

Highlights
-
•
We cloned a DENV-2 infectious cDNA into a BAC under the control of the CMV promoter.
-
•
We assessed the production of infectious particles.
-
•
We rescued infectious viruses after serial passages in C6/36 cells.
-
•
Parental and recombinant viruses were similar in plaque and syncytia phenotypes.
Keywords: Infectious cDNA, Dengue virus, Flavivirus, Bacterial artificial chromosome, Eukaryotic promoter, Reverse genetics
Abstract
Major progress in Dengue virus (DENV) biology has resulted from the use of infectious clones obtained through reverse genetics. The construction of these clones is commonly based on high- or low-copy number plasmids, yeast artificial chromosomes, yeast-Escherichia coli shuttle vectors, and bacterial artificial chromosomes (BACs). Prokaryotic promoters have consistently been used for the transcription of these clones. The goal of this study was to develop a novel DENV infectious clone in a BAC under the control of the cytomegalovirus immediate-early promoter and to generate a virus with the fusion envelope-green fluorescent protein in an attempt to track virus infection. The transfection of Vero cells with a plasmid encoding the DENV infectious clone facilitated the recovery of infectious particles that increased in titer after serial passages in C6/36 cells. The plaque size and syncytia phenotypes of the recombinant virus were similar to those of the parental virus. Despite the observation of autonomous replication and the detection of low levels of viral genome after two passages, the insertion of green fluorescent protein and Renilla luciferase reporter genes negatively impacted virus rescue. To the best of our knowledge, this is the first study using a DENV infectious clone under the control of the cytomegalovirus promoter to facilitate the recovery of recombinant viruses without the need for in vitro transcription. This novel molecular clone will be useful for establishing the molecular basis of replication, assembly, and pathogenesis, evaluating potential antiviral drugs, and the development of vaccine candidates for attenuated recombinant viruses.
1. Introduction
Dengue has become the main arthropod-borne viral disease in several tropical and subtropical countries, causing more than 50–100 million cases and 500,000 hospitalizations annually around the world (Guzman et al., 2010). Disease manifestations cover a wide spectrum ranging from dengue with or without warning signs to severe dengue, in which plasma leakage, hemorrhage and organ impairment can lead to death (TDR/WHO, 2009). The disease is caused by one of four Dengue virus (DENV) serotypes (DENV-1 to -4), which are enveloped, single-stranded and positive-sense RNA viruses belonging to the Flavivirus genus within the Flaviviridae family (Lindenbach et al., 2007). These viruses possess a short genome of approximately 10.7 kb with a type 1 cap structure (m7GpppAmpN2) at the 5′end, two highly structured untranslated regions (UTRs) at the 5′ and 3′ ends, a long open reading frame (ORF) encoding a single viral polyprotein and lacking a polyadenylate tail at the 3′ end. After proteolytic processing, three structural and seven non-structural proteins are generated from the viral polyprotein (Bartenschlager and Miller, 2008).
Several cis-acting elements at the 5′UTR [stem-loops SLA and SLB, upstream the AUG region (5′UAR)], at the capsid-coding region [downstream AUG region (5′DAR), capsid-hairpin (cHP), 5′ cyclization sequence (5′CS), downstream 5′CS (dCS), CCR1] and at the 3′UTR [highly variable region (HVR), semi-variable region (SVR), pseudoknots (PK2 and PK1), 3′CS, 3′DAR, 3′UAR and 3′ stem-loop (3′SL)] are critical for virus replication, translation or assembly (Friebe and Harris, 2010, Friebe et al., 2012, Groat-Carmona et al., 2012, Paranjape and Harris, 2010).
The role of the DENV proteins and the UTRs of the RNA genome in viral genome replication (Lindenbach et al., 2007), post-translational cleavage (Falgout et al., 1991), virion morphogenesis [reviewed in (Murray et al., 2008)], regulation of the host immune response (Munoz-Jordan et al., 2003, Munoz-Jordan et al., 2005) and virus entry into susceptible cells through receptor-mediated endocytosis (Crill and Roehrig, 2001, Hsieh et al., 2011) and membrane fusion for the release of virions into the cytoplasm (Modis et al., 2004) have been partially clarified.
Although there is not an effective and available antiviral therapy or vaccine and many aspects of the DENV biology remain unclear, the major progress during the last two decades in the study of this virus, particularly in identifying cis-acting elements regulating translation and replication, has resulted from the use of infectious clones and replicons obtained through reverse genetics (Paranjape and Harris, 2010). This technology has also been important in identifying and evaluating adaptive mutations and virulence determinants, due to the possibility of generating mutations in specific viral genes or RNA elements, whose effect on viral RNA replication and pathogenesis can be assessed in vitro and in vivo, respectively (Clyde et al., 2008, Filomatori et al., 2011, Friebe and Harris, 2010, Grant et al., 2011, Iglesias et al., 2011, Leardkamolkarn et al., 2012, Lodeiro et al., 2009, Yu et al., 2008). Infectious clones and derived replicons lacking structural components necessary for virus assembly and propagation have also shown potential for the development and characterization of live-attenuated viruses as vaccine candidates (Huang et al., 2003, Kelly et al., 2010, Zhu et al., 2007), genetic vaccines (Pang et al., 2001a) and high-throughput platforms for screening of chemical antiviral compounds (Chao et al., 2012, Hsu et al., 2012, Leardkamolkarn and Sirigulpanit, 2012, Qing et al., 2010, Zou et al., 2011) and siRNAs (Ng et al., 2007). In addition, infectious clones with reporter genes fused to some of the structural proteins become a powerful tool in cell biology of viral infection for subcellular localization of viral proteins and for tracking the viral particles during the entry, maturation and exit steps of the vital cycle (Brandenburg and Zhuang, 2007).
Common strategies for the construction of infectious cDNAs from DENV genomes through reverse genetics have been based on standard high-copy and low-copy number plasmids (Gualano et al., 1998, Kinney et al., 1997, Lai et al., 1991, Sriburi et al., 2001). However, the high instability of the full-length clones leading to rearrangements and unsuccessful clone rescue has been a common problem when using some of these backbones (Suzuki et al., 2007). To overcome this problem, yeast artificial chromosomes and yeast-Escherichia coli shuttle vectors have been successfully used with higher stability (Kelly et al., 2010, Kelly et al., 2011, Polo et al., 1997, Pu et al., 2011, Puri et al., 2000). More recently, DENV-1 and -2 infectious cDNAs have been cloned into bacterial artificial chromosomes (BACs) (de Borba et al., 2012, Pierro et al., 2006, Suzuki et al., 2007), a system facilitating the stable cloning of inserts up to 300 kb, as there are typically only one or two copies of the BAC per cell (Kim et al., 1996, Shizuya et al., 1992). However, these DENV infectious clones have been constructed under the control of the prokaryotic T7 promoter, requiring in vitro transcription (Run-off) and subsequent transfection of the synthesized RNAs. One decade ago, the first stable cloning of the transmissible gastroenteritis virus (TGEV) under the transcriptional control of the eukaryotic cytomegalovirus (CMV) immediate-early promoter was demonstrated (Almazan et al., 2000). TGEV, as a member of the coronavirus genus, has one of the largest RNA genomes of approximately 30 kb in length, thus demonstrating the functional stable cloning of that genome into infectious cDNAs (Lai, 2000) and the opportunity for DNA-launched transcription from an eukaryotic promoter, avoiding expensive in vitro transcription and the subsequent manipulation of transcribed RNAs for transfection.
In the present study, we developed a new infectious cDNA for DENV-2, stably cloned into the BAC system under the control of the CMV promoter. The generated infectious cDNA was successfully rescued and constitutes a useful tool for improving our knowledge of the role of structural and non-structural proteins during the life cycle of DENV, as well as the role of naturally occurring mutations generated in circulating strains during epidemic spread, because of the possibility of evaluating their direct effect on virus replication, assembly and pathogenesis, through the use of isogenic variants with punctual mutations. Additionally, EGFP-bearing clone of DENV-2 was constructed, which expanded our knowledge of the constrictions of the viral genome for the generation of reporter viruses.
2. Materials and methods
2.1. Cells and viruses
The sequence of the DENV type 2 New Guinea C prototype strain (DENV-2 NGC), originally isolated from a clinical case of dengue fever in New Guinea in 1944 (GenBank accession number: EU854293), was used to assemble the infectious clone. African Green Monkey kidney (Vero) cells were obtained from the American Type Culture Collection (ATCC number: CCL-81) and maintained as low passage cultures (up to passage 10) in Dulbecco's Modified Eagle Medium (DMEM) (Gibco BRL, Carlsbad, CA, USA), supplemented with 2–10% heat-inactivated fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin and incubated at 37 °C, 5% CO2 atmosphere and 95% relative humidity. C6/36 cells, derived from whole larvae of Aedes albopictus (ATCC number: CRL-1660), were cultured in Leibovitz's L-15 medium (Gibco BRL, Carlsbad, CA, USA), supplemented with 2–10% FBS and 20 mg/mL tryptose phosphate broth and incubated at 28 °C, 5% CO2 atmosphere and 95% relative humidity. A viral stock of DENV-2 NGC M2 was produced by inoculating C6/36 cells at a multiplicity of infection (MOI) of 0.01 plaque-forming units (PFU)/cell. The supernatants were collected at 96 h postinfection (hpi), cleared from cell debris through low-speed centrifugation, aliquoted and stored at −80 °C. For virus titration using a plaque assay, Vero cells were infected with serially diluted virus supernatants, incubated at 37 °C with an overlay containing carboxymethylcellulose, fixed with paraformaldehyde and stained with Crystal Violet at 9 days post-infection (dpi), as previously described (Martinez-Gutierrez et al., 2011). Because of the small plaque size phenotype of the parental DENV-2 NGC M2 strain and the derived infectious clone, the plaques were counted and captured using an inverted light microscope with minimal magnification (4× objective). The ImagePro Plus 3.1 software (Media Cybernetics, Silver Springs, MD, USA) was used to delimitate the plaques, and their diameters were estimated by ruling the images with a scale obtained from the microscope configuration.
2.2. Plasmids and bacterial strains
The plasmids pBeloBAC11 (Kim et al., 1996, Shizuya et al., 1992) and pBAC-TGEV5′-3′ (St-Jean et al., 2006), kindly provided by Dr. Luis Enjuanes (Centro Nacional de Biotecnología CNB-CSIC, Madrid, Spain), were used for assembling the infectious DENV cDNA clone flanked by the cytomegalovirus (CMV) immediate-early promoter, the Hepatitis Delta Virus Ribozyme (HDV-RZ) and the bovine growth hormone (BGH) termination and polyadenylation signal sequences. BAC propagation and manipulation was performed in E. coli DH10B (Gibco BRL, Carlsbad, CA, USA) as previously described (Almazan et al., 2008). The enhanced green fluorescence protein (EGFP) and synthetic Renilla Luciferase (hRLuc) genes were amplified from plasmids pEGFP-N2 (BD Biosciences Clontech, Palo Alto, CA, USA) and psiCHECK-2 (Promega Corp., Madison, WI, USA), respectively. Plasmid DNA purification was performed using QIAprep Spin Miniprep and QIAGEN Large-Construct Kits (QIAGEN GmbH, Hilden, Germany). QIAquick PCR purification kit (Qiagen®, Chatsworth, CA, USA) was used to purify highly specific PCR products. Restriction endonuclease-digested vectors and inserts exceeding 4 kb were purified through QIAEX II gel extraction kit (Qiagen®, Chatsworth, CA, USA).
2.3. Viral RNA extraction and cDNA synthesis
Viral RNA was isolated from supernatants of DENV-infected C6/36 cells using QIAamp Viral RNA minikit (Qiagen®, Chatsworth, CA, USA), according to the manufacturer's instructions. SuperScript® III First-Strand Synthesis System (Life Technologies Corp., Carlsbad, CA, USA) was used to generate cDNAs, although some modifications were implemented depending on the specific downstream requirements. Viral RNA (∼1 μg) was mixed with 10 to 50 ng of random hexamers or 10 pmol of a gene-specific primer, 1 mM of each dNTP and DEPC-treated water to a final volume of 10 μL, incubated at 65 °C for 5 min and immediately transferred to an ice–water bath for 2 min. After mixing with the other RT components, final concentrations were: 1X RT buffer, 0.5 to 2.5 ng/μL random hexamers or 0.5 μM gene-specific primer, 5 mM MgCl2, 0.5 mM each dNTP, 10 mM DTT, 40 units RNAseOUT™ and 200 units SuperScript III® RT. Finally, 1 μL of RNAse H was added to each reaction and incubated at 37 °C for 20 min. Lower concentration of random hexamers during cDNA synthesis allowed the amplification of larger DNA fragments (approximately 4 kb in length). Additionally, annealing of a specific primer on the last 27 nucleotides of the viral RNA genome during cDNA synthesis permitted amplification of the 3′ end of DENV genome.
2.4. Cloning strategy for construction of a DENV infectious cDNA under the control of the CMV promoter
The BAC clone of the full-length DENV genome under the control of the CMV promoter (pBAC-DENV-FL) was generated through PCR amplification, overlapping PCR and the sequential cloning of the amplified products as shown in Fig. 1 . The first step was the selection of naturally occurring unique recognition sites in the DENV genome spaced at approximately 3 to 4 kb. The selected Sph I (at position 1385), Nsi I (at position 4701) and Pml I (at position 8834) restriction sites were absent in pBeloBAC11. The second and third steps were the generation (PCR 1, 2, 5, and 6, overlapping PCR 1 and 2) and cloning of the fusion fragments (Cloning 1 and 2). The fusion of the CMV promoter sequence to the DENV 5′UTR and the partial structural region (nt 1 to 1384) was performed through overlapping PCR containing both fragments in equimolecular quantities (up to 100 ng of the larger PCR product) and forward (CMV5) and reverse (DEN2L3) primers of the first and second fragment to be fused, respectively. Thermal conditions included an optimal annealing temperature for the overlapping region, and initial amplification (1 to 10 cycles) in the absence of primers to allow the annealing and extension of the single-stranded templates. Cloning 1 facilitated the insertion of a multiple cloning site containing the restriction sites Sph I, Nsi I, Pml I and Hind III, used in the subsequent assembly. The last part of the non-structural region and the 3′UTR (nt 8835 to 10723) fused to HDV-RZ and BGH termination and polyadenylation signals to stabilize the mRNA transcribed from the CMV promoter and for proper processing to generate the DENV 3′ end, was obtained by overlapping PCR using the primers DEN2L5 and BGH3 and cloned in the previously described construct by using Pml I and Hind III (Cloning 2). The fourth and fifth steps involved the assembly of the full-length DENV infectious clone through amplification of two internal genome fragments [PCR 3 comprising the remaining structural region and the partial non-structural region (nt 1385 to 4701) and PCR 4 spanning most of the non-structural region (nt 4702 to 8834)], which were independently cloned into pBeloBAC11, sequenced and subsequently used in the corresponding cloning step (Cloning 3 and 4). The oligonucleotides used for PCR amplification and sequencing (Table 1 ) were designed using PrimerSelect module from LaserGene® version 7.2.1 (DNASTAR Inc. 2007; Madison, WI, USA.). Oligonucleotide BGH3 was designed to eliminate the natural Sph I restriction site at the termination and polyadenylation signals of BGH gene to avoid interference with the cloning strategy. The designs were assessed to verify the secondary structure at the 5′- and 3′-UTRs, and the absence of changes in the ORF or stop codons in the coding region. All fragments were amplified through PCR using the Platinum® Pfx DNA polymerase (Life Technologies Corp., Carlsbad, CA, USA). For cloning, the digested plasmids were dephosphorylated by using shrimp alkaline phosphatase (Promega Corp., Madison, WI, USA), ligated at different equimolecular quantities through T4 DNA ligase (New England BioLabs Inc., Ipswich, MA, USA) and used to transform electrocompetent DH10B as previously described (Almazan et al., 2008). Depending on the construct, the identification of recombinant plasmids was performed by blue/white colony screening, restriction analysis or PCR, and sequencing of at least five clones using the corresponding sequencing oligonucleotides (Suppl. Tables 1–4) before the next cloning step.
Fig. 1.
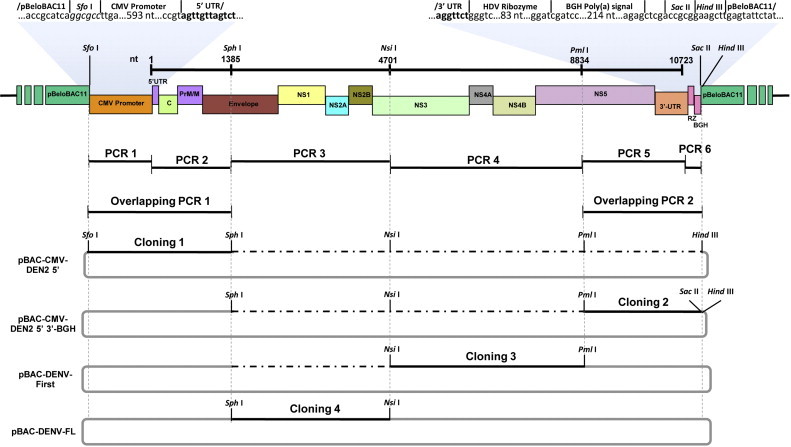
Strategy to assemble a DENV infectious cDNA clone as a BAC: A four-step cloning strategy, in which the overlapping cDNA fragments, termed overlapping PCR 1, PCR 2, PCR 3 and PCR 4, were sequentially cloned into the plasmid pBeloBAC11 and used to generate the infectious clone pBAC-DENV-FL. This plasmid includes the CMV promoter precisely joined to the DENV genome, followed by the hepatitis delta virus ribozyme (RZ) and the bovine growth hormone termination and polyadenylation sequences (BGH). The genomic positions of the relevant restriction sites used for the assembly of the full-length cDNA clone are illustrated at the top.
Table 1.
Oligonucleotides used for the amplification, cloning and quantification of partial DENV genome fragments.
| Oligonucleotide name | Orientation | Restriction endonucleases | Amplicon name | Sequence (5′ → 3′)a, b |
|---|---|---|---|---|
| CMV5 | F | Sfo I | CMV, CMV-DEN2 5′ | aatgatggcgccttgacattgattattgactagttattaatag |
| CMV3 | R | CMV | ggtccacgtagactaacaactacggttcactaaacgagctctgc | |
| DEN2L5 | F | DEN2 5′ | agttgttagtctacgtggaccgacaaagacag | |
| DEN2L3 | R | Hind III; Pml I; Nsi I; Sph I | DEN2 5′, CMV-DEN2 5′ | cgcgaagctttggccacgtgcaggatgcatgactgcatgctcttcccctgagtgaggtg |
| DEN2R5 | F | Pml I | DEN2 3′, DEN2 3′-RZ-BGH | gaagtcggcacgtgaggctgttgaagatagt |
| 3UTR-RZ-R | R | DEN2 3′ | atgccgacccagaacctgttgattcaacagcaccatt | |
| RZ-F | F | RZ-BGH | aacaggttctgggtcggcatggcatctccacctcctcg | |
| BGH3 | R | Hind III; Sac II | RZ-BGH, DEN2 3′-RZ-BGH | gccctaagcttccgcggtcgagctctccccaAcatgcctgctattgtcc |
| DENV1385-4701F | F | Sfo I | 1385-4701, 1385-ENV | aatgatggcgccggaatacaccattgtgataac |
| DENV1385-4701R | R | Sph I | 1385-4701, NS1-4701 | gaagagcatgctggttcaatcctctttcctttat |
| DENV4702-8834F | F | Sph I | 4702-8834 | gaagagcatgcccagatcggagccggagtttacaa |
| DENV4702-8834R | R | Sph I | 4702-8834 | gaagagcatgcaccagctcccaaaacctactatct |
| NS1-F | F | Sfo I; Mlu I | NS1-4701 | aatgatggcgccgcgcacgcgtacgctgtatttgggagttat |
| ENV-R | R | Mlu I | 1385-ENV | gcgcacgcgtggcctgcaccataactcccaaat |
| PEGFP-F | F | Mlu I | EGFP | gcgcacgcgtatggtgagcaagggcgaggag |
| PEGFP-R | R | Mlu I | EGFP | gcgcacgcgtcttgtacagctcgtccat |
| hRLuc-F | F | Mlu I | hRLuc | gcgcacgcgtatggcttccaaggtgta |
| hRLuc-R | R | Mlu I | hRLuc | gcgcacgcgttaactgctcgttcttcag |
| NS5_F | F | NS5 | tcacaccatttccatgagttaatca | |
| NS5_R | R | NS5 | cgggctctaccaatcagttca | |
| NS5_DENV (probe) | F | FAM-ccgcgtacttgtagttccatgcagaaacc |
The underlined sequences correspond to recognition sites for the restriction endonucleases used in the cloning strategy.
The boldface sequences correspond to oligonucleotide regions that hybridize with the DENV genome.
The uppercase letter in the BGH3 sequence corresponds to a punctual mutation introduced to eliminate the Sph I restriction site. F: forward; R: reverse; FAM: 6-carboxyfluorescein.
2.5. Insertion of EGFP and hRLuc reporter genes between viral E and NS1 genes into the DENV infectious cDNA
The EGFP and hRLuc genes were cloned into the envelope-nonstructural 1 (E-NS1) intergenic region of the DENV infectious cDNA as shown in Fig. 4. The recognition peptide for cleavage through the cellular signalase present in the lumen of the endoplasmic reticulum was conserved in the 5′ end of the NS1 gene (Chambers et al., 1990), while the recognition sequence at the 3′ end of the E gene was not conserved, facilitating the expression of the reporters as fusion proteins with E protein (E-EGFP and E-hRLuc) after polyprotein processing. The cloning was performed in three steps. The first step involved the selection of the appropriate restriction site (Mlu I) for insertion of the reporter gene. The second step involved the PCR amplification of the 1385-ENV, EGFP/hRLuc and NS1-4701 fragments containing the Sph I, Mlu I, or Nsi I at the ends (PCR1 to 3). The third step involved the sequential cloning (Cloning 1 to 3) and replacement of the DENV1385-4701 fragment in the pBAC-DENV-FL using Sph I and Nsi I restriction sites (Subcloning 1) to obtain pBAC-DENV-FL-GFP or pBAC-DENV-FL-LUC. The oligonucleotides for the amplification and sequential insertion of EGFP and hRLuc reporter genes are shown in Table 1. The oligonucleotides used for sequencing are shown in Suppl. Tables 5–7.
Fig. 4.
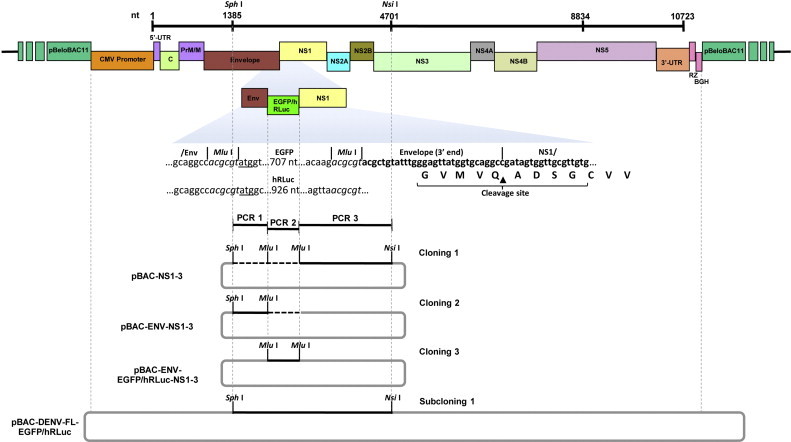
Strategy for the construction of EGFP- and hRLuc-bearing DENV infectious clones: A three-step cloning strategy, in which three overlapping cDNA fragments (PCR1 to PCR3) were sequentially cloned into the plasmid pBeloBAC11 and used to generate the plasmid pBAC-ENV-GFP/hRLuc-NS1-3. After verifying the nucleotide sequence, the Sph I-Nsi I fragment was cloned into pBAC-DENV-FL to generate pBAC-DENV-FL-GFP and pBAC-DENV-FL-LUC. The genetic structure of the DENV infectious clones expressing EGFP or hRLuc, the position of relevant restriction sites and the cleavage site for host cell signalase are illustrated.
2.6. Transfection and recovery of infectious viruses from the cDNA clones
Vero cells grown to 90% confluence in 24-well plates were transfected with 2 μg of the plasmid pBAC-DENV-FL, pBAC-DENV-FL-GFP or pBAC-DENV-FL-LUC, using 2 μL of Lipofectamine™ 2000 (Life Technologies Corp., Carlsbad, CA, USA) according to manufacturer's instructions. After an incubation period of 6 h at 37 °C, the transfection media were replaced and cells were incubated at 37 °C for 48 h. Cell supernatants were harvested and passaged twice on fresh C6/36 cells, and the recovered viruses of all three passages were analyzed as described below.
2.7. Analysis of viral RNA synthesis by two-step RT-qPCR
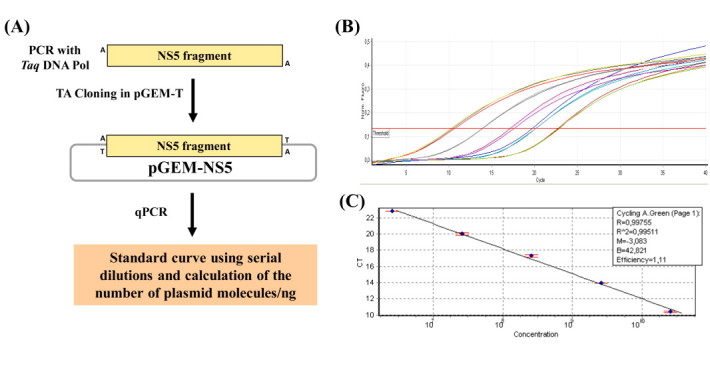
Viral RNAs from transfected/infected cell culture supernatants and intracellular viral RNAs were extracted with the QIAamp Viral RNA minikit and the RNeasy minikit (Qiagen®, Chatsworth, CA, USA), respectively, according to the manufacturer's instructions. All RNAs were treated with RQ1 RNase-free DNase I (Promega Corp., Madison, WI, USA) before use as templates for cDNA synthesis. For reverse transcription, the GoScript reverse transcriptase (Promega Corp., Madison, WI, USA) was used according to the manufacturer's instructions. The oligonucleotides NS5_F and NS5_R and the TaqMan® MGB™probe NS5_DENV, used for quantitative amplification through qPCR, were designed using the Primer Express® v2.0 software (Applied Biosystems, Foster City, CA, USA) (Table 1). The Rotor-Gene® Q Real-Time PCR system (Qiagen®, Chatsworth, CA, USA) was used for amplification and subsequent quantitative analyses. For the relative quantification of the viral RNA levels in the cellular lysates, the 2−ΔΔCT method (Livak and Schmittgen, 2001), normalized to the endogenous 18S ribosomal RNA (Applied Biosystems, Foster City, CA, USA) reference gene, was used. For the absolute quantification of viral RNA copies in the cell culture supernatants, a standard plasmid was constructed by cloning the DENV NS5 region, comprising the target for oligonucleotides and probe hybridization into the pGEM®-T Easy vector system (Promega Corp., Madison, WI, USA) (Suppl. Fig. 1).
2.8. Immunofluorescence and confocal microscopy
At different times post-transfection, Vero cells grown on coverslips in 24-well plates were fixed with paraformaldehyde 3.8% in cytoskeletal buffer for 30 min at 37 °C and treated as previously described (Gallego-Gomez et al., 2003, Martinez-Gutierrez et al., 2011). Cell cultures transfected with EGFP-containing constructs were stained with Hoechst (nuclei), while the other cells were stained with the 4G2 mouse anti-E (flavivirus) monoclonal antibody (Henchal et al., 1982) or the anti-E (DENV-2) polyclonal antibody (2133) (kindly provided by Dr. Eva Harris). The 4G2 monoclonal antibodies were collected from a hybridoma cell line (ATCC number: HB-112) cultured in ATCC Hybri-Care, supplemented with 10% FBS, incubated at 37 °C in a 5% CO2 atmosphere and 95% relative humidity. The images were obtained using a Spinning Disk Confocal microscope IX-81 (Olympus Corporation, Tokyo, Japan) equipped with a IX2-DSU (disk scanning unit) module (Olympus Europa Holding GmbH, Hamburg, Germany) and a digital CCD camera ORCA-R 2 (Hamamatsu Photonics K. K., Japan) and analyzed using Xcellence Pro 1.2 software (Olympus Soft Imaging Solutions GmbH, Münster, Germany).
2.9. Renilla luciferase activity assay
Vero cells cultured in 6-well plates and transfected with pBAC-DENV-FL-LUC were lysed with 500 μL of lysis buffer from the Renilla Luciferase assay system (Promega Corp., Madison, WI, USA) at 24 and 48 h post-transfection (hpt) according to the manufacturer's instructions. Subsequently, a 20-μL aliquot of the lysate was mixed with 100 μL of the Assay Reagent, and the luminescence was quantified in a GloMax96 microplate luminometer (Promega Corp., Madison, WI, USA).
2.10. Statistical analysis
For quantitative analyses, the experiments were performed independently three times, using three replicates for each experimental point. The results were expressed as the means ± standard error of the mean (SEM). Student's t-test (two-tailed) was used to determine the significance of the mean differences.
3. Results
3.1. Successful construction of a DENV infectious cDNA clone as a bacterial artificial chromosome under the control of the CMV promoter
Using the cloning strategy depicted in Fig. 1, an infectious clone was successfully constructed. The sequential cloning of the four fragments resulted in the generation of the constructs pBAC-CMV-DEN2-5′, pBAC-CMV-DEN2-5′3′-BGH, pBAC-DENV-First and finally the DENV infectious clone pBAC-DENV-FL. After elimination of one non-synonymous mutation at the NS5 gene (nt 9152) and two mutations at the 3′ UTR (nt 10316 and 10420) (data not shown), three synonymous mutations at the E and NS3 genes (nt 1215, 2040 and 5067) and one non-synonymous mutation at the NS3 gene (nt 5846) remained in the full-length clone compared with the parental DENV-2 NGC M2 strain (Table 2 ). Despite the potentially negative effect of the non-synonymous mutation, the presence of this mutation in several sequenced clones of pBAC-DENV4702-8834 suggested that the change was present at a high frequency in the mutant swarm of the parental strain and did not result from an error introduced during PCR amplification or plasmid propagation in E. coli.
Table 2.
Synonymous and non-synonymous mutations present in the DENV full-length infectious clones respect to the parental strain.
| Strain/construct |
Promoter |
Positiona |
|||
| 1215 Envelope |
2040 Envelope |
5067 NS3 |
5846 NS3b |
||
| DENV2 NGC_M2 | N/A | Lys (− − A) | Glu (− − G) | Ile (− − U) | Ile (− U − ) |
| pBAC-DENV-FL | CMV | Lys → Lys (− − A →− − G) | Glu → Glu (− − G →− − A) | Ile → Ile (− − U →− − A) | Ile → Thr (− U− → −C −) |
| pBAC-DENV-FL-GFP | CMV | Lys → Lys (− − A → − − G) | – | Ile → Ile (− − U →− − A) | Ile → Thr (− U− → − C − |
| pBAC-DENV-FL-LUC | CMV | Lys → Lys (− − A →− − G) | – | Ile → Ile (− − U →− − A) | Ile → Thr (−U − → − C−) |
Positions with respect to the reference strain DENV-2 NGC (GenBank accession number: M29095.1). Specific changes in the nucleotide sequence and codon position are detailed in parenthesis.
Non-synonymous mutation present in more than one partial clone (pBAC-DENV4702-8834 clones 2 and 4) before assembly of the full-length construct. N/A: not applicable.
3.2. Rescue of infectious DENV from the cDNA clone in Vero and C6/36 cells
To recover infectious virus, Vero cells were transfected with the DENV cDNA clone. At **48 hpt the cell supernatants were collected and used to inoculate C6/36 mosquito cell cultures. Three serial passages of the C6/36 culture supernatants were performed to increase the virus titer. The rescued recombinant full-length DENV (rDENV-FL) induced a clear cytopathic effect in C6/36 cells, characterized by syncytia formation, visible from the first passage (Fig. 2A). All synonymous and non-synonymous mutations that characterized the infectious clone remained after three passages in C6/36 cells, as corroborated through sequencing (data not shown), thereby acting as genetic markers for the identification of rDENV-FL and differentiation from the parental strain. To evaluate the phenotypic properties of the rescued virus, the cytopathic effect (Fig. 2B) and plaque size phenotype (Fig. 2C) were analyzed in Vero cells and compared with those of the parental virus. The cytopathic effect, characterized by the presence of rounded cells in the supernatants, was visible after 7 dpi (Fig. 2B), and both viruses induced the formation of small plaques with diameters ± standard deviations of 0.355 ± 0.168 and 0.404 ± 0.043 for the parental M2 and rDENV-FL strains, respectively (Fig. 2D), without statistically significant differences. These results demonstrate the successful rescue of rDENV-FL in C6/36 cells and the attenuated phenotype of this virus in Vero cells, as expected from the parental strain.
Fig. 2.
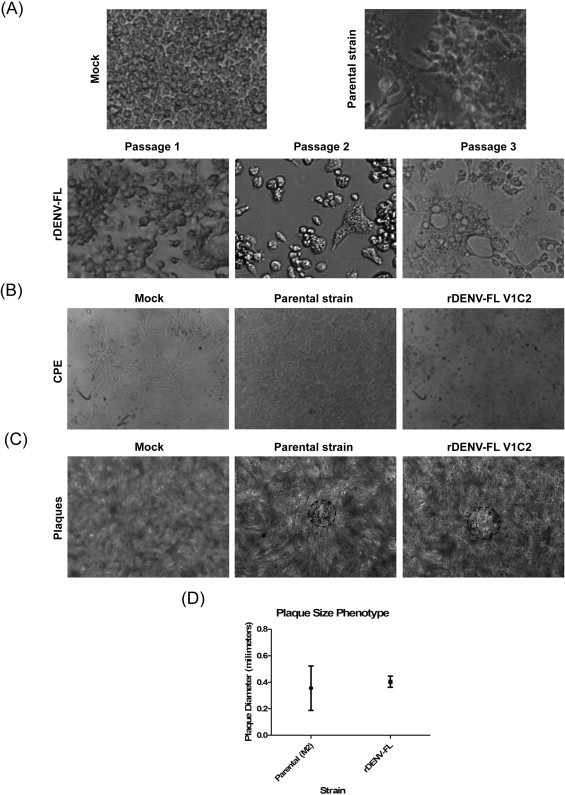
Phenotypic characterization of the rDENV-FL virus: (A) cytopathic effect (CPE) in C6/36 cells. C6/36 cells were mock infected, infected with the parental virus or infected during three passages with the rDENV-FL and subsequently analyzed for the induction of CPE through light microscopy. (B) CPE in Vero cells. Monolayers of Vero cells were mock infected, infected with the parental virus or the rDENV-FL and subsequently analyzed for the induction of CPE through light microscopy. (C) and (D) Plaque size phenotype. The plaque sizes of the rDENV-FL and the parental virus were analyzed as described in Section 2. Circles were drawn to delimitate the plaques and estimate the diameter, which were expressed in millimeters as the mean ± standard deviation. V1C2 corresponds to the history of the virus rescue (in this case, one passage in Vero cells and two passages in C6/36 cells).
In spite of having phenotypic evidence of a viral infection, the identity of the rescued virus was further confirmed through the detection of the DENV envelope protein in infected C6/36 cells by immunofluorescence. As expected, only rDENV-FL-infected cells were positive for the specific staining of the DENV envelope protein using a polyclonal antibody (Fig. 3A). Finally, viral RNA levels from infected cell supernatants were quantified, and a corresponding increase similar to that previously observed in cytopathicity in C6/36 cells after serial passage was confirmed as a significant increase in the total number of viral RNA copies, primarily for passages 2 and 3 (p < 0.01) (Fig. 3B). These results confirm the presence of the specific DENV antigen and the presence and increase in DENV genomes after serial passages of the rDENV-FL in C6/36 cells.
Fig. 3.
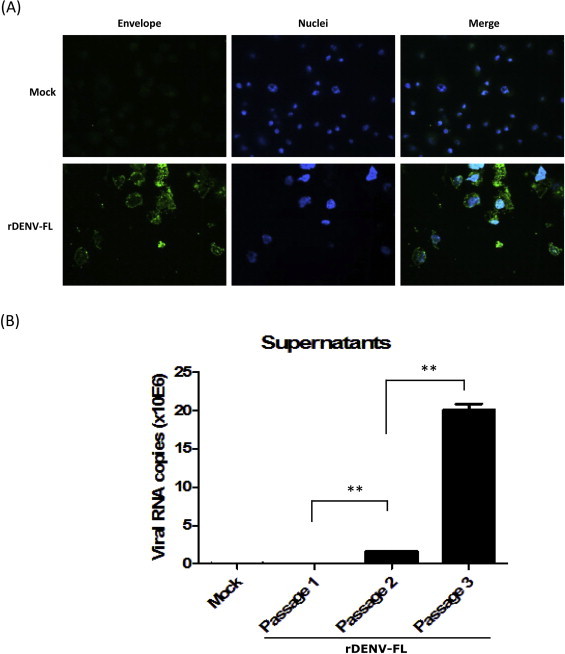
Specific detection of rDENV-FL virus: (A) Envelope protein detection. C6/36 cells were mock infected or infected with rDENV-FL passage 2 and subsequently the envelope protein expression was analyzed through indirect immunofluorescence using the polyclonal antibody 2133 and a secondary antibody conjugated to Alexa Fluor® 488. Nuclei were stained with Hoechst. (B) Viral genome detection. Supernatants of the serial passages of rDENV-FL in C6/36 cells were used for viral RNA extraction and RT-qPCR quantification. The error bars represent the standard error of the mean (SEM). **p < 0.01.
3.3. The expression of EGFP as a fusion protein with the viral envelope protein could affect the assembly or exit of the viral progeny
To construct a useful tool for the study of the cell biology of DENV infection, to track the envelope protein and viral particles, the reporter EGFP gene was fused to the 3′ end of the viral envelope gene, as depicted in Fig. 4 (see Section 2 for details). The EGFP reporter gene was successfully inserted into the intergenic region E-NS1, facilitating the elimination of the synonymous mutation located at position 2040 in the previously obtained infectious clone (Table 2). After transfection of the EGFP-bearing DENV recombinant clone (pBAC-DENV-FL-GFP) in Vero cells and two passages in C6/36 cells, no cytopathic effect was observed (data not shown), suggesting that the recombinant virus was not successfully rescued. To further evaluate the rescue, RNA was extracted from cell culture supernatants at different passages of the virus and measured through RT-qPCR. Despite a moderate increase in the viral RNA levels at passage 2 in C6/36 cells (p < 0.05) (Fig. 5A), viral RNA levels were lower than those obtained for the DENV infectious clone without a reporter (Fig. 3B). Indeed, EGFP-positive cells were not detected in cell cultures inoculated with the supernatant from this passage (data not shown), indicating that no infectious virus was successfully rescued. To rule out a defect in the transcription from the CMV promoter or translation from the proper ORF, the expression of EGFP in Vero cells transfected with the pBAC-DENV-FL-GFP construct was evaluated. EGFP-positive cells were detected at 24 hpt (Fig. 5B), suggesting that viral polyprotein, including the reporter EGFP, were properly translated. Finally, to assess the ability of the CMV-transcribed RNAs to autonomously replicate and generate negative-sense viral RNAs from the viral RNA-dependent RNA Polymerase and non-structural proteins activity, RT-qPCR for detecting negative-sense viral RNAs was performed and significant amounts of DENV negative-sense RNA were detected at 24 hpt (Fig. 5C).
Fig. 5.
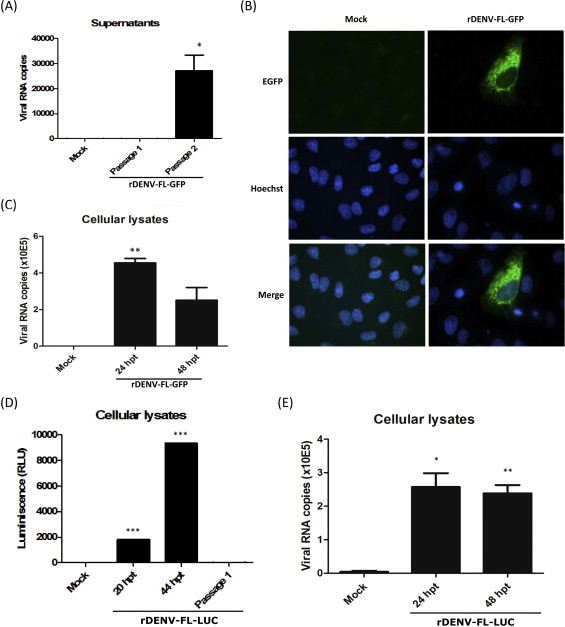
Characterization of the rDENV-FL-EGFP and rDENV-FL-LUC: (A) Viral genome quantification. The supernatants from passages 1 and 2 of the rDENV-FL-EGFP in C6/36 cells were used for viral RNA extraction and RT-qPCR quantification. (B) EGFP expression. Vero cells were mock transfected or transfected with the plasmid pBAC-DENV-FL-EGFP, fixed at 24 hpt, stained with Hoechst and visualized through confocal microscopy. (C) and (E) Viral genome replication analysis. Cellular lysates from Vero cells transfected with (C) pBAC-DENV-FL-EGFP or (E) pBAC-DENV-FL-LUC were collected at 24 and 48 hpt and used for total RNA extraction and RT-qPCR quantification of negative strand of the viral RNA. (D) Luciferase activity. Cellular lysates from Vero cells transfected with the plasmid pBAC-DENV-FL-LUC were collected at 20 and 44 hpt, and after the first passage in Vero cells, the luciferase activity was quantified. The values are given in Relative Luciferase Units (RLU). The error bars represent the SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To evaluate whether the negative effect can be attributed to the nature of the EGFP gene, as a result of the production of a non-functional fusion protein by incorrect folding or improper subcellular localization, another reporter gene (hRLuc) was cloned and evaluated in the same manner. This new molecular construct (pBAC-DENV-FL-LUC) exhibited the same mutation pattern as pBAC-DENV-FL-GFP (Table 2). The ability of the clone to transcribe from the CMV promoter and translate from the correct ORF was demonstrated through the presence of luciferase activity (Fig. 5D). The viral RNA and replicase complex activity was demonstrated through the presence of significant amounts of negative-sense viral RNA strands (Fig. 5E). However, a total absence of the cytopathic effect in Vero and C6/36 cells (data not shown) and a failure to recover infectious particles after one passage (Fig. 5D) was demonstrated.
4. Discussion
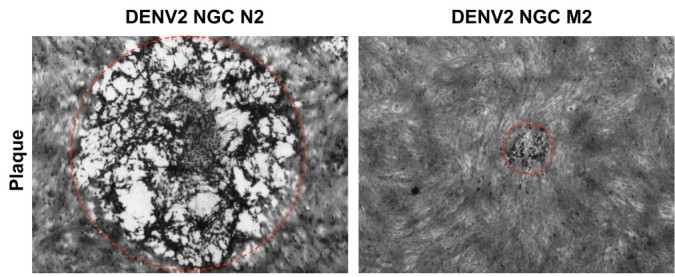
The development of reverse genetics systems for several RNA viruses has become an invaluable tool for multiple purposes. DENV research has greatly benefited from these developments in several ways, ranging from the study of gene and protein function to vaccine generation and antiviral screening (Chao et al., 2012, Grant et al., 2011, Hsu et al., 2012, Huang et al., 2003, Kelly et al., 2010, Ng et al., 2007, Qing et al., 2010, Zhu et al., 2007, Zou et al., 2011). The rational design and construction of the DENV infectious clone as a BAC under the control of the CMV promoter described in the present study facilitated the successful rescue of DENV infectious particles, leading to plaque formation with phenotypic features similar to the parental strain. The standard deviation of the plaque diameter for rDENV-FL was smaller than that for the parental strain (DENV-2 NGC M2), reflecting the clonal origin of the rDENV-FL strain and suggesting its convenience for studying the effect of single mutations, where highly homogeneous viral populations are required. The parental strain showed a very attenuated phenotype, with plaque diameters significantly smaller than those of a highly adapted DENV strain (called DENV-2 NGC N2) (Suppl. Fig. 2). The parental (M2) and highly adapted (N2) DENV-2 NGC strains belong to the same original clinical isolation (New Guinea C); however, different passage histories, genetic drift and selection has led to sequence differences and extremely different phenotypes. The parental strain was chosen for cloning because of its smaller plaque size and the biological safety concerns for the potential derivation of a novel viral vector from the DENV infectious clone. The dramatic plaque size phenotype differences between these two closely related DENV strains reflect the usefulness of the infectious clone for studying virulence determinants and pathogenesis.
Despite the presence of a candidate for donor splicing site at position 3395 (NS1 gene) in the full-length DENV genome (Suppl. Tables 8 and 9), a commonly high number of false-positive splice sites predictions (Brunak et al., 1991) and the existence of viable DENV replicons under the control of the CMV promoter, including the entire NS1 gene (Ansarah-Sobrinho et al., 2008, Leardkamolkarn et al., 2012, Lee et al., 2010, Pang et al., 2001b), are consistent with this site not being strictly used for splicing. The sequencing of several fragments amplified from the viral RNA extracted after serial passages in C6/36 cells demonstrated the stability and integrity of these viral genome regions. The functionality of the rDENV-FL was evidenced by productive infection when the supernatants of transfected/infected cells were used to inoculate fresh cellular cultures. Although some DNA-launched replicons of DENV under the control of the CMV promoter have been previously described (Ansarah-Sobrinho et al., 2008, Leardkamolkarn and Sirigulpanit, 2012, Leardkamolkarn et al., 2012, Lee et al., 2010, Pang et al., 2001b), to the best of our knowledge, this study presents the first DENV infectious clone under the control of the CMV promoter for the recovery of recombinant viruses without the need for in vitro transcription.
In addition to the DENV infectious clone, we generated an EGFP-bearing molecular clone (pBAC-DENV-FL-GFP), expressing EGFP as a fusion protein with the viral envelope protein in an attempt to generate a clone in which the early (entry and membrane fusion) and late (assembly of new viral particles) steps and subcellular localization of the envelope protein during infection could be tracked. Fusion proteins are crucial for deciphering viral intracellular trafficking during the early and late steps of infection (Brandenburg and Zhuang, 2007). However, in the present study, the fusion of the EGFP protein had a profoundly negative effect in virus rescue. Although autonomous viral genome replication was demonstrated for this clone after transfection, the detection of viral RNA (low levels) at passage 2 in C6/36 cells was not supported by the presence of the reporter protein. This disparity could be explained by the higher sensibility of the RT-qPCR to detect low levels of RNA molecules and the potential presence of a very low number of undetected fluorescent (EGFP) cells in those cultures at passage 2. The results suggest a negative impact of the fused EGFP in viral function (packaging, exit, or infectivity of the viral progeny). Undetectable or minimal levels of infectious virus in the supernatants of transfected cells have been previously observed when the reporter GFP gene was inserted into the anchored-C region of a DENV-2 infectious clone, and a significant defect in virus infectivity through the insertion of the reporter gene has also been suggested (Leardkamolkarn et al., 2012).
Because of the size and potential spatial constrictions of the core structure, our initial design did not contemplate the fusion of the reporter to the capsid protein, but to the carboxyl-terminus of the envelope protein, which has a major size and dynamic structure. A previous study using Yellow fever virus showed the successful insertion of the EGFP reporter gene in the E-NS1 intergenic region through the duplication of the stem-anchor region of the E protein (Bonaldo et al., 2007). Although that design avoided the negative effects associated with flavivirus E-NS1 separation, as the carboxyl-terminus of the envelope protein serves as a signal sequence required for the production of glycosylated NS1 protein (Falgout et al., 1989), the duplication of the ER signal peptidase recognition site at E-NS1 resulted in the separation of the E and EGFP proteins and was therefore not useful for protein and/or virion tracking.
Recently, an infectious clone of DENV-2, expressing the EGFP reporter gene, was successfully constructed (Schoggins et al., 2012). This molecular construct was a powerful tool for cell-based screening assays to identify antiviral molecules and their mechanisms of action. However, the EGFP protein was not fused to any viral structural protein and therefore, this EGFP-bearing DENV recombinant clone facilitated neither the tracking of individual particles outside the cell nor the examination the subcellular localization of any viral structural protein during the early and late steps of the DENV life cycle. Future attempts to mark at least one viral structural protein for tracking the course of infection should evaluate other positions for fusion of the reporter gene, including the carboxyl-terminus of the prM/M protein and the amino-terminal of the envelope protein.
Similarly, only two DENV infectious clones expressing the luciferase gene have been previously reported as excellent alternatives for quantitative analysis in the high-throughput screening of antiviral compounds (Zou et al., 2011) and bioluminescence imaging of DENV infection in vivo (Schoggins et al., 2012). These reporter-bearing infectious clones contain the 5′UTR, the sequence encoding the 38 N-terminal amino acids of the capsid protein, the hRLuc reporter and the FMDV2A sequence, followed by the full-length DENV ORF, which includes the complete capsid gene. The results from the present study, with a molecular clone constructed using a different strategy (pBAC-DENV-FL-LUC), confirm the correct expression of luciferase and the replicase complex function after transfection. The non-proportional increase between the Relative Luciferase Units and the negative-sense viral RNA could be explained by the principle of the methods, stability of the reporter protein or apoptotic cell death of the transfected cells at 36 hpt and later, leading to viral RNA decline, as it was observed for other constructs (DENV-derived replicons) (unpublished data). However, the absence of cytopathic effects and luciferase activity after one passage suggests that alterations of the carboxyl-terminus of the envelope protein through fusion with the reporter hRLuc negatively impacted virus morphogenesis or the infectivity of the viral progeny, consistent with the data obtained for the EGFP-bearing clone. Despite the negative impact on the production of virions, these results help to delimit future studies concerning the fusion of reporters to DENV structural genes.
Taken together, the results shown in the present study demonstrate the successful construction of a novel CMV-driven infectious clone of DENV-2 in a BAC for the effective recovery of infectious particles displaying phenotypic characteristics similar to the parental strain. This infectious clone will overcome the costs and degradation risks associated with in vitro transcription and RNA manipulation, and this clone will also serve as a backbone to assess the potential positions for fusion with any reporter gene. In addition, this construct will be useful for a number of applications in the study of DENV biology, molecular pathogenesis, evolution and the development of viral vectors.
5. Conclusions
We successfully cloned and expressed the whole DENV-2 genome as an infectious BAC under the control of the CMV promoter. The use of this promoter allows the generation of infectious viruses avoiding the limitations related to in vitro RNA synthesis and RNA transfection methods. This infectious full-length cDNA clone is a useful tool that could be implemented in the study of DENV biology, antiviral screening and vaccine development.
Acknowledgments
This study was financially supported through funding from the Administrative Department of Science, Technology and Innovation–Colciencias (Colombia) grants 111545921525 and 111554531621; and the Ministry of Science and Innovation of Spain (MCINN) (BIO2010-16075). JCGG received a possition for the Full-Time Professor Program 2012-3 of the Medicine Faculty. JUC was recipient of a Doctoral Fellowship from Colciencias.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.virusres.2013.12.001.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- Almazan F., Gonzalez J.M., Penzes Z., Izeta A., Calvo E., Plana-Duran J., Enjuanes L. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(10):5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan F., Galan C., Enjuanes L. Engineering infectious cDNAs of coronavirus as bacterial artificial chromosomes. Methods in Molecular Biology. 2008;454:275–291. doi: 10.1007/978-1-59745-181-9_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansarah-Sobrinho C., Nelson S., Jost C.A., Whitehead S.S., Pierson T.C. Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology. 2008;381(1):67–74. doi: 10.1016/j.virol.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R., Miller S. Molecular aspects of dengue virus replication. Future Microbiology. 2008;3(2):155–165. doi: 10.2217/17460913.3.2.155. [DOI] [PubMed] [Google Scholar]
- Bonaldo M.C., Mello S.M., Trindade G.F., Rangel A.A., Duarte A.S., Oliveira P.J., Freire M.S., Kubelka C.F., Galler R. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virology Journal. 2007;4:115. doi: 10.1186/1743-422X-4-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburg B., Zhuang X. Virus trafficking—learning from single-virus tracking. Nature Reviews Microbiology. 2007;5(3):197–208. doi: 10.1038/nrmicro1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunak S., Engelbrecht J., Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. Journal of Molecular Biology. 1991;220(1):49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- Chambers T.J., Hahn C.S., Galler R., Rice C.M. Flavivirus genome organization, expression, and replication. Annual Review of Microbiology. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- Chao B., Tong X.K., Tang W., Li D.W., He P.L., Garcia J.M., Zeng L.M., Gao A.H., Yang L., Li J., Nan F.J., Jacobs M., Altmeyer R., Zuo J.P., Hu Y.H. Discovery and optimization of 2,4-diaminoquinazoline derivatives as a new class of potent dengue virus inhibitors. Journal of Medicinal Chemistry. 2012;55(7):3135–3143. doi: 10.1021/jm2015952. [DOI] [PubMed] [Google Scholar]
- Clyde K., Barrera J., Harris E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology. 2008;379(2):314–323. doi: 10.1016/j.virol.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crill W.D., Roehrig J.T. Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells. Journal of Virology. 2001;75(16):7769–7773. doi: 10.1128/JVI.75.16.7769-7773.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Borba L., Strottmann D.M., de Noronha L., Mason P.W., Dos Santos C.N. Synergistic interactions between the NS3(hel) and E proteins contribute to the virulence of dengue virus type 1. PLoS Neglected Tropical Diseases. 2012;6(4):e1624. doi: 10.1371/journal.pntd.0001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falgout B., Chanock R., Lai C.J. Proper processing of dengue virus nonstructural glycoprotein NS1 requires the N-terminal hydrophobic signal sequence and the downstream nonstructural protein NS2a. Journal of Virology. 1989;63(5):1852–1860. doi: 10.1128/jvi.63.5.1852-1860.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falgout B., Pethel M., Zhang Y.M., Lai C.J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. Journal of Virology. 1991;65(5):2467–2475. doi: 10.1128/jvi.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filomatori C.V., Iglesias N.G., Villordo S.M., Alvarez D.E., Gamarnik A.V. RNA sequences and structures required for the recruitment and activity of the dengue virus polymerase. The Journal of Biological Chemistry. 2011;286(9):6929–6939. doi: 10.1074/jbc.M110.162289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P., Harris E. Interplay of RNA elements in the dengue virus 5′ and 3′ ends required for viral RNA replication. Journal of Virology. 2010;84(12):6103–6118. doi: 10.1128/JVI.02042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P., Pena J., Pohl M.O., Harris E. Composition of the sequence downstream of the dengue virus 5′ cyclization sequence (dCS) affects viral RNA replication. Virology. 2012;422(2):346–356. doi: 10.1016/j.virol.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Gomez J.C., Risco C., Rodriguez D., Cabezas P., Guerra S., Carrascosa J.L., Esteban M. Differences in virus-induced cell morphology and in virus maturation between MVA and other strains (WR, Ankara, and NYCBH) of vaccinia virus in infected human cells. Journal of Virology. 2003;77(19):10606–10622. doi: 10.1128/JVI.77.19.10606-10622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant D., Tan G.K., Qing M., Ng J.K., Yip A., Zou G., Xie X., Yuan Z., Schreiber M.J., Schul W., Shi P.Y., Alonso S. A single amino acid in nonstructural protein NS4B confers virulence to dengue virus in AG129 mice through enhancement of viral RNA synthesis. Journal of Virology. 2011;85(15):7775–7787. doi: 10.1128/JVI.00665-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groat-Carmona A.M., Orozco S., Friebe P., Payne A., Kramer L., Harris E. A novel coding-region RNA element modulates infectious dengue virus particle production in both mammalian and mosquito cells and regulates viral replication in Aedes aegypti mosquitoes. Virology. 2012;432(2):511–526. doi: 10.1016/j.virol.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualano R.C., Pryor M.J., Cauchi M.R., Wright P.J., Davidson A.D. Identification of a major determinant of mouse neurovirulence of dengue virus type 2 using stably cloned genomic-length cDNA. The Journal of General Virology. 1998;79(Pt 3):437–446. doi: 10.1099/0022-1317-79-3-437. [DOI] [PubMed] [Google Scholar]
- Guzman M.G., Halstead S.B., Artsob H., Buchy P., Farrar J., Gubler D.J., Hunsperger E., Kroeger A., Margolis H.S., Martinez E., Nathan M.B., Pelegrino J.L., Simmons C., Yoksan S., Peeling R.W. Dengue: a continuing global threat. Nature Reviews Microbiology. 2010;8(12 Suppl):S7–S16. doi: 10.1038/nrmicro2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchal E.A., Gentry M.K., McCown J.M., Brandt W.E. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. The American Journal of Tropical Medicine and Hygiene. 1982;31(4):830–836. doi: 10.4269/ajtmh.1982.31.830. [DOI] [PubMed] [Google Scholar]
- Hsieh S.C., Zou G., Tsai W.Y., Qing M., Chang G.J., Shi P.Y., Wang W.K. The C-terminal helical domain of dengue virus precursor membrane protein is involved in virus assembly and entry. Virology. 2011;410(1):170–180. doi: 10.1016/j.virol.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y.C., Chen N.C., Chen P.C., Wang C.C., Cheng W.C., Wu H.N. Identification of a small-molecule inhibitor of dengue virus using a replicon system. Archives of Virology. 2012;157(4):681–688. doi: 10.1007/s00705-012-1224-z. [DOI] [PubMed] [Google Scholar]
- Huang C.Y., Butrapet S., Tsuchiya K.R., Bhamarapravati N., Gubler D.J., Kinney R.M. Dengue 2 PDK-53 virus as a chimeric carrier for tetravalent dengue vaccine development. Journal of Virology. 2003;77(21):11436–11447. doi: 10.1128/JVI.77.21.11436-11447.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias N.G., Filomatori C.V., Gamarnik A.V. The F1 motif of dengue virus polymerase NS5 is involved in promoter-dependent RNA synthesis. Journal of Virology. 2011;85(12):5745–5756. doi: 10.1128/JVI.02343-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E.P., Polo S., Sun W., Falgout B. Evolution of attenuating mutations in dengue-2 strain S16803 PDK50 vaccine and comparison of growth kinetics with parent virus. Virus Genes. 2011;43(1):18–26. doi: 10.1007/s11262-011-0602-z. [DOI] [PubMed] [Google Scholar]
- Kelly E.P., Puri B., Sun W., Falgout B. Identification of mutations in a candidate dengue 4 vaccine strain 341750 PDK20 and construction of a full-length cDNA clone of the PDK20 vaccine candidate. Vaccine. 2010;28(17):3030–3037. doi: 10.1016/j.vaccine.2009.10.084. [DOI] [PubMed] [Google Scholar]
- Kim U.J., Birren B.W., Slepak T., Mancino V., Boysen C., Kang H.L., Simon M.I., Shizuya H. Construction and characterization of a human bacterial artificial chromosome library. Genomics. 1996;34(2):213–218. doi: 10.1006/geno.1996.0268. [DOI] [PubMed] [Google Scholar]
- Kinney R.M., Butrapet S., Chang G.J., Tsuchiya K.R., Roehrig J.T., Bhamarapravati N., Gubler D.J. Construction of infectious cDNA clones for dengue 2 virus: strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology. 1997;230(2):300–308. doi: 10.1006/viro.1997.8500. [DOI] [PubMed] [Google Scholar]
- Lai C.J., Zhao B.T., Hori H., Bray M. Infectious RNA transcribed from stably cloned full-length cDNA of dengue type 4 virus. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(12):5139–5143. doi: 10.1073/pnas.88.12.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M. The making of infectious viral RNA: no size limit in sight. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(10):5025–5027. doi: 10.1073/pnas.97.10.5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leardkamolkarn V., Sirigulpanit W. Establishment of a stable cell line coexpressing dengue virus-2 and green fluorescent protein for screening of antiviral compounds. Journal of Biomolecular Screening. 2012;17(3):283–292. doi: 10.1177/1087057111426903. [DOI] [PubMed] [Google Scholar]
- Leardkamolkarn V., Sirigulpanit W., Chotiwan N., Kumkate S., Huang C.Y. Development of Dengue type-2 virus replicons expressing GFP reporter gene in study of viral RNA replication. Virus Research. 2012;163(2):552–562. doi: 10.1016/j.virusres.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.C., Lin Y.L., Liao J.T., Su C.M., Lin C.C., Lin W.P., Liao C.L. Utilizing liver-specific microRNA-122 to modulate replication of dengue virus replicon. Biochemical and Biophysical Research Communications. 2010;396(3):596–601. doi: 10.1016/j.bbrc.2010.04.080. [DOI] [PubMed] [Google Scholar]
- Lindenbach D.B., Thiel H.-J., Rice C.M. Flaviviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., editors. Fields Virology. fifth ed. Wolkers Kluwer/Lippincott Williams and Wilkins; Philadelphia, PA: 2007. pp. 1101–1152. [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lodeiro M.F., Filomatori C.V., Gamarnik A.V. Structural and functional studies of the promoter element for dengue virus RNA replication. Journal of Virology. 2009;83(2):993–1008. doi: 10.1128/JVI.01647-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Gutierrez M., Castellanos J.E., Gallego-Gomez J.C. Statins reduce dengue virus production via decreased virion assembly. Intervirology. 2011;54(4):202–216. doi: 10.1159/000321892. [DOI] [PubMed] [Google Scholar]
- Modis Y., Ogata S., Clements D., Harrison S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427(6972):313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- Munoz-Jordan J.L., Laurent-Rolle M., Ashour J., Martinez-Sobrido L., Ashok M., Lipkin W.I., Garcia-Sastre A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. Journal of Virology. 2005;79(13):8004–8013. doi: 10.1128/JVI.79.13.8004-8013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Jordan J.L., Sanchez-Burgos G.G., Laurent-Rolle M., Garcia-Sastre A. Inhibition of interferon signaling by dengue virus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(24):14333–14338. doi: 10.1073/pnas.2335168100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C.L., Jones C.T., Rice C.M. Architects of assembly: roles of Flaviviridae non-structural proteins in virion morphogenesis. Nature reviews: Microbiology. 2008;6(9):699–708. doi: 10.1038/nrmicro1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C.Y., Gu F., Phong W.Y., Chen Y.L., Lim S.P., Davidson A., Vasudevan S.G. Construction and characterization of a stable subgenomic dengue virus type 2 replicon system for antiviral compound and siRNA testing. Antiviral Research. 2007;76(3):222–231. doi: 10.1016/j.antiviral.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Pang X., Zhang M., Dayton A.I. Development of dengue virus replicons expressing HIV-1 gp120 and other heterologous genes: a potential future tool for dual vaccination against dengue virus and HIV. BMC Microbiology. 2001;1:28. doi: 10.1186/1471-2180-1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang X., Zhang M., Dayton A.I. Development of dengue virus type 2 replicons capable of prolonged expression in host cells. BMC Microbiology. 2001;1:18. doi: 10.1186/1471-2180-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paranjape S.M., Harris E. Control of dengue virus translation and replication. Current Topics in Microbiology and Immunology. 2010;338:15–34. doi: 10.1007/978-3-642-02215-9_2. [DOI] [PubMed] [Google Scholar]
- Pierro D.J., Salazar M.I., Beaty B.J., Olson K.E. Infectious clone construction of dengue virus type 2, strain Jamaican 1409, and characterization of a conditional E6 mutation. The Journal of General Virology. 2006;87(Pt 8):2263–2268. doi: 10.1099/vir.0.81958-0. [DOI] [PubMed] [Google Scholar]
- Polo S., Ketner G., Levis R., Falgout B. Infectious RNA transcripts from full-length dengue virus type 2 cDNA clones made in yeast. Journal of Virology. 1997;71(7):5366–5374. doi: 10.1128/jvi.71.7.5366-5374.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu S.Y., Wu R.H., Yang C.C., Jao T.M., Tsai M.H., Wang J.C., Lin H.M., Chao Y.S., Yueh A. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. Journal of Virology. 2011;85(6):2927–2941. doi: 10.1128/JVI.01986-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri B., Polo S., Hayes C.G., Falgout B. Construction of a full length infectious clone for dengue-1 virus Western Pacific, 74 strain. Virus Genes. 2000;20(1):57–63. doi: 10.1023/a:1008160123754. [DOI] [PubMed] [Google Scholar]
- Qing M., Liu W., Yuan Z., Gu F., Shi P.Y. A high-throughput assay using dengue-1 virus-like particles for drug discovery. Antiviral Research. 2010;86(2):163–171. doi: 10.1016/j.antiviral.2010.02.313. [DOI] [PubMed] [Google Scholar]
- Schoggins J.W., Dorner M., Feulner M., Imanaka N., Murphy M.Y., Ploss A., Rice C.M. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(36):14610–14615. doi: 10.1073/pnas.1212379109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shizuya H., Birren B., Kim U.J., Mancino V., Slepak T., Tachiiri Y., Simon M. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(18):8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriburi R., Keelapang P., Duangchinda T., Pruksakorn S., Maneekarn N., Malasit P., Sittisombut N. Construction of infectious dengue 2 virus cDNA clones using high copy number plasmid. Journal of Virological Methods. 2001;92(1):71–82. doi: 10.1016/s0166-0934(00)00277-9. [DOI] [PubMed] [Google Scholar]
- St-Jean J.R., Desforges M., Almazan F., Jacomy H., Enjuanes L., Talbot P.J. Recovery of a neurovirulent human coronavirus OC43 from an infectious cDNA clone. Journal of Virology. 2006;80(7):3670–3674. doi: 10.1128/JVI.80.7.3670-3674.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R., de Borba L., Duarte dos Santos C.N., Mason P.W. Construction of an infectious cDNA clone for a Brazilian prototype strain of dengue virus type 1: characterization of a temperature-sensitive mutation in NS1. Virology. 2007;362(2):374–383. doi: 10.1016/j.virol.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TDR/WHO . World Health Organization; Geneva, Switzerland: 2009. Dengue Guidelines for Diagnosis, Treatment, Prevention and Control. [PubMed] [Google Scholar]
- Yu L., Nomaguchi M., Padmanabhan R., Markoff L. Specific requirements for elements of the 5″ and 3′ terminal regions in flavivirus RNA synthesis and viral replication. Virology. 2008;374(1):170–185. doi: 10.1016/j.virol.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Qin C., Chen S., Jiang T., Yu M., Yu X., Qin E. Attenuated dengue 2 viruses with deletions in capsid protein derived from an infectious full-length cDNA clone. Virus Research. 2007;126(1-2):226–232. doi: 10.1016/j.virusres.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Zou G., Xu H.Y., Qing M., Wang Q.Y., Shi P.Y. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antiviral Research. 2011;91(1):11–19. doi: 10.1016/j.antiviral.2011.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.