

Highlights
-
•
TGEV induced cell cycle arrest at S and G2/M phase in PK-15 and ST cells.
-
•
p53 might play key roles in mediation of TGEV-induced cell cycle arrest.
-
•
The host cells staying at S or G2/M phase is beneficial for TGEV replication.
Keywords: TGEV, Cell cycle arrest, p53 pathway, Virus replication
Abstract
p53 signaling pathway plays an important role in the regulation of cell cycle. Our previous studies have demonstrated that TGEV infection induces the activation of p53 signaling pathway. In this study we investigated the effects of TGEV infection on the cell cycle of host cells and the roles of p53 activation in this process. The results showed that TGEV infection induced cell cycle arrest at S and G2/M phases in both asynchronous and synchronized PK-15 and ST cells, while UV-inactivated TGEV lost the ability of induction of cell cycle arrest. TGEV infection promoted p21 accumulation, down-regulated cell cycle-regulatory proteins cyclins B1, cdc2, cdk2 and PCNA. Further studies showed that inhibition of p53 signaling could attenuate the TGEV-induced S- and G2/M-phase arrest by reversing the expression of p21 and corresponding cyclin/cdk. In addition, TGEV infection of the cells synchronized in various stages of cell cycle showed that viral genomic RNA and subgenomic RNA, and virus titer were higher in the cells released from S-phase- or G2/M phase-synchronized cells than that in the cells released from the G0/G1 phase-synchronized or asynchronous cells after 18 h p.i. Taken together, our data suggested that TGEV infection induced S and G2/M phase arrest in host cells, which might provide a favorable condition for viral replication.
1. Introduction
Viral infection could activate a variety of signal transduction pathways to induce subversion of the host cell cycle, which plays important roles in the viral life cycle by facilitating the replication of progeny virus after viral infection (Davy and Doorbar, 2007). A variety of RNA viruses have been shown to induce G0/G1, S or G2/M arrest in infected cells. For example, Human immunodeficiency virus (HIV)-infected T lymphocytes isolated from patients are arrested in G2/M (Zimmerman et al., 2006); Influenza A virus A/WSN/33 (H1N1) infection results in G0/G1-phase accumulation of infected cells, which increase viral protein expression and progeny virus production (He et al., 2010); Hepatitis C virus (HCV) NS2 protein induces cell cycle arrest in the S-phase in mammalian cells to facilitate HCV viral replication (Yang et al., 2006). In term of coronaviruses, murine coronavirus mouse hepatitis virus (MHV) has been shown to induce G0/G1 arrest (Chen and Makino, 2004); SARS-CoV nucleocapsid (N) protein can disrupt cytokinesis and block S-phase progression in mammalian cells (Surjit et al., 2006); Bronchitis virus (IBV) infection can induce G2/M phase arrest to facilitate viral replication (Dove et al., 2006). Transmissible gastroenteritis virus (TGEV) infection has been shown to alter some cell signaling pathways implicated in cell cycle regulation in our previous study (Huang et al., 2013). However, the effects of TGEV infection on the cell cycle of host cells and the significance of cell cycle regulation in TGEV replication need to be further investigated.
The cell-cycle progression is tightly regulated through a complex network of cell-cycle regulatory molecules. CyclinD-cdk2 complex and cyclinE-cdk4 complex regulate cell cycle progression in the G0/G1 phase. CyclinA-cdk2 complex regulates cell cycle progression in the S phase. CyclinB-cdc2 complex is a crucial regulator for progression through late G2 and early M (Malumbres and Barbacid, 2009). These cell-cycle regulatory molecules have been shown to be regulated by some upstream pathways like p53 signaling. p53 signaling can control the expression of p21, which directly bind to some cdk-cyclin complexes to inhibit their kinases activity (He et al., 2005). Our previous studies have demonstrated that TGEV infection induced the activation of p53 signaling pathway to regulate cell growth and apoptosis (Huang et al., 2013). In this study, we further investigated the effects of TGEV infection on the cell cycle of host cells, the roles of p53 signaling activation in regulation of cell cycle progression in TGEV-infected cells, and the significance of cell cycle regulation in TGEV replication. Results showed that that TGEV infection could perturb the progression of cell cycle and facilitate virus gene replication.
2. Materials and methods
2.1. Viruses and cells
PK-15 cells (ATCC, CCL-33) and ST cells (ATCC, CRL-1746) were grown in Dulbecco Minimal Essential Medium (D-MEM) (Gibco BRL, MD, US) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), 100 IU of penicillin and 100 μg of streptomycin per ml, at 37 °C in a 5% CO2 atmosphere incubator. The TGEV Shaanxi strain was isolated from intestinal tract contents of TGEV-infected piglets in Shaanxi Province of China and propagated in PK-15 cells and ST cells (Ding et al., 2011). Virus titers determined by 50% tissue culture infective doses (TCID50) as described previously (Reed and Muench, 1938).
2.2. Cell cycle analysis by flow cytometry
Cell cycle analysis was measured by propidium iodide staining. Briefly, cells were fixed in 70% ethanol for 30 min at 4 °C. After several washes with phosphate-buffered saline (PBS), the cell pellets were resuspended in 0.5 ml PBS containing 0.1% Triton X-100 (Sigma–Aldrich, US), 20 μg /ml RNase A (Sigma) and 10 μg/ml propidium iodide (Sigma) for 30 min, prior to FACS analysis (Beckman Coulter, Inc. Fullerton, CA, US). At least 15 000 nuclei were counted for each sample.
2.3. BrdU incorporation and flow cytometry analysis
The thymidine analog bromodeoxyuridine (BrdU) (Sigma) is incorporated into actively replicating DNA and thus accurately determines the proportion of cells in S phase. Briefly, 10 μM BrdU was added to cell medium and incubated at 37 °C for 30 min to allow BrdU incorporation. Cells were collected and then fixed in 70% ethanol. Then cells were pelleted and incubated in 2 M HCl in PBS at 37 °C for 30 min. After incubating in wash buffer and pelleting, the cell pellet was resuspended in 0.1 M sodium borate. Samples were then pelleted before addition of 100 μl of anti-BrdU antibody (Cell Signalling Technology) and incubated for 60 min at room temperature. Samples were then incubated by fluorescein isothiocyanate (FITC)-labeled antibody for 30 min in the dark. Samples were stained by PI before analyzed using a FACS Calibur analyzer.
2.4. G0/G1, G1/S, and G2/M synchronization in PK-15 and ST cells
PK-15 and ST cells were synchronized at G0/G1 phase using serum deprivation by maintenance of cells in DMEM containing no FBS supplementation for 48 h. Synchronized cells were mock infected or infected with 0.5 MOI of TGEV. After 1 h of virus adsorption, cells were treated with medium containing 10% FBS and harvested at various times post infection (p.i.) for cell cycle analysis.
PK-15 and ST cells were synchronized at the G1/S phase border using double-thymidine treatment by incubation for 12 h in maintenance media supplemented with 2 mM thymidine (Sigma). Cells were then washed three times with PBS and incubated for 10 h in maintenance media, followed by additional 12 h incubation in maintenance media supplemented with 2 mM thymidine. Then, synchronized cells were mock infected or infected with 0.5 MOI of TGEV.
PK-15 and ST cells were synchronized at G2/M phase using nocodazole (Sigma) treatment by incubation of cells in maintenance media supplemented with 100 ng/ml nocodazole for 16 h. Cells were washed three times with PBS, and then infected with 0.5 MOI of TGEV. At indicated times p.i., cells were processed for RT-PCR and flow cytometric analysis.
2.5. Western blot analysis
Cell extracts were prepared as described previously (Ding et al., 2012). Protein concentrations were measured using BCA Protein Assay Reagent (Pierce, Rockford, IL, US). Equivalent amounts of proteins were loaded and electrophoresed on 12% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE). Subsequently, proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore Corp, Atlanta, GA, US). The membranes were blocked with 5% nonfat dry milk at room temperature for 1 h, and then incubated with indicated primary antibodies over night at 4 °C, followed by HRP-conjugated secondary antibodies at room temperature for 1 h. The signal was detected using ECL reagent (Pierce, Rockford, IL, US).
2.6. RNA analysis by real-time RT-PCR
Total RNA extraction and reverse transcription were performed as described (Ding et al., 2012). Quantitative analysis of genomic RNA (gRNA) and subgenomic mRNAs (sgRNAs) from TGEV-derived replicons was performed by real-time RT-PCR using Bio-Rad iQ5 Real Time PCR System. The primers for qRT-PCR in this study were described in previous study (Dufour et al., 2011). Reactions were carried out in 25 μl volume containing 1× SYBR Premix Ex TaqTM II (Takara, Dalian, China), sense and anti-sense primers and target cDNA. The relative quantification of gene expression was analyzed by the two-ddCt method.
2.7. Inhibitor treatments
Pifithrin-α (PTF-α) was purchased from Sigma and stored as a 50 mM stock solution in DMSO. To reduce the activation of p53, PTF-α (20 μM) was diluted in cell culture medium without serum and added to cultures 1 h prior to infection. Inhibitor was not included in the virus inoculum. After 1 h of TGEV adsorption, the virus inoculum was removed and fresh basal medium containing fresh inhibitor was added to the culture. At indicated times, cells were harvested and correlative indicators were detected.
2.8. Statistical analysis
Data are mean ± SEM of three independent experiments. Results were analyzed by one-way analysis of variance (ANOVA). For each assay, student's t-test was used for statistical comparison. A value of P < 0.05 was considered significant.
3. Results
3.1. TGEV-infected cells accumulated at S and G2/M phase of the cell cycle
To investigate the influence of TGEV infection on cell cycle progression, mock infected or TGEV-infected asynchronously growing PK-15 and ST cells were harvested at different times p.i., and cell cycle profiles were detected by flow cytometry. Representative cell cycle histograms and profiles in PK-15 and ST cells were presented in Fig. 1A and B, respectively. In TGEV-infected PK-15 cells, there was a significant increase in the proportion of cells in the S phases and G2/M phases of the cell cycle from 6 h p.i. and 12 h p.i., respectively, and continued to increase with infection time, when compared to mock-infected cells (Fig. 1A). In TGEV-infected ST cells, the proportion of cells in the G2/M phases and S phases significantly increased from 12 h p.i. and 18 h p.i., respectively, when compared to mock-infected cells (Fig. 1B). These results suggest that TGEV infection induced the arrest of the cell cycle in the S and G2/M phase.
Fig. 1.
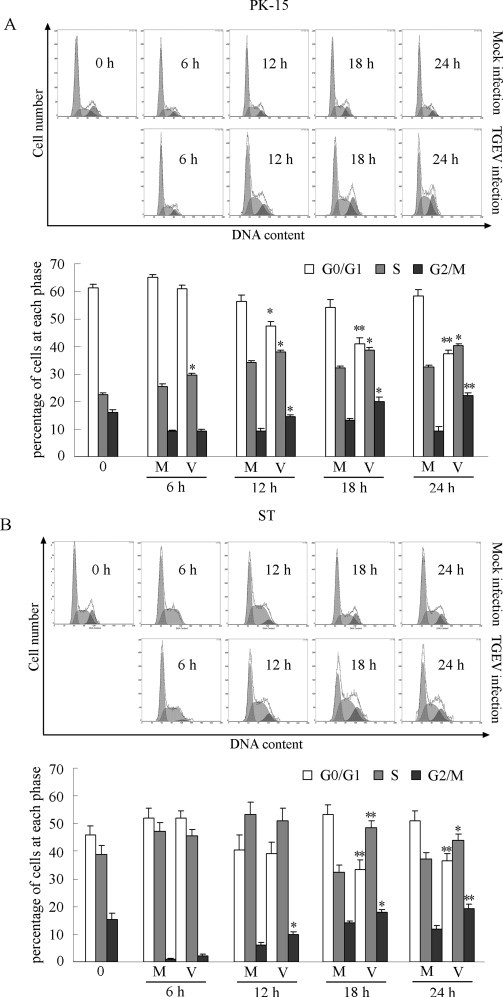
TGEV infection induced the subversion of cell cycle in asynchronously growing cells. PK-15 (A) and ST (B) cells were mock infected or infected with TGEV at an MOI of 0.5. At the indicated times, cells were collected and stained with propidium iodide for cell cycle analysis using flow cytometry. The data are from one of three experiments. The histograms were analyzed to determine the percentage of cells in each phase of the cell cycle. The results are shown as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 versus mock infection.
In TGEV-infected cells, TGEV infectious levels were evaluated by detection of the gRNA and sgRNAs (N sgRNA, M sgRNA and ORF 7 sgRNA) of TGEV using qRT-PCR. Results showed that the levels of TGEV gRNA increased significantly from 12 h p.i. in PK-15 (Fig. 2A, left panel) and ST cells (Fig. 2B, left panel). The sgRNA levels of N, M, ORF 7 could not been detected at 0 h p.i., detected at 6 h p.i., significantly increased at 12 h p.i., and continued to increase with following infectious time in TGEV-infected cells (Fig. 2A and B, right panel). Moreover, the titers of TGEV were determined at various times postinoculation. Results showed that little infectious virus was detected at 0 and 6 h p.i., thereafter the virus titer showed a rapid increase between 12 and 18 h p.i. and then reached a plateau (Fig. 2C).
Fig. 2.
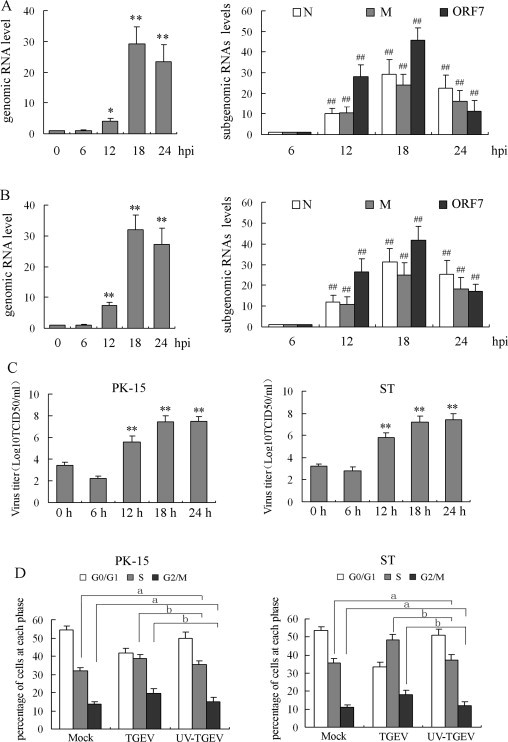
TGEV-induced cell cycle arrest is dependent on TGEV replication. PK-15 cells (A) and ST cells (B) were mock infected or infected with 0.5 MOI of TGEV. At the indicated times, cells were collected and examined by qRT-PCR for gRNA (left panel) and sgRNAs (right panel) of TGEV. Values are shown as the mean ± SEM. **P < 0.01 versus TGEV infected cells for 0 h (left panel). ##P < 0.01 versus TGEV infected cells for 6 h (right panel). (C) Cells were treated as in (A), total virus at indicated times was harvested by freezing and thawing cells three times and the viral titers were shown as log10TCID50/ml. **P < 0.01 versus TGEV infected cells for 0 h. (D) Cell cycle profiles were measured at 18 h p.i. by flow cytometry. Histograms were analyzed to determine the percentage of cells in each phase of the cell cycle. The results are shown as mean ± SEM of three independent experiments. (a) P > 0.05 versus mock infection; (b) P < 0.05 versus TGEV infection.
To further determine whether the TGEV-induced S and G2/M arrest requires virus replication, UV-inactivated TGEV was inoculated in PK-15 and ST cells, and cell cycle profiles were measured at 18 h p.i. by flow cytometry. Results showed that cell cycle arrest were not observed in cells infected with UV-inactivated TGEV, while there was no significant difference between mock-infected cells and cells treated with UV-inactivated TGEV (Fig. 2D), suggesting that viral replication was required for induction of cell cycle arrest in TGEV-infected cells.
3.2. TGEV infection of quiescent cells induces cell cycle arrest
To further confirm that TGEV replication caused S and G2/M cell cycle arrest, we infected serum-starved quiescent cells with TGEV and examined cell cycle progression after serum stimulation. As shown in Fig. 3A, prior to infection, approximately 85% of serum-starved cells were arrested at the G0/G1 phase in PK-15 cells. At 18, 24 and 30 h p.i., the cells population at G0/G1-phase significantly decreased, while the cells population at S-phase dramatically increased in TGEV-infected cells compared to that in mock-infected cells, and apparent increase of G2/M-phase cells was observed at 30 h p.i. (Fig. 3A). Similar feature were also seen in quiescent ST cells after infection (Fig. 3B). These results suggest that TGEV-infected cells exhibited S-phase delay and G2/M-phase arrest.
Fig. 3.
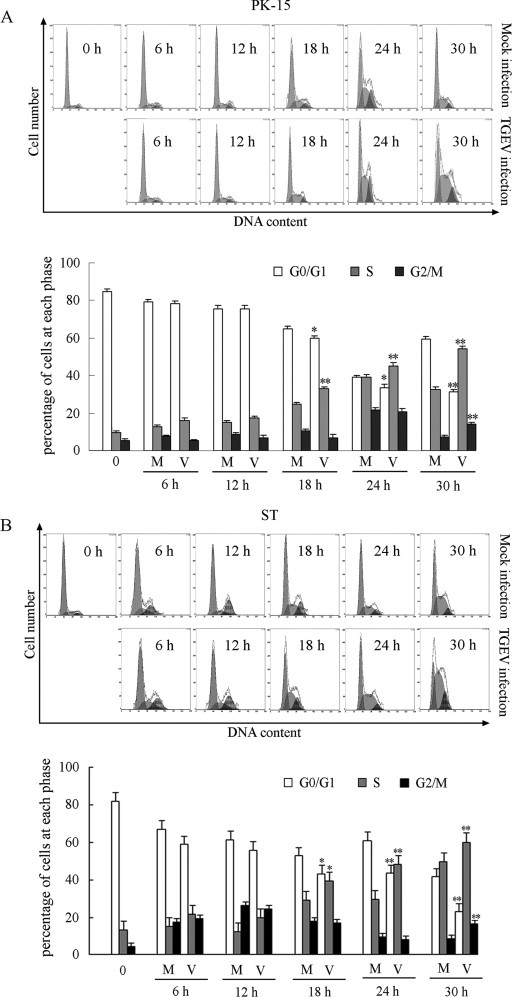
TGEV infection induced quiescent PK-15 and ST cells to accumulate in S and G2/M phase. Serum-starved PK-15 cells (A) and ST cells (B) were mock infected or infected with 0.5 MOI of TGEV. After 1 h of virus adsorption, medium containing 10% FBS was added to the cells, and cell cycle profiles at the indicated times were determined by FACS analysis. The data are from one of three experiments. The histograms were analyzed to determine the percentage of cells in each phase of the cell cycle. The results are shown as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 versus mock infection.
To determine whether TGEV replication may induce the release of some soluble factors to affect the cell cycle progression of uninfected cells, we used high MOIs of TGEV to infect serum-starved quiescent PK-15 cells and detect cell cycle profiles. The results showed that S-phase delay induced by 1 and 5 MOI of TGEV occurred at 18 h p.i., and the number of S-phase arrested cells induced by 1 MOI and 5 MOI of TGEV were approximately two and four folds as many as that by 0.5 MOI TGEV, respectively (Fig. 4A). However, the S-phase and G2/M-phase arrested cells number induced by 1 MOI and 5 MOI of TGEV were not significantly higher than that by 0.5 MOI TGEV at 24 h p.i. (Fig. 4A). These results suggested that TGEV replication just affected the cell cycle progression of infected cells, and that over 1 MOI of TGEV infection would interfere the progression of cell cycle due to the involvement of cell apoptosis.
Fig. 4.
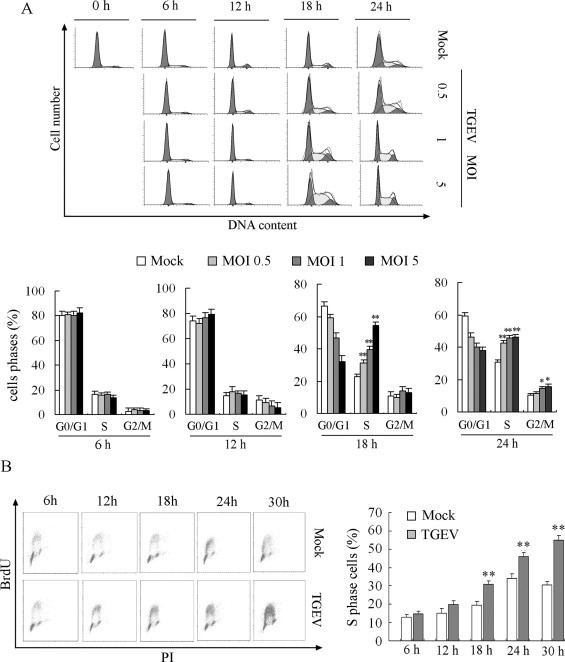
Effects of high MOIs of TGEV on cell cycle progression. (A) Serum-starved PK-15 cells were mock infected or infected with 0.5, 1 and 5 MOI of TGEV. Cell cycle profiles at the indicated times were determined by FACS analysis. The data are from one of three experiments (upper panel). The histograms were analyzed to determine the percentage of cells in each phase of the cell cycle. The results are shown as mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 versus mock infection. (B) Serum-starved PK-15 cells were mock infected or infected with 0.5 MOI of TGEV. Cells were co-stained with BrdU and PI and analyzed using flow cytometry. The histograms were analyzed to determine the percentage of cells in S phase of the cell cycle. S phase, upper gate; G1, lower left; G2, lower right. The results are shown as mean ± SEM of three independent experiments. **P < 0.01 versus mock infection.
To further confirm the changes of TGEV-infected cells in the S phase of cell cycle, we used BrdU incorporation assay to accurately determine the cell number in S phase. Results showed that the proportion of cells in S phases significantly increased at 18 h p.i., and further increased with infection time, when compared to mock-infected cells (Fig. 4B), which were in consistent with the results of PI staining.
3.3. Cdk2, cdc2 and cyclin B1 decreased in TGEV-infected cells
To determine the molecular mechanism underlying the TGEV-induced cell cycle arrest, we detected the levels of associated cell cycle regulatory factors at 0, 4, 8, 12, 18 and 24 h p.i. in TGEV-infected cells. Results showed that cdc2, acting as a main coordinator complexed with cyclin A and cyclin B to advance the cell cycle into M phase, began to decrease at 8 h p.i., and continued to decrease with infection time in infected cells compared to that in mock-infected cells (Fig. 5A), suggesting that the cell cycle might be arrested at G2/M phase in TGEV-infected cells. In addition, the levels of cdk2 significantly decreased from 4 h p.i. in TGEV-infected cells compared to mock-infected cells, but cdk4 and cdk6 did not show significant difference between in mock-infected and TGEV-infected PK-15 cells (Fig. 5A, left panel). The levels of cyclin B1 began to significantly decrease at 12 h p.i. in TGEV-infected PK-15 cells compared to mock-infected cells, but cyclin A, cyclin D and cyclin E did not show significant difference between mock-infected and TGEV-infected PK-15 cells (Fig. 5B, left panel). In parallel experiments, Western blot analysis showed similar or analogous results in the expression of these cdks and cyclins in TGEV-infected ST cells (Fig. 5A and B, right panel). Among them, the reduction of cyclin B1 was more significant in TGEV-infected ST cells than in TGEV-infected PK-15 cells when compared with mock infection. Taken together, these results suggest that TGEV infection lead to an accumulation of cells in the S and G2/M phases of the cell cycle through decreasing certain cell cycle factors that regulate the proceeding of S and G2/M phase.
Fig. 5.

Effect of TGEV infection on the levels of cell cycle-associated proteins. (A and B) PK-15 cells (left panel) and ST cells (right panel) were mock-infected (M) or infected with 0.5 MOI of TGEV (V). At the indicated times, cells were lysed and equal amounts of proteins from the samples were tested by western blot analysis. The same membranes were also probed with β-actin as a loading control. The numbers below the proteins indicated the relative folds of mock infection after normalized to β-actin.
3.4. p53 and p21 mediated TGEV-induced cell cycle arrest
Our previous study showed that TGEV infection induced the accumulation and activation of p53 in cells. Here, we examined the levels of p53 and p21 in 0.5 MOI TGEV-infected cells. Western blot analysis showed that 0.5 MOI of TGEV infection could also increase the protein level of p53 and induce phosphorylation of p53 at serine 15 and 20 in PK-15 and ST cells (Fig. 6A). Consequently, the level of p21 increased in TGEV infected PK-15 and ST cells from 8 h p.i., and significantly increased at 12 h p.i., while proliferating-cell nuclear antigen (PCNA), a mediator involved in p21-regulated DNA replication within S phase, decreased accordingly in TGEV-infected PK-15 and ST cells (Fig. 6A). These results suggest that TGEV infection could up-regulate p21 expression to regulate cell cycle through activation of p53 signaling.
Fig. 6.
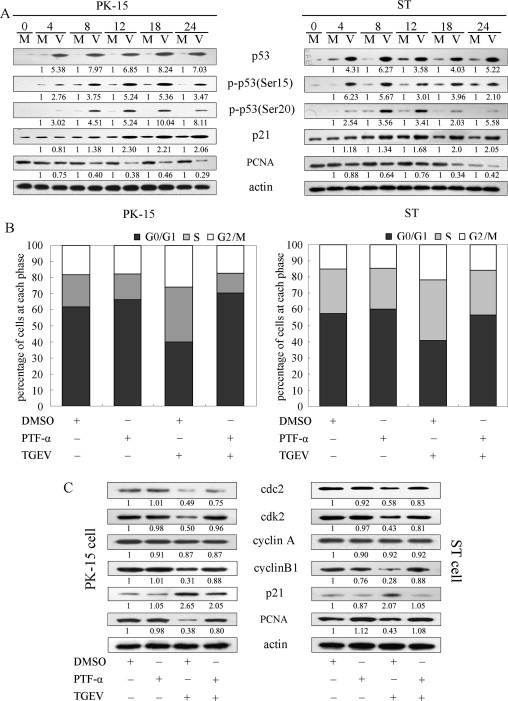
Roles of p53 and p21 in TGEV-induced cell cycle arrest. (A) PK-15 cells (left panel) and ST cells (right panel) were mock infected (M) or infected with 0.5 MOI of TGEV (V). Cells were collected at indicated times and subjected to western blot analysis. The numbers below the proteins indicated the relative folds of mock infection after normalized to β-actin. (B) Effects of PTF-α (p53 inhibitor) on the changes of cell cycle progression induced by TGEV infection. PK-15 cells (left panel) and ST cells (right panel) were pretreated with PTF-α for 1 h, and then co-incubated with TGEV for 18 h. Cells were collected and stained with propidium iodide for FACS analysis. (C) Effect of PTF-α on expression of p21, cdks and cyclins induced by TGEV infection. Cells were treated as in (B), and then collected and subjected to western blot analysis.
To further determine the roles of p53 in TGEV-induced cell cycle arrest, we investigated the effects of PFT-α, a specific inhibitor of p53 that does not affect the mRNA levels of TGEV genes (Huang et al., 2013), on the cell cycle profiles and the expression of p21, cdks and cyclins in TGEV-infected PK-15 and ST cells. As shown in Fig. 6B, pre-incubation of PK-15 and ST cells with PFT-α attenuated cell cycle arrest at S and G2/M phase induced by TGEV infection. In addition, cdc2, cdk2 and cyclin B1 expression increased in TGEV-infected PK-15 and ST cells with PFT-α compared to that with DMSO (Fig. 6C). As expected, pre-incubation of PK-15 and ST cells with PFT-α also attenuated p21 up-regulation and PCNA reduction induced by TGEV infection (Fig. 6C). These results suggest that p53 and p21 might play key roles in mediation of TGEV-induced cell cycle arrest.
3.5. Effects of cell cycle arrest at S and G2/M phases on TGEV replication
Since the proportion of cells in the S and G2/M phases was greater in TGEV infected cells compared to mock-infected cells, to investigate the effects of cell accumulation at S and G2/M phases on TGEV replication, synchronized cells in G2/M phase, G0/G1 phase, G1/S phase or asynchronous cells were simultaneously released from their different phase of cell cycle, and then either mock infected or infected with 0.5 MOI of TGEV, cell cycle profiles were determined by flow cytometry and TGEV gRNA and sgRNAs levels were determined by qRT-PCR at 18 h p.i. Results showed that using serum deprivation treatments, over 85% of PK-15 and 82% of ST cells were synchronized at the G0 phase (Fig. 7A). Using nocodazole treatments, over 90% of PK-15 cells and 80% of ST cells were synchronized at the G2/M phase (Fig. 7B). Using double-thymidine treatment, approximately 80% of cells were synchronized at the G1/S phase border in PK-15 and ST cells (Fig. 7C). In the cells released from the G0-phase-synchronized cells, the proportion of S phase cells was greater in TGEV infected cells compared to mock-infected cells at 18 h p.i. (Fig. 7A), which was in consistent with the results above. In the cells released from the G1/S and G2/M-phase-synchronized cells, the cells population at G2/M phase significantly increased in TGEV-infected cells compared to that in mock-infected cells at 18 h p.i. (Fig. 7B and C).
Fig. 7.
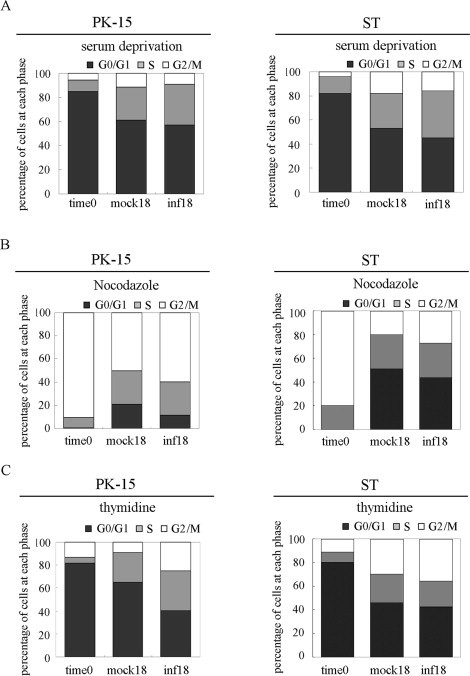
S and G2/M phases synchronization treatment. (A) Serum-starved PK-15 cells (left panel) and ST cells (right panel) were mock infected or infected with TGEV. Cell cycle profiles at 18 h p.i. were determined by FACS analysis. (B) Asynchronously growing PK-15 cells (left panel) and ST cells (right panel) were treated with DMSO or nocodazole for 16 h. Following synchronization (time 0) cells were released from block and simultaneously mock-infected and infected with TGEV. Cell cycle profiles were analyzed at 18 h p.i. (C) Cell cycle profiles of PK-15 cells (left panel) and ST cells (right panel) synchronized at the G1/S phase border using double-thymidine treatment. Following synchronization (time 0) cells were released from block and simultaneously mock infected and infected with TGEV, and cell cycle profiles were analyzed at 18 h p.i.
Real-time RT-PCR analysis of the gRNA and sgRNAs of TGEV showed that the replication levels of gRNA and the synthesis of sgRNA were higher in the cells released from G2/M or G1/S-synchronized cells than that in the cells released from G0/G1 phase-synchronized or asynchronous cells (Fig. 8A). In addition, the titers of the virus also were higher in the cells released from G2/M or G1/S-synchronized cells than that in the cells released from G0/G1 phase-synchronized or asynchronous cells (Fig. 8B). These results suggest that TGEV induction of host cell staying at S and G2/M phase might be beneficial for virus replication.
Fig. 8.
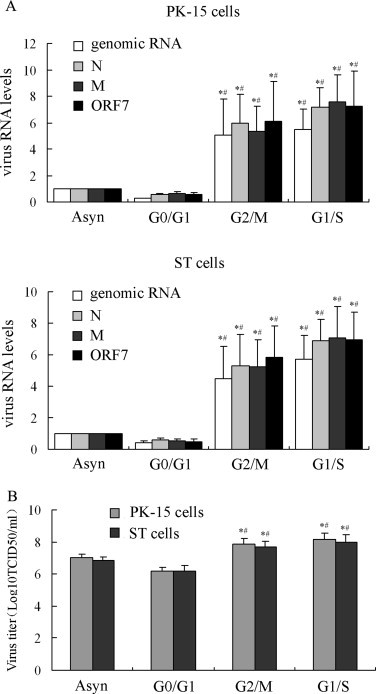
Effects of cell cycle arrest at S and G2/M phases on TGEV replication. (A) TGEV gRNA and sgRNAs levels. TGEV gRNA and sgRNAs levels were determined at 18 h p.i. by qRT-PCR in the cells released from G1/S, G2/M, G0/G1-synchronized cells and asynchronous replicating cells. *P < 0.05 versus the RNA levels of TGEV in G0/G1-released cells. #P < 0.05 versus RNA levels of TGEV in asynchronous cells. (B) The titers of TGEV in different treated cells. Total virus at 18 h p.i. was harvested by freezing and thawing cells three times and the viral titers were shown as log10TCID50/ml. Values are shown as the mean ± SEM. *P < 0.05 versus the titer of TGEV in G0/G1-released cells. #P < 0.05 versus the titer of TGEV in asynchronous cells.
4. Discussion
Cell cycle manipulation is crucial event occurred in viruses-infected cells (Davy and Doorbar, 2007, Kannan et al., 2011). In this study we demonstrated that TGEV-infected cells accumulated in the S and G2/M phase of the cell cycle, and the effect was not cell type specific and could be reproduced in synchronously replicating cells. The cell arrest at the S and G2/M phases was dependent upon TGEV replication and was controlled by viral modulation of certain cyclins/cdks.
Many viruses employ a variety of mechanisms to utilize or manipulate the cell cycle pathways of infected cells (Chowdhury et al., 2003, De Bolle et al., 2004, Li et al., 2007). Throughout the cell cycle, progression is regulated by a series of cyclins and cdks (King and Cidlowski, 1998). The G2/M transition in the cell cycle is positively controlled by complex of cdc2 (CDK1) and cyclin B. Members of the CDKI family of proteins (CDK inhibitors) bind to the CDK/cyclin complexes and inhibit the kinase activity, to regulate the G2/M-phase transition (King and Cidlowski, 1998). Cyclin A-CDK2 complex is the main cyclin-CDK complex in the S phase, and the activity of the complex is required for S phase transition and control of DNA replication (Elledge, 1996). It is possible that the regulation of S and/or G2/M phase is one of the crucial mechanisms that viruses employed to manipulate the cell cycle (Li et al., 2011, Martin-Lluesma et al., 2008). In TGEV-infected cells, we observed that a significant decrease of cdk2, cyclin B1 and cdc2, involved in S phase and G2/M phase transition, which regulated the cell cycle to arrest at the S and G2/M phases.
p53 is a key element in the induction of cell cycle arrest in viruses infected cells (Casavant et al., 2006). As a DNA damage response protein, p53 transcriptionally activates numerous genes involved in DNA repair and cell cycle arrest (Vogelstein et al., 2000), and p53-dependent arrest of cells at G1/S or G2/M phase is an important component of the cellular response to genotoxic stress including virus infection (Casavant et al., 2006, De Bolle et al., 2004). The first transcriptional target of p53 was p21, a CKI of the Cip/Kip family, which bridges the function of p53 with the cell cycle and plays important roles in regulation of cell cycle progression or arrest (He et al., 2005). The p21 is recognized to be an important mediator to delay transit from G1 to S phase and/or from G2 to M phase, thus preventing the effects of DNA damage on gene functions (Abbas and Dutta, 2009). p21 has also been shown to regulate DNA replication within the S phase in vitro, by binding to proliferating-cell nuclear antigen (PCNA)/cdk complexes and by dissociating cyclin A/cdk2/p107/E2F transitional complexes (Abbas and Dutta, 2009). In this study, we showed that TGEV infection induced p53 and p21 accumulation and PCNA decrease. Furthermore, PFT-α, a specific inhibitor of p53, prevented the arrest of the cell cycle in TGEV-infected cells. The observation that TGEV infection induced cell cycle arrest at S and G2/M phases in both PK-15 and ST cells virtually required the possible involvement of p53 in these processes. This observation is not consistent with the findings in other coronavirus-infectious bronchitis virus, which induced cell cycle arrest at both S and G2/M phases independent of p53 (Li et al., 2007), suggesting that p53 might play different roles in viruses-infected cells.
Some viruses can regulate the cell cycle progression through interaction between virus and host cells in order to induce the cell cycle arrest and provide an advantageous environment for viral replication (Dove et al., 2006, Li et al., 2007). Infectious bronchitis virus (IBV) could increase protein accumulation and progeny virus production in cells enriched in the G2/M phase of the cell cycle (Li et al., 2007). Human immunodeficiency virus (HIV) infection also increased the transduction of HIV in the G2/M phase compared to other stages of the cell cycle (Groschel and Bushman, 2005). Many substrates (i.e., nucleotides) in S phase are abundantly available for viral replication (Yang et al., 2006). Furthermore, there is evidence that one consequence of G2/M arrest is to establish a pseudo-S phase state for viruses to replicate of their genomes (Belyavskyi et al., 1998). To specifically investigate whether a particular stage of the cell cycle favored TGEV replication, we synchronized cells in various stages of the cell cycle and compared efficiencies of virus infection. Results showed that TGEV replication levels, sgRNA synthesis were greater in the cells released from G1/S phase or G2/M phase of the cell cycle after 18 h p.i., when compared to either cells released from the G0/G1 phase or asynchronous cells. Our results also provided evidences that TGEV titer in the cells released from G1/S phase or G2/M phase synchronized cells was higher than that in cells released from G0/G1 phase synchronized cells. This might be that cells synchronized at G0/G1 phase need longer time to reenter cell cycle progression than synchronized cells at the G1/S phase, which support the hypothesis that S phase state was beneficial for virus replication. Therefore, we favor the hypothesis that TGEV induces S and G2/M phase cell cycle arrest in order to provide a more favorable condition for virus production.
In conclusion, our study revealed that TGEV infection induced the host cells to arrest at S and G2/M phases of the cell cycle, which were responsible by the certain key players in the S and G2/M transition. Moreover, the cell cycle arrest induced by TGEV infection might provide a more favorable condition for viral replication.
Contributors and authorship
Li Ding and Yong Huang designed the experiments and interpreted the data and wrote the article. Li Ding performed the experiments with assistance and advice from Meiling Dai, Xiaomin Zhao, Qian Du, Feng Dong, Lili Wang, Ruichao Huo, Wenlong Zhang and Xingang Xu. Yong Huang and Dewen Tong revised the manuscript. All authors have read the manuscript and approved to submit it to your journal.
Conflict of interest
There is no conflict of interest of any authors in relation to the submission.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 31072108/C1802), the Doctoral Program of Higher Education of China (Grant No. 20110204110014), and the Scientific Research Program of Northwest A&F University (No. Z111021103).
References
- Abbas T., Dutta A. p21 in cancer: intricate networks and multiple activities. Nature Reviews Cancer. 2009;9(6):400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyavskyi M., Braunagel S.C., Summers M.D. The structural protein ODV-EC27 of Autographa californica nucleopolyhedrovirus is a multifunctional viral cyclin. Proceedings of the National Academy of Sciences. 1998;95(19):11205–11210. doi: 10.1073/pnas.95.19.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casavant N., Luo M., Rosenke K., Winegardner T., Zurawska A., Fortunato E. Potential role for p53 in the permissive life cycle of human cytomegalovirus. Journal of Virology. 2006;80(17):8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-J., Makino S. Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. Journal of Virology. 2004;78(11):5658–5669. doi: 10.1128/JVI.78.11.5658-5669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury I.H., Wang X.F., Landau N.R., Robb M.L., Polonis V.R., Birx D.L., Kim J.H. HIV-1 Vpr activates cell cycle inhibitor p21/Waf1/Cip1: a potential mechanism of G2/M cell cycle arrest. Virology. 2003;305(2):371–377. doi: 10.1006/viro.2002.1777. [DOI] [PubMed] [Google Scholar]
- Davy C., Doorbar J. G2/M cell cycle arrest in the life cycle of viruses. Virology. 2007;368(2):219–226. doi: 10.1016/j.virol.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bolle L., Hatse S., Verbeken E., De Clercq E., Naesens L. Human herpesvirus 6 infection arrests cord blood mononuclear cells in G2 phase of the cell cycle. FEBS Letters. 2004;560(1):25–29. doi: 10.1016/S0014-5793(04)00035-3. [DOI] [PubMed] [Google Scholar]
- Ding L., Chen G.D., Xu X.G., Tong D.W. Isolation and identification of porcine transmissible gastroenteritis virus Shaanxi strain and clone and sequence analysis of its gene. Chinese Journal of Veterinary Medicine. 2011;47(10):9–12. [Google Scholar]
- Ding L., Xu X., Huang Y., Li Z., Zhang K., Chen G., Yu G., Wang Z., Li W., Tong D. Transmissible gastroenteritis virus infection induces apoptosis through FasL-and mitochondria-mediated pathways. Veterinary Microbiology. 2012;158(1):12–22. doi: 10.1016/j.vetmic.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove B., Brooks G., Bicknell K., Wurm T., Hiscox J.A. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. Journal of Virology. 2006;80(8):4147–4156. doi: 10.1128/JVI.80.8.4147-4156.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour D., Mateos-Gomez P.A., Enjuanes L., Gallego J., Sola I. Structure and functional relevance of a transcription-regulating sequence involved in coronavirus discontinuous RNA synthesis. Journal of Virology. 2011;85(10):4963–4973. doi: 10.1128/JVI.02317-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge S.J. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274(5293):1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Groschel B., Bushman F. Cell cycle arrest in G2/M promotes early steps of infection by human immunodeficiency virus. Journal of Virology. 2005;79(9):5695–5704. doi: 10.1128/JVI.79.9.5695-5704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G., Siddik Z.H., Huang Z., Wang R., Koomen J., Kobayashi R., Khokhar A.R., Kuang J. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene. 2005;24(18):2929–2943. doi: 10.1038/sj.onc.1208474. [DOI] [PubMed] [Google Scholar]
- He Y., Xu K., Keiner B., Zhou J., Czudai V., Li T., Chen Z., Liu J., Klenk H.-D., Shu Y.L. Influenza A virus replication induces cell cycle arrest in G0/G1 phase. Journal of Virology. 2010;84(24):12832–12840. doi: 10.1128/JVI.01216-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Ding L., Li Z., Dai M., Zhao X., Li W., Du Q., Xu X., Tong D. Transmissible gastroenteritis virus infection induces cell apoptosis via activation of p53 signaling. Journal of General Virology. 2013;94:1807–1817. doi: 10.1099/vir.0.051557-0. [DOI] [PubMed] [Google Scholar]
- Kannan R.P., Hensley L.L., Evers L.E., Lemon S.M., McGivern D.R. Hepatitis C virus infection causes cell cycle arrest at the level of initiation of mitosis. Journal of Virology. 2011;85(16):7989–8001. doi: 10.1128/JVI.00280-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K., Cidlowski J. Cell cycle regulation and apoptosis 1. Annual Review of Physiology. 1998;60(1):601–617. doi: 10.1146/annurev.physiol.60.1.601. [DOI] [PubMed] [Google Scholar]
- Li F.Q., Tam J.P., Liu D.X. Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology. 2007;365(2):435–445. doi: 10.1016/j.virol.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Gu B., Zhou F., Chi J., Wang F., Peng G., Xie F., Qing J., Feng D., Lu S. Human herpesvirus 6 suppresses T cell proliferation through induction of cell cycle arrest in infected cells in the G2/M phase. Journal of Virology. 2011;85(13):6774–6783. doi: 10.1128/JVI.02577-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M., Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature Reviews Cancer. 2009;9(3):153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Martin-Lluesma S., Schaeffer C., Robert E.I., Van Breugel P.C., Leupin O., Hantz O., Strubin M. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology. 2008;48(5):1467–1476. doi: 10.1002/hep.22542. [DOI] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty per cent endpoints. American Journal of Epidemiology. 1938;27(3):493–497. [Google Scholar]
- Surjit M., Liu B., Chow V.T., Lal S.K. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. Journal of Biological Chemistry. 2006;281(16):10669–10681. doi: 10.1074/jbc.M509233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B., Lane D., Levine A.J. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Yang X.-J., Liu J., Ye L., Liao Q.-J., Wu J.-G., Gao J.-R., She Y.-L., Wu Z.-H., Ye L.-B. HCV NS2 protein inhibits cell proliferation and induces cell cycle arrest in the S-phase in mammalian cells through down-regulation of cyclin A expression. Virus Research. 2006;121(2):134–143. doi: 10.1016/j.virusres.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Zimmerman E.S., Sherman M.P., Blackett J.L., Neidleman J.A., Kreis C., Mundt P., Williams S.A., Warmerdam M., Kahn J., Hecht F.M. Human immunodeficiency virus type 1 Vpr induces DNA replication stress in vitro and in vivo. Journal of Virology. 2006;80(21):10407–10418. doi: 10.1128/JVI.01212-06. [DOI] [PMC free article] [PubMed] [Google Scholar]