

Abstract
Infectious pancreatic necrosis virus (IPNV) is a major pathogen in the aquaculture industry worldwide. Factors contributing to IPNV pathogenicity are yet poorly understood. Indications of IPNV being able to evade or counteract innate host defense come from its lack of ability to induce strong type I interferon (IFN) responses in cell culture. We show here that addition of salmon rIFN-α1 to cells prior to IPNV infection halts the viral protein synthesis and prevents processing of pVP2 into mature VP2. Furthermore, compared to pre-treatment with IFN-α1 the antiviral state in cells infected with IPNV prior to IFN-treatment, was antagonized by IPNV, as detected by higher viral titers, faster viral protein synthesis and also by reduced Mx expression. The longer headstart the virus gets, the more prominent is the weakening of IFN signaling. IPNV VP4 and VP5 inhibit IFN-induced expression from the Mx promoter, indicating that these proteins contribute to the antagonistic effect.
Keywords: IPNV, Birnavirus, IFN, Fish, Antagonism, VP2-processing
1. Introduction
Infectious pancreatic necrosis virus (IPNV) causes infectious pancreatic necrosis (IPN), an acute and serious disease with high mortality in salmonid fish worldwide. Restrain of this disease is of major economical importance for the aquaculture industry. IPNV, like infectious bursal disease virus (IBDV) is a member of the family Birnaviridae (Dobos et al., 1979, ICTVdB, 2009). It is the prototype species of the genus Aquabirnavirus (Hill and Way, 1995) and a common pathogenic microorganism in aquatic fauna world wide. IPNV is a non-enveloped virion with a 60 nm icosahedral structure and a bi-segmented double-stranded RNA (dsRNA) genome (Dobos et al., 1977, Duncan and Dobos, 1986). Segment B of the genome encodes the RNA-dependent RNA polymerase (RdRp) VP1, whereas segment A has two overlapping open reading frames (ORFs) of which the smaller ORF encodes the non-structural protein VP5 and the larger ORF encodes a 106 kDa polyprotein. This large polyprotein is cotranslationally processed through the proteolytic activity of VP4 to generate pVP2 (precursor of VP2), VP4 and VP3 (Duncan et al., 1987, Petit et al., 2000). The pVP2 is further processed into a mature structural VP2 and three peptides which are associated with the virus particle (Galloux et al., 2004). Provirions are particles with incomplete assembly, which first occur in the infected cells 8 h p.i. and harbor both pVP2 and mature VP2. Upon provirion maturation, infectious particles (virions) are formed 2–4 h later. A population of purified mature virions does not harbor the precursor form of VP2 (Villanueva et al., 2004). In the closely related avibirnavirus, IBDV, two of the peptides cleaved off of pVP2 are shown to be essential for virus viability (Da Costa et al., 2002). To establish infections in vivo, viruses must not only enter host cells, replicate its genome, synthesize viral proteins and assemble new infectious particles; they must also face powerful immune defense mechanisms of the host cells. In order to replicate efficiently it is likely that IPNV, like many other viruses have evolved strategies to avoid elements of the host immune responses.
The type I interferon (IFN) system has a crucial role in the first line of defense against virus infections whereby IFN-α/β are induced by virus entry and is secreted to protect non-infected cells from virus attack. IFN genes have been cloned from a variety of fish species including salmonids (Robertsen et al., 2003, Zou et al., 2007) and also several IFN-inducible genes are known to be present in fish, among them Mx (Robertsen, 2008). Once secreted, IFNs activate the Janus-activated kinase 1 (JAK1) and tyrosine kinase 2 (TYK2) by binding to the IFN-α/β receptor. The signal is cascaded further by tyrosine phosphorylation of the signal transducers and activators of transcription 1 and 2 (STAT1 and STAT2). The STATs heterodimerize and translocate into the nucleus, where they associate with nuclear protein IRF9, and this complex activates the transcription of large number of genes known as IFN-stimulated genes (ISGs) (Garcia-Sastre and Biron, 2006). There are many pathways leading to production of IFN and subsequently many intervening pathways leading to the activation of ISGs. Therefore, in general it seems difficult for a virus to use one single mechanism to inhibit the IFN antiviral response, and a variety of mechanisms have evolved in viruses to evade host defense. It is shown that many viruses dedicate parts of their genome to encode gene products able to counteract components of the IFN pathways (Garcia-Sastre and Biron, 2006). The strategies could be either to antagonize IFN induction, IFN signaling, expression or action of ISGs (for a review, see Randall and Goodbourn, 2008).
IPNV is known to be sensitive to the antiviral action of IFNs, demonstrated by efficiently inhibited growth in tissue culture by exogenously added salmon IFN-α1 (Robertsen et al., 2003). Furthermore, the salmon Mx protein has been shown to directly inhibit viral protein synthesis (Larsen et al., 2004). We and others have noticed, however, that IPNV is able to inhibit IFN signaling in host cells when treated with IFN after infection (Collet et al., 2007, Jørgensen et al., 2007), suggesting that IPNV have evolved mechanisms to overcome the IFN responses. To gain a better understanding of the molecular mechanisms by which IFN controls IPNV infection we addressed the two following questions; when IFN induces an antiviral state within a cell, what effect does this have on IPNV replication and how is IPNV able to circumvent the IFN response? Attempting to answer these questions several assays were applied to examine the effects of type I IFN on IPNV replication. Moreover, the effects of IFN were studied under conditions where virus replication already had been established.
2. Materials and methods
2.1. Cell cultures and virus
Chinook salmon embryo cells (CHSE-214) were grown as monolayers at 20 °C, 5.0% CO2 in Eagle minimal essential medium with GlutaMAX (EMEM+GlutaMAX, Invitrogen) supplemented with 100 μg/ml streptomycin, 60 μg/ml penicillin, 1% nonessential amino acids and 8% fetal bovine serum (FBS, Euroclone). For infection experiments and Western analyses, CHSE-214 cells were seeded into 24-well plates (2 × 105 cells/well) and grown to 80% confluence prior to infection and incubated at 17.5 °C, 5.0% CO2 during infection. The transgenic cell-line CHSE-Mx10, derived from CHSE-214 cells, featuring the endogenously expressed luciferase reporter gene under control of the rainbow trout Mx promoter, was used for luciferase assays (Jørgensen et al., 2007). These cells were grown under the same conditions as the CHSE-214 cells.
TO-cells, originating from Atlantic salmon head kidney (Wergeland and Jakobsen, 2001), were grown as monolayers at 20 °C, 5.0% CO2 in Eagle minimal essential medium with GlutaMAX (EMEM+GlutaMAX, Invitrogen) supplemented with 100 μg/ml streptomycin, 60 μg/ml penicillin, 1% nonessential amino acids and 5% fetal bovine serum (FBS, Euroclone). These cells were used for co-transfection experiments followed by luciferase analyses.
IPNV of the N1 strain, serotype Sp (Christie et al., 1988), was used in this study. The experiments were performed with a multiplicity of infection (MOI) of 2 or 10 infectious particles/cell in CHSE-214 or CHSE-Mx10 cells. After absorption of the virus for 3–4 h in serum free culture medium, the medium containing virus was carefully removed from the cells. The infection was then carried out at 17.5 °C in the presence of 2% FBS and cells harvested at different time points. Propagation and titration of virus were performed as described in Pedersen et al. (2007).
2.2. Cloning of viral protein expression constructs
Regions encoding the individual viral proteins VP1, VP2, VP3, VP4 and VP5 were amplified from cDNA or plasmid DNA by PCR and inserted into pENTR/D-TOPO vector (Invitrogen) as described in (Pedersen et al., 2007). Inserts were further transferred to Gateway compatible eukaryotic expression vectors with a CMV promoter, either pDEST12.2 (Invitrogen) or pDEST-myc (provided by Dr. T. Lamark, University of Tromsø) for transfection in cells.
2.3. Co-transfection
For transfection TO-cells were seeded into 24-well plates with a density of 2 × 105 cells/well and grown to 80–90% confluence. The transfection reagent FuGENE HD (Roche Applied Science) was used according to the manufacturer's protocol. A total of 0.6 μg of plasmid DNA was mixed with 1.25 μl FuGENE HD in 50 μl EMEM and incubated 15 min before added to the cells with medium containing 2% FBS. The cells were harvested for luciferase assay 48 h after transfection. Co-transfections were performed using the pGL3-Basic-PrMx1 plasmid containing a luciferase reporter gene under control of the Mx promoter (Collet and Secombes, 2001) combined with a plasmid expressing β-galactosidase under the control of an actin promoter for estimation of transfection efficiency. These two plasmids were co-transfected together with the eukaryotic expression vector pDEST 12.2 (Invitrogen) or pDEST-myc expressing the individual IPN virus proteins (VP1, VP2, VP3, VP4, VP5 or inverted VP4).
2.4. IFN stimulation of cells
Recombinant Atlantic salmon IFN-α1 was produced in HEK293 cells as described elsewhere (Robertsen et al., 2003). The salmon IFN-α1 used in this study had a titer of 24,237 U/ml as estimated by the formula given by Renault et al. (1991). IFN-α1 was administered to the cells at a concentration of 80 U/ml in EMEM containing 2% FBS at different time points prior to, and after IPNV infection or after co-transfection. A dose dependent IFN-α1 response, with an optimum at 80 U/ml, is earlier reported for the CHSE cell-line (Jørgensen et al., 2007).
2.5. Luciferase assay
CHSE-Mx10 cells, or transiently transfected TO-cells were lysed in 50 μl lysis buffer with DTT, from the Dual-Light Assay kit (Applied Biosystems). Buffer B and buffer A were added to 20 μl of the lysate (according to the manufacturer's protocol) and the luciferase and β-galactosidase activity (Martin et al., 1996) was measured in a Luminoscan RT luminometer (Labsystems OY). All samples for the luciferase assay were set up in triplicate and the results given as relative light units (RLU). For the transiently transfected cells the transfection efficiency of the reporter gene was normalized by dividing RLU values with values of β-galactosidase expression. Here, the mean RLU values were transformed and presented as fold increase or percent luciferase induction.
2.6. Gel electrophoresis, Western blotting and antibodies
CHSE-214 cells were lysed in 50 μl sodium dodecyl sulfate (SDS) sample buffer (160 mM Tris–HCl [pH 6.8], 10% β-mercaptoethanol, 2% SDS, 20% glycerol, 0.1% bromophenol blue), transferred from the well into a microcentrifuge tube and boiled for 5 min. Twenty microliter of the samples were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting using the Invitrogen NuPAGE system. Precast 4–12% gradient NuPAGE Novex Bis–Tris gels were used with MES buffer. Gel electrophoresis, blotting, blocking and antibody incubation were performed as described by the manufacturer. Primary antibodies used in this study were the monoclonal antibodies against VP2 (αVP2, 1:1000 dilution) (kindly provided by K.E. Christie, Intervet Norbio), the polyclonal antibodies against VP1 (αVP1, 1:1000 dilution), VP3 (αVP3, 1:4000 dilution) (produced as described in (Pedersen et al., 2007), a polyclonal Mx antibody (α-Mx, 1:1000 dilution) (Trobridge et al., 1997) and the polyclonal eEF2 antibody (α-eEF2, 1:1000 dilution (Hansen et al., 2008) (Cell Signaling Technology). Goat anti-rabbit-horseradish peroxidase (HRP) antibody or goat anti-mouse-HRP antibody (Santa Cruz Biotechnology) diluted 1:25,000 were used as secondary antibodies. Detection was performed by using SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology Inc.). Stripping of the membranes was performed in 0.2 M NaOH for 10 min followed by washing, blocking and new antibody incubation.
2.7. Radio-labeling and autoradiography
For in vivo labeling of proteins, confluent monolayers of CHSE-214 cells in a 6-well plate were infected with IPNV strain N1 at a MOI of 2 or mock-infected and treated with IFN-α1 at different time points. At 10, 12, 24, 48, 72 or 96 h post-infection (p.i.), cells were starved of methionine and cysteine for 1 h in EMEM medium lacking these amino acids (Gibco/BRL) and pulse-labeled for 3 h with 20–50 μCi/ml l-[35S]methionine/cysteine (Pro-mix l-[35S] in vitro cell labeling mix, Amersham) in methionine- and cysteine-free EMEM medium. Radio-labeled samples were analyzed by SDS-PAGE and visualized by autoradiography. Dried gels were exposed to Kodak BioMax MS film over night.
2.8. Immunoprecipitation (IP) analyses
Radio-labeled CHSE-214 cells were washed twice with ice-cold PBS and harvested in HA-lysis buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 2 mM EDTA, 1 mM EGTA, 1% Triton X-100) with a protease inhibitor cocktail added (Complete EDTA-free, Roche). Cell lysates were incubated on ice for 15 min and cleared by centrifugation for 15 min at 18,000 × g in a microfuge. Lysates were then subjected to IP with a mixture of the antibodies αVP1, αVP2 and αVP3 together with pre-blocked Protein A/G PLUS-Agarose beads (Santa Cruz biotechnology). The beads were then washed four times with HA-lysis buffer, and all traces of buffer removed with a pipette tip before elution in 50 μl 2× SDS sample buffer. Eluted proteins were subjected to SDS-PAGE and visualized by autoradiography.
2.9. Statistics
The group means were compared by one-way ANOVA, followed by Tuckey's multiple comparison test for differences between the control group and the treated groups. Statistical analyses were performed using the GraphPad Prism4 software (GraphPad Software Inc). The value of p < 0.05 was considered to be significant.
3. Results
A time-course study was conducted, where CHSE-214 or CHSE-Mx10 cells were infected with IPNV at different time points prior to, and after treatment with type I IFN. Cells were harvested for multiple assays over the time-course in order to try to determine at which level(s) IPNV inhibits IFN signal transduction and at which level IFN inhibits viral protein synthesis.
3.1. Antiviral effects of type I IFN on IPNV replication
Viral titers were determined from cell supernatants harvested 24, 48 and 72 h p.i. Consistent with earlier observations (Jensen and Robertsen, 2002), treatment with IFN-α1 24 h prior to IPNV infection resulted in no detectable infectious particles at 24 h p.i., and at 48 h p.i. the virus titers were reduced by 10 orders of magnitude compared to those in untreated cells (Fig. 1 ). However, treatment of the cells with IFN-α1 after the viral infection had started, diminished the inhibitory effect of the IFN on viral synthesis. IFN-treatment 4 h p.i. still gave a substantial reduction of 8 orders of magnitude, while for cells treated with IFN-α1 at 10 h p.i. there was a less pronounced antiviral effect (5 orders of magnitude reduction at 48 h p.i.). Moreover, at 72 h p.i. the IPNV titers in the non-treated cells and the cells treated with IFN-α1 at 10 h p.i. were nearly the same.
Fig. 1.
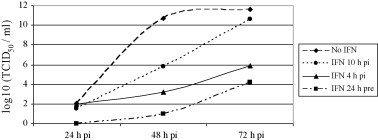
Analysis of virus growth upon type I IFN-treatment. CHSE-Mx10 cells untreated, or treated with recombinant salmon IFN-α1 24 h prior to infection, 4 h p.i. or 10 h p.i., were infected with IPNV (N1) (MOI 2) in four parallel wells. Supernatants were harvested at indicated times and virus titers were determined as described in Section 2. These data represent one of two repeated experiments which gave reproducible results.
3.2. IFN-α1 treatment delays viral maturation
To determine the presence of viral proteins in lysates from cells subjected to different IFN-α1 treatments, the proteins synthesized at 10, 12, 24, 48, 72 and 96 h p.i., respectively, were metabolically labeled with [35S]methionine/cysteine. The IPNV proteins were immunoprecipitated with a mixture of three IPNV specific antibodies, αVP1, αVP2 and αVP3, in order to eliminate the background of simultaneously synthesized cellular proteins. First we examined the effects of administering IFN-α1 to the cells 24 h prior to IPNV infection. In infected cell lysates without IFN-treatment, the first viral proteins were detected at 12 h p.i. (Fig. 2A). By contrast, no viral proteins were detected in the IFN-treated cells up to 24 h, while at later time points (48–96 h) bands representing VP1, pVP2 and VP3 were also detected upon IFN-treatment (Fig. 2B). This implies that IFN-α1 pre-treatment delays synthesis of viral proteins. Interestingly, another observation was made, that even though the viral proteins were present in the IFN pre-treated cells at 48 h p.i., the pVP2 (precursor of VP2) was not processed into the mature form of VP2. In earlier studies by us and others, two bands representing the non-processed pVP2 (56 kDa) and the mature VP2 (49 kDa), respectively, are detected at 48 h p.i. (Pedersen et al., 2007, Villanueva et al., 2004), as seen in infected cells without IFN-treatment (Fig. 3A). Apparently, the maturation of pVP2 into VP2 was inhibited when IFN was added prior to, or at an early stage of infection. Even in IFN pre-treated cells incubated up to 96 h before labeling and harvesting the VP2 maturation was still inhibited (Fig. 2B). Fig. 2C shows that upon pre-treatment with IFN the cellular protein synthesis is maintained up to 96 h p.i. even though a substantial quantity of viral proteins is present in the cell lysate from 48 h on.
Fig. 2.
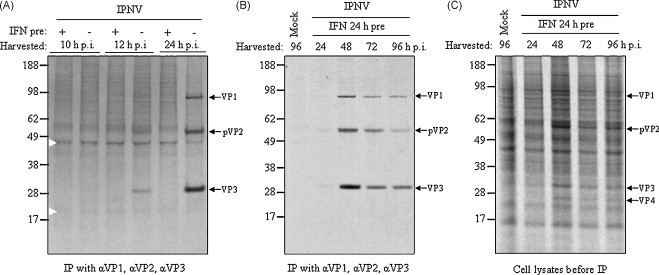
Effects of type I IFN pre-treatment on IPNV protein synthesis. CHSE-214 cells were subjected to treatment with recombinant salmon IFN-α1 24 h prior to infection with IPNV (MOI 2) or left untreated as –IFN control. At indicated times post-infection the proteins synthesized were metabolically labeled with [35S]methionine/cysteine and harvested. (A and B) Subsequently, IPNV proteins were immunoprecipitated (IP) with a pool of three IPNV specific antibodies; αVP1, αVP2, αVP3, and analyzed by SDS-PAGE and autoradiography. The arrowheads indicate unidentified host cell proteins. (C) The same cellular lysates were also analyzed by SDS-PAGE and autoradiography before being subjected to IP.
Fig. 3.
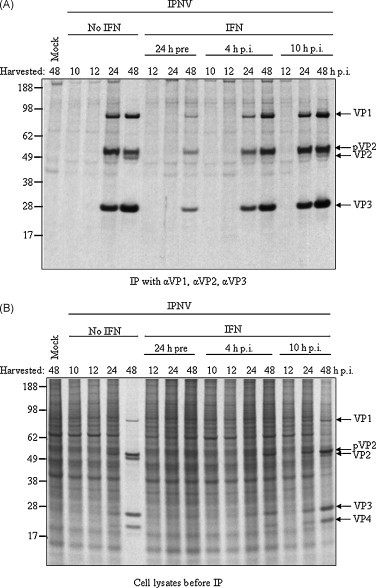
Analysis of virus protein synthesis by type I IFN-treatment prior to or after IPNV infection. CHSE-214 cells were infected with IPNV (MOI 2) and subjected to treatment with recombinant salmon IFN-α1 24 h prior to infection, IFN-α1 4 h p.i., or 10 h p.i. or left untreated. At indicated times post-infection the proteins synthesized were metabolically labeled with [35S]methionine/cysteine and subsequently (A) IPNV proteins were immunoprecipitated with a mixture of three IPNV specific antibodies; αVP1, αVP2, αVP3, and analyzed by SDS-PAGE and autoradiography. (B) The labeled proteins present in the total extracts used in (A). These data represent one of a series of repeated experiments which generated reproducible data.
We next examined the effects of type I IFN under conditions where virus replication already have been established, by adding the IFN-α1 to the cells 4 and 10 h following infection. In the non-treated cells strong bands representing VP1, pVP2 and VP3 were detected at 24 h p.i., and at 48 h p.i. also a band representing processed VP2 was seen (Fig. 3). By treating the infected cells with IFN-α1 4 h p.i., this maturation process was somewhat delayed, and in these cells the processed form of VP2 was not detected. Treatment with IFN-α1 as late as 10 h p.i. had a more subtle impact on this process and there was no obvious reduction in the levels of VP1, pVP2, VP2 and VP3 compared to the infected non-treated cells (Fig. 3A). To confirm that the difference in viral protein expression was not an artifact of the immunoprecipitation step, total cell extracts were also examined. Results presented in Fig. 3B confirmed that IFN pre-treatment significantly inhibited the expression of the viral proteins, and processed VP2 was only found in the untreated cells and when IFN was added 10 h p.i. Moreover, infected cells without IFN-treatment were completely taken over by virus at 48 h p.i., whereas with IFN-treatment, even as late as 10 h p.i., the cellular protein synthesis was maintained even though viral proteins were abundant at 48 h p.i. The infections were done with IPNV MOI 2, and also with MOI 10, where 99% of the cells should be synchronously infected. Both MOIs gave similar results (results of MOI 10 are not shown). The observations were made repeatedly in our experiments.
3.3. IPNV infection post-IFN-treatment inhibits IFN-mediated responses
As shown here the inhibitory effects of IFN on IPNV replication were less robust if the cells were treated after IPNV infection as opposed to pre-treatment with IFN. This suggested that IPNV encodes proteins that interfere with the establishment of an antiviral state normally induced by IFN, or counteract the antiviral activity of ISGs. The Mx gene is commonly used as a marker for interferon-mediated gene regulation and we therefore chose to test the effects of IPNV on IFN-mediated gene regulation by measuring Mx expression. Our results showed that Mx induction in IPNV infected cells treated with IFN post-infection was at a lower rate compared to cells pre-treated with IFN (Fig. 4). In cells where IFN-α1 was added at 4 h p.i. and incubated with the cells for 20 h, we found a reduction in Mx-levels compared to the non-infected cells (Fig. 4 , lanes 5 and 4 upper panel). This effect vanished over time (48 and 72 h p.i.). For the cells treated with IFN 10 h p.i. almost no Mx expression was detected at 24 h p.i. (Fig. 4, lane 7 upper panel). Twenty-four hours later, at 48 h p.i., the Mx was still down-regulated, but at 72 h p.i. there were only marginal differences in Mx-levels between infected and uninfected cells, suggesting that the antiviral response at late time points was balanced in the favor of the host. Elongation factor eEF2 was used as a loading control in this experiment. Cytopathic effects were observed 72 h p.i. in cells that did not receive IFN-treatment or had been treated with IFN late (10 h p.i.) in the infection process. Consistent with earlier data (Collet et al., 2007, Jensen and Robertsen, 2002) the Western blots also showed that IPNV is unable to initiate expression of the antiviral Mx protein by itself in CHSE cells (Fig. 4, lane 8). Uninfected cells were included as a negative control and showed no onset of Mx (Fig. 4, lane 1).
Fig. 4.
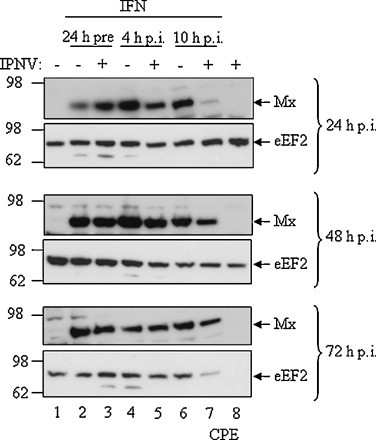
Expression levels of the antiviral protein Mx in CHSE-214 cells upon type I IFN-treatment prior to or after IPNV infection. CHSE-214 cells were left untreated or were treated with recombinant salmon IFN-α1 at 24 h prior to IPNV infection, or at 4 and 10 h p.i. Lysates were harvested at indicated times post-infection and analyzed by SDS-PAGE and Western blotting with a polyclonal antibody directed towards salmon Mx. The membranes were stripped and reprobed with an antibody towards the housekeeping gene eEF2. The experiment was repeated twice with the same results.
To explore whether IPNV also is able to interfere with the regulation of ISGs, the cell-line CHSE-Mx10 containing the luciferase reporter gene under the control of the IFN-induced rainbow trout Mx promoter were used. The trout Mx promoter contains an ISRE-element, which is also found in other IFN-induced genes in fish (Collet and Secombes, 2001). As shown here IPNV (MOI 2 and 10) was unable to activate this reporter gene by itself (Fig. 5A and B). Stimulation with IFN-α1 24 h prior to infection resulted in a more than 6-fold induction of luciferase activity from the Mx-promoter plasmid, which was the same level of luciferase induction seen in the mock-infected cells treated the same way. This was the case for cells harvested both at 24 and 48 h p.i. (Fig. 5A and B). When IFN-α1 was added to the cells 4 h p.i. a modest, non-significant reduction in luciferase activity was detected at both sampling time points compared to the mock-infected cells. By allowing the infection to proceed for 10 h before IFN was added, the inhibiting effect of IPNV was more pronounced and at 48 h p.i. there was a significant difference (p < 0.001) in luciferase activity between mock-infected and infected cells (Fig. 5B). Indeed, luciferase activity at MOI 10 was almost comparable to mock-infected non-treated cells. The constitutively expressed elongation factor eEF2, detected by Western blotting, served as a loading control (Fig. 5C and D).
Fig. 5.
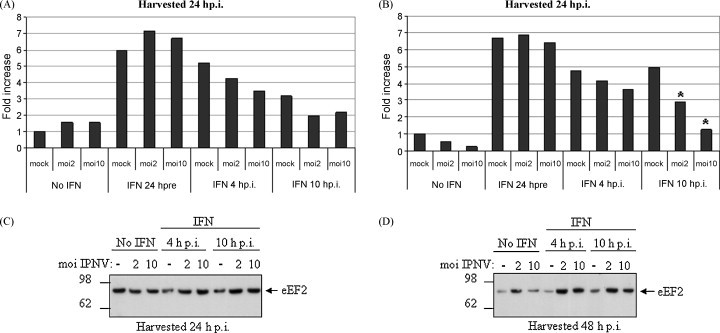
Type I IFN-treatment after IPNV infection inhibits the induction of expression from an IFN-responsive promoter. CHSE-Mx10 cells containing a transgenic luciferase reporter gene under the control of the rainbow trout Mx promoter were used. The cells were left untreated or were treated with IFN-α1 24 h prior to IPNV infection, treated with IFN-α1 4 h p.i. or 10 h p.i. The cells were infected with IPNV at MOI 2 and 10. All treatments were set up in triplicates. (A) The cells were harvested for luciferase assay at 24 h p.i. and (B) at 48 h p.i. The results are presented as fold increase in expression of the luciferase reporter gene. The asterisks mark significant reduction in luciferase induction for the IPNV infected cells compared to their mock-infected controls. These data represent one of three repeated experiments which generated reproducible data. (C and D) Cell lysates used in the luciferase assay in A and B were subjected to Western blotting with an eEF2 antibody as a loading control to estimate the levels of total protein present in each sample.
3.4. An antagonistic effect of IPNV-encoded proteins on type I IFN?
In order to point to which specific component of the IPN virus is responsible for the antagonistic effect observed, a transient co-transfection with constructs of the individual viral proteins together with the Mx-promoter-luciferase reporter was applied. A plasmid expressing β-galactosidase under the control of an actin promoter was also included for estimation of transfection efficiency. The results shown in Fig. 6 are an average of several independent experiments done in TO-cells. The level of luciferase activated by IFN-α1 and the empty vector (pDEST12.2 or pDEST-myc) was set to 100%. A significant reduction in expression from the Mx promoter was seen in TO-cells transfected with the viral protease VP4 (76.6% reduction on average) and also with the non-structural protein VP5 (50.8% reduction on average). The same experiment performed in CHSE-214 cells gave similar results (data not shown). This observation makes VP4 and VP5 the most probable candidates responsible of interfering with the IFN-signaling pathway in salmon. In the presence of both VP4 and VP5 the induction of the reporter gene was reduced to a level similar to that of VP4 alone, indicating no synergy effect between the two proteins. An apparent variation in the base levels of luciferase expression from the unstimulated cells was observed. Transfection with the empty vector and VP1 construct always gave slightly higher luciferase expression than the other constructs, whereas VP4 and VP5 gave the lowest luciferase expression (Fig. 6B). Independent expression studies (Western blots) were performed in order to make sure that transient transfected cells indeed expressed the viral proteins (results not shown). VP4 was tagged with c-Myc in order to detect the protein as no VP4 antibody was available. Accordingly, the Mx-promoter-luciferase assay was performed both with c-Myc-tagged constructs, and untagged constructs with the same results. Additionally a reverse-orientation construct with VP4 gave the same result as the empty vector control (results not shown).
Fig. 6.
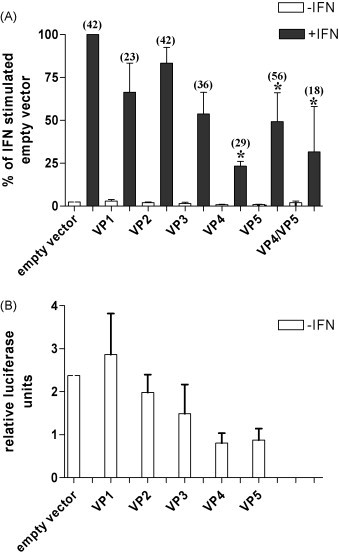
Transiently transfected VP4 and VP5 inhibit the induction of expression from an IFN-responsive promoter. Cells were transiently co-transfected with a luciferase reporter gene under the control of the rainbow trout Mx promoter together with a β-galactosidase expressing plasmid and vectors (pDEST12.2 or pDEST-myc) expressing individual IPNV proteins in addition to an empty vector control (in triplicates). The cells were left untreated (white bars) or were treated with IFN-α1 (black bars) 24 h post-transfection. At 48 h the cells were harvested for measurement of luciferase and β-galactosidase activities. (A) The results from at least three independent experiments in TO-cells were merged and are presented as percent increase in luciferase induction for each construct, where the IFN-stimulated empty vector was set to 100%. The asterisks mark significant differences in the IFN-induced luciferase values when comparing each of the viral constructs with the empty vector control. The stimulation indexes (fold increase = stimulated/unstimulated) are shown in parentheses above each bar. (B) The luciferase expression base levels (–IFN) were variable, here visualized by an enlarged scale of the values in (A).
4. Discussion
Aquatic birnaviruses like IPNV have a bi-segmented genome encoding five different proteins. These proteins, with the exception of VP5, are required for replication of the viral genetic material and assembly of new viral particles before its release from the infected cell. In order for the virus to survive and spread, viral proteins may interact with the host machinery to minimize activation of antiviral responses in infected cells. Hence, the outcome of IPNV infection reflects the ability of the virus to redirect cellular processes in favor of virus propagation while avoiding cellular antiviral responses. So far, no proteins encoded by birnaviruses have been reported to have such effect. In the present study we have initiated work aimed to understand the mechanisms IPNV employs for suppressing cellular antiviral responses and the nature of cellular responses that can interfere with IPNV replication.
Our study demonstrates that type I IFN-treatment prior to IPNV infection is interfering with viral protein synthesis. Viral protein synthesis was altered in terms of reduction in synthesis of VP1, VP2 and VP3 as seen by metabolic labeling of the viral proteins at different time points. Inhibition of IPNV protein synthesis has also previously been observed as a consequence of the IFN-induced Mx protein being expressed from a transgenic cell-line 48 h p.i. (Larsen et al., 2004). Other ISGs are also likely to participate in inhibition of IPNV replication. A natural consequence of the reduction in viral protein synthesis is a delay of particle assembly and the overall frequency of viral replication. In addition to a generally reduced synthesis of all viral proteins upon pre-treatment with IFN, the pVP2 remained unprocessed even at 96 h p.i. This lack of processing must be regarded crucial for the viral particle assembly considering VP2 being the outer structural capsid protein. The cellular defense mechanisms induced by pre-treatment with IFN, target pVP2 directly or indirectly and result in stalling the particles in a provirion state. Villanueva et al. (2004) showed the involvement of the VP4 Ser-Lys protease in the proteolytic process of maturation from provirions to infectious virions. The four cleavage sites in the pVP2 specific domain (amino acid stretch from 443 to 508) share the similarity of the defined cleavage motif [S/T]XA↓A (Galloux et al., 2004, Petit et al., 2000), suggesting that VP4 is involved in the maturation of VP2 by cleavage at all these positions. Hence the viral protease VP4 could be a target for the IFN system in order to inhibit maturation of infectious particles. IFN introduced at an early point after infection (4 h p.i.) also delays the processing of pVP2 into VP2. This suggest that an effector molecule activated relatively early upon IFN signaling is involved in this observed delay.
With an IPNV infection preceding IFN-treatment, the virus is interfering with the cells’ ability to establish an antiviral effect. When IFN was added 4 h p.i. we observed a modest reduction of viral protein synthesis and as a consequence a reduction in IPNV titers consistent with moderately reduced Mx expression. When adding IFN as late as 10 h p.i. the IPNV titers were reduced even less (as compared to IFN added 4 h p.i.), while no inhibition of viral protein synthesis was detected, which could be a consequence of the significant reduction in Mx expression levels. We claim that, the down-regulation of Mx protein synthesis is probably specific and representative for IFN-induced genes, since protein synthesis in general seemed not affected. A general down-regulation of cellular gene-expression as a consequence of viral cytopathic effect will occur later in the infection progress, after viral propagation. These observations illustrate a battle between virus and host defense which is balanced delicately. As shown in Fig. 3B cellular protein synthesis is maintained even when IPNV proteins are abundant. And in Figs. 4 and 5D we show that housekeeping genes are highly expressed even though Mx is down-regulated. Because of the ability of virus proteins, such as SV5V protein (Andrejeva et al., 2002), to target the IFN-signaling pathway, the host might have evolved optional pathways leading to activation of antiviral genes. Thus, although the virus is able to block IFN signaling, such a mechanism will result only in a partially ability to overcome the IFN response, as seen in the assays conducted in this study.
In addition to this study, earlier work has demonstrated that IPNV is unable to activate antiviral genes like Mx (Collet et al., 2007, Jensen and Robertsen, 2002). When ISGs are directly activated in salmonid cell-lines by stimuli such as poly I:C or type I IFN, they seem to be suppressed upon IPNV infection (Collet et al., 2007, Jørgensen et al., 2007). The mechanisms the virus employs to secure its own survival still needs to be elucidated, but our results indicate that the virus harbors one or several proteins or molecules with an antagonistic effect on the cells’ innate immune response. In the reporter gene assay shown in Fig. 6 the most potential candidate molecules for counteracting the IFN response on the Mx promoter were VP4 and VP5. These two proteins reduced the IFN-induced expression from the Mx promoter significantly, and more than the other proteins tested. The co-expression of both VP4 and VP5 did not further inhibits the induction of luciferase activity suggesting no co-operative or synergistic effects. VP4 and VP5 might use different strategies to circumvent the IFN response. VP5 is synthesized and expressed early (at 3 h p.i. and disappears later, after 10 h p.i. (Hong et al., 2002, Magyar and Dobos, 1994)), whereas VP4 is more abundantly expressed at later timepoints and has a prolonged expression (Fig. 3B). By simultaneously over-expressing both proteins, the partial inhibitory effect of each protein was not reinforced. Transfection with the complete ORF expressing segment A had a similar impact on Mx expression as VP4 and VP5 had individually (results not shown). The base levels of luciferase expression without IFN-stimulation varied with the different constructs being introduced into the cells. This could be due to endogenous expression of IFN, possibly induced by the transfection procedure. It was apparent that this sparse IFN expression also was affected by, and down-regulated by the viral proteins, especially by the VP4 and VP5 proteins which repeatedly gave lower values than the empty vector and VP1 (Fig. 6B). As a consequence of the variable base levels, a calculation of stimulation indexes (fold increase of stimulated/unstimulated values) gravely affected the results. When comparing the stimulation indexes of the viral proteins to that of the empty vector control, none of the values differed significantly from the control. Such calculation will in this case mask the actual inhibitory effect of viral proteins observed both on endogenous and externally administered IFN.
VP4 is a Ser-Lys protease and VP5 a nonstructural protein, which function is so far unraveled. Interestingly, other RNA viruses have been shown to utilize their viral encoded protease to inhibit host defense responses. Proteases like Npro of classical swine fever virus (CSFV) and papain-like protease (PLpro) of severe acute respiratory coronavirus (SARS-CoV) both inhibit the induction of type I IFN (Devaraj et al., 2007, Ruggli et al., 2005). Npro by transcriptional inhibition of interferon regulatory factor 3 (IRF3) (La Rocca et al., 2005), and PLpro by inhibition of phosphorylation and thereby nuclear translocation of IRF3 (Devaraj et al., 2007). These are mechanisms targeting the initiation of IFN production. In this study we determine that with type I IFN supplemented to the cells after a viral infection has begun, the cells are unable to establish a complete antiviral state as efficiently as if pre-treated with IFN. This suggests that IPNV has approaches for counterattack on cellular defense at stages in the IFN signaling pathways, in addition to being a poor inducer of IFN production.
The nonstructural VP5 of IPNV has been extensively studied, but has not yet revealed its function. It has been shown to be dispensable for viral replication both in vitro and in vivo (Santi et al., 2005b). Studies by Hong et al. (2002) showed that ectopically expressed VP5 delays apoptosis, while Santi et al. (2005a) failed to show anti-apoptotic activity by VP5. Studies of Norwegian field isolates have revealed heterogeneity in the open reading frame encoding VP5 (Santi et al., 2005b), and while some strains encode the full-length 15-kDa protein, other strains have mutations causing earlier stop-codons, though encoding truncated forms of VP5. By using reverse genetics Santi et al. (2005b) showed that recombinant VP5 deficient viruses are as virulent as the wild-type IPNV. Assuming that inhibition of IFN signaling is required for viral replication and pathogenicity in the host, the results from Santi et al. are inconsistent with the observations of VP5 described in the present study. Alternatively, this suggests that VP5's ability to inhibit IFN signaling is not absolutely required, or that other IPNV-encoded proteins, like VP4, will compensate or overlap its function. To our knowledge it has not been investigated whether VP5 deficient strains differ in their ability to regulate type I IFN responses upon infection compared to wild-types, which is an interesting question that needs to be addressed in future studies.
Cellular proteins which activate ISGs, including proteins in the JAK/STAT pathway are prime candidates for viral inhibition. In our work treatment with type I IFN soon after infection (4 h p.i.) has a better antiviral effect than treatment at a later point (10 h p.i.). Unpublished data from our group show neither up-regulation nor a significant down-regulation of STAT1 upon IPNV infection in CHSE-214 or TO-cells. Even though STAT1 levels remain stable upon infection, it is not yet determined whether the salmon STAT1 is being activated by phosphorylation or able to relocalize into the nucleus. Nipah virus (Paramyxoviridae) V protein inhibits IFN responses by forming high-molecular-weight complexes with both STAT1 and STAT2. This result in inhibited phosphorylation and accumulation of STAT in the cytoplasm in cells expressing the Nipah virus V protein (Rodriguez et al., 2002). Our data indicate a race between virus replication and IFN signaling and situate the antagonistic property of IPNV early in the type I IFN-signaling pathway. Further studies are clearly needed in order to identify how the IPNV proteins VP4 and VP5 and host molecules interact in the JAK/STAT pathway. Understanding the mechanisms underlying the inhibition of the IFN signaling is essential, since the outcome of a viral infection, in terms of pathogenicity and persistence, is influenced by its actions on the IFN system.
Acknowledgements
This work was supported by the National Program for Research in Functional Genomics in Norway (FUGE) of the Research Council of Norway (grant no. 159326/S10). We thank Karen E. Christie, Intervet Norbio, for providing the IPNV N1 strain and the monoclonal antibody specific to VP2. We also thank Dr. Ingrid Skjæveland, Norwegian College of Fishery Science, for critical reading of the manuscript.
References
- Andrejeva J., Young D.F., Goodbourn S., Randall R.E. Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons. J. Virol. 2002;76(5):2159–2167. doi: 10.1128/jvi.76.5.2159-2167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie K.E., Havarstein L.S., Djupvik H.O., Ness S., Endresen C. Characterization of a new serotype of infectious pancreatic necrosis virus isolated from Atlantic salmon. Arch. Virol. 1988;103(3–4):167–177. doi: 10.1007/BF01311090. [DOI] [PubMed] [Google Scholar]
- Collet B., Munro E.S., Gahlawat S., Acosta F., Garcia J., Roemelt C., Zou J., Secombes C.J., Ellis A.E. Infectious pancreatic necrosis virus suppresses type I interferon signalling in rainbow trout gonad cell line but not in Atlantic salmon macrophages. Fish Shellfish Immunol. 2007;22(1-2):44–56. doi: 10.1016/j.fsi.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Collet B., Secombes C.J. The rainbow trout (Oncorhynchus mykiss) Mx1 promoter. Structural and functional characterization. Eur. J. Biochem. 2001;268(6):1577–1584. [PubMed] [Google Scholar]
- Da Costa B., Chevalier C., Henry C., Huet J.C., Petit S., Lepault J., Boot H., Delmas B. The capsid of infectious bursal disease virus contains several small peptides arising from the maturation process of pVP2. J. Virol. 2002;76(5):2393–2402. doi: 10.1128/jvi.76.5.2393-2402.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.T.K., Baker S.C., Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282(44):32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobos P., Hallett R., Kells D.T., Sorensen O., Rowe D. Biophysical studies of infectious pancreatic necrosis virus. J. Virol. 1977;22(1):150–159. doi: 10.1128/jvi.22.1.150-159.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobos P., Hill B.J., Hallett R., Kells D.T., Becht H., Teninges D. Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. J. Virol. 1979;32(2):593–605. doi: 10.1128/jvi.32.2.593-605.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R., Dobos P. The nucleotide sequence of infectious pancreatic necrosis virus (IPNV) dsRNA segment A reveals one large ORF encoding a precursor polyprotein. Nucleic Acids Res. 1986;14(14):5934. doi: 10.1093/nar/14.14.5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R., Nagy E., Krell P.J., Dobos P. Synthesis of the infectious pancreatic necrosis virus polyprotein, detection of a virus-encoded protease, and fine structure mapping of genome segment A coding regions. J. Virol. 1987;61(12):3655–3664. doi: 10.1128/jvi.61.12.3655-3664.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloux M., Chevalier C., Henry C., Huet J.C., Costa B.D., Delmas B. Peptides resulting from the pVP2 C-terminal processing are present in infectious pancreatic necrosis virus particles. J. Gen. Virol. 2004;85(Pt 8):2231–2236. doi: 10.1099/vir.0.80012-0. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A., Biron C.A. Type 1 interferons and the virus–host relationship: a lesson in detente. Science. 2006;312(5775):879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Hansen T.E., Puntervoll P., Seternes O.M., Jorgensen J.B. Atlantic salmon possess three mitogen activated protein kinase kinase 6 paralogs responding differently to stress. FEBS J. 2008;275(19):4887–4902. doi: 10.1111/j.1742-4658.2008.06628.x. [DOI] [PubMed] [Google Scholar]
- Hill B.J., Way K. Serological classification of infectious pancreatic necrosis (IPN) virus and other aquatic birnaviruses. Annu. Rev. Fish Dis. 1995;5:55–77. [Google Scholar]
- Hong J.R., Gong H.Y., Wu J.L. IPNV VP5, a novel anti-apoptosis gene of the Bcl-2 family, regulates Mcl-1 and viral protein expression. Virology. 2002;295(2):217–229. doi: 10.1006/viro.2001.1336. [DOI] [PubMed] [Google Scholar]
- ICTVdB. The Universal Virus Database of the International Committee on Taxonomy of Viruses, version 4. http://www.ncbi.nlm.nih.gov/ICTVdb/ICTVdB/.
- Jensen I., Robertsen B. Effect of double-stranded RNA and interferon on the antiviral activity of Atlantic salmon cells against infectious salmon anemia virus and infectious pancreatic necrosis virus. Fish Shellfish Immunol. 2002;13(3):221–241. doi: 10.1006/fsim.2001.0397. [DOI] [PubMed] [Google Scholar]
- Jørgensen J.B., Johansen A., Hegseth M.N., Zou J., Robertsen B., Collet B., Secombes C.J. A recombinant CHSE-214 cell line expressing an Mx1 promoter-reporter system responds to both interferon type I and type II from salmonids and represents a versatile tool to study the IFN-system in teleost fish. Fish Shellfish Immunol. 2007;23(6):1294–1303. doi: 10.1016/j.fsi.2007.07.008. [DOI] [PubMed] [Google Scholar]
- La Rocca S.A., Herbert R.J., Crooke H., Drew T.W., Wileman T.E., Powell P.P. Loss of interferon regulatory factor 3 in cells infected with classical swine fever virus involves the N-terminal protease, N-pro. J. Virol. 2005;79(11):7239–7247. doi: 10.1128/JVI.79.11.7239-7247.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen R., Rokenes T.P., Robertsen B. Inhibition of infectious pancreatic necrosis virus replication by atlantic salmon Mx1 protein. J. Virol. 2004;78(15):7938–7944. doi: 10.1128/JVI.78.15.7938-7944.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magyar G., Dobos P. Evidence for the detection of the infectious pancreatic necrosis virus polyprotein and the 17-kDa polypeptide in infected cells and of the NS protease in purified virus. Virology. 1994;204(2):580–589. doi: 10.1006/viro.1994.1572. [DOI] [PubMed] [Google Scholar]
- Martin C.S., Wight P.A., Dobretsova A., Bronstein I. Dual luminescence-based reporter gene assay for luciferase and β-galactosidase. Biotechniques. 1996;21:520–524. doi: 10.2144/96213pf01. [DOI] [PubMed] [Google Scholar]
- Pedersen T., Skjesol A., Jørgensen J.B. VP3, a structural protein of infectious pancreatic necrosis virus, interacts with RNA-dependent RNA polymerase VP1 and with double-stranded RNA. J. Virol. 2007;81(12):6652–6663. doi: 10.1128/JVI.02831-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit S., Lejal N., Huet J.C., Delmas B. Active residues and viral substrate cleavage sites of the protease of the birnavirus infectious pancreatic necrosis virus. J. Virol. 2000;74(5):2057–2066. doi: 10.1128/jvi.74.5.2057-2066.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall R.E., Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008;89(1):1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Renault T., Torchy C., de Kinkelin P. Spectrophotometric method for titration of trout interferon, and its application to rainbow trout fry experimentally infected with viral haemorrhagic septicaemia virus. Dis. Aquat. Organ. 1991;10:23–29. [Google Scholar]
- Robertsen B. Expression of interferon and interferon-induced genes in salmonids in response to virus infection, interferon-inducing compounds and vaccination. Fish Shellfish Immunol. 2008;24(5):351–357. doi: 10.1016/j.fsi.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Robertsen B., Bergan V., Rokenes T., Larsen R., Albuquerque A. Atlantic salmon interferon genes: cloning, sequence analysis, expression, and biological activity. J. Interferon Cytokine Res. 2003;23(10):601–612. doi: 10.1089/107999003322485107. [DOI] [PubMed] [Google Scholar]
- Rodriguez J.J., Parisien J.P., Horvath C.M. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 2002;76(22):11476–11483. doi: 10.1128/JVI.76.22.11476-11483.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggli N., Bird B.H., Liu L., Bauhofer O., Tratschin J.-D., Hofmann M.A. Npro of classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-[alpha]/[beta] induction. Virology. 2005;340(2):265–276. doi: 10.1016/j.virol.2005.06.033. [DOI] [PubMed] [Google Scholar]
- Santi N., Sandtro A., Sindre H., Song H., Hong J.R., Thu B., Wu J.L., Vakharia V.N., Evensen O. Infectious pancreatic necrosis virus induces apoptosis in vitro and in vivo independent of VP5 expression. Virology. 2005;342(1):13–25. doi: 10.1016/j.virol.2005.07.028. [DOI] [PubMed] [Google Scholar]
- Santi N., Song H., Vakharia V.N., Evensen O. Infectious pancreatic necrosis virus VP5 is dispensable for virulence and persistence. J. Virol. 2005;79(14):9206–9216. doi: 10.1128/JVI.79.14.9206-9216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge G.D., Chiou P.P., Leong J.A. Cloning of the rainbow trout (Oncorhynchus mykiss) Mx2 and Mx3 cDNAs and characterization of trout Mx protein expression in salmon cells. J. Virol. 1997;71:5304–5311. doi: 10.1128/jvi.71.7.5304-5311.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva R.A., Galaz J.L., Valdes J.A., Jashes M.M., Sandino A.M. Genome assembly and particle maturation of the birnavirus infectious pancreatic necrosis virus. J. Virol. 2004;78(24):13829–13838. doi: 10.1128/JVI.78.24.13829-13838.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wergeland H., Jakobsen R. A salmonid cell line (TO) for production of infectious salmon anaemia virus (ISAV) Dis. Aquat. Organ. 2001;44(3):183–190. doi: 10.3354/dao044183. [DOI] [PubMed] [Google Scholar]
- Zou J., Tafalla C., Truckle J., Secombes C.J. Identification of a second group of type I IFNs in fish sheds light on IFN evolution in vertebrates. J. Immunol. 2007;179(6):3859–3871. doi: 10.4049/jimmunol.179.6.3859. [DOI] [PubMed] [Google Scholar]