

Abstract

Computer-assisted drug design (CADD) methods have greatly contributed to the development of new drugs. Among CADD methodologies, virtual screening (VS) can enrich the compound collection with molecules that have the desired physicochemical and pharmacophoric characteristics that are needed to become drugs. Many free tools are available for this purpose, but they are difficult to use and do not have a graphical user interface. Furthermore, several free tools must be used to carry out the entire VS process, requiring the user to process the results of one software program so that they can be used in another program, adding a potential source of human error. Moreover, some software programs require knowledge of advanced computational skills, such as programming languages. This context has motivated us to develop Molecular Architect (MolAr). MolAr is a workflow with a simple and intuitive interface that acts in an integrated and automated form to perform the entire VS process, from protein preparation (homology modeling and protonation state) to virtual screening. MolAr carries out VS through AutoDock Vina, DOCK 6, or a consensus of the two. Two case studies were conducted to demonstrate the performance of MolAr. In the first study, the feasibility of using MolAr for DNA–ligand systems was assessed. Both AutoDock Vina and DOCK 6 showed good results in performing VS in DNA–ligand systems. However, the use of consensus virtual screening was able to enrich the results. According to the area under the ROC curve and the enrichment factors, consensus VS was better able to predict the positions of the active ligands. The second case study was performed on 8 targets from the DUD-E database and 10 active ligands for each target. The results demonstrated that using the final ligand conformation provided by AutoDock Vina as an input for DOCK 6 improved the DOCK 6 ROC curves by up to 42% in VS. These case studies demonstrated that MolAr is capable conducting the VS process and is an easy-to-use and effective tool. MolAr is available for download free of charge at http: //www.drugdiscovery.com.br/software/.
Introduction
The drug design process aims to identify bioactive compounds to assist in the treatment of diseases. The development of a new drug has an average cost of $2.6 billion1 and can take 12–14 years.2Figure 1 shows a summary of the developmental process of a new drug, which starts with the identification of molecular targets for a given compound and is followed by their validation. Next, virtual screening (VS) can be used to identify active drug candidates (hit identification), and biologically active compounds are transformed into appropriate drugs by improving their physicochemical compositions (lead optimization). Finally, optimized leads undergo preclinical and clinical trials before they are approved for use by regulatory bodies.3
Figure 1.

Drug design process.
One way to minimize costs and time in the drug development process is making use of computer-aided drug design (CADD) methodologies.4 CADD is a fast and valid methodology that is used for researching new compounds with pharmacological potential.5,6 CADD allows many molecules to be analyzed in a short time and enables the simulation and prediction of several essential factors, such as toxicity, activity, bioavailability, and efficacy, even before the compound is submitted to in vitro testing.5
In this context, VS is used to identify new hits in large compound libraries. VS uses computational methods to identify promising bioactive substances.7
The use of virtual screening in drug development, however, has some drawbacks. There are several advantages and disadvantages to be considered:
-
1Advantages
-
a.Virtual screening of millions of small compounds can be performed computationally in a short amount of time, minimizing the timeline and the total cost of developing new drugs.
-
b.The ligand molecules used in VS do not need to exist physically. Thus, a molecule can be screened before it is synthesized. If VS demonstrates that a molecule is not a good candidate, there is no need to synthesize it.
-
c.There are several free and proprietary tools available to assist in VS.
-
a.
-
2.Disadvantages
-
a.Some VS tools work best in specific cases.8 Thus, the result may be different, depending on the tool used.
-
b.It is difficult to set the parameters of the ligand–receptor binding interactions. Therefore, it is challenging to predict the correct binding position of the compounds.
-
c.VS can generate false positives and false negatives; thus, it can discard promising ligands or indicate a compound as an active ligand that will prove to be inactive in a subsequent stage of development.
-
a.
Despite its disadvantages, VS is a widely used tool in drug design and has been used extensively in recent years,7,9−14 which indicates that although there are disadvantages, the reduced time and cost enabled by VS is useful and promising for the development of new drugs.
One of the most widely used VS techniques is structure-based drug design (SBDD).15 SBDD attempts to predict the best binding orientation with the best binding affinity and/or free energy of two molecules to form a stable complex, but it requires knowledge of the 3D structure of the target to predict the interactions between the target and the ligand.16
The availability of a 3D structure of a molecular target is essential for performing VS, but these structures are difficult to obtain experimentally. Additionally, the atomic coordinates of highly flexible loops in available structures are often poorly described by experimental methods. As a result, there are often gaps in the structure, and these may be near the binding site. Predicting the 3D structure of a protein from its amino acid sequence can be accomplished by homology modeling (HM). HM predicts a protein structure based on the general observation that proteins with similar sequences have similar structures.17 Consequently, minor changes in the sequence will result in only small changes in the 3D structure.18
VS would be more effective if it took the different protonation states of the ligand and the molecular target into consideration since the proteins around the active site may influence the local pH.19 However, the position of hydrogen atoms cannot be determined experimentally by X-ray crystallography.20 Most ligand–receptor interactions are pH-dependent, and the protonation states of the molecules must be appropriately assigned.21 Adjusting the protonation state of ligands and targets can be done manually, but since structure databases contain thousands of compounds, this preparation should be done automatically.
After performing virtual screening, the next challenge of this method is to differentiate compounds that are active against the target from those that are inactive (false positives).22 Thus, VS tools must have ways to assist their users to distinguish false positives from true positives. The ROC curve and the area under the ROC curve (AUC-ROC)23 are widely used for this purpose. The AUC value can vary between 0 and 1; if the AUC is above 0.70, then the software program used is satisfactorily separating active from inactive ligands.24
Consensus virtual screening (CVS) has been used to increase the accuracy of VS studies and reduce the number of false positives obtained in virtual screening.25−31 The main idea of this technique is that the combination of two different approaches in VS is better than the application of a single approach alone.30 CVS is a relatively recent technique, and although it has not yet been widely applied in the literature,27 it has presented some promising results.25,28,30,31 However, CVS is difficult to apply as the use of more than one approach involves handling entries in different formats and using various software programs. Hence, if CVS could be performed automatically, this technique could be more widely applied.
Due to the importance of CADD in modern drug development, many software programs have been developed to perform particular operations throughout the VS process.2,32−34 In general, the existing free software programs do not perform the whole VS process in an automated way, making it necessary to combine several free software programs, such as Pymol,35 MGLTools,36 MODELLER,37 PROPKA,38 Chimera,39 AutoDock Vina,40 and DOCK 6.41 Thus, a large effort is required from researchers, which makes VS more prone to human errors. Another problem of using free software is the complexity of their interfaces. Sometimes, the software has no graphical interface, and it must be used by entering command lines (e.g., AutoDock Vina40 and DOCK 642), making it very difficult to use for those who are not comfortable with command-line programs. In some cases, such as MODELLER,37 the program is even more difficult to use because the user needs prior knowledge in computer programming languages.
Therefore, developing an easy-to-use software program would have a great value for the VS process since automated procedures ensure that the methods can be easily reproduced, excluding the variability of a manual process performed by a human. This type of program would allow the results of VS to be evaluated and compared with greater reliability and accuracy.
This paper presents Molecular Architect (MolAr), a workflow composed of a set of integrated tools that carries out the VS process. MolAr does not require the researcher to have advanced computational skills (e.g., installation of tools and libraries, need for prior knowledge in computer programming languages), thus facilitating the execution of VS simulations through simplifying and automating the process. The application of MolAr is expected to decrease human error in VS and execute the procedures at a greater speed due to the simplified, integrated, and automated VS process. In addition, MolAr implements a consensus virtual screening approach between AutoDock Vina and DOCK 6, and it was evaluated in two case studies demonstrating that it was able to achieve satisfactory results when performing in silico simulations. MolAr represents promising VS software. MolAr has an easy-to-use interface and does not require multiple software programs to be run from the command line to perform the VS process.
Methods
MolAr is a workflow composed of a set of tools that act in an integrated manner to carry out the entire virtual screening process, from protein preparation (homology modeling, necessary asymmetry, protonation) to virtual screening. MolAr was developed using the high-level programming languages Java and Python.
MolAr has intuitive interfaces that negate the need for advanced computing knowledge, making it accessible to a wider range of people. Figure 2 shows the virtual screening workflow using MolAr. First, after MolAr initialization, if it is necessary to perform homology modeling, then MolAr uses MODELLER to perform this task. The protonation state of the protein can then be adjusted using PROPKA38 (targets) and Open Babel43 (ligands), in which the pH can be defined by the user. Finally, VS can be performed. MolAr can be applied in three different ways: using AutoDock Vina,40 DOCK 6,42 or through a consensus virtual screening (CVS) that uses a combination of the AutoDock Vina and DOCK 6 results.
Figure 2.

MolAr workflow.
MolAr was evaluated and validated at the pharmaceutical chemistry laboratory of the Federal University of São João del-Rei. In the first stage of the tests, only researchers from the pharmaceutical chemistry laboratory used the program. In this way, the execution was carried out in a more controlled environment. Problems that arose during the execution were solved and served as an opportunity to improve the platform. Next, two case studies were performed using MolAr; these will be discussed in this paper. The first case study investigated the feasibility of using MolAr for DNA–ligand systems. In this case study, both AutoDock Vina and DOCK 6 presented good reliability when considering the AUC-ROC values, but the use of consensus virtual screening was able to enrich these results. The second case study demonstrated that applying DOCK 6 using the final ligand conformation provided by AutoDock Vina improved DOCK 6 performance during VS and consequently improved the VS results. The simulations using the amber score were performed in 3000 steps and 100 energy minimization cycles, and the ligand conformational searching was enabled. In the simulations, the ligand was allowed to move during scoring. In the AutoDock Vina simulations, the dimensions of the box was 20 on the x, y, and z axes, and the exhaustiveness parameter was set to 24 to provide a better docking result.44 MolAr refined the ligands through MOPAC201645 using the Parametric Method 7 (PM7) and EF routine to search for the structure of local minimum. The configuration files used in the simulations, containing all the applied parameters, can be accessed in the supporting materials. There are Windows and Linux versions of MolAr, and it is freely available for download at http://www.drugdiscovery.com.br/.
Results and Discussion
It is necessary to install several software programs to perform VS when using free software, and many of them are difficult to install and/or configure. MolAr automates the installation and configuration process of all software programs it requires to execute VS. These are Open JDK, which contains the necessary infrastructure to develop and run Java applications, Python, MOPAC2016,45 MODELLER,37 Procheck,46 Pymol,35 Jmol,47 Pdb2pqr,38 MPI,48 AutoDock Vina 1.1.2,40 DOCK 6,42 Sphgen (available for download at http://dock.compbio.ucsf.edu/Contributed_Code/sphgen_cpp.htm), AutoDocktools,36 Chimera,39 AmberTools,49 Open Babel,43 MGLTools,36 and some software installed by DOCK 6 (DMS, Grid, and Showbox). Thus, MolAr can facilitate VS experiments by automating the process.
There are software packages similar to MolAr. However, they have limitations that MolAr does not have, such as lack of support in the free version (PyRx50), an inability to perform consensus virtual screening (EasyVS51 and Raccoon244), and limits on the number of ligands (DockThor52,53 limits guest users to 100 structures and approved project users are limited to 1000 structures). Moreover, none of these software packages can perform homology modeling, whereas MolAr uses MODELLER54 for this step.
MolAr has three main menus: target builder, docking, and tools.
Target Builder Menu
The target builder (TB) menu contains features related to predicting the 3D structure of a protein from its amino acid sequence (homology modeling). MolAr uses MODELLER55 to perform homology modeling. Although MODELLER is quite complete, it is necessary for one to know the Python programming language to perform the modeling. Moreover, because it is necessary to know the 3D structure of a protein to realize VS, it would be beneficial if homology modeling could be performed by the same tool used to perform VS. In this way, the user could build the target protein from its amino acid sequence or build gap regions of a target protein whose 3D structure already exists prior to performing VS.
MolAr renders MODELLER easier to use and eliminates the need for knowing Python.
Other software also facilitates the use of MODELLER, such as the Modweb webserver.55 However, this software specializes in homology modeling, while MolAr allows the user to perform several virtual screening steps in a single tool.
MolAr carries out the homology modeling process in 15 steps (Figure 3).
Figure 3.

Target builder workflow.
After initiating the homology modeling process (step 1 in Figure 3), MolAr checks which data type the researcher has provided. If the user entered the PDB code, MolAr will download the FASTA file corresponding to that code. If the user entered the PDB file, MolAr will convert the PDB file to a FASTA file. If the system performed steps 2 or 3 or if the user entered the FASTA sequence, MolAr will convert the FASTA file to a file in the alignment format used by MODELLER. MolAr identifies the templates to be used in the homology modeling process (if the user has not specified them) and automatically downloads the PDB files corresponding to the selected templates. MolAr then identifies the similarities between the selected templates to perform multiple alignments of them. Next, the protein is aligned to the multiple templates selected, and models are built by MODELLER in parallel. The DOPE score and the RMSD between the generated model and the templates are then calculated. MolAr generates the Ramachandran plot for each model using Procheck;46 finally, a window with the results is shown to the user.
In the homology modeling process, it is important to know whether the developed model is of sufficient quality. Thus, for each model built, MolAr displays the value of the RMSD relative to the target template and allows the visualization of the Ramachandran plot (generated using Procheck46). In this way, the user can decide which model is the best. The results screen also allows the results to be ordered in ascending or descending order.
Occasionally, an available 3D structure contains gaps. These residues are documented in the PDB file, but during X-ray crystallography, it was not possible to determine their atomic coordinates. The MolAr missing residues option was created to address this problem by applying homology modeling. If there are gaps in the target protein’s PDB file, MolAr can still try to fill them in by making changes only in the gap region, which causes fewer changes in the pre-existing 3D structure than if homology modeling was applied for the entire protein.
Docking Menu
The docking menu has features that execute the molecular docking and virtual screening procedures. The developed platform allows the realization of virtual screening through AutoDock Vina,40 DOCK 641 or a consensus between them.
AutoDock Vina and DOCK 6 were chosen to integrate MolAr because both tools are free and effective. Several recent studies have demonstrated the effectiveness of AutoDock Vina and DOCK 6 in the development of new drugs. Among them, Lagarde et al.56 validated the use of AutoDock Vina in the development of cancer drugs, Shukla et al.57 used AutoDock Vina to identify a potential anti-fasciolid compound, and Kondratyev and Zakharova58 used AutoDock Vina to simulate and analyze the interactions of peroxiredoxin 6 with captopril, unithiol, succimer, cystamine, and three cysteine-containing peptides, ECECE, KCKCK, and ACC. DOCK 641 is an enhanced version of DOCK 5 with additional sampling, scoring, and optimization features, fixed bugs, and the ability to conduct RNA compatibility testing. Holden et al.59 employed DOCK 6.5 in the successful discovery of novel HIVgp41 inhibitors. Nunes et al.12 used DOCK 6 to select a compound that was active and selective against Plasmodium falciparum. These results were later confirmed by in vitro assays, indicating that DOCK 6 correctly identified the compound. In addition, DOCK 6 has been integrated with AmberTools and is one of the few free software packages with graphics processing unit (GPU) implementation for AMBER scoring and PBSA/GBSA calculations for target–ligand60 complexes, which allows for faster calculation of the AMBER scoring function. MolAr allows the user to apply these features during docking. Allen et al.41 described additional cases of the successful use of DOCK, such as in the discovery of a new amidohydrolase,61 thiamine synthase phosphate.62
MolAr has two integrated databases: the Our Own Molecular Target (OOMT)7 and the Brazilian Malaria Molecular Target (BRAMMT) databases.12 The OOMT database comprises various receptors from the Protein Data Bank (PDB) and includes specific targets for cancer, dengue, and malaria. The BRAMMT database comprises receptors for P. falciparum. The MolAr docking menu contains three submenus: Octopus, DOCK 6, and consensus virtual screening.
Octopus Submenu
The Octopus submenu63 performs VS using AutoDock Vina.40 Octopus carries out the VS process using AutoDock Vina in two different forms depending on which menu option is chosen: with previous execution of MOPAC201645 (a semi-empirical quantum chemistry program) or without running MOPAC2016. By implementing MOPAC2016, the net atomic charge for each atom in each molecule is calculated, avoiding massive work by the user. Next, the ligands, in PDB file format, are refined through MOPAC2016 using Parametric Method 7 (PM7)64 and the EF routine65 to search for the structure of the local minimum. An overview of the Octopus workflow can be seen in Figure 4.
Figure 4.

Octopus workflow.
First, directories of the ligands and targets are chosen. The ligands must be in the PDB format, and files in the target directory must be in the AutoDock Vina format. If the user chooses to refine the ligands, MolAr will perform the refinement using MOPAC2016.45 Next, ligands are converted from PDB to PDBQT file format while assigning the rotatable bonds and the Gasteiger–Marsili net atomic charges.66 Only those hydrogens on polar atoms (oxygen and nitrogen) are kept, while other hydrogens atoms are removed. A visual inspection of the geometries of the ligands can then be performed through PyMOL.35 In the next step, docking is carried out using AutoDock Vina, which runs until all the ligands have been docked to a set of targets. Finally, the target–ligand binding energies for the complex are generated. The standard crystallographic values for the binding energies between the ligand and target are also displayed.
Database Manager Option of Octopus Submenu
The Database Manager option of the Octopus submenu is used to manage the Octopus database. This functionality allows the user to create a new database and verify that the target databases used by Octopus are correct. Target databases are constantly changing as new molecules are inserted or targets are modified. If the database updates do not follow a standard, VS can fail, and precious time is spent trying to identify and correct the problem. To solve this, the Database Manager was developed. This feature corrects the format and any inconsistences in the databases to be used by Octopus. The Database Manager tool has three basic functions: creation of a new database, fixing problems in an existing database, and editing the data stored in an existing database. If there are any missing or incorrect data in the database, it is possible to alter this manually.
Dock 6 Submenu
The DOCK 6 program was created in the 1980s by Irwin and Kuntz’s group at the Pharmaceutical Chemistry Laboratory of the University of California and was the first docking program.41,67 In our method, a graphical interface was developed to enable VS using DOCK 6 with a minimal amount of user effort. The user does not have to intervene much in the process, and it is expected that fewer human errors will be committed because the workflow is automatic. Figure 5 illustrates the execution of the DOCK 6 workflow performed by MolAr.
Figure 5.

DOCK 6 workflow.
In the workflow presented in Figure 5, the user initiates re-docking or VS with DOCK 6. It is necessary to first prepare the molecular target and the ligand (add hydrogens and calculate charges). MolAr performs this task automatically using Chimera.39 Thereafter, the binding site is prepared using the DMS program to calculate the target surface to which the solvent will have access. Next, spheres representing the binding site are created using the sphgen_cpp program. This program defines the volume or space within the binding site where the drug will interact. Its purpose is to generate a grid of sphere centers that reflects the shape of the active site. Next, the box (which is the cubic region where DOCK 6 will perform the docking) is generated using the Showbox program. After box generation, the grid energies are calculated using the grid program. Next, docking is conducted by DOCK 6. If the user is performing VS and if there are additional ligands, docking will be performed again with all ligands. If VS is not required or if there are no more targets, then the result screen will be shown. Finally, the user can save the results and close the DOCK 6 results screen.
Consensus Virtual Screening (CVS)
MolAr implements CVS between DOCK 6 and AutoDock Vina. Figure 6 shows the workflow of the CVS approach implemented by MolAr. First, VS using AutoDock Vina is performed as described in Figure 4. The AutoDock Vina output (pdbqt file) is then converted to the DOCK 6 format by MolAr and the Open Babel43 program. Next, VS is performed using DOCK 6, as described in Figure 5. During the consensus, AutoDock Vina is executed first followed by DOCK 6. This is because DOCK 6 allows docking using the amber score. If DOCK 6 were executed before AutoDock Vina during the consensus, the gains obtained from the molecular dynamics performed by DOCK 6 with the amber score would be lost. Before displaying the results, the AutoDock Vina and DOCK 6 results are merged. Finally, the CVS score is calculated and displayed to the user.
Figure 6.

Consensus virtual screening workflow.
The scoring function results displayed by AutoDock Vina and DOCK 6 are normalized to values between 0 and 10. The CVS score calculated by MolAr corresponds to the average between these two values. Thus, the CVS scoring function is calculated according to the following equation:
In the consensus approach, as an input to DOCK 6, MolAr uses the resulting ligand conformation after performing docking with AutoDock Vina, thus aiming to achieve better results with DOCK 6 since it starts from a conformation already optimized by AutoDock Vina. The final ligand pose selected by consensus virtual screening is the pose defined by DOCK 6.
Tools Menu
MolAr is integrated with a set of tools to support the realization of the entire VS process. With these tools, it is possible to visualize the 3D structure of a molecule (using Jmol47 and PyMol35), to analyze the quality of a structure (through the RMSD calculation and the Ramachandran plot generated by Procheck46), and to adjust the protonation state (using PROPKA38). MolAr can generate the ROC curve and the AUC-ROC of a given VS result to verify whether VS can separate two potential compounds by pressing the ROC curve command from the Tools Menu.
Case Studies
The software described herein was evaluated in two case studies developed in the Pharmaceutical Chemistry Laboratory of the Federal University of São João del-Rei:
-
I.
Investigation of the best in silico model for DNA–ligand systems.
-
II.
Evaluation of the CVS approach implemented by MolAr.
In the following subsections, we describe the above case studies.
Investigation of the Best In Silico Model for DNA–Ligand Systems
DNA is a common target in the treatment of several genetic diseases, most notably cancer, due to its importance in the cell cycle.68 However, drugs that interact with DNA are often highly toxic due to the low selectivity between the DNA of normal and abnormal cells; therefore, this strategy is used a last resort, motivating the development of new drugs targeting DNA.69
Molecular docking is widely validated for protein–ligand systems, and despite recent works that have used the method to model DNA–ligand complexes, there is not yet a consensus regarding the best in silico model for nucleic acids. DNA has several unique properties, such as a high charge density and high flexibility.68 Holt et al.70 showed that molecular docking techniques can be successfully extended to include nucleic acid targets. Other docking studies have also been carried out on this topic. Evans and Neidle63 showed the utility of using DOCK and AutoDock software to predict poses in DNA–ligand complexes. Ricci and Netz71 used AutoDock 4.0 to perform a docking study using two ligands and four distinct DNA receptors. The authors demonstrated that this approach could be used in DNA–ligand complexes because the predicted binding mode corresponded to the experimentally suggested mode. Fong and Wong72 evaluated four different scoring functions (AutoDock, ASP@GOLD, ChemScore@GOLD, and GoldScore@GOLD) for DNA–ligand complexes and concluded that DNA–ligand complex evaluation using docking can obtain good results. Moreover, they demonstrated that the use of more than one scoring function improves the results. Srivastava et al.73 validated the use of docking approaches and molecular dynamics in DNA–ligand complexes. They presented a systematic computational analysis of 57 DNA ligands using four popular docking protocols (GOLD, Glide, CDOCKER, and AutoDock) and concluded that the GOLD and Glide protocols were very reliable when modeling nucleic acid–ligand complexes.
To compare recent in silico models of DNA–ligand systems, MolAr was used to evaluate the different approaches used by AutoDock Vina40 and DOCK 642 as well as the combination of these two (i.e., consensus virtual screening, which intends to improve the reliability of VS results by using a combination of results of different VS approaches). ROC curve analysis was used to compare and validate these approaches. The active compounds were selected from the research of Srivastava et al.73 They were used to compare several molecular docking approaches using 57 crystal structures of DNA–ligand complexes with known minor groove binders as ligands.73 To perform our study, we selected the four most active ligands from Srivastava et al.,73 the DNA model related to them, and 50 decoys for each ligand that were obtained using the DUD-E service.99 VS was performed for 204 ligands (four active ligands and 200 decoys). The target was 1VZK (a thiophene-based diamidine that strongly binds the minor groove at AT sites), which was the same as that used by Srivastava et al.73Figure 7 presents the 3D structure of the target 1VZK and the interaction in 2D with its crystallographic ligand (D1B).
Figure 7.

1VZK: (a) 3D structure view. (b) Interactions in 2D.
Three virtual screenings were carried out: (1) Minimization of the ligands with the MOPAC program followed by virtual screening with AutoDock Vina, (2) virtual screening with DOCK 6, and (3) CVS between approaches (1) and (2). The AUC-ROC was used to assess whether CVS (DOCK 6 plus AutoDock Vina) could increase the reliability of the docking.
Results of the Case Study to Find the Best In Silico Model for DNA–Ligand Systems
All configurations resulted in excellent AUC-ROC values (Figure 8), showing that AutoDock Vina, DOCK 6, and the consensus docking approach proposed by MolAr were able to differentiate active compounds from inactive compounds. An AUC-ROC greater than 0.7 means that the software program was able to differentiate active ligands from inactive ligands.24 In this case study, the AUC-ROC values were 0.98, 0.88, and 0.99 for AutoDock Vina, DOCK 6, and CVS, respectively (Figure 8).
Figure 8.
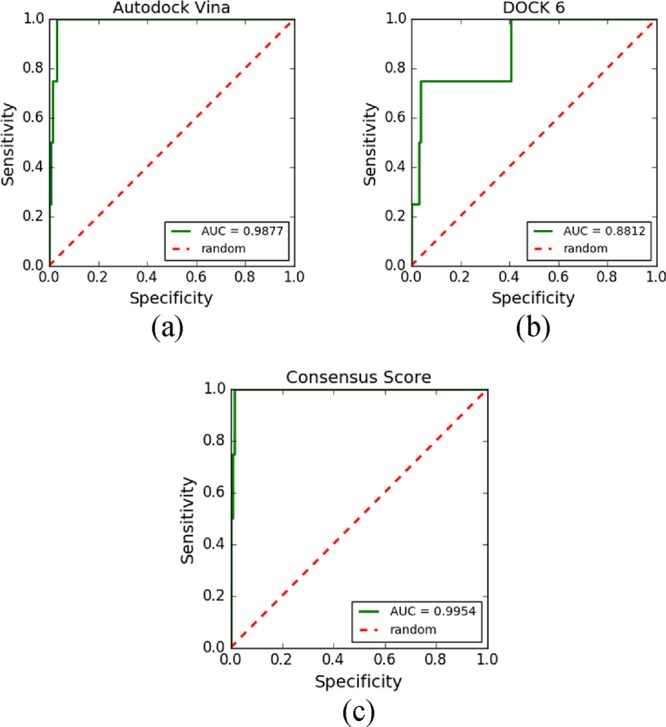
ROC curves obtained after performing VS with (a) AutoDock Vina, (b) DOCK 6, and (c) CVS.
The AUC values remained consistent in all ROC curves. AutoDock Vina (Figure 8a) showed better results than DOCK 6 (Figure 8b), probably due to differences in the search algorithms and scoring functions. The AutoDock Vina search algorithm relies on random changes of conformation and is able to search outside of local sites of minimum energy, while DOCK 6 uses an anchor-and-grow search algorithm. In addition, AutoDock Vina and DOCK 6 use different scoring functions. Scoring functions are the main reason for the failure or success of docking tools because they are responsible for predicting the binding affinity between a target and its candidate ligand.20 AutoDock Vina uses empirical scoring functions to classify ligands, while DOCK 6 uses scoring functions based on the force field to classify the compounds. These differences in scoring function are probably the main reason for the best result being obtained by AutoDock Vina.
CVS carries out VS using AutoDock Vina first and then performs a second VS using DOCK 6 as a refining step. The resulting ROC curve of the CVS approach (Figure 8c) showed even better results for the AUC-ROC.
In addition to the AUC-ROC, the enrichment factor (EF) was calculated to verify the VS performance. Lätti, Niinivehmas, and Pentikäinen23 stated that using the AUC-ROC along with the EF provide a good idea of the quality of the approach used to separate true positives from false positives. The enrichment factor (EF) consists of the number of active compounds found in relation to the number of active compounds that would be found after a random search.74 EFs are often calculated against a given percentage of the database. For example, EF10% represents the value obtained when 10% of the database is screened. EFs can be defined by the following formula:
The EF results are summarized in Table 1. DOCK 6 achieved EF1% = 25, EF2% = 12.5, and EF5% = 15, while AutoDock Vina achieved 25, 25, and 15, and consensus VS achieved 50, 25, and 20, respectively. These results show that consensus VS has a clear advantage compared to DOCK 6 and AutoDock Vina.
Table 1. Enrichment Factors for DOCK 6, AutoDock Vina, and the Consensus between Them.
| EF | DOCK 6 | AutoDock Vina | Consensus |
|---|---|---|---|
| 1% | 25 | 25 | 50 |
| 2% | 12.5 | 25 | 25 |
| 5% | 15 | 15 | 20 |
| 10% | 7.5 | 7.5 | 10 |
Finally, it is important to note that even though DOCK 6 had, in general, worse results than AutoDock Vina when considering the AUC-ROC, this program contributed to improving the results (Table 2). For example, the position of active ligands between all molecules in virtual screening can be checked using DOCK 6, AutoDock Vina, and Consensus.
Table 2. Position of Active Ligands Identified by DOCK 6, AutoDock Vina, and Consensus.
| DOCK
6 |
AutoDock
Vina |
Consensus |
||||
|---|---|---|---|---|---|---|
| active | energy | position | energy | position | consensus score | position |
| 121d | –57.61 | 1th | –8.9 | 10th | 1.97 | 2th |
| 1eel | –45.30 | 8th | –9.7 | 4th | 1.46 | 1th |
| 2dnd | –42.83 | 10th | –8.4 | 21th | 2.79 | 8th |
| 127d | –27.77 | 71th | –11.5 | 2th | 2.76 | 7th |
In Table 2, the active ligand with the highest energy according to DOCK 6 (127d) was in position 71. In Octopus, the active ligand with the highest energy was in position 21 (2dnd). In the consensus between them, the active ligand with the highest energy was identified in position 8. The result was improved with CVS because the active ligand with the highest energy identified by DOCK 6 (127d) was the one with the lowest energy in AutoDock Vina. The same was true for AutoDock Vina’s worst active ligand (2dnd). In DOCK 6, it had the 10th lowest energy among all ligands. Thus, when MolAr calculated the CVS between DOCK 6 and AutoDock Vina, the result was improved.
It can be concluded that even with DOCK 6 and AutoDock Vina showing different results, the combination of these results may, in principle, be closer to the true answer than that of only one of these programs alone. When analyzing the AUC-ROC curves resulting from the CVS, the result reliability was increased, giving a better prediction of the active ligand positions.
Evaluation of the Consensus Virtual Screening Approach Implemented by MolAr
In CVS, MolAr uses the resulting ligand conformation after docking with AutoDock Vina as input for DOCK 6. Thus, it is expected that when starting from a ligand conformation already evaluated and optimized by AutoDock Vina, DOCK 6 will achieve better results still.
It was necessary to verify whether this approach would have the opposite effect. Therefore, eight targets from the DUD database were used, and 10 active ligands were selected for each target. The DUD database contains a set of 102 targets and 22,886 active compounds for these targets, with an average of 224 active ligands per target. In the consensus setting, in addition to AutoDock Vina VS, a VS was performed using DOCK 6 with Grid Score using amber. This choice promises better results, as it performs some molecular dynamics simulations during docking using DOCK 6; however, it leads to a much longer execution time than GridScore flex without amber. In addition to performing VS of the active ligands, it was necessary to generate decoys for the chosen ligands to carry out VS and plot the ROC curves.
Thus, a subset of the DUD38 targets was selected. DUD38 is a subset of DUD that can be subdivided into six target families: metalloenzymes (4), nuclear hormone receptors (8), kinases (9), folate enzymes (2), serine proteases (2), and a diverse family called other enzymes (13). We used a total of eight targets to perform the evaluation. For each target, we selected 10 active ligands, and we used DUD to generate 50 decoys for each ligand. The VS was performed for 510 ligands for each one of the eight targets (10 active ligands and 500 decoys). The ligands were different for each target and were taken from DUD-E. The targets were chosen primarily by considering their resolution in the PDB. Homology modeling was performed to reconstruct gap regions (such as loop regions) for all chosen targets. Table 3 lists the chosen targets, and Chart 1 shows the interactions of crystallographic ligands with the targets used in this case study.
Table 3. A Subset of Targets Chosen from DUD38.
| family | PDB code |
|---|---|
| kinase | 1H00 (CDK2 in complex with a disubstituted 4,6-bis-anilino pyrimidine CDK4 inhibitor) |
| 2QD9 (P38 alpha MAP kinase inhibitor based on heterobicyclic scaffolds) | |
| metalloenzyme | 3BKL (testis ACE co-crystal structure with ketone ACE inhibitor kAW) |
| nuclear hormone receptor | 2AM9 (crystal structure of human androgen receptor ligand-binding domain in complex with testosterone) |
| 3KBA (progesterone receptor bound to sulfonamide pyrrolidine partial agonist) | |
| folate enzyme | 3NXO (preferential selection of isomer binding from chiral mixtures: alternate binding modes observed for the E- and Z-isomers of a series of 5-substituted 2,4-diaminofuro[2,3-d]pyrimidines as ternary complexes with NADPH and human dihydrofolate reductase) |
| serine protease | 2AYW (solution structure of Drosophila melanogaster SNF RBD2) |
| other | 1XL2 (HIV-1 protease in complex with pyrrolidinmethanamine) |
Chart 1. Interactions of Crystallographic Ligands with the Targets Used in This Case Study. The Hydrogens Were Omitted for Better Visualization.

After choosing the targets, it was necessary to choose which active ligands would be used for each target. We then used MolAr to perform virtual screening using AutoDock Vina for each of the chosen targets and for all DUD active ligands for each target. The 10 active ligands with the best energy were chosen for each target. This preselection of active ligands was necessary because it is impracticable to perform VS in the chosen configuration for all active ligands of each target and their respective decoys to generate the ROC curve since we performed molecular dynamics simulations (3000 steps) using the amber score scoring function in the consensus experiments. The results were improved by considering the ligand movement in molecular dynamics steps during the docking process. Consequently, the computational cost of execution was much longer than when using a scoring function that does not perform the dynamics, such as the grid score. Because AutoDock Vina had a shorter runtime than DOCK 6, it was chosen to perform these tests (in general, for the selected targets, this step required approximately 1 day for each).
CVS was performed for all targets, and another VS using only DOCK 6 was carried out to compare the influence of using the AutoDock Vina output as input for DOCK 6; the results obtained by DOCK 6 used the original ligands and their decoys. On average, due to the configuration chosen for the tests, the CVS for each target required 2 weeks to be performed. Some targets, such as metalloenzymes, VS took up to 30 days for VS of a single target.
Figure 9 outlines the experiment performed in this case study.
Figure 9.

DUD-E experimental workflow.
Results of the Case Study Evaluating the CVS Approach Implemented by MolAr
In this experiment, we performed CVS and VS using only DOCK 6 to compare the performance of DOCK 6 using the final ligand conformations defined by AutoDock Vina with the performance of DOCK 6 using the original conformations.
There was an improvement in the ROC curve of up to 42% (3BKL protein) when using the CVS approach, which demonstrates that it can lead to significant gains. The table above shows the results of the experiments performed, and except for the 3KBA protein, there was an improvement in the ROC curve when executing DOCK 6 based on the final ligand conformation defined by AutoDock Vina compared to the execution of DOCK 6 using the original ligand conformation. The 3KBA protein showed the same AUC-ROC in both scenarios. Table 4 summarizes the results.
Table 4. Comparison between Running DOCK 6 Using Ligand Conformations Provided by AutoDock Vina vs Running DOCK 6 Using Original Ligands.
| AUC-ROC |
||||
|---|---|---|---|---|
| family | PDB code | Dock 6 | Dock 6 after AutoDock Vina | AUC-ROC curve improvement (%) |
| kinase | 1H00 | 0.85 | 0.96 | 13 |
| 2QD9 | 0.48 | 0.59 | 23 | |
| metalloenzyme | 3BKL | 0.57 | 0.81 | 42 |
| nuclear hormone receptor | 2AM9 | 0.35 | 0.46 | 31 |
| 3KBA | 0.56 | 0.56 | 0 | |
| folate enzyme | 3NX0 | 0.63 | 0.80 | 27 |
| serine protease | 2AYW | 0.90 | 0.95 | 6 |
| other enzymes | 1XL2 | 0.63 | 0.76 | 21 |
These data show that the CVS approach increases the reliability of the tests performed by DOCK 6; according to the value of the AUC-ROC, this approach tends to decrease the number of false negatives identified by DOCK 6.
The EF results are summarized in Table 5. Regarding the targets 2QD9, 2AM9, and 3KBA, the AUC-ROC in the best case (DOCK 6 after AutoDock Vina) was less than 0.7, and the EF values were 0. An AUC-ROC of less than 0.7 means that the software program was not able to differentiate active ligands from inactive ligands.24 The AUC-ROC and EF values of these proteins are consistent as they indicate that there was no improvement over a random choice of elements. In the experiments of the other five targets (1H00, 3BKL, 3NX0, 2AYW, and 1XL2), the EF values indicated that the consensus approach in which DOCK 6 uses the selected conformations provided by AutoDock Vina improved the DOCK 6 performance for four of them (1H00, 3BKL, 3NX0, and 2AYW). For target 1XL2, the consensus approach gave the same EF values as was obtained by DOCK 6. Interestingly, the 3BKL protein is a metalloprotein, and the metal (zinc) is in the binding site. A recent study demonstrated that DOCK 6 failed in these situations.75 However, in the study by Çınaroǧlu and Timuçin,75 the scoring function used was the grid score. In the current paper, the AMBER scoring function was used in consensus docking between AutoDock Vina and DOCK 6. The force field FF09 used by the AMBER scoring function was parameterized for zinc ions.76 We were able to achieve good results (AUC-ROC of 0.81), which could be a starting point for further studies to verify whether this improvement can be reproduced for other metalloproteins.
Table 5. Enrichment Factors EF1%, EF2%, EF5%, and EF10% for DOCK 6 Experiments Using Original Ligands and Using the Ligand Conformations Provided by AutoDock Vina.
| EF for
DOCK 6 experiments using original ligands |
EF for
DOCK 6 experiments using ligand conformations provided by AutoDock
Vina |
|||||||
|---|---|---|---|---|---|---|---|---|
| EF | EF1% | EF2% | EF5% | EF10% | EF1% | EF2% | EF5% | EF10% |
| 1H00 | 0 | 0 | 6 | 5 | 20 | 20 | 12 | 6 |
| 2QD9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3BKL | 10 | 10 | 4 | 5 | 10 | 15 | 6 | 6 |
| 2AM9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3KBA | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3NX0 | 0 | 0 | 2 | 3 | 0 | 5 | 6 | 5 |
| 2AYW | 10 | 5 | 10 | 5 | 10 | 5 | 12 | 6 |
| 1XL2 | 10 | 5 | 4 | 2 | 10 | 5 | 4 | 2 |
Conclusions
This paper presents MolAr, a new software program that aims to assist in the VS process. One of the main contributions of our work is the development of software that is composed of several integrated tools to facilitate the VS process. MolAr automates the VS process, minimizing the need for human interference and thereby reducing the chances of error. MolAr allows researchers to perform docking, and the program is easy to use and does not require advanced computing knowledge. A new consensus virtual screening approach between DOCK 6 and AutoDock Vina has been developed, allowing the user to easily perform this task. The software programs used by MolAr to perform VS are all free for academic use.
Finally, two case studies were performed with MolAr. The first case investigated the use of virtual screening in DNA–ligand systems. The results demonstrated that both AutoDock Vina and DOCK 6 presented good reliability when considering the AUC-ROC values. Furthermore, although the results were excellent, the CVS approach further increased the reliability of VS. The combined approach increased the reliability of the AUC-ROC values compared with applying the VS tools separately. The identification of active ligands was also improved. AutoDock Vina, which identified active ligands better than DOCK 6 when performing VS, identified all active ligands in the top 22, while in the CVS approach used by MolAr, all active ligands were identified in the top 8. Notably, using MolAr to perform CVS has the advantage that MolAr carries out the CVS process automatically. Thus, the user does not have to worry about the various steps required to perform CVS between AutoDock Vina and DOCK 6, such as preparing the ligands, converting the output generated by AutoDock Vina to the DOCK 6 format, and comparing the results generated by the two tools (which can be difficult because they are in different units). The final case study validated the CVS strategy implemented by MolAr, where AutoDock Vina is executed first, and the resulting ligand conformations are used as input for the VS carried out by DOCK 6. This strategy improved the AUC values by up to 42%. Thus, the use of the final conformation determined by AutoDock Vina as input for the virtual screening performed by DOCK 6 is a good strategy to improve the reliability of the screening performed by DOCK 6.
This study demonstrated that MolAr was able to not only perform in silico simulations correctly but also could achieve satisfactory results. MolAr automates the installation of the many software programs required in the virtual screening process, contributing to the reduction of time and errors that may occur. MolAr represents a promising framework, with easy-to-use interfaces, and eliminates the need to use multiple command-line programs. MolAr has Linux and Windows versions, and it is freely available for download at http://www.drugdiscovery.com.br/.
Additional features will be added to MolAr in the future, including the implementation of artificial intelligence techniques; binding site prediction; ab initio prediction methods for use in homology modeling; molecular dynamics simulations; automation of other validation methods, such as enrichment factors and BedROC;77 generation of ligand tautomers for use in VS simulations; and the addition of other open-source virtual screening tools.
Acknowledgments
Funding sources for this project include FAPEMIG (APQ-02742-17 and APQ-00557-14), CNPq (449984/2014-1), and UFSJ/PPGBiotec. A.G.T. and L.C.A. are grateful to CNPq (305117/2017-3) and CAPES for their research fellowships. The authors would like to thank the Federal University of São João del-Rei (UFSJ) and the Federal Center for Technological Education of Minas Gerais (CEFET-MG) for providing the physical infrastructure.
Author Contributions
All authors contributed to writing the manuscript, and all authors approved the final version of the manuscript.
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001, FAPEMIG (APQ-00557-14), CNPq (449984/2014-1), CNPq Universal (426261/2018-6), PPGCF, and PPGBiotec/UFSJ. A.G.T. received a fellowship from CNPq (305117/2017-3).
The authors declare no competing financial interest.
References
- Leelananda S. P.; Lindert S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. 10.3762/bjoc.12.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maithri G.; Manasa B.; Vani S. S.; Narendra A.; Harshita T. Computational Drug Design and Molecular Dynamic Studies-A Review. Int J Biomed Data Min. 2016, 06, 1–7. 10.4172/2090-4924.1000123. [DOI] [Google Scholar]
- Kong D. X.; Guo M. Y.; Xiao Z. H.; Chen L. L.; Zhang H. Y. Historical variation of structural novelty in a natural product library. Chem. Biodiversity 2011, 8, 1968–1977. 10.1002/cbdv.201100156. [DOI] [PubMed] [Google Scholar]
- Oglic D.; Oatley S. A.; Macdonald S. J. F.; et al. Active Search for Computer-aided Drug Design. Mol. Inf. 2018, 37, 1–15. 10.1002/minf.201700130. [DOI] [PubMed] [Google Scholar]
- Ferreira R. S.; Glaucius O.; Andricopulo A. D. Integração das técnicas de triagem virtual e triagem biológica automatizada em alta escala: oportunidades e desafios em P&D de fármacos. Quim. Nova 2011, 34, 1770–1778. 10.1590/S0100-40422011001000010. [DOI] [Google Scholar]
- Ripphausen P.; Nisius B.; Bajorath J. J. State-of-the-art in ligand-based virtual screening. Drug Discovery Today 2011, 16, 372–376. 10.1016/j.drudis.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Carregal A. P.; Maciel F. V.; Carregal J. B.; dos Reis Santos B.; da Silva A. M.; Taranto A. G. Docking-based virtual screening of Brazilian natural compounds using the OOMT as the pharmacological target database. J. Mol. Model. 2017, 23, 111. 10.1007/s00894-017-3253-8. [DOI] [PubMed] [Google Scholar]
- Lionta E.; Spyrou G.; Vassilatis D.; Cournia Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. 10.2174/1568026614666140929124445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surabhi S.; Singh B. COMPUTER AIDED DRUG DESIGN: AN OVERVIEW. J Drug Deliv Ther. 2018, 8, 504–509. 10.22270/jddt.v8i5.1894. [DOI] [Google Scholar]
- Dutkiewicz Z.; Mikstacka R. Structure-Based Drug Design for Cytochrome P450 Family 1 Inhibitors. Bioinorg. Chem. Appl. 2018, 2018, 1–21. 10.1155/2018/3924608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójcikowski M.; Ballester P. J.; Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Nat. Publ. Gr. 2017, 7, 1–10. 10.1038/srep46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes R. R.; Fonseca A. L. D.; Pinto A. C. D. S.; Maia E. H. B.; Silva A. M. D.; Varotti F. D. P.; Taranto A. G. Brazilian malaria molecular targets (BraMMT): selected receptors for virtual high-throughput screening experiments. Mem. Inst. Oswaldo. Cruz. 2019, 114, 1–10. 10.1590/0074-02760180465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugumbate G.; Mendes V.; Blaszczyk M.; et al. Target identification of Mycobacterium tuberculosis phenotypic hits using a concerted chemogenomic, biophysical, and structural approach. Front Pharmacol. 2017, 8, 681. 10.3389/fphar.2017.00681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter K. A.; Cohen D. S.; Jarrell J. T.; Huang X. Deep learning and virtual drug screening. Future Med. Chem. 2018, 10, 2557–2567. 10.4155/fmc-2018-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y.; Ding Y.; Feinstein W. P.; et al. Geauxdock: Accelerating Structure-Based Virtual Screening With Heterogeneous Computing. PLoS One 2016, 11, e0158898 10.1371/journal.pone.0158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Alnammi M.; Ericksen S. S.; et al. Practical Model Selection for Prospective Virtual Screening. J. Chem. Inf. Model. 2019, 59, 282–293. 10.1021/acs.jcim.8b00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasotto C. N.; Phatak S. S. Homology modeling in drug discovery: current trends and applications. Drug Discovery Today 2009, 14, 676–683. 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Hillisch A.; Pineda L. F.; Hilgenfeld R. Utility of homology models in the drug discovery process. Drug Discovery Today 2004, 9, 659–669. 10.1016/S1359-6446(04)03196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Brink T.; Exner T. E. pKa based protonation states and microspecies for protein–ligand docking. J. Comput.-Aided Mol. Des. 2010, 24, 935–942. 10.1007/s10822-010-9385-x. [DOI] [PubMed] [Google Scholar]
- ten Brink T.; Exner T. E. Influence of Protonation, Tautomeric, and Stereoisomeric States on Protein–Ligand Docking Results. J. Chem. Inf. Model. 2009, 49, 1535–1546. 10.1021/ci800420z. [DOI] [PubMed] [Google Scholar]
- Petukh M.; Stefl S.; Alexov E. The role of protonation states in ligand-receptor recognition and binding. Curr. Pharm. Des. 2013, 19, 4182–4190. 10.2174/1381612811319230004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awuni Y.; Mu Y. Reduction of false positives in structure-based virtual screening when receptor plasticity is considered. Molecules 2015, 20, 5152–5164. 10.3390/molecules20035152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lätti S.; Niinivehmas S.; Pentikäinen O. T. Rocker: Open source, easy-to-use tool for AUC and enrichment calculations and ROC visualization. Aust. J. Chem. 2016, 8, 45. 10.1186/s13321-016-0158-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza A.; Wei N.-N.; Zhan C.-G. Ligand-Based Virtual Screening Approach Using a New Scoring Function. J. Chem. Inf. Model. 2012, 52, 963–974. 10.1021/ci200617d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliebrahimi S.; Kouhsari S. M.; Ostad S. N.; Arab S. S.; Karami L. Identification of Phytochemicals Targeting c-Met Kinase Domain using Consensus Docking and Molecular Dynamics Simulation Studies. Cell Biochem. Biophys. 2018, 135–145. 10.1007/s12013-017-0821-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuccinardi T.; Poli G.; Romboli V.; Giordano A.; Martinelli A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. 10.1021/ci500424n. [DOI] [PubMed] [Google Scholar]
- Poli G.; Martinelli A.; Tuccinardi T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 167–173. 10.1080/14756366.2016.1193736. [DOI] [PubMed] [Google Scholar]
- Park H.; Eom J. W.; Kim Y. H. Consensus scoring approach to identify the inhibitors of AMP-activated protein kinase α2 with virtual screening. J. Chem. Inf. Model. 2014, 54, 2139–2146. 10.1021/ci500214e. [DOI] [PubMed] [Google Scholar]
- Kukol A. Consensus virtual screening approaches to predict protein ligands. Eur. J. Med. Chem. 2011, 46, 4661–4664. 10.1016/j.ejmech.2011.05.026. [DOI] [PubMed] [Google Scholar]
- Houston D. R.; Walkinshaw M. D. Consensus docki, ng: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. 10.1021/ci300399w. [DOI] [PubMed] [Google Scholar]
- Chermak E.; De Donato R.; Lensink M. F.; et al. Introducing a clustering step in a consensus approach for the scoring of protei,n-protein docking models. PLoS One 2016, 11, 1–15. 10.1371/journal.pone.0166460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter M. Challenges in Docking: Mini Review. JSM Chem. 2016, 4, 1025. [Google Scholar]; https://www.jscimedcentral.com/Chemistry/chemistry-4-1025.pdf
- Chaudhary K. K.; Mishra N. A Review on Molecular Docking : Novel Tool for Drug Discovery. JSM Chem. 2016, 4, 1–4. [Google Scholar]
- Yuriev E.; Ramsland P. A. Latest developments in molecular docking: 2010-2011 in review. J. Mol. Recognit. 2013, 26, 215–239. 10.1002/jmr.2266. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.. The PyMOL Molecular Graphics System, Version 1.8. Schrädinger LLC: 2002:http://www.pymol.org, 10.1038/hr.2014.17. [DOI]
- Morris G. M.; Huey R.; Lindstrom W.; et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šali A.MODELLER: A Program for Protein Structure Modeling Release 9.12, r9480. Rockefeller Univ.2013:779–815. http://scholar.google.com/scholar?hl=en&btnG=Search&q=intitle:MODELLER+A+Program+for+Protein+Structure+Modeling#6.
- Dolinsky T. J.; Czodrowski P.; Li H.; et al. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen W. J.; Balius T. E.; Mukherjee S.; et al. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozell S. R.; Mukherjee S.; Balius T. E.; Roe D. R.; Case D. A.; Rizzo R. C. Evaluation of DOCK 6 as a pose generation and database enrichment tool. J. Comput.-Aided Mol. Des. 2012, 26, 749–773. 10.1007/s10822-012-9565-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An Open chemical toolbox. Aust. J. Chem. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli S.; Huey R.; Pique M. E.; Sanner M. F.; Goodsell D. S.; Olson A. J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 5, 905–919. 10.1038/nbt.3121.ChIP-nexus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. J. P.. MOPAC2016TM; Stewart SCC, 2016. http://openmopac.net/MOPAC2016.html.
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Herráez A. Biomolecules in the computer: Jmol to the rescue. Biochem. Mol. Biol. Educ. 2006, 34, 255–261. 10.1002/bmb.2006.494034042644. [DOI] [PubMed] [Google Scholar]
- Graham R. L., Shipman G. M., Barrett B. W., Castain R. H., Bosilca G., Lumsdaine A.. Open MPI: A high-performance, heterogeneous MPI. In: Proceedings - IEEE International Conference on Cluster Computing, ICCC; IEEE: Barcelona, Spain, 2006, 1-9, 10.1109/CLUSTR.2006.311904. [DOI]
- Salomon-ferrer R.; Case D. A.; Walker R. C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. 10.1002/wcms.1121. [DOI] [Google Scholar]
- Dallakyan S.; Olson A. J.. Small-Molecule Library Screening by Docking with PyRx. In: Chemical Biology; Humana Press: New York, NY, Vol 1263, 2015:243–250, 10.1007/978-1-4939-2269-7_19 [DOI] [PubMed] [Google Scholar]
- Veloso W. N. P.; Silveira C. H. Pires DEV. EasyVS – Virtua0l Screening in just a few clicks. https://easyvs.unifei.edu.br/. Accessed December 13, 2019.
- de Magalhães C. S.; Almeida D. M.; Barbosa H. J. C.; Dardenne L. E. A dynamic niching genetic algorithm strategy for docking highly flexible ligands. Inf Sci (Ny) 2014, 289, 206–224. 10.1016/j.ins.2014.08.002. [DOI] [Google Scholar]
- da Silva E. K., Almeida D. M., Barbosa H. J. C., Dardenne L. E., Custódio F. L., Guedes I. A.. DockThor: A receptor-ligand docking program. https://dockthor.lncc.br/v2/. Accessed December 13, 2019.
- Webb B., Sali A.. Comparative Protein Structure Modeling Using MODELLER. In: Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA: 2016. 5.6.1–5.6.37 [DOI] [PMC free article] [PubMed]
- Eswar N.; Webb B.; Marti-Renom M. A.; et al. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Protein Sci. 2007, 50, 2.9.1–2.9.31. 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- Lagarde N.; Goldwaser E.; Pencheva T.; et al. A free web-based protocol to assist structure-based virtual screening experiments. Int J Mol Sci. 2019, 20, 1–15. 10.3390/ijms20184648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R.; Shukla H.; Kalita P.; et al. Identification of potential inhibitors of Fasciola gigantica thioredoxin1: computational screening, molecular dynamics simulation, and binding free energy studies. J. Biomol. Struct. Dyn 2017, 36, 2147–2162. 10.1080/07391102.2017.1344141. [DOI] [PubMed] [Google Scholar]
- Kondratyev M. S.; Zakharova E. V. Virtual Screening of Thiol Peroxiredoxin 6 Reducers. Biophysics 2018, 63, 669–674. 10.1134/S0006350918050123. [DOI] [Google Scholar]
- Holden P. M.; Kaur H.; Goyal R.; Gochin M.; Rizzo R. C. Footprint-based identification of viral entry inhibitors targeting HIVgp41. Bioorg. Med. Chem. Lett. 2012, 22, 3011–3016. 10.1016/j.bmcl.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto-Martênez F. D.; Arciniega M.; Medina-Franco J. L. Molecular docking: current advances and challenges. TIP Rev Espec en Ciencias Quêmico-Biológicas 2018, 21, 65–87. [Google Scholar]
- Hermann J. C.; Marti-Arbona R.; Fedorov A. A.; et al. Structure-based activity prediction for an enzyme of unknown function. Nature 2007, 448, 775–779. 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare G.; Kar R.; Tyagi A. K. Identification of inhibitors against Mycobacterium tuberculosis Thiamin phosphate synthase, an important target for the development of Anti-TB drugs. PLoS One 2011, 6, e22441. 10.1371/journal.pone.0022441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia E. H. B.; Campos V. A.; Santos R.; et al. Octopus : a platform for the virtual high-throughput screening of a pool of compounds against a set of molecular targets. J. Mol. Model. 2017, 23–26. 10.1007/s00894-016-3184-9. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. P. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. J. P.MOPAC2012; Stewart SCC, 2012. http://openmopac.net.
- Gasteiger J.; Marsili M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. 10.1016/0040-4020(80)80168-2. [DOI] [Google Scholar]
- Kuntz I. D.; Blaney J. M.; Oatley S. J.; Langridge R.; Ferrin T. E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. 10.1016/0022-2836(82)90153-X. [DOI] [PubMed] [Google Scholar]
- Gilad Y.; Senderowitz H. Docking studies on DNA intercalators. J. Chem. Inf. Model. 2013, 54, 96–107. 10.1021/ci400352t. [DOI] [PubMed] [Google Scholar]
- Silverman R. B.. The Organic Chemistry of Drug Design and Drug Action: Second Edition. 2nd ed.; Elsevier: Evanston: 2004, 10.1016/C2009-0-22222-7. [DOI] [Google Scholar]
- Holt P. A.; Chaires J. B.; Trent J. O. Molecular docking of intercalators and groove-binders to nucleic acids using autodock and surflex. J. Chem. Inf. Model. 2008, 48, 1602–1615. 10.1021/ci800063v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci C. G.; Netz P. A. Docking studies on DNA-ligand interactions: Building and application of a protocol to identify the binding mode. J. Chem. Inf. Model. 2009, 49, 1925–1935. 10.1021/ci9001537. [DOI] [PubMed] [Google Scholar]
- Fong P.; Wong H.-K. Evaluation of Scoring Function Performance on DNA-ligand Complexes. Open Med Chem J. 2019, 13, 40–49. 10.2174/1874104501913010040. [DOI] [Google Scholar]
- Srivastava H. K.; Chourasia M.; Kumar D.; Sastry G. N. Comparison of computational methods to model DNA minor groove binders. J. Chem. Inf. Model. 2011, 51, 558–571. 10.1021/ci100474n. [DOI] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55 (14), 6582–6594. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truchon J. F.; Bayly C. I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. 10.1021/ci600426e. [DOI] [PubMed] [Google Scholar]
- Çınaroǧlu S. S.; Timuçin E. Comparative Assessment of Seven Docking Programs on a Nonredundant Metalloprotein Subset of the PDBbind Refined. J. Chem. Inf. Model. 2019, 59, 3846–3859. 10.1021/acs.jcim.9b00346. [DOI] [PubMed] [Google Scholar]
- Case D. A., Ben-Shalom I. Y., Brozell S. R., et al. Amber 2018. San Francisco; 2018. [Google Scholar]
- Zhao W.; Hevener K. E.; White S. W.; Lee R. E.; Boyett J. M. A statistical framework to evaluate virtual screening. BMC Bioinformatics. 2009, 10, 225. 10.1186/1471-2105-10-225. [DOI] [PMC free article] [PubMed] [Google Scholar]