

Highlights
-
•
Novel viruses were identified in feces from cattle by metagenomics approach.
-
•
These viruses had a standard picornavirus genome organization.
-
•
They are related to unclassified Chinese picornaviruses from bat, cat and dog.
-
•
These viruses were detected in 23% (20/87) feces from cattle in Hokkaido in Japan.
Keywords: Bovine, Feces, Japan, Metagenomics, Novel picornavirus
Abstract
We identified novel viruses in feces from cattle with diarrhea collected in 2009 in Hokkaido Prefecture, Japan, by using a metagenomics approach and determined the (near) complete sequences of the virus. Sequence analyses revealed that they had a standard picornavirus genome organization, i.e. 5′ untranslated region (UTR) - L- P1 (VP4- VP3- VP2- VP1) - P2 (2A- 2B- 2C) - P3 (3A- 3B- 3C-3D) - 3′UTR- poly(A). They are closely related to other unclassified Chinese picornaviruses; bat picornaviruses group 1–3, feline picornavirus, and canine picornavirus, sharing 45.4–51.4% (P1), 38.0–44.9% (P2), and 49.6–53.3% (P3) amino acid identities, respectively. The phylogenetic analyses and detailed genome characterization showed that they, together with the unclassified Chinese picornaviruses, grouped as a cluster for the P1, 2C, 3CD and VP1 coding regions. These viruses had conserved features (e.g. predicted protein cleavage sites, presence of a leader protein, 2A, 2C, 3C, and 3D functional domains), suggesting they have a common ancestor. Reverse-transcription-PCR assays, using specific primers designed from the 5′UTR sequence of these viruses, showed that 23.0% (20/87) of fecal samples from cattle with diarrhea were positive, indicating the prevalence of these picornavirus in the Japanese cattle population in Hokkaido Prefecture. However, further studies are needed to investigate the pathogenic potential and etiological role of these viruses in cattle.
1. Introduction
Viruses in the family Picornaviridae are small, icosahedral and nonenveloped, with a polyadenylated, single-stranded, positive-sense RNA genome and these viruses are currently divided into 29 officially recognized genera: Aphthovirus, Aquamavirus, Avihepatovirus, Avisivirus, Cardiovirus, Cosavirus, Dicipivirus, Enterovirus, Erbovirus, Gallivirus, Hepatovirus, Hunnivirus, Kobuvirus, Kunsagivirus, Megrivirus, Mischivirus, Mosavirus, Oscivirus, Parechovirus, Pasivirus, Passerivirus, Rosavirus, Sakobuvirus, Salivirus, Sapelovirus, Senecavirus, Sicinivirus, Teschovirus and Tremovirus (Knowles et al., 2012, Adams et al., 2013) plus several candidate genera. Recently, a number of novel picornaviruses were discovered from different hosts (Boros et al., 2012, Pankovics et al., 2012, Reuter et al., 2012, Sauvage et al., 2012, Boros et al., 2013, Liao et al., 2014) and the list of picornaviruses is rapidly expanding (http://www.picornaviridae.com). In the Asia-Pacific region, several new picornaviruses, including three bat picornaviruses, one feline picornavirus, and one canine picornavirus were discovered in Hong Kong (Lau et al., 2011, Lau et al., 2012, Woo et al., 2012). However, their clinical relevance remains to be elucidated. Picornaviruses can cause various symptoms including diarrhea and they are widely distributed in animals (Whitton et al., 2005). In the course of surveillance for unexplained bovine diarrhea, using a metagenomics approach, we identified some novel picornaviruses. The present study describes the identification of these new picornaviruses (termed bovine Japanese picornaviruses) from fecal samples of calves with diarrhea in Hokkaido Prefecture, Japan and the genomic characterization of these viruses.
2. Material and methods
2.1. Sample collection of bovine feces and preparation
Forty fecal samples obtained from cattle with diarrhea in Hokkaido Prefecture in 2009 were analyzed using deep sequencing. The most common bovine viral pathogens inducing diarrhea, including group A, B, and C rotaviruses, bovine torovirus, and bovine coronavirus had been ruled out as the cause of the diarrhea by using reverse transcription (RT)-PCR diagnostic assays (20%; 10 of 50 samples). Samples were collected directly from the rectums to prevent contamination from the environment or between samples. Fecal samples were diluted 1:9 (w/v) in sterile phosphate-buffered saline with 1% antibiotics, and centrifuged at 10,000 × g for 10 min followed by filtration through a 0.22 μm filter. The filtrates were stored at −80 °C until used for genomic analysis and virus isolation.
2.2. Viral RNA extraction, cDNA library construction, and deep sequencing
Viral RNAs were extracted from the filtrates using TRIzol® LS Reagent (Life technologies, Carlsbad, CA, USA) and treated with DNase I (Takara Bio, Shiga, Japan). cDNA libraries for deep sequencing were constructed from RNA using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s guidelines. Whole genome sequencing was determined by deep sequencing and the rapid amplification cDNA end method (RACE) (Roche Diagnostics GmbH, Mannheim, Germany) and 5′-Full RACE Core Set (Takara Bio). After assessing the library quality and quantity on a Bioanalyzer® and Qubit® 2.0 Fluorimeter (Invitrogen, Carlsbad, CA, USA), deep sequencing was conducted by using a MiSeq bench-top sequencer (Illumina, San Diego, CA, USA) using 51 nucleotide (nt) single reads. The runs were performed three times. Sequence data analysis was performed using the MiSeq Reporter v1 (Illumina) to generate FASTQ formatted sequence data. Trimmed reads were assembled into contigs using de novo assembly in CLC Genomics Workbench 6.0 (CLC bio, Aarhus, Denmark).
2.3. Genome analysis
Viral sequence searches for assembled contigs were performed with the BLAST program in the National Center for Biotechnology Information non-redundant nucleotide database. Nucleotide sequences were aligned using ClustalW (Thompson et al., 1997), and phylogenetic analysis was performed by the maximum likelihood method using MEGA5.22 (Tamura et al., 2011). The trees were statistically supported by bootstrapping with 1000 replicates (Felsenstein, 1985). Sequence similarity calculations were performed by using CLC Genomics Workbench 6.0 (CLC bio). Hypothetical polyprotein cleavage sites in the picornavirus were predicted using the NetPicoRNA program (Blom et al., 1996). Secondary structure elements within the 5′UTR were predicted using Mfold (Zuker, 2003).
2.4. RT-PCR for 5′UTR
RT-PCR was performed with PrimeScript™ One Step RT-PCR Kit Ver.2 (TaKaRa Bio) using RNA extracted from fecal samples. The primers F (5′- CTT TTT CCC CCT CTT GYA AC -3′) and R (5′- TTA GCC GCA TTC AGG GKC CTG GAG -3′) were designed using the nucleotide sequence from the 5′UTR of the bovine Japanese picornaviruses. The production and amplification of the cDNA fragment was achieved under the following conditions: 50 °C for 30 min and 94 °C for 2 min, followed by 40 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min, and a final extension at 72 °C for 5 min. The RT-PCR products were electrophoresed on a 2% agarose gel and purified.
2.5. Accession numbers of Japanese bovine picornavirus nucleotide sequences
The nucleotide sequences of the bovine Japanese picornavirus (Bo-11-39/2009/JPN; Bo-11-39, Bo-12-3/2009/JPN; Bo-12-3, Bo-12-7/2009/JPN; Bo-12-7, Bo-12-11/2009/JPN; Bo-12-11, Bo-12-38/2009/JPN; Bo-12-38) were deposited in the DNA Data Bank of Japan, the DDBJ/EMBL/GenBank, under accession numbers LC006971 and LC036579 to LC036582.
3. Results
3.1. Identification of novel picornaviruses by deep sequencing
In the course of surveillance for unexplained bovine diarrhea using a metagenomics approach, we found 18 large sequence contigs that had a length of approximately 6000–8000 nucleotides (nt) from 14 samples. Ten, two and one sequence contigs showed high sequence homology to published bovine kobuvirus, bovine astrovirus and enterovirus F genomes respectively using nucleotide BLAST searches. Five of 18 sequence contigs, that had a length of approximately 7500 nt, were derived from five calves with diarrhea kept on five dairy farms and exhibited similarity to nucleotide sequences of other unclassified picornavirus genomes. Three of the five samples had more than two contigs (two samples: unclassified picornavirus and bovine kobuvirus, one sample; unclassified picornavirus, bovine kobuvirus and bovine astrovirus). Since the nucleotide sequence homology of the five contigs to other picornavirus genomes was rather low (<50%), we performed further analyses of these five contigs which were termed Bo-11-39, Bo-12-3, Bo-12-7, Bo-12-11, and Bo-12-38.
3.2. Genome characterization of the bovine Japanese picornavirus
The complete genome length of Bo-11-39 was 7570 nt with G + C content of 0.41, excluding the poly(A) tail. We determined nearly complete genome sequences (including the complete open reading frames) of Bo-12-3, Bo-12-7, Bo-12-11 and Bo-12-38 but could not completely determine the sequence due to insufficient amount of samples. The genome organization of each is similar to that of other picornaviruses. They include a single large open reading frame (ORF) containing 6915 nt (Bo-11-38), or 7023 nt (Bo-12-3, 7, 11 and 38), which encodes a polyprotein precursor of 2305 amino acids (aa) (Bo-11-39), or 2341 aa (Bo-12-3, 7, 11 and 38), flanked by the 5'UTR and the 3'UTR with a poly(A) tail. Cleavage sites within the polyprotein were predicted by NetPicoRNA analysis and alignments with other picornaviruses. The junction sites appeared to be mainly Gln(Q)/Gly(G), except for predicted cleavage sites of the L/VP4 (Arg(R)/Gly(G), Bo-11-38 and Thr(T)/Gly(G), Bo-12-3, 7, 11 and 38), VP4/VP2 (Lys(K)/Ser(S)), and VP1/2A (Asp(D)/ Gly(G), Bo-12-3, 7, 11 and 38), respectively (Fig. 1 ). The coding region consists of a leader protein (L), a structural protein region P1 (833 aa), and the non-structural protein regions P2 (645 aa in Bo-11-39 and 654 aa in Bo-12-3, 7, 11 and 38), and P3 (768 aa in Bo-11-39 and 793 aa in Bo-12-3, 7, 11 and 38). Pair-wise alignment of the amino acid sequences of Bo-11-39 and Bo-12-3, 7, 11 and 38 with those of other picornaviruses revealed that they shared higher sequence identities with bovine enterovirus 1 (P1: 36.1% and 34.8%; P2: 30.0% and 29.0%; P3: 43.2% and 42.0%) and porcine sapelovirus 1 (P1: 38.9% and 39.5%; P2: 30.5% and 28.0%; P3: 45.9% and 44.0%) than with other representative viruses from the recognized genera within the Picornaviridae. However, the highest sequence identities were found with unclassified Chinese picornaviruses; bat picornavirus group 1–3 (P1: 45.4–48.4%; P2: 40.0–42.5%; P3: 49.6–55.2%), feline picornavirus (P1: 49.3–51.4%; P2: 41.4–44.9%; P3: 51.5–52.3 %), and canine picornavirus (P1: 48.8–50.3%; P2: 38.0–40.7%; P3: 53.3%) (Table 1 ). These viruses are clearly very different from all previously described picornaviruses.
Fig. 1.
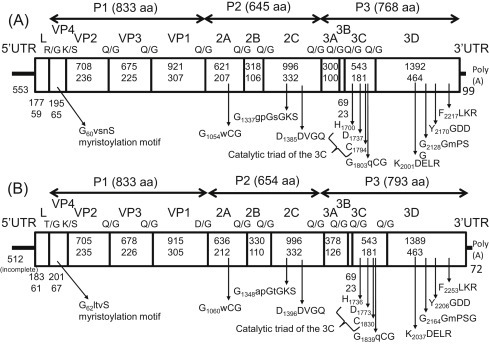
Genome organization of Bo-11-39/2009/JPN (A) and Bo-12-3, 7, 11 and 38/2009/JPN (B) with predicted cleavage sites and conserved picornavirus motifs. The ORF is flanked by the 5′-UTR and the 3′-UTR with a poly(A) tail. P1 (VP4, VP2, VP3, and VP1) includes the virus capsid proteins, while P2 (2A, 2B, and 2C) and P3 (3A, 3B, 3C, and 3D) are nonstructural proteins. Nucleotide lengths (upper) and amino acid lengths (lower) are shown for each portion of the polyprotein coding region. The positions of the amino acid motifs are indicated with the first position of the motif.
Table 1.
Genome features and pairwise amino acid identity (%) comparisons of bovine Japanese picornavirus to other picornaviruses.
| Picornavirus | Virus | DDBJ/EMBL/GenBank | Genomic feature | Pairwise amino acid identity (%) |
Pairwise amino acid identity (%) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bo-11-39/2009/JPN |
Bo-12-3, 7, 11 and 38/2009/JPN |
|||||||||
| Genus | Accession no. | Size (nt) | G + C content | P1 | P2 | P3 | P1 | P2 | P3 | |
| Aphthovirus | Foot-and-mouth disease virus | AF308157 | 8119 | 0.55 | 18.2 | 16.1 | 18.9 | 19.2 | 16.3 | 19.1 |
| Aquamavirus | Aquamavirus A | EU142040 | 6693 | 0.44 | 8.9 | 9.8 | 16.4 | 9.7 | 9.9 | 17.5 |
| Avihepatovirus | Duck hepatitis virus 1 | DQ226541 | 7690 | 0.43 | 8.7 | 8.7 | 17.9 | 9.3 | 9.0 | 16.0 |
| Avisivirus | Avisivirus A | KC465954 | 7532 | 0.45 | 8.2 | 9.4 | 15.9 | 9.1 | 8.6 | 15.4 |
| Cardiovirus | Encephalomyocarditis virus 1 | M81861 | 7835 | 0.49 | 18.4 | 13.9 | 19.1 | 20.7 | 14.8 | 19.1 |
| Cosavirus | Cosavirus A | FJ438902 | 7632 | 0.44 | 17.5 | 14.3 | 21.6 | 19.3 | 14.8 | 20.0 |
| Dicipivirus | Cadicivirus A | JN819202 | 8755 | 0.42 | 17.4 | 12.4 | 20.7 | 16.3 | 11.9 | 21.8 |
| Enterovirus | Bovine enterovirus 1 | D00214 | 7414 | 0.49 | 36.1 | 30.0 | 43.2 | 34.8 | 29.0 | 42.0 |
| Erbovirus | Equine rhinitis B virus 2 | X96871 | 8828 | 0.49 | 18.7 | 13.7 | 19.3 | 19.9 | 14.6 | 21.3 |
| Gallivirus | Gallivirus A | JQ691613 | 8496 | 0.48 | 11.4 | 11.5 | 20.8 | 12.4 | 12.4 | 22.0 |
| Hepatovirus | Hepatitis A virus 1 | M14707 | 7478 | 0.38 | 11.5 | 8.9 | 17.4 | 12.8 | 10.0 | 17.6 |
| Hunnivirus | Hunnivirus A | JQ941880 | 7583 | 0.46 | 19.9 | 15.1 | 22.2 | 20.9 | 14.9 | 22.5 |
| Kobuvirus | Bovine kobuvirus U-1 | AB084788 | 8374 | 0.54 | 15.0 | 11.7 | 21.8 | 15.6 | 11.9 | 21.7 |
| Megrivirus | Turkey hepatitis virus 1 | HM751199 | 9075 | 0.46 | 14.4 | 12.4 | 20.5 | 14.5 | 12.7 | 21.1 |
| Mischivirus | Mischivirus A | JQ814851 | 8457 | 0.48 | 18.0 | 14.9 | 20.8 | 18.6 | 15.0 | 19.8 |
| Mosavirus | Mosavirus A2 | KF958461 | 8398 | 0.45 | 19.7 | 15.1 | 20.7 | 21.2 | 16.3 | 21.2 |
| Oscivirus | Oscivirus A | GU182408 | 7625 | 0.47 | 16.2 | 12.0 | 20.9 | 17.0 | 11.3 | 22.4 |
| Parechovirus | Human parechovirus 2 | AJ005695 | 7348 | 0.39 | 10.0 | 11.3 | 16.4 | 10.7 | 10.5 | 17.9 |
| Pasivirus | Swine pasivirus 1 | JQ316470 | 6,896 | 0.43 | 10.3 | 10.7 | 16.4 | 10.3 | 10.7 | 16.9 |
| Passerivirus | Passerivirus A1 | GU182406 | 8019 | 0.58 | 14.9 | 11.7 | 22.2 | 15.9 | 12.3 | 22.5 |
| Rosavirus | Rosavirus M-7 | JF973686 | 8724 | 0.52 | 17.4 | 11.5 | 20.8 | 16.6 | 12.3 | 22.9 |
| Salivirus | Salivirus 1 | GQ179640 | 7982 | 0.56 | 16.3 | 11.7 | 19.5 | 16.8 | 12.0 | 21.1 |
| Sapelovirus | Porcine sapelovirus 1 | AF406813 | 7491 | 0.41 | 38.9 | 30.5 | 45.9 | 39.5 | 28.0 | 44.0 |
| Senecavirus | Seneca vally virus 1 | DQ641257 | 7310 | 0.51 | 19.0 | 14.9 | 20.3 | 19.7 | 13.3 | 19.0 |
| Teschovirus | Porcine teschovirus 1 | AJ011380 | 7117 | 0.45 | 19.3 | 15.6 | 23.1 | 19.0 | 13.6 | 23.7 |
| Tremovirus | Avian encephalomyelitis virus 1 | AJ225173 | 7055 | 0.45 | 11.6 | 8.3 | 19.1 | 12.4 | 9.7 | 19.2 |
| Unclassified | Bat picornavirus group 1 | HQ595340 | 7737 | 0.45 | 48.2 | 41.2 | 55.2 | 47.3 | 40.8 | 51.0 |
| Unclassified | Bat picornavirus group 2 | HQ595342 | 7677 | 0.43 | 45.9 | 40.0 | 54.9 | 45.4 | 40.5 | 51.0 |
| Unclassified | Bat picornavirus group 3 | HQ595344 | 7749 | 0.50 | 48.4 | 40.3 | 53.3 | 47.7 | 42.5 | 49.6 |
| Unclassified | Feline picornavirus | JN572117 | 7415 | 0.50 | 51.4 | 41.1 | 51.5 | 49.3 | 44.9 | 52.3 |
| Unclassified | Canine picornavirus | JN831356 | 7948 | 0.41 | 50.3 | 38.0 | 53.3 | 48.8 | 40.7 | 53.3 |
| Bo-11-39/2009/JPN | LC006971 | 7570 | 0.41 | – | – | – | 54.9 | 48.1 | 55.9 | |
| Bo-12-3/2009/JPN | LC036579 | 7610 (incomplete) | 0.41 | 54.9 | 48.1 | 55.9 | – | – | – | |
3.3. Analyses of the 5′UTR and 3′UTR
The complete 5'UTR of Bo-11-39 and near complete 5′UTR of Bo-12-3, 7, 11 and 38 had 38.5–41.7% and 38.5–41.9% identities to those of bat picornavirus group 3 and canine picornavirus, which have a type I internal ribosomal entry site (IRES). In contrast, the only 20.1–24.2% nt identities were found with the bat picornavirus group 1 and 2, and feline picornavirus sequences, which have type IV IRES elements (Table 2 ). Within the picornavirus family it is now known that there are four classes of IRES element which vary in size and properties (Belsham, 2009). Secondary structure prediction of the 5′-UTR of Bo-11-39 using the Mfold program revealed a stable stem-loop structure at the extreme 5′ end of the genome, many picornaviruses have such a feature (Fig. 2 (a)). Bat picornavirus group 3 has a section of the 5′-UTR with high nucleotide identity to the corresponding region of poliovirus (Lau et al., 2011). Similarly, the 5′-UTR of Bo-11-39 and Bo-12-3, 7, 11 and 38 also has a region (position from nt 353 to nt 527 in Bo-11-39 and nt 317 to nt 490 in Bo-12-3, 7, 11 and 38) with high similarity to that of human enteroviruses (76–81%) including poliovirus (75–81%). A potential structures for domain V of the Bo-11-39 IRES, which by analogy to the poliovirus IRES, should bind to eIF4G (de Breyne et al., 2009) is shown in Fig. 2(b). Both the base of the domain and the apex were extremely similar to other type I IRES elements and the domain is followed by a pyrimidine rich tract and then a short stem loop (domain VI), which contains an AUG and this is probably the site at which ribosomes start scanning although protein synthesis starts at the next AUG (at nt 554) (Fig. 2(c)). We predict that the translation initiation sites of Bo-11-39 and Bo-12-3, 7, 11 and 38 are at position nt 554 and nt 513, respectively, which is contained in the sequence GCCA554UGU in Bo-11-39 (Fig. 2(c)) and AACA513UGC in Bo-12-3, 7, 11 and 38. The initiation codon is not in an optimal Kozak context (Kozak, 1986) RNNAUGG with one nucleotide substitution (from G to U after AUG (RNNAUGU) in Bo-11-39 and G to C after AUG (RNNAUGC) in Bo-12-3, 7, 11 and 38). The 3′-UTR of bovine Japanese picornavirus was not closely related to any nucleotide sequences in the DDBJ/EMBL/GenBank database. There is less than 22.8% sequence identity within the 3′UTR between them and the unclassified Chinese picornaviruses.
Table 2.
Pairwise nucleotide and amino acid identity (%) of the Bo-11-39/2009/JPN and Bo-12-3/2009/JPN to Chinese picornaviruses.
| Pairwise nucleotide (5′UTR and 3′UTR), and amino acid (L, VP1-4, 2A-C, and 3A-D) identity (%) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bo-11-39/2009/JPN |
Bo-12-3/2009/JPNa |
||||||||||
| Bat-1(NC16A) | Bat-2(MH9F) | Bat-3(TLC5F) | Feline(356F) | Canine(325F) | Bo-12-3 | Bat-1(NC16A) | Bat-2(MH9F) | Bat-3(TLC5F) | Feline(356F) | Canine(325F) | |
| 5′UTR | 22.2 | 22.2 | 41.7 | 20.1 | 41.9 | 57.5 | 24.2 | 24.1 | 38.5 | 23.3 | 38.5 |
| L | 10.5 | 10.7 | 12.9 | 7.6 | 9.5 | 15.6 | 14.7 | 16.0 | 28.6 | 10.7 | 17.5 |
| VP4 | 51.5 | 44.1 | 51.5 | 50.8 | 52.9 | 60.3 | 50.0 | 41.2 | 50.8 | 49.3 | 52.2 |
| VP2 | 58.7 | 55.1 | 59.2 | 61.6 | 60.2 | 55.2 | 54.0 | 49.8 | 52.9 | 57.2 | 56.7 |
| VP3 | 51.3 | 51.3 | 53.0 | 52.0 | 55.4 | 59.0 | 50.9 | 51.1 | 53.3 | 54.9 | 54.7 |
| VP4 | 38.6 | 37.1 | 37.4 | 45.7 | 41.4 | 49.2 | 39.1 | 37.5 | 37.6 | 42.5 | 38.7 |
| 2A | 24.5 | 23.8 | 25.0 | 24.1 | 17.0 | 35.6 | 23.0 | 25.9 | 17.4 | 21.9 | 23.4 |
| 2B | 34.2 | 35.8 | 36.0 | 34.2 | 35.4 | 37.2 | 30.0 | 32.5 | 35.7 | 33.3 | 26.5 |
| 2C | 58.2 | 56.9 | 56.9 | 54.6 | 56.8 | 60.1 | 60.5 | 57.3 | 60.4 | 60.5 | 60.7 |
| 3A | 29.3 | 30.4 | 28.6 | 24.3 | 22.5 | 36.7 | 34.1 | 39.1 | 29.2 | 36.1 | 33.1 |
| 3B | 41.7 | 41.7 | 41.7 | 37.5 | 37.5 | 50.0 | 37.5 | 45.8 | 29.2 | 45.8 | 33.3 |
| 3C | 49.7 | 49.2 | 48.1 | 52.5 | 52.5 | 56.0 | 48.1 | 48.1 | 48.1 | 49.2 | 47.0 |
| 3D | 64.4 | 63.7 | 63.1 | 60.3 | 61.7 | 61.4 | 58.0 | 56.3 | 57.3 | 58.5 | 62.2 |
| 3'UTR | 16.2 | 18.5 | 18.4 | 22.8 | 16.0 | 26.7 | 14.8 | 18.4 | 14.2 | 20.0 | 13.3 |
5′UTR of Bo-12-3/2009/JPN is incomplete.
Fig. 2.
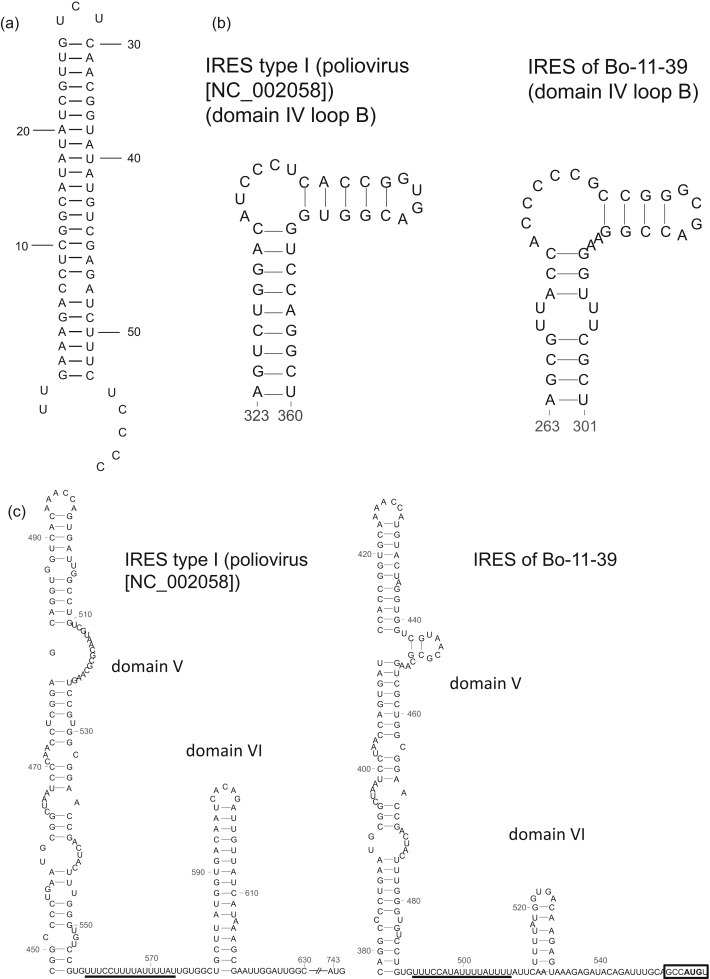
Secondary structure prediction of elements within the 5′-UTR of Bo-11-39/2009/JPN generated using the Mfold program. (a) A stable stem-loop structure at the extreme 5' end of the 5′UTR (3-53 nt). (b) A conserved structure (loop B) of type I IRES (poliovirus [NC_002058]) and Bo-11-39 within the domain IV. (c) The domain V and VI of the IRES of type I IRES (poliovirus [NC_002058]) and Bo-11-39. The domain V, by analogy to the poliovirus IRES, should bind to eIF4G (Bo-11-39 position 378-488 nt). A short stem loop (analogous to domain VI in other type 1 IRES elements [Bo-11-39 position 513-531 nt]) contains an AUG and this is probably the site at which ribosomes start scanning. The pyrimidine tract is underlined. The AUG start codon is in boldface within the sequence GCCA554UGU and is included within the rectangle.
3.4. Analyses of coding regions
For the coding regions, the predicted leader (L) protein of Bo-11-39 and Bo-12-3, 7, 11 and 38 are 59 and 61 aa long, respectively, and exhibit very low aa identities (less than 11.6%) to those of other representative picornaviruses, including unclassified Chinese picornaviruses. These L proteins are very short compared to viruses within the Aphthovirus and Erbovirus genera. The putative zing finger motifs which play a role in viral genome translation contained in the Cardiovirus genus were not found in the L proteins of the new bovine viruses (Chen et al., 1995, Dvorak et al., 2001). The function of these bovine Japanese picornavirus L proteins remains to be identified.
The P1 region of the genomes of Bo-11-39 and Bo-12-3, 7, 11 and 38 encode the capsid proteins VP4, VP2, VP3, and VP1. Myristic acid is covalently linked to a glycine at the amino terminus of VP4 of most picornaviruses (Chow et al., 1987). A potential myristoylation motif (GxxxT/S) is present in VP4 of Bo-11-39 (G62vsnS) and Bo-12-3, 7, 11 and 38 (G62ltvS) (Fig. 1).
The P2 region of the genomes of Bo-11-39 and Bo-12-3, 7, 11 and 38 encode the nonstructural proteins 2A, 2B, and 2C. The conserved GxCG motif, which is considered to form part of the active site of the protease (Gorbalenya et al., 1989a), in the 2A protein of Bo-11-39 (G1056wCG) and Bo-12-3, 7, 11 and 38 (G1060wCG) suggests that the 2A protein of these viruses cleaves at its own N-terminus, i.e. the VP1/2A junction. The conserved H-box/NC motif involved in cell proliferation control (Hughes and Stanway, 2000) was not present in the 2A. Similar to all the other picornaviruses, the NTP-binding motif GXXGXGKS (Gorbalenya et al., 1989b) was found in the 2C of Bo-11-39 and Bo-12-3, 7, 11 and 38 as G1337gpGsGKS and G1348apGtGKS, respectively. The DDLXQ motif is believed to be important for putative helicase activity (Gorbalenya et al., 1989a, Gorbalenya et al., 1990) but the third amino acid of this motif in the 2C of Bo-11-39 and Bo-12-3, 7, 11 and 38 was substituted by Val (D1385DVGQ and D1396DVGQ).
The P3 genome region of Bo-11-39 Bo-12-3, 7, 11 and 38 encodes the non-structural proteins 3A, 3B, 3Cpro, and 3Dpol. The conserved catalytic triad of H-D/E-C was seen in 3Cpro of Bo-11-39 (H1700-D1737-C1794) and Bo-12-3, 7, 11 and 38 (H1736D1773-C1830). The conserved GxCG motif was also found in 3Cpro of Bo-11-39 (G1803qCG) and Bo-12-3, 7, 11 and 38 (G1839qCG), as with other picornaviruses. Two highly conserved motifs of RNA-dependent RNA polymerases (Kamer and Argos, 1984), K2001DELR, G2128GmPSG, Y2170GDD, and F2217LKR (Bo-11-39) and K2037DELR, G2164GmPSG, Y2206GDD, and F2253LKR (Bo-12-3, 7, 11 and 38), were also present in the 3Dpol of these viruses.
3.5. Phylogenetic analyses
Phylogenetic trees were constructed from the complete amino acid cording sequences for P1, 2C, 3CD, and VP1 of bovine Japanese picornaviruses together with representative strains of the different genera and some unclassified picornaviruses. Bo-11-39 and Bo-12-3, 7, 11 and 38 were closely related to each other and formed a distinct cluster in the 3CD and VP1 trees with high bootstrap support (98% and 79%). The bovine Japanese picornaviruses were related to enteroviruses, Sapeloviruses, and unclassified Chinese picornaviruses and formed an independent cluster with unclassified Chinese picornaviruses supported by high bootstrap value (76%–99%) in all four phylogenetic trees (Fig. 3 ).
Fig. 3.
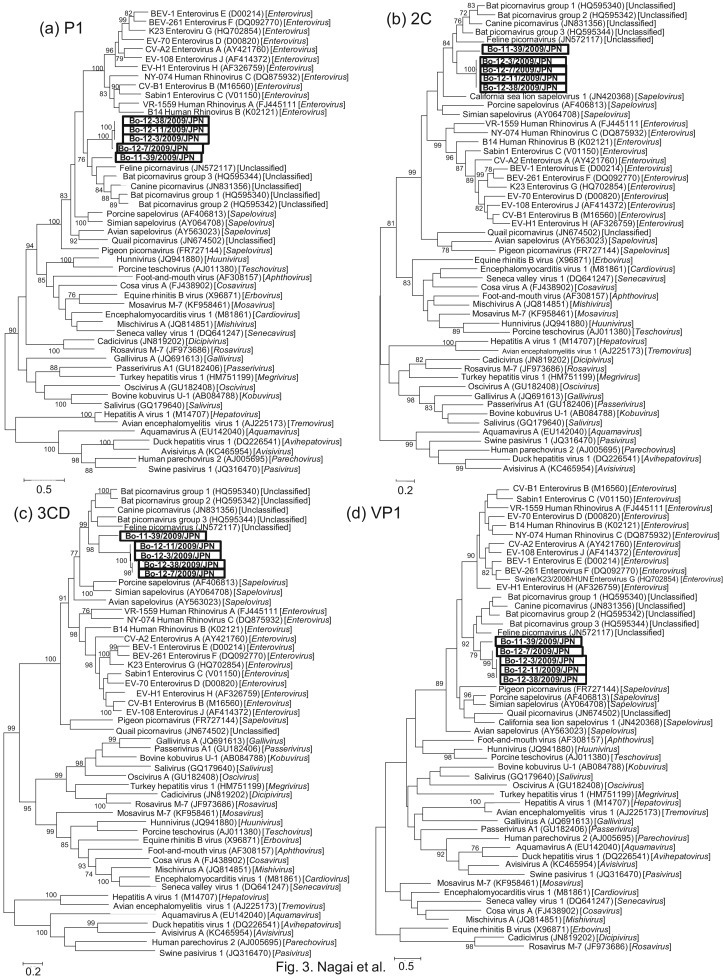
Phylogenetic analyses based on the complete amino acid sequences of P1 (a), 2C (b), 3CD (c), and VP1 (d) of Bo-11-39/2009/JPN, Bo-12-3/2009/JPN, Bo-12-7/2009/JPN, Bo-12-11/2009/JPN, and Bo-12-38/2009/JPN (black open box) with reference picornaviruses obtained from the DDBJ/EMBL/GenBank database. Phylogenetic trees were constructed using the maximum likelihood method in MEGA5.22 with bootstrap values (1000 replicates) above 70 being shown. The scale bar indicates amino acid substitutions per site.
3.6. Prevalence investigation of bovine Japanese picornavirus using RT-PCR
The prevalence of bovine Japanese picornaviruses in Hokkaido Prefecture in 2009 was studied using RT-PCR assays performed using specific primers designed from the 5′UTR sequences. Fecal samples were collected from 87 cattle with diarrhea (calves: 47; heifers: 14; cows: 26) from 72 farms. Using standard diagnostic assays, the most common bovine viral pathogens that induce diarrhea were ruled out as being responsible. However, amplicons of the expected size (326 base pairs) consistent with the presence of the bovine Japanese picornaviruses were obtained from twenty of these samples (from 15 calves (32%), a heifer (7%) and four cows (15%)) from nineteen different farms.
4. Discussion
In recent years, the advances in the metagenomic field have permitted the definition of new viral genomic sequences for clinical and epidemiological research (Beerenwinkel et al., 2012, Mokili et al., 2011, Rosario and Breitbart, 2011). Deep sequencing technologies have facilitated virus discovery due to the potential to detect viruses having an unknown genetic background. In this study, we have discovered a new picornavirus from cattle using metagenomic technology. We tried to isolate viruses using primary bovine testis cells, primary bovine fetal muscles cells, the Madin–Darby Bovine Kidney cells, Vero cells, baby hamster kidney cells, cloned porcine kidney cells, and HRT-18 cells. Neither cytopathic effect nor the expected RT-PCR products from RNA extracted from cell culture supernatants were observed in any of the inoculated cells, indicating a lack of virus replication in these cell lines. However, using RNA-seq based strategy with DNase treatment before construction of cDNA libraries, we could obtain sufficient viral genomic sequences of RNA viruses from fecal samples not only for picornaviruses but also, in earlier studies, for other viruses such as rotavirus (Minami-Fukuda et al., 2013, Masuda et al., 2014, Nagai et al., 2015).
Genomic characterization showed that these bovine Japanese picornaviruses had a genome organization similar to other picornaviruses, i.e. the presence of single large open reading frame flanked by UTRs. The 5′UTR of bovine Japanese picornavirus begins with a stem loop structure of about 50 nt, which is different from entero-/rhinoviruses that have a cloverleaf structure. However these viruses appear to have a type I IRES with a predicted domain IV (including a loop B), plus the domains V and VI including the eIF4G binding site and an AUG codon respectively, the latter is preceded by a polypyrimidine tract. Comparisons with representative strains of the different defined genera in the family Picornaviridae revealed that amino acid identities in the P1, P2, and P3 regions were up to 39.5, 30.5, and 45.9%, respectively. The Picornaviridae Study Group of the International Committee on Taxonomy of Viruses (http://www.picornastudygroup.com/definitions/genus_definition.htm) has proposed that different picornavirus genera are defined by amino acid identity in the P1, P2, and P3 regions being <40, <40, and <50%, respectively. Based on these criteria, bovine Japanese picornaviruses do not fall into the current official genera. On the other hand, bovine Japanese picornaviruses shared 45.4 to 51.4% (P1), 38.0 to 44.9% (P2), and 49.6 to 55.2% (P3) amino acid identities with certain unclassified Chinese picornaviruses; bat picornaviruses group 1-3 (Lau et al., 2011), feline picornavirus (Lau et al., 2012), and canine picornavirus (Woo et al., 2012), respectively. Furthermore, phylogenetic analyses demonstrated that bovine Japanese picornaviruses formed a cluster with unclassified Chinese picornaviruses in P1, 2C, 3CD, and VP1 coding regions, respectively, suggesting that bovine Japanese picornaviruses may be classified together with the currently unclassified Chinese picornaviruses into a novel genus in the family Picornaviridae.
Detailed analyses showed that bovine Japanese picornaviruses possessed conserved characteristic motifs, similar to unclassified Chinese picornaviruses (e.g. protein cleavage sites, presence of a leader protein, 2A, 2C, 3C, and 3D functional domains), with minor variance. Genomic comparison of bovine Japanese picornaviruses with unclassified Chinese picornaviruses revealed that although amino acid identities in L, 2A, 2B, and 3A were low (<40%), amino acid identities in the other regions were higher and indicated that these viruses were moderately related to each other. Phylogenetic analysis of the VP1 coding regions indicated that bovine Japanese picornaviruses and unclassified Chinese picornaviruses formed a cluster with high bootstrap support, even though VP1 is the most diverse capsid protein of picornaviruses (Oberste et al., 1999, Rossmann et al., 1985). These findings suggest that they have a common ancestor. Bovine Japanese picornaviruses seem most related to bat picornavirus group 3 and canine picornaviruses which also contain a type I IRES. RNA viruses have a remarkable potential to evolve due to high mutation and recombination rates (Cook et al., 2013). Bovine Japanese picornaviruses may be derived from the unclassified Chinese picornavirus and have evolved independently in cattle in Japan.
Finally, RT-PCR assays identified the presence of RNA from bovine Japanese picornaviruses in 15 calves (32%), one heifer (7%), and four cows (15%) with diarrhea from 19 farms. These finding indicates that these viruses are circulating in Hokkaido Prefecture in Japan. The potential transmission of these viruses to other domestic animals or human should be clarified. Since we did not perform analyses using fecal samples of cattle without diarrhea, the association of bovine Japanese picornaviruses with bovine diarrhea is unclear. To clarify whether they are associated or causal for bovine diarrhea, further studies are needed.
Acknowledgement
This work was supported by the Grants from the Ministry of Health, Labor and Welfare of Japan.
References
- Adams M.J., King A.M.Q., Carstens E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch. Virol. 2013;158:2023–2030. doi: 10.1007/s00705-013-1688-5. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N., Günthard H.F., Roth V., Metzner K.J. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front Microbiol. 2012;3:329. doi: 10.3389/fmicb.2012.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham G.J. Divergent picornavirus IRES elements. Virus Res. 2009;139:183–192. doi: 10.1016/j.virusres.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Blom N., Hansen J., Blaas D., Brunak S. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci. 1996;5:2203–2216. doi: 10.1002/pro.5560051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros Á., Nemes C., Pankovics P., Kapusinszky B., Delwart E., Reuter G. Identification and complete genome characterization of a novel picornavirus in turkey (Meleagris gallopavo) J. Gen. Virol. 2012;93:2171–2182. doi: 10.1099/vir.0.043224-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros A., Nemes C., Pankovics P., Kapusinszky B., Delwart E., Reuter G. Genetic characterization of a novel picornavirus in turkeys (Meleagris gallopavo) distinct from turkey galliviruses and megriviruses and distantly related to the members of the genus Avihepatovirus. J. Gen. Virol. 2013;94:1496–1509. doi: 10.1099/vir.0.051797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.H., Kong W.P., Roos R.P. The leader peptide of Theiler’s murine encephalomyelitis virus is a zinc-binding protein. J. Virol. 1995;69:8076–8078. doi: 10.1128/jvi.69.12.8076-8078.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow M., Newman J.F., Filman D., Hogle J.M., Rowlands D.J., Brown F. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature. 1987;327:482–486. doi: 10.1038/327482a0. [DOI] [PubMed] [Google Scholar]
- Cook S., Chung B.Y., Bass D., Moureau G., Tang S., McAlister E., Culverwell C.L., Glücksman E., Wang H., Brown T.D., Gould E.A., Harbach R.E., de Lamballerie X., Firth A.E. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS One. 2013;8:e80720. doi: 10.1371/journal.pone.0080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Breyne S., Yu Y., Unbehaun A., Pestova T.V., Hellen C.U. Direct functional interaction of initiation factor eIF4G with type 1 internal ribosomal entry sites. Proc. Natl. Acad. Sci. U.S.A. 2009;106:9197–9202. doi: 10.1073/pnas.0900153106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak C.M., Hall D.J., Hill M., Riddle M., Pranter A., Dillman J., Deibel M., Palmenberg A.C. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection, and affects the efficiency of genome translation. Virology. 2001;290:261–271. doi: 10.1006/viro.2001.1193. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Donchenko A.P., Blinov V.M., Koonin E.V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Lett. 1989;243:103–114. doi: 10.1016/0014-5793(89)80109-7. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989;17:4713–4730. doi: 10.1093/nar/17.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Wolf Y.I. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 1990;262:145–148. doi: 10.1016/0014-5793(90)80175-i. [DOI] [PubMed] [Google Scholar]
- Hughes P.J., Stanway G. The 2A proteins of three diverse picornaviruses are related to each other and to the H-rev107 family of proteins involved in the control of cell proliferation. J. Gen. Virol. 2000;81:201–207. doi: 10.1099/0022-1317-81-1-201. [DOI] [PubMed] [Google Scholar]
- Kamer G., Argos P. Primary structural comparison of RNA-dependent polymerases from plant, animal and bacterial viruses. Nucleic Acids Res. 1984;12:7269–7282. doi: 10.1093/nar/12.18.7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles N.J., Hovi T., Hyypiä T., King A.M.Q., Lindberg A.M., Pallansch M.A., Palmenberg A.C., Simmonds P., Skern T., Stanway G., Yamashita T., Zell R., 2012. Picornaviridae. In: Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. Ed: King, A.M.Q., Adams, M.J., Carstens, E.B. and Lefkowitz, E.J. San Diego, Elsevier, pp 855–880.
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- Lau S.K., Woo P.C., Lai K.K., Huang Y., Yip C.C., Shek C.T., Lee P., Lam C.S., Chan K.H., Yuen K.Y. Complete genome analysis of three novel picornaviruses from diverse bat species. J. Virol. 2011;85:8819–8828. doi: 10.1128/JVI.02364-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Woo P.C., Yip C.C., Choi G.K., Wu Y., Bai R., Fan R.Y., Lai K.K., Chan K.H., Yuen K.Y. Identification of a novel feline picornavirus from the domestic cat. J. Virol. 2012;86:395–405. doi: 10.1128/JVI.06253-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Q., Zheng L., Yuan Y., Shi J., Zhang D. Genomic characterization of a novel picornavirus in Pekin ducks. Vet. Microbiol. 2014;172:78–91. doi: 10.1016/j.vetmic.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Masuda T., Nagai M., Yamasato H., Tsuchiaka S., Okazaki S., Katayama Y., Oba M., Nishiura N., Sassa Y., Omatsu T., Furuya T., Koyama S., Shirai J., Taniguchi K., Fujii Y., Todaka R., Katayama K., Mizutani T. Identification of novel bovine group A rotavirus G15P[14] strain from epizootic diarrhea of adult cows by de novo sequencing using a next-generation sequencer. Vet. Microbiol. 2014;171:66–73. doi: 10.1016/j.vetmic.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami-Fukuda F., Nagai M., Takai H., Murakami T., Ozawa T., Tsuchiaka S., Okazaki S., Katayama Y., Oba M., Nishiura N., Sassa Y., Omatsu T., Furuya T., Koyama S., Shirai J., Tsunemitsu H., Fujii Y., Katayama K., Mizutani T. Detection of bovine group a rotavirus using rapid antigen detection kits, rt-PCR and next-generation DNA sequencing. J. Vet. Med. Sci. 2013;75:1651–1655. doi: 10.1292/jvms.13-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokili J.L., Rohwer F., Dutilh B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2011;2:63–77. doi: 10.1016/j.coviro.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M., Shimada S., Fujii Y., Moriyama H., Oba M., Katayama Y., Tsuchiaka S., Okazaki S., Omatsu T., Furuya T., Koyama S., Shirai J., Katayama K., Mizutani T. H2 genotypes of G4P[6], G5P[7], and G9[23] porcine rotaviruses show super-short RNA electropherotypes. Vet. Microbiol. 2015;176:250–256. doi: 10.1016/j.vetmic.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Oberste M.S., Maher K., Kilpatrick D.R., Pallansch M.A. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999;73:1941–1948. doi: 10.1128/jvi.73.3.1941-1948.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankovics P., Boros A., Reuter G. Novel picornavirus in domesticated common quail (Coturnix coturnix) in Hungary. Arch. Virol. 2012;157:525–530. doi: 10.1007/s00705-011-1192-8. [DOI] [PubMed] [Google Scholar]
- Reuter G., Pankovics P., Knowles N.J., Boros Á. Two closely related novel picornaviruses in cattle and sheep in Hungary from 2008 to 2009, proposed as members of a new genus in the family Picornaviridae. J. Virol. 2012;86:13295–13302. doi: 10.1128/JVI.01142-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario K., Breitbart M. Exploring the viral world through metagenomics. Curr. Opin Virol. 2011;1:289–297. doi: 10.1016/j.coviro.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Rossmann M.G., Arnold E., Erickson J.W., Frankenberger E.A., Griffith J.P., Hecht H.J., Johnson J.E., Kamer G., Luo M., Mosser A.G., Rueckert R.R., Sherry B., Vriend G. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature. 1985;317:145–153. doi: 10.1038/317145a0. [DOI] [PubMed] [Google Scholar]
- Sauvage V., Ar Gouilh M., Cheval J., Muth E., Pariente K., Burguiere A., Caro V., Manuguerra J.C., Eloit M. A member of a new Picornaviridae genus is shed in pig feces. J. Virol. 2012;86:10036–10046. doi: 10.1128/JVI.00046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S., 1 MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton J.L., Cornell C.T., Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 2005;3:765–776. doi: 10.1038/nrmicro1284. [DOI] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Choi G.K., Yip C.C., Huang Y., Tsoi H.W., K Yuen. Complete genome sequence of a novel picornavirus, canine picornavirus, discovered in dogs. J. Virol. 2012;86:3402–3403. doi: 10.1128/JVI.07228-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic. Acids. Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]