

Abstract
Mucosal immunity establishes the first line of defence against pathogens entering the body via mucosal surfaces. Besides eliciting both local and systemic immunity, mucosal vaccination strategies that are non-invasive in nature may increase patient compliance and reduce the need for vaccine application by trained personnel. A relatively new concept is mucosal immunization using DNA vaccines. The advantages of DNA vaccines, such as the opportunity to combine the genetic information of various antigen epitopes and stimulatory cytokines, the enhanced stability and ease of production make this class of vaccines attractive and suitable for mucosal application. In contrast to the area of intranasal vaccination, only a few recent studies have focused on pulmonary immunization and the involvement of the pulmonary immune system in eliciting protective immune responses against inhaled pathogens. This review focuses on DNA vaccine delivery to the lung as a promising approach to prevent pulmonary-associated diseases caused by inhaled pathogens. Attractive immunological features of the lung as a site for immunization, the mechanisms of action of DNA vaccines and the pulmonary application of such vaccines using novel delivery systems will be discussed. We also examine pulmonary diseases prone to prevention or therapeutical intervention by application of DNA vaccines.
Keywords: Pulmonary immunization, Airborne pathogens, DNA vaccines, Delivery systems
1. Introduction
Mucosal vaccine delivery embraces appealing characteristics for new immunization strategies against invading pathogens. The majority of human pathogens enter the body via mucosal surfaces in contact with the environment in the nose, lungs and the gastrointestinal tract. Mucosal vaccination at these sites can result in prevention of pathogen entry into the systemic circulation by local immune responses or in induction of systemic immunity and prevention of infection spread. In addition, there are many practical reasons that make mucosal vaccination strategies favourable in comparison to injected vaccines. Easier and less painful administration, reduced risk associated with injected vaccines and possible elimination of the cold chain are only some of the benefits associated with mucosal vaccines.
The pulmonary epithelium itself has a crucial role in host defence against inhaled pathogens since it presents a physical barrier including the mucociliary escalator, which in concert with the secreted antimicrobial agents that are present in the mucus layer covering the airway epithelium prevents colonization of microorganisms. Additionally, the lung has many attractive immunological properties. Organized lymphoid follicles, known as the broncho-alveolar lymphoid tissue (BALT) and local antigen-presenting cells (APCs) located ideally to sample antigens entering the airways are found in the lung.
DNA vaccines constitute an exciting new approach in vaccine development. The vaccine construct is created by insertion of a DNA encoding the desired antigen into a bacterial plasmid vector. The extent to which the plasmid DNA is able to transfect cells is dependent on the application route and the delivery system used. The encoded protein is then expressed in the transfected cells in vivo and consequently, an immune response is elicited to the expressed antigen [1].
In this manuscript we present the rationale and potential of pulmonary DNA immunization. We describe the major features of pulmonary immunology and give an overview of DNA vaccines in general and in association with pulmonary pathologies. In addition, we discuss the main work performed so far in the field of pulmonary DNA vaccine delivery and argue that direct vaccine application to the lower respiratory tract will be beneficial in protecting against inhaled pathogens causing life-threatening diseases.
2. Pulmonary immunology
2.1. The respiratory epithelium and its role in host defence
The respiratory tract continues beyond the larynx as the trachea, which then divides into two primary bronchi. Further branching from the bronchi are the bronchioles, terminal bronchioles and in the further branching of the respiratory bronchioles, alveolar ducts and alveoli where the exchange of oxygen and CO2 with the blood occurs [2]. The proximal airways represent a pseudostratified columnar epithelium, composed of ciliated cells, basal cells and smaller number of goblet cells (Fig. 1 ). Goblet cells in the superficial epithelium and mucous cells in the submucosal glands secrete mucus into the respiratory lining. In the more distal airways the epithelium becomes less multilayered, glands are less frequent and cells become less columnar. The alveolar surface is composed mainly of type I and type II pneumocytes that function as gas-exchange pneumocytes and surfactant secreting cells, respectively [2], [3].
Fig. 1.
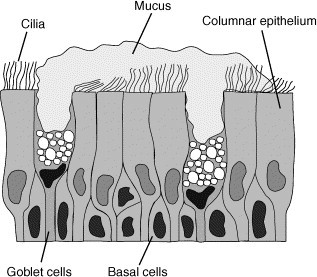
The proximal airways represent a pseudostratified columnar epithelium, composed of ciliated cells, basal cells and mucous secreting goblet cells. Particles and pathogens are trapped in the mucus. The mucus–particulate complexes are propelled by the beating cilia to the glottis where they are cleared from the airways by swallowing.
The initial barrier to prevent pathogen entry into the airways is formed by epithelial cells that effectively separate the luminal surface from the basolateral surface. In the proximal airways, large particles (> 5 μm in diameter) are trapped in the mucus and cleared by the mucociliary escalator and by coughing. Additional defence mechanisms are necessary to maintain lung sterility since most of the pathogens are smaller than 5 μm. The cells lining the upper and lower respiratory tract were shown to produce a variety of molecules that are involved in inflammatory and immune responses [3], [4], [5], [6]. By secreting these mediators, the pulmonary epithelium is capable of recruiting and activating cells of the innate immune system, killing pathogens and initiating an adaptive immune response. The airways have a constant antimicrobial environment provided by both constitutively expressed chemical defences and inducible chemical defences which are enhanced in response to infection.
The secreted molecules in the lung can be categorized into antimicrobial substances, chemotactic mediators and inflammatory mediators. Antimicrobial products are secreted from secretory cells in the mucosa and possess direct antimicrobial activity or facilitate the elimination of infectious pathogens by phagocytes. Defensins [7], [8], [9], cathelicidins [10], [11], [12] and collectins [13], [14], [15] are the principal antimicrobial peptides families that are expressed in the respiratory tract. Another important molecule with antimicrobial function secreted from pulmonary epithelial cells is immunoglobulin A (IgA). It is synthesized in the lamina propria as a dimer that specifically binds to a secretory component on the basolateral surface of the epithelial cells and is internalized as a complex. The complex is then transported to the apical surface of the epithelial cells and released in the airways where it can neutralize viruses and block the entrance of bacterial agents into the body [16], [17]. Other secreted products associated with antimicrobial function in the lungs are lysozyme, lactoferrin, secretory leukocyte proteinase inhibitor (SLPI), phospholipase A2 (PLA2), transferrin and immunoglobulin G (IgG). Chemotactic agents are induced in response to a variety of stimuli and recruit inflammatory cells to the lung. Immune cells are recruited to the infected area by release of cytokines like IL-1, IL-5, IL-6, IL-8 and granulocyte/macrophage colony-stimulating factor (GM-CSF) as well as chemokines and expression of adhesion molecules by airway epithelial cells. Important inflammatory mediators that were shown to play an important role in pulmonary host defence include the cytokines TNF-α, IL-12, IL-10, interferon gamma (IFN-γ) and GM-CSF. The inflammatory reaction will stay localized within the infected tissue as long as no pathological changes occur. Lung injury may result in cytokine leakage to the circulation and a systemic cytokine response [4], [5], [6].
Pulmonary epithelial cells can act as antigen-presenting and immunoregulatory cells of the lung. Bronchial epithelial cells were shown to express HLA-DR class II molecules on their surface as well as HLA-DR mRNA [18]. These cells are able to stimulate T cells and the expression of MHC class II molecules is modulated by immune mediators like IFN-γ [19]. Nevertheless, human bronchial epithelial cells do not express the co-stimulatory molecules CD80 and CD86 that are required for efficient APC function [20]. By contrast, in addition to MHC class II molecules, type II alveolar epithelial cells can also express co-stimulatory molecules and these are able to deliver co-stimulatory signals to T cells [21]. This shows another role for pulmonary epithelial cells as antigen-presenting and immunoregulatory cells in the respiratory tract.
2.2. Pulmonary phagocytes
Phagocytic cells like macrophages and dendritic cells (DCs) are essential for protection against inhaled pathogens. They play a role in both innate and adaptive immunity of the respiratory tract and a failure to function results in immunodeficiency and disease.
2.2.1. Pulmonary macrophages
Macrophages are a versatile population of cells that have many functions in the human body. Although showing large diversity, it is clear that these cells have a role in protection and preservation of tissue integrity. These cells remove debris generated by the host, protect from infectious pathogens using their microbicidal capacity, promote specific acquired immunity by their antigen presentation capabilities and regulate inflammation and effector mechanisms by secreting soluble mediators [22], [23].
Pulmonary macrophages are bone marrow derived cells that differentiate from blood monocytes after migrating to the lung. After their recruitment from the capillaries mediated by chemotactic factors, they populate the different lung compartments and differentiate into mature lung macrophages. Macrophages can be found in the pleura, interstitium, alveoli and in the pulmonary blood vessels, and are able to migrate from one lung compartment to the other. Subtle differences in cell physiology, membrane antigen expression and function have been observed between macrophages in the different lung compartments. These differences should probably be attributed to the response of the cells to their location and micromilieu rather than to fundamental differences in their development [23], [24].
Macrophages are an essential element of human innate immunity to inhaled pathogens. Together with other components of the innate immune response, such as pulmonary surfactant, mucus secretion, mucociliary clearance and antimicrobial peptides, macrophages are able to control infection in the lungs, which are constantly exposed to the environment. Elimination of any potential infectious agents is achieved by phagocytosis and subsequent digestion [23], [24]. Macrophages have enhanced phagocytic capabilities through three groups of membrane receptors that facilitate opsonization: Fc receptors (FcγRI, FcγRII, FcγRIII for different IgG subclasses, FcR for IgE and IgA), complement receptors (CR1, CR3, CR4) and lectin receptors (mannose receptor) [25], [26]. Scavenger receptors (SRs) and CD14 on macrophage cell surfaces are associated with phagocytosis of apoptotic cells [27], and pattern-recognition receptors such as Toll-like receptors (TLRs) are associated with macrophage stimulation after ligation by their microbial ligands [28]. Following phagocytosis, activated macrophages start to release microbicidal enzymes, reactive oxygen intermediates and T cell stimulatory and pro-inflammatory cytokines. The success of this non-specific pathogen elimination process is dependent on the pathogen type and load. A small number of microorganisms (∼ 105) can be eliminated by alveolar macrophages alone while higher inocula (∼ 108) will require activation of B and T cells for successful clearance. The macrophage's ability to eliminate a specific pathogen is also dependent on the microorganism's species. While Staphylococcus aureus is phagocytosed and killed intracellularly by alveolar macrophages, Mycobacteria spp., Listeria spp. and Legionella pneumophila are readily phagocytosed but no intracellular killing occurs, allowing these pathogens to reside and multiply in non-activated macrophages [25]. The decreased susceptibility to infection by these pathogens could be due to interference with TLR and IFN-γ-mediated activation pathways in macrophages [29], [30]. More recently it was suggested that human-macrophage plasticity has a critical effect on pathogen survival and alternative activation modes of macrophages can lead to an anti-inflammatory phenotype and persistence of chronic infection [31].
The macrophage population of the lung also participates in inducing pulmonary acquired immune responses. Macrophages are able to phagocytose, process and present antigens to stimulate T cells. Primary stimulation of T cell clones within the pulmonary lymphoid tissue is induced when alveolar macrophages migrate to bronchial lymph nodes and home to T cell paracortical areas [23], [32]. Pulmonary macrophages also have an effector function in acquired immunity since they are responsive to T cell derived cytokines and respond by increased microbicidal activity. Macrophages also have a regulatory function in acquired immunity. Since the lung is constantly exposed to harmful pathogens, unregulated acquired immune responses can result in a constant state of inflammation and damage to normal tissue and function. The macrophage populations in the lung are heterogeneous and some of the cells exhibit characteristics of suppressive cells [23], [33]. The relative proportions of inductive or suppressive macrophages developing from monocytes can be regulated by T cell cytokines (IFN-γ and IL-4 promote the inductive macrophages while IL-10 promotes the development of suppressive cells). Even after maturation, macrophages are able to switch phenotype and function in response to cytokine signals, which is an ideal characteristic for a cell population that is migrating constantly through the different environments within the lung.
2.2.2. Dendritic cells
Dendritic cells (DCs) are a part of the innate defence system and are initiators and modulators for specific and non-specific immunity. These cells are professional antigen-presenting cells and are present in most tissues. DCs are potent stimulators of T cells and are specialized in uptake, transport, processing and presentation of antigens [34], [35], [36].
DCs originate from bone marrow haematopoietic stem cells. Circulating precursor DCs enter tissues where they reside as immature DCs with high phagocytic capacity. The phagocytic capacity of immature DCs is established by several mechanisms: phagocytosis of particles and microbes, macropinocytosis and receptor-mediated endocytosis. The last two mechanisms are extremely efficient in antigen sampling and require very small concentrations of the antigen in order to activate DCs. Upon stimulation, immature DCs capture the antigen, migrate to the draining lymphoid organs and differentiate into mature DCs. Maturation of DCs results in a decreased capacity to take up antigens and increased presentation capabilities. Mature DCs can be distinguished from immature DCs by the loss of receptors that are involved in antigen uptake and increased expression of MHC class II molecules, co-stimulatory molecules like CD40, CD58, CD80, CD86 and the maturation marker CD83 [37], [38], [39], [40], [41]. Following migration, DCs present their previous environment to T cells by displaying the processed antigen's peptides using MHC class-I and MHC class-II molecules, leading to cellular and humoral immune responses [38], [42].
In the lung, the most pronounced populations of DCs were found in the epithelial linings of the conducting airways. Additional populations exist in the submucosa below the airway epithelium, within alveolar septal walls and on the alveolar surface. It was previously shown that airway epithelial DC turnover time is 2–3 days. Under local stress (e.g. inflammatory state), the turnover of pulmonary DCs further accelerates, indicating their importance in local antigen surveillance [43], [44]. Local antigenic challenge will result in higher density of DCs in the airways and increased expression of activation markers [45], [46]. The level of mature DCs migrating to the peribronchial lymph nodes is increased during the first 24 h of inflammation. However, this response quickly wanes and the respiratory DCs are refractory to further migration in spite of continued inflammatory state. The induction of this refractory state also results in transient inhibition of DC migration in response to a second pulmonary stimulus [47].
Danger signals from microbial agents can be recognized by DC receptors. Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) are the main DC receptors to be engaged in the direct recognition of characteristic molecular patterns of antigens. Recognition of pathogen-associated molecular patterns like bacterial nucleic acid, lipoproteins and lipopolysaccharide (LPS) by TLRs triggers intracellular signalling that result in DC maturation. This includes upregulation of co-stimulatory molecules, MHC molecules and expression of pro-inflammatory cytokines and ultimately leads to T cell activation. By contrast, CLRs recognize specific carbohydrate structures (pathogen cell-wall components) and internalize the pathogens for degradation in the lysosomal compartment to enhance processing and presentation by DCs. Nevertheless, one should remember that DCs act as “sentinels” in the body and hence are sampling both self- and non-self-antigens. It is becoming evident that some of the CLRs, such as dectin-1, DC-SIGN and mannose receptor, may inhibit stimulatory signals. It has been proposed that TLRs and CLRs intersect and dictate the balance between immune-activation and tolerance [48], [49], [50]. It was also suggested that some pathogens such as HIV and Mycobacterium tuberculosis target DC-SIGN to seek immune escape. The “misuse” of DC-SIGN results in pathogen survival by mechanisms that circumvent antigen processing or alter TLR-mediated signalling and skew T cell responses [51], [52], [53].
DC activation of T cells can result in immunity or tolerance, in activation of effector T cells or regulatory T cells and in different cytokine secretion profiles of T cells, polarizing Th1 and Th2 responses [35], [54]. In addition to the interactions with T cells in the lymphoid organs, DCs interact with other cells like B cells and natural killer (NK) cells. The different and sometimes opposing roles performed by DCs are not likely to be carried out by the same cell and therefore different subtypes of DCs were identified. These subsets differ in their phenotype, localization and function. The two subsets effecting Th1/Th2 balance are plasmacytoid DCs (also known as the DC2 subset) localized in T cell zones of lymphoid tissues and are associated with Th2 responses. The other subset is myeloid DCs (DC1 subset) localized in the interstitium and in germinal centers of various tissues and is associated with Th1 responses [55], [56], [57], [58], [59]. The development of two functionally different sublineages could be explained by two models (Fig. 2 ): a single lineage that depends on local environmental signals (the functional plasticity model) or two different lineages that became committed at early developmental stage (the specialized lineage model). It seems that reality is a mixture of both models and there is functional plasticity influenced by environmental cytokines in both DCs and their precursors [35], [38], [60]. It is clear by now that several factors control the T helper balance of the immune response initiated by DCs: the DC lineage (DC1/DC2), the maturation signal and the inflammatory mediators present in the microenvironment [61], [62], [63], [64]. The “default” T helper response at mucosal surfaces is Th2, which might be due to an inherent property of the resident DC and macrophage populations. Induction of Th1 responses in the lungs will require exposure of DCs to appropriate stimulus and high level production of IL-12 [35], [65], [66], [67], [68].
Fig. 2.
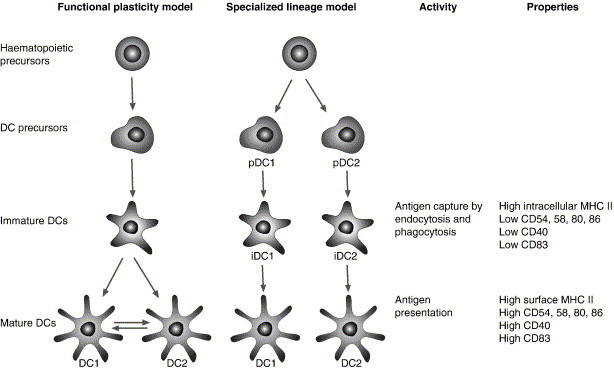
Maturation steps of DCs and the different models for the origin of the functionally distinct DC subsets, DC1 and DC2. Functionally distinct DCs can originate from a single lineage, which has different activation states that are dependent entirely on the local environment signals (the functional plasticity model) or from completely separate developmental lineages where the signals effect lineage segregation earlier in the developmental pathway and the DCs precursors are already separated and functionally committed (the specialized lineage model). Although distinct in the immunological outcome by skewing Th1 and Th2 responses, both subsets become good antigen-presenting cells upon maturation and increase surface expression of co-stimulatory molecules (modified from [35]).
2.3. Toll-like receptors
The innate immune system can sense pathogenic microorganisms through TLRs, a major class of pattern-recognition receptors (PRRs) that recognizes pathogen-associated molecular patterns (PAMPs). TLRs were discovered in the fruitfly Drosophila and are conserved throughout evolution [69], [70], [71], [72]. It was shown that drosophila, mouse, human and several more species possess a similar number of TLRs [73], [74]. Eleven human TLRs have now been identified that are able to recognize structurally unrelated ligands [75], [76], [77], [78]. TLR-activating ligands (summarized in Table 1 ) include lipopolysaccharide (LPS), bacterial lipoproteins, unmethylated CpG oligodeoxynucleotides, double stranded RNA (dsRNA), and others [79], [80], [81]. Not all TLRs are expressed on the cell surface, a feature that is in accordance with the nature of the different TLRs ligands. TLR4 was found to be localized at the cell membrane, while TLR9 and TLR3 are localized intracellularly. LPS is present in the cell wall of the pathogen and is therefore recognized upon first contact with the host cell by TLR4 located on the cell surface. However, as bacterial DNA resides inside the bacteria, endo-lysosomal degradation of the pathogen's cell wall is required before it is available for recognition by TLR9 present in the endosomal compartment of APCs [82], [83], [84]. An exception to LPS response is intestinal epithelial cells where TLR4 is localized in the Golgi apparatus [85].
Table 1.
Toll-like receptors and their ligands
| Receptor | Ligand | Origin |
|---|---|---|
| TLR1 | Triacyl lipopeptides | Bacteria, Mycobacteria |
| Soluble factors | Neisseria meningitidis | |
| TLR2 | Lipoprotein/lipopeptides | Different pathogens |
| Peptidoglycan | Gram-positive bacteria | |
| Lipoteichoic acid | Gram-positive bacteria | |
| Lipoarabinomannan | Mycobacteria | |
| Phenol-soluble modulin | Staphylococcus epidermidis | |
| Glycoinositolphospholipids | Trypanosoma cruzi | |
| Glycolipids | Treponema maltophilum | |
| Porins | Neisseria | |
| Zymosan | Fungi | |
| Atypical LPS | Leptospira interrogans | |
| Atypical LPS | Porphyromonas gingivalis | |
| Heat-shock protein 70 | Host | |
| TLR3 | Double-stranded RNA | Viruses |
| TLR4 | LPS | Gram-negative bacteria |
| Taxol | Plants | |
| Fusion protein | Respiratory syncytial virus | |
| Envelope protein | Mouse mammary-tumor virus | |
| Heat-shock protein 60 | Chlamydia pneumoniae | |
| Heat-shock protein 70 | Host | |
| Type III repeat extra domain A of fibronectin | Host | |
| Oligosaccharides of hyaluronic acid | Host | |
| Polysaccharide fragments of heparan sulphate | Host | |
| Fibrinogen | Host | |
| TLR5 | Flagellin | Bacteria |
| TLR6 | Diacyl lipopeptides | Mycoplasma |
| Lipoteichoic acid | Gram-positive bacteria | |
| Zymosan | Fungi | |
| TLR7 | Imidazoquinoline | Synthetic compounds |
| Loxoribine | Synthetic compounds | |
| Bropirimine | Synthetic compounds | |
| Single-stranded RNA | Viruses | |
| TLR8 | Imidazoquinoline | Synthetic compounds |
| Single-stranded RNA | Viruses | |
| TLR9 | CpG-containing DNA | Bacteria and viruses |
| TLR10 | Unknown | Unknown |
| TLR11 | Unknown | Uropathogenic bacteria |
TLRs are expressed in cells that are located and involved in the first line of defence like mucosal epithelial cells, macrophages, DCs, dermal endothelial cells and neutrophils [86]. Higher levels of TLR mRNA were shown to be expressed in tissues that are exposed to the external environment like the lung and the gastrointestinal tract, as well as at immunologically active sites such as peripheral blood leukocytes and the spleen [87]. Different cells can show distinct sets of TLR expression leading to differences in the reactivity to microbial molecules. This suggests that cells have undergone a distinct evolutionary development pathway, enabling them to recognize different pathogens. In DCs, the expression pattern has widely been explored and it was found that monocytes (DC1s) preferentially express TLRs 1, 2, 4, 5 and 8 and plasmacytoid DCs (DC2s) express TLR7 and TLR9 [88], [89].
The role of TLRs in pulmonary infections and lung disease is being intensively investigated (reviewed in Ref. [90]). Recently, expression of all known TLRs was reported in an airway epithelial cell line and in primary bronchial epithelial cells, however, TLRs 2, 3, 5 and 6 were the most highly expressed [91]. The interaction between inhaled pathogens and innate immunity is mediated by TLRs expressed on pulmonary epithelial cells lining the airways and the network of APCs present in the lungs. The most explored interaction is that of pulmonary pathogens with TLRs 2 and 4. Human airway epithelial cells express TLR2 and their activation enhances the host defence by increasing expression of the antimicrobial peptide human β-defensin 2 [92], [93]. TLR4 was shown to be constitutively expressed in human alveolar and bronchial epithelial cells and despite its intracellular localization, lung epithelial cells responded to LPS [94]. Respiratory syncytial virus was shown to upregulate TLR4 mRNA and protein expression and increase TLR4 localization on the membrane of airway epithelial cells. This increased interaction between LPS and TLR4 expressed on lung epithelial cells can promote airway inflammation response during RSV infection [95]. In bacterial infection, TLR4 is associated with recognition of gram-negative bacterial LPS while TLR2 has the capacity to recognize major cell-wall constituents of gram-positive bacteria. Despite this generally accepted model, involvement of both receptors in the immune response against the gram-positive bacteria Streptococcus pneumoniae was investigated and confirmed. A point of debate is to which extent TLR2 and TLR4 are involved in antibacterial host defence, and contradictory in vivo results were obtained, likely due to differences in the bacterial challenge models [96], [97], [98], [99]. Determining the roles of TLRs in host defence against M. tuberculosis has also focused mainly on TLR2 and 4 [100]. It was suggested that TLR2 has an important function in immunity to tuberculosis [101], [102], however, TLR4 did not have a significant role in immunity to tuberculosis in the mouse model [103], [104], [105]. Although evidence was collected to show involvement of TLRs in immunity to pulmonary pathogens, future work should define more clearly the extent to which the host defence against these pathogens is dependent on TLR signalling, including the bacterial pathogen and load involved.
2.4. Bronchus-associated lymphoid tissue
The BALT is the mucosal-associated lymphoid tissue (MALT) of the respiratory tract. It is a secondary lymphoid tissue comprised of non-encapsulated accumulation of lymphoid tissue. Specialized epithelial cells with microfolds on their luminal side, also known as M cells, are the principal site for sampling of mucosal antigens and transferring antigens unchanged to the underlying dome area that contains many APCs and T cells (Fig. 3 ). Stimulation by an antigen will result in generation of secretory IgA antibodies, which are able to cross epithelial membranes and help in the prevention of future entry of pathogens through the mucosal site. Furthermore, lymphocytes that were stimulated by antigens in the mucosal inductive site migrate via the regional lymph nodes and the thoracic duct to the blood stream and from there to other mucosal effector sites. This migration leads to IgA production at other mucosal sites of the MALT (gut, nasal, and genitourinary-associated lymphoid tissues) and was termed the common mucosal immune system (CMIS) [106], [107], [108], [109]. The CMIS appears to have organ selectivity as enhanced memory is seen at the site of mucosal priming compared to that of distant mucosal sites [110]. Nevertheless, the role of IgA should not be overemphasized in the discussion of respiratory immunity since T cell responses are of prominent importance in initiating local immune responses in the lung and induction of cytotoxic T cell responses are necessary for elimination of viral and bacterial infections [111], [112], [113].
Fig. 3.
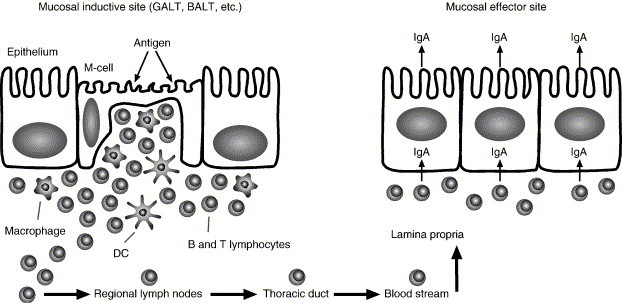
Schematic representation of the epithelium present in the mucosal-associated lymphoid tissue (MALT) and the common mucosal immune system (CMIS). Particulate antigens are endocytosed by the M cells that transfer the antigens to the underlying lymphoid tissue where they are processed by macrophages and DCs. Antigen-activated lymphocytes will migrate via the regional lymph nodes, thoracic duct and the blood stream to the lamina propria of other mucosal effector sites and induce IgA production at the mucosal surfaces.
The BALT was first described as follicular aggregates in the bronchial wall of rabbits. It is located mainly at branching sites such as the bifurcations of the bronchial tree. It was found that the respiratory epithelium overlaying the bronchial lymphoid structures contains M cells that are flattened and devoid of cilia [114], [115], [116], [117]. The BALT shows high variability between species and although constitutive structures are seen in rabbits and rats, this is not the case in humans [118], [119]. Studies of human BALT showed that BALT is not a constitutive structure in lungs of healthy adults and BALT observed in younger adults (until the age of 20) consists of smaller follicles than those described, e.g. in rabbits [120], [121]. It was previously discussed that detection of BALT in the human adult lung is associated with infection or inflammation [121]. Moyron-Quiroz et al. [122] had shown that influenza infection resulted in the formation of inducible bronchus-associated lymphoid tissue (iBALT) in mice lacking lymphoid organs (spleen, lymph nodes and Peyer's patches). The iBALT was capable of priming influenza-specific T and B cells that were able to clear the virus and facilitate host survival. Although iBALT resembles constitutive BALT, it is not induced by an antigen-independent developmental pathway, its size varies widely and it can be found in different pulmonary compartments than the constitutive BALT [122]. These findings point out that although constitutive BALT is not found in human adults, this tissue can be induced by local infection. However, observations in animal models may not directly be extrapolated to the human situation.
3. DNA vaccines
The concept of DNA vaccines arose from the observation that intramuscular injection of plasmid DNA containing reporter genes resulted in protein expression in the muscle that could be detected even 2 months after injection [123]. Hence, it was suggested that administration of a plasmid DNA encoding an antigenic protein could result in the in situ expression of the antigen, followed by antigen-specific immunity. In the following years, many plasmid DNA vectors encoding different bacterial, viral, parasitic and cancer antigens were constructed and tested for their immunogenicity and protective efficacy in animal models and primates [124], [125], [126]. Recently it was also suggested that these vaccines can serve not only for prevention purposes but also as therapeutic vaccines in chronic infections where they might restore immune control and prevent severe complication of the disease [127].
DNA vaccines hold many distinct advantages in comparison to recombinant proteins or inactivated pathogens: (I) The production is easy and similar between different plasmids, making up-scaling more simple and economical. (II) The DNA is stable at higher temperatures, a property increasing shelf-life and facilitating transport and distribution of such vaccines [128]. (III) Different antigens can be encoded in the same vector, leading to expression of multiple antigenic proteins in one vaccination. And finally (IV) these vaccines have the advantage of inducing a strong cellular immunity with a preference to cytotoxic T lymphocyte (CTL) and T helper type 1 (Th1) T cell responses.
Although the immunological benefits of DNA vaccines are well established, three safety issues were evaluated in preclinical animal models and have to be evaluated in DNA vaccination studies. The main safety issue is the risk of chromosomal integration into the host genome that might result in activation of oncogenes or disruption of tumor suppressor genes. The theoretical chance of an integration event is low and in mice there was no evidence of integration at a sensitivity level of 1–7.5 integrations in 150,000 nuclei, which was calculated to be 3 orders of magnitude lower than the spontaneous mutation frequency [129]. Nevertheless, this safety concern needs to be addressed in future clinical trials to assure the same result in human subjects. The second safety issue is the possibility of plasmid DNA to elicit anti-DNA antibodies. However, no anti-DNA antibodies were detected in sera from immunized mice and it was suggested that proper purification from E. coli would most likely prevent pathogenic anti-DNA antibody production [130], [131]. The third safety concern is the development of tolerance to the encoded antigen. This issue will need to be addressed in the context of the age of the vaccinated individual since there is a high chance of inducing tolerance by immunizing newborns with an immature immune system [126], [131].
Today we have the possibility to sequence the complete genomes of pathogens in a relatively short period of time and to identify most antigens of a pathogen and test its ability to induce immunity. This new genome-based approach to vaccine development was termed “reverse vaccinology” [132]. This approach enables prediction of new potential antigens and their testing much faster in comparison to conventional vaccinology. The most recent example was the SARS virus where the viral genome was sequenced 1 month after it was first suggested that a coronavirus was involved in the disease [133]. This approach will undeniably increase the amount of potential new DNA vaccines against pathogens causing a wide range of diseases.
3.1. DNA vectors for vaccine use
Plasmid vectors intended for use as DNA vaccines need to have certain essential elements (Fig. 4 ): (I) a bacterial backbone with an origin of replication (ORI) (usually from E. coli) that facilitates amplification of large quantities of DNA for purification. Obviously, this should not be active in mammalian cells in vivo in order to reduce the possibility of plasmid integration into the host genome. (II) A prokaryotic marker gene such as an antibiotic resistance gene to facilitate selection of organisms carrying the plasmid. (III) A strong eukaryotic promoter to drive the expression of the antigenic gene (usually from cytomegalovirus (CMV) or Simian virus 40 (SV40)). (IV) A transcription terminator to ensure that the mRNA is appropriately terminated, such as polyA signal from bovine growth hormone (BGH). (V) DNA sequence encoding the antigen of interest. (VI) The addition of a mammalian signal sequence may be desirable to facilitate protein secretion that may be required for efficient antibody production or presentation by MHC class II molecules [1], [131], [134].
Fig. 4.
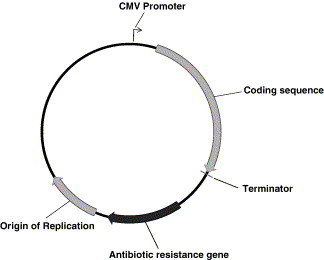
Schematic representation of a DNA vaccine. Plasmid constructs intended for immunization are bacterially derived and contain the genetic sequence of a desired antigen. The vector requires a strong bacterial origin of replication, an antibiotic selection marker, a strong eukaryotic promoter (usually a strong viral promoter like pCMV) and a transcription terminator.
3.2. Immunology of DNA vaccines
DNA immunization results in antigen expression in vivo and generation of both humoral and cellular immune responses. The major advantage of DNA vaccines is their ability to generate MHC class I-restricted CTL responses. This was demonstrated by intramuscular injection of plasmid DNA encoding influenza A nucleoprotein [135], HIV Gag and Env antigens [136], [137], [138], hepatitis B virus surface and core antigens [139], [140], M. tuberculosis antigen 85A and heat shock protein 65 [141], [142], among others. Activation of CTLs after DNA immunization can occur in two ways: (I) by APCs that were directly transfected by the DNA; (II) by cross-priming, in which non-APCs initially produce the protein encoded by the DNA vaccine and then deliver the antigen to a professional APC for priming of MHC class I restricted CTL responses. Although non-APCs (e.g. myocytes) take up and produce protein more than other cell types after DNA administration, they usually lack the co-stimulatory signals needed for the CTL activation process [143], [144], [145]. In the case of type II alveolar cells, it was shown they are able to express co-stimulatory molecules and deliver co-stimulatory signals to T cells therefore present an additional mechanism for activating CTL responses in the lung (Fig. 5 ) [21].
Fig. 5.
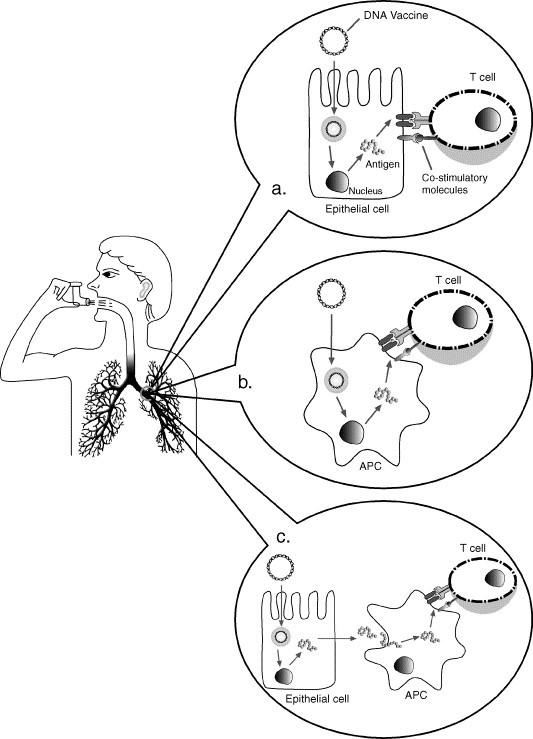
CD8+ cytolytic T lymphocytes (CTL) activation mechanisms after pulmonary DNA immunization. Type II alveolar cells were found to express co-stimulatory molecules and were able to deliver co-stimulatory signals to T cells, therefore could lead to CTL activation (a). The other CTL activation mechanisms involve direct transduction of professional APCs after DNA immunization (b) and cross-priming, where the respiratory epithelium is transfected, produces the antigen and then transfer it to professional APCs, which are directly responsible for activation of CTL responses (c).
Intramuscular DNA vaccination can further initiate T helper responses. Bacterial plasmid DNA contains unmethylated CpG motifs that are not prevalent in vertebrate genomic DNA and are able to stimulate APCs via TLR9. These CpG motifs can trigger the production of pro-inflammatory cytokines such as IL-12, and therefore, the generation of Th1 responses may be a general property of DNA vaccines. Nevertheless, the route of vaccination and the vaccine carrier has a pronounced effect on the nature of the immune response elicited (reviewed in [146]). As an example, the application of DNA vaccines by ‘gene gun’ resulted in a Th2 biased response. The gene gun application system uses vaccine coated gold particles that are introduced intradermally by ballistic action. It was shown that the Th2 response was a result of the mode of application and not a result of the different site of vaccination (skin vs. muscle). A possible explanation is that the gene gun delivers the DNA directly into the cells, bypassing the surface interaction of APCs with the CpG motifs. The ability of DNA vaccines to preferentially induce Th1 responses may be particularly important for preventing intracellular infections that require Th1 immunity to optimize the pathogen's eradication [124], [125].
DNA immunization was shown to induce strong humoral immune responses to a wide variety of antigens in animals and in human subjects. The antibody response is increased in a dose–response manner, however, once the optimal dose is given, a plateau in the antibody response is reached, and no significant effect is achieved after increasing the dose further or giving multiple injections. Since DNA vaccination generally enhances Th1 cytokine production, the subclass of antibodies generated will be biased toward IgG2a production. Also, as was seen for the T helper responses, the route of DNA vaccination can bias toward IgG1 production (gene gun). In some cases, antibodies have contributed to protection against challenge with the relevant pathogen, indicating that the antigens could generate neutralizing antibodies [124], [125].
3.3. DNA vaccines for pulmonary pathologies
Many antigens from pulmonary pathogens were identified as candidate vaccines by genome-based or classical approaches. The corresponding DNA vaccines were synthesized and evaluated for immunogenicity and protective efficacy. In the following section we discuss the possible benefits of pulmonary vaccination for three pathogens causing severe diseases in the airways: M. tuberculosis, respiratory syncytial virus (RSV) and the SARS-associated coronavirus (SARS-CoV).
3.3.1. M. tuberculosis
One third of the world population is infected with the M. tuberculosis bacillus [147]. Only 5–10% of the infected people develop an active disease during their lifetime since in normal healthy persons, the immune system is able to control the disease. However, the bacilli can stay dormant in the body and in an immunosuppressed state, the chances of developing an active disease increase. The disease is spread by individuals with active pulmonary tuberculosis by air. If left untreated, a sick person infects on average 10–15 people every year by creating aerosol droplets containing the bacilli (by coughing, sneezing etc.). Overall, 2 million people die every year from tuberculosis. A relieve of this situation seems far-off, especially with the emergence of multi-drug resistant tuberculosis (MDR-TB) and the deadly combination of AIDS and tuberculosis [147], [148]. The “Stop TB initiative” is a world-embracing program that is committed to diagnosis, treatment and future prevention of tuberculosis. Using the Directly Observed Therapy Short-course program (DOTS; an internationally recommended TB control strategy), tuberculosis is better controlled in different areas in the world, however, it is clear that without immunization it will be difficult to stop the transmission of the disease [149], [150]. A preventative vaccine will best control a disease like this, however, the only vaccine available for tuberculosis is the Bacille Calmette-Guerin (BCG), an attenuated strain of Mycobacterium bovis that was shown to have a protective effect against meningeal tuberculosis in children. However, the BCG fails to prevent pulmonary tuberculosis in adults [151]. Many efforts are directed at finding new candidate vaccines which will provide broad protection against disease and infection, induce lifelong immunological memory, would be safe, stable and inexpensive [149], [152].
Mycobacterial infections require cell-mediated immunity and both CD4+ and CD8+ T cells are essential for optimal responses. Since DNA vaccines can induce strong cellular immunity, many preclinical studies were performed using different encoding antigens [153]. The first reports of DNA vaccines against tuberculosis showing immunogenicity and protective efficacy in mice were performed using the antigen 85A (Ag85A) and 65 kDa heat shock protein (hsp65) from M. tuberculosis [141], [142]. Many other antigens such as ESAT-6 [154], [155], [156], Mtb 8.4 [157], 19 kDa lipoptotein [158], [159], MPT64 [155], [156], Ag85B [155], [160], putative phosphate transport receptors (PstS) [161], [162] and more [153] were also studied and provided different degrees of immunogenicity and protection. Most of these vaccines encode mycobacterial proteins that are secreted in mycobacterial culture filtrate or are exposed on the mycobacterial cell-wall surface. The availability of the complete M. tuberculosis genome [163] makes tuberculosis DNA vaccines popular for research since open reading frames (ORF) presenting potential antigens can be immediately used without the need to express and purify the protein. The general belief is that a successful tuberculosis DNA or protein vaccine will require a “cocktail” of immunogens [164].
3.3.2. Respiratory syncytial virus
RSV is the most common cause of severe respiratory disease in infants, leading to hospitalization of 2% of children in their first year of life. It accounts for approximately 70% of all cases of viral bronchiolitis in infants, and causes the highest morbidity in infants 2–4 months old. The disease is transmitted by respiratory secretions and by direct contact with contaminated surfaces [165], [166], [167]. Re-infection with RSV is common during life, however, it results in mild courses of upper respiratory tract infection and is life threatening only in cases of immunocompromised individuals.
A complete immune response to RSV includes antibody production, helper T cell response and CD8+ CTLs. Although antibodies to most RSV proteins will be generated in RSV infection, protection is incomplete and re-infection is possible even in the presence of high levels of virus neutralizing antibodies. Nevertheless, local antibodies in the respiratory tract were found to be related to protection: neutralizing antibodies were found to correlate with relative protection and local production of IgA concurs with virus clearance. Maternal antibodies appear to protect newborns against infection, however, their concentration decreases during the first 6 months of life where the disease peaks in infants. RSV infection is associated with a balanced type 1 and type 2 T helper response that is important for coordination of protective and immunopathogenic responses [166], [167]. The promise that DNA vaccines hold for preventing RSV is that the encoded antigen might be expressed for prolonged periods of time, immunizing the infant at the appropriate time point when the maternal antibodies concentration decreases under protection levels [168]. Most of the vaccination studies so far were performed in rodents and focused on two glycoproteins that are localized on the surface of the virus. The F protein is responsible for fusion of the viral envelope with the host cell membrane and is highly conserved between RSV subtypes while the G protein mediates attachment to the host cell surface and is responsible for the antigenic diversity between the RSV subtypes. DNA vaccines encoding RSV-G protein induced balanced systemic and pulmonary Th1/Th2 responses, neutralizing antibodies and protection against RSV infection of the lower respiratory tract of both mice and rats [169], [170]. RSV-F protein encoding vector showed to induce high neutralizing antibodies titers, CTL responses, protection against intranasal challenge of live RSV and high IFN-γ expression in the lungs after challenge [171]. Furthermore, it was shown that addition of CD40 ligand to DNA vaccines encoding RSV-F and G antigens can enhance viral clearance and some parameters of the immune response to RSV challenge [172].
3.3.3. Severe acute respiratory syndrome coronavirus
The SARS outbreak of an atypical pneumonia was first reported in the Guangdong Province in the south of China in November 2002. By July 2003, WHO had recorded over 8000 cases of SARS worldwide and more than 800 deaths [173], [174]. Viral isolation and serological assays confirmed that SARS-CoV was the primary infectious agent, its genome was sequenced and it was defined as a new coronavirus that is not a member of any of the three coronavirus groups known so far [175], [176]. The route of transmission has not been completely established yet, but it was immediately observed that health-care workers in close contact with SARS patients are at high risk, probably as a result of aerosol-generating procedures [177], [178], [179]. Airborne transmission seems possible and supported by a case of community SARS outbreak in a Hong Kong housing complex and the observation that a face mask is the most efficient precaution for medical staff protection [180]. There is a need for better understanding of the exact transmission mechanism by droplets and a definition whether SARS is obligatory, preferentially or opportunistically airborne transmitted [181].
Although SARS emerged only 2 years ago, data concerning vaccine candidates are already starting to accumulate. The most promising vaccine candidates are the two structural proteins: the nucleocapside protein (N) and the spike protein (S). It was shown that 89% of a group of patients with SARS produced antibodies to the N protein while less than 63% produced antibodies to the S protein [182]. In another study using neutralization assay with pseudotype virus, it was shown that SARS-CoV elicited a strong humoral neutralizing response to the S protein [183]. Many efforts are directed at finding specific domains and epitopes of the S protein that will induce strong production of anti SARS-CoV neutralizing antibodies and will advance the development of vaccines and therapeutics against SARS [184], [185], [186], [187]. DNA vaccine encoding the S protein of the SARS-CoV induces neutralizing antibodies, T cell response and protective immunity in mice. The protection mechanism was not dependent on T cell immunity since T cell depletion did not affect vaccine-induced protection and furthermore, adoptive T cell transfer did not reduce pulmonary viral replication in the recipient animals. In contrast, passive transfer of purified IgG from immunized mice provided protection similar to that observed after DNA vaccination [188]. DNA immunization using N protein encoding plasmid resulted in specific anti-N antibodies and specific CTL response [189], [190]. Since in some models it was shown that only the S protein was protective [191], future experiments should detect the best vaccine candidate or prove that a combination of these structural antigens will induce the best viral protection.
4. Pulmonary delivery of DNA vaccines
4.1. Pulmonary application of vaccines
Mucosal immunization is a promising and rational strategy to combat infectious agents that enter the body via mucosal surfaces. The site of antigen encounter will determine the immune response and circulating antigen-specific T cells that are primed in a mucosal tissue have increased homing commitment to the original mucosal site [192]. Furthermore, vaccination at one mucosal inductive site can induce immune responses in distant effector mucosal sites in the common mucosal immune system after secondary exposure to the antigen [193]. Therefore, the lung should be the preferred vaccination site for pathogens entering the body via the airways and inducing pulmonary disease.
Most of the pulmonary immunization strategies described to date involve nasal application due to the ease of administration and the belief that this vaccination site can stimulate respiratory mucosal immunity by interacting with the nasal-associated lymphoid tissue (NALT). Although it can induce both local and systemic immune responses, the main mucosal effect remains in the upper airways, which may lead to insufficient respiratory immunity against pulmonary pathogens. Using a murine model of respiratory reovirus infection it was shown that stimulation of NALT alone did not induce an optimal antibody response and effective immunity was only achieved by a combination of upper and lower respiratory tract infection [194]. A combined approach using intranasal and intratracheal administration simultaneously was recently used in African green monkeys where an attenuated parainfluenza virus expressing the SARS coronavirus spike protein was administered once and induced detectable neutralising antibodies against SARS coronavirus in serum [195]. Furthermore, a comparison between upper respiratory tract immunization (intranasal) and lower respiratory tract immunization (intratracheal) using inactivated influenza virus and a prototype split-subunit vaccine showed that intratracheal immunization was more effective in inducing local and systemic immune responses [196]. Another comparison between intratracheal, intranasal and intramuscular delivery of microencapsulated mixed Yersinia pestis subunit vaccines (V and F1 subunits) showed that intratracheal immunization resulted in higher immune responses in the respiratory tract, dominated by local IgG to both antigens [197].
Inhaled vaccines are poorly investigated so far despite reported success in several disease models. Efficient immune responses were demonstrated for tuberculosis [198], [199], [200], [201], measles [202] and influenza [203] emphasizing the great potential of deep lung immunization for prevention of pulmonary infections. The measles vaccine is the most explored for pulmonary administration. The WHO has been focusing on the development of aerosol measles vaccine for several years and is supporting efforts to change the administration route of the vaccine [204]. The main reasons for this are the ability for mass vaccination campaigns, the possible administration by non-medical personnel and most importantly, the ability to eliminate transmission of diseases like HIV, hepatitis B and other blood-borne pathogens by poor practice of injection safety procedures. Dry powder formulations of measles vaccine for pulmonary aerosol delivery appear to be a viable approach, retaining the vaccine's potency and eliminating the cold chain requirement during immunization [205], [206]. Future studies with other vaccines aiming to eradicate respiratory diseases are needed to compare current application methods for vaccines with the pulmonary route.
Although a promising approach, pulmonary immunization presents several disadvantages. The first disadvantage is the possibility of hypersensitivity responses to the vaccine preparation. Therefore, immunization studies should check specific immune responses to the vaccine and determine local hypersensitivity responses or lung pathologies that resulted from the vaccine and/or its formulation. Another immunological concern in pulmonary immunization is the development of mucosal tolerance as a response to antigen deposition in the lung. It was established that soluble antigens delivered to the respiratory mucosa without inflammatory signals or sensitised T cells, induced tolerance after systemic challenge [207]. Finally, a practical disadvantage is the small volume of liquid-formulations that can be applied in small animal models, especially in mice that are the best immunologically characterized animal serving as model for many investigated diseases. This argument, however, may be mute in the case of vaccines given as dry powder formulations.
4.2. Pulmonary DNA immunization
Pulmonary mucosal immunization employing DNA vaccines is a rather new, yet in our opinion promising vaccination approach, presenting many appealing properties. In addition to the practical issues mentioned above for mucosal vaccine delivery in general, this approach will enable vaccination of immunocompromised individuals since there is no threat of virulence as encountered by live attenuated vaccines [208]. Inhalation of DNA vaccines was recently described using plasmids encoding ovalbumin, hepatitis B surface antigen [209] and HLA-A*0201-restricted T cell epitopes of M. tuberculosis [210] and were shown to increase immunity as measured by antibodies and cytokine production. It was suggested that the immune response is highly dependent on the encoded antigen and it should be further investigated whether this application method holds an advantage in comparison to the common vaccination procedure by intramuscular injection. Furthermore, the mechanism of immune induction should be explored with an emphasis on the interaction of the antigens with pulmonary DCs, DCs migration to other lymphoid organs and the migration of immune cells to the lung after pulmonary immunization. The role of the BALT as a part of the CMIS should also be defined in the context of pulmonary DNA vaccination in order to clarify whether the lung can influence immunity in other mucosal effectors sites. Since IgA is not always present in broncho-alveolar lavage [209], its role in pulmonary immunity and disease protection should be elucidated.
4.3. Delivery systems for DNA vaccines
The goals of DNA delivery systems are to achieve long-term expression of the antigen encoded in the DNA vaccine, protect DNA from enzymatic degradation and enhance the immune response. Although DNA vaccines showed good potency in preclinical animal models, results in primates and in ensuing phase I clinical trials in humans were disappointing [211], emphasizing the need to develop delivery applications and strategies to increase the immune response (reviewed in [146]).
DNA carriers are categorized into two groups: viral and non-viral vectors. Each group presents different characteristics and has its own advantages and drawbacks.
4.3.1. Viral vectors
Viruses are appealing vectors because of their natural ability to incorporate DNA into the host genome. In order to deliver DNA for therapeutic purposes, viral carriers were modified to eliminate induction of pathogenic effects and ensure safe transgene expression. The main disadvantage of viral vectors is that they are detected by the immune system and induce an immune response directed against them. This does not allow application of repeated doses, which are usually needed for gene therapy and genetic immunization [212], [213]. Viral DNA vaccine delivery research was performed mainly with adenoviruses and poxviruses (vaccinia viruses).
Replication-incompetent adenoviral vectors expressing HIV antigens were shown to be efficient in inducing humoral and cellular immune responses. Best results were usually achieved by DNA vaccine priming and adenoviral boost [214]. Intranasal immunization with adenoviral vectors induced greater mucosal IgA responses against HIV antigens and recombinant adenovirus expressing Ag85A of M. tuberculosis provided potent protection in a tuberculosis challenge model after intranasal administration [215], [216]. However, detection of infection in the central nervous system could limit the utility of this route of delivery of adenoviral vectors [215].
Highly attenuated vaccinia viral vectors (modified vaccinia Ankara; MVA) were used for HIV antigens [217], multi-epitope construct derived from Plasmodium falciparum antigens [218], cancer-associated antigens [219] and Ag85A of M. tuberculosis [220]. These vectors presented the highest immunogenicity when given as a booster dose and induced efficient protection against challenge in animal models. MVA vectors carrying cancer and P. falciparum antigens presented no serious adverse effect in clinical trials with human subjects [218], [219].
4.3.2. Non-viral vectors
Although non-viral vectors does not present the disadvantages demonstrated by viral vectors, they are less efficient in inducing high transgene expression levels. These carriers can encapsulate, complex or adsorb the DNA when complexation and adsorption are usually based on electrostatic forces between negatively charged DNA and positively charged carrier. The carriers are usually able to protect the DNA from nuclease degradation and, depending on the system, present controlled release profiles. These carriers can vary considerably, integrating targeting moieties, endosomal disruptive agents and nuclear localization signals NLS in order to improve tissue targeting and intracellular delivery (Fig. 6 ) [221], [222]. Essential qualities of the delivery systems are biocompatibility to enable multiple administrations, low toxicity and efficient gene delivery so sufficient levels of antigen will be produced to result in a protective immune response. Most of the described synthetic vectors for DNA vaccine delivery are lipid or polymer based.
Fig. 6.
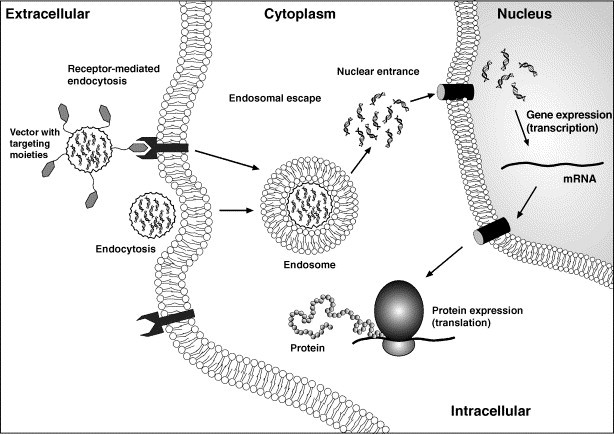
Schematic illustration of the internalization and intracellular trafficking of plasmid DNA delivered in a non-viral carrier system.
4.3.2.1. Lipid-based DNA delivery systems
Lipid-based delivery systems are cationic lipids with a positive charge that is pH independent. Positively charged lipids interact with negatively charged DNA to form complexes [223]. In lipid-based formulations, DNA can be complexed to the cationic moieties or entrapped within the aqueous phase of liposomes by a dehydration–rehydration procedure [224]. Liposomal complexes (lipoplexes) were evaluated with M. tuberculosis and anthrax encoded antigens and were shown to improve immunogenicity and protective efficacy [225], [226]. Mannosylated liposomes were synthesized to target APCs in vivo and enhanced the immune response after complexation of plasmid encoding the model antigen, ovalbumin [227]. Entrapment of DNA into the aqueous compartment of the liposomes was shown to be more effective than complexation and resulted in better immune responses against hepatitis B surface antigen [224], [228]. It is assumed that liposomes improve immunogenicity of DNA vaccines by facilitating uptake by APCs [229].
Emulsions are another delivery system derived from lipids that could deliver DNA vaccines efficiently. The surface charge of an oil-in-water (O/W) submicron emulsion, based on the potent squalene adjuvant MF59, was cationized using 1, 2-Dioleoyl-3-methylammonium-propane (DOTAP). Immunization with a DNA vaccine encoding HIV Gag protein adsorbed to the positively charged emulsion resulted in significantly enhanced immune responses in mice and in rabbits [230].
4.3.2.2. Polymer-based DNA delivery systems
These systems include various polymers that are positively charged at physiological pH to be able to complex DNA. Naturally and synthetically derived polymers can be manipulated by chemical modifications to achieve cell targeting and higher transfection efficiency [231]. Some qualities that are of major importance for future use in humans are biocompatibility, biodegradability and low toxicity.
Chitosan is the deacetylated form of the naturally originated polysaccharide, chitin, and has been extensively explored as a pharmaceutical excipient and as a delivery vehicle [232]. Chitosan polymers can vary in their molecular weight, viscosity and degree of deacetylation. The solubility of chitosan is highly dependent on the degree of deacetylation and the pH. In pH values lower than 6.5 chitosan is positively charged and is able to form complexes with anions (Fig. 7a). It is biodegradable, biocompatible and presents an excellent toxicity profile. Furthermore, chitosan was shown to be mucoadhesive and enhance mucosal absorption by opening tight junctions, therefore it is favourable for vaccine delivery into mucosal tissues. Oral delivery of chitosan nanoparticles loaded with DNA encoding peanut allergy antigen generated protective immunity [233]. Additionally, these nanoparticles enhanced immune responses when given intranasally with a cocktail of plasmid DNAs encoding RSV antigens [234] and a lyophilized formulation of DNA encoding a CTL epitope from M2 protein of RSV complexed to chitosan induced significant reduction of viral load in lungs of intranasally immunized mice [235]. Oral delivery of chitosan nanoparticles loaded with DNA encoding GRA1 protein of Toxoplasma gondii presented enhanced immunogenicity [236]. These studies point out chitosan as an efficient carrier for DNA vaccines.
Fig. 7.
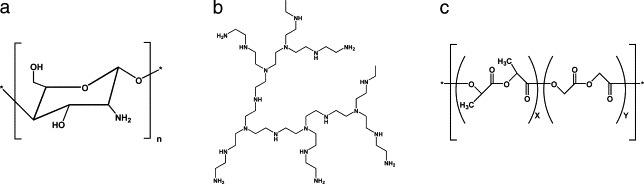
Molecular structures of chitosan (a), branched PEI (b) and Poly (d,l-lactide-co-glycolide) (PLGA) polymers (c).
Polyethyleneimines (PEI) are polymers of various molecular weights with the highest cationic charge density potential where every third atom is an amine group that can be protonated (Fig. 7b). These polymers were first shown to be efficient gene delivery systems in 1995 [237]. DNA encoding human growth hormone (hGH) and complexed to PEI induced high levels of antibodies after a single administration [238]. Gene expression analysis demonstrated that PEI is able to activate the immune system without DNA and activates genes involved in cellular processes like cell-cycle regulation, oncogenesis and differentiation [239]. This evidence suggests careful monitoring of toxic effects when immunizing with PEIs.
Poly (d,l-lactide-co-glycolide) (PLGA) polymers are one of the most commonly investigated materials for delivery of therapeutics (Fig. 7c). They are biocompatible, biodegradable and present low toxicity [240]. PLGA microparticles and nanoparticles were described to efficiently encapsulate plasmid DNA [241], [242]. Encapsulated DNA plasmid in PLGA microparticles was shown to elicit protective immune responses against tumors [243] and stimulate durable T cell responses to tumor-associated antigen cytochrome P450 1B1 [244]. Release of DNA was improved by addition of poly-β amino ester to the PLGA microparticles and enhanced antigen-specific rejection of tumor cells in vivo compared to conventional PLGA microparticles [245]. Cationic PLGA microparticles were designed to enable adsorption of DNA to the surface instead of encapsulation [246]. PLGA microparticles bearing the cationic agent cetyltrimethylammonium bromide (CTAB) enhanced immune responses to HIV-1 Gag protein after intramuscular and intranasal administration in mice [230], [247]. Potent responses were also achieved for DNA encoding E1E2 envelope proteins of hepatitis C virus [248]. These delivery systems are currently evaluated in clinical trials with genetic vaccines and immunotherapeutic agents (DNA encoding HPV antigenic epitopes encapsulated in PLGA microparticles, Zycos/MGI Pharma, http://www.zycos.com; DNA encoding HIV clade B Gag and Env antigens adsorbed to PLGA microparticles, NIAID, http://www.aidsinfo.nih.gov/clinical_trials).
Pulmonary delivery of DNA vaccines using carrier systems is rarely explored and most of the existing data describes optimization of formulations for aerosol gene delivery and evaluates gene expression in the lungs [249], [250], [251], [252], [253], [254], [255], [256]. In addition to inducing transfection of cells in vitro and lung tissue in vivo, carriers for pulmonary genetic vaccine delivery will have to be explored with DNA encoding pulmonary antigens in the relevant animal models and prove immunogenicity and protective efficacy. In vivo results regarding immunogenicity in the lung after pulmonary administration of DNA with a carrier system exist only for PEI–DNA polyplexes [238] and chitosan nanoparticles [210]. Further development of pulmonary compatible formulations and novel delivery systems for DNA vaccines will enable prevention of life-threatening diseases caused by respiratory pathogens.
5. Concluding remarks
In this era where new pathogens are genetically sequenced in a few months and their antigenic moieties can be detected quickly, DNA vaccines constitute a promising next generation of protective agents against life-threatening viruses and bacteria. Pulmonary DNA vaccination holds a promise for inducing local and systemic immune responses and should promote protection against inhaled airborne pathogens. In this review we presented the potential of the lung as a vaccination site that is specialized in antigen sampling and initiation of immunity using its epithelial cells, immune cells and local lymphoid tissue. Further evaluation of DNA vaccines against airborne pathogens is needed to determine their protective efficacy. In addition, new non-toxic pulmonary adjuvants and delivery systems should be characterized and investigated in vivo. In order to perform challenge studies where airborne pathogens are given as an aerosol, collaborative work with the appropriate biohazard facilities authorized to execute such experiments will be beneficial and enable the researchers to study the full potential of pulmonary applied vaccines.
References
- 1.Hanlon L., Argyle D. The science of DNA vaccination. Infect. Dis. Rev. 2001;3:2–12. [Google Scholar]
- 2.Bloom W., Fawcett D. Respiratory system. In: Fawcett, editor. A Textbook of Histology. Chapman & Hall; New York: 1994. pp. 704–727. [Google Scholar]
- 3.Rennard S.I., West W.W., Robbins R.A. Epithelial cells in host defense. In: Stockley R.A., editor. Pulmonary Defences. John Wiley & Sons Ltd; Chichester: 1997. pp. 163–178. [Google Scholar]
- 4.Diamond G., Legarda D., Ryan L.K. The innate immune response of the respiratory epithelium. Immunol. Rev. 2000;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- 5.Zhang P., Summer W.R., Bagby G.J., Nelson S. Innate immunity and pulmonary host defense. Immunol. Rev. 2000;173:39–51. doi: 10.1034/j.1600-065x.2000.917306.x. [DOI] [PubMed] [Google Scholar]
- 6.Bals R., Hiemstra P.S. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004;23:327–333. doi: 10.1183/09031936.03.00098803. [DOI] [PubMed] [Google Scholar]
- 7.McCray P.B., Jr., Bentley L. Human airway epithelia express a beta-defensin. Am. J. Respir. Cell Mol. Biol. 1997;16:343–349. doi: 10.1165/ajrcmb.16.3.9070620. [DOI] [PubMed] [Google Scholar]
- 8.Singh P.K., Jia H.P., Wiles K., Hesselberth J., Liu L., Conway B.A., Greenberg E.P., Valore E.V., Welsh M.J., Ganz T., Tack B.F., McCray P.B., Jr. Production of beta-defensins by human airway epithelia. Proc. Natl. Acad. Sci. U. S. A. 1998;95:14961–14966. doi: 10.1073/pnas.95.25.14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat. Rev., Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 10.Larrick J.W., Hirata M., Balint R.F., Lee J., Zhong J., Wright S.C. Human CAP18: a novel antimicrobial lipopolysaccharide-binding protein. Infect. Immun. 1995;63:1291–1297. doi: 10.1128/iai.63.4.1291-1297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zanetti M., Gennaro R., Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995;374:1–5. doi: 10.1016/0014-5793(95)01050-o. [DOI] [PubMed] [Google Scholar]
- 12.Bals R., Wang X., Zasloff M., Wilson J.M. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc. Natl. Acad. Sci. U. S. A. 1998;95:9541–9546. doi: 10.1073/pnas.95.16.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright J.R. Immunomodulatory functions of surfactant. Physiol. Rev. 1997;77:931–962. doi: 10.1152/physrev.1997.77.4.931. [DOI] [PubMed] [Google Scholar]
- 14.Mason R.J., Greene K., Voelker D.R. Surfactant protein A and surfactant protein D in health and disease. Am. J. Physiol. 1998;275:L1–L13. doi: 10.1152/ajplung.1998.275.1.L1. [DOI] [PubMed] [Google Scholar]
- 15.Hickling T.P., Clark H., Malhotra R., Sim R.B. Collectins and their role in lung immunity. J. Leukoc. Biol. 2004;75:27–33. doi: 10.1189/jlb.0703304. [DOI] [PubMed] [Google Scholar]
- 16.Bienenstock J. Mucosal immunological protection mechanisms in the airways. Eur. J. Respir. Dis., Suppl. 1986;147:62–71. [PubMed] [Google Scholar]
- 17.Nicod L.P. Pulmonary defence mechanisms. Respiration. 1999;66:2–11. doi: 10.1159/000029329. [DOI] [PubMed] [Google Scholar]
- 18.Rossi G.A., Sacco O., Balbi B., Oddera S., Mattioni T., Corte G., Ravazzoni C., Allegra L. Human ciliated bronchial epithelial cells: expression of the HLA-DR antigens and of the HLA-DR alpha gene, modulation of the HLA-DR antigens by gamma-interferon and antigen-presenting function in the mixed leukocyte reaction. Am. J. Respir. Cell Mol. Biol. 1990;3:431–439. doi: 10.1165/ajrcmb/3.5.431. [DOI] [PubMed] [Google Scholar]
- 19.Kalb T.H., Chuang M.T., Marom Z., Mayer L. Evidence for accessory cell function by class II MHC antigen-expressing airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 1991;4:320–329. doi: 10.1165/ajrcmb/4.4.320. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka H., Maeda K., Nakamura Y., Azuma M., Yanagawa H., Sone S. CD40 and IFN-gamma dependent T cell activation by human bronchial epithelial cells. J. Med. Investig. 2001;48:109–117. [PubMed] [Google Scholar]
- 21.Zissel G., Ernst M., Rabe K., Papadopoulos T., Magnussen H., Schlaak M., Muller-Quernheim J. Human alveolar epithelial cells type II are capable of regulating T cell activity. J. Investig. Med. 2000;48:66–75. [PubMed] [Google Scholar]
- 22.Bowden D.H. The alveolar macrophage. Environ. Health Perspect. 1984;55:327–341. doi: 10.1289/ehp.8455327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poulter L.W. Pulmonary macrophages. In: Stockley R.A., editor. Pulmonary Defences. John Wiley & Sons Ltd; Chichester: 1997. pp. 77–92. [Google Scholar]
- 24.Gordon S.B., Read R.C. Macrophage defences against respiratory tract infections. Br. Med. Bull. 2002;61:45–61. doi: 10.1093/bmb/61.1.45. [DOI] [PubMed] [Google Scholar]
- 25.Lohmann-Matthes M.L., Steinmuller C., Franke-Ullmann G. Pulmonary macrophages. Eur. Respir. J. 1994;7:1678–1689. [PubMed] [Google Scholar]
- 26.Linehan S.A., Martinez-Pomares L., Gordon S. Macrophage lectins in host defence. Microbes Infect. 2000;2:279–288. doi: 10.1016/s1286-4579(00)00300-2. [DOI] [PubMed] [Google Scholar]
- 27.Aderem A., Underhill D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 28.Janeway C.A., Jr., Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 29.Nau G.J., Richmond J.F., Schlesinger A., Jennings E.G., Lander E.S., Young R.A. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 2002;99:1503–1508. doi: 10.1073/pnas.022649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ting L.M., Kim A.C., Cattamanchi A., Ernst J.D. Mycobacterium tuberculosis inhibits IFN-gamma transcriptional responses without inhibiting activation of STAT1. J. Immunol. 1999;163:3898–3906. [PubMed] [Google Scholar]
- 31.Verreck F.A., de Boer T., Langenberg D.M., Hoeve M.A., Kramer M., Vaisberg E., Kastelein R., Kolk A., de Waal-Malefyt R., Ottenhoff T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4560–4565. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thepen T., Claassen E., Hoeben K., Breve J., Kraal G. Migration of alveolar macrophages from alveolar space to paracortical T cell area of the draining lymph node. Adv. Exp. Med. Biol. 1993;329:305–310. doi: 10.1007/978-1-4615-2930-9_51. [DOI] [PubMed] [Google Scholar]
- 33.Holt P.G., Oliver J., Bilyk N., McMenamin C., McMenamin P.G., Kraal G., Thepen T. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J. Exp. Med. 1993;177:397–407. doi: 10.1084/jem.177.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 35.Shortman K., Liu Y.J. Mouse and human dendritic cell subtypes. Nat. Rev., Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 36.Steinman R.M. Some interfaces of dendritic cell biology. APMIS. 2003;111:675–697. doi: 10.1034/j.1600-0463.2003.11107802.x. [DOI] [PubMed] [Google Scholar]
- 37.Steinman R.M., Inaba K. Myeloid dendritic cells. J. Leukoc. Biol. 1999;66:205–208. doi: 10.1002/jlb.66.2.205. [DOI] [PubMed] [Google Scholar]
- 38.Banchereau J., Briere F., Caux C., Davoust J., Lebecque S., Liu Y.J., Pulendran B., Palucka K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 39.Kruse M., Rosorius O., Kratzer F., Bevec D., Kuhnt C., Steinkasserer A., Schuler G., Hauber J. Inhibition of CD83 cell surface expression during dendritic cell maturation by interference with nuclear export of CD83 mRNA. J. Exp. Med. 2000;191:1581–1590. doi: 10.1084/jem.191.9.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peebles R.S., Jr., Graham B.S. Viruses, dendritic cells and the lung. Respir. Res. 2001;2:245–249. doi: 10.1186/rr63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujii S., Liu K., Smith C., Bonito A.J., Steinman R.M. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J. Exp. Med. 2004;199:1607–1618. doi: 10.1084/jem.20040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lambrecht B.N., Prins J.B., Hoogsteden H.C. Lung dendritic cells and host immunity to infection. Eur. Respir. J. 2001;18:692–704. [PubMed] [Google Scholar]
- 43.Holt P.G., Haining S., Nelson D.J., Sedgwick J.D. Origin and steady-state turnover of class II MHC-bearing dendritic cells in the epithelium of the conducting airways. J. Immunol. 1994;153:256–261. [PubMed] [Google Scholar]
- 44.Holt P.G., Stumbles P.A., McWilliam A.S. Functional studies on dendritic cells in the respiratory tract and related mucosal tissues. J. Leukoc. Biol. 1999;66:272–275. doi: 10.1002/jlb.66.2.272. [DOI] [PubMed] [Google Scholar]
- 45.Schon-Hegrad M.A., Oliver J., McMenamin P.G., Holt P.G. Studies on the density, distribution, and surface phenotype of intraepithelial class II major histocompatibility complex antigen (Ia)-bearing dendritic cells (DC) in the conducting airways. J. Exp. Med. 1991;173:1345–1356. doi: 10.1084/jem.173.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McWilliam A.S., Napoli S., Marsh A.M., Pemper F.L., Nelson D.J., Pimm C.L., Stumbles P.A., Wells T.N., Holt P.G. Dendritic cells are recruited into the airway epithelium during the inflammatory response to a broad spectrum of stimuli. J. Exp. Med. 1996;184:2429–2432. doi: 10.1084/jem.184.6.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Legge K.L., Braciale T.J. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–277. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 48.Figdor C.G., van Kooyk Y., Adema G.J. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev., Immunol. 2002;2:77–84. doi: 10.1038/nri723. [DOI] [PubMed] [Google Scholar]
- 49.Geijtenbeek T.B., van Vliet S.J., Engering A., t Hart B.A., van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu. Rev. Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- 50.Netea M.G., van der Graaf C., Van der Meer J.W., Kullberg B.J. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J. Leukoc. Biol. 2004;75:749–755. doi: 10.1189/jlb.1103543. [DOI] [PubMed] [Google Scholar]
- 51.van Kooyk Y., Geijtenbeek T.B. DC-SIGN: escape mechanism for pathogens. Nat. Rev., Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- 52.Kwon D.S., Gregorio G., Bitton N., Hendrickson W.A., Littman D.R. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity. 2002;16:135–144. doi: 10.1016/s1074-7613(02)00259-5. [DOI] [PubMed] [Google Scholar]
- 53.Tailleux L., Schwartz O., Herrmann J.L., Pivert E., Jackson M., Amara A., Legres L., Dreher D., Nicod L.P., Gluckman J.C., Lagrange P.H., Gicquel B., Neyrolles O. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 2003;197:121–127. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinman R.M., Hawiger D., Nussenzweig M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 55.Pulendran B. Modulating vaccine responses with dendritic cells and Toll-like receptors. Immunol. Rev. 2004;199:227–250. doi: 10.1111/j.0105-2896.2004.00144.x. [DOI] [PubMed] [Google Scholar]
- 56.Reis e Sousa C. Toll-like receptors and dendritic cells: for whom the bug tolls. Semin. Immunol. 2004;16:27–34. doi: 10.1016/j.smim.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Kapsenberg M.L. Dendritic-cell control of pathogen-driven T cell polarization. Nat. Rev., Immunol. 2003;3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 58.Moser M., Murphy K.M. Dendritic cell regulation of TH1–TH2 development. Nat. Immunol. 2000;1:199–205. doi: 10.1038/79734. [DOI] [PubMed] [Google Scholar]
- 59.Rissoan M.C., Soumelis V., Kadowaki N., Grouard G., Briere F., de Waal Malefyt R., Liu Y.J. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999;283:1183–1186. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- 60.Despars G., O'Neill H.C. A role for niches in the development of a multiplicity of dendritic cell subsets. Exp. Hematol. 2004;32:235–243. doi: 10.1016/j.exphem.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 61.Mazzoni A., Segal D.M. Controlling the Toll road to dendritic cell polarization. J. Leukoc. Biol. 2004;75:721–730. doi: 10.1189/jlb.1003482. [DOI] [PubMed] [Google Scholar]
- 62.Dabbagh K., Lewis D.B. Toll-like receptors and T-helper-1/T-helper-2 responses. Curr. Opin. Infect. Dis. 2003;16:199–204. doi: 10.1097/00001432-200306000-00003. [DOI] [PubMed] [Google Scholar]
- 63.Barton G.M., Medzhitov R. Control of adaptive immune responses by Toll-like receptors. Curr. Opin. Immunol. 2002;14:380–383. doi: 10.1016/s0952-7915(02)00343-6. [DOI] [PubMed] [Google Scholar]
- 64.Pasare C., Medzhitov R. Toll-like receptors and acquired immunity. Semin. Immunol. 2004;16:23–26. doi: 10.1016/j.smim.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 65.Stumbles P.A., Thomas J.A., Pimm C.L., Lee P.T., Venaille T.J., Proksch S., Holt P.G. Resting respiratory tract dendritic cells preferentially stimulate T helper cell type 2 (Th2) responses and require obligatory cytokine signals for induction of Th1 immunity. J. Exp. Med. 1998;188:2019–2031. doi: 10.1084/jem.188.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holt P.G., Stumbles P.A. Characterization of dendritic cell populations in the respiratory tract. J. Aerosol Med. 2000;13:361–367. doi: 10.1089/jam.2000.13.361. [DOI] [PubMed] [Google Scholar]
- 67.Iwasaki A., Kelsall B.L. Mucosal immunity and inflammation: I. Mucosal dendritic cells: their specialized role in initiating T cell responses. Am. J. Physiol. 1999;276:G1074–G1078. doi: 10.1152/ajpgi.1999.276.5.G1074. [DOI] [PubMed] [Google Scholar]
- 68.Lambrecht B.N., Hammad H. Taking our breath away: dendritic cells in the pathogenesis of asthma. Nat. Rev., Immunol. 2003;3:994–1003. doi: 10.1038/nri1249. [DOI] [PubMed] [Google Scholar]
- 69.Hashimoto C., Hudson K.L., Anderson K.V. The Toll gene of Drosophila, required for dorsal–ventral embryonic polarity, appears to encode a transmembrane protein. Cell. 1988;52:269–279. doi: 10.1016/0092-8674(88)90516-8. [DOI] [PubMed] [Google Scholar]
- 70.Lemaitre B., Nicolas E., Michaut L., Reichhart J.M., Hoffmann J.A. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 71.Takeda K., Kaisho T., Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 72.Lemaitre B. The road to Toll. Nat. Rev., Immunol. 2004;4:521–527. doi: 10.1038/nri1390. [DOI] [PubMed] [Google Scholar]
- 73.Imler J.L., Zheng L. Biology of Toll receptors: lessons from insects and mammals. J. Leukoc. Biol. 2004;75:18–26. doi: 10.1189/jlb.0403160. [DOI] [PubMed] [Google Scholar]
- 74.Vasselon T., Detmers P.A. Toll receptors: a central element in innate immune responses. Infect. Immun. 2002;70:1033–1041. doi: 10.1128/IAI.70.3.1033-1041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aderem A., Ulevitch R.J. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 76.Akira S., Takeda K. Toll-like receptor signalling. Nat. Rev., Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 77.Akira S. Mammalian Toll-like receptors. Curr. Opin. Immunol. 2003;15:5–11. doi: 10.1016/s0952-7915(02)00013-4. [DOI] [PubMed] [Google Scholar]
- 78.Abreu M.T., Arditi M. Innate immunity and toll-like receptors: clinical implications of basic science research. J. Pediatr. 2004;144:421–429. doi: 10.1016/j.jpeds.2004.01.057. [DOI] [PubMed] [Google Scholar]
- 79.Sieling P.A., Modlin R.L. Toll-like receptors: mammalian “taste receptors” for a smorgasbord of microbial invaders. Curr. Opin. Microbiol. 2002;5:70–75. doi: 10.1016/s1369-5274(02)00288-6. [DOI] [PubMed] [Google Scholar]
- 80.Underhill D.M., Ozinsky A. Toll-like receptors: key mediators of microbe detection. Curr. Opin. Immunol. 2002;14:103–110. doi: 10.1016/s0952-7915(01)00304-1. [DOI] [PubMed] [Google Scholar]
- 81.Underhill D.M. Toll-like receptors: networking for success. Eur. J. Immunol. 2003;33:1767–1775. doi: 10.1002/eji.200324037. [DOI] [PubMed] [Google Scholar]
- 82.Ahmad-Nejad P., Hacker H., Rutz M., Bauer S., Vabulas R.M., Wagner H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 2002;32:1958–1968. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 83.Hacker H., Mischak H., Miethke T., Liptay S., Schmid R., Sparwasser T., Heeg K., Lipford G.B., Wagner H. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J. 1998;17:6230–6240. doi: 10.1093/emboj/17.21.6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matsumoto M., Funami K., Tanabe M., Oshiumi H., Shingai M., Seto Y., Yamamoto A., Seya T. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 2003;171:3154–3162. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- 85.Hornef M.W., Frisan T., Vandewalle A., Normark S., Richter-Dahlfors A. Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J. Exp. Med. 2002;195:559–570. doi: 10.1084/jem.20011788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Armant M.A., Fenton M.J. Toll-like receptors: a family of pattern-recognition receptors in mammals. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-8-reviews3011. (REVIEWS3011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zarember K.A., Godowski P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 88.Kadowaki N., Ho S., Antonenko S., Malefyt R.W., Kastelein R.A., Bazan F., Liu Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Visintin A., Mazzoni A., Spitzer J.H., Wyllie D.H., Dower S.K., Segal D.M. Regulation of Toll-like receptors in human monocytes and dendritic cells. J. Immunol. 2001;166:249–255. doi: 10.4049/jimmunol.166.1.249. [DOI] [PubMed] [Google Scholar]
- 90.Basu S., Fenton M.J. Toll-like receptors: function and roles in lung disease. Am. J. Physiol., Lung Cell. Mol. Physiol. 2004;286:L887–L892. doi: 10.1152/ajplung.00323.2003. [DOI] [PubMed] [Google Scholar]
- 91.Sha Q., Truong-Tran A.Q., Plitt J.R., Beck L.A., Schleimer R.P. Activation of airway epithelial cells by toll-like receptor agonists. Am. J. Respir. Cell Mol. Biol. 2004;31:358–364. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- 92.Hertz C.J., Wu Q., Porter E.M., Zhang Y.J., Weismuller K.H., Godowski P.J., Ganz T., Randell S.H., Modlin R.L. Activation of Toll-like receptor 2 on human tracheobronchial epithelial cells induces the antimicrobial peptide human beta defensin-2. J. Immunol. 2003;171:6820–6826. doi: 10.4049/jimmunol.171.12.6820. [DOI] [PubMed] [Google Scholar]
- 93.Wang X., Zhang Z., Louboutin J.P., Moser C., Weiner D.J., Wilson J.M. Airway epithelia regulate expression of human beta-defensin 2 through Toll-like receptor 2. FASEB J. 2003;17:1727–1729. doi: 10.1096/fj.02-0616fje. [DOI] [PubMed] [Google Scholar]
- 94.Guillot L., Medjane S., Le-Barillec K., Balloy V., Danel C., Chignard M., Si-Tahar M. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J. Biol. Chem. 2004;279:2712–2718. doi: 10.1074/jbc.M305790200. [DOI] [PubMed] [Google Scholar]
- 95.Monick M.M., Yarovinsky T.O., Powers L.S., Butler N.S., Carter A.B., Gudmundsson G., Hunninghake G.W. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J. Biol. Chem. 2003;278:53035–53044. doi: 10.1074/jbc.M308093200. [DOI] [PubMed] [Google Scholar]
- 96.Kadioglu A., Andrew P.W. The innate immune response to pneumococcal lung infection: the untold story. Trends Immunol. 2004;25:143–149. doi: 10.1016/j.it.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 97.Malley R., Henneke P., Morse S.C., Cieslewicz M.J., Lipsitch M., Thompson C.M., Kurt-Jones E., Paton J.C., Wessels M.R., Golenbock D.T. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. U. S. A. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Branger J., Knapp S., Weijer S., Leemans J.C., Pater J.M., Speelman P., Florquin S., van der Poll T. Role of Toll-like receptor 4 in gram-positive and gram-negative pneumonia in mice. Infect. Immun. 2004;72:788–794. doi: 10.1128/IAI.72.2.788-794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Knapp S., Wieland C.W., van't Veer C., Takeuchi O., Akira S., Florquin S., van der Poll T. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J. Immunol. 2004;172:3132–3138. doi: 10.4049/jimmunol.172.5.3132. [DOI] [PubMed] [Google Scholar]
- 100.Krutzik S.R., Modlin R.L. The role of Toll-like receptors in combating mycobacteria. Semin. Immunol. 2004;16:35–41. doi: 10.1016/j.smim.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 101.Stenger S., Modlin R.L. Control of Mycobacterium tuberculosis through mammalian Toll-like receptors. Curr. Opin. Immunol. 2002;14:452–457. doi: 10.1016/s0952-7915(02)00355-2. [DOI] [PubMed] [Google Scholar]
- 102.Drennan M.B., Nicolle D., Quesniaux V.J., Jacobs M., Allie N., Mpagi J., Fremond C., Wagner H., Kirschning C., Ryffel B. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am. J. Pathol. 2004;164:49–57. doi: 10.1016/S0002-9440(10)63095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reiling N., Holscher C., Fehrenbach A., Kroger S., Kirschning C.J., Goyert S., Ehlers S. Cutting edge: toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J. Immunol. 2002;169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 104.Shim T.S., Turner O.C., Orme I.M. Toll-like receptor 4 plays no role in susceptibility of mice to Mycobacterium tuberculosis infection. Tuberculosis (Edinb.) 2003;83:367–371. doi: 10.1016/s1472-9792(03)00071-4. [DOI] [PubMed] [Google Scholar]
- 105.Kamath A.B., Alt J., Debbabi H., Behar S.M. Toll-like receptor 4-defective C3H/HeJ mice are not more susceptible than other C3H substrains to infection with Mycobacterium tuberculosis. Infect. Immun. 2003;71:4112–4118. doi: 10.1128/IAI.71.7.4112-4118.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nagler-Anderson C. Man the barrier! Strategic defences in the intestinal mucosa. Nat. Rev., Immunol. 2001;1:59–67. doi: 10.1038/35095573. [DOI] [PubMed] [Google Scholar]
- 107.MacDonald T.T. The mucosal immune system. Parasite Immunol. 2003;25:235–246. doi: 10.1046/j.1365-3024.2003.00632.x. [DOI] [PubMed] [Google Scholar]
- 108.Azzali G. Structure, lymphatic vascularization and lymphocyte migration in mucosa-associated lymphoid tissue. Immunol. Rev. 2003;195:178–189. doi: 10.1034/j.1600-065x.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 109.Lugton I. Mucosa-associated lymphoid tissues as sites for uptake, carriage and excretion of tubercle bacilli and other pathogenic mycobacteria. Immunol. Cell Biol. 1999;77:364–372. doi: 10.1046/j.1440-1711.1999.00836.x. [DOI] [PubMed] [Google Scholar]
- 110.Pierce N.F., Cray W.C., Jr. Determinants of the localization, magnitude, and duration of a specific mucosal IgA plasma cell response in enterically immunized rats. J. Immunol. 1982;128:1311–1315. [PubMed] [Google Scholar]
- 111.Bienenstock J., McDermott M.R., Befus A.D. The significance of bronchus-associated lymphoid tissue. Bull. Eur. Physiopathol. Respir. 1982;18:153–177. [PubMed] [Google Scholar]
- 112.Bienenstock J. The lung as an immunologic organ. Annu. Rev. Med. 1984;35:49–62. doi: 10.1146/annurev.me.35.020184.000405. [DOI] [PubMed] [Google Scholar]
- 113.Bienenstock J. Bronchus-associated lymphoid tissue. Int. Arch. Allergy Appl. Immunol. 1985;76(Suppl. 1):62–69. doi: 10.1159/000233736. [DOI] [PubMed] [Google Scholar]
- 114.Bienenstock J., Johnston N., Perey D.Y. Bronchial lymphoid tissue: I. Morphologic characteristics. Lab. Invest. 1973;28:686–692. [PubMed] [Google Scholar]
- 115.Bienenstock J., Johnston N., Perey D.Y. Bronchial lymphoid tissue: II. Functional characteristics. Lab. Invest. 1973;28:693–698. [PubMed] [Google Scholar]
- 116.Pankow W., von Wichert P. M cell in the immune system of the lung. Respiration. 1988;54:209–219. doi: 10.1159/000195527. [DOI] [PubMed] [Google Scholar]
- 117.Gebert A., Pabst R. M cells at locations outside the gut. Semin. Immunol. 1999;11:165–170. doi: 10.1006/smim.1999.0172. [DOI] [PubMed] [Google Scholar]
- 118.Sminia T., van der Brugge-Gamelkoorn G.J., Jeurissen S.H. Structure and function of bronchus-associated lymphoid tissue (BALT) Crit. Rev. Immunol. 1989;9:119–150. [PubMed] [Google Scholar]
- 119.Pabst R., Gehrke I. Is the bronchus-associated lymphoid tissue (BALT) an integral structure of the lung in normal mammals, including humans? Am. J. Respir. Cell Mol. Biol. 1990;31:131–135. doi: 10.1165/ajrcmb/3.2.131. [DOI] [PubMed] [Google Scholar]
- 120.Hiller A.S., Tschernig T., Kleemann W.J., Pabst R. Bronchus-associated lymphoid tissue (BALT) and larynx-associated lymphoid tissue (LALT) are found at different frequencies in children, adolescents and adults. Scand. J. Immunol. 1998;47:159–162. doi: 10.1046/j.1365-3083.1998.00276.x. [DOI] [PubMed] [Google Scholar]
- 121.Tschernig T., Pabst R. Bronchus-associated lymphoid tissue (BALT) is not present in the normal adult lung but in different diseases. Pathobiology. 2000;68:1–8. doi: 10.1159/000028109. [DOI] [PubMed] [Google Scholar]
- 122.Moyron-Quiroz J.E., Rangel-Moreno J., Kusser K., Hartson L., Sprague F., Goodrich S., Woodland D.L., Lund F.E., Randall T.D. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat. Med. 2004;10:927–934. doi: 10.1038/nm1091. [DOI] [PubMed] [Google Scholar]
- 123.Wolff J.A., Malone R.W., Williams P., Chong W., Acsadi G., Jani A., Felgner P.L. Direct gene transfer into mouse muscle in vivo. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 124.Donnelly J.J., Ulmer J.B., Shiver J.W., Liu M.A. DNA vaccines. Annu. Rev. Immunol. 1997;15:617–648. doi: 10.1146/annurev.immunol.15.1.617. [DOI] [PubMed] [Google Scholar]
- 125.Gurunathan S., Klinman D.M., Seder R.A. DNA vaccines: immunology, application, and optimization. Annu. Rev. Immunol. 2000;18:927–974. doi: 10.1146/annurev.immunol.18.1.927. [DOI] [PubMed] [Google Scholar]
- 126.Mor G. Plasmid DNA: a new era in vaccinology. Biochem. Pharmacol. 1998;55:1151–1153. doi: 10.1016/s0006-2952(97)00529-7. [DOI] [PubMed] [Google Scholar]
- 127.Autran B., Carcelain G., Combadiere B., Debre P. Therapeutic vaccines for chronic infections. Science. 2004;305:205–208. doi: 10.1126/science.1100600. [DOI] [PubMed] [Google Scholar]
- 128.Whalen R.G. DNA vaccines for emerging infectious diseases: what if? Emerg. Infect. Dis. 1996;2:168–175. doi: 10.3201/eid0203.960302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nichols W.W., Ledwith B.J., Manam S.V., Troilo P.J. Potential DNA vaccine integration into host cell genome. Ann. N. Y. Acad. Sci. 1995;772:30–39. doi: 10.1111/j.1749-6632.1995.tb44729.x. [DOI] [PubMed] [Google Scholar]
- 130.Xiang Z.Q., Spitalnik S.L., Cheng J., Erikson J., Wojczyk B., Ertl H.C. Immune responses to nucleic acid vaccines to rabies virus. Virology. 1995;209:569–579. doi: 10.1006/viro.1995.1289. [DOI] [PubMed] [Google Scholar]
- 131.Montgomery D.L., Ulmer J.B., Donnelly J.J., Liu M.A. DNA vaccines. Pharmacol. Ther. 1997;74:195–205. doi: 10.1016/s0163-7258(97)82003-7. [DOI] [PubMed] [Google Scholar]
- 132.Adu-Bobie J., Capecchi B., Serruto D., Rappuoli R., Pizza M. Two years into reverse vaccinology. Vaccine. 2003;21:605–610. doi: 10.1016/s0264-410x(02)00566-2. [DOI] [PubMed] [Google Scholar]
- 133.Rappuoli R., Covacci A. Reverse vaccinology and genomics. Science. 2003;302:602. doi: 10.1126/science.1092329. [DOI] [PubMed] [Google Scholar]
- 134.Donnelly J.J., Ulmer J.B. DNA vaccines for viral diseases. Braz. J. Med. Biol. Res. 1999;32:215–222. doi: 10.1590/s0100-879x1999000200010. [DOI] [PubMed] [Google Scholar]
- 135.Ulmer J.B., Donnelly J.J., Parker S.E., Rhodes G.H., Felgner P.L., Dwarki V.J., Gromkowski S.H., Deck R.R., DeWitt C.M., Friedman A. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259:1745–1749. doi: 10.1126/science.8456302. [DOI] [PubMed] [Google Scholar]
- 136.Shiver J.W., Davies M.E., Perry H.C., Freed D.C., Liu M.A. Humoral and cellular immunities elicited by HIV-1 vaccination. J. Pharm. Sci. 1996;85:1317–1324. doi: 10.1021/js9600991. [DOI] [PubMed] [Google Scholar]
- 137.Shiver J.W., Davies M.E., Yasutomi Y., Perry H.C., Freed D.C., Letvin N.L., Liu M.A. Anti-HIV env immunities elicited by nucleic acid vaccines. Vaccine. 1997;15:884–887. doi: 10.1016/s0264-410x(96)00251-4. [DOI] [PubMed] [Google Scholar]
- 138.Barouch D.H., Craiu A., Santra S., Egan M.A., Schmitz J.E., Kuroda M.J., Fu T.M., Nam J.H., Wyatt L.S., Lifton M.A., Krivulka G.R., Nickerson C.E., Lord C.I., Moss B., Lewis M.G., Hirsch V.M., Shiver J.W., Letvin N.L. Elicitation of high-frequency cytotoxic T-lymphocyte responses against both dominant and subdominant simian–human immunodeficiency virus epitopes by DNA vaccination of rhesus monkeys. J. Virol. 2001;75:2462–2467. doi: 10.1128/JVI.75.5.2462-2467.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schirmbeck R., Bohm W., Ando K., Chisari F.V., Reimann J. Nucleic acid vaccination primes hepatitis B virus surface antigen-specific cytotoxic T lymphocytes in nonresponder mice. J. Virol. 1995;69:5929–5934. doi: 10.1128/jvi.69.10.5929-5934.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kuhrober A., Wild J., Pudollek H.P., Chisari F.V., Reimann J. DNA vaccination with plasmids encoding the intracellular (HBcAg) or secreted (HBeAg) form of the core protein of hepatitis B virus primes T cell responses to two overlapping Kb- and Kd-restricted epitopes. Int. Immunol. 1997;9:1203–1212. doi: 10.1093/intimm/9.8.1203. [DOI] [PubMed] [Google Scholar]
- 141.Huygen K., Content J., Denis O., Montgomery D.L., Yawman A.M., Deck R.R., DeWitt C.M., Orme I.M., Baldwin S., D'Souza C., Drowart A., Lozes E., Vandenbussche P., Van Vooren J.P., Liu M.A., Ulmer J.B. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine. Nat. Med. 1996;2:893–898. doi: 10.1038/nm0896-893. [DOI] [PubMed] [Google Scholar]
- 142.Tascon R.E., Colston M.J., Ragno S., Stavropoulos E., Gregory D., Lowrie D.B. Vaccination against tuberculosis by DNA injection. Nat. Med. 1996;2:888–892. doi: 10.1038/nm0896-888. [DOI] [PubMed] [Google Scholar]
- 143.Liu M.A. DNA vaccines: a review. J. Intern. Med. 2003;253:402–410. doi: 10.1046/j.1365-2796.2003.01140.x. [DOI] [PubMed] [Google Scholar]
- 144.Srivastava I.K., Liu M.A. Gene vaccines. Ann. Intern. Med. 2003;138:550–559. doi: 10.7326/0003-4819-138-7-200304010-00011. [DOI] [PubMed] [Google Scholar]
- 145.Sbai H., Schneider J., Hill A.V., Whalen R.G. Role of transfection in the priming of cytotoxic T cells by DNA-mediated immunization. Vaccine. 2002;20:3137–3147. doi: 10.1016/s0264-410x(02)00251-7. [DOI] [PubMed] [Google Scholar]
- 146.Alpar H.O., Bramwell V.W. Current status of DNA vaccines and their route of administration. Crit. Rev. Ther. Drug Carr. Syst. 2002;19:307–383. doi: 10.1615/critrevtherdrugcarriersyst.v19.i45.20. [DOI] [PubMed] [Google Scholar]
- 147.WHO, Tuberculosis, Fact Sheet No. 104. http://www.who.int/mediacentre/factsheets/fs104/en/ (2004).
- 148.Schluger N.W., Rom W.N. The host immune response to tuberculosis. Am. J. Respir. Crit. Care Med. 1998;157:679–691. doi: 10.1164/ajrccm.157.3.9708002. [DOI] [PubMed] [Google Scholar]
- 149.WHO, Stop TB initiative, http://www.stoptb.org/stop.tb.initiative/ (2004).
- 150.Brennan M.J. A new generation of tuberculosis vaccines. In: de Quadros C.A., editor. Vaccines: Preventing Disease and Protecting Health. Pan American Health Organization; Washington, DC: 2004. pp. 177–182. [Google Scholar]
- 151.Orme I.M. The search for new vaccines against tuberculosis. J. Leukoc. Biol. 2001;70:1–10. [PubMed] [Google Scholar]
- 152.Andersen P. TB vaccines: progress and problems. Trends Immunol. 2001;22:160–168. doi: 10.1016/s1471-4906(01)01865-8. [DOI] [PubMed] [Google Scholar]
- 153.Huygen K. On the use of DNA vaccines for the prophylaxis of mycobacterial diseases. Infect. Immun. 2003;71:1613–1621. doi: 10.1128/IAI.71.4.1613-1621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lowrie D.B., Tascon R.E., Bonato V.L., Lima V.M., Faccioli L.H., Stavropoulos E., Colston M.J., Hewinson R.G., Moelling K., Silva C.L. Therapy of tuberculosis in mice by DNA vaccination. Nature. 1999;400:269–271. doi: 10.1038/22326. [DOI] [PubMed] [Google Scholar]
- 155.Kamath A.T., Feng C.G., Macdonald M., Briscoe H., Britton W.J. Differential protective efficacy of DNA vaccines expressing secreted proteins of Mycobacterium tuberculosis. Infect. Immun. 1999;67:1702–1707. doi: 10.1128/iai.67.4.1702-1707.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li Z., Howard A., Kelley C., Delogu G., Collins F., Morris S. Immunogenicity of DNA vaccines expressing tuberculosis proteins fused to tissue plasminogen activator signal sequences. Infect. Immun. 1999;67:4780–4786. doi: 10.1128/iai.67.9.4780-4786.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Coler R.N., Campos-Neto A., Ovendale P., Day F.H., Fling S.P., Zhu L., Serbina N., Flynn J.L., Reed S.G., Alderson M.R. Vaccination with the T cell antigen Mtb 8.4 protects against challenge with Mycobacterium tuberculosis. J. Immunol. 2001;166:6227–6235. doi: 10.4049/jimmunol.166.10.6227. [DOI] [PubMed] [Google Scholar]
- 158.Erb K.J., Kirman J., Woodfield L., Wilson T., Collins D.M., Watson J.D., LeGros G. Identification of potential CD8+ T cell epitopes of the 19 kDa and AhpC proteins from Mycobacterium tuberculosis. No evidence for CD8+ T cell priming against the identified peptides after DNA-vaccination of mice. Vaccine. 1998;16:692–697. doi: 10.1016/s0264-410x(97)00253-3. [DOI] [PubMed] [Google Scholar]
- 159.Yeremeev V.V., Lyadova I.V., Nikonenko B.V., Apt A.S., Abou-Zeid C., Inwald J., Young D.B. The 19-kD antigen and protective immunity in a murine model of tuberculosis. Clin. Exp. Immunol. 2000;120:274–279. doi: 10.1046/j.1365-2249.2000.01212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lozes E., Huygen K., Content J., Denis O., Montgomery D.L., Yawman A.M., Vandenbussche P., Van Vooren J.P., Drowart A., Ulmer J.B., Liu M.A. Immunogenicity and efficacy of a tuberculosis DNA vaccine encoding the components of the secreted antigen 85 complex. Vaccine. 1997;15:830–833. doi: 10.1016/s0264-410x(96)00274-5. [DOI] [PubMed] [Google Scholar]
- 161.Tanghe A., Lefevre P., Denis O., D'Souza S., Braibant M., Lozes E., Singh M., Montgomery D., Content J., Huygen K. Immunogenicity and protective efficacy of tuberculosis DNA vaccines encoding putative phosphate transport receptors. J. Immunol. 1999;162:1113–1119. [PubMed] [Google Scholar]
- 162.Romano M., Denis O., D'Souza S., Wang X.M., Ottenhoff T.H., Brulet J.M., Huygen K. Induction of in vivo functional Db-restricted cytolytic T cell activity against a putative phosphate transport receptor of Mycobacterium tuberculosis. J. Immunol. 2004;172:6913–6921. doi: 10.4049/jimmunol.172.11.6913. [DOI] [PubMed] [Google Scholar]
- 163.Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E., III, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Barrell B.G. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 164.McMurray D.N. Recent progress in the development and testing of vaccines against human tuberculosis. Int. J. Parasitol. 2003;33:547–554. doi: 10.1016/s0020-7519(03)00061-4. [DOI] [PubMed] [Google Scholar]
- 165.Dudas R.A., Karron R.A. Respiratory syncytial virus vaccines. Clin. Microbiol. Rev. 1998;11:430–439. doi: 10.1128/cmr.11.3.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Brandenburg A.H., Neijens H.J., Osterhaus A.D. Pathogenesis of RSV lower respiratory tract infection: implications for vaccine development. Vaccine. 2001;19:2769–2782. doi: 10.1016/s0264-410x(00)00536-3. [DOI] [PubMed] [Google Scholar]
- 167.Openshaw P.J. Potential therapeutic implications of new insights into respiratory syncytial virus disease. Respir. Res. 2002;3:S15–S20. doi: 10.1186/rr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Crowe J.E., Jr. Respiratory syncytial virus vaccine development. Vaccine. 2001;20(Suppl. 1):S32–S37. doi: 10.1016/s0264-410x(01)00287-0. [DOI] [PubMed] [Google Scholar]
- 169.Li X., Sambhara S., Li C.X., Ettorre L., Switzer I., Cates G., James O., Parrington M., Oomen R., Du R.P., Klein M. Plasmid DNA encoding the respiratory syncytial virus G protein is a promising vaccine candidate. Virology. 2000;269:54–65. doi: 10.1006/viro.2000.0186. [DOI] [PubMed] [Google Scholar]
- 170.Miller M., Cho J.Y., Baek K.J., Castaneda D., Nayar J., Rodriguez M., Roman M., Raz E., Broide D.H. Plasmid DNA encoding the respiratory syncytial virus G protein protects against RSV-induced airway hyperresponsiveness. Vaccine. 2002;20:3023–3033. doi: 10.1016/s0264-410x(02)00217-7. [DOI] [PubMed] [Google Scholar]
- 171.Li X., Sambhara S., Li C.X., Ewasyshyn M., Parrington M., Caterini J., James O., Cates G., Du R.P., Klein M. Protection against respiratory syncytial virus infection by DNA immunization. J. Exp. Med. 1998;188:681–688. doi: 10.1084/jem.188.4.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Harcourt J.L., Brown M.P., Anderson L.J., Tripp R.A. CD40 ligand (CD154) improves the durability of respiratory syncytial virus DNA vaccination in BALB/c mice. Vaccine. 2003;21:2964–2979. doi: 10.1016/s0264-410x(03)00119-1. [DOI] [PubMed] [Google Scholar]
- 173.Navas-Martin S.R., Weiss S. Coronavirus replication and pathogenesis: implications for the recent outbreak of severe acute respiratory syndrome (SARS), and the challenge for vaccine development. J. Neurovirology. 2004;10:75–85. doi: 10.1080/13550280490280292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Christian M.D., Poutanen S.M., Loutfy M.R., Muller M.P., Low D.E. Severe acute respiratory syndrome. Clin. Infect. Dis. 2004;38:1420–1427. doi: 10.1086/420743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 177.Li T.S., Buckley T.A., Yap F.H., Sung J.J., Joynt G.M. Severe acute respiratory syndrome (SARS): infection control. Lancet. 2003;361:1386. doi: 10.1016/S0140-6736(03)13052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Benitez M.A. Hong Kong bears brunt of latest outbreak. Lancet. 2003;361:1018. doi: 10.1016/S0140-6736(03)12844-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hawkey P.M., Bhagani S., Gillespie S.H. Severe acute respiratory syndrome (SARS): breath-taking progress. J. Med. Microbiol. 2003;52:609–613. doi: 10.1099/jmm.0.05321-0. [DOI] [PubMed] [Google Scholar]
- 180.Seto W.H., Tsang D., Yung R.W., Ching T.Y., Ng T.K., Ho M., Ho L.M., Peiris J.S. Effectiveness of precautions against droplets and contact in prevention of nosocomial transmission of severe acute respiratory syndrome (SARS) Lancet. 2003;361:1519–1520. doi: 10.1016/S0140-6736(03)13168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Roy C.J., Milton D.K. Airborne transmission of communicable infection—the elusive pathway. N. Engl. J. Med. 2004;350:1710–1712. doi: 10.1056/NEJMp048051. [DOI] [PubMed] [Google Scholar]
- 182.Leung D.T., Tam F.C., Ma C.H., Chan P.K., Cheung J.L., Niu H., Tam J.S., Lim P.L. Antibody response of patients with severe acute respiratory syndrome (SARS) targets the viral nucleocapsid. J. Infect. Dis. 2004;190:379–386. doi: 10.1086/422040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nie Y., Wang G., Shi X., Zhang H., Qiu Y., He Z., Wang W., Lian G., Yin X., Du L., Ren L., Wang J., He X., Li T., Deng H., Ding M. Neutralizing antibodies in patients with severe acute respiratory syndrome-associated coronavirus infection. J. Infect. Dis. 2004;190:1119–1126. doi: 10.1086/423286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hua R., Zhou Y., Wang Y., Hua Y., Tong G. Identification of two antigenic epitopes on SARS-CoV spike protein. Biochem. Biophys. Res. Commun. 2004;319:929–935. doi: 10.1016/j.bbrc.2004.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhou T., Wang H., Luo D., Rowe T., Wang Z., Hogan R.J., Qiu S., Bunzel R.J., Huang G., Mishra V., Voss T.G., Kimberly R., Luo M. An exposed domain in the severe acute respiratory syndrome coronavirus spike protein induces neutralizing antibodies. J. Virol. 2004;78:7217–7226. doi: 10.1128/JVI.78.13.7217-7226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Choy W.Y., Lin S.G., Chan P.K., Tam J.S., Lo Y.M., Chu I.M., Tsai S.N., Zhong M.Q., Fung K.P., Waye M.M., Tsui S.K., Ng K.O., Shan Z.X., Yang M., Wu Y.L., Lin Z.Y., Ngai S.M. Synthetic peptide studies on the severe acute respiratory syndrome (SARS) coronavirus spike glycoprotein: perspective for SARS vaccine development. Clin. Chem. 2004;50:1036–1042. doi: 10.1373/clinchem.2003.029801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang H., Wang G., Li J., Nie Y., Shi X., Lian G., Wang W., Yin X., Zhao Y., Qu X., Ding M., Deng H. Identification of an antigenic determinant on the S2 domain of the severe acute respiratory syndrome coronavirus spike glycoprotein capable of inducing neutralizing antibodies. J. Virol. 2004;78:6938–6945. doi: 10.1128/JVI.78.13.6938-6945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yang Z.Y., Kong W.P., Huang Y., Roberts A., Murphy B.R., Subbarao K., Nabel G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428:561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhu M.S., Pan Y., Chen H.Q., Shen Y., Wang X.C., Sun Y.J., Tao K.H. Induction of SARS-nucleoprotein-specific immune response by use of DNA vaccine. Immunol. Lett. 2004;92:237–243. doi: 10.1016/j.imlet.2004.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim T.W., Lee J.H., Hung C.F., Peng S., Roden R., Wang M.C., Viscidi R., Tsai Y.C., He L., Chen P.J., Boyd D.A., Wu T.C. Generation and characterization of DNA vaccines targeting the nucleocapsid protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004;78:4638–4645. doi: 10.1128/JVI.78.9.4638-4645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Buchholz U.J., Bukreyev A., Yang L., Lamirande E.W., Murphy B.R., Subbarao K., Collins P.L. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc. Natl. Acad. Sci. U. S. A. 2004;101:9804–9809. doi: 10.1073/pnas.0403492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kantele A., Zivny J., Hakkinen M., Elson C.O., Mestecky J. Differential homing commitments of antigen-specific T cells after oral or parenteral immunization in humans. J. Immunol. 1999;162:5173–5177. [PubMed] [Google Scholar]
- 193.Ogra P.L., Faden H., Welliver R.C. Vaccination strategies for mucosal immune responses. Clin. Microbiol. Rev. 2001;14:430–445. doi: 10.1128/CMR.14.2.430-445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Thompson A.H., McRoberts J.G., Crowe S.R., London L., London S.D. Optimal induction of upper respiratory tract immunity to reovirus 1/L by combined upper and lower respiratory tract inoculation. Vaccine. 1999;17:1404–1415. doi: 10.1016/s0264-410x(98)00382-x. [DOI] [PubMed] [Google Scholar]
- 195.Bukreyev A., Lamirande E.W., Buchholz U.J., Vogel L.N., Elkins W.R., St. Claire M., Murphy B.R., Subbarao K., Collins P.L. Mucosal immunisation of African green monkeys (Cercopithecus aethiops) with an attenuated parainfluenza virus expressing the SARS coronavirus spike protein for the prevention of SARS. Lancet. 2004;363:2122–2127. doi: 10.1016/S0140-6736(04)16501-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Smith D.J., Bot S., Dellamary L., Bot A. Evaluation of novel aerosol formulations designed for mucosal vaccination against influenza virus. Vaccine. 2003;21:2805–2812. doi: 10.1016/s0264-410x(03)00224-x. [DOI] [PubMed] [Google Scholar]
- 197.Eyles J.E., Spiers I.D., Williamson E.D., Alpar H.O. Analysis of local and systemic immunological responses after intra-tracheal, intranasal and intra-muscular administration of microsphere co-encapsulated Yersinia pestis sub-unit vaccines. Vaccine. 1998;16:2000–2009. doi: 10.1016/s0264-410x(98)00089-9. [DOI] [PubMed] [Google Scholar]
- 198.Rosenthal S.R., McEnery J.T., Raisys N. Aerogenic BCG vaccination against tuberculosis in animal and human subjects. J. Asthma Res. 1968;5:309–323. doi: 10.3109/02770906809100348. [DOI] [PubMed] [Google Scholar]
- 199.Orme I.M., Collins F.M. Aerogenic vaccination of mice with Mycobacterium bovis BCG. Tubercle. 1986;67:133–140. doi: 10.1016/0041-3879(86)90007-3. [DOI] [PubMed] [Google Scholar]
- 200.Reuman P.D., Ayoub E.M. Intrapulmonary BCG cell wall vaccination in mice. Microbios. 1987;49:7–15. [PubMed] [Google Scholar]
- 201.Lagranderie M., Ravisse P., Marchal G., Gheorghiu M., Balasubramanian V., Weigeshaus E.H., Smith D.W. BCG-induced protection in guinea pigs vaccinated and challenged via the respiratory route. Tuber. Lung Dis. 1993;74:38–46. doi: 10.1016/0962-8479(93)90067-8. [DOI] [PubMed] [Google Scholar]
- 202.Dilraj A., Cutts F.T., de Castro J.F., Wheeler J.G., Brown D., Roth C., Coovadia H.M., Bennett J.V. Response to different measles vaccine strains given by aerosol and subcutaneous routes to schoolchildren: a randomised trial. Lancet. 2000;355:798–803. doi: 10.1016/s0140-6736(99)95140-1. [DOI] [PubMed] [Google Scholar]
- 203.Waldman R.H., Mann J.J., Small P.A., Jr. Immunization against influenza. Prevention of illness in man by aerosolized inactivated vaccine. JAMA. 1969;207:520–524. doi: 10.1001/jama.207.3.520. [DOI] [PubMed] [Google Scholar]
- 204.WHO, Initiative for vaccine research (IVR), Measles. http://www.who.int/vaccine_research/diseases/measles/en/ (2004).
- 205.LiCalsi C., Christensen T., Bennett J.V., Phillips E., Witham C. Dry powder inhalation as a potential delivery method for vaccines. Vaccine. 1999;17:1796–1803. doi: 10.1016/s0264-410x(98)00438-1. [DOI] [PubMed] [Google Scholar]
- 206.LiCalsi C., Maniaci M.J., Christensen T., Phillips E., Ward G.H., Witham C. A powder formulation of measles vaccine for aerosol delivery. Vaccine. 2001;19:2629–2636. doi: 10.1016/s0264-410x(00)00503-x. [DOI] [PubMed] [Google Scholar]
- 207.Stumbles P.A., Upham J.W., Holt P.G. Airway dendritic cells: co-ordinators of immunological homeostasis and immunity in the respiratory tract. APMIS. 2003;111:741–755. doi: 10.1034/j.1600-0463.2003.11107806.x. [DOI] [PubMed] [Google Scholar]
- 208.McCluskie M.J., Davis H.L. Mucosal immunization with DNA vaccines. Microbes Infect. 1999;1:685–698. doi: 10.1016/s1286-4579(99)80070-7. [DOI] [PubMed] [Google Scholar]
- 209.Lombry C., Marteleur A., Arras M., Lison D., Louahed J., Renauld J.C., Preat V., Vanbever R. Local and systemic immune responses to intratracheal instillation of antigen and DNA vaccines in mice. Pharm. Res. 2004;21:127–135. doi: 10.1023/b:pham.0000012160.00222.55. [DOI] [PubMed] [Google Scholar]
- 210.Bivas-Benita M., van Meijgaarden K.E., Franken K.L., Junginger H.E., Borchard G., Ottenhoff T.H., Geluk A. Pulmonary delivery of chitosan-DNA nanoparticles enhances the immunogenicity of a DNA vaccine encoding HLA-A*0201-restricted T cell epitopes of Mycobacterium tuberculosis. Vaccine. 2004;22:1609–1615. doi: 10.1016/j.vaccine.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 211.MacGregor R.R., Boyer J.D., Ugen K.E., Lacy K.E., Gluckman S.J., Bagarazzi M.L., Chattergoon M.A., Baine Y., Higgins T.J., Ciccarelli R.B., Coney L.R., Ginsberg R.S., Weiner D.B. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: safety and host response. J. Infect. Dis. 1998;178:92–100. doi: 10.1086/515613. [DOI] [PubMed] [Google Scholar]
- 212.Bessis N., GarciaCozar F.J., Boissier M.C. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004;11(Suppl. 1):S10–S17. doi: 10.1038/sj.gt.3302364. [DOI] [PubMed] [Google Scholar]
- 213.El-Aneed A. An overview of current delivery systems in cancer gene therapy. J. Control. Release. 2004;94:1–14. doi: 10.1016/j.jconrel.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 214.Shiver J.W., Fu T.M., Chen L., Casimiro D.R., Davies M.E., Evans R.K., Zhang Z.Q., Simon A.J., Trigona W.L., Dubey S.A., Huang L., Harris V.A., Long R.S., Liang X., Handt L., Schleif W.A., Zhu L., Freed D.C., Persaud N.V., Guan L., Punt K.S., Tang A., Chen M., Wilson K.A., Collins K.B., Heidecker G.J., Fernandez V.R., Perry H.C., Joyce J.G., Grimm K.M., Cook J.C., Keller P.M., Kresock D.S., Mach H., Troutman R.D., Isopi L.A., Williams D.M., Xu Z., Bohannon K.E., Volkin D.B., Montefiori D.C., Miura A., Krivulka G.R., Lifton M.A., Kuroda M.J., Schmitz J.E., Letvin N.L., Caulfield M.J., Bett A.J., Youil R., Kaslow D.C., Emini E.A. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415:331–335. doi: 10.1038/415331a. [DOI] [PubMed] [Google Scholar]
- 215.Lemiale F., Kong W.P., Akyurek L.M., Ling X., Huang Y., Chakrabarti B.K., Eckhaus M., Nabel G.J. Enhanced mucosal immunoglobulin A response of intranasal adenoviral vector human immunodeficiency virus vaccine and localization in the central nervous system. J. Virol. 2003;77:10078–10087. doi: 10.1128/JVI.77.18.10078-10087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wang J., Thorson L., Stokes R.W., Santosuosso M., Huygen K., Zganiacz A., Hitt M., Xing Z. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 2004;173:6357–6365. doi: 10.4049/jimmunol.173.10.6357. [DOI] [PubMed] [Google Scholar]
- 217.Amara R.R., Villinger F., Altman J.D., Lydy S.L., O'Neil S.P., Staprans S.I., Montefiori D.C., Xu Y., Herndon J.G., Wyatt L.S., Candido M.A., Kozyr N.L., Earl P.L., Smith J.M., Ma H.L., Grimm B.D., Hulsey M.L., McClure H.M., McNicholl J.M., Moss B., Robinson H.L. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Vaccine. 2002;20:1949–1955. doi: 10.1016/s0264-410x(02)00076-2. [DOI] [PubMed] [Google Scholar]
- 218.Moorthy V.S., Imoukhuede E.B., Keating S., Pinder M., Webster D., Skinner M.A., Gilbert S.C., Walraven G., Hill A.V. Phase 1 evaluation of 3 highly immunogenic prime-boost regimens, including a 12-month reboosting vaccination, for malaria vaccination in Gambian men. J. Infect. Dis. 2004;189:2213–2219. doi: 10.1086/421118. [DOI] [PubMed] [Google Scholar]
- 219.Liu M., Acres B., Balloul J.M., Bizouarne N., Paul S., Slos P., Squiban P. Gene-based vaccines and immunotherapeutics. Proc. Natl. Acad. Sci. U. S. A. 2004;101(Suppl. 2):14567–14571. doi: 10.1073/pnas.0404845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.McShane H., Pathan A.A., Sander C.R., Keating S.M., Gilbert S.C., Huygen K., Fletcher H.A., Hill A.V. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat. Med. 2004;10:1240–1244. doi: 10.1038/nm1128. [DOI] [PubMed] [Google Scholar]
- 221.Niidome T., Huang L. Gene therapy progress and prospects: nonviral vectors. Gene Ther. 2002;9:1647–1652. doi: 10.1038/sj.gt.3301923. [DOI] [PubMed] [Google Scholar]
- 222.Roth C.M., Sundaram S. Engineering synthetic vectors for improved DNA delivery: insights from intracellular pathways. Annu. Rev. Biomed. Eng. 2004;6:397–426. doi: 10.1146/annurev.bioeng.6.040803.140203. [DOI] [PubMed] [Google Scholar]
- 223.Hobson P., Barnfield C., Barnes A., Klavinskis L.S. Mucosal immunization with DNA vaccines. Methods. 2003;31:217–224. doi: 10.1016/s1046-2023(03)00139-7. [DOI] [PubMed] [Google Scholar]
- 224.Gregoriadis G., Bacon A., Caparros-Wanderley W., McCormack B. A role for liposomes in genetic vaccination. Vaccine. 2002;20(Suppl. 5):B1–B9. doi: 10.1016/s0264-410x(02)00514-5. [DOI] [PubMed] [Google Scholar]
- 225.D'Souza S., Rosseels V., Denis O., Tanghe A., De Smet N., Jurion F., Palfliet K., Castiglioni N., Vanonckelen A., Wheeler C., Huygen K. Improved tuberculosis DNA vaccines by formulation in cationic lipids. Infect. Immun. 2002;70:3681–3688. doi: 10.1128/IAI.70.7.3681-3688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hermanson G., Whitlow V., Parker S., Tonsky K., Rusalov D., Ferrari M., Lalor P., Komai M., Mere R., Bell M., Brenneman K., Mateczun A., Evans T., Kaslow D., Galloway D., Hobart P. A cationic lipid-formulated plasmid DNA vaccine confers sustained antibody-mediated protection against aerosolized anthrax spores. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13601–13606. doi: 10.1073/pnas.0405557101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hattori Y., Kawakami S., Suzuki S., Yamashita F., Hashida M. Enhancement of immune responses by DNA vaccination through targeted gene delivery using mannosylated cationic liposome formulations following intravenous administration in mice. Biochem. Biophys. Res. Commun. 2004;317:992–999. doi: 10.1016/j.bbrc.2004.03.141. [DOI] [PubMed] [Google Scholar]
- 228.Gregoriadis G., Saffie R., de Souza J.B. Liposome-mediated DNA vaccination. FEBS Lett. 1997;402:107–110. doi: 10.1016/s0014-5793(96)01507-4. [DOI] [PubMed] [Google Scholar]
- 229.Perrie Y., Frederik P.M., Gregoriadis G. Liposome-mediated DNA vaccination: the effect of vesicle composition. Vaccine. 2001;19:3301–3310. doi: 10.1016/s0264-410x(00)00432-1. [DOI] [PubMed] [Google Scholar]
- 230.Ott G., Singh M., Kazzaz J., Briones M., Soenawan E., Ugozzoli M., O'Hagan D.T. A cationic sub-micron emulsion (MF59/DOTAP) is an effective delivery system for DNA vaccines. J. Control. Release. 2002;79:1–5. doi: 10.1016/s0168-3659(01)00545-4. [DOI] [PubMed] [Google Scholar]
- 231.Thomas M., Klibanov A.M. Non-viral gene therapy: polycation-mediated DNA delivery. Appl. Microbiol. Biotechnol. 2003;62:27–34. doi: 10.1007/s00253-003-1321-8. [DOI] [PubMed] [Google Scholar]
- 232.Illum L. Chitosan and its use as a pharmaceutical excipient. Pharm. Res. 1998;15:1326–1331. doi: 10.1023/a:1011929016601. [DOI] [PubMed] [Google Scholar]
- 233.Roy K., Mao H.Q., Huang S.K., Leong K.W. Oral gene delivery with chitosan–DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat. Med. 1999;5:387–391. doi: 10.1038/7385. [DOI] [PubMed] [Google Scholar]
- 234.Kumar M., Behera A.K., Lockey R.F., Zhang J., Bhullar G., De La Cruz C.P., Chen L.C., Leong K.W., Huang S.K., Mohapatra S.S. Intranasal gene transfer by chitosan–DNA nanospheres protects BALB/c mice against acute respiratory syncytial virus infection. Hum. Gene Ther. 2002;13:1415–1425. doi: 10.1089/10430340260185058. [DOI] [PubMed] [Google Scholar]
- 235.Iqbal M., Lin W., Jabbal-Gill I., Davis S.S., Steward M.W., Illum L. Nasal delivery of chitosan–DNA plasmid expressing epitopes of respiratory syncytial virus (RSV) induces protective CTL responses in BALB/c mice. Vaccine. 2003;21:1478–1485. doi: 10.1016/s0264-410x(02)00662-x. [DOI] [PubMed] [Google Scholar]
- 236.Bivas-Benita M., Laloup M., Versteyhe S., Dewit J., De Braekeleer J., Jongert E., Borchard G. Generation of Toxoplasma gondii GRA1 protein and DNA vaccine loaded chitosan particles: preparation, characterization, and preliminary in vivo studies. Int. J. Pharm. 2003;266:17–27. doi: 10.1016/s0378-5173(03)00377-6. [DOI] [PubMed] [Google Scholar]
- 237.Boussif O., Lezoualc'h F., Zanta M.A., Mergny M.D., Scherman D., Demeneix B., Behr J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Densmore C.L., Orson F.M., Xu B., Kinsey B.M., Waldrep J.C., Hua P., Bhogal B., Knight V. Aerosol delivery of robust polyethyleneimine–DNA complexes for gene therapy and genetic immunization. Molec. Ther. 2000;1:180–188. doi: 10.1006/mthe.1999.0021. [DOI] [PubMed] [Google Scholar]
- 239.Regnstrom K., Ragnarsson E.G., Koping-Hoggard M., Torstensson E., Nyblom H., Artursson P. PEI—a potent, but not harmless, mucosal immuno-stimulator of mixed T-helper cell response and FasL-mediated cell death in mice. Gene Ther. 2003;10:1575–1583. doi: 10.1038/sj.gt.3302054. [DOI] [PubMed] [Google Scholar]
- 240.Luo D., Saltzman W.M. Synthetic DNA delivery systems. Nat. Biotechnol. 2000;18:33–37. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- 241.Ando S., Putnam D., Pack D.W., Langer R. PLGA microspheres containing plasmid DNA: preservation of supercoiled DNA via cryopreparation and carbohydrate stabilization. J. Pharm. Sci. 1999;88:126–130. doi: 10.1021/js9801687. [DOI] [PubMed] [Google Scholar]
- 242.Perez C., Sanchez A., Putnam D., Ting D., Langer R., Alonso M.J. Poly(lactic acid)-poly(ethylene glycol) nanoparticles as new carriers for the delivery of plasmid DNA. J. Control. Release. 2001;75:211–224. doi: 10.1016/s0168-3659(01)00397-2. [DOI] [PubMed] [Google Scholar]
- 243.McKeever U., Barman S., Hao T., Chambers P., Song S., Lunsford L., Hsu Y.Y., Roy K., Hedley M.L. Protective immune responses elicited in mice by immunization with formulations of poly(lactide-co-glycolide) microparticles. Vaccine. 2002;20:1524–1531. doi: 10.1016/s0264-410x(01)00509-6. [DOI] [PubMed] [Google Scholar]
- 244.Luby T.M., Cole G., Baker L., Kornher J.S., Ramstedt U., Hedley M.L. Repeated immunization with plasmid DNA formulated in poly(lactide-co-glycolide) microparticles is well tolerated and stimulates durable T cell responses to the tumor-associated antigen cytochrome P450 1B1. Clin. Immunol. 2004;112:45–53. doi: 10.1016/j.clim.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 245.Little S.R., Lynn D.M., Ge Q., Anderson D.G., Puram S.V., Chen J., Eisen H.N., Langer R. Poly-beta amino ester-containing microparticles enhance the activity of nonviral genetic vaccines. Proc. Natl. Acad. Sci. U. S. A. 2004;101:9534–9539. doi: 10.1073/pnas.0403549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Singh M., Briones M., Ott G., O'Hagan D. Cationic microparticles: a potent delivery system for DNA vaccines. Proc. Natl. Acad. Sci. U. S. A. 2000;97:811–816. doi: 10.1073/pnas.97.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Singh M., Ugozzoli M., Briones M., Kazzaz J., Soenawan E., O'Hagan D.T. The effect of CTAB concentration in cationic PLG microparticles on DNA adsorption and in vivo performance. Pharm. Res. 2003;20:247–251. doi: 10.1023/a:1022327305369. [DOI] [PubMed] [Google Scholar]
- 248.O'Hagan D.T., Singh M., Dong C., Ugozzoli M., Berger K., Glazer E., Selby M., Wininger M., Ng P., Crawford K., Paliard X., Coates S., Houghton M. Cationic microparticles are a potent delivery system for a HCV DNA vaccine. Vaccine. 2004;23:672–680. doi: 10.1016/j.vaccine.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 249.Schwarz L.A., Johnson J.L., Black M., Cheng S.H., Hogan M.E., Waldrep J.C. Delivery of DNA–cationic liposome complexes by small-particle aerosol. Hum. Gene Ther. 1996;7:731–741. doi: 10.1089/hum.1996.7.6-731. [DOI] [PubMed] [Google Scholar]
- 250.Deshpande D., Blezinger P., Pillai R., Duguid J., Freimark B., Rolland A. Target specific optimization of cationic lipid-based systems for pulmonary gene therapy. Pharm. Res. 1998;15:1340–1347. doi: 10.1023/a:1011933117509. [DOI] [PubMed] [Google Scholar]
- 251.Densmore C.L., Giddings T.H., Waldrep J.C., Kinsey B.M., Knight V. Gene transfer by guanidinium-cholesterol: dioleoylphosphatidyl–ethanolamine liposome–DNA complexes in aerosol. J. Gene Med. 1999;1:251–264. doi: 10.1002/(SICI)1521-2254(199907/08)1:4<251::AID-JGM43>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 252.Koping-Hoggard M., Tubulekas I., Guan H., Edwards K., Nilsson M., Varum K.M., Artursson P. Chitosan as a nonviral gene delivery system. Structure–property relationships and characteristics compared with polyethylenimine in vitro and after lung administration in vivo. Gene Ther. 2001;8:1108–1121. doi: 10.1038/sj.gt.3301492. [DOI] [PubMed] [Google Scholar]
- 253.Okamoto H., Nishida S., Todo H., Sakakura Y., Iida K., Danjo K. Pulmonary gene delivery by chitosan–pDNA complex powder prepared by a supercritical carbon dioxide process. J. Pharm. Sci. 2003;92:371–380. doi: 10.1002/jps.10285. [DOI] [PubMed] [Google Scholar]
- 254.Stern M., Ulrich K., Geddes D.M., Alton E.W. Poly(d,l-lactide-co-glycolide)/DNA microspheres to facilitate prolonged transgene expression in airway epithelium in vitro, ex vivo and in vivo. Gene Ther. 2003;10:1282–1288. doi: 10.1038/sj.gt.3301994. [DOI] [PubMed] [Google Scholar]
- 255.Bivas-Benita M., Romeijn S., Junginger H.E., Borchard G. PLGA–PEI nanoparticles for gene delivery to pulmonary epithelium. Eur. J. Pharm. Biopharm. 2004;58:1–6. doi: 10.1016/j.ejpb.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Bivas-Benita M., Oudshoorn M., Romeijn S., van Meijgaarden K., Koerten H., van der Meulen H., Lambert G., Ottenhoff T., Benita S., Junginger H., Borchard G. Cationic submicron emulsions for pulmonary DNA immunization. J. Control. Release. 2004;100:145–155. doi: 10.1016/j.jconrel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 257.Takeuchi O., Sato S., Horiuchi T., Hoshino K., Takeda K., Dong Z., Modlin R.L., Akira S. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 258.Wyllie D.H., Kiss-Toth E., Visintin A., Smith S.C., Boussouf S., Segal D.M., Duff G.W., Dower S.K. Evidence for an accessory protein function for Toll-like receptor 1 in anti-bacterial responses. J. Immunol. 2000;165:7125–7132. doi: 10.4049/jimmunol.165.12.7125. [DOI] [PubMed] [Google Scholar]
- 259.Aliprantis A.O., Yang R.B., Mark M.R., Suggett S., Devaux B., Radolf J.D., Klimpel G.R., Godowski P., Zychlinsky A. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 260.Takeuchi O., Hoshino K., Kawai T., Sanjo H., Takada H., Ogawa T., Takeda K., Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 261.Schwandner R., Dziarski R., Wesche H., Rothe M., Kirschning C.J. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 1999;274:17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 262.Means T.K., Wang S., Lien E., Yoshimura A., Golenbock D.T., Fenton M.J. Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 1999;163:3920–3927. [PubMed] [Google Scholar]
- 263.Hajjar A.M., O'Mahony D.S., Ozinsky A., Underhill D.M., Aderem A., Klebanoff S.J., Wilson C.B. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J. Immunol. 2001;166:15–19. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- 264.Coelho P.S., Klein A., Talvani A., Coutinho S.F., Takeuchi O., Akira S., Silva J.S., Canizzaro H., Gazzinelli R.T., Teixeira M.M. Glycosylphosphatidylinositol-anchored mucin-like glycoproteins isolated from Trypanosoma cruzi trypomastigotes induce in vivo leukocyte recruitment dependent on MCP-1 production by IFN-gamma-primed-macrophages. J. Leukoc. Biol. 2002;71:837–844. [PubMed] [Google Scholar]
- 265.Opitz B., Schroder N.W., Spreitzer I., Michelsen K.S., Kirschning C.J., Hallatschek W., Zahringer U., Hartung T., Gobel U.B., Schumann R.R. Toll-like receptor-2 mediates Treponema glycolipid and lipoteichoic acid-induced NF-kappaB translocation. J. Biol. Chem. 2001;276:22041–22047. doi: 10.1074/jbc.M010481200. [DOI] [PubMed] [Google Scholar]
- 266.Massari P., Henneke P., Ho Y., Latz E., Golenbock D.T., Wetzler L.M. Cutting edge: immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J. Immunol. 2002;168:1533–1537. doi: 10.4049/jimmunol.168.4.1533. [DOI] [PubMed] [Google Scholar]
- 267.Werts C., Tapping R.I., Mathison J.C., Chuang T.H., Kravchenko V., Saint Girons I., Haake D.A., Godowski P.J., Hayashi F., Ozinsky A., Underhill D.M., Kirschning C.J., Wagner H., Aderem A., Tobias P.S., Ulevitch R.J. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat. Immunol. 2001;2:346–352. doi: 10.1038/86354. [DOI] [PubMed] [Google Scholar]
- 268.Hirschfeld M., Weis J.J., Toshchakov V., Salkowski C.A., Cody M.J., Ward D.C., Qureshi N., Michalek S.M., Vogel S.N. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect. Immun. 2001;69:1477–1482. doi: 10.1128/IAI.69.3.1477-1482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Underhill D.M., Ozinsky A., Hajjar A.M., Stevens A., Wilson C.B., Bassetti M., Aderem A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 270.Asea A., Rehli M., Kabingu E., Boch J.A., Bare O., Auron P.E., Stevenson M.A., Calderwood S.K. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 271.Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 272.Poltorak A., He X., Smirnova I., Liu M.Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 273.Kawasaki K., Akashi S., Shimazu R., Yoshida T., Miyake K., Nishijima M. Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J. Biol. Chem. 2000;275:2251–2254. doi: 10.1074/jbc.275.4.2251. [DOI] [PubMed] [Google Scholar]
- 274.Kurt-Jones E.A., Popova L., Kwinn L., Haynes L.M., Jones L.P., Tripp R.A., Walsh E.E., Freeman M.W., Golenbock D.T., Anderson L.J., Finberg R.W. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 275.Rassa J.C., Meyers J.L., Zhang Y., Kudaravalli R., Ross S.R. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. U. S. A. 2002;99:2281–2286. doi: 10.1073/pnas.042355399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Bulut Y., Faure E., Thomas L., Karahashi H., Michelsen K.S., Equils O., Morrison S.G., Morrison R.P., Arditi M. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J. Immunol. 2002;168:1435–1440. doi: 10.4049/jimmunol.168.3.1435. [DOI] [PubMed] [Google Scholar]
- 277.Ohashi K., Burkart V., Flohe S., Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 278.Vabulas R.M., Ahmad-Nejad P., Ghose S., Kirschning C.J., Issels R.D., Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J. Biol. Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 279.Okamura Y., Watari M., Jerud E.S., Young D.W., Ishizaka S.T., Rose J., Chow J.C., Strauss J.F., III The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 280.Termeer C., Benedix F., Sleeman J., Fieber C., Voith U., Ahrens T., Miyake K., Freudenberg M., Galanos C., Simon J.C. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Johnson G.B., Brunn G.J., Kodaira Y., Platt J.L. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J. Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 282.Smiley S.T., King J.A., Hancock W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 283.Hayashi F., Smith K.D., Ozinsky A., Hawn T.R., Yi E.C., Goodlett D.R., Eng J.K., Akira S., Underhill D.M., Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 284.Takeuchi O., Kawai T., Muhlradt P.F., Morr M., Radolf J.D., Zychlinsky A., Takeda K., Akira S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 285.Ozinsky A., Underhill D.M., Fontenot J.D., Hajjar A.M., Smith K.D., Wilson C.B., Schroeder L., Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. U. S. A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Heil F., Ahmad-Nejad P., Hemmi H., Hochrein H., Ampenberger F., Gellert T., Dietrich H., Lipford G., Takeda K., Akira S., Wagner H., Bauer S. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur. J. Immunol. 2003;33:2987–2997. doi: 10.1002/eji.200324238. [DOI] [PubMed] [Google Scholar]
- 287.Hemmi H., Kaisho T., Takeuchi O., Sato S., Sanjo H., Hoshino K., Horiuchi T., Tomizawa H., Takeda K., Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 288.Heil F., Hemmi H., Hochrein H., Ampenberger F., Kirschning C., Akira S., Lipford G., Wagner H., Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 289.Lund J.M., Alexopoulou L., Sato A., Karow M., Adams N.C., Gale N.W., Iwasaki A., Flavell R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Diebold S.S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 291.Jurk M., Heil F., Vollmer J., Schetter C., Krieg A.M., Wagner H., Lipford G., Bauer S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 292.Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 293.Zhang D., Zhang G., Hayden M.S., Greenblatt M.B., Bussey C., Flavell R.A., Ghosh S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]