

Abstract

We previously developed a biobetter version of rhIFN-β (R27T) that possesses an additional glycosylation site compared with rhIFN-β 1a. Herein, we characterized N-glycosylation heterogeneity of R27T, which includes both N-glycan site occupancy heterogeneity (macro-heterogeneity) and complexity of carbohydrate moieties (micro-heterogeneity). N-glycan site occupancy manifested as distinct differences in size and isoelectric point. The analysis of complex carbohydrate moieties of R27T involved the common biopharmaceutical glycosylation critical quality attributes such as core fucosylation, antennary composition, sialylation, N-acetyllactosamine extensions, linkages, and overall glycan profiles using weak anion-exchange and hydrophilic interaction high-performance liquid chromatography with 2-aminobenzoic acid-labeled N-glycans. The double-glycosylated form accounted for approx. 94% R27T, while the single-glycosylated form accounted for 6% R27T. N-glycans consisted of a mixture of bi-, tri-, and tetra-antennary glycans, some with N-acetyllactosamine extensions, but neither outer arm fucose nor α-galactose was detected. Sialic acid major variants, N-acetyl- and N-glycolyl-neuraminic acid, were more abundant in R27T than in Rebif. The major N-glycan, accounting for ∼42% of total N-glycans, had a di-sialylated, core-fucosylated bi-antennary structure.
Introduction
Several immunomodulatory treatments have been approved by the US Food and Drug Administration and European Medicines Agency for relapsing-remitting forms of multiple sclerosis (RRMS).1−4 Among them, recombinant human interferon-β (rhIFN-β) has long been used as an effective first therapeutic intervention and a disease-modifying therapy for RRMS.5−7 Although almost three decades have passed since the introduction of rhIFN-β therapies, they remain important for the management of MS because of their good long-term safety profile and cost efficacy.8−10 However, there are direct and indirect limitations for clinical use, including the need for frequent injections and high immunogenicity and aggregation propensity, the latter of which is responsible for the therapeutic effect of the protein.11−13 To address these issues, in our previous study, an additional single-glycosylation site was introduced at amino acid 25 in rhIFN-β 1a, resulting in R27T in which Arg at position 27 is mutated to Thr.14 The additional glycosylation site increases the half-life and in vitro biological activity, as well as thermostability, allowing less-frequent dosing.14,15
As observed for R27T, glyco-engineering of therapeutic proteins can enhance in vivo activities by improving pharmacokinetic properties, solubility, thermal stability, and protease resistance, and reducing the immunogenicity, all of which may improve clinical outcomes.16−21 However, despite its importance, it is very difficult to accurately determine the influence of glycosylation on glycoproteins because of its inherent complexity. Potential glycosylation sites such as Asn residues within Asn-X-Ser/Thr consensus sequences are not always occupied by oligosaccharides in mammalian cells because a consensus sequence alone is essential but not sufficient for N-linked protein glycosylation, resulting in site occupancy heterogeneity (macro-heterogeneity).22,23 In addition, the composition of attached oligosaccharides can also vary considerably, although a core pentasaccharide unit (Man3GlcNAc2) is typically linked to an asparagine (Asn) residue via a chitobiose (GlcNAc2).24 The glycan structure can be highly variable in terms of the antennary structure, monosaccharide composition, and sialylation depending on the host cell type, cell culture, and manufacturing conditions.25−28 This results in inherent structural complexity and variability (micro-heterogeneity).22 Since the complexity of glycosylation heterogeneity makes it difficult to understand structure–function relationships, it is imperative to characterize glycosylation variability. Herein, we performed a comprehensive characterization of the main R27T glycoforms. Glycosylation analysis can identify glycosylation parameters that may influence drug safety and efficacy profiles via critical quality attributes (CQAs). In the present study, we investigated common biopharmaceutical glycosylation CQAs including site occupancy heterogeneity, core fucosylation, antennary composition, sialylation, N-acetyllactosamine extensions, linkages, and overall glycan profiles of the R27T glycan.
Results and Discussion
N-Glycosylation Site Occupancy Heterogeneity
We analyzed three batches of R27T purification products and purification intermediates. We defined proteins separated by hydrophobicity over four column steps into the main target (R27T) and intermediate (shoulder) peaks (Figure S1). Each peak was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and isoelectric focusing (IEF) with Rebif, a single-glycosylated rhIFN-β, as a reference (Figure 1A). The main R27T peaks revealed a protein of ∼25–26 kDa with an isoelectric point (pI) of ∼5.3–6.9 and a smear pI of 7.5, 8.0, and 8.3 on IEF gels, while Rebif ran as a 22.5 kDa product with major pI values of 8.0 and 8.3. The intermediate peaks separated from the major peaks yielded a broad medium-sized band that ran between the R27T and Rebif bands, and its pI bands were more basic than those of R27T, and some were within the main pI range of Rebif, indicating a mixture of single- and double-glycosylated proteins. We attempted to quantitate species with one or two glycosylation sites to analyze the glycosylation site occupancy heterogeneity. We used a microchip assay and capillary IEF (cIEF) to separate and quantitate species based on size and pI differences (Figure 1B,C). We were able to correlate the size with glycosylation occupancy, although there was considerable size variation (∼15.2 kDa for no glycans, 21.9–34.8 kDa for one glycosylation, and 41.6–45.7 kDa for two glycosylations, Table S1) compared with qualitative analysis due to limitations of the capillary analysis methods with glycoproteins.29 There were also clear differences in pI (∼7.19–7.97 for one glycan and 5.67–7.09 for two glycans). Based on size differences, we found that R27T was made up of 94% of the double-glycosylated form and 6% of the single-glycosylated form (Figure 1B). For the R27T intermediate, the ratio was 5, 77, and 18% nonglycosylated, single-glycosylated, and double-glycosylated, respectively. By contrast, the Rebif reference protein displayed 100% single-glycosylation. These results were supported by cIEF, although there was a slight difference in ratios (94.1% double-glycosylation and 1.9% single-glycosylation for R27T, and 84.5% single-glycosylation and 12.2% double-glycosylation for the R27T intermediate) because of the more direct effect of the number of sialic acid rather than the number of glycosylation (Figure 1C). The inclusion of the 6% single-glycosylated form as a macro-heterogeneity with the 94% double-glycosylation form of R27T did not have a significant influence on the specific activity, as discussed in our previous study.14 Thus, the presence of 6% single-glycosylated R27T could be considered a control factor for more homogeneous molecular entities and an example of macro-heterogeneity, but such products would not fail to meet the quality criteria.
Figure 1.

N-glycosylation site occupancy heterogeneity reflects the R27T size and pI differences. (A) SDS-PAGE and IEF analysis of glycoproteins. Lanes 2–4 show the main target peaks from R27T purification, and lanes 5–6 show intermediate peaks comprising mainly non- or partially glycosylated protein. Lane 7 is the singly glycosylated Rebif reference. (B) Microchip analysis (C) cIEF analysis.
Sialic Acid Content and Variation
Because sialic acids are terminal, negatively charged components that can determine the serum half-life and influence the immunogenicity and biological activity, they are typically considered to be glycosylation CQAs. The abundance and terminal capping ratio of sialic acids on galactose are very important because they impact the function and biological half-life of biopharmaceuticals.30−32
Overall charge profiles for R27T was shown according to the number of sialic acids (Figure S2). The 2S component was the most abundant (35.6%) followed by 1S (26.1%), 3S (21.7%), and 4S (16.6%). The absolute quantitation of sialic acid residues of R27T was also measured as the molar ratio of sialic acid per R27T molecule (Table 1). The sialic acid content of R27T (2.81 mol/mol protein) was, as expected, much greater (3-fold) than that of Rebif (0.77 mol/mol protein), and the value was consistent with that in our previous study.14 Sufficient sialylation can prolong the half-life, as demonstrated in our previous study.14 Sufficient sialylation could be the mean galactose capping ratio with sialic acids as well as the absolute amount of sialylation. The galactose capping ratio could have direct effect on the glycoprotein half-life because neutral glycans without sialic acid are cleared by asialoglycoprotein receptors in the liver.30−32 The galactose monosaccharide analysis was performed to investigate the sialic acid capping ratio and evaluate the quality of R27T and Rebif (Table 1). The terminal sialic acid capping ratio on galactose was ∼0.28–0.31 for both R27T and Rebif samples. Considering the pharmacokinetic improvement from our previous study,14 these capping ratios were somewhat lower than expected. It could cause some errors in the ratio between galactose and sialic acid because they are separated from the glycan under different conditions. Therefore, the absolute value of capping ratio does not absolutely reflect the degree of capping. It just means that R27T has a capping ratio similar to that of Rebif, which is known to be clinically effective and safe.
Table 1. Sialylation Content and Variation.
| sialylation contents and variation | R27T | Rebif |
|---|---|---|
| Neu5Ac (mol/mol protein) | 2.75 | 0.77 |
| Neu5Gc (mol/mol protein) | 0.06 | |
| total sialic acid content (mol/mol protein) | 2.81 | 0.77 |
| galactose (mol/mol protein) | 10.00 | 2.48 |
| capping ratio | 0.28 | 0.31 |
Another important aspect of sialylation is variation, which mainly includes N-acetyl-neuraminic acid (Neu5Ac) and N-glycolyl-neuraminic acid (Neu5Gc). Because Neu5Gc cannot be synthesized in humans, it may be recognized as a foreign epitope with an immunogenic potential.33,34 Numerous studies have illustrated the necessity to minimize the relative content of Neu5Gc sialic acids in glycoprotein biopharmaceuticals.33 Both sialic acid major variants, Neu5Ac and Neu5Gc, were more abundant in R27T samples (2.75, 0.06 mol/mol protein, respectively) than in Rebif (0.77 mol/mol protein, not detected) (Table 1). The relative Neu5Gc percentage of the total sialylation for R27T was 2.1%, but this was not detected for Rebif. In Chinese hamster ovary (CHO) cells, ∼3% of sialic acids were identified as Neu5Gc for the recombinant plasminogen activator, follicle-stimulating hormone, and erythropoietin (EPO).34 In addition, EPO containing 1% Neu5Gc induces a negligible immunogenic response, whereas EPO with ∼7% Neu5Gc elicits a clinically significant immunogenic response.35 However, because of differences in products, as well as dosage and frequency for clinical use, it is difficult to generalize criteria values for the Neu5Gc content in biopharmaceutical products. Various factors can be conveniently used to minimize or maintain Neu5Gc levels below 1% during the manufacturing of R27T.36
Antennary Structures Based on Exoglycosidase Sequencing
Detailed structures including core fucosylation, antennary composition, N-acetyllactosamine extensions, and linkages were identified by exoglycosidase sequencing. The results of hydrophilic interaction chromatography (HILIC)–high-performance liquid chromatography (HPLC) analysis revealed 22 peaks (Figure S3, Table S2). The exoglycosidase treatment facilitated a more accurate analysis of the N-glycan antennary structure of R27T (Figure 2). A summary of the results of exoglycosidase sequencing of N-glycans is included in Table S3. Data from sialidase (a368S), fucosidase (a34F, a6F), and galactosidase (a36G, b4G) exoglycosidase digestion can be used to quantify different types of antennary structures present within samples. A combination of enzymes can determine the structures down to the core A2, A3, A3′, and A4 structures, for which the glucose unit (GU) values are documented (Table S3). Digestion with α-galactosidase (a36G) or α-3/4 fucosidase (a34F) generated identical chromatograms, indicating that neither outer arm fucose nor α-galactose moieties were present. Comparison of digestion with sialidase (a368S) alone and digestion with sialidase (a368S) plus both fucosidases (a34F and a6F) revealed that most peaks moved by ∼0.4 GU units, in line with the loss of a core α1-6 fucose component. This means that core-fucosylated structures accounted for 94% of the total. A comparison of digestion with sialidase (a368S) plus fucosidases (a34F and a6F) and digestion with sialidase (a368S) plus fucosidases (a34F and a6F) plus beta-galactosidase (b4G) revealed the core antennary structures (A2, A3, A3, and A4) for some peaks. However, a number of peaks did not digest and reveal the core structures until N-acetylglucosaminidase (sph) was also added presumably because they contained N-acetyllactosamine extension, which can be added to any antennae, resulting in diverse isomeric structures. The bi-antennary structures were the most abundant. The N-glycosylation of R27T consists of a mixture of bi- (42.5%), tri- (17.2%), and tetra-antennary glycosylation (8.8%), some of which have N-acetyllactosamine extensions (29.3%). Most glycosylation in R27T (∼94%) also have a core-fucosylated structure (Table 2). This could play a crucial role in the stabilization of the R27T structure because the core fucose structure of the rhIFN-β carbohydrate may help to stabilize the rhIFN-β structure.37 In these structures, galactose α1-3 linked to β-galactose was not observed. This is also very important because the presence of α-galactose could lead to potential adverse reactions, immunogenic potential, and neutralization of the drug by anti-α-galactose antibodies.38
Figure 2.
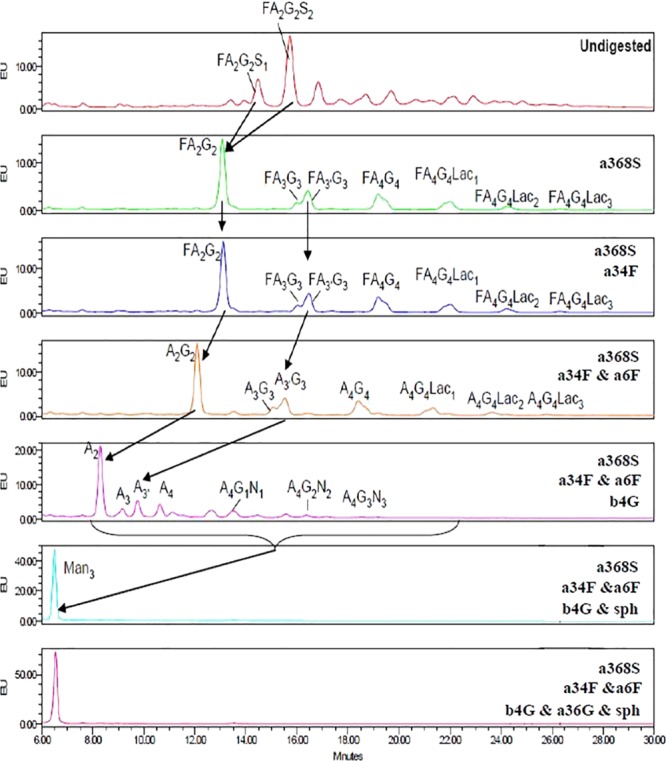
Exoglycosidase digestion sequencing of the R27T N-glycan by HILIC–HPLC arrows indicate digestion. The following exoglycosidases were used: sialidase (a368S; specific for α2-3, -6, -8, and -9 sialic acids); beta-galactosidase (b4G; specific for β1-4 galactose); α-galactosidase (a36G; specific for α1-3/6 galactose); fucosidase (a34F; specific for α1-3 and -4 fucose); fucosidase (a6F; specific for α1-6 > 2 fucose); N-acetylglucosaminidase (sph; specific for β-GlcNAc). Glycan structures were allocated by a combination of elution position (expressed as GU value) and subsequent GU values of peaks following digestion with specific exoglycosidases.
Table 2. Summary of Average Values for the Relative Percentage of Different Antennary Structures.
| structure | relative percentage (%) |
|---|---|
| % A2 | 42.5 |
| % A3 | 6.3 |
| % A3′ | 10.9 |
| % A4 | 8.8 |
| % Fuc | 94 |
| % Lac | 29.3 |
N-Glycan Structure Determination
We performed glycan structure profiling of R27T. Herein, two strategies for comprehensive N-glycan profiling, fluorescence profiling by WAX/HILIC–HPLC, in which HPLC database is publicly available,39−41 and matrix-assisted laser desorption ionization-mass spectrometry (MALDI-MS), were employed in this study.
N-Glycan Profiles Based on WAX- and HILIC–HPLC
Because sialic acid is the most common CQA for biopharmaceutical products, it can be useful to separate N-glycans on the basis of charge by WAX–HPLC, enabling more accurate relative quantification of multisialylated structures. R27T glycans were separated on a WAX–HPLC column, and 17 peak fractions were collected (Figure S4A). Fractions were combined (F2 + F3, F4 + F5, and F7 + F8) when peaks were not clearly separated. Each fraction containing zero (F1), one (F4 + F5), two (F7 + F8), three (F9), or four (F10 – 13) sialic acids was then analyzed by HILIC–HPLC (Figure S4B). Fractions 2, 3, 6, and 14–17 did not contain any glycans. Fractions containing charged glycans were then digested with sialidase and rerun on the HILIC–HPLC column (Figure 3). The structures were identified from the GU values in relation to neutral structures identified by exoglycosidase sequencing. Fraction 1 contained neutral glycans (bi-antennary structures A2G2 and FA2G2). Fraction 4 + 5 contained mono-sialylated, core-fucosylated bi-, tri-, and tetra-antennary (with zero, one, or two N-acetyllactosamine repeats) glycans. Fraction 7 + 8 contained di-sialylated glycans, and both major peaks (GU 8.4 and 8.9) digested to FA2G2 at GU 7.5. α2-3-linked sialic acid adds less to the GU value than α2-6-linked sialic acid. Thus, we can conclude that the largest peak (GU 8.4) includes an α2-3-linked sialic acid while the second peak (GU 8.9) has an α2-6-linked sialic acid. A small proportion of tri-antennary glycans were also di-sialylated. Fraction 9 contained tri-sialylated, core-fucosylated bi-, tri-, and tetra-antennary glycans with zero, one, or two N-acetyllactosamine repeats. Both forms of tri-antennary glycans were identified with sialidase and fucosidase treatment: A3G3 (GU 8.35), in which the third GlcNAc is β1-4-linked to the 3-linked mannose, and A3′G3 (GU 8.54), in which the third GlcNAc is β1-6-linked to the 6-linked mannose. There were also some tri-sialylated tetra-antennary glycans without core fucose. Fraction 10 contained tetra-sialylated, core-fucosylated tetra-antennary glycans with two or three N-acetyllactosamine repeats. Fraction 11 contained tetra-sialylated, core-fucosylated tetra-antennary glycans with two N-acetyllactosamine repeats. Both peaks showed the same GU value after the removal of sialic acids, suggesting that the second smaller peak contained glycans with α2-6-linked sialic acid(s). Fraction 12 contained tetra-sialylated, core-fucosylated tetra-antennary glycans with one N-acetyllactosamine repeat, as well as tetra-sialylated tetra-antennary glycans with and without core fucose. Fraction 13 contained tetra-sialylated, core-fucosylated tetra-antennary glycans. All peaks digested to the same GU value after the removal of sialic acids, suggesting that the smaller peaks contained glycans with α2-6-linked sialic acid(s). The N-acetyllactosamine extension adds approximately the same value to the GU value as the addition of an extra antenna (both are Gal–GlcNAc).
Figure 3.

HILIC–HPLC profiles of R27T WAX fractions before and after sialidase digestion.
A summary of the identified glycans matched to peaks for the whole pool is given in Table 3 and Figure S5. The GU values changed slightly over time (this is normal for sialylated glycans); hence, peaks in fractions were matched to a profile of the whole undigested pool run at the same time. The major N-glycan, which accounts for ∼42% of total N-glycans, is a di-sialylated, core-fucosylated bi-antennary structure. However, R27T also exhibited considerable variability in its glycoprofile, with tri- and tetra-antennary glycans, as well as bi-antennary glycan forms being detected. Surprisingly, rhIFN-β containing a larger proportion of higher antennary glycoforms showed more sustained bioactivity over time.42 Indeed, in our previous study, R27T exhibited more prolonged signaling than the mono-glycosylated rhIFN-β, with altered receptor-binding kinetics.15 A larger portion of higher antennary components in R27T may therefore influence the cellular signaling effects.
Table 3. N-Glycan Profilesa.

Number of sialic acids is 0S (gray), 1S (green), 2S (blue), 3S (brown), and 4S (red). * structures contains α2-6 linked sialic acids.
Identification of N-Glycan Structures Using MALDI-MS
MALDI spectra from permethylated N-glycans were consistent with most glycans being core-fucosylated bi-antennary types (FA2G2; H5N4F1) with zero, one, or two sialic acids (Figure 4). Masses corresponding to the core-fucosylated tri-antennary glycans (FA3G3; H6N5F1) with one, two, or three sialic acids were also detected. We also acquired positive control data from the N-glycan of fetuin, and no peaks were detected in the negative controls (data not shown). Annotation of the R27T N-glycan structures revealed complex glycans with variable degrees of core fucosylation, galactosylation, and sialylation. The major di-sialylated, core-fucosylated bi-antennary structure was consistent with the 2-AB labeling HPLC results.
Figure 4.
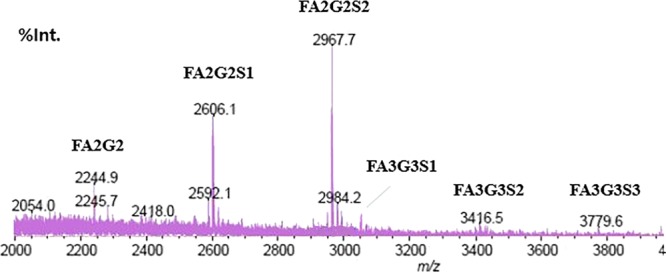
MALDI-MS spectra from permethylated R27T. m/z [M + Na]+.
Conclusions
We found the glycosylation heterogeneity of hyperglycosylated recombinant human interferon-β (rhIFN-β), R27T. The glycosylation site occupancy showed approx. 94% double-glycosylated form R27T with 6% single-glycosylated R27T. R27T N-glycans were a micro-heterogeneous mixture of bi-, tri-, and tetra-antennary glycans, some with lactosamine extensions, but neither outer arm fucose nor α-galactose was detected. Sialic acid major variants, Neu5Ac and Neu5Gc, were more abundant in R27T than in Rebif. The major N-glycan, accounting for ∼42% of total N-glycans, had a di-sialylated, core-fucosylated bi-antennary structure. These findings could assist the development of second-generation rhIFN-β products and help us better understand glycosylation CQAs and their biological significance.
Experimental Section
rhIFN-β Preparation
R27T was produced from the CHO cell line by ABION Inc. (Seoul, Republic of Korea). For purification, R27T was applied to an affinity column to capture the target protein, and the eluent from the affinity column was sequentially loaded onto ion exchange and C4 RP-HPLC columns. Most single- and double-glycosylated R27T molecules were separated by the RP-HPLC step from which the main peak contained mainly two-glycosylated forms, while the right shoulder (the intermediate peak) contained a considerable amount of single-glycosylated R27T. Major and intermediate peaks separated by RP-HPLC were concentrated and loaded onto a size exclusion column. The Rebif control was purchased from Merck KGaA (Hesse, Germany).
SDS-PAGE and Microchip Analysis
Samples (0.8 mg/mL, 30 μL) were denatured at 100 °C for 3 min after mixing with reducing buffers. Separation took place at 100 V for 30 min within the stacking gel, and 130 V for 2 h within the 12% TRIS-glycine mini gel. Coomassie Blue staining was used for protein visualization, and the quantitative microchip analysis based on the protein size was performed using a Protein 230 kit (Agilent Technologies, Waldbronn, Germany) following the manufacturer’s protocol on an Agilent 2100 Bioanalyzer (Agilent Technologies).
IEF and cIEF Analysis
IEF was used to confirm the pI of proteins and was performed using a pH 3–10 IEF gel (Invitrogen, CA, USA) with 18 μg samples. Coomassie Blue staining was used for visualization. To quantitate each pI band, we performed cIEF using a neutral capillary (50 μm internal diameter; total length, 30.2 cm; effective length, 20 cm, Beckman Coulter, Brea, CA, USA) on a PA800 plus CE instrument (Beckman Coulter, Brea, CA, USA). Samples (5 mg/mL, 10 μL) were prepared and mixed with 240 μL of master mix, and 200 μL was loaded onto the sample tray. UV detection was performed at 280 nm.
N-Glycan Release and Labeling
Triplicate aliquots of samples and single aliquots of negative control buffers were analyzed following the release protocol. Triplicate 100 μg aliquots of samples and equivalent volumes of buffers were reduced and alkylated, then separated by SDS-PAGE. Samples were washed before overnight incubation with PNGase F, and the released glycans were eluted, converted to aldoses, filtered, dried, and resuspended in 20 μL of water. After drying using an evaporator, 2-AB glycan labeling reagent (LT-KAB-A2 and a LudgerTag 2-AB glycan labeling kit; Ludger, UK) was added to samples and incubated at 65 °C for 3 h in a heat block. Once the reaction was complete, samples were purified using LudgerClean S Glycan Cleanup Cartridges (LC-S-A6, Ludger) to yield labeled glycans. Drying using an evaporator and redissolving in water gave the final 2-AB-labeled samples for clean-up and analysis.
Sialic Acid Release and Labeling
A 1,2-diamino-4,5-methylenedioxybenzene (DMB) sialic acid release and Labeling Kit (Ludger) was used to analyze the sialic acid content. A sample volume corresponding to 50–500 μg of product protein in a 1.5 mL screw-cap vial was dried in a vacuum centrifuge, and sialic acids were released by treating the dried sample with 25 μL of 2 M acetic acid for 2 h at 80 °C. After transferring 5 μL of the hydrolyzed sample to another tube, a 25 μL aliquot of LudgerTag DMB reagent was added, and the sample was heated for 3 h at 50 °C in the dark. The derivatization reaction was then quenched by the addition of 475 μL of water.
Galactose Release and Labeling
A sample volume corresponding to 50 μg of product protein in a 1.5 mL screw-cap vial was dried in a vacuum centrifuge (EYELA, Japan). Galactose were released by acid-treating the dried sample. For galactose hydrolysis, 200 μL of 2 M trifluoroacetic acid (TFA) was added, and reaction mixtures were incubated at 100 °C for 3 h, then dried in a vacuum centrifuge. Hydrolyzed samples were derivatized with a Labeling Kit (Ludger, USA) according to the manufacturer’s instruction. After dissolving hydrolyzed samples in 50 μL of sodium acetate, 50 μL of LudgerTag 2-AB and 50 μL of cyanoborohydride were added, and the sample was heated for 45 min at 80 °C. The derivatization reaction was stopped, and samples were serial diluted with BPT (a purified water-based solvent containing 0.2% butylamine, 0.5% phosphoric acid, and 1% tetrahydrofuran) before injection into HPLC.
WAX–HPLC Detection of Differently Charged Glycans
WAX chromatography separation of the 2-AB-labeled glycans was performed using a LudgerSep-C3 column (7.5 × 75 mm) at a column temperature of 35 °C with a sample volume of 25 μL. The gradient involved 100% A for 5 min, followed by 100–96% A over 5–21 min, 96–75% A over 21–61 min, 75–60% A over 61–72 min, and 60% A for 72–75 min, all at a flow rate of 0.4 mL/min, before washing at 100% A for 76–90 min at 0.8 mL/min. Solvent A was 20% acetonitrile in water, and solvent B was 500 mM ammonium acetate: 20% acetonitrile. The HPLC system consisted of a Waters Alliance 2795 separation unit and a Waters 2475 fluorescence detector (λex = 330 nm, λem = 420 nm) controlled by Empower 2.
Determining Sialic Acid Content and Variation
A DMB sialic acid release and Labeling Kit (Ludger) was used to analyze the sialic acid content. A sample volume corresponding to 50–500 μg of product protein in a 1.5 mL screw-cap vial was dried in a vacuum centrifuge, and sialic acids were released by treating the dried sample with 25 μL of 2 M acetic acid for 2 h at 80 °C. After transferring 5 μL of hydrolyzed sample to another tube, a 25 μL aliquot of LudgerTag DMB reagent was added, and the sample was heated for 3 h at 50 °C in the dark. The derivatization reaction was then quenched by the addition of 475 μL of water. A monosaccharide release and Labeling Kit (Ludger) was also used to analyze the galactose content. A sample volume corresponding to 50 μg of product protein in a 1.5 mL screw-cap vial was dried in a vacuum centrifuge (EYELA, Japan). Monosaccharides were released by acid-treating the dried sample. For neutral-sugar hydrolysis, 200 μL of 2 M TFA was added, and for amino-sugar hydrolysis, 200 μL of 6 M HCl was added, and reaction mixtures were incubated at 100 °C for 3 h, then dried in a vacuum centrifuge. Hydrolyzed samples were derivatized with a Labeling Kit (Ludger) according to the LT-MONO-96-GUIDE. After dissolving hydrolyzed samples in 50 μL of sodium acetate, 50 μL of LudgerTag 2-AA (2-AB) and 50 μL of cyanoborohydride were added, and the sample was heated for 45 min at 80 °C. The derivatization reaction was stopped, and samples were serial diluted with BPT (a purified water-based solvent containing 0.2% butylamine, 0.5% phosphoric acid, and 1% tetrahydrofuran) before injection.
The UPLC analysis was performed within 24 min of the sample preparation on an Agilent 1290 Infinity II System equipped with a fluorescence detector (excitation = 373 nm, emission = 448 nm). A 5 μL sample was injected onto a LudgerSep uR2 column (LS-UR2-2.1 × 100 for sialic acid, LS-UR2-2.1 × 50 for galactose). Each sample was applied after equilibrating the column with acetonitrile/methanol/water (9:7:84%) at 0.25 mL/min (for sialic acid) or BPT solvent at 30 °C.
Determining the Antennary Structure by Exoglycosidase Sequencing
The 2-AB-labeled glycans derived from R27T were incubated overnight at 37 °C with various exoglycosidase enzymes in 50 mM sodium acetate of pH 5.5. The following exoglycosidases were used: sialidase (QABio, USA) from Arthrobacter ureafaciens (a368S); specific for α2-3, -6, -8, and -9 sialic acids; beta-galactosidase (QABio) from Streptococcus pneumoniae (b4G; specific for β1-4 galactose); α-galactosidase (Sigma, USA) from green coffee bean (a36G; specific for α1-3/6 galactose); fucosidase (QABio) from Almond meal (a34F; specific for α1-3 and -4 fucose); fucosidase (Sigma, USA) from Bovine kidney (a6F; specific for α1-6 > 2 fucose); and N-acetylglucosaminidase (QABio) from S. pneumoniae (sph; specific for β-GlcNAc). Enzymes were removed by a protein-binding membrane, and samples were analyzed by HILIC–HPLC using an LSN2-40m-Nlink-35%_G100 and a LudgerSep-N2 column (LS-N24.6 × 150) on a Waters 2795 HPLC instrument linked to a 2475 fluorescence detector controlled by Empower software version 2, build 2154. Buffer A was 50 mM ammonium formate made from Ludger stock buffer # LS-BUFAMMFORM-2M-50ML, and Buffer B was acetonitrile (190 far-UV/gradient quality; Romil #H049). Samples were injected in 35% aqueous buffer/65% acetonitrile (35 μL samples +65 μL of acetonitrile), and the injection volume was 25 μL.
Determining N-Glycan Structures by WAX/HILIC–HPLC with Fluorescence Labeling of Glycans
The 2-AB-labeled glycans derived from R27T were separated using a LudgerSep-C3 WAX–HPLC column with a manual fraction collection of the separated peaks. The collected fractions were desalted by the evaporation of the HPLC buffer in a centrifugal evaporator followed by the addition of 1 mL aliquots of water to each fraction and subsequent centrifugal evaporation of the water. This process was repeated until no salt deposit was visible. Half of each fraction was analyzed directly by LudgerSep-N2 HILIC–HPLC, and the remaining half was digested with sialidase and then analyzed by LudgerSep-N2 HILIC–HPLC. For sialidase digestion, aliquots of 2-AB-labeled glycan fractions were incubated overnight at 37 °C with Sialidase (QABio) from A. ureafaciens (a368S; specific for a2-3, -6, -8, and -9 sialic acids) in 50 mM sodium acetate pH 5.5. Enzyme was removed using a protein-binding membrane before analysis by LudgerSep-N2 HILIC–HPLC.
Glucose Unit Allocation with the Glycosylation Structure
Waters GPC software with a cubic spline fit was used to allocate GU values to peaks. 2-AB-labeled glucose homopolymer (Ludger product CAB-GHP-30, 2-AB glucose homopolymer ladder) was used as a system suitability standard as well as an external calibration standard for GU allocation (the system is deemed to be within specifications if the peak width at half height for GU10 is less than 0.4 min).
Determining N-Glycan Structures by HILIC–MS
Aliquots (1 μL) of the unlabeled portion of N-glycans released from samples were taken for the permethylation analysis along with negative and positive controls (buffer, water, and fetuin). The unlabeled glycan samples were permethylated using methyl iodide in a dimethylsulfoxide/NaOH suspension for in-solution permethylation of glycans. The permethylated glycans were obtained from solution by extracting with chloroform, and samples were run on a Shimadzu Axima Biotech Resonance MALDI-TOF-MS using 10 mg/mL 2,5-dihydroxybenzoic acid in acetonitrile as the matrix. The AXIMA Resonance is a unique hybrid MALDI mass spectrometer designed for the structural determination of biomolecules by fragmentation using MS/MS and MSn. The ion-to-trap introduction sequence was optimized with Rapid-RF Start-Up and helium Hypercool. The peaks are labeled with the most abundant peak in the molecular ion cluster.
Acknowledgments
We would like to thank ABION Inc (Seoul, Republic of Korea) for providing R27T and Rebif and Tae Won Yun from the LOGONE Bio Convergence Research Foundation for help with illustrations.
Glossary
Glycosylation Structure Abbreviations
- F at the start of the abbreviation
a core fucose
- Ax
number of antennae (GlcNAc) on the trimannosyl core
- A2
bi-antennary with both GlcNAcs β1-2-linked
- A3
tri-antennary with a GlcNAc linked via β1-2 to mannose, and the third GlcNAc linked via β1-4 to the α1-3-linked mannose
- A3′
tri-antennary with two GlcNAc units linked via β1-2 to mannose, and the third GlcNAc linked via β1-6 to the α1-6-linked mannose
- A4
GlcNAcs linked as in A3 with an additional GlcNAc linked via β1-6 to α1-6 mannose
- Gx
number (x) of linked β-galactose on antennae
- Sx
number (x) of sialic acids linked to galactose
- Lacx
number (x) of N-acetyllactosamine (Gal–GlcNAc) extensions
- Nx
number of terminal GlcNAc units following removal of Gal from a Lac
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b04385.
Flow chart of R27T purification; differentially charged glycans derived from R27T; HILIC–HPLC profiles of R27T samples; HILIC–HPLC profiles of R27T WAX fractions; overlayed HILIC–HPLC profiles of R27T WAX fractions quantitation of N-glycosylation site occupancy using microchip; HILIC–HPLC data for R27T; and exoglycosidase digestion data for R27T (PDF)
The authors declare the following competing financial interest(s): Kyoung Song and Young Kee Shin currently hold stock and Na Young Kim hold stock options in ABION Inc.
Supplementary Material
References
- Diebold M.; Derfuss T. Immunological treatment of multiple sclerosis. Semin. Hematol. 2016, 53, S54–S57. 10.1053/j.seminhematol.2016.04.016. [DOI] [PubMed] [Google Scholar]
- Goodin D. S.; Frohman E. M.; Garmany G. P. Jr.; Halper J.; Likosky W. H.; Lublin F. D.; Silberberg D. H.; Stuart W. H.; van den Noort S. Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology 2002, 58, 169–178. 10.1212/wnl.58.2.169. [DOI] [PubMed] [Google Scholar]
- Madsen C. The innovative development in interferon beta treatments of relapsing-remitting multiple sclerosis. Brain Behav. 2017, 7, e00696 10.1002/brb3.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Wu N.; Watson C. Multiple sclerosis patients who are stable on interferon therapy show better outcomes when staying on same therapy than patients who switch to another interferon. Clin. Outcomes Res. 2018, Volume 10, 723–730. 10.2147/ceor.s163907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L.; Freedman M. S.; Polman C. H.; Edan G.; Hartung H.-P.; Miller D. H.; Montalbán X.; Barkhof F.; Radü E.-W.; Bauer L.; Dahms S.; Lanius V.; Pohl C.; Sandbrink R.; Group B. S. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet 2007, 370, 389–397. 10.1016/s0140-6736(07)61194-5. [DOI] [PubMed] [Google Scholar]
- Kappos L.; Polman C. H.; Freedman M. S.; Edan G.; Hartung H. P.; Miller D. H.; Montalban X.; Barkhof F.; Bauer L.; Jakobs P.; Pohl C.; Sandbrink R. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology 2006, 67, 1242–1249. 10.1212/01.wnl.0000237641.33768.8d. [DOI] [PubMed] [Google Scholar]
- Borden E. C.; Sen G. C.; Uze G.; Silverman R. H.; Ransohoff R. M.; Foster G. R.; Stark G. R. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu L.; Constantinescu C. S.; Tanasescu R. Recent developments in interferon-based therapies for multiple sclerosis. Expet Opin. Biol. Ther. 2018, 18, 665–680. 10.1080/14712598.2018.1462793. [DOI] [PubMed] [Google Scholar]
- Castro-Borrero W.; Graves D.; Frohman T. C.; Flores A. B.; Hardeman P.; Logan D.; Orchard M.; Greenberg B.; Frohman E. M. Current and emerging therapies in multiple sclerosis: a systematic review. Ther. Adv. Neurol. Disord. 2012, 5, 205–220. 10.1177/1756285612450936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini C.; Ruggieri S. Emerging oral drugs for relapsing-remitting multiple sclerosis. Expet Opin. Emerg. Drugs 2011, 16, 697–712. 10.1517/14728214.2011.642861. [DOI] [PubMed] [Google Scholar]
- Grossberg S. E.; Oger J.; Grossberg L. D.; Gehchan A.; Klein J. P. Frequency and magnitude of interferon beta neutralizing antibodies in the evaluation of interferon beta immunogenicity in patients with multiple sclerosis. J. Interferon Cytokine Res. 2011, 31, 337–344. 10.1089/jir.2010.0038. [DOI] [PubMed] [Google Scholar]
- Hartung H.-P.; Munschauer F. 3rd; Schellekens H. Significance of neutralizing antibodies to interferon beta during treatment of multiple sclerosis: expert opinions based on the Proceedings of an International Consensus Conference. Eur. J. Neurol. 2005, 12, 588–601. 10.1111/j.1468-1331.2005.01104.x. [DOI] [PubMed] [Google Scholar]
- van Beers M. M. C.; Jiskoot W.; Schellekens H. On the role of aggregates in the immunogenicity of recombinant human interferon beta in patients with multiple sclerosis. J. Interferon Cytokine Res. 2010, 30, 767–775. 10.1089/jir.2010.0086. [DOI] [PubMed] [Google Scholar]
- Song K.; Yoon I.-S.; Kim N. A.; Kim D.-H.; Lee J.; Lee H. J.; Lee S.; Choi S.; Choi M.-K.; Kim H. H.; Jeong S. H.; Son W. S.; Kim D.-D.; Shin Y. K. Glycoengineering of interferon-beta 1a improves its biophysical and pharmacokinetic properties. PLoS One 2014, 9, e96967 10.1371/journal.pone.0096967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Son W. S.; Yang H. B.; Rajasekaran N.; Kim S. S.; Hong S.; Choi J. S.; Choi J. Y.; Song K.; Shin Y. K. A Glycoengineered interferon-beta mutein (R27T) generates prolonged signaling by an altered receptor-binding kinetics. Front. Pharmacol. 2019, 9, 1568. 10.3389/fphar.2018.01568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaderi D.; Zhang M.; Hurtado-Ziola N.; Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol. Genet. Eng. Rev. 2012, 28, 147–176. 10.5661/bger-28-147. [DOI] [PubMed] [Google Scholar]
- Hossler P.; Khattak S. F.; Li Z. J. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 2009, 19, 936–949. 10.1093/glycob/cwp079. [DOI] [PubMed] [Google Scholar]
- Walsh G.; Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 2006, 24, 1241–1252. 10.1038/nbt1252. [DOI] [PubMed] [Google Scholar]
- Sareneva T.; Pirhonen J.; Cantell K.; Julkunen I. N-glycosylation of human interferon-gamma: glycans at Asn-25 are critical for protease resistance. Biochem. J. 1995, 308, 9–14. 10.1042/bj3080009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A.; Morrison S. L. Effect of glycosylation on antibody function: implications for genetic engineering. Trends Biotechnol. 1997, 15, 26–32. 10.1016/s0167-7799(96)10062-7. [DOI] [PubMed] [Google Scholar]
- Solá R. J.; Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs 2010, 24, 9–21. 10.2165/11530550-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Li W.; Lu H.; Liu Y. Quantification of N-glycosylation site occupancy status based on labeling/label-free strategies with LC-MS/MS. Talanta 2017, 170, 509–513. 10.1016/j.talanta.2017.04.053. [DOI] [PubMed] [Google Scholar]
- Jenkins N.; Parekh R. B.; James D. C. Getting the glycosylation right: implications for the biotechnology industry. Nat. Biotechnol. 1996, 14, 975–981. 10.1038/nbt0896-975. [DOI] [PubMed] [Google Scholar]
- Barry C. S.; Cocinero E. J.; Çarçabal P.; Gamblin D. P.; Stanca-Kaposta E. C.; Remmert S. M.; Fernández-Alonso M. C.; Rudić S.; Simons J. P.; Davis B. G. “Naked” and hydrated conformers of the conserved core pentasaccharide of N-linked glycoproteins and its building blocks. J. Am. Chem. Soc. 2013, 135, 16895–16903. 10.1021/ja4056678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J. H.; Zhang F.; Ermonval M.; Linhardt R. J.; Sharfstein S. T. The effects of culture conditions on the glycosylation of secreted human placental alkaline phosphatase produced in Chinese hamster ovary cells. Biotechnol. Bioeng. 2008, 100, 1178–1192. 10.1002/bit.21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra-Arteaga A.; Shuler M. L. Influence of culture medium supplementation of tobacco NT1 cell suspension cultures on the N-glycosylation of human secreted alkaline phosphatase. Biotechnol. Bioeng. 2007, 97, 1585–1593. 10.1002/bit.21344. [DOI] [PubMed] [Google Scholar]
- Joosten C. E.; Shuler M. L. Effect of culture conditions on the degree of sialylation of a recombinant glycoprotein expressed in insect cells. Biotechnol. Prog. 2003, 19, 739–749. 10.1021/bp0201049. [DOI] [PubMed] [Google Scholar]
- Wurm F. M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. 10.1038/nbt1026. [DOI] [PubMed] [Google Scholar]
- Engel N.; Weiss V. U.; Wenz C.; Rüfer A.; Kratzmeier M.; Glück S.; Marchetti-Deschmann M.; Allmaier G. Challenges of glycoprotein analysis by microchip capillary gel electrophoresis. Electrophoresis 2015, 36, 1754–1758. 10.1002/elps.201400510. [DOI] [PubMed] [Google Scholar]
- Achord D. T.; Brot F. E.; Sly W. S. Inhibition of the rat clearance system for agalacto-orosomucoid by yeast mannans and by mannose. Biochem. Biophys. Res. Commun. 1977, 77, 409–415. 10.1016/s0006-291x(77)80213-1. [DOI] [PubMed] [Google Scholar]
- Dobryszycka W.; Kukral J. C. Metabolic studies on sialic acid-free haptoglobin. Arch. Immunol. Ther. Exp. 1970, 18, 527–36. [PubMed] [Google Scholar]
- Morell A. G.; Irvine R. A.; Sternlieb I.; Scheinberg I. H.; Ashwell G. Physical and chemical studies on ceruloplasmin. V. Metabolic studies on sialic acid-free ceruloplasmin in vivo. J. Biol. Chem. 1968, 243, 155–9. [PubMed] [Google Scholar]
- Ghaderi D.; Taylor R. E.; Padler-Karavani V.; Diaz S.; Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat. Biotechnol. 2010, 28, 863–867. 10.1038/nbt.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokke C. H.; Bergwerff A. A.; van Dedem G. W. K.; van Oostrum J.; Kamerling J. P.; Vliegenthart J. F. G. Sialylated carbohydrate chains of recombinant human glycoproteins expressed in Chinese hamster ovary cells contain traces of N-glycolylneuraminic acid. FEBS Lett. 1990, 275, 9–14. 10.1016/0014-5793(90)81427-p. [DOI] [PubMed] [Google Scholar]
- Noguchi A.; Mukuria C. J.; Suzuki E.; Naiki M. Immunogenicity of N-glycolylneuraminic acid-containing carbohydrate chains of recombinant human erythropoietin expressed in Chinese hamster ovary cells. J. Biochem. 1995, 117, 59–62. 10.1093/oxfordjournals.jbchem.a124721. [DOI] [PubMed] [Google Scholar]
- Borys M. C.; Dalal N. G.; Abu-Absi N. R.; Khattak S. F.; Jing Y.; Xing Z.; Li Z. J. Effects of culture conditions on N-glycolylneuraminic acid (Neu5Gc) content of a recombinant fusion protein produced in CHO cells. Biotechnol. Bioeng. 2010, 105, 1048–1057. 10.1002/bit.22644. [DOI] [PubMed] [Google Scholar]
- Karpusas M.; Whitty A.; Runkel L.; Hochman P. The structure of human interferon-beta: implications for activity. Cell. Mol. Life Sci. 1998, 54, 1203–1216. 10.1007/s000180050248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eon-Duval A.; Broly H.; Gleixner R. Quality attributes of recombinant therapeutic proteins: an assessment of impact on safety and efficacy as part of a quality by design development approach. Biotechnol. Prog. 2012, 28, 608–622. 10.1002/btpr.1548. [DOI] [PubMed] [Google Scholar]
- Royle L.; Campbell M. P.; Radcliffe C. M.; White D. M.; Harvey D. J.; Abrahams J. L.; Kim Y.-G.; Henry G. W.; Shadick N. A.; Weinblatt M. E.; Lee D. M.; Rudd P. M.; Dwek R. A. HPLC-based analysis of serum N-glycans on a 96-well plate platform with dedicated database software. Anal. Biochem. 2008, 376, 1–12. 10.1016/j.ab.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Royle L.; Radcliffe C. M.; Dwek R. A.; Rudd P. M. Detailed structural analysis of N-glycans released from glycoproteins in SDS-PAGE gel bands using HPLC combined with exoglycosidase array digestions. Methods Mol. Biol. 2006, 347, 125–43. 10.1385/1-59745-167-3:125. [DOI] [PubMed] [Google Scholar]
- Campbell M. P.; Royle L.; Radcliffe C. M.; Dwek R. A.; Rudd P. M. GlycoBase and autoGU: tools for HPLC-based glycan analysis. Bioinformatics 2008, 24, 1214–1216. 10.1093/bioinformatics/btn090. [DOI] [PubMed] [Google Scholar]
- Mastrangeli R.; Rossi M.; Mascia M.; Palinsky W.; Datola A.; Terlizzese M.; Bierau H. In vitro biological characterization of IFN-beta-1a major glycoforms. Glycobiology 2015, 25, 21–29. 10.1093/glycob/cwu082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.