

Highlights
► Overexpression of HCV p7 protein induces apoptosis in Huh7.5 cells. ► p7-induced apoptosis involves both the intrinsic and extrinsic pathways. ► p7-induced apoptosis is independent of its ion channel activity. ► p7 is functionally similar to the well-characterized M2 protein of influenza A virus but they differ in their activation of autophagic cell-death.
Keywords: HCV, p7 protein, Cell death, Ion channel activity
Abstract
Most viruses encode proteins that modulate cell-death signaling by the host. For hepatitis C virus (HCV) infection, apoptosis and other forms of cell-death have been observed in vitro and in vivo but the detailed understanding of this intricate viral-host interplay is unclear. This study examined the role played by the HCV p7 protein in the induction of cell-death. By measuring caspase-3/7 activation and cleavage of endogenous PARP, two hallmarks of apoptosis, the overexpression of p7 protein was shown to induce apoptosis in Huh7.5 cells. Furthermore, p7-induced apoptosis is caspase-dependent and involves both the intrinsic and extrinsic pathways. Similar to the M2 protein of influenza A virus, p7-induced apoptosis is independent of its ion channel activity. Coimmunoprecipitation experiments further showed that both M2 and p7 interact with the essential autophagy protein Beclin-1. However, only the M2 protein could cause an increase in the level of LC3-II, which is an indicator of autophagic activity. Thus, although the p7 protein is functionally similar to the well-characterized M2 protein, they differ in their activation of autophagic cell-death. Taken together, these results shed more light on the relationship between the HCV p7 ion channel protein and cell-death induction in host cells.
1. Introduction
Hepatitis C virus (HCV), a small positive strand RNA virus that belongs to the Hepacivirus genus in the Flaviviridae family, is one of the major causes of liver disease such as cirrhosis, steatosis, and hepatocellular carcinoma (Chen and Morgan, 2006). In fact, HCV infection is the most frequent indication for liver transplantation in developed countries (Brown, 2005). Currently, about 130–170 million or 3% of the world population are estimated to be infected with HCV; with about 3–4 million new infections per year (WHO, 2012). HCV has a high degree of genetic diversity, so it is classified phylogenetically into six major genotypes and several subtypes (Simmonds, 2004). The HCV genome of ∼9.6 kb encodes a unique open reading frame that is translated into a precursor polyprotein of ∼3000 residues. Co- and post-translational processing of the polyprotein by both viral and cellular proteases yields three structural (core, E1 and E2), six non-structural proteins (NS2–NS5), and a small membrane ion channel protein (p7) (Lin et al., 1994); the protein of interest in this study.
The HCV p7 protein, a small (63 amino acid) hydrophobic protein located between the structural and nonstructural region of HCV, is not clearly identified as structural or nonstructural protein, although it has been shown to be a transmembrane protein (reviewed in Khaliq et al., 2011). HCV p7 is classified as a viroporin, a group of small hydrophobic proteins encoded by a variety of RNA viruses that oligomerize to form pores (ion channels) in host-cell membranes through which viruses can enter/exit as well as contribute to virus assembly and pathology of disease by altering membrane permeability and disrupting ion homeostasis in cells (Gonzalez and Carrasco, 2003). The precise role of p7 in the HCV life-cycle has been hard to define. However, some recent papers have implicated p7 in the assembly and release of virus particles, mostly acting in concert with other viral proteins (reviewed in Khaliq et al., 2011, Steinmann and Pietschmann, 2010), and in a genotype-specific manner (Steinmann et al., 2007). For example, a strain-specific tripartite relationship between core, p7 and NS2 has been reported to be responsible for modulating the subcellular localization of core (Boson et al., 2011) and NS2 (Tedbury et al., 2011), which is independent of p7's ion channel activity. Similarly, it has been shown that mutations in the p7 protein, either singly or in combination with E2 glycoprotein enhances several-fold production of infectious virus particles in cell culture (Kim et al., 2011). Thus, p7 unlike other viroporins such as M2 of influenza A virus and Vpu of HIV-1, is absolutely essential for HCV replication in vitro (Brohm et al., 2009, Steinmann et al., 2007).
Cell death regulation is an important determinant in the survival of most viruses, therefore, many viruses encode proteins that interfere with cell death signaling pathways, skewing it in their favor (Chen et al., 2006). For HCV, both proapoptotic and prosurvival properties have been attributed to different HCV proteins (reviewed in Aweya and Tan, 2011, Fischer et al., 2007). HCV p7 like other members of the viroporins (e.g., HIV-1 Vpu, human T-cell lymphotropic virus-1 p13II, hepatitis B virus X protein, and influenza virus PB1 ORF2), might be targeted to the mitochondrial membranes where it modulates apoptosis by altering the mitochondrial membrane permeability. However, there is currently limited information on the apoptotic activity of p7 except that reported by Madan et al. (2008) who demonstrated that the p7 protein of genotype 1b HCV induces caspase-dependent apoptosis via the mitochondria in baby hamster kidney cells. Since the genotype 2a JFH1 strain is the only one that can replicate efficiently in permissive cells, such as Huh7.5 cells, without adaption, this study sought to examine how p7 protein of this strain modulates cell-death and how similar it is to the M2 ion channel protein of influenza A virus, which is a well characterized viroporin. We were able to show that HCV p7 protein induces caspase-dependent apoptosis which is independent of its ion channel activity, and although p7 protein shares a couple of functional properties with M2 protein of influenza A virus, they seem to differ in their induction of autophagic cell death.
2. Materials and methods
2.1. Cell culture and cell lines
Huh7.5 cells (subclone of the Huh-7 human hepatoma cell line; Apath, Brooklyn, NY) and 293FT cells (human embryonic kidney cell line with the temperature sensitive gene for SV40 T-antigen; Invitrogen, Karlsruhe, Germany) were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (HyClone, Utah, USA), nonessential amino acids and antibiotics (10 units/ml penicillin and 10 μg/ml streptomycin) (Invitrogen, Carlsbad, CA). All cells were maintained in a 37 °C incubator with 5% CO2.
2.2. Plasmid construction
Expression plasmids for the HCV p7 proteins were generated by PCR cloning using Titanium Taq DNA polymerase (Clontech Laboratories Inc., Palo Alto, CA). Two plasmids containing the full-length HCV genome of the genotype 1b (S1) cloned in Singapore ((Soo et al., 2002) GenBank accession number AF356827) and genotype 2a JFH-1 strain ((Wakita et al., 2005), GenBank accession number AB047639) were used as templates. The PCR products were digested with restriction enzymes BamHI and XhoI and then ligated into the pXJ40flag vector which is a flag-tagged plasmid derived from pXJ40 (Xiao et al., 1991). The pXJ40flag vector was used so that a flag epitope is fused to the N-terminus of the p7 protein to allow the comparison of protein expression levels with an anti-flag antibody. Similarly, the p7 mutants were generated by 2 rounds of PCR: the first round of PCR was used to generate 5′ and 3′ fragments of p7 containing the appropriate alanine or glutamine substitutions; these were then amplified into full-length p7 using end primers for the p7 gene and cloned into pXJ40flag vector using BamHI and XhoI sites. The M2 gene of influenza A virus (A/Puerto Rico/8/34/Mount Sinai/Wi (H1N1), GenBank accession number AY768951.1) was produced by gene synthesis (GenScript USA Inc., Piscataway, NJ, USA) and cloned into the pXJ40flag vector in a similar manner. All sequences were confirmed by sequencing performed by the DNA Sequencing Facility at the Department of Microbiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
2.3. Transient transfections and Western blot analysis
Transient transfections of cells were performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Approximately 24 h after transfection, the cells were harvested by scrapping into the media, spun down in a bench-top centrifuge and washed twice with cold PBS. The cell pellets were then resuspended in RIPA buffer (50 mM Tris (pH 8.0), 150 mM NaCl, 0.5% NP40, 0.5% deoxycholic acid, 0.005% SDS and 1 mM phenylmethylsulfonyl fluoride) and subjected to freeze–thawing five times before being spun down at 13,000 rpm to remove cellular debris. The cell lysate was then used for Western blot analysis and quantification of apoptosis (see next section below).
Primary antibodies used in the study included anti-actin monoclonal, anti-flag monoclonal and polyclonal (Sigma, St. Louis, MO), anti-poly[ADP-ribose] polymerase [PARP] polyclonal, and anti-LC3B polyclonal (Cell Signaling Technology Inc., Beverly, MA) antibodies. Secondary antibodies used included horseradish peroxidase (HRP)-conjugated goat anti-mouse, and goat anti-rabbit antibodies (Pierce, Rockford, IL).
2.4. Apo-One fluorometric and TUNEL assay
Apoptosis was quantified in the form of caspase-3/7 activation using the Apo-One fluorometric assay system from Promega Corporation (Madison, WI) according to the manufacturer's protocol. To further confirm the induction of apoptosis by cells overexpressing p7 protein, the Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was carried out after 24 h of transfection using the DeadEnd Fluorometric TUNEL system (Promega, Madison, WI) according to the manufacturer's protocol. Briefly, cells were plated in Lab-Tek™ Chamber Slides, transfected with plasmids expressing p7 protein followed by treatment with cell permeable caspase inhibitors as described below. After 24 h, cells were fixed in 4% paraformaldehyde at 4 °C for 25 min. Fixed cells were then permeabilized in 0.2% Triton X-100 and labeled with fluorescein-12-dUTP using terminal deoxynucleotidyl transferase. After rinsing with PBS, slides were mounted with VECTASHIELD® Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA). The TUNEL-positive cells (green fluorescence) corresponding to the nuclei location (DAPI) were captured with Olympus FluoView FV1000 (Olympus, Japan) laser scanning confocal microscope using a 60x/1.45 oil objective, with 543 nm HeNe laser as the excitation source.
2.5. Treatment of cells with caspase inhibitors and ammonium chloride
Cells were transfected (as above) followed by treatment with 2.5 μM of caspase-3 (Z-DEVD-fmk), caspase-8 (Z-IETD-fmk), caspase-9 (Z-LEHD-fmk) or a negative caspase inhibitor (Z-FA-fmk) (BD Biosciences). Briefly, 6 h post-transfection, the transfection media was changed and replinished with fresh media plus 2.5 μM of the respective cell permeable caspase inhibitors which are reported to have a very short half-life (Ekert et al., 1999, Garcia-Calvo et al., 1998). After about 24 h post-transfection, the cells were harvested by scrapping into the media, spun down in a bench-top centrifuge and washed three times with 1× cold PBS to remove any residual inhibitor found in the media. The cell pellets were then resuspended in RIPA buffer and subjected to freeze–thawing five times before being spun down at 13,000 rpm to clarify the lysate. Subsequently, the lysate was analyzed using the Apo-One fluorometric assay as described above.
In order to block autophagosome formation, cells were treated with ammonium chloride (BDH, AnalaR). Briefly, the spent media from an overnight transient transfection was replaced with complete media containing 2.7 mg/ml of ammonium chloride and then incubated further for 2.5 h. Cells were then washed twice with 1× cold PBS, lysed directly in 2× Laemmli's sodium dodecyl sulfate (SDS) buffer and the cell lysate subjected to SDS-polyacrylamide gel electrophoresis, followed by Western blot analysis to determine the conversion of LC3-I to LC3-II.
2.6. Coimmunoprecipitation experiments
For the coimmunoprecipitation experiments, each 6 cm dish of cells was resuspended in 400 μl of RIPA buffer and subjected to freeze–thawing five times. Anti-Beclin-1 rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was added to the lysates containing 200 μg of total protein and allowed to mix for 1 h at 4 °C. Protein A agarose beads (Roche, Indianapolis, IN) were added, and the mixture was subjected to end-over-end mixing at 4 °C overnight. Beads were washed four times with cold RIPA buffer, and then 15 μl of Laemmli's SDS buffer was added and the samples were boiled at 100 °C for 5 min to release the immunocomplexes. Samples were separated by SDS-polyacrylamide gel electrophoresis and subjected to Western blot analysis as described above.
2.7. Membrane permeabilization assay
Permeability of the plasma membrane of cells expressing p7 or mutants to hygromycin B (a translation inhibitor) was a modification of the method described by Liao et al. (2006). Briefly, 293FT cells in 6 cm dishes were transfected with plasmids as described above, followed by treatment of the cells with different concentrations of hygromycin B (Roche) for 30 min at 6 h post-transfection. After 24 h post-transfection, the cells were harvested, lysed in RIPA buffer and subjected to freeze–thaw five times. The cell extracts were then spun at 13,000 rpm for 20 min at 4 °C to clarify the lysate. The proteins were immunoprecipitated with anti-Flag beads (Sigma) overnight at 4 °C, and washed three times with RIPA buffer. The samples were then analyzed by 15% SDS-PAGE.
2.8. Immunofluorescence assay
For indirect immunofluorescence staining, transfected Huh 7.5 cells grown on coverslips were fixed with 4% paraformaldehyde in PBS for 15 min. Fixed cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min, blocked with 1% bovine serum albumin (BSA) in PBS for 30 min and incubated with primary antibodies (anti-flag polyclonal (Sigma)) for 1 h. After washing, cells were incubated with Alexa-Fluor-564-conjugated goat anti-rabbit IgG secondary antibodies (Invitrogen) for 1 h. Images were captured with Olympus BX60 fluorescence microscope (Olympus, Japan) using a 100× oil objective.
2.9. Quantification of autoradiographs and statistical analysis
ImageJ open-source gel analysis software (Rasband) was used for the quantification of the intensities of specific bands on autoradiographs. All experiments were repeated at least three times. The mean data is presented. Statistical analysis was performed using the Student's t-test. p < 0.05 was considered significant.
3. Results
3.1. HCV p7 protein induces apoptosis in Huh7.5 cells
To determine if genotype 2a HCV p7 protein can induce apoptosis in host cells, Huh7.5 cells, which are highly permissive for HCV replication, were transiently transfected with a cDNA expression plasmid containing the genotype 2a (JFH-1 strain) p7. For comparison, Huh7.5 cells were also transfected with genotype 1b (S1) HCV p7 protein, the empty vector or an expression plasmid containing the Bax gene, which is a well-known apoptosis inducer that acts via the mitochondria. About 24 h post-transfection, the cells were harvested, lysed and the cell lysate was used for Western blot analysis and measurement of the activation of caspase-3/7, which is a hallmark of apoptosis. As shown in Fig. 1 A, cells overexpressing genotype 1b p7 (abbreviated p7-1b), genotype 2a p7 (abbreviated p7-2a), or Bax have significantly higher level of caspase-3/7 activity than the vector control cells. Similarly, the cleavage of endogenous poly(ADP-ribose) polymerase (PARP), a substrate of activated caspase-3/7, was clearly observed in cells overexpressing p7-1b, p7-2a and Bax but there was less cleavage in cells transfected with the empty vector (Fig. 1B, middle). Western blot analysis showed the expression of flag-tagged protein (Fig. 1B, top) and endogenous actin (Fig. 1B, bottom). The rest of the experiments were performed using only the genotype 2a p7 (hereafter p7).
Fig. 1.
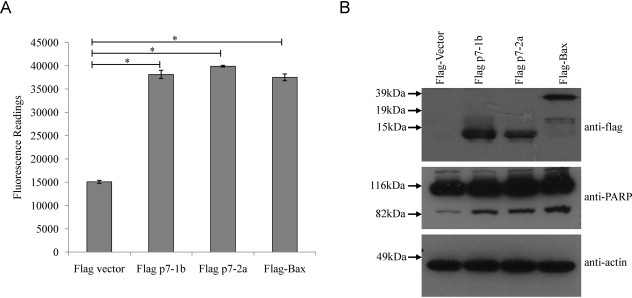
Induction of apoptosis by overexpression of HCV genotype 1b (S1) and genotype 2a p7 in Huh7.5 cells. (A) Apo-One fluorometric assay system from Promega Corporation (Madison, WI) was used to measure the activation of caspase-3/7, which is a hallmark of apoptosis, in Huh7.5 cells that were transfected with empty vector, p7 of genotype 1b (p7-1b), p7 of genotype 2a (p7-2a) and Bax. All experiments were performed in triplicate, and the average values with standard deviations are plotted. (B) Western blot analysis was performed to determine the expression levels of the flag-tagged proteins in the different samples used in (A) (top), the cleavage of endogenous PARP (middle), and the levels of endogenous actin as a loading control of total cell lysates used (bottom). p-Values indicated by asterisks are considered statistically significant, p < 0.01 (*).
3.2. HCV p7-induced apoptosis is caspase-dependent
To determine if p7-induced apoptosis is caspase-dependent, Huh7.5 cells were transiently transfected with the p7 plasmid, followed by treatment with cell permeable pan-caspase inhibitor (zVAD-fmk) or an irrelevant peptide (zFA-fmk) 6 h post-transfection. The level of apoptosis induced, as measured by caspase-3/7 activity, was observed to be significantly inhibited by the pan-caspase inhibitor but not the irrelevant peptide, suggesting the involvement of a caspase-dependent pathway in p7-induced apoptosis (Fig. 2 A). Consistently, there was only a low level of cleaved PARP, as detected by Western blot analysis, in the p7-overexpressing cells treated with zVAD-fmk treated cells (Fig. 2B, middle). However, a high level of cleaved PARP was observed in the p7-overexpressing cells that were treated with zFA-fmk (Fig. 2A, densitometry values below). To confirm the involvement of caspases in p7-induced apoptosis, the terminal uridine nick 3′ end-labeling (TUNEL) assay was performed. Cells overexpressing p7 and treated with the irrelevant peptide (zFA-fmk) had more TUNEL-positive cells than those treated with the pan-caspase inhibitor (zVAD-fmk) (Fig. 4, top right panel).
Fig. 2.
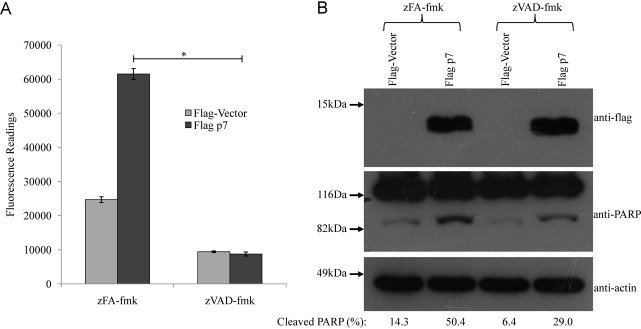
HCV p7 induces apoptosis via caspase-dependent pathways. (A) Apo-One fluorometric assay system from Promega Corporation (Madison, WI) was used to measure the activation of caspase-3/7 in Huh7.5 cells that were transfected with empty vector or flag-tagged p7. The cells were treated with an irrelevant peptide, zFA-fmk (columns 1 and 2) or a pan-caspase inhibitor, zVAD-fmk (columns 3 and 4). All experiments were performed in triplicate, and the average values with standard deviations are plotted. (B) Western blot analysis was performed to determine the expression levels of the flag-tagged proteins in the different samples used in (A) (top), the cleavage of endogenous PARP (middle), and the levels of endogenous actin as a loading control of total cell lysates used (bottom). Percentage cleaved-PARP values are shown below the blots. p-Values indicated by asterisks are considered statistically significant, p < 0.01 (*).
Fig. 4.
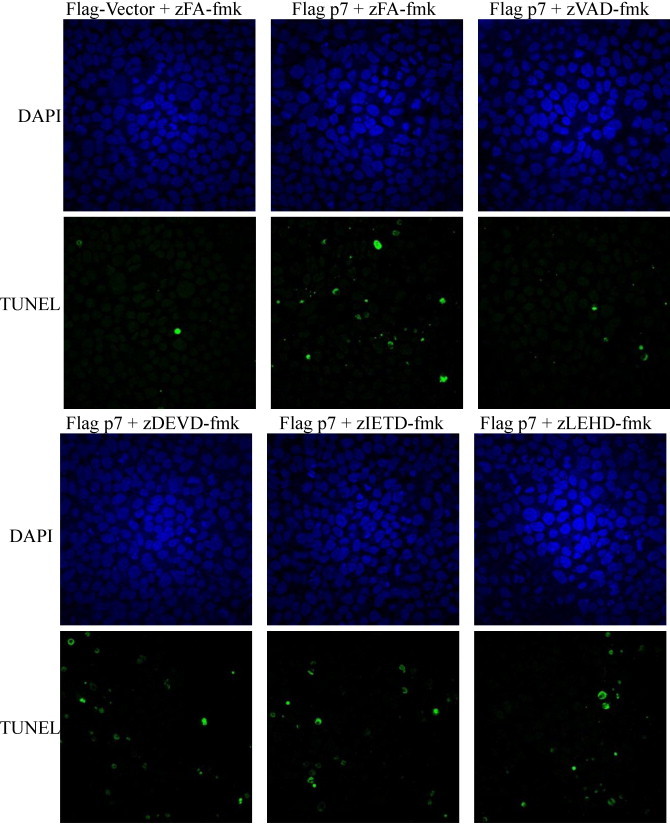
Detection of HCV p7 induced apoptotic cell by TUNEL assay. The DeadEnd™ Fluorometric TUNEL System from Promega Corporation (Madison, WI) was used to measure the activation of caspase-3/7 in Huh7.5 cells that were transiently transfected with empty vector or flag-tagged p7. The cells were treated with an irrelevant peptide (zFA-fmk) or caspase-3, caspase-8 and caspase-9 inhibitors (zDEVD-fmk, zIETD-fmk and zLEHD-fmk). At 24 h post transfection, the cells were fixed and permeabilized, and TUNEL assay was performed.
3.3. HCV p7 protein induces apoptosis via both the intrinsic and extrinsic pathways
Since p7 induces apoptosis via caspase-dependent pathway, the next step was to determine which of the caspase-dependent pathways are involved. To examine this, cell permeable non-toxic peptide caspase inhibitors that bind irreversibly to activated caspases and which have a short half-life (Ekert et al., 1999, Garcia-Calvo et al., 1998) and specific for different caspases, namely caspase-3 (zDEVD-fmk), caspase-8 (zIETD-fmk) and caspase-9 (zLEHD-fmk), were added to the cells 6 h post-transfection. As shown in Fig. 3 A, the level of p7-induced apoptosis after about 24 h post-transfection, as reflected by the caspase-3/7 activity in the cell lysate, was significantly inhibited by all three caspase inhibitors, but not by the irrelevant peptide (zFA-fmk). However, high levels of cleaved PARP were still observed in cells treated with the caspase inhibitors despite the low caspase-3/7 activity (Fig. 3B, middle). This could possibly be due to the cleavage of PARP by caspase-independent events (Yang et al., 2004) and other caspases in the absence of caspase-3 or caspase-7 activation. Indeed, it has been reported that PARP can be cleaved by other proteases and protease-like molecules as well as several caspases (Fernandes-Alnemri et al., 1995, Germain et al., 1999, Lippke et al., 1996, Malireddi et al., 2010, Muzio et al., 1996, Orth et al., 1996, Tewari et al., 1995) albeit at lower efficiency than by caspase-3 or -7. Similarly, the number of TUNEL-positive cells was not much different between cells treated with the irrelevant peptide (zFA-fmk) and the three caspase inhibitors (Fig. 4 , lower panel). One could therefore speculate that, probably because p7-induced PARP cleavage and DNA fragmentation are late apoptosis events (Collins et al., 1997, Duriez and Shah, 1997), they could not be completely blocked when a caspase inhibitor that is specific for only one of the caspases is used. Nevertheless, our results showed that p7-induced apoptosis is indeed caspase-dependent and involves activation of both caspase-8 and -9 which are initiator caspases of the death receptor (extrinsic) and mitochondria-mediated (intrinsic) pathways, respectively, and found upstream of caspase-3/7, the executioner caspases.
Fig. 3.
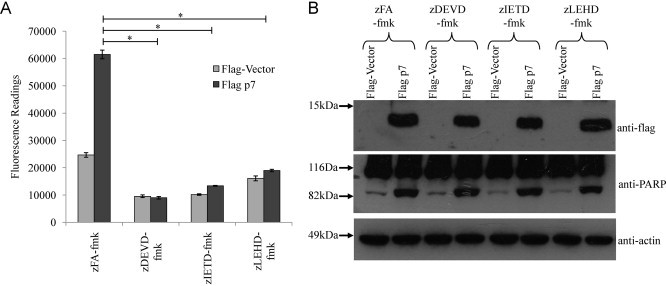
HCV p7 induced apoptosis is via both the intrinsic and extrinsic apoptosis pathways. (A) Apo-One fluorometric assay system from Promega Corporation (Madison, WI) was used to measure the activation of caspase-3/7 in Huh7.5 cells that were transiently transfected with empty vector or flag-tagged p7. The cells were treated with an irrelevant peptide (zFA-fmk) or caspase-3, caspase-8 and caspase-9 inhibitors (zDEVD-fmk, zIETD-fmk and zLEHD-fmk). All experiments were performed in triplicate, and the average values with standard deviations are plotted. (B) Western blot analysis was performed to determine the expression levels of the flag-tagged proteins in the different samples used in (A) (top), the cleavage of endogenous PARP (middle), and the levels of endogenous actin as a loading control of total cell lysates used (bottom). p-Values indicated by asterisks are considered statistically significant, p < 0.01 (*).
3.4. HCV p7-induced apoptosis does not depend on its ion channel activity
The HCV p7 protein possesses ion channel activity and is thus classified in the family of viral ion channel proteins known as viroporins (Griffin et al., 2003, Pavlovic et al., 2003). Moreover, H17, located within the amino-terminal transmembrane domain of p7, has been implicated in p7's ion channel function, and has also been shown to share functional similarity with H37 of the influenza A virus M2 ion channel protein (StGelais et al., 2009). However, the p7 of genotype 2a has an additional H31, which is found in only a few other genotypes and thought to partly influence the gating function of p7 (Brohm et al., 2009, Montserret et al., 2010). Thus, in order to express p7 lacking ion channel activity and to eliminate the probable influence of H31, both H17 and H31 were substituted with A to generate a double substitution mutant p7H17AH31A. Using the membrane permeabilization assay (see Section 2 for details), the overexpression of p7 resulted in the permeabilization of the cell membrane, as the translation of p7 protein was clearly inhibited in the presence of Hygromycin B (Fig. 5 A, left panel). In contrast, the overexpression of p7H17AH31A seems to have little effect on membrane permeability as the addition of Hygromycin B did not inhibit the translation of p7H17AH31A to a great extent (Fig. 5A, right panel).
Fig. 5.
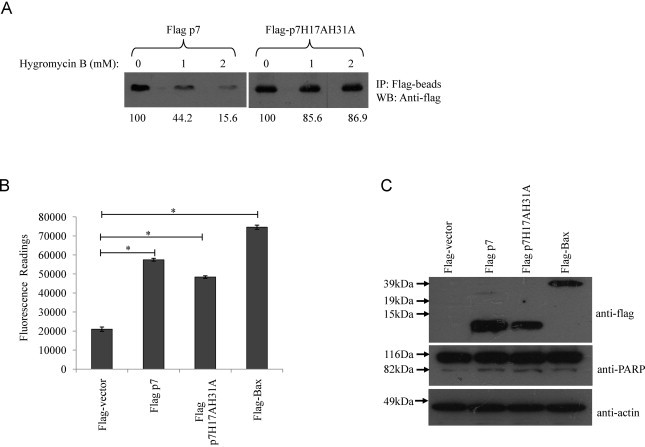
HCV p7 induced apoptosis is independent of its ion channel activity. (A) Modification of membrane permeability by HCV p7 protein. 293FT cells expressing wild-type p7 protein or the mutant (p7H17A31A) were treated with 0, 1, and 2 mM of hygromycin B for 30 min at 6 h post-transfection (lanes 1, 2, and 3). Cell lysates were prepared and the expression of the flag-tagged proteins was detected by immunoprecipitation with anti-Flag antibody under mild washing conditions. Proteins were separated by SDS-PAGE and visualized by autoradiography. The percentages of p7 and p7H17AH31A proteins detected in the presence of hygromycin B were determined by densitometry and indicated at the bottom. (B) Apo-One fluorometric assay system from Promega Corporation was used to measure the activation of caspase-3/7 in Huh7.5 cells 24 h post-transfection with flag-vector, wild-type p7, p7H17AH31A substitution mutant and the known apoptosis inducer, Bax. (B) Western blot analysis was performed to determine the expression levels of the different flag-tagged proteins (top) and the cleavage of endogenous PARP, which is a substrate of activated caspase-3, from 116 to 83 kDa (middle). Equal sample loading was verified by the detection of endogenous actin (bottom). p-Values indicated by asterisks are considered statistically significant, p < 0.01 (*).
When the level of apoptosis of cells overexpressing p7 and the double mutant was subsequently measured, it was observed that both the wild-type p7 protein and the p7H17AH31A mutant induced significantly higher caspase-3/7 activity than the vector (Fig. 5B). It therefore appears that p7-induced apoptosis is independent of its ion channel activity. In addition, the cleavage of endogenous PARP, a substrate of activated caspase-3/7, was clearly observed in Huh7.5 cells overexpressing either the wild-type p7 protein or the p7H17AH31A mutant (Fig. 5C, middle).
3.5. Mutation of the conserved dibasic amino acids in the cytoplasmic domain of p7 protein affects its apoptosis activity
Apart from the histidine residues in p7, there are two highly conserved basic amino acids (K/R33 and R35) in the cytoplasmic domain of p7 protein. When these dibasic residues in p7 are substituted by A or Q, the ion channel activity of p7 is lost and the production of infectious particles is reduced (Griffin et al., 2004, Steinmann et al., 2007). To determine if these amino acid residues are essential for the apoptotic activity of p7 protein, substitution mutants were generated in the background of genotype 2a HCV p7 protein. Using the membrane permeabilization assay, we demonstrated that these mutants lacked ion channel activity (results not shown). However, compared to the wild-type p7 protein, overexpression of the mutants had a significantly lower level of caspase-3/7 activity (Fig. 6 A). Both single and double substitution mutants were expressed at higher levels than the wild-type p7 protein, indicating that the lack of apoptosis is not a consequence of lower protein expression levels (Fig. 6B, top). As it has been reported that these residues (R33 and R35) are important in maintaining the topology of p7 (Steinmann et al., 2007), the mutants could not induce much apoptosis as the wild-type p7 probably because they are orientated wrongly in the host membranes.
Fig. 6.
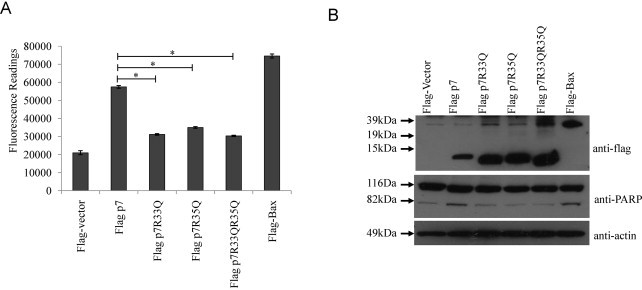
HCV p7 induced apoptosis is dependent on the cellular topology of p7 protein. (A) Apo-One fluorometric assay system from Promega Corporation was used to measure the activation of caspase-3/7 in Huh7.5 cells 24 h post-transfection with flag-vector, wild-type p7, substitution mutants (containing either R to A or changes) and the known apoptosis inducer, Bax. (B) Western blot analysis was performed to determine the expression levels of the different flag-tagged proteins (top) and the cleavage of endogenous PARP, which is a substrate of activated caspase-3, from 116 to 83 kDa (middle). Equal sample loading was verified by the detection of endogenous actin (bottom). p-Values indicated by asterisks are considered statistically significant, p < 0.01 (*).
3.6. HCV p7 protein does not induce autophagic cell death but can interact with Beclin-1
Based on the above characterization of p7, it seems that p7, like M2 protein, can induce caspase-dependent apoptosis, but this is independent of ion channel activity. M2 has also been shown to block autophagosome fusion with lysosomes and resulting in cell death (Gannage et al., 2009). Thus, to examine if p7 protein behaves in the same manner as M2, Huh7.5 cells were transfected with plasmids for expressing p7 and M2 and their effects on the conversion of microtubule-associated protein 1 light chain 3 (LC3) from isomer I to isomer II (LC3-I to LC3-II) was analyzed using Western blot. However, M2 failed to block macroautophagy probably due to the low transfection efficiency in Huh7.5 cells (data not shown). Hence, the experiment was repeated using the highly transfectable 293FT cells (Invitrogen) which is derived from human embryonal kidney cells transformed with the SV40 large T antigen to allow very high levels of protein expression from vectors containing the SV40 origin (http://products.invitrogen.com/ivgn/product/R70007). The results showed that both p7 and M2 induce apoptosis in 293FT cells when overexpressed (Fig. 7 A and B), which is consistent with p7 induction of apoptosis in Huh7.5 cells (see above) and M2 in BHK cells (Madan et al., 2008). Interestingly, the levels of activated caspase-3/7 as well as cleaved PARP were much higher in the cells over-expressing p7 compared to those over-expressing M2. In lane 2, the major band for M2 is ∼20 kDa, which is the expected molecular weight, but there are also some higher molecular weight bands. These are likely to be oligomers of M2 which are resistant to boiling, 20 mM dithiothreitol, and 1% SDS (which are found in the Laemmli SDS loading buffer) as we have previously observed the same phenomenon with another transmembrane protein (Tan et al., 2007).
Fig. 7.
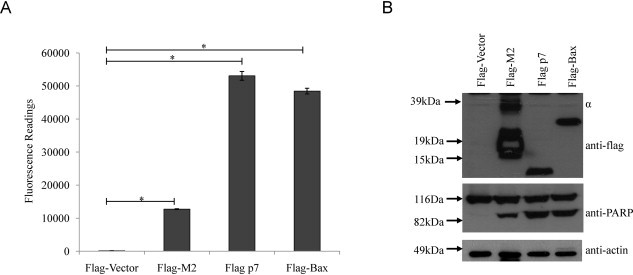
HCV p7 induces a higher level of apoptosis than M2 of influenza A virus. (A) Apo-One fluorometric assay system from Promega Corporation was used to measure the activation of caspase-3/7 in 293FT cells 24 h post-transfection with flag-vector, flag-p7, flag-M2 and the known apoptosis inducer, Bax. (B) Western blot analysis was performed to determine the expression levels of the different flag-tagged proteins (top) and the cleavage of endogenous PARP, which is a substrate of activated caspase-3/7 (middle). Equal sample loading was verified by the detection of endogenous actin (bottom). The multiple bands (indicated by α) in lane 2 are oligomers of M2. p-Values indicated by asterisks are considered statistically significant, p < 0.01 (*).
Consistent with the previous study (Gannage et al., 2009), the level of LC3-II in M2 overexpressing cells was much higher than cells transfected with the empty vector (Fig. 8 A (middle), lanes 2 and 4). After performing four independent experiments and using densitometry to quantify the intensities of LC3-I and II, the conversion to LC3-II (i.e. LC3-II/LC3-I + LC3-II) was found to be significantly higher (63%) in the cells overexpressing M2 as compared to the cells transfected with the empty vector (49%). In contrast, the level of LC3-II (the autophagic marker) in p7 overexpressing cells (41%) was not significantly different from the cells transfected with the empty vector (Fig. 8B). Untransfected cells untreated and treated with ammonium chloride, which is an inhibitor of autophagosome formation, served as negative and positive controls, respectively. As the percentage of conversion to LC-II in the M2 overexpressing cells (63%) was statistically significant as in the case of the ammonium chloride-treated cells (87%), the assay was deemed sensitive enough to detect autophagic cell death. The expression of flag-tagged protein was detected by Western blot analysis (Fig. 8A, top) and the level of endogenous actin (Fig. 8A, bottom) was used as the loading control of the total proteins in the cell lysates. In addition to LC3-II as an autophagosome marker, we also analyzed the accumulation of GFP-LC3 puncta (Bampton et al., 2005, Mizushima, 2009). Huh7.5 cells were transiently transfected with GFP-LC3 and M2 or p7 expression plasmids. Twenty-four hours post transfection; we observed that only cells overexpressing M2 and GFP-LC3 showed an increased accumulation of LC3 puncta by immunofluorescence microscopy analysis (Fig. 9 A).
Fig. 8.
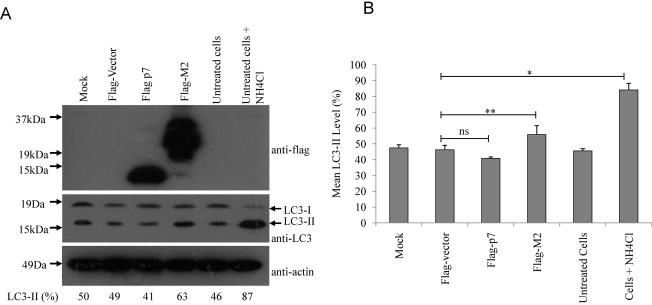
HCV p7 does not induce accumulation of autophagosomes. (A) Measurement of the conversion of LC3-I to LC3-II was used as an autophagic marker. 293FT cells, mock transfected, or transiently transfected with flag vector, flag p7, flag-M2 and untransfected cells untreated or treated with ammonium chloride were analyzed by Western blot analysis using anti-LC3 antibody (middle). Similarly, the expression levels of the different flag-tagged proteins were determined (top). Equal sample loading was verified by the detection of endogenous actin (bottom). The percentage conversion of LC3-I to LC3-II (i.e. LC3-II/LC3-I + LC3-II) is calculated after densitometry was used to quantify the intensities of LC3-I and II in the autoradiographs. Percentage LC3-II values are shown below the blots. (B) The mean LC3-II values quantified for four independent experiments were computed after densitometry and are plotted with standard deviations (error bars). p-Values indicated by asterisks are considered statistically significant, p < 0.05 (**), p < 0.01 (*) and no significant difference (ns).
Fig. 9.
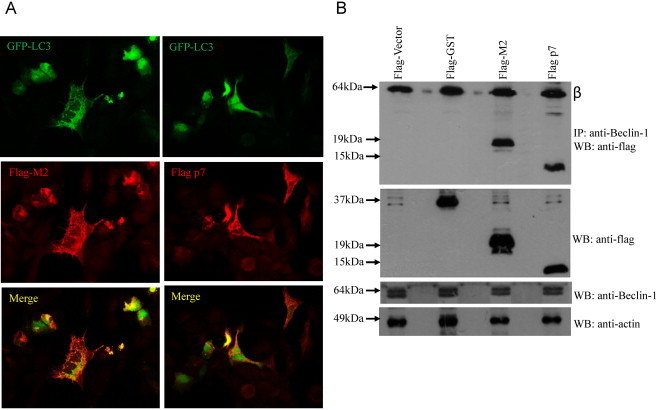
Interaction of M2 and p7 proteins with endogenous Beclin-1 and their effects on the accummulation of GFP-LC3 puncta. (A) Immunofluorescence analysis was performed on 293FT cells transfected with FLAG-tagged GFP-LC3 and expression plasmids encoding either FLAG-tagged M2 or p7. Twenty-four hours after transfection, cells expressing GFP-LC3 (green) and p7 or M2 (red) were analyzed by fluorescence microscopy. Merged images are shown below. (B) Co-immunoprecipitation of M2 and p7 proteins with endogenous Beclin-1. 293FT cells were transfected with flag-vector, flag-GST (negative control), flag-M2 and flag-p7. The cells were harvested at about 24 h post-transfection, lysed, and subjected to IP with anti-Beclin-1 antibody and protein A agarose beads. The amount of flag-tagged proteins that co-immunoprecipitated (IP) with Beclin-1 was determined by Western blot analysis (WB) with an anti-flag antibody (top). The amounts of flag-tagged proteins and endogenous Beclin-1 in the lysates before IP were determined by subjecting aliquots of the lysates to Western blot analysis (middle and bottom). The protein marked with Greek letter beta (β) represents the heavy chain of the antibody used for IP.
To further examine the difference in the ability of p7 and M2 to induce autophagic cell death, co-immunoprecipitation experiment was next performed to determine if they can interact with Beclin-1, which is a key component of the molecular machinery of macroautophagy (reviewed in Kang et al., 2011) and also constitutes a major target for manipulation of autophagy by viruses (Dai et al., 2012, Munz, 2011). As shown in the top panel of Fig. 9B, both flag-M2 and flag-p7, but not by an irrelevant protein, glutathione S-transferase (GST), bound to the endogenous Beclin-1 protein. The expression levels of the flag-tagged proteins in the cell lysates were similar (Fig. 9B, middle) and endogenous Beclin-1 expression was not affected by the expression of the flagged tag proteins (Fig. 9B, bottom). Hence, it appears that the binding of p7 to Beclin-1 is not sufficient to interfere with the autophagic process.
4. Discussion
Currently, apoptosis or caspase-dependent cell death is the most well characterized cell death pathway, although there is increasing evidence that other non-apoptotic or caspase-independent cell death pathways are involved in programmed cell death (Hetz et al., 2005). Autophagic cell death and programmed necrosis (necroptosis) are cell death mechanisms which are also important in viral infections and are thought to be exploited by viruses for their survival (Edinger and Thompson, 2004, Park et al., 2012). There is also some crosstalk or interrelationship between the different cell death mechanisms which are exploited by some viruses for their survival in the host, as reported for influenza A virus which determines the death of its host by inducing apoptosis and blocking autophagy (Gannage et al., 2009). With some recent studies suggesting that HCV induces caspase-independent cell death such as autophagy (reviewed in Dreux and Chisari, 2011), it is possible that HCV, like influenza A virus, might also be exploiting the interrelationship between apoptosis and non-apoptotic cell death for its survival in the host. Incidentally, the protein of interest in this study, p7 protein, happens to share some functional relationship with influenza A virus M2 protein (Mihm et al., 2006), so it is tempting to speculate that p7 and M2 might play similar roles in cell death modulation.
Currently, the apoptotic activity of p7 of different HCV genotypes has not been reported except that of genotype 1b (Madan et al., 2008). Since the genotype 2a JFH1 strain is the only one that can replicate efficiently in permissive cells, such as Huh7.5 cells, without adaption, this study focuses on characterizing the cell death pathways activated by the p7 protein of the JFH-1 strain. To our knowledge, this is the first time that the p7 protein of genotype 2a has been shown to induce apoptosis to a similar level as that of genotype 1b p7 (Fig. 1). As for genotype 1b p7, the result is similar to the observation by Madan et al. (2008) except that Huh7.5 cells were used in this study while BHK cells were used in the previous study. Further characterization of the apoptosis induced by the p7 protein of genotype 2a reveals that p7-induced apoptosis was efficiently blocked by a pan-caspase inhibitor as well as inhibitors specific for caspases 3, 8 or 9 (Fig. 2, Fig. 3). In mammalian cells, apoptosis can be induced via two major pathways; the death receptor pathway (extrinsic pathway), triggered by the binding of FasL to Fas (CD95), with caspase-8 as the initiator caspase; and the mitochondria-mediated pathway (intrinsic pathway), induced by the mitochondria in response to DNA damage, oxidative stress and viral proteins, with caspase-9 as the initiator caspase (Kumar, 2007). Thus, our results confirm that p7-induced apoptosis is indeed caspase-dependent and involves both the intrinsic and extrinsic apoptosis pathways.
Given that some previous studies have reported that ions channels play a key role in the regulation of cell physiology and in the induction of apoptotic cell death (Burg et al., 2006, Lang et al., 2003, Szabo et al., 2004), it was important to also determine whether this property of p7 has a link with its apoptotic activity. Although, details and the exact function of most ion channels in apoptosis mediation are currently unknown, it is believed that some viruses employ the control of ion channel homeostasis to promote viral persistence (Mankouri et al., 2009). A recent study has demonstrated a relationship between ion channel activity and apoptosis by showing that the caspase-dependent apoptosis induced by the SARS-coronavirus 3a protein (which possesses ion channel activity) is abolished when its ion channel activity is blocked (Chan et al., 2009). In the case of p7, substitution of the two histidine residues (H17 and H31), which have been implicated in the gating function of p7 (Brohm et al., 2009, Montserret et al., 2010), with alanine, did not reduce its apoptotic activity greatly (Fig. 5B), although the resultant double mutant had lost most of its ion channel activity (Fig. 5A). It thus suggests that the caspase-dependent apoptosis induced by p7 protein is independent of its ion channel function. Nevertheless, p7 has to be properly folded and maintain the correct topology in the cell membranes in order to induce apoptosis as single or double substitution of the dibasic amino acid residues R33 and R35, to glutamine (Q) in the background of genotype 2a HCV p7 protein significantly reduced the level of apoptosis induced by p7 (Fig. 6) and to function as an ion channel. This observation is in line with a previous report that mutating the dibasic motif of p7 can substantially impact on the polyprotein processing and thus distort the topology of p7 protein (Steinmann et al., 2007).
Recent studies (reviewed in Eisenberg-Lerner et al., 2009, Gordy and He, 2012, Zhou et al., 2011) have shown that there seems to exist a connection between pathways that regulate autophagy and apoptosis, with a number of molecules including Bcl-2 family proteins, Beclin-1, caspase-8, caspase-9, phosphoinositide-3-kinase class I, and ceramide implicated in the control of both apoptosis and autophagy. Apart from apoptotic or caspase-dependent cell death, HCV is reported to induce non-apoptotic cell death such as autophagy (reviewed in Dreux and Chisari, 2011). In one such report, when the autophagy machinery of HCV-infected hepatocytes was interrupted, it resulted in the induction of apoptosis (Shrivastava et al., 2011). Thus, it is believed that HCV exploits the autophagy pathway for its propagation and survival in the host, although the biological significance of this in the lifecycle HCV is still unclear (Taguwa et al., 2011). Similarly, the pro-apoptotic M2 ion channel protein of influenza A virus is reported to cause accumulation of autophagosomes by blocking their fusion with lysosomes which thus leads to apoptosis of the infected cells (Gannage et al., 2009). Since both p7 and M2 are viroporins and are known to induce caspase-dependent cell death, several experiments were therefore conducted to determine if the p7 protein behaves like M2 protein of influenza A virus. Our results showed that unlike M2, the overexpression of p7 did not significantly increase the conversion of LCI to LCII (Fig. 8) or the accumulation of GFP-LC3 puncta (Fig. 9A) suggesting that probably p7 was not involved in autophagosome maturation despite the fact that both flag-tagged M2 and p7 could interact with endogenous Beclin-1 in co-immunoprecipitation experiment (Fig. 9B). Since a number of viral proteins including M2 have been reported to interact with Beclin-1 and thus interfere with the class III PI3K (UVRAG-Beclin-1-hVps34) complex's function in autophagosome maturation (Dai et al., 2012, He and Levine, 2010, Munz, 2011), it is therefore tempting to speculate that probably M2 and p7 bind to different domains in the class III PI3K complex, with only M2 able to interact with and inhibit autophagosome maturation (Gannage et al., 2010).
Taken together, these results indicate that HCV p7 protein induces caspase-dependent apoptosis which is independent of its ion channel activity. Although p7 and M2 share some functional properties in terms of ion channel and pro-apoptotic activity, they seem to differ in their abilities to induce autophagic cell death. Further studies are underway in our laboratory to identify residues or motifs that are responsible for the apoptosis activity of p7. The HCVcc system will then be used to generate mutant virus containing mutation(s) that abolishes p7 pro-apoptotic activity and establish the functional significance of the apoptosis activity of p7 during HCV infection. In addition, identification of these residues will allow the study of the interaction between HCV p7 and other viral and host factors in cell death signaling during an infection.
Acknowledgements
We thank T. Wakita for the JFH-1 construct and Megha Haridas Upadya for technical assistance in the fluorescence and confocal microscopy work. This work was supported by a NUHS Cross Department Collaborative Grant [R-182-000-171-733] to S. G. Lim and Y.-J. Tan, and a NUS/NUHS start-up grant [R-182-000-156-720 and R-182-000-156-133] to Y.-J. Tan. The GFP-LC3 expression vector was a kind gift from T. Yoshimori.
References
- Aweya J.J., Tan Y.J. Modulation of programmed cell death pathways by the hepatitis C virus. Frontiers in Bioscience. 2011;16:608–618. doi: 10.2741/3709. [DOI] [PubMed] [Google Scholar]
- Bampton E.T., Goemans C.G., Niranjan D., Mizushima N., Tolkovsky A.M. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1(1):23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Boson B., Granio O., Bartenschlager R., Cosset F.L. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathogen. 2011;7(7):e1002144. doi: 10.1371/journal.ppat.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohm C., Steinmann E., Friesland M., Lorenz I.C., Patel A., Penin F., Bartenschlager R., Pietschmann T. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. Journal of Virology. 2009;83(22):11682–11693. doi: 10.1128/JVI.00691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R.S. Hepatitis C and liver transplantation. Nature. 2005;436(7053):973–978. doi: 10.1038/nature04083. [DOI] [PubMed] [Google Scholar]
- Burg E.D., Remillard C.V., Yuan J.X. K+ channels in apoptosis. Journal of Membrane Biology. 2006;209(1):3–20. doi: 10.1007/s00232-005-0838-4. [DOI] [PubMed] [Google Scholar]
- Chan C.M., Tsoi H., Chan W.M., Zhai S., Wong C.O., Yao X., Chan W.Y., Tsui S.K., Chan H.Y. The ion channel activity of the SARS-coronavirus 3a protein is linked to its pro-apoptotic function. International Journal of Biochemistry and Cell Biology. 2009;41(11):2232–2239. doi: 10.1016/j.biocel.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.L., Morgan T.R. The natural history of hepatitis C virus (HCV) infection. International Journal of Medical Sciences. 2006;3(2):47–52. doi: 10.7150/ijms.3.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.B., Seo S.Y., Kirsch D.G., Sheu T.T., Cheng W.C., Hardwick J.M. Alternate functions of viral regulators of cell death. Cell Death and Differentiation. 2006;13(8):1318–1324. doi: 10.1038/sj.cdd.4401964. [DOI] [PubMed] [Google Scholar]
- Collins J.A., Schandi C.A., Young K.K., Vesely J., Willingham M.C. Major DNA fragmentation is a late event in apoptosis. Journal of Histochemistry and Cytochemistry. 1997;45(7):923–934. doi: 10.1177/002215549704500702. [DOI] [PubMed] [Google Scholar]
- Dai J.P., Li W.Z., Zhao X.F., Wang G.F., Yang J.C., Zhang L., Chen X.X., Xu Y.X., Li K.S. A drug screening method based on the autophagy pathway and studies of the mechanism of evodiamine against influenza A virus. PLoS One. 2012;7(8):e42706. doi: 10.1371/journal.pone.0042706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreux M., Chisari F.V. Impact of the autophagy machinery on hepatitis C virus infection. Viruses. 2011;3(8):1342–1357. doi: 10.3390/v3081342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duriez P.J., Shah G.M. Cleavage of poly(ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochimie et Biologie CellulaireBiochemistry and Cell Biology. 1997;75(4):337–349. [PubMed] [Google Scholar]
- Edinger A.L., Thompson C.B. Death by design: apoptosis, necrosis and autophagy. Current Opinion in Cell Biology. 2004;16(6):663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Eisenberg-Lerner A., Bialik S., Simon H.U., Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death and Differentiation. 2009;16(7):966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- Ekert P.G., Silke J., Vaux D.L. Caspase inhibitors. Cell Death and Differentiation. 1999;6(11):1081–1086. doi: 10.1038/sj.cdd.4400594. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T., Litwack G., Alnemri E.S. Mch2, a new member of the apoptotic Ced-3/Ice cysteine protease gene family. Cancer Research. 1995;55(13):2737–2742. [PubMed] [Google Scholar]
- Fischer R., Baumert T., Blum H.E. Hepatitis C virus infection and apoptosis. World Journal of Gastroenterology. 2007;13(36):4865–4872. doi: 10.3748/wjg.v13.i36.4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannage M., Dormann D., Albrecht R., Dengjel J., Torossi T., Ramer P.C., Lee M., Strowig T., Arrey F., Conenello G., Pypaert M., Andersen J., Garcia-Sastre A., Munz C. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host & Microbe. 2009;6(4):367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannage M., Ramer P.C., Munz C. Targeting Beclin 1 for viral subversion of macroautophagy. Autophagy. 2010;6(1):166–167. doi: 10.4161/auto.6.1.10624. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson E.P., Leiting B., Ruel R., Nicholson D.W., Thornberry N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. Journal of Biological Chemistry. 1998;273(49):32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Germain M., Affar E.B., D’Amours D., Dixit V.M., Salvesen G.S., Poirier G.G. Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. Journal of Biological Chemistry. 1999;274(40):28379–28384. doi: 10.1074/jbc.274.40.28379. [DOI] [PubMed] [Google Scholar]
- Gonzalez M.E., Carrasco L. Viroporins. FEBS Letters. 2003;552(1):28–34. doi: 10.1016/s0014-5793(03)00780-4. [DOI] [PubMed] [Google Scholar]
- Gordy C., He Y.W. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012;3(1):17–27. doi: 10.1007/s13238-011-1127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin S.D., Beales L.P., Clarke D.S., Worsfold O., Evans S.D., Jaeger J., Harris M.P., Rowlands D.J. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Letters. 2003;535(1-3):34–38. doi: 10.1016/s0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- Griffin S.D.C., Harvey R., Clarke D.S., Barclay W.S., Harris M., Rowlands D.J. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. Journal of General Virology. 2004;85:451–461. doi: 10.1099/vir.0.19634-0. [DOI] [PubMed] [Google Scholar]
- He C., Levine B. The Beclin 1 interactome. Current Opinion in Cell Biology. 2010;22(2):140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C.A., Torres V., Quest A.F. Beyond apoptosis: nonapoptotic cell death in physiology and disease. Biochimie et Biologie CellulaireBiochemistry and Cell Biology. 2005;83(5):579–588. doi: 10.1139/o05-065. [DOI] [PubMed] [Google Scholar]
- Kang R., Zeh H.J., Lotze M.T., Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death and Differentiation. 2011;18(4):571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq S., Jahan S., Hassan S. Hepatitis C virus p7:molecular function and importance in hepatitis C virus life cycle and potential antiviral target. Liver International. 2011;31(5):606–617. doi: 10.1111/j.1478-3231.2010.02442.x. [DOI] [PubMed] [Google Scholar]
- Kim C.S., Keum S.J., Jang S.K. Generation of a cell culture-adapted hepatitis C virus with longer half life at physiological temperature. PLoS One. 2011;6(8):e22808. doi: 10.1371/journal.pone.0022808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. Caspase function in programmed cell death. Cell Death and Differentiation. 2007;14(1):32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- Lang F., Lang K.S., Wieder T., Myssina S., Birka C., Lang P.A., Kaiser S., Kempe D., Duranton C., Huber S.M. Cation channels, cell volume and the death of an erythrocyte. Pflugers Archiv. 2003;447(2):121–125. doi: 10.1007/s00424-003-1150-8. [DOI] [PubMed] [Google Scholar]
- Liao Y., Yuan Q., Torres J., Tam J.P., Liu D.X. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology. 2006;349(2):264–275. doi: 10.1016/j.virol.2006.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Lindenbach B.D., Pragai B.M., McCourt D.W., Rice C.M. Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two distinct E2-specific products with different C termini. Journal of Virology. 1994;68(8):5063–5073. doi: 10.1128/jvi.68.8.5063-5073.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippke J.A., Gu Y., Sarnecki C., Caron P.R., Su M.S. Identification and characterization of CPP32/Mch2 homolog 1, a novel cysteine protease similar to CPP32. Journal of Biological Chemistry. 1996;271(4):1825–1828. doi: 10.1074/jbc.271.4.1825. [DOI] [PubMed] [Google Scholar]
- Madan V., Castello A., Carrasco L. Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell Microbiology. 2008;10(2):437–451. doi: 10.1111/j.1462-5822.2007.01057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malireddi R.K., Ippagunta S., Lamkanfi M., Kanneganti T.D. Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. Journal of Immunology. 2010;185(6):3127–3130. doi: 10.4049/jimmunol.1001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri J., Dallas M.L., Hughes M.E., Griffin S.D., Macdonald A., Peers C., Harris M. Suppression of a pro-apoptotic K+ channel as a mechanism for hepatitis C virus persistence. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(37):15903–15908. doi: 10.1073/pnas.0906798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihm U., Grigorian N., Welsch C., Herrmann E., Kronenberger B., Teuber G., von Wagner M., Hofmann W.P., Albrecht M., Lengauer T., Zeuzem S., Sarrazin C. Amino acid variations in hepatitis C virus p7 and sensitivity to antiviral combination therapy with amantadine in chronic hepatitis C. Antiviral Therapy. 2006;11(4):507–519. [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods in Enzymology. 2009;452:13–23. doi: 10.1016/S0076-6879(08)03602-1. [DOI] [PubMed] [Google Scholar]
- Montserret R., Saint N., Vanbelle C., Salvay A.G., Simorre J.P., Ebel C., Sapay N., Renisio J.G., Bockmann A., Steinmann E., Pietschmann T., Dubuisson J., Chipot C., Penin F. NMR structure and ion channel activity of the p7 protein from hepatitis C virus. Journal of Biological Chemistry. 2010;285(41):31446–31461. doi: 10.1074/jbc.M110.122895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz C. Beclin-1 targeting for viral immune escape. Viruses. 2011;3(7):1166–1178. doi: 10.3390/v3071166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M., Chinnaiyan A.M., Kischkel F.C., O’Rourke K., Shevchenko A., Ni J., Scaffidi C., Bretz J.D., Zhang M., Gentz R., Mann M., Krammer P.H., Peter M.E., Dixit V.M. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85(6):817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Orth K., Chinnaiyan A.M., Garg M., Froelich C.J., Dixit V.M. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. Journal of Biological Chemistry. 1996;271(28):16443–16446. [PubMed] [Google Scholar]
- Park J., Kang W., Ryu S.W., Kim W.I., Chang D.Y., Lee D.H., Park do Y., Choi Y.H., Choi K., Shin E.C., Choi C. Hepatitis C virus infection enhances TNFalpha-induced cell death via suppression of NF-kappaB. Hepatology. 2012;56(3):831–840. doi: 10.1002/hep.25726. [DOI] [PubMed] [Google Scholar]
- Pavlovic D., Neville D.C., Argaud O., Blumberg B., Dwek R.A., Fischer W.B., Zitzmann N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):6104–6108. doi: 10.1073/pnas.1031527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband, W.S., ImageJ. U. S. National Institutes of Health, Bethesda, Maryland, USA (accessed from <http://imagej.nih.gov/ij/>), 1999–2012.
- Shrivastava S., Raychoudhuri A., Steele R., Ray R., Ray R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology. 2011;53(2):406–414. doi: 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. Genetic diversity and evolution of hepatitis C virus—15 years on. Journal of General Virology. 2004;85(Pt 11):3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- Soo H.M., Garzino-Demo A., Hong W., Tan Y.H., Tan Y.J., Goh P.Y., Lim S.G., Lim S.P. Expression of a full-length hepatitis C virus cDNA up-regulates the expression of CC chemokines MCP-1 and RANTES. Virology. 2002;303(2):253–277. doi: 10.1006/viro.2002.1617. [DOI] [PubMed] [Google Scholar]
- Steinmann E., Penin F., Kallis S., Patel A.H., Bartenschlager R., Pietschmann T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathogen. 2007;3(7):e103. doi: 10.1371/journal.ppat.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann E., Pietschmann T. Hepatitis C virus P7-A viroporin crucial for virus assembly and an emerging target for antiviral therapy. Viruses-Basel. 2010;2(9):2078–2095. doi: 10.3390/v2092078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- StGelais C., Foster T.L., Verow M., Atkins E., Fishwick C.W., Rowlands D., Harris M., Griffin S. Determinants of hepatitis C virus p7 ion channel function and drug sensitivity identified in vitro. Journal of Virology. 2009;83(16):7970–7981. doi: 10.1128/JVI.00521-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I., Adams C., Gulbins E. Ion channels and membrane rafts in apoptosis. Pflugers Archiv. 2004;448(3):304–312. doi: 10.1007/s00424-004-1259-4. [DOI] [PubMed] [Google Scholar]
- Taguwa S., Kambara H., Fujita N., Noda T., Yoshimori T., Koike K., Moriishi K., Matsuura Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. Journal of Virology. 2011;85(24):13185–13194. doi: 10.1128/JVI.06099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y.X., Tan T.H., Lee M.J., Tham P.Y., Gunalan V., Druce J., Birch C., Catton M., Fu N.Y., Yu V.C., Tan Y.J. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the Bcl-XL protein. Journal of Virology. 2007;81(12):6346–6355. doi: 10.1128/JVI.00090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedbury P., Welbourn S., Pause A., King B., Griffin S., Harris M. The subcellular localization of the hepatitis C virus non-structural protein NS2 is regulated by an ion channel-independent function of the p7 protein. Journal of General Virology. 2011;92:819–830. doi: 10.1099/vir.0.027441-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari M., Quan L.T., O’Rourke K., Desnoyers S., Zeng Z., Beidler D.R., Poirier G.G., Salvesen G.S., Dixit V.M. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81(5):801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Krausslich H.G., Mizokami M., Bartenschlager R., Liang T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nature Medicine. 2005;11(7):791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, 2012. Hepatitis C. Fact sheet No. 164, July 2012 ed., World Health Organization.
- Xiao J.H., Davidson I., Matthes H., Garnier J.M., Chambon P. Cloning, expression, and transcriptional properties of the human enhancer factor TEF-1. Cell. 1991;65(4):551–568. doi: 10.1016/0092-8674(91)90088-g. [DOI] [PubMed] [Google Scholar]
- Yang Y., Zhao S., Song J. Caspase-dependent apoptosis and -independent poly(ADP-ribose) polymerase cleavage induced by transforming growth factor beta1. International Journal of Biochemistry and Cell Biology. 2004;36(2):223–234. doi: 10.1016/s1357-2725(03)00215-2. [DOI] [PubMed] [Google Scholar]
- Zhou F., Yang Y., Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS Journal. 2011;278(3):403–413. doi: 10.1111/j.1742-4658.2010.07965.x. [DOI] [PubMed] [Google Scholar]