. Eur. J. Pediatr. 2008;67:75–80. doi: 10.1007/s00431-007-0429-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R., Montalto-Morrison C., Masters P.S. Genetic analysis of determinants for spike glycoprotein assembly into murine coronavirus virions: distinct roles for charge-rich and cysteine-rich regions of the endodomain. J. Virol. 2004;78:9904–9917. doi: 10.1128/JVI.78.18.9904-9917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn S., Collisson E.W., Machamer C.E. Contribution of trafficking signals in the cytoplasmic tail of the infectious bronchitis virus spike protein to virus infection. J. Virol. 2005;79:13209–13217. doi: 10.1128/JVI.79.21.13209-13217.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q., Deng Y., Liu J., van der Hoek L., Berkhout B., Lu M. Core structure of S2 from the human coronavirus NL63 spike glycoprotein. Biochemistry. 2006;45:15205–15215. doi: 10.1021/bi061686w. [DOI] [PubMed] [Google Scholar]