

Highlights
-
•
IPNV genes preVP2, VP3, VP4 and VP5 inhibited activation of the IFNa1 promoter.
-
•
The viral protease VP4 was the most potent inhibitor of IFN induction.
-
•
IFN antagonism by VP4 is independent of its protease activity.
-
•
The RNA polymerase VP1 activated the IFNa1 promoter.
Keywords: IPNV, Interferon, Antagonism, IPS-1, IRF1, IRF3
Abstract
Infectious pancreatic necrosis virus (IPNV) is one of the major viral pathogens causing disease in farmed Atlantic salmon worldwide. In the present work we show that several of the IPN proteins have powerful antagonistic properties against type I IFN induction in Atlantic salmon. Each of the five IPNV genes cloned into an expression vector were tested for the ability to influence activation of the Atlantic salmon IFNa1 promoter by the interferon promoter inducing protein one (IPS-1) or interferon regulatory factors (IRF). This showed that preVP2, VP3 and VP5 inhibited activation of both promoters, while VP4 only antagonized activation of the IFNa1 promoter. The viral protease VP4 was the most potent inhibitor of IFN induction, apparently targeting the IRF1 and IRF3 branch of the signaling cascade. VP4 antagonism is independent of its protease activity since the catalytically dead mutant VP4K674A inhibited activation of the IFNa1 promoter to a similar extent as wild type VP4. In contrast to the other IPNV proteins, the RNA-dependent RNA polymerase VP1 activated the IFNa1 promoter. The ability to activate the IFN response was disrupted in the mutant VP1S163A, which has lost the ability to produce dsRNA. VP1 also exhibited synergistic effects with IRF1 and IRF3 in inducing an IFNa1-dependent antiviral state in cells. Taken together these results suggest that IPNV has developed multiple IFN antagonistic properties to prevent IFN-induction by VP1 and its dsRNA genome.
1. Introduction
Infectious pancreatic necrosis virus (IPNV) causes disease and mortality in hatchery-reared salmonid fry and in Atlantic salmon postsmolts after transfer to the sea (Hill and Way, 1995, Jarp et al., 1995, Smail et al., 1992). Elucidation of how IPNV interacts with the innate immune system of Atlantic salmon is important to understand its pathogenic properties. IPNV belongs to the genus Aquabirnavirus of the Birnaviridae family. The genome consists of two double-stranded (ds) RNA segments, packed in a non-enveloped single-shelled icosahedrical capsid (Coulibaly et al., 2005, Dobos, 1976, Pous et al., 2005). Segment B encodes VP1, which is a 94 kDa RNA dependent RNA polymerase (RdRp) found both in a free and a genome-linked form in the virion (Dobos, 1995). Segment A contains a single large open reading frame (ORF) that encodes a polyprotein, which is co-translationally cleaved by the non-structural protease VP4 to generate the mature structural proteins VP2 and VP3 (Duncan et al., 1987). VP2 is the outer capsid protein while VP3 is found in the inner surface of the viral capsid. VP3 has recently been shown to interact with VP1 and with double-stranded RNA (Pedersen et al., 2007). In addition, segment A contains a small overlapping ORF encoding VP5, which is a non-structural protein with undefined properties, proposed to have an anti-apoptotic function and to be of importance for virulence (Santi et al., 2005). The size of VP5 varies depending on the isolate, from 15 kDa to 3.3 kDa, but some isolates lack the VP5 reading-frame altogether (Heppell et al., 1995, Skjesol et al., 2011).
To establish a systemic infection, viruses have developed a wide range of mechanisms to evade and subvert the type I interferon (IFN) system, which plays a crucial role in the innate immunity against viruses of vertebrates (van den Broek et al., 1995). Type I IFNs are induced upon recognition of viral RNA by the RNA helicases RIG-I and MDA5 in the cytoplasm, and by the toll-like receptors TLR3 and TLR7, which are embedded in the membrane of endosomes (Arpaia and Barton, 2011, Takeuchi and Akira, 2007). Upon release, IFNs induce a range of antiviral proteins both in non-infected and infected cells preventing further virus infection. Among the antiviral proteins are Mx, ISG15, viperin and PKR (Chin and Cresswell, 2001, Liu et al., 2011, Samuel, 2001). Targeting initiation of transcription of IFN and IFN-induced genes are frequently used strategies for viruses to establish an infection in the host (Randall and Goodbourn, 2008). IPNV VP4 and VP5 have been shown to inhibit IFN-induced activation of the Mx promoter, but the effect of IPNV proteins on induction of type I IFN is unknown (Skjesol et al., 2009).
Atlantic salmon possesses at least four type I IFN subtypes, IFNa, IFNb, IFNc and IFNd, which show large differences in sequence and expression properties (Svingerud et al., 2012). IFNa1, IFNb and IFNc all show potent antiviral activity against IPNV and they induce Mx protein, which inhibits IPNV replication (Rokenes et al., 2007, Svingerud et al., 2012). In this work we chose to study the influence of IPNV proteins on activation of the Atlantic salmon IFNa1 promoter since IFNa1 is induced in most cells upon stimulation with dsRNA, while IFNb and IFNc are mainly induced in lymphoid organs (Sun et al., 2009, Svingerud et al., 2012). Salmon IFNa1 shows expression properties strikingly similar to mammalian IFNβ, which is induced through the RIG-I/MDA5 and TLR3 pathways (Bergan et al., 2006, Sun et al., 2009, Svingerud et al., 2012). Similar to the human IFNβ promoter, the salmon IFNa1 promoter has one NFκB-binding site and two IRF binding sites (Bergan et al., 2006).
Teleost fish possess all key components of the RIG-I/MDA5 signaling pathway, including RIG-I, MDA5, IPS-1, MITA, TBK1 and IRFs (Bergan et al., 2010, Biacchesi et al., 2009, Chang et al., 2011, Feng et al., 2011, Holland et al., 2008, Lauksund et al., 2009, Ohtani et al., 2011, Ohtani et al., 2012, Simora et al., 2010, Su et al., 2010, Sun et al., 2011, Zou et al., 2009). In mammals, RIG-I and MDA5 interact with the adaptor protein IPS-1 upon binding viral RNA (Berke and Modis, 2012, Jiang et al., 2011), which activates the transcription factors IRF-3, IRF-7 and NFκB, resulting in start of IFNβ transcription (Randall and Goodbourn, 2008). In Atlantic salmon, IPS-1, IRF1 and IRF3 are strong inducers of the IFNa1 promoter (Bergan et al., 2010, Lauksund et al., 2009).
In a previous work, we noticed that IPNV infection failed to induce the Atlantic salmon IFNa1 promoter (Bergan et al., 2006). To elucidate the reason why the virus does not induce a successful IFN response, we here investigated the effect of the individual IPNV proteins on IFNa1 induction. The present work demonstrates that IPNV has developed multiple mechanisms to inhibit induction of IFNa1 transcription. Surprisingly, however, we found that VP1 strongly activated IFNa1 transcription, which may in part explain why the virus needs potent IFN-antagonistic properties.
2. Materials and methods
2.1. Cells and viruses
Atlantic salmon TO cells (Wergeland and Jakobsen, 2001) were obtained from Dr. Heidrunn Wergeland (University of Bergen, Bergen, Norway). TO cells were cultivated at 20 °C in L-15 medium (Gibco, Life technologies) supplemented with 1% non-essential amino acids, 100 μg/ml streptomycin, 100 units/ml penicillin (Life technologies) and 8% fetal bovine serum (FBS) superior (Biochrom AG). IPNV serotype Sp N1 (Christie et al., 1988) was propagated in CHSE cells and viral titer was determined to be 1 × 107 in CHSE cells based on the TCID50 method (42). The virus was stored at −80 °C until use (Reed and Muench, 1938).
2.2. Infection of TO cells by IPNV
TO cells were seeded in 24-well plates and 100% confluent cells were infected with 10 MOI of IPNV or left untreated (control). The cells (treated in triplicates) were harvested after 6, 12, 24 and 48 h.
2.3. Neon transfections and luciferase assays
TO cells were split 2:3 the day before transfection. The cells were transfected at 80% confluence using the 10 μl Neon transfection system (Life technologies) with buffer R and at a pulse voltage of 110, pulse width 30 ms, 2 pulses. For each transfection 450,000 cells were used. For each well a total of 500 ng luciferase vector and test vectors were used, and 50 ng of the pGL74 Renilla luciferase vector was included as a transfection control. The expression vectors used are shown in Table 1 . After transfection the cells were seeded in 24-well plates with L-15 medium with 12% FBS, without antibiotics, and incubated at 20 °C. Cells were harvested in 50 μl passive lysis buffer and 10 μl of the sample was measured according to the dual-luciferase reporter assay system protocol (Promega). Results are shown as relative light units (RLU) of test reporter (firefly luciferase) over control reporter (Renilla luciferase).
Table 1.
Plasmid constructs.
| Plasmid | Reference |
|---|---|
| pDESTmycVPl | Skjesol et al. (2009) |
| pDESTmycpreVP2 | Skjesol et al. (2009) |
| pDESTmycVP3 | Skjesol et al. (2009) |
| pDESTmycVP4 | Skjesol et al. (2009) |
| pDESTmycVP5 | Skjesol et al. (2009) |
| pIPS-1 | Lauksund et al. (2009) |
| pIRFIHA | Bergan et al. (2010) |
| pIRF3HA | Bergan et al. (2010) |
| pA1(-202) | Bergan et al. (2006) |
| pDESTmycVP1S163A | This study |
| pDESTmycVP4K674A | This study |
| pVP1flag | This study |
| pVP4flag | This study |
| pVP1S163Aflag | This study |
| pVP4K674Aflag | This study |
| pGL3basic | Promega corporation |
| pGL74 | Promega corporation |
| pcDNA3.1 | Invitrogen – Life technologies |
| pcDNA3.3 | Invitrogen – Life technologies |
2.4. Antiviral assay and IFN neutralization
TO cells were transfected in 10 μl Neon transfection reactions as described in the previous section, resuspended in 400 μl growth media without antibiotics and transferred to 4 wells in a 96-well plate containing 100 μl growth media without antibiotics. Control cells were treated the same way without plasmid present. The cells were incubated at 20 °C for 72 h prior to infection. Cells were infected with IPNV at a multiplicity of infection (MOI) of 0.1 in L-15 without supplements for 1 h before changing to growth media with 2% FBS. The supernatants from the transfected cells were harvested and stored at −80 °C until use in antiviral and antibody neutralization assay. For the IFN neutralization assay, TO cells were seeded in 96 well plates at a density of 2 × 104 cells per well. The supernatant was divided in two, and one part was pre-incubated with IFNa1 antibody at a 1:100 dilution for 1 h at 37 °C. Untreated supernatant was also incubated for 1 h at 37 °C. The cells were incubated with 100 μl of either untreated or neutralized supernatant for 24 h, and subsequently infected with IPNV at a MOI of 0.1 as previously described. When complete cytopathogenic effect (CPE) was observed in control cells four days after infection, cell survival was determined by crystal violet staining as described in (Lauksund et al., 2009). Viral titration was determined in TO cells seeded in 96 well plates at a density of 2 × 104 cells per well. Calculation of viral titer was determined by the TCID50 method (Reed and Muench, 1938).
2.5. Site-directed mutagenesis
Mutations were introduced to the VP1 and VP4 genes by site-directed mutagenesis using the GeneArt® Site-Directed Mutagenesis System from Invitrogen and the following primers:
VP1S163Afwd: 5′-CTGCAATACGGGTCCGGCGCCTACTCAGGACAACTC-3′ and VP1S163Arev: 5′-GAGTTGTCCTGAGTAGGCGCCGGACCCGTATTGCAG-3′;
VP4K674Afwd: 5′-GCGGTGTAGACATCGCAGCCATCGCAGCCCATGAAC-3′ and VP4K674Arev: 5′-GTTCATGGGCTGCGATGGCTGCGATGTCTACACCGC-3′.
Mutagenesis was performed according to the specifications in the kit.
2.6. Subcloning
Plasmids containing the VP1, VP1S163A, VP4 and VP4K674A gene sequences were used as templates and new constructs with Flag tag were amplified by PCR with primers which included a Flag tag sequence (DYKDDDDK) in the N-terminal of each ORF segment. These PCR products were cloned into a pcDNA3.3 plasmid (Invitrogen). The following primer sequences were used:
VP1fwd, 5′-CACCATGGACTACAAAGACGATGACGACAAGATGTCGGACATCTTCAATTC-3′ and VP1rev, 5′-TCAGTTTCTTCTCTGCTTCTCCCGACG-3′;
VP4fwd, 5′-CACCATGGACTACAAAGACGATGACGACAAGAGCGGAGGGCCCGACGGAAA-3′ and VP4rev, 5′-TCATGCATTTGATGCCATCAGCTCTCCCAGGTACT-3′.
2.7. Relative quantitative real time PCR (qPCR) analysis
RNA was isolated from neon-transfected or infected TO-cells by using an RNeasy Mini Kit (Qiagen, Santa Clarita, CA). RNA integrity was verified by 1% agar gel electrophoresis, Quantity of RNA was assessed with the NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Criterion to include RNA samples was 260/280 nm absorbance ratio of 1.9–2.2. 0.2 μg of DNase-treated total RNA was reverse-transcribed to cDNA with oligo(dT) primers in a 20 μl reaction (QuantiTect® Reverse Transcription Kit (QIAGEN)). For PCR primer efficiency, a ten-fold dilution series with eight measuring points from a random pool of cDNA was used. The efficiencies were calculated by the formula: efficiency (%) = (10(−1/slope) − 1)*100. PCR primer efficiencies (between 2.04 and 2.22) were close to 100% allowing use of the 2−ΔΔ C T method for calculation of relative gene expression (43). The qPCR assays were carried out with the ABI PRISM 7700 Sequence Detection System using 2× Fast SYBR® Green Master Mix (Applied Biosystems) in a 15 μl reaction volume, with 6 μl 1:10 diluted cDNA, and primer concentrations of 0.50 μM. PCRs were run in triplicates in 96-well optical plates system under the following conditions: 95 °C for 5 min (pre-incubation), 95 °C for 5 s, 60 °C for 15 s, 72 °C for 15 s (45 cycles) and continuous increase from 65 °C to 97 °C with standard ramp rate (melting curve). 18S used as a reference gene showed stable expression in control and test samples according to the BestKeeper software (Pfaffl et al., 2002). The following primers were used for the relative quantification: IFNa, 5′-TGCAGTATGCAGAGCGTGTG-3′ and 5′-TCTCCTCCCATCTGGTCCAG-3′; IFNb, 5′-TGCATTGGAGGCTATGCGATAT-3′ and 5′-TTCCCAAACACCACCTACGACA-3′; IFNc, 5′-ATGTATGATGGGCAGTGTGG-3′ and 5′-CCAGGCGCAGTAACTGAAAT-3′; 18S, 5′-TTGCCGCTAGAGGTGAAATT-3′ and 5′-GCAAATGCTTTCGCTTTCG-3′; EF1α, 5′-TGCCCCTCCAGGATGTCTAC-3′ and 5′-CACGGCCCACAGGTACTG-3′; VP2, 5′-GCCAAGATGACCCAGTCCAT-3′ and 5′-TGACAGCTTGACCCTGGTGAT-3′.
2.8. Western blotting
TO cells were split 2:3 the day before transfection. The cells were transfected at 80% confluence using the 10 μl Neon transfection system (Life technologies) with buffer R and at a pulse voltage of 1200, pulse width 20 ms, 2 pulses. 450,000 cells were used for each transfection. For each well a total of 350 ng plasmid were used. After transfection the cells were seeded in 48-well plates in triplicates with MEM growth medium containing 12% FBS without antibiotics, and incubated with CO2 at 20 °C. Cells were harvested 48 h post transfection in 40 μl 2× SDS buffer. 15–20 μl of the samples were loaded in each well of a precast 4–12% gradient NuPAGE Novex Bis-Tris gel and subjected to SDS–polyacrylamide gel electrophoresis (SDS–PAGE) with 1× MOPS (Invitrogen) for 45 min at 200 V and 120 mA. The markers MagicMark™ XP (Invitrogen) and SeeBlue Plus2 Prestained (Invitrogen) were simultaneously loaded for molecular weight estimations. Western blotting of the separated proteins to a polyvinylidene difluoride (PVDF) membrane (Millipore) was performed using the Invitrogen NuPAGE system according to the manufacturer's instructions. An anti-Flag antibody (1:3000 dilution) (Sigma) for detection of Flag-tagged proteins were used and goat anti-mouse-HRP antibody (1:5000) (Santa Cruz Biotechnology) as a secondary antibody.
2.9. Statistical analyses
Two-sided unpaired Student t test was used to calculate statistics, where p ≤ 0.05 was considered to indicate a statistically significant difference.
3. Results
3.1. IPNV infection does not induce IFNa transcription in cell culture
To study the effect of IPNV infection on the IFN response, TO-cells were infected with IPNV at an MOI of 10, and expression of IFN transcripts and VP2 transcripts were measured after 6–48 h (Fig. 1 ). The cells showed beginning CPE at 48 h and strong CPE at 72 h after infection. The IFNa expression showed relatively small changes in infected cells compared to uninfected control cells, where only a minor increase in IFNa transcripts was observed at 6 h (1.5 times) (Fig. 1A). As expected, the expression of the viral protein VP2 showed a strong increase throughout the 48 h infection period (Fig. 1B). IPNV does thus not induce IFNa during infection of TO cells, which supports that the virus may express IFN antagonizing proteins.
Fig. 1.
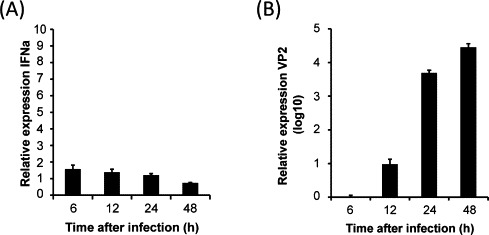
Expression of IFNa and VP2 in IPNV infected TO cells measured by qPCR. Quantitative PCR showing expression of IFNa (A) and VP2 (B) in TO cells infected with IPNV (MOI 10). Gene expression was normalized against EF1α. Expression was determined relative to transcript levels in uninfected TO cells (=1) for IFNa, and relative to the 6 h infection time point for VP2 (mean Ct = 31). Values are mean ± SD (N = 3).
3.2. IPNV preVP2, VP3, VP4 and VP5 proteins inhibit IFNa1 promoter activation while VP1 activates the IFNa1 promoter
To measure the effect of IPNV proteins on IFNa1 promoter activation, we used a previously described reporter gene construct where the Atlantic salmon IFNa1 minimal promoter region controls expression of a luciferase gene (Bergan et al., 2006). As activator of the IFNa1 promoter, we used Atlantic salmon IPS-1, which is known to activate the IFNa1 promoter upon overexpression (Lauksund et al., 2009). IPS-1 is a key adapter protein in the RIG-I/MDA5 signaling pathway, which controls type I IFN expression in most cells. TO-cells were co-transfected with the reporter plasmid and a plasmid containing one of the IPNV genes and a plasmid expressing IPS-1. Luciferase activity in the cells was measured at 48 h after transfection. The results showed that the viral proteins preVP2, VP3, VP4 and VP5 inhibited IPS-1 mediated activation of the IFNa1 promoter (Fig. 2A). The strongest inhibitory effect was observed with VP4, which virtually abolished promoter activation. In contrast, VP1 surprisingly increased activation of the IFNa1 promoter when co-transfected with IPS-1. To find out if VP1 could activate the IFNa1 promoter by itself, individual VP constructs were co-transfected with IFNa1 promoter construct into TO cells and assayed for luciferase expression. As shown in Fig. 2B, VP1 expression alone did indeed activate the IFNa1 promoter while preVP2-VP5 gave a reduction in promoter activity compared with the transfection control.
Fig. 2.
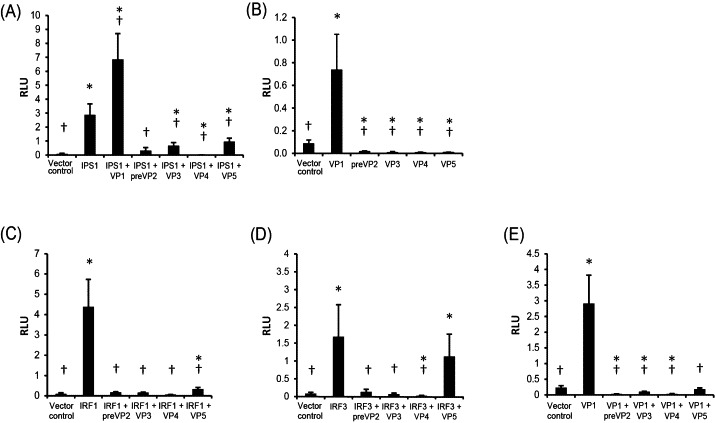
Effect of IPNV proteins on activation of the Atlantic salmon IFNa1 promoter. In all experiments TO-cells were co-transfected with four expression constructs; a plasmid containing the IFNa1 promoter fused to a luciferase gene, a plasmid containing the Renilla luciferase gene as a transfection control, a plasmid containing an IPNV gene (VP1-VP5), and a control plasmid or a plasmid containing a gene encoding a signaling protein in the IFNa1 induction pathway. Samples were harvested 48 h after transfection and analyzed for promoter activation by measuring luciferase activity. Results are presented as mean relative light units (RLU) ± SD (N = 3). (A) Effect of IPNV proteins on IPS-1 mediated activation of the promoter. Significant differences (p < 0.05) from vector control and IPS-1 indicated by * and †, respectively. (B) Effect of the individual IPNV proteins on promoter activation. Significant difference (p < 0.05) from vector control and IPS-1 indicated by * and †, respectively. (C) Effect of IPNV proteins on IRF1 mediated activation of the promoter. Significant differences (p < 0.05) from vector control and IRF-1 indicated by * and †, respectively. (D) Effect of IPNV proteins on IRF3 mediated activation of the promoter. Significant differences (p < 0.05) from vector control and IRF-3 indicated by * and †, respectively. (E) Effect of IPNV proteins on VP1 mediated activation of the promoter. Significant differences (p < 0.05) from vector control and VP1 indicated by * and †, respectively.
Since activation of type I IFN transcription through the RIG-I/MDA5 pathway ultimately results in activation of IRFs, we next studied the effect of IPNV proteins on IRF1 and IRF3 mediated activation of the IFNa1 promoter. Overexpression of these IRFs has previously been shown to result in strong activation of the IFNa1 promoter (Bergan et al., 2010). In these experiments VP constructs were co-transfected with the promoter construct and an expression plasmid encoding either IRF1 or IRF3. The results showed that preVP2, VP3, and VP4 strongly inhibited both IRF1 and IRF3 mediated activation of the IFNa1 promoter. VP5 had inhibitory effect against IRF1 mediated activation of the promoter, but did not cause significant inhibition of IRF3 mediated activation of the IFNa1 promoter (Fig. 2C and D).
Taken together, the data showed that preVP2, VP3, VP4 and VP5 were all able to inhibit IPS-1, IRF1 and IRF3 mediated activation of the IFNa1 promoter. Since we observed IFNa1-promoter activating properties of VP1, we also wanted to examine if VP2-VP5 had inhibitory effect against this activation. Co-transfection of TO-cells with IFNa1 promoter construct, VP1 and each of the other IPNV genes was performed. The results showed that indeed, VP2–VP5 were also able to inhibit VP1 mediated activation of the IFNa1 promoter (Fig. 2E).
3.3. Synergistic effects of VP1 and IRFs on induction of IFNa1
VP1 and IRF1 or VP1 and IRF3 showed strong synergistic effects in activation of the IFNa1 promoter. Transfecting TO cells with a combination of IRF1 and VP1 plasmids increased the activation capacity of VP1 12 times, while combining IRF3 with VP1 increased the activation capacity of VP1 by 10.5 times (Fig. 3A and B).
Fig. 3.
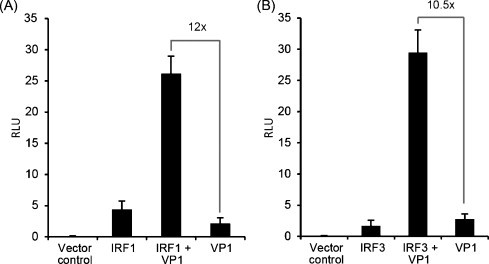
Co-stimulatory effects of VP1 and IRFs on activation of the Atlantic salmon IFNa1 promoter. TO-cells were co-transfected with a plasmid containing the minimal IFNa1 promoter fused to a luciferase gene along with an expression construct for Renilla luciferase as a transfection control. In addition, expression constructs for IRF1 or IRF3, VP1, and VP1 in combination with IRF1 or IRF3 were included. Results are presented as mean relative light units (RLU) ± SD (N = 3). (A) Effects of IRF1, VP1, a combination of VP1 and IRF1 and vector control. IRF1 + VP1 is significantly different from VP1 and IRF1 (p < 0.05). (B) Effects of IRF3, VP1, a combination of VP1 and IRF3 and a vector control. IRF3 + VP1 is significantly different from VP1 and IRF3 (p < 0.05). All groups are different from vector control in A and B (p < 0.05).
Since overexpression of VP1 activated the IFNa1 promoter, we wanted to study whether VP1 was able to induce transcription of IFN either alone or in combination with IRF1 and IRF3. Accordingly, TO-cells were transfected with plasmids containing VP1 alone or VP1 together with plasmids containing IRF1 or IRF3. Expression of IFNa in response to IPS-1 plasmid was used as a positive control and preVP2 was included as a negative control. After 24 h the cells were harvested, and analyzed for IFNa, IFNb and IFNc transcripts. Transfection with IPS-1 resulted in a 6-fold up-regulation of IFNa transcripts compared with the control (p < 0.001) (Fig. 4 ). Up-regulation of IFNa was hardly detectable after transfection with VP1, IRF1 or IRF3 alone whereas the combination of VP1 with either IRF1 or IRF3 showed a 6–8 times up-regulation of IFNa transcript levels. The preVP2 plasmid showed no significant up-regulation of IFNa1 transcripts (p = 0.8). None of the cell groups showed IFNb transcripts or up-regulation of IFNc transcripts (data not shown).
Fig. 4.
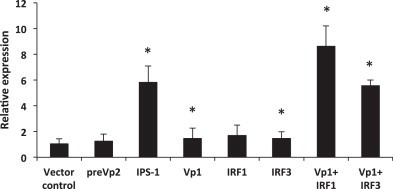
IFNa expression in TO cells in response to overexpression of VP1 in combination with IRF1 or IRF3. Cells were transfected with expression constructs for VP1, IRF1 and IRF3 in combination with plasmid control or VP1 in combination with IRF1 or IRF3. Cells transfected with IPS-1 or preVP2 were used as positive and negative control. RNA was extracted after 24 h and IFNa expression was determined by qPCR relative to transcript levels in plasmid control transfected cells (=1). Gene expression was normalized against 18S rRNA. Values are mean ± SD (N = 3). Asterisks denote significant differences from vector control (p < 0.05). Cells transfected with VP1 + IRF1 were significantly different from cells transfected with IRF1 + vector control and from VP1 + vector control (p < 0.005). Cells transfected with VP1 + IRF3 were significantly different from cells transfected with IRF3 + vector control and from VP1 + vector control (p < 0.005).
3.4. Antiviral effect induced by VP1 and IRF1/IRF3 is due to IFNa
Since VP1 together with IRF1/IRF3 activated IFNa transcription, we wanted to test their ability to induce antiviral activity in TO cells. The cells were transfected with plasmids expressing VP1, IRF1, IRF3 alone or VP1 in combination with the IRFs, or transfected with plasmid without insert for infected and uninfected controls. After 72 h, the cell supernatants were harvested for antiviral assay of IFN while the cells were infected with IPNV for direct measurement of antiviral activity on the transfected cells. When the infected control cells had reached full CPE, the supernatants were harvested for viral titration and the remaining cell layer was stained with crystal violet for measurement of cell survival. The assay showed significant increased cell survival for all plasmid transfections, but cells transfected with combination of VP1 and IRF1/IRF3 plasmids showed highest survival (Fig. 5A). This pattern of antiviral activity was also observed measured as reduction in virus titers as shown by the TCID50 calculations shown under the corresponding bars of OD550 measurements (Fig. 5A).
Fig. 5.
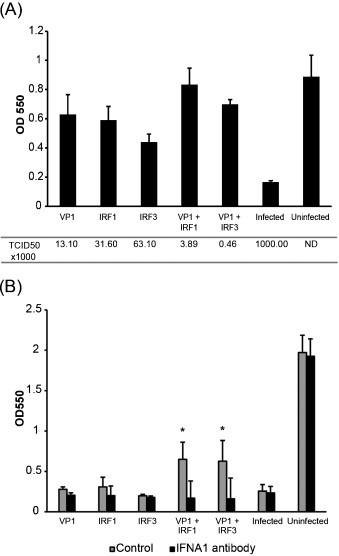
Antiviral activity of VP1, IRF1 and IRF3 against IPNV. (A) TO-cells were transfected with expression constructs for VP1, IRF1, IRF3 alone or VP1 in combination with IRF1 or IRF3, or empty vector for control samples. Three days after transfection, supernatants from the transfected cells were harvested and kept for use in the IFN neutralization assay shown in (B) while the cells were infected with IPNV (MOI 0.1). When full CPE was observed after 4 days, the supernatants were harvested for viral titration, and the surviving cell layer was stained with crystal violet. Cell survival was determined by measuring the absorbance at 550 nm. Viral titers in medium supernatants from the different treatment groups were determined by the TCID50 method. All groups are different from infected control (p < 0.05). (B) Measurement of IFN activity in cell supernatants described in (A) in the presence or absence of neutralizing antibody against IFNa1. TO-cells were treated with supernatants for 24 h and then infected with IPNV as described for (A). At full CPE, cell survival was determined by staining cells with crystal violet and measuring absorbance at 550 nm. Asterisks denote significant differences from infected vector control (p < 0.05).
We next studied if IFN was secreted by cells transfected with the different plasmid combinations. For this purpose the supernatant from the transfected cells was added to new TO cells in the presence or absence of IFNa1 antibody. The cells were incubated for 24 h for induction of antiviral proteins and infected with IPNV. Cell survival was measured by crystal violet staining when the infected control cells had reached full CPE. As shown in Fig. 5B, only supernatants from cells transfected with combinations of VP1 and IRFs gave significant protection of cells. The antiviral activity of these supernatants was completely blocked by pre-treatment with IFNa1 antibody.
3.5. The ability of VP1 to activate the IFNa1 promoter is dependent on an intact serine in position 163
VP1 is the IPNV polymerase and has been demonstrated to produce RNA from both viral and non-viral sources in vitro (Graham et al., 2011). It can thus be hypothesized that VP1 might stimulate IFNs through synthesis of dsRNA. We wanted to test this hypothesis by transfecting TO cells with a mutant VP1 that was defect in RNA polymerase activity. For this purpose we generated a VP1 sequence that was mutated in position 163 since this mutant (VP1S163A) has been shown neither to be able to guanylylate VP1 nor to be able to produce dsRNA (Petit et al., 2000).
VP1S163A was co-transfected into TO-cells together with the IFNa1-promoter construct to see if the mutant VP1 was able to activate the promoter (Fig. 6 ). Wild-type VP1 was included as a positive control. The results showed that the S163A mutation completely abolished VP1s ability to activate the IFNa1 promoter. The VP1 and VP1S163A proteins were expressed at comparable levels when detected with an anti-flag antibody (Fig. S2).
Fig. 6.
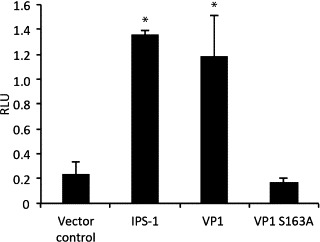
Effect of the VP1 mutant S163A on activation of the IFNa1 promoter. TO-cells were co-transfected with a plasmid containing the minimal IFNa1 promoter fused to a luciferase gene along with an expression construct for Renilla luciferase as a transfection control. In addition expression constructs for VP1, the mutant VP1S163A (without polymerase activity), or vector control were included. Results are presented as mean relative light units (RLU) ± SD (N = 3). Asterisks denote groups significantly different from cells transfected with vector control (p ≤ 0.05).
3.6. Antagonistic effect of VP4 is independent of an intact proteolytic active site
The observation that VP4 was able to inhibit IPS-1, IRF1 and IRF3 mediated activation of the IFNa1 promoter, indicated that VP4 acted on the IRFs or on signaling members involved in IRF-phosphorylation. An obvious question was if the IFN antagonistic effect of VP4 was due to its protease activity (Duncan et al., 1987). First we studied whether VP4 might degrade IRF1 and IRF3. However, no degradation of the IRFs could be detected in a Western blot following co-transfection of VP4 with the IRF1 or IRF3 (data not shown). We then wanted to test if IFN antagonistic activity of VP4 was dependent of its protease activity by creating a protease-dead mutant of VP4. Since mutations in the site of the lysine general base K674 have been shown to be even more efficient in abolishing protease activity than mutating the reactive serine residue (Petit et al., 2000), this site was chosen for a single amino acid mutation. This mutation has previously shown to abolish the protease activity of VP4 with all tested mutations (Petit et al., 2000). The expressed VP4 and VP4K674A proteins were present in comparable amounts when detected with an anti-flag antibody (Fig. S2). The constructed VP4K674A was found to be just as efficient as the wild type VP4 in inhibition of IPS1-mediated IFNa1 promoter activation in a co-transfection assay (Fig. 7 ). This suggests that the IFN antagonistic property of VP4 is independent of its protease activity. We then looked for potential non-covalent binding of VP4 to the IRFs. However, we did not observe any binding of IRF1/3 to VP4 in a co-immunoprecipitation assay (data not shown). As there are an abundance of proteins and regulators involved in the IRF branch of the signaling cascade, there are many other potential targets for inhibition. Unfortunately, far from all of the potential signaling members have been cloned in Atlantic salmon.
Fig. 7.
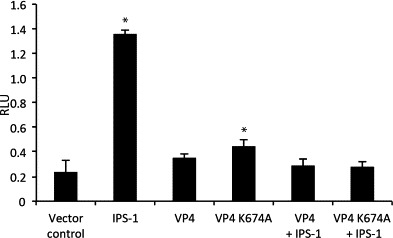
Effect of the VP4 mutant K674A on activation of the IFNa1 promoter. TO-cells were co-transfected with a plasmid containing the minimal IFNa1 promoter fused to a luciferase gene along with an expression construct for Renilla luciferase as a transfection control. In addition expression constructs for VP4, the mutant VP4K674A (without protease activity), or a vector control were included. Results are presented as mean relative light units (RLU) ± SD (N = 3). Asterisks denote groups significantly different from cells transfected with vector control (p ≤ 0.05).
4. Discussion
IPNV replication is strongly inhibited in salmon cells treated by type I IFN (Robertsen et al., 2003, Svingerud et al., 2012). The virus must therefore be dependent on inhibition of IFN induction during infection. This is supported by the observation that IPNV infection of TO cells only resulted in very small changes in the amounts of IFNa transcripts (Fig. 1). Moreover, the present work demonstrates that IPNV encodes proteins with powerful IFN antagonistic properties where all of the proteins encoded by IPNV segment B were able to inhibit induction of IFNa1 through the RIG-I/MDA5 signaling pathway. Overexpression of preVP2, VP3, VP4 and VP5 all inhibited IPS-1 mediated activation of the IFNa1 promoter. While VP4 virtually abolished IFNa1 promoter activation, preVP2, VP3 and VP5 all displayed a strong albeit not complete inhibition. These viral proteins are likely to act on signaling factors downstream of IPS-1 since they all inhibited activation of the IFNa1 promoter mediated by overexpression of IRF1 and IRF3. The mechanisms of inhibition observed by preVP2, VP3 and VP5 remain to be elucidated. The dsRNA binding ability of VP3 has previously been proposed as a mechanism for avoiding antiviral host responses (Pedersen et al., 2007). Binding dsRNA is, however, not likely to be the main mechanism of antagonism observed in the present studies, as the assays are based on overexpression of proteins from DNA vectors, and the protein levels of the transfection control, Renilla luciferase, were not affected by the presence of VP3. It is thus possible that VP3 has multiple IFN antagonistic properties, a well-known trait in viruses. One of the most studied viral proteins with multiple functions as an IFN antagonist is the non-structural NS1 protein of influenza A viruses. In addition to being important for enhancing viral mRNA translation, NS1 is able to ablate the host innate immune system both by limiting the induction of IFN by blocking RIG-I activation, and by directly inhibiting antiviral proteins such as PKR and OAS/RNaseL. NS1 also inhibits export of mRNA from the nucleus (reviewed in Hale et al., 2008).
Since VP4 possesses protease activity, it was suspected that it might cleave members of the RIG-I/MDA5-signaling pathway similar to several other viral proteases. For instance the picornavirus 3Cpro cleaves RIG-I (Barral et al., 2009), and the NS3/4A serine protease of hepatitis C virus, the NS3/4A protease of GB virus B and the 3ABC of hepatitis A virus all cleaves IPS-1 (Chen et al., 2007, Li et al., 2005, Yang et al., 2007). However, VP4 mutated to abolish protease activity (K674A), retained its ability to inhibit IPS-1 mediated activation of the IFNa1 promoter. Neither could we detect any degradation products of IRF1 or IRF3 when co-transfected into cells with VP4. The protease activity of VP4 is thus not likely to be important for its IFNa antagonistic activity in the RIG-I/MDA5 pathway downstream of IPS-1. As interactions between VP4 and IRF1 or IRF3 were not observed, VP4 is likely to act on other signaling members involved in activation of the IRFs. Potentially VP4 might prevent phosphorylation of the IRFs. Other possibilities are that VP4 might modify the function of the IRFs by promoting or reversing ubiquitination, SUMOylation or ISGylation on the members of the signaling cascade, similar modifications have been shown of other antagonistic viral proteins such as influenza A virus NS1, murine hepatitis virus NSP3 and ebolavirus VP35 (Gack et al., 2009, Kubota et al., 2009, Zheng et al., 2008). It has been shown that the IFN antagonistic activity of the papain-like protease (PLP) of the human coronavirus NL63 is independent of its enzymatic activity (Clementz et al., 2010). The PLP is both a protease and a deubiquitinase, able to process both K-48 and K-63 linked polyubiquitin chains. Ablating these functions either by mutation or by adding a protease inhibitor did not inhibit the antagonistic nature of the protein. Also the protease of the murine hepatitis virus acts as an IFN antagonist, and even if the antagonistic activity is partly dependent on the deubiquitinase activity, the protease defective mutants still retained some antagonistic activity (Zheng et al., 2008). Like these viral proteases, the IFN antagonistic effect of IPNV VP4 seems to be independent of an intact catalytic activity.
The birnavirus infectious bursal disease virus (IBDV) is structurally related to IPNV, but causes acute immunosuppressive disease in birds due to infection of B cells (Mahgoub et al., 2012). Even if the immunosuppressive nature of IBDV has been well established, the molecular basis for this mechanism is not fully understood. It was recently reported that the VP4 protease of IBDV is inhibiting IFN expression by interacting with the glucocorticoid-induced leucine zipper (GILZ) (Li et al., 2013). GILZ has previously been shown to bind both NF-κB and IRF3, inhibiting gene transcription directed by these transcription factors (Ogawa et al., 2005, Reily et al., 2006). Whether IPNV VP4 utilizes a similar mechanism as the IBDV VP4 in IFN suppression, is a subject for future studies.
A surprising finding in the present work was the IFN inducible properties of the viral polymerase VP1. Overexpression of VP1 alone potently activated the IFNa1 promoter and increased activation mediated by overexpression of IRF1 and IRF3. Moreover, overexpression of VP1 in combination with IRF1 or IRF3 up-regulated IFNa transcription and increased antiviral activity against IPNV in TO cells. The antiviral activity from the transfected cells with VP1 in combination with IRF1 or IRF3 was due to IFNa since the antiviral activity of the supernatants could be completely negated by the addition of IFNa1 antibody. The IFNa inducing properties of VP1 may be due to synthesis of RNA since it has been demonstrated that VP1 produces RNA from both viral and non-viral sources in vitro (Graham et al., 2011). This hypothesis is supported by the fact that one single amino acid substitution in position 163 abolished the ability of VP1 to activate IFNa1 promoter activation. The VP1S163A mutant has been shown neither to be able to guanylylate nor to produce dsRNA (Xu et al., 2004). Even though the 163 site is not the actual guanylylation site (Graham et al., 2011), this mutant is deficient in producing dsRNA (Xu et al., 2004). RNA synthesized by VP1 may be recognized by RIG-I or MDA5 and thus trigger activation of the IFNa1 promoter. During IPNV infection both viral dsRNA and non-specific RNA species formed by VP1 may thus potentially be recognized by RIG-I/MDA5 risking activation of IFN transcription. IPNV may thus have developed multiple IFN antagonizing mechanisms to avoid triggering of IFN synthesis by VP1 since this virus is highly sensitive to the antiviral activities induced by type I IFNs in cells. dsRNA could not be detected by a dsRNA antibody after transfecting cells with VP1 (data not shown), but antibody binding might be dependent of sizes of dsRNA molecules that are larger than the ones produced by VP1. The concentration of such RNA molecules is also likely to be low, since VP1 is highly dependent on an IPNV specific primer molecule to start synthesis of dsRNA (Dobos, 1995).
In conclusion, Atlantic salmon cells seem to have the ability to establish an antiviral state by recognizing dsRNA produced by the IPNV VP1. To overcome this potential barrier of infection, IPNV has developed multiple mechanisms for inhibition of IFNa induction by targeting the RIG-I/MDA5 pathway.
Conflict of interest
There are no actual or potential conflicts of interest.
Acknowledgements
We thank Professor Jorunn Jørgensen at the University of Tromsø, who kindly provided the IPNV vector constructs. This work was in part supported by the Aquaculture Program of the Research Council of Norway (Grant 185217).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.virusres.2014.11.018.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- Arpaia N., Barton G.M. Toll-like receptors: key players in antiviral immunity. Curr. Opin. Virol. 2011;1:447–454. doi: 10.1016/j.coviro.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral P.M., Sarkar D., Fisher P.B., Racaniello V.R. RIG-I is cleaved during picornavirus infection. Virology. 2009;391:171–176. doi: 10.1016/j.virol.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergan V., Kileng O., Sun B., Robertsen B. Regulation and function of interferon regulatory factors of Atlantic salmon. Mol. Immunol. 2010;47:2005–2014. doi: 10.1016/j.molimm.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Bergan V., Steinsvik S., Xu H., Kileng O., Robertsen B. Promoters of type I interferon genes from Atlantic salmon contain two main regulatory regions. FEBS J. 2006;273:3893–3906. doi: 10.1111/j.1742-4658.2006.05382.x. [DOI] [PubMed] [Google Scholar]
- Berke I.C., Modis Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012;31:1714–1726. doi: 10.1038/emboj.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacchesi S., LeBerre M., Lamoureux A., Louise Y., Lauret E., Boudinot P., Bremont M. Mitochondrial antiviral signaling protein plays a major role in induction of the fish innate immune response against RNA and DNA viruses. J. Virol. 2009;83:7815–7827. doi: 10.1128/JVI.00404-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M., Collet B., Nie P., Lester K., Campbell S., Secombes C.J., Zou J. Expression and functional characterization of the RIG-I-like receptors MDA5 and LGP2 in Rainbow trout (Oncorhynchus mykiss) J. Virol. 2011;85:8403–8412. doi: 10.1128/JVI.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Benureau Y., Rijnbrand R., Yi J., Wang T., Warter L., Lanford R.E., Weinman S.A., Lemon S.M., Martin A., Li K. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J. Virol. 2007;81:964–976. doi: 10.1128/JVI.02076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K.C., Cresswell P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 2001;98:15125–15130. doi: 10.1073/pnas.011593298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie K.E., Havarstein L.S., Djupvik H.O., Ness S., Endresen C. Characterization of a new serotype of infectious pancreatic necrosis virus isolated from Atlantic salmon. Arch. Virol. 1988;103:167–177. doi: 10.1007/BF01311090. [DOI] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K., Li K., Mesecar A.D., Baker S.C. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulibaly F., Chevalier C., Gutsche I., Pous J., Navaza J., Bressanelli S., Delmas B., Rey F.A. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell. 2005;120:761–772. doi: 10.1016/j.cell.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Dobos P. Size and structure of the genome of infectious pancreatic necrosis virus. Nucleic Acids Res. 1976;3:1903–1924. doi: 10.1093/nar/3.8.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobos P. Protein-primed RNA synthesis in vitro by the virion-associated RNA polymerase of infectious pancreatic necrosis virus. Virology. 1995;208:19–25. doi: 10.1006/viro.1995.1125. [DOI] [PubMed] [Google Scholar]
- Duncan R., Nagy E., Krell P.J., Dobos P. Synthesis of the infectious pancreatic necrosis virus polyprotein, detection of a virus-encoded protease, and fine structure mapping of genome segment A coding regions. J. Virol. 1987;61:3655–3664. doi: 10.1128/jvi.61.12.3655-3664.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H., Liu H., Kong R., Wang L., Wang Y., Hu W., Guo Q. Expression profiles of carp IRF-3/-7 correlate with the up-regulation of RIG-I/MAVS/TRAF3/TBK1, four pivotal molecules in RIG-I signaling pathway. Fish Shellfish Immunol. 2011;30:1159–1169. doi: 10.1016/j.fsi.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Gack M.U., Albrecht R.A., Urano T., Inn K.S., Huang I.C., Carnero E., Farzan M., Inoue S., Jung J.U., Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham S.C., Sarin L.P., Bahar M.W., Myers R.A., Stuart D.I., Bamford D.H., Grimes J.M. The N-terminus of the RNA polymerase from infectious pancreatic necrosis virus is the determinant of genome attachment. PLoS Pathog. 2011;7:e1002085. doi: 10.1371/journal.ppat.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale B.G., Randall R.E., Ortin J., Jackson D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Heppell J., Tarrab E., Berthiaume L., Lecomte J., Arella M. Characterization of the small open reading frame on genome segment A of infectious pancreatic necrosis virus. J. Gen. Virol. 1995;76:2091–2096. doi: 10.1099/0022-1317-76-8-2091. [DOI] [PubMed] [Google Scholar]
- Hill B.J., Way K. Serological classification of infectious pancreatic necrosis (IPN) virus and other aquatic birnaviruses. Ann. Rev. Fish Dis. 1995;5:55–77. [Google Scholar]
- Holland J.W., Bird S., Williamson B., Woudstra C., Mustafa A., Wang T., Zou J., Blaney S.C., Collet B., Secombes C.J. Molecular characterization of IRF3 and IRF7 in rainbow trout, Oncorhynchus mykiss: functional analysis and transcriptional modulation. Mol. Immunol. 2008;46:269–285. doi: 10.1016/j.molimm.2008.08.265. [DOI] [PubMed] [Google Scholar]
- Jarp J., Gjevre A.G., Olsen A.B., Bruheim T. Risk-factors for furunculosis, infectious pancreatic necrosis and mortality in post-smolt of Atlantic salmon, Salmo salar L. J. Fish Dis. 1995;18:67–78. [Google Scholar]
- Jiang F., Ramanathan A., Miller M.T., Tang G.Q., Gale M., Jr., Patel S.S., Marcotrigiano J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T., Matsuoka M., Chang T.H., Bray M., Jones S., Tashiro M., Kato A., Ozato K. Ebolavirus VP35 interacts with the cytoplasmic dynein light chain 8. J. Virol. 2009;83:6952–6956. doi: 10.1128/JVI.00480-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauksund S., Svingerud T., Bergan V., Robertsen B. Atlantic salmon IPS-1 mediates induction of IFNa1 and activation of NF-kappaB and localizes to mitochondria. Dev. Comp. Immunol. 2009;33:1196–1204. doi: 10.1016/j.dci.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Li X.D., Sun L., Seth R.B., Pineda G., Chen Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Wang Y., Li X., Li X., Cao H., Zheng S.J. Critical roles of glucocorticoid-induced leucine zipper in infectious bursal disease virus (IBDV)-induced suppression of type I interferon expression and enhancement of IBDV growth in host cells via interaction with VP4. J. Virol. 2013;87:1221–1231. doi: 10.1128/JVI.02421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.Y., Sanchez D.J., Cheng G. New developments in the induction and antiviral effectors of type I interferon. Curr. Opin. Immunol. 2011;23:57–64. doi: 10.1016/j.coi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahgoub H.A., Bailey M., Kaiser P. An overview of infectious bursal disease. Arch. Virol. 2012;157:2047–2057. doi: 10.1007/s00705-012-1377-9. [DOI] [PubMed] [Google Scholar]
- Ogawa S., Lozach J., Benner C., Pascual G., Tangirala R.K., Westin S., Hoffmann A., Subramaniam S., David M., Rosenfeld M.G., Glass C.K. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani M., Hikima J., Hwang S.D., Morita T., Suzuki Y., Kato G., Kondo H., Hirono I., Jung T.S., Aoki T. Transcriptional regulation of type I interferon gene expression by interferon regulatory factor-3 in Japanese flounder, Paralichthys olivaceus. Dev. Comp. Immunol. 2012;36:697–706. doi: 10.1016/j.dci.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Ohtani M., Hikima J., Kondo H., Hirono I., Jung T.S., Aoki T. Characterization and antiviral function of a cytosolic sensor gene, MDA5, in Japanese flounder, Paralichthys olivaceus. Dev. Comp. Immunol. 2011;35:554–562. doi: 10.1016/j.dci.2010.12.013. [DOI] [PubMed] [Google Scholar]
- Pedersen T., Skjesol A., Jorgensen J.B. VP3, a structural protein of infectious pancreatic necrosis virus, interacts with RNA-dependent RNA polymerase VP1 and with double-stranded RNA. J. Virol. 2007;81:6652–6663. doi: 10.1128/JVI.02831-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit S., Lejal N., Huet J.C., Delmas B. Active residues and viral substrate cleavage sites of the protease of the birnavirus infectious pancreatic necrosis virus. J. Virol. 2000;74:2057–2066. doi: 10.1128/jvi.74.5.2057-2066.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M.W., Horgan G.W., Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pous J., Chevalier C., Ouldali M., Navaza J., Delmas B., Lepault J. Structure of birnavirus-like particles determined by combined electron cryomicroscopy and X-ray crystallography. J. Gen. Virol. 2005;86:2339–2346. doi: 10.1099/vir.0.80942-0. [DOI] [PubMed] [Google Scholar]
- Randall R.E., Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 1938;27:493–497. [Google Scholar]
- Reily M.M., Pantoja C., Hu X., Chinenov Y., Rogatsky I. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J. 2006;25:108–117. doi: 10.1038/sj.emboj.7600919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertsen B., Bergan V., Rokenes T., Larsen R., Albuquerque A. Atlantic salmon interferon genes: cloning, sequence analysis, expression, and biological activity. J. Interferon Cytokine Res. 2003;23:601–612. doi: 10.1089/107999003322485107. [DOI] [PubMed] [Google Scholar]
- Rokenes T.P., Larsen R., Robertsen B. Atlantic salmon ISG15: expression and conjugation to cellular proteins in response to interferon, double-stranded RNA and virus infections. Mol. Immunol. 2007;44:950–959. doi: 10.1016/j.molimm.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Samuel C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi N., Sandtro A., Sindre H., Song H., Hong J.R., Thu B., Wu J.L., Vakharia V.N., Evensen O. Infectious pancreatic necrosis virus induces apoptosis in vitro and in vivo independent of VP5 expression. Virology. 2005;342:13–25. doi: 10.1016/j.virol.2005.07.028. [DOI] [PubMed] [Google Scholar]
- Simora R.M., Ohtani M., Hikima J., Kondo H., Hirono I., Jung T.S., Aoki T. Molecular cloning and antiviral activity of IFN-beta promoter stimulator-1 (IPS-1) gene in Japanese flounder, Paralichthys olivaceus. Fish Shellfish Immunol. 2010;29:979–986. doi: 10.1016/j.fsi.2010.08.012. [DOI] [PubMed] [Google Scholar]
- Skjesol A., Skjaeveland I., Elnaes M., Timmerhaus G., Fredriksen B.N., Jorgensen S.M., Krasnov A., Jorgensen J.B. IPNV with high and low virulence: host immune responses and viral mutations during infection. Virol J. 2011;8:396. doi: 10.1186/1743-422X-8-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjesol A., Aamo T., Hegseth M.N., Robertsen B., Jorgensen J.B. The interplay between infectious pancreatic necrosis virus (IPNV) and the IFN system: IFN signaling is inhibited by IPNV infection. Virus Res. 2009;143:53–60. doi: 10.1016/j.virusres.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smail D.A., Bruno D.W., Dear G., Mcfarlane L.A., Ross K. Infectious pancreatic necrosis (IPN) virus Sp serotype in farmed Atlantic salmon Salmo salar L., post-smolts associated with mortality and clinical disease. J. Fish Dis. 1992;15:77–83. [Google Scholar]
- Su J., Huang T., Dong J., Heng J., Zhang R., Peng L. Molecular cloning and immune responsive expression of MDA5 gene, a pivotal member of the RLR gene family from grass carp Ctenopharyngodon idella. Fish Shellfish Immunol. 2010;28:712–718. doi: 10.1016/j.fsi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Sun B., Robertsen B., Wang Z., Liu B. Identification of an Atlantic salmon IFN multigene cluster encoding three IFN subtypes with very different expression properties. Dev. Comp. Immunol. 2009;33:547–558. doi: 10.1016/j.dci.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Sun F., Zhang Y.B., Liu T.K., Shi J., Wang B., Gui J.F. Fish MITA serves as a mediator for distinct fish IFN gene activation dependent on IRF3 or IRF7. J. Immunol. 2011;187:2531–2539. doi: 10.4049/jimmunol.1100642. [DOI] [PubMed] [Google Scholar]
- Svingerud T., Solstad T., Sun B., Nyrud M.L., Kileng O., Greiner-Tollersrud L., Robertsen B. Atlantic salmon type I IFN subtypes show differences in antiviral activity and cell-dependent expression: evidence for high IFNb/IFNc-producing cells in fish lymphoid tissues. J. Immunol. 2012;189:5912–5923. doi: 10.4049/jimmunol.1201188. [DOI] [PubMed] [Google Scholar]
- Takeuchi O., Akira S. Recognition of viruses by innate immunity. Immunol. Rev. 2007;220:214–224. doi: 10.1111/j.1600-065X.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- van den Broek M.F., Muller U., Huang S., Zinkernagel R.M., Aguet M. Immune defence in mice lacking type I and/or type II interferon receptors. Immunol. Rev. 1995;148:5–18. doi: 10.1111/j.1600-065x.1995.tb00090.x. [DOI] [PubMed] [Google Scholar]
- Wergeland H.I., Jakobsen R.A. A salmonid cell line (TO) for production of infectious salmon anaemia virus (ISAV) Dis. Aquat. Organ. 2001;44:183–190. doi: 10.3354/dao044183. [DOI] [PubMed] [Google Scholar]
- Xu H.T., Si W.D., Dobos P. Mapping the site of guanylylation on VP1, the protein primer for infectious pancreatic necrosis virus RNA synthesis. Virology. 2004;322:199–210. doi: 10.1016/j.virol.2004.01.024. [DOI] [PubMed] [Google Scholar]
- Yang Y., Liang Y., Qu L., Chen Z., Yi M., Li K., Lemon S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D., Chen G., Guo B., Cheng G., Tang H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008;18:1105–1113. doi: 10.1038/cr.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J., Chang M., Nie P., Secombes C.J. Origin and evolution of the RIG-I like RNA helicase gene family. BMC Evol. Biol. 2009;9:85. doi: 10.1186/1471-2148-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.