

Abstract
The elimination of numerous endogenous compounds and xenobiotics via glucuronidation by uridine-5′-diphosphate glycosyltransferase enzymes (UGTs) is an essential process of the body’s chemical defense system. UGTs have distinct but overlapping substrate preferences, but the molecular basis for their substrate specificity remains poorly understood. Three-dimensional protein structures can greatly enhance our understanding of the interactions between enzymes and their substrates, but because of the inherent difficulties in purifying and crystallizing integral endoplasmic reticulum membrane proteins, no complete mammalian UGT structure has yet been produced. To address this problem, we have created a homology model of UGT1A6 using I-TASSER to explore, in detail, the interactions of human UGT1A6 with its substrates. Ligands were docked into our model in the presence of the cosubstrate uridine-5′-diphosphate-glucuronic acid, interacting residues were examined, and poses were compared to those cocrystallized with various plant and bacterial glycosyltransferases (GTs). Our model structurally resembles other GTs, and docking experiments replicated many of the expected UGT-substrate interactions. Some bias toward the template structures’ protein–substrate interactions and binding preferences was evident.
Introduction
Uridine-5′-diphosphate glycosyltransferases (UGTs) belong to the glycosyltransferase (GT) superfamily and are responsible for the metabolism and detoxification of numerous natural compounds and xenobiotics.1 They make up a family of phase 2 conjugation enzymes that act to catalyze the covalent addition of sugars to lipophilic substrates and have been estimated to perform approximately 35% of all phase 2 conjugation reactions in the human body.2,3 In humans, the UGTs are divided into four subfamilies: UGT1, 2, 3, and 8, with the most important drug-conjugating UGTs belonging to the 1 and 2 subfamilies.4,5 The UGT1A subfamily is composed of 13 members generated from alternate splicing of a single gene on chromosome 2q37, four of which are nonfunctional pseudogenes. Exons 2–5 are common to all UGT1 members, with exon 1 and its associated promoter region unique to each member.6 The UGT2 subfamily is located on chromosome 4q13 and is divided into the 2A and 2B subfamilies containing 3 and 12 (7 functional) genes, respectively. The UGT3 subfamily contains two genes encoded on chromosome 5p13 that are expressed at low levels in the body and are primarily involved in the endogenous metabolism rather than the xenobiotic metabolism.2,7,8 The UGT8 subfamily consists of a single gene encoding uridine-5′-diphosphate (UDP)-galactose ceramide galactosyltransferase on chromosome 4q26 that catalyzes the formation of galactosylceramides, but it is also able to catalyze the galactosidation of bile acids.2,9−11
UGTs are widely expressed throughout the body, with the majority of the 1A and 2B enzymes highly expressed in the liver. The UGT1A and UGT2B enzymes use UDP-glucuronic acid (UDPGA) as their preferred cosubstrate and sugar donor, with each member of the family having different but overlapping preferred substrates.12 UGT1A6 principally glucuronidates small phenolic substances using UDPGA as a donor, including a number of phenol containing chemical classes such as flavonoids, stilbenes, and coumarins.12 Other phenolic compounds such as acetaminophen, 1-naphthol, and 4-nitrophenol are also metabolized by UGT1A6.13−15 Many compounds are substrates for multiple UGTs; however, some are preferentially glucuronidated using a single isoform, for example, bilirubin (UGT1A1) and serotonin (UGT1A6).16,17
There is extensive evidence, suggesting that UGTs form quaternary structures. Multiple studies have demonstrated homodimerization of individual isoforms, including the entire UGT1A family, UGT2B1, and UGT2B7.18−22 Heterodimerization between different UGT1A enzymes, as well as between UGT1A subfamily members and both UGT2B1 and UGT2B7 has also been observed.18,23,24 A putative dimerization region has been identified in UGT2B7, with a signature sequence FPPSYVPVVMS (residues 189–199) that is able to stabilize protein–protein interactions via proline brackets and π–π interactions.22 The analogous region in the UGT1A subfamily also contains a dimerization signature motif (PXPXSY[V/I]PXXX), with approximately half of the UGT1A subfamily containing an isoleucine at the seventh position (UGT1A3-6), and the remainder having a valine at this site. In UGT1A6, the dimerization motif is PDPVSYIPRCY (residues 186–196). Oligomerization has been shown to influence substrate specificities, reaction rates, and the end products. For example, UGT1A1 dimers produce bilirubin monoglucuronide, but the homotetramer forms bilirubin diglucuronide.25 Heteromerization of UGT2B7 and UGT1A6 decreased glucuronidation of zidovudine and morphine in Sf9 cells but increased serotonin glucuronidation.26 Although UGT1A1 is the only UGT to efficiently glucuronidate bilirubin, heteromers of UGT1A1 and UGT1A4 or UGT1A1 and UGT1A6 have been shown to increase formation of bilirubin glucuronide compared to UGT1A1 alone.27 Additionally, heteromers of UGT1A4 and UGT1A6 increase trifluoperazine and serotonin glucuronide formation compared to UGT1A4 or UGT1A6 alone.27 In addition to the interactions between UGTs, there is also some evidence that UGTs are able to directly interact with other proteins, including cytochromes P450, thereby affecting their functions.28,29
GTs are currently classified into 106 families on the basis of amino acid similarity and the sugar donor ligand type.30,31 Despite low levels of sequence identity, GTs share a high degree of structural conservation and fall into GT-A, GT-B, or GT-A-like fold families.32,33 The GT-A fold consists of an α/β/α sandwich which resembles a Rossman fold, contains a DxD motif, and requires a divalent metal cation (commonly Mn2+) that is important for donor ligand binding. The GT-B fold comprises two α/β/α Rossman-like folds that associate to form a catalytic cleft at their interface and lacks both the DxD motif and requirement for a divalent metal. The GT-A-like fold is made up of an α/β/α sandwich with a different β-sheet arrangement and lacks the DxD motif.32 Unlike bacterial, fungal, and plant GTs studied and crystallized to date, mammalian UGTs are membrane proteins with a single transmembrane (TM) domain anchoring them into the membrane of the endoplasmic reticulum (ER). The bulk of the protein resides within the ER lumen (and likely interacts with the membrane itself) with only a short C-terminal region present on the cytoplasmic side. Purifying and crystallizing membrane-bound proteins are inherently difficult, and therefore, there are no complete X-ray crystal structures of any mammalian UGTs available. The crystal structure of the C-terminal UDPGA binding domain of the human UGT2B7 was published in 2007 and, until very recently, was the only human UGT structure available.33,34 This structure confirmed that human UGTs belong to the GT-B fold family of GTs and that they show a high degree of structural similarity to other plant, bacterial, and fungal GT crystal structures. In 2020, a second partial crystal structure for a human UGT was released, the C-terminal UDPGA binding domain of the human UGT2B15, whose structure closely matches that of UGT2B7.35
X-ray structures can provide numerous insights into protein functions, associations, and substrate selectivity. In the absence of a full crystal structure, homology modeling approaches can generate 3D protein structures to assist in answering questions about protein structure–function relationships. Several attempts have been made to produce UGT homology models, with models of UGT1A1, 1A3, 1A8, 1A9, 1A10, 2B7, and the C-terminal domains of UGT1, UGT2A1/2A2, UGT2A3, UGT2B4, UGT2B10, UGT2B11, UGT2B15, UGT2B17, and UGT2B28 having been produced.36,37 Despite the enzyme’s importance in drug metabolism, there has been only a single UGT1A6 homology model produced to date, which was used to assess inhibitory properties of a number of compounds against UGT1A6, with support vector machine classification and 3D quantitative structure–activity relationship methods used to build predictive models of glucuronidation inhibition.38
In this study, we aimed at producing a new homology model of UGT1A6 to further examine features involved in the enzyme function and substrate binding. We chose to model UGT1A6 because of the enzyme’s critical role in metabolism of a wide array of endogenous and exogenous compounds, including many popular analgesics, as well as the relative paucity of information available about the UGT1A6 structure and key residues important for substrate binding and specificity. By producing this model, we hope to obtain additional information about its structure and key features to better understand the role of this enzyme in the glucuronidation of endogenous compounds and xenobiotics. To achieve this, we generated our initial homology model of UGT1A6 using the online I-TASSER (Iterative Threading ASSEmbly Refinement) server, with additional refinements and energetic minimizations performed using the BIOVIA Discovery Studio. The UGT1A6 co-substrate, UDPGA, as well as a number of compounds known to be metabolized by UGT1A6, the template GTs, or not metabolized by UGT1A6 as negative controls, were docked into the completed model and key binding residues examined. Mutated versions of UGT1A6 with important binding residues converted to alanine were produced and docked with the cosubstrate and substrate molecules to investigate the importance of these residues. Molecular dynamics simulations were conducted on the model docked with both UDPGA and the top scoring substrate.
Results and Discussion
UGTs are structurally and functionally complex, with high levels of similarity in the amino acid sequence and function. Despite this, various isoforms have very different, but overlapping substrate specificities because of small differences in the composition of the N-terminal substrate binding domain. Because of the difficulty in generating membrane protein crystal structures, no complete crystal structures of mammalian UGTs have been produced to date. We produced a homology model of UGT1A6 using I-TASSER, a hierarchical approach to the protein structure and function prediction, which has been ranked as the top server for protein structure prediction in the Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP) since 2006.
UGT1A6 Model Generation
The I-TASSER server produced five potential models for each of the four input conditions: with or without the 26-amino acid N-terminal signal peptide (normally absent in the mature enzyme) and with or without the specification of the UGT2B7 C-terminal crystal structure as a guiding template (Figure S1). I-TASSER evaluated each model and provided a confidence score (C-score) to estimate the model quality based on the significance of threading template alignments and convergence parameters of structure assembly simulations, typically ranging from −5 to 2, with higher scores indicating increased confidence in the model. The expected template modeling score (TM-Score) and root-mean-square deviation (rmsd) of atomic positions values were also calculated for the model with the highest C-score.39 Models with TM-Scores greater than 0.5 are considered to have correct topology, whereas models with scores less than 0.17 are due to random similarities. The no-signal peptide and with a guiding template (NSWT) and with signal peptide and no guiding template (WSNT) models had the highest C-score (−1.65) and TM-score (0.51 ± 0.15), with predicted rmsd values of 11.2 ± 4.6 and 11.4 ± 4.5 Å, respectively (Table S1).
The normalized discrete optimized protein energy (DOPE) score and the verify score for each model were calculated (Table S2). Positive DOPE scores indicate a poor model quality, with scores lower than −1, indicating that the model is likely accurate. Verify scores higher than the expected high score indicate that the structure is likely to be correct, and proteins with scores that are between the high and low reference values may have some of the structure being incorrect. The NSWT model had the best normalized DOPE (−0.60) and verify scores (173.76), although neither were as good as those of the partial human UGT2B7 (2O6L, DOPE: −1.99, verify: 161.43) or the Medicago truncatula UGT71G1 (2ACW, DOPE: −2.12, verify: 256.69) crystal structures. Structural alignments with 30 GT crystal structures indicated that the models generated using the UGT2B7 crystal structure as a guiding template [with signal peptide and with a guiding template (WSWT) and NSWT] had the smallest rmsds from these structures (Table S3).
The major C-terminal and N-terminal domains are connected by a central linker, and two C-terminal envelope helices that fold back over both domains, which in mammalian UGTs, connect with the TM domain (Figure 1). The C-terminal domain was broadly conserved between models, with the largest divergences observed between the NSWT and no signal peptide and no guiding template (NSNT) models. Specifically, K316 in helix Cα1 differed by 2.8 Å between models, and the T337 residue located between the Cβ2 and Cβ3 sheets differed by 4.2 Å in these models. The inclusion or exclusion of the N-terminal signal peptide had minor effects on the C-terminal domain, showing only small differences in some of the flexible loop regions. A high degree of variability was observed at the C-terminal TM helix and envelope helix (EH) 2.
Figure 1.

Structure of UGT1A6. (A) Final refined homology model of UGT1A6. Helices are shown as spirals, and β-sheets are shown as broad ribbons. The C-terminal domain (blue) is shown at the top, with the N-terminal domain (green) shown at the bottom of the image. The envelope helices (magenta) are shown to the left, with the TM domain (purple) at the bottom. The putative dimerization domain (PDPVSYIPRCY) of UGT1A6 is highlighted in orange. (B) Schematic diagram of the UGT1A6 structure. Colors are the same as in (A), with α-helices shown as cylinders and β-sheets shown as arrows. The predicted Nα2 and Nα6 helices were not present in the final model and have been rendered as transparent cylinders in the diagram as a reference as to where they would be expected to form.
The N-terminal domain was more variable between models (Figure 2). Although key catalytic residues such as H38 and D150 differed by a maximum of 1.4 and 2.3 Å between the NSNT and NSWT structures, respectively, other regions differed more substantially. The expected Nα2 helix was not present in the NSWT/WSWT models, replaced by a disordered loop, but was present in the NWNT/WSNT models. The Nα3-1 helix extended for only 8 amino acids in the NSWT model, but 14 residues in the WSWT model and 17 residues in the NSNT model. The positions of the Nα3-1, Nα3-2, and Nα3-3 helices also differed substantially between the four models, with the Nα3-3 helix being absent in the NSWT model. The Nα5-1 and Nα5-4 helices were absent in the NSNT and WSNT models, and the Nα5-3 helix in these models was in a substantially different location compared to the NSWT and WSWT models, the F226 residue being located 11.9 Å apart in these models. A number of additional differences were seen in the loop regions, or helices within larger loop regions, and may be due to the modeling process rather than the inclusion or exclusion of the signal peptide.
Figure 2.

Comparison of initial I-TASSER generated homology models. The models generated without the use of the 2B7 crystal as a guide are shown in teal (without the signal peptide, NSNT) and green (with the signal peptide, WSNT). The models generated using the 2B7 structure as a guide are shown in blue (with the signal peptide, WSWT) and purple (without the signal peptide, NSWT). (A) Key catalytic residues H38 and D150 in the NSNT and NSWT models differ by only a minute amount between the models. (B) Nα2 helix is not present in the NSWT and WSWT models but does appear in the NSNT and WSNT models. (C) Orientation, length, and positioning of the Nα3 helices differed substantially between models. (D) Nα5 helices also were substantially different between models, with the Nα5-2 positions of the NSWT/WSWT and NSNT/WSNT in different locations and orientations, and the Nα5-3 helix shifted down in the NSNT/WSNT models compared to the NSWT/WSWT model, as demonstrated by the position of the highlighted F226 residue (yellow).
Based on these results, the NSWT model was selected for further refinement and docking studies. Following refinements, the model had a normalized DOPE score of −0.891 and a verify score of 176.15 (high: 231.22, low: 104.05). The final model had an ERRAT score of 88.76, and 94.5% of residues were within the outer bounds of the Ramachandran plot (Figure S2). Because of the fact that there are no GT crystal structures available containing the C-terminal TM domain that anchors mammalian UGTs into the ER membrane, it was not possible to properly constrain the models for the presence of the membrane, and the TM and C-terminal domains could not be accurately localized. Compared to the template UGT2B7 structure, the final model had an rmsd of 1.71 Å over the aligned sequence, with only short loop regions deviating by more than 2 Å from the template (Figure S3). In contrast, when the final structure was compared to the two other template structures (2ACV and 5TME), there was a greater deviation between the model and the templates, principally in various loops and regions connecting the α-helices and β-sheets (Figure S4). The C-terminal UDPGA binding domain was generally similar with a large number of conserved amino acids, whereas the N-terminal domain was more variable with fewer conserved residues. The putative dimerization region corresponding to residues P186–Y196 in UGT1A6 (PDPVSYIPRCY) was present on the exterior surface of the model in a flexible loop region (Figure 1).22
Our best model was generated using plant GTs—the M. truncatula triterpene UDP-glucosyltransferase and an Oryza sativa UDP-glucosyltransferase—with the C-terminal region guided using the human UGT2B7 X-ray crystal structure. Although there were some differences between the models produced using the four differing input conditions, only minute differences in the loop and helix placement were seen in the highly conserved C-terminal sugar binding domain, although the TM and EH regions of the C-terminal domain were more variable. This is not unexpected because all of the template GTs are cytosolic proteins, and the human UGT2B7 C-terminal fragment was crystallized without the TM domain; thus, there are no templates onto which the extreme C-terminal end of the protein can be based. Because of the lack of template structures, the TM region must be modeled ab initio each time, resulting in highly variable placement of the residues. This also limits our ability to examine interactions of the model with the ER membrane, as it cannot be properly localized in this space. The N-terminal domain is responsible for binding the enzyme’s substrate, and because of the variety of substrates bound using the template structures, it is not surprising that this region was also quite variable. Many regions in the N-terminal domain consist of loops or helices within larger loop regions and are inherently disordered, making it difficult to crystallize or model these areas. It is likely that in the native state, these regions are flexible, allowing binding of different substrates, with the protein conformation shifting, depending on which substrate is bound.
UGT1A6 is strongly associated with the luminal ER membrane surface. In addition to being targeted to the ER via the N-terminal signal peptide, UGT1A6 possesses an internal ER-targeting signal located between E141 and D240 that is sufficient to allow entry and association with the ER membrane in the absence of the N-terminal signal peptide and the C-terminal TM and cytoplasmic domains.40 This region roughly corresponds to Nβ4 through Nα5-3 regions of the protein, with much of it buried in the protein core. Because of the lipophilic nature of many of the drugs that UGTs metabolize, it has been proposed that hydrophobic helices may serve as routes to allow for the lipophilic aglycone substrates to reach the active site following diffusion through the lipid bilayer. Examining the hydrophobicity of each residue of the region in the context of our homology models shows that while the inner regions of the protein surface helices and loops are hydrophobic, the external, lumen-facing sides are generally hydrophilic. It is possible that these residues are able to interact with the hydrophilic phosphate head groups of the lipid bilayer to stabilize the protein on the membrane surface. Other exterior α helices, including the Nα3-3 helix and the C-terminal EH1 helix, possess larger hydrophobic regions flanked by more hydrophilic residues, allowing for the possibility that they embed more deeply into the lipid bilayer to firmly anchor the protein to the luminal ER surface. However, similar regions are present in the soluble plant and bacterial GT proteins that lack the TM domain found in mammalian UGTs, so the exact role of these regions and nature of the protein–membrane interaction remain unclear.
Comparison of our final UGT1A6 model with the UGT1A6 model produced by Ghemtio et al. (2014) showed a high degree of alignment in the C-terminal region, with both of the models having been produced using the UGT2B7 (2O6L) C-terminal crystal structure as a guide for the C-terminal end (Figure S5).38 The rmsds between the models increased as they near EH2, the TM domain not being present in the Ghemtio et al. model. Although the M. truncatula UGT71G1 (2ACV) and the O. sativa subsp. Japonica UGT79 GT (5TME) were used as the templates for building both models, Ghemtio et al. also used the M. truncatula UGT78G1 (3HBF), UGT85H2 (2PQ6), and Vitis vinifera UFGT (2C1X) crystal structures. The N-terminal region showed a greater degree of variability between the models, particularly in the Nα2–Nα3-1 and Nα5-2–Nβ7 regions, which correspond to regions with poor structural homology to the template structures in both our model and the Ghemtio et al. model (Figure S5).
Molecular Docking
The most likely UDPGA binding pose was determined via a comparison of 24 GT crystal structures that had been crystallized with their cosubstrate or cosubstrate analogue [UDP, UDP-glucose, UDP-2-deoxy-2-fluoro-α-d-glucose (U2F), or thymidine-5′-diphosphate (TDP)] present in the cosubstrate binding site to determine which residues were important for interacting with the constituent groups of the nucleotides (Table S4). Structural alignment of all structures with the UGT1A6 homology model allowed for the determination of which UGT1A6 residues would likely be important in binding UDPGA, with the caveat that a number of residues that are known to interact with the cosubstrate in other GT structures fall within flexible loop regions of the UGT1A6 model, making exact determination of the corresponding interacting residues difficult.
Residues W353 and L354 (all positions listed relative to the UGT1A6 sequence) interacted with the pyrimidine group in the vast majority of all templates (70.8 and 95.8%, respectively), with R335 (33.5%), P355 (25%), and Q356 (25%) also being fairly common. Additional residues were found to interact with pyrimidine in one or more models, specifically S37 (20.8%), R282 (8.3%), K283 (4.2%), S305 (16.7%), and N357 (12.5%). The most common residue to interact with the (deoxy)ribose ring was E379 (66.7%), with S37 (8.3%), L40 (4.2%), and N357 (12.5%) also interacting in some structures. The α phosphate group commonly interacted with G376 (95.8%), H375 (83.3%), and H371 (62.5%), with some structures also showing interactions with S37 (37.5%), G307 (4.2%), S308 (4.2%), G373 (16.7%), and S374 (25%). The residues interacting with β phosphate were located in a loop region of the 1A6 protein: S308 (87.5%) and S37 (79.2%). G36 (20.8%), G307 (16.7%), M309 (8.3%), G373 (12.5%), H375 (4.2%), G376 (4.2%), and F393 (16.7%). Only five of the crystal structures contained a cosubstrate with a hexose moiety, but S374, D395, and Q396 interacted with this group in all five structures. H38 interacted with the group in the three structures, and G36 and R172 had interactions in one structure each.
UDPGA was docked into the cosubstrate binding site using CDOCKER, with the top pose having a negative CDOCKER energy score of 59.9849 kcal mol–1. This score, which is used to rank the binding of compounds, consists of the CHARMm receptor–ligand interaction energy (H-bonds, van der Waals, and electrostatic interactions) and the internal ligand strain energy. BIOVIA Discovery Studio reports the negative of the CDOCKER energy score determined using the algorithm; thus, a higher score indicates poses that are more favorable to binding. Many of the interactions expected from analysis of other GT structures were observed in this model. Apart from van der Waals interactions between UDPGA and nearby residues, the majority of interactions were either classical or nonclassical hydrogen bonds (Figure 3). The commonly observed interactions between the pyrimidine group, W353 (hydrophobic π–π stacking interaction), and L354 (hydrogen bonding) were recapitulated in our model. Classical hydrogen bonding interactions were also seen between the pyrimidine and N357, Q356, and R335, with nonclassical hydrogen bonds to E379 and P355. A hydrophobic π–alkyl interaction was also present between K281 and the pyrimidine ring. Residues G276–S286 are located in the flexible linker joining the N- and C-terminal domains which could allow for variability in the residues available to interact with the cosubstrate, depending on the conformation of the enzyme at the time of crystallization or modeling. Although the expected Q356 interaction with the ribose group was seen, the expected interaction with E379 was not observed, the hydroxyl groups of the ribose being rotated away from the glutamate residue in this pose. As expected, hydrogen bond interactions between G376, H375, G373, H371, S308, G307, R172, S37, and the α and/or β phosphate groups were seen. An electrostatic interaction between R256 and α phosphate, not seen in other GT structures, was also observed. The conserved Q396 interaction with the hexose group was observed, but no direct interaction was observed between this group and S374. Hydrogen bond interactions between the hexose and S308 and R172 were present, as was an electrostatic interaction with F393. Although D395 and the hexose group interacted in our model, it was as an unfavorable donor–donor interaction between the protein backbone and the protonated glucuronic acid backbone. As this docking algorithm is not able to account for receptor flexibility, or for changes in the local micro-environment, it is possible that in vivo interactions are slightly different, with additional water bonding and side-chain flexibility allowing for more favorable interactions.
Figure 3.
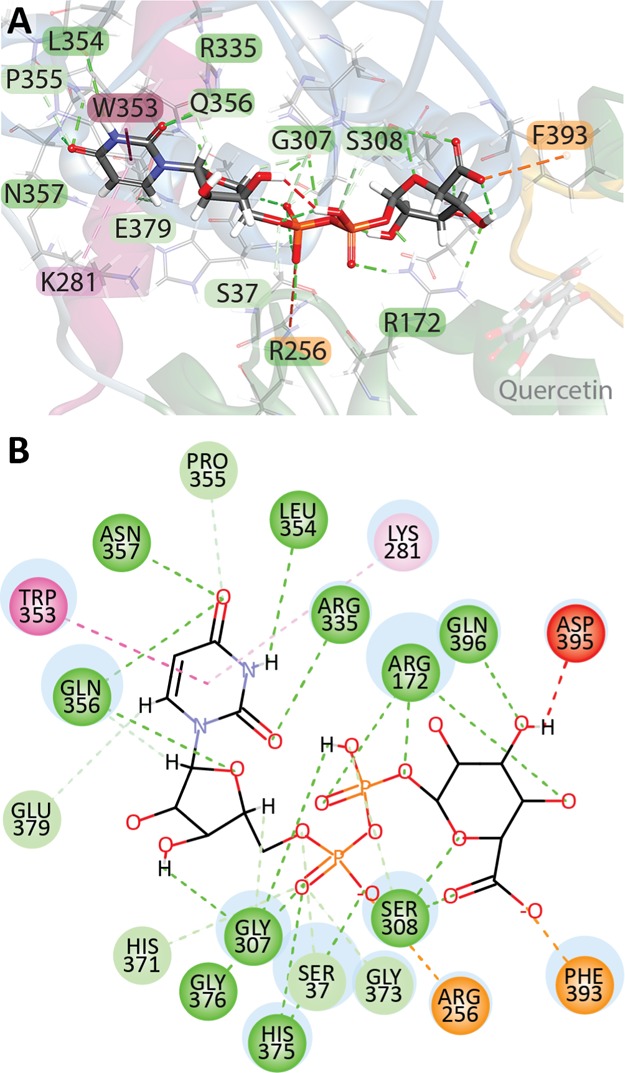
UDPGA interacts with multiple residues in the C-terminal domain as well as three residues in the N-terminal domain (S37, R172, and R256). (A) UDPGA docked into the cleft located between the C-terminal (blue ribbons) and N-terminal domains with several interacting residues indicated. (B) 2D schematic diagram of all non-van der Waals UDPGA–UGT1A6 interactions. Classical hydrogen bonds are shown in dark green, nonclassical hydrogen bonds, in which the donor is a polarized carbon atom, are shown in pale green, electrostatic interactions are shown in orange, hydrophobic interactions in pink, with π–π stacking interactions in dark pink and π–alkyl interactions in light pink, and unfavorable donor–donor interactions are shown in red.
In silico mutagenesis was performed to eliminate a number of amino acids that were identified as important for binding or may be of physiological importance (Table S5). When residues S37, G376, or W353 were changed to the nonpolar alanine, normal UDPGA docking was abolished, with the majority of poses having UDPGA bound in the reverse orientation, with the UDP group closest to the substrate binding site, and rmsds greater than 10 Å compared to the pose in the wild-type protein (Figure S6). Mutation of residues H38, R172, S308, and H375 also negatively affected UDPGA binding, resulting in nonstandard ligand conformations. Mutation of residues D150, T181, R184, L354, or F393 had no effect on UDPGA binding, with similar interactions between the enzyme and cosubstrate seen in the wild-type enzyme, and rmsds of <0.4 Å.
Docking the cosubstrate UDPGA into our structure identified many of the same interactions in the C-terminal domain that have been previously identified as being crucial for sugar binding in other GTs and UGT homology models. Interaction with these residues, many of which are highly conserved and previously identified as being required for glucuronidation, such as H371 and E379, indicated that our model was a reasonable approximation of the UGT structure. In silico mutagenesis of multiple binding residues confirmed the importance of these amino acids for UDPGA binding, with normal binding being abolished or negatively impacted when these residues were lost. Accurately modeling the N-terminal region of UGTs is far more difficult because of higher variability in both the sequence and substrate specificity.
Ligands were docked into the model in the presence of the cosubstrate UDPGA using CDOCKER. All compounds, with the exception of bilirubin diglucuronide, one of our negative controls, were able dock into the model. Bilirubin is metabolized exclusively by UGT1A1, and the fully glucuronidated diglucuronide is produced by a UGT1A1 homotetramer and is highly unlikely to be bound by UGT1A6. The top five docked ligands quercetin, kaempferol, apigenin, prunetin, and galangin, were all flavonoids (Figure 4, Table S6). It is known that the scores provided by docking algorithms are affected by the ligand size and flexibility, with larger molecules often receiving higher scores, although the exact degree of effect varies between different docking algorithms.41−43 We compared the -CDOCKER energy scores obtained with the compound molecular weight, molecular volume, number of rotatable bonds, number of hydrogen donors and acceptors, total number of atoms, and the number of O and N atoms present (Table S7). When all compounds were evaluated, as the -CDOCKER energy score increased, there was a negative correlation with the total number of atoms and a slight negative correlation with molecular weight and volume. When restricted to just those compounds that can be metabolized using UGT1A6 (27 compounds, Table S6), there is a slight negative correlation between -CDOCKER energy, molecular volume, total number of atoms, the number of rotatable bonds, as well as a slight positive correlation with the number of H acceptors. If only those compounds with known Km values are examined (13 compounds), only a slight negative correlation between -CDOCKER energy and both total number of atoms and molecular volume remains.
Figure 4.

Top scoring compounds were all small flavonoids with a limited number of direct interactions. (A) 3D view of quercetin docked into the receptor (-CDOCKER energy: 73.3885 kcal mol–1), (B) kaempferol (72.648 kcal mol–1), (C) apigenin (64.9334 kcal mol–1), (D) prunetin (61.2897 kcal mol–1), (E) galangin (60.0664 kcal mol–1). Interaction colors are as shown in Figure 3.
Several residues frequently interacted favorably with the bound substrate. In 96% of compounds, K228 interacted with the substrate predominantly through ionic or hydrogen-bond interactions, with cis-resveratrol and codeine being the only compounds that did not interact with this residue. Other residues commonly involved in substrate binding included R172 (58% of compounds), R184 (53%), F393 (22%), and H180 (20%). Approximately, 46% of compounds also formed direct interactions with the co-substrate UDPGA.
To determine if the obtained poses resembled those seen in actual GTs, the substrate-GT interactions in several available GT crystal structures were examined for quercetin (PDB: 2C9Z), kaempferol (PDB: 2C1Z, 4REL), and 2,4,5-trichlorophenol (TCP) (PDB: 2VCE). All three compounds were docked in the same general area; however, the orientation of the substrate and the interacting residues were quite different (Figure 5).
Figure 5.

Comparison of three docked substrates and the same substrates cocrystallized in plant GTs. Residue-substrate interactions are colored as shown in Figure 3. (A) Quercetin (QUE) docked with UDPGA in UGT1A6 (B) Quercetin cocrystallized with UDP in the V. vinifera flavonoid 3-O glucosyltransferase UFGT (PDB: 2C9Z). (C) Chemical structure of quercetin/kaempferol with common glucuronidation sites indicated with arrows. The circled 3′-OH group is absent in kaempferol. (D) Kaempferol (KMP) docked into UGT1A6 with UDPGA. (E) Kaempferol cocrystallized with U2F in the V. vinifera UFGT (PDB: 2C1Z). F. Kaempferol crystallized in the Clitoria ternatea anthocyanidin GT (PDB: 4REL) in the absence of a cosubstrate. (G) TCP docked in UDPGA. (H) TCP cocrystallized with U2F in the Arabidopsis thaliana hydroquinone glucosyltransferase UGT72B1 (PDB: 2VCE). (I) Chemical structure of TCP with the glucuronidation site indicated with an arrow.
Although some of the docked compounds had hydroxyl groups in close proximity to both the UDPGA and H38 residues to make glucuronidation possible, the docked compounds had greater distances between their hydroxyl groups and catalytic H38 (average 8.97 ± 2.63 Å, n = 27) than was observed in the plant GT crystal structures, in which the closest hydroxyl group was an average of 3.38 ± 1.24 Å (n = 7, Table S8). Similarly, the distance between the closest hydroxyl and C1′ of glucuronic acid was 9.64 ± 1.63 Å (n = 27) in the docked compounds but only 4.02 ± 0.34 Å in the two plant structures that were crystalized with both a substrate and cosubstrate (UDP-glucose) or cosubstrate analogue (U2F). In 2C9Z, which has UDP bound in the cosubstrate binding region, all interactions were either conventional (S18, Q84, H150, and Q188) or nonclassical (H20 and S146) hydrogen bonds, whereas in our model, quercetin docked in a different orientation with an rmsd of 7.2869 Å and primarily used charged interactions between itself and R172, H180, R184, and K228, which are not the equivalent residues to 2C9Z when the structures are aligned (Table S9). The distances between the Nε2 of the catalytic H38 residue and the closest normally glucuronidated OH group of quercetin also differed, being 5.046 Å in our model but only 3.387 Å from the equivalent H20 residue in 2C9Z. Similarly, our docked kaempferol was bound in a substantially different orientation compared to 2C1Z (rmsd 7.265 Å) but only in a moderately different position compared to 4REL (rmsd 3.888 Å). In both 2C1Z and 4REL, most binding interactions were either π–π or conventional hydrogen bonds, whereas in our model, only charged interactions were seen. Distances between the atoms involved in the glucuronidation reaction were again larger in our model compared to either 2C1Z or 4REL, with 7.455 Å between the C1′ of the glucuronic acid group and 3-OH of kaempferol in our model, but only 4.263 Å in 2C1Z, and 4.588 Å between the 3-OH and Nε2 of H38 in UGT1A6 compared with 2.696 and 2.492 Å in 2C1Z and 4REL, respectively. Although UGT1A6 is able to glucuronidate the 3-OH position of kaempferol, the 7-OH position is preferentially glucuronidated in humans, indicating that the binding pocket in our model may more closely resemble the plant templates than the human enzyme.44 Comparison of TCP binding showed only a single structurally overlapping interaction between UGT1A6 and 2VCE, a π–π interaction between F393 (A387 in 2VCE) and the TCP ring. There were a total of 10 π–π or π–alkyl bonds in 2VCE, as well as a conventional hydrogen bond to the cocrystallized U2F. Comparatively, in UGT1A6, TCP (rmsd 3.989 Å), exhibited only a single π–π (F393), π–alkyl bond (L225), and electrostatic interaction (K228). The distance from the C1′ and 1-OH was 9.557 Å in UGT1A6 and 3.779 Å in 2VCE and 8.552 Å between the 1-OH and Nε2 of the catalytic H38 in UGT1A6, and 2.324 Å in 2VCE.
In silico mutagenesis was performed on commonly interacting, or otherwise important, residues to examine their importance in substrate binding. Specifically, H38, D150, R172A, H180A, T181A, R184A, R184S, K228A, and F393A were produced. None of the mutations made had a major effect on which compounds had the highest docking scores (Table S10). Quercetin, kaempferol, and apigenin were always in the top three bound compounds, with galangin or prunetin generally as either the fourth or fifth. This varied only in the R172A and K228A mutations where the fifth best compound was genistein (ranked eighth in the wild-type docking) and in the R184A and R184S mutations where dopamine and norepinephrine had the fourth and fifth best docking scores, respectively. Similarly, although there were a number of alterations in the number and type of interactions with the surrounding residues, the majority of interactions with each compound were the same as were seen when docking into the nonmutated model. The R172A and K228A mutants showed the largest change in the compound binding location within the substrate binding pocket with the top five docked compounds all having rmsds > 2 Å compared to the WT docked poses (Figure S7). Mutation of R172 caused the appearance of unfavorable anion–anion interactions between UDPGA and both quercetin and kaempferol, with quercetin changing its binding position such that the 3′-OH was closer to the glucuronic acid C1′ than the 3-OH, as seen in the wild-type structure. Quercetin is not a preferred substrate of UGT1A6, only 3% being glucuronidated, and is preferentially glucuronidated in the order 4′-OH (32%) > 7-OH (30%) > 3′-OH (22%) > 3-OH (16%).45 Despite this, neither the 4′-OH nor 7-OH positions were ever in the closest position to the catalytic H38 or glucuronic acid C1′; instead, the 3′-OH or 3-OH tended to have the closest proximity in the wild-type and all mutants. Although the identity of the residues interacting with apigenin did not change, its binding position altered such that the 7-OH was closer to the glucuronic acid. Genistein also moved closer to the glucuronic acid group and catalytic H38, as well as showing a substantial change in the identity of interacting residues and the type of interactions seen, with more π–π and π–alkyl interactions observed in the mutant. The K228A mutation also resulted in the appearance of unfavorable anion–anion interactions between UDPGA and quercetin, apigenin, and genistein. Kaempferol was docked in a substantially different position such that interactions with UDPGA were not possible (Figure S7). Substrates docked into our model generally showed plausible interactions and conformations; however, when compared to the plant GT templates, the docked substrates were bound further away from the catalytically important H38 residue and UDPGA, which would negatively affect glucuronidation. In silico mutagenesis of nine residues identified as potentially being important for substrate binding had a relatively little effect on substrate binding, and the loss of one interacting residue was not sufficient to completely disrupt binding, the other interactions still allowing binding in a similar position.
UGT1A6 is subject to several genetic polymorphisms, and examination of the T181A and R184 mutations present in the UGT1A6*2 (S7A, T181A, R184A), UGT1A6*4 (S7A, R184A), and UGT1A6*5 (T181A) genetic polymorphisms revealed a minimal effect on substrate binding. The S7 residue, located in the ER targeting sequence, is not present in our model, but examination of both T181A and R184S showed only mild effects on the binding of the top compounds (Table S10). We specifically examined the binding of 4-nitrophenol, serotonin, and acetaminophen, as the T181A and R184S mutations are known to affect the metabolism of these compounds in recombinant cell lines, with R184S lowering the Km and T181A increasing the Vmax, however, only when both S7A and T181A mutations were present in the gene.46 Although these mutations are known to affect glucuronidation in combination, on their own, they have little effect, with only serotonin moving closer to H38 and the glucuronic acid group in the T181A mutation (Figure S8). It has been hypothesized that T181 may act to enhance catalysis via active site interactions, but not directly influencing substrate affinity, whereas R184 may play a role in defining substrate specificity.46 This matches our modeling data which showed relatively minor effects on the binding of any substrates. The loss of R172 and K228 had the greatest effect on substrate binding, but their loss was not necessarily negative. Apigenin, prunetin, and genistein were bound in more favorable positions for glucuronidation in the R172 mutant, and both quercetin and galangin were bound in positions closer to UDPGA and the catalytic H38 residue in the K228 mutant. This may indicate that protein flexibility is important in substrate binding. The docking algorithm held both the sidechains and main chain protein backbone relatively inflexible during binding, mimicking a lock and key model of substrate binding. However, many drug-metabolizing enzymes, such as the cytochromes P450 and the sulfotransferases, show a great deal of flexibility in their protein structures, allowing for the metabolism of a diverse range of substrates.4 It is well-known that the binding of substrate triggers a conformational change in many enzymes, and this is also the case in other human GTs, where binding of the co-substrate prompts an induced fit conformational change in the enzyme structure.47−49 In GT-A type inverting GTs, such as β4Gal-T1, GnT I, GlcAT-I, and GlcAT-P, the binding of a divalent metal cation and the sugar donor co-substrate prompts restructuring of a nearby flexible loop region that acts to cover the buried donor, creating a region that is more favorable for binding the acceptor substrate.49 Despite the high level of structural conservation among the GT superfamily, this particular loop is not present in the GT-B type UGT2B7 or plant GT template structures, but other flexible linker regions are present. It is reasonable to postulate that some of these regions of UGTs may undergo a conformational change upon cosubstrate binding, allowing increased solvent accessibility to the substrate binding pocket and allowing for the formation of a more optimized substrate binding site. Structural analyses of various plant GTs indicate that the flexible loops in both the N- and C-terminal regions, as well as the loop linking the two domains may also play a role in the binding of both the sugar donor and acceptor molecules.50 It is possible that the lack of a bound cosubstrate in the partial human UGT2B7 crystal structure used as a guide during model creation could have adversely affected the substrate binding site in our model, but both other crystal structure templates had UDP bound in the cosubstrate binding site, potentially mitigating this issue. Although it is possible that binding of the substrate may induce further conformational changes in the enzyme, because of the relatively solvent accessible location of the substrate binding site, and analyses of structurally similar plant GTs, it is unlikely that any major conformational changes would be required to allow for substrate binding.49,51 It is possible that additional allosteric interactions between UGT1A6 and other compounds may also affect substrate binding. Calmodulin has been shown to allosterically enhance the glycosylation activity of the woodland strawberry Fragaria vesca FvUGT1 toward its major substrate, the anthocyanin pelargonidin, and a number of endogenous, and exogenous compounds have been shown to allosterically affect mammalian UGT metabolism.52,53 Additionally, UGT homo- and hetero-oligomers have been shown to change substrate specificity and enzyme kinetics, but the mechanisms by which these effects are accomplished is still unclear, and it is possible that allosteric interactions between UGTs may play a role in substrate binding and metabolism.
Poses with the highest -CDOCKER energy scores were compared with available enzyme kinetic parameters for these compounds. Previously reported kinetic data for 13 compounds is shown in Table S6, and the average value for each compound (μM) was compared to the CDOCKER scores. No correlation was observed between binding scores and enzyme kinetic data (Figure 6).
Figure 6.

Comparison of the average Km equivalent (μM) with the -CDOCKER score for 13 compounds with available enzyme kinetics data. There was no correlation between the enzyme kinetics data and -CDOCKER energy scores for these compounds, 4-MU, 4-methylumbelliferone.
Although it is tempting to seek a correlation between binding affinity and substrate docking scores, there is no guarantee that binding affinity is an accurate measure of glucuronidation activity. As has been seen in other studies, Km values do not necessarily correlate well with Vmax or Clint values, and there is little reason to believe that docking scores would necessarily correlate any better.54 The UGT-substrate interaction is complex, with many compounds having multiple sites of glucuronidation with different affinities, as well as altered affinities and kinetics based on whether the UGT has formed a homo- or hetero-oligomer. There are multiple viable poses in which a compound can bind the active site, this being how compounds such as quercetin, kaempferol, or cis-resveratrol can have glucuronide added to multiple possible hydroxyl groups, the hydroxyl closest to the catalytic site being glucuronidated. A particular hydroxyl group is usually preferred over the others, likely due to optimal binding interactions, but the other sites may be used depending on the local micro-environment, protein conformation, post-translational modifications, oligomerization, and cosubstrate binding status at the time of substrate binding.
The compounds with the highest docking scores were all flavonoids, and although it is possible for UGT1A6 to metabolize certain flavonoids, they are not the preferred substrate. Typically, UGTs 1A1, 1A3, 1A7, 1A8, 1A9, 1A10, and 2B7 are the major contributors to flavone glucuronidation in humans.12 Scoring functions are typically biased toward molecules with higher molecular weights generally because of the fact that they offer a larger surface area for protein interaction and increased opportunities for hydrogen bonding.41,55 We did not see a correlation between increased molecular weight and higher docking scores, rather there was a slight correlation between smaller ligands and increased docking scores, and the accuracy of the CDOCKER algorithm is minimally affected by molecular weight and ligand flexibility.43 The preference towards smaller ligands makes sense physiologically, as it has been hypothesized that the UGT1A6 substrate binding pocket may be smaller and more closed than in plant and bacterial UGTs because of its preference for metabolizing relatively small molecules such as simple phenols.4
Our homology model was principally derived from two plant GTs, possibly explaining the high docking scores for plant UGT substrates. Quercetin is the preferred substrate of the M. truncatula triterpene/flavonoid GT UGT71G1, which glycosylates the 3′-OH preferentially, followed by 3-OH, and 4′-OH, with the 7-OH and 5-OH positions also glycosylated to a small extent.56 The other template, the O. sativa trichothecene GT OsUGT79 preferentially glycosylates trichothecene mycotoxins such as deoxynivalenol, and has no activity toward flavonoids such as quercetin.57,58 Despite the high degree of structural conservation, particularly around the active site, small changes in the amino acid composition result in substantially different substrate specificities and the regioselectivity of the glycosylation site. It seems likely that if other GT structures were chosen as initial templates, the resulting models would be biased toward resembling those enzymes, with similar preferred substrates.
Unfortunately, no complete mammalian GT crystal structures are available, with only plant and bacterial GTs crystalized to date. The bias toward resembling the template structure is not unique to our model. Docking the 56 prepared compounds into the Ghemtio et al. model (2014), which was produced using five plant GT templates, gave similar results to our model, with all of the top 5 docked compounds being flavonoids, despite their poor metabolism by UGT1A6 (Table S11). As in our model, bilirubin diglucuronide failed to dock, as did biochannin A, oleandomycin, irinotecan, and bilirubin monoglucuronide, all compounds that are not metabolized by UGT1A6. The residues that interacted with the docked compounds differed substantially between the two models, as did the distances between the catalytic H38 residue and the C1′ of the cosubstrate. This was not entirely unexpected as the N-terminal domains of the two models differed substantially (Figure S5). Using plant GTs as the primary templates for the structure, the UGT1A6 models may have been forced into a non-native conformation, increasing the size and shape of the binding pocket and allowing for preferred docking of larger substrates, thus causing the docking position of small phenolic molecules to be less favorable and farther away from the catalytic site. This may also explain the relatively minor effects of mutations in the substrate binding site on substrate binding. This is a concern for any UGT homology models that might be used for drug discovery or substrate specificity studies. Results more likely reflect the plant UGT templates than the actual human UGTs when using homology models, leading to the failure of in silico-led drug discovery studies because of inherent bias. Only the future production and description of a true mammalian UGT structure can substantially move structure-based design and modeling efforts forward in this field.
Molecular Dynamics Simulations
The aim of this study was to analyze the importance of residues in the NTD of UGT1A6 for substrate specificity. In order to better understand the UGT1A6 structure and ligand interactions, it is important to consider not only the static structure but also the dynamic state and internal motions that could occur under physiological conditions. To examine the role of substrate binding residues, the best docked pose of quercetin, the top scoring compound, in the UGT1A6 model was subjected to molecular dynamics simulations for an in-depth binding interaction study, with the stability of the binding conformation in the dynamic state analyzed. The rmsd of the protein backbone, bound cosubstrate UDPGA, and bound substrate were plotted and analyzed with respect to the initial model (Figure 7). The model backbone showed a gradual increase in rmsd over the run time, with the rmsd between the initial and final structure after 50 ns being 5.795 Å (Table S12). The largest and most frequent changes in rmsd from the average model over time (root-mean-square fluctuation, RMSF) were in the N-terminal regions, primarily in the solvent accessible regions of the protein, with some short C-terminal regions also showing some fluctuations (Figure S9). UDPGA fluctuated within its binding pocket, with 2 Å fluctuations occurring between 4 and 12 ns, stabilizing at about 18 ns but fluctuating further between 33 and 50 ns. Quercetin showed smaller fluctuations, appearing to stabilize within a range of 1 Å after 17 ns.
Figure 7.
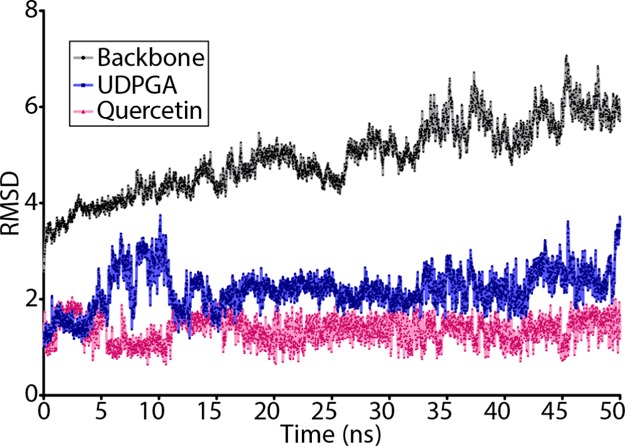
rmsd of the UGT1A6 model and bound cosubstrate and substrate over the course of the molecular dynamics simulation. The UGT1A6 protein backbone rmsd is shown in black, UPDGA in blue, and quercetin in pink.
To assess the secondary structure’s stability, the simulation trajectory was examined every 5 ns from 0 to 50 ns, showing that the overall structure appeared to spread out compared to the initial state (Figure S10). The majority of the CTD remained relatively stable during the simulation run, with an rmsd of 4.515 Å from the initial model. The loop linking the NTD and CTD moved substantially over the run, as did the Cα0 helix, the linker having an rmsd of 8.327 Å by the end of the run. The Cα0 helix had a large rmsd of 8.225 Å at the starting Q287 residue, but by the final residue, G297, it differed by only 2.328 Å. The Cβ2–Cα3 region also differs after 50 ns, the location of the Cβ3 sheet and Cα3 helix altered in the 50 ns model, with Cα3 shifting upward and increasing in length by two amino acids (Q356–H362) in the final structure (Figure S11). This shift altered the positions of several key UDPGA binding residues, including W353 (rmsd 9.927 Å), affecting UDPGA binding. UDPGA moved 6.895 Å relative to its initial position, preventing the formation of the highly conserved π–π stacking interaction between W353 and the uridine group of UDPGA (Figure S12). The loss of this interaction is part of a substantial shift in the positioning of the UDPGA molecule, contributing to the further decrease in protein–UDPGA interactions. Because of the large number of other changes in the protein structure, and interacting residues, it is difficult to determine the significance of the loss of this specific interaction, although given its conserved nature, the loss is likely detrimental. The uridine ring previously had 8 interactions with the UGT1A6 protein, but after simulation, only 3 interactions to T337, R335, and Y336 remained, plus an additional three water–hydrogen bonds. Following the simulation, the previously unfavorable D395 interaction with the glucuronic acid disappeared, leaving seven water–hydrogen interactions and a hydrogen bond/electrostatic interaction with R172 as the only interaction between the protein and the UDPGA glucuronic acid group (Table S13).
The NTD exhibited significant structural deviation from the initial model by the end of the simulation (Figure S13). The Nβ2–Nβ3 region differed by 6.132 Å, causing a deviation in the location of Nβ2, the Nα2 loop, and Nβ3. The Nα3 helices also differed substantially by the end of the simulation, Na3-1 was shortened by four amino acids (N96–E101 compared to Y91–E101 initially; rmsd 8.936 Å). The Nα3-2 helix had the highest deviation by the end of the simulation (rmsd 9.647 Å), mostly because of being lengthened by three amino acids and being shifted down compared to the initial structure. The Nα3-3 helix was essentially missing in the initial structure, but by the end of the simulation, a clear helix had formed immediately following the Nα3-2 helix (rmsd 6.041 Å). The region surrounding the Nα5-2 helix, which includes the dimerization domain, also showed a high level of variation (rmsd 9.903 Å). The starting model had an initial helix (R194–K198) followed by a loop connecting to a second helical turn (S206–A210), but all helical structures were abolished by the end of the 50 ns of simulation. The Nα5-3 helix (E218–V236) following the simulation was longer than in the initial model (E218–E230), but the model converged at the Nα5-4 helix (rmsd 1.951 Å). Although the externally facing regions showed a high level of variance, the internal Nβ4–Nβ5 region (F144–R172) showed relatively little deviation within the structure (rmsd 1.691 Å), despite the lengthening of the Nβ4 sheet (F144–D150) in the final model compared to the initial structure (L147–D150).
The position of the docked quercetin molecule shifted by 6.895 Å relative to its initial position, causing a shift in the interacting residues (Figure S14 and Table S14). By the end of the simulation, none of the original interactions had been preserved from the initial state, three different residue interactions, and 10 water–hydrogen interactions appearing because of the shift in the quercetin position within the substrate binding pocket. The distances between the potentially glucuronidated hydroxyls of quercetin and both the catalytic Nε2 residue of H38, and the C1′ of the glucuronic acid group, increased over the course of the simulation (Figure S15). The distance between quercetin and the glucuronic acid C1′ was particularly large, ranging from 16.670 to 22.626 Å by the end of the simulation compared to the initial measurements of 7.513–11.743 Å (Table S15).
Molecular dynamics simulations revealed a large amount of flexibility in the UGT1A6 protein backbone, as well as in the bound cosubstrate and substrate. This flexibility was seen in both the NTD and CTD and affected the interactions of both UDPGA and quercetin with the protein. A high degree of flexibility in substrate binding is physiologically consistent, as UGT1A6 can produce multiple glucuronidated forms of quercetin, with specific hydroxyl groups being preferred over others.45 In order for these various products to be formed, it is necessary for the substrate to be able to bind the enzyme in a variety of different orientations, moving the target hydroxyl group closest to the catalytic H38 residue and the UDPGA sugar group. Unlike the cytosolic plant GTs, the human UGT1A6 is intimately associated with the ER membrane.40 It is likely that this association confers some degree of additional stability or constraint to the protein structure, and simulating protein movement in the absence of this influence may not accurately reflect physiologically relevant conformational changes. Some of the observed shifts in protein helices, sheets, and loops may not occur when the protein is anchored in the membrane, and the highly conserved CTD would probably show a smaller degree of conformational change. Protein–protein interactions in UGT homo- and hetero-oligomers may also provide additional protein stability, but it is currently infeasible to model these interactions in silico because of the localization of the dimerization motif within a variable loop region, which is inherently disordered and substantially more difficult to model compared to more ordered α-helix or β-sheet secondary structures. The majority of protein–protein docking algorithms available are not optimized for detecting associations between disordered loop regions, and elucidation of these interactions will need to await the production of a complete crystal structure. Because of the high level of structural conservation between GTs in the C-terminal sugar binding domain, one might expect to see fewer shifts in protein interactions and helix positions than were observed following the molecular dynamics simulations. The high degree of flexibility observed in the protein structure does lend support to the idea that the protein may undergo a conformational shift upon cosubstrate and/or substrate binding, as it is at least able to make a number of energetically favorable conformational changes. The lack of true system stabilization following 50 ns of simulation may be due to the absence of appropriate membrane or protein–protein interactions. Although it is possible that the system would stabilize with additional simulation time, any increased biological relevance of the resulting structure would be questionable. This is because as simulations progressed, the derived structure increasingly deviated, particularly in the highly conserved C-terminal domain, which differed from the UGT2B7 crystal structure by 4.672 Å (Table S12). Similarly, most conserved interactions between the cosubstrate and protein were abolished with extended simulation time, and the substrate and cosubstrate became so far apart as to make transfer of the glucuronide (the biological imperative of this enzyme) impossible.
Post-translational modifications may also contribute to protein flexibility, substrate binding, and metabolism. Multiple UGTs have been shown to be phosphorylated by protein kinase C (PKC) and inhibition of PKC resulting in significant loss of activity.59 UGT1A6 is predicted to have four PKC sites (S/TXK/R) at S41, S50, T74, and S206, as well as one tyrosine phosphorylation site at Y191, with PKC-δ specifically implicated in UGT1A6 phosphorylation.60 The mechanism by which phosphorylation affects protein function is currently unclear, although it has been suggested that it may stabilize the protein, slowing protein degradation, and affect both catalysis and substrate specificity.59,60 Glycosylation plays a role in UGT folding and has been shown to affect enzyme activity.61,62 Although UGT1A6 possesses two glycosylation sites at N294 and N346, it is unclear how glycosylation specifically affects the UGT function. Both phosphorylation and glycosylation may play important roles in protein stability and function, but it is not possible to account for their effects in current homology models, which likely affects the results obtained.
Conclusions
Because of extensive functional and structural conservation within the GT superfamily, homology modeling may provide insight into the structure and function of UGTs in the absence of high-resolution crystal structures. We have produced a UGT1A6 model that closely resembles the crystal structures of other plant, bacterial, and fungal GTs, which is able to bind the cosubstrate UDPGA as well as many compounds known to be metabolized by UGT1A6, with the majority of expected interactions represented. This, and other UGT homology models, have a number of limitations. Because of the small number of available GT templates, all UGT homology models are based on the same pool of plant and bacterial GTs, and our data indicate that this biases the model toward closely resembling the template structures, particularly in their substrate specificities, which makes interpretation with respect to human and other mammalian systems more difficult. The limitations of currently available homology modeling software, and biological information, make modeling ER membrane constraints difficult at best, so the inherent instability of loops and disordered regions of the protein cannot be accurately predicted. Post-translational modifications, and protein–protein interactions (for which there is increasing evidence in the literature18−29,59−63) also cannot be accurately predicted nor their effects on substrate binding quantified. Despite these limitations, our model was able to recapitulate many of the expected UGT–substrate and UGT–cosubstrate interactions, highlighting a number of residues that may be critical for binding. Because of the dynamic nature of the protein, it is expected that many of the sidechains, loops, helices, and sheets can move to allow for better access and binding of both co-substrates and substrates, as well as to allow for homo- and hetero-oligomerization. This was supported by the high level of protein flexibility observed in our molecular dynamics simulations. Elucidating how these regions and interactions affect substrate binding and enzyme kinetics is a difficult problem, and will likely not be solved even with the elucidation of the UGT1A6 crystal structure, as crystals cannot capture the dynamic movement of the protein. It is possible that with multiple crystal structures produced with bound substrates, we will be able to obtain a more clear picture of how the loops and helices move to accommodate different substrates, although careful biochemical experiments will also be necessary to elucidate dynamic processes. With these data, and advances in structural modeling and docking algorithms, it may be possible to construct new models and molecular dynamics simulations to better understand the parameters informing enzyme specificity and kinetics.
Computational Methods
In Silico Modeling of UGT1A6
Homology models of the human UGT1A6 protein (GenBank accession #: NP_001063.2, Uniprot: P19224) were produced using the I-TASSER server.64−66 UGT1A6 possesses a 26-amino acid N-terminal signal peptide which acts to target the immature protein to the ER.40 Once inserted into the ER, this signal sequence is removed to form the mature protein, and it is thus unlikely to contribute to the function of the mature protein. We produced four initial models: with and without this targeting sequence, using the human UGT2B7 C-terminal end crystal structure (PDB: 2O6L) as a template to guide the I-TASSER modeling (WSNT); with the signal peptide and with the UGT2B7 crystal template (WSWT); without the signal peptide and without the UGT2B7 template (NSNT); and without the signal peptide and with the UGT2B7 template (NSWT).34
I-TASSER begins modeling of the submitted sequence by automatically retrieving templates with similar folds or secondary structures from the PDB database using LOMETS (Table S16).67 It then assembles continuous fragments taken from the PDB templates into full-length models using Monte Carlo simulations with unaligned regions built using ab initio modeling. The clustering algorithm SPICKER is then used to identify low free-energy states, and then, the fragment assembly simulation is repeated using the SPICKER cluster results with spatial restraints obtained from LOMETS and the template PDB structures.68 The final full-atomic models are built from selected I-TASSER decoys via optimization of the hydrogen bond network. Models produced without the use of UGT2B7 were constructed using four crystal templates: 5GL5 (Saccharomyces cerevisiae ugt51), 2IYA (Streptomyces antibioticus OleI), 2IYF (S. antibioticus OleD), and 2PQ6 (M. truncatula UGT85H2). The WSNT model also used 4M83 (an alternative model of the data from 2IYF). The models produced using UGT2B7 as a guide were primarily based on 2ACV (M. truncatula UGT71G1) and 5TME (O. sativa subsp. Japonica UGT79) pdb structures.
The I-TASSER server automatically constructed five models for each set of input conditions, and the top 10 structural analogues for each set of models were determined (Table S17). Structural alignments with other GT crystal structures available in the RCSB database were produced to evaluate differences between the homology models and available crystal structures (Table S18).69 The first model for each input condition, which generally corresponded to the structure with the highest C-score and largest number of decoys made, was imported into BIOVIA Discovery Studio 4.5 (Dassault Systèmes, Courbevoie, France) for continued analysis. The models for all four input conditions were assessed using the tools Verify Protein (MODELER) to generate DOPE scores, and Verify Protein (Profiles-3D) to evaluate the compatibility of an amino acid sequence within a 3D protein structure.
The model with the best scores (no signal peptide, using the UGT2B7 guide structure) was selected to undergo further energetic minimizations using CHARMm.70 Two rounds of smart minimization (1000 steps of steepest descent with a root mean square (rms) gradient tolerance of 3, followed by conjugate gradient minimization) were performed followed by further two rounds of adopted basis Newton–Raphson minimization. The model then underwent side-chain refinement using the ChiRotor algorithm for CHARMm energy minimization.71 CHARMm-based loop refinement was performed on loop regions not present in UGT2B7 that had substantial structural variation compared with other aligned GTs, specifically amino acids F182–L217 and G275–S286; F182–L217 refinement was split into four groups for optimization because of algorithm constraints on the loop length: F182–D201, T197–A210, R208–P219, and M203–L217.72 Loop refinement was followed by a final round of side-chain optimization and smart minimization. All minimizations were performed using a generalized Born with molecular volume implicit solvent model. The side-chain rotamer for W353 was manually adjusted to match the rotation of the homologous tryptophan residue present in multiple crystal structures cocrystalized with UDP, UDP-glucose, U2F, or TDP, as the specific ring–ring interactions are important in UDPGA binding.36 The final model was evaluated using the SAVES server (http://servicesn.mbi.ucla.edu/SAVES/) to determine the ERRAT score, with BIOVIA used to generate a Ramachandran plot.73 Structural alignments with other GT crystal structures available in the RCSB database were produced to evaluate differences between the homology models and available crystal structures, and to identify important residues.69
In Silico Mutagenesis
To examine the importance of specific residues to cosubstrate and substrate binding, selected residues were mutated from their initial composition to alanine, and the resulting mutant structure used for additional molecular docking studies. A total of 15 mutant structures were made, 6 of which were used to specifically investigate cosubstrate binding, 5 of which were used to examine substrate binding, and the remaining 4 used to examine binding of both (Table S19). Residues were selected based on observed cosubstrate and substrate binding in other GT crystal structures (Table S4), as well as those identified in our own docking experiments.
Molecular Docking
Structures of all ligands selected for docking were obtained from PubChem. A total of 56 compounds were selected for docking comprising 13 compounds known to be metabolized by UGT1A6 with available Km or S50 values, 14 compounds that are poorly metabolized by UGT1A6, 7 compounds whose ability to be metabolized by UGT1A6 are unknown, and 22 compounds that either cannot be metabolized by UGT1A6 or are metabolized at levels below the level of detection (Table S6). Of these, approximately half are substrates for plant UGTs, including quercetin and kaempferol, substrates bound in several available GT crystal structures. With the exception of three compounds (irinotecan, warfarin, and tamoxifen), which require additional processing before they are able to be glucuronidated, or bilirubin diglucuronide, which is already fully glucuronidated and cannot be further glucuronidated, all the other included compounds can be metabolized by UGT1A6, other human UGTs, plant GTs, or bacterial GTs (oleandomycin). All ligands were prepared for docking in BIOVIA Discovery Studio 4.5 and canonical tautomers generated. Cosubstrate and substrate docking spheres in the UGT1A6 homology model were defined using the locations of the bound co-substrate or substrate molecules present in GT crystal structures following alignment with our final UGT1A6 model. The UDPGA docking sphere was defined using the locations of bound UDP-2-deoxy-2-fluoro-α-d-glucose or UDP-glucose present in available GT crystal structures, and the substrate binding sphere was defined using the locations of several bound planar molecules (Table S8). The final cosubstrate docking sphere had a radius of 9.46 Å, whereas the substrate docking sphere had a 7.80 Å radius.
Both the cosubstrate UDPGA and the substrate ligands were docked into the wild-type and mutant UGT1A6 models using CDOCKER, a grid-based molecular docking method that allows for flexible ligands to be docked into a rigid protein receptor.74 The substrate ligands were only docked into the models in the presence of docked UDPGA. Ten random ligand conformations were generated for each compound by translating the ligand center within the receptor docking sphere and performing a series of 1000 random rotations. Each orientation was then subjected to 2000 rounds of simulated annealing molecular dynamics where the temperature is increased to 700 K followed by cooling to the target temperature of 300 K, ending with final minimization of the ligand within the rigid receptor.
The prepared substrate ligands were also docked into the UGT1A6 model produced by Ghemtio et al. (2014). This model contains U2F in the cosubstrate binding position, and the ligands were docked into this model using the same settings as described above, with the substrate binding sphere placed in the same location as in our UGT1A6 model.
Molecular Dynamics Simulations
Molecular dynamics simulations were performed on the enzyme complexed with both UDPGA and quercetin, the ligand with the highest docking score, to analyze the stability of binding conformation using BIOVIA Discovery Studio 4.5.
Terminal residues G469–H532 of the models corresponding to the C-terminal TM domain, which did not align with any of the template structures, were removed to reduce the computational burden, as they were not implicated in the cosubstrate or substrate docking and physiologically would not be available to interact with ligands because of their position within the ER membrane. Models and docked ligands were typed with a CHARMM36 forcefield, then solvated using an explicit periodic boundary with an orthorhombic cell shape and a minimum of 7 Å from the periodic boundary. The completely solvated system contained 37,373 atoms including water molecules (10,030 atoms), cosubstrate, ligand, receptor, and counter-ions (28 sodium, 27 chloride). Minimization of the protein–ligand complex was carried out in two steps: a maximum of 1000 steps of steepest descent with an rms gradient of 1.0, followed by a maximum of 2000 steps of adopted basis Newton–Raphson with a 0.1 rms gradient. The system was gradually heated from 50 to 310 K over 50 ps using 2 fs time steps, followed by equilibration for 20 ps at 310 K. The final production phase was performed using constant temperature dynamics using the Berendsen weak coupling method (NVT) at a temperature of 310 K for a total of 50 ns. The step size was kept at 2 fs, with trajectories saved every 10 ps. During the run, SHAKE constraints were applied to all bonds containing hydrogen atoms. The nonbond lower cut-off distance was set to 8 Å, the higher cut-off distance set to 12 Å, and the nonbond list radius set to 14 Å. Trajectory analysis was performed using the trajectory analysis tool in BIOVIA Discovery Studio.
Acknowledgments
This study was supported in part by the Natural Sciences and Engineering Research Council of Canada, grant 17-003808 to A.C.C.
Glossary
Abbreviations
- C-score
confidence score
- DOPE
discrete optimized protein energy
- ER
endoplasmic reticulum
- GT
glycosyltransferase
- I-TASSER
Iterative Threading ASSEmbly Refinement
- NSNT
no signal peptide and no guiding template
- NSWT
no signal peptide and with a guiding template
- PKC
protein kinase C
- TCP
2,4,5-trichlorophenol
- TDP
thymidine-5′-diphosphate
- U2F
uridine-5′-diphosphate-2-fluoro-α-d-glucose
- UDP
uridine-5′-diphosphate
- UDPGA
uridine-5′-diphosphate-glucuronic acid
- UGT
uridine-5′-diphosphate-glycosyltransferases
- WSNT
with signal peptide and no guiding template
- WSWT
with signal peptide and with a guiding template
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00205.
SMILES of all chemicals used in docking experiments (XLSX)
Cover sheet; substrate_docking; UGT1A6_UGT2B7_alignment; UGT1A6_all_alignments; UGT1A6_homology_model; alignments_for_threading; and I-TASSER aligned models (ZIP)
Five models produced by I-TASSER; Ramachandran plot; alignment of the final UGT1A6 model; structural alignments of the final UGT1A6 model; comparison of UDPGA top docking positions; comparison of the docking of selected substrates; comparison of the docking of three small planar phenolic compounds; RMSF for each residue; snapshots from the molecular dynamics simulation trajectory; shifts in the CTD; changes in the UDPGA–UGT1A6 interacting residues; shifts in the NTD; changes in the quercetin–UGT1A6 interacting residues; distances between the Nε2 group of the catalytic H38, the glucuronic acid C1′, and the potentially glucuronidated hydroxyl groups of the bound quercetin ligand; confidence scores for each model; DOPE and verify protein scores for I-TASSER-produced models; rmsd between UGT1A6 models and GT crystal structures; residues important for cosubstrate binding in multiple GT structures; UDPGA docking site mutagenesis; substrates, docking scores, and enzyme kinetic data; correlation of ligand properties with the -CDOCKER energy score; crystal structures; comparison of substrate binding residues; interactions with selected bound substrates; docking scores and residue distances; change in structures over the course of molecular dynamics simulations; change in UDPGA interacting residues over time; change in quercetin interacting residues over time; distances between potential quercetin glucuronidation sites and important enzyme and cosubstrate atoms over time; top 10 I-TASSER threading alignments; top 10 identified structural analogues; GT crystal structures; and mutant UGT1A6 structures (PDF)
Author Contributions
Study design: A.D.S. and M.W.H.C.; performed experiments/modeling: A.D.S.; data interpretation: A.D.S., B.D.G.P., A.C.C., and M.W.H.C.; wrote and/or edited manuscript: A.D.S., B.D.G.P., A.C.C., and M.W.H.C. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
Authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Oda S.; Fukami T.; Yokoi T.; Nakajima M. A Comprehensive Review of UDP-Glucuronosyltransferase and Esterases for Drug Development. Drug Metab. Pharmacokinet. 2015, 30, 30–51. 10.1016/j.dmpk.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Meech R.; Miners J. O.; Lewis B. C.; Mackenzie P. I. The Glycosidation of Xenobiotics and Endogenous Compounds: Versatility and Redundancy in the UDP Glycosyltransferase Superfamily. Pharmacol. Ther. 2012, 134, 200–218. 10.1016/j.pharmthera.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Evans W. E.; Relling M. V. Pharmacogenomics: Translating Functional Genomics into Rational Therapeutics. Science 1999, 286, 487–491. 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- Dong D.; Ako R.; Hu M.; Wu B. Understanding Substrate Selectivity of Human UDP-Glucuronosyltransferases through QSAR Modeling and Analysis of Homologous Enzymes.. Xenobiotica 2012, 42, 808–820. 10.3109/00498254.2012.663515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Coughtrie M. Revisiting the Latency of Uridine Diphosphate-Glucuronosyltransferases (UGTs)—How Does the Endoplasmic Reticulum Membrane Influence Their Function?. Pharmaceutics 2017, 9, 32. 10.3390/pharmaceutics9030032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie P. I.; Walter Bock K.; Burchell B.; Guillemette C.; Ikushiro S.-i.; Iyanagi T.; Miners J. O.; Owens I. S.; Nebert D. W. Nomenclature Update for the Mammalian Udp Glycosyltransferase (Ugt) Gene Superfamily. Pharmacogenet. Genomics 2005, 15, 677–685. 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- Meech R.; Mackenzie P. I. UGT3A: Novel UDP-Glycosyltransferases of the UGT Superfamily. Drug Metab. Rev. 2010, 42, 45–54. 10.3109/03602530903205823. [DOI] [PubMed] [Google Scholar]
- Meech R.; Rogers A.; Zhuang L.; Lewis B. C.; Miners J. O.; Mackenzie P. I. Identification of Residues That Confer Sugar Selectivity to UDP-Glycosyltransferase 3A (UGT3A) Enzymes. J. Biol. Chem. 2012, 287, 24122–24130. 10.1074/jbc.M112.343608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio A.; Binczek E.; Le Beau M. M.; Fernald A. A.; Stoffel W. The Human Gene CGT Encoding the UDP-Galactose Ceramide Galactosyl Transferase (Cerebroside Synthase): Cloning, Characterization, and Assignment to Human Chromosome 4, Band Q26. Genomics 1996, 34, 69–75. 10.1006/geno.1996.0242. [DOI] [PubMed] [Google Scholar]
- Sprong H.; Kruithof B.; Leijendekker R.; Slot J. W.; van Meer G.; van der Sluijs P. UDP-Galactose:Ceramide Galactosyltransferase Is a Class I Integral Membrane Protein of the Endoplasmic Reticulum. J. Biol. Chem. 1998, 273, 25880–25888. 10.1074/jbc.273.40.25880. [DOI] [PubMed] [Google Scholar]
- Meech R.; Mubarokah N.; Shivasami A.; Rogers A.; Nair P. C.; Hu D. G.; McKinnon R. A.; Mackenzie P. I. A Novel Function for UDP Glycosyltransferase 8: Galactosidation of Bile Acids. Mol. Pharmacol. 2015, 87, 442–450. 10.1124/mol.114.093823. [DOI] [PubMed] [Google Scholar]
- Wu B.; Kulkarni K.; Basu S.; Zhang S.; Hu M. First-Pass Metabolism via UDP-Glucuronosyltransferase: A Barrier to Oral Bioavailability of Phenolics. J. Pharm. Sci. 2011, 100, 3655–3681. 10.1002/jps.22568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners J. O.; Bowalgaha K.; Elliot D. J.; Baranczewski P.; Knights K. M. Characterization of Niflumic Acid as a Selective Inhibitor of Human Liver Microsomal UDP-Glucuronosyltransferase 1A9: Application to the Reaction Phenotyping of Acetaminophen Glucuronidation. Drug Metab. Dispos. 2011, 39, 644–652. 10.1124/dmd.110.037036. [DOI] [PubMed] [Google Scholar]
- Soars M. G.; Ring B. J.; Wrighton S. A. The Effect of Incubation Conditions on the Enzyme Kinetics of Udp-Glucuronosyltransferases. Drug Metab. Dispos. 2003, 31, 762–767. 10.1124/dmd.31.6.762. [DOI] [PubMed] [Google Scholar]
- Luukkanen L.; Taskinen J.; Kurkela M.; Kostiainen R.; Hirvonen J.; Finel M. Kinetic Characterization of the 1a Subfamily of Recombinant Human Udp-Glucuronosyltransferases. Drug Metab. Dispos. 2005, 33, 1017–1026. 10.1124/dmd.105.004093. [DOI] [PubMed] [Google Scholar]
- Bosma P. J.; Seppen J.; Goldhoorn B.; Bakker C.; Elferink R. P. O.; Chowdhury J. R.; Chowdhury N. R.; Jansen P. L. Bilirubin UDP-Glucuronosyltransferase 1 Is the Only Relevant Bilirubin Glucuronidating Isoform in Man. J. Biol. Chem. 1994, 269, 17960–17964. [PubMed] [Google Scholar]
- Krishnaswamy S.; Duan S. X.; Von Moltke L. L.; Greenblatt D. J.; Sudmeier J. L.; Bachovchin W. W.; Court M. H. Serotonin (5-Hydroxytryptamine) Glucuronidation in Vitro : Assay Development, Human Liver Microsome Activities and Species Differences. Xenobiotica 2003, 33, 169–180. 10.1080/0049825021000048809. [DOI] [PubMed] [Google Scholar]
- Operaña T. N.; Tukey R. H. Oligomerization of the UDP-Glucuronosyltransferase 1A Proteins: Homo- and Heterodimerization Analysis by Fluorescence Resonance Energy Transfer and Co-Immunoprecipitation. J. Biol. Chem. 2007, 282, 4821–4829. 10.1074/jbc.M609417200. [DOI] [PubMed] [Google Scholar]
- Ghosh S. S.; Sappal B. S.; Kalpana G. V.; Lee S. W.; Chowdhury J. R.; Chowdhury N. R. Homodimerization of Human Bilirubin-Uridine-Diphosphoglucuronate Glucuronosyltransferase-1 (UGT1A1) and Its Functional Implications. J. Biol. Chem. 2001, 276, 42108–42115. 10.1074/jbc.M106742200. [DOI] [PubMed] [Google Scholar]
- Kurkela M.; García-Horsman J. A.; Luukkanen L.; Mörsky S.; Taskinen J.; Baumann M.; Kostiainen R.; Hirvonen J.; Finel M. Expression and Characterization of Recombinant Human UDP-Glucuronosyltransferases (UGTs). UGT1A9 Is More Resistant to Detergent Inhibition than Other UGTs and Was Purified as an Active Dimeric Enzyme. J. Biol. Chem. 2003, 278, 3536–3544. 10.1074/jbc.M206136200. [DOI] [PubMed] [Google Scholar]
- Meech R.; Mackenzie P. I. UDP-Glucuronosyltransferase, the Role of the Amino Terminus in Dimerization. J. Biol. Chem. 1997, 272, 26913–26917. 10.1074/jbc.272.43.26913. [DOI] [PubMed] [Google Scholar]
- Lewis B. C.; Mackenzie P. I.; Miners J. O. Homodimerization of UDP-Glucuronosyltransferase 2B7 (UGT2B7) and Identification of a Putative Dimerization Domain by Protein Homology Modeling. Biochem. Pharmacol. 2011, 82, 2016–2023. 10.1016/j.bcp.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Ikushiro S.-i.; Emi Y.; Iyanagi T. Protein–Protein Interactions between UDP-Glucuronosyltransferase Isozymes in Rat Hepatic Microsomes. Biochemistry 1997, 36, 7154–7161. 10.1021/bi9702344. [DOI] [PubMed] [Google Scholar]
- Fujiwara R.; Nakajima M.; Oda S.; Yamanaka H.; Ikushiro S.-i.; Sakaki T.; Yokoi T. Interactions between Human UDP-Glucuronosyltransferase (UGT) 2B7 and UGT1A Enzymes. J. Pharm. Sci. 2010, 99, 442–454. 10.1002/jps.21830. [DOI] [PubMed] [Google Scholar]
- Peters W. H.; Jansen P. L.; Nauta H. The Molecular Weights of UDP-Glucuronyltransferase Determined with Radiation-Inactivation Analysis. A Molecular Model of Bilirubin UDP-Glucuronyltransferase. J. Biol. Chem. 1984, 259, 11701–11705. [PubMed] [Google Scholar]
- Kurkela M.; Patana A.-S.; Mackenzie P. I.; Court M. H.; Tate C. G.; Hirvonen J.; Goldman A.; Finel M. Interactions with Other Human UDP-Glucuronosyltransferases Attenuate the Consequences of the Y485D Mutation on the Activity and Substrate Affinity of UGT1A6. Pharmacogenet. Genomics 2007, 17, 115–126. 10.1097/FPC.0b013e328011b598. [DOI] [PubMed] [Google Scholar]
- Fujiwara R.; Nakajima M.; Yamanaka H.; Katoh M.; Yokoi T. Interactions between Human UGT1A1, UGT1A4, and UGT1A6 Affect Their Enzymatic Activities. Drug Metab. Dispos. 2007, 35, 1781–1787. 10.1124/dmd.107.016402. [DOI] [PubMed] [Google Scholar]
- Ishii Y.; Koba H.; Kinoshita K.; Oizaki T.; Iwamoto Y.; Takeda S.; Miyauchi Y.; Nishimura Y.; Egoshi N.; Taura F.; et al. Alteration of the Function of the UDP-Glucuronosyltransferase 1A Subfamily by Cytochrome P450 3A4: Different Susceptibility for UGT Isoforms and UGT1A1/7 Variants. Drug Metab. Dispos. 2014, 42, 229–238. 10.1124/dmd.113.054833. [DOI] [PubMed] [Google Scholar]
- Fujiwara R.; Itoh T. Extensive Protein-Protein Interactions Involving UDP-Glucuronosyltransferase (UGT) 2B7 in Human Liver Microsomes. Drug Metab. Pharmacokinet. 2014, 29, 259–265. 10.2133/dmpk.DMPK-13-RG-096. [DOI] [PubMed] [Google Scholar]
- Campbell J. A.; Davies G. J.; Bulone V.; Henrissat B. A Classification of Nucleotide-Diphospho-Sugar Glycosyltransferases Based on Amino Acid Sequence Similarities. Biochem. J. 1997, 326, 929–939. 10.1042/bj3260929u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P. M.; Deleury E.; Davies G. J.; Henrissat B. An Evolving Hierarchical Family Classification for Glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. 10.1016/S0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- Breton C.; Šnajdrová L.; Jeanneau C.; Koča J.; Imberty A. Structures and Mechanisms of Glycosyltransferases. Glycobiology 2006, 16, 29R–37R. 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- Radominska-Pandya A.; Bratton S. M.; Redinbo M. R.; Miley M. J. The Crystal Structure of Human UDP-Glucuronosyltransferase 2B7 C-Terminal End Is the First Mammalian UGT Target to Be Revealed: The Significance for Human UGTs from Both the 1A and 2B Families. Drug Metab. Rev. 2010, 42, 133–144. 10.3109/03602530903209049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miley M. J.; Zielinska A. K.; Keenan J. E.; Bratton S. M.; Radominska-Pandya A.; Redinbo M. R. Crystal Structure of the Cofactor-Binding Domain of the Human Phase II Drug-Metabolism Enzyme UDP-Glucuronosyltransferase 2B7. J. Mol. Biol. 2007, 369, 498–511. 10.1016/j.jmb.2007.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Zhu L.; Qu W.; Wu F.; Hu M.; Xie W.; Liu Z.; Wang C. Insight into Tartrate Inhibition Patterns in Vitro and in Vivo Based on Cocrystal Structure with UDP-Glucuronosyltransferase 2B15. Biochem. Pharmacol. 2020, 172, 113753. 10.1016/j.bcp.2019.113753. [DOI] [PubMed] [Google Scholar]
- Nair P. C.; Meech R.; Mackenzie P. I.; McKinnon R. A.; Miners J. O. Insights into the UDP-Sugar Selectivities of Human UDP-Glycosyltransferases (UGT): A Molecular Modeling Perspective. Drug Metab. Rev. 2015, 47, 1–11. 10.3109/03602532.2015.1071835. [DOI] [PubMed] [Google Scholar]
- Fujiwara R.; Yokoi T.; Nakajima M. Structure and Protein–Protein Interactions of Human UDP-Glucuronosyltransferases. Front. Pharmacol. 2016, 7, 388. 10.3389/fphar.2016.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghemtio L.; Soikkeli A.; Yliperttula M.; Hirvonen J.; Finel M.; Xhaard H. SVM Classification and CoMSIA Modeling of UGT1A6 Interacting Molecules. J. Chem. Inf. Model. 2014, 54, 1011–1026. 10.1021/ci400577a. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Skolnick J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins: Struct., Funct., Bioinf. 2004, 57, 702–710. 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- Ouzzine M.; Magdalou J.; Burchell B.; Fournel-Gigleux S. An Internal Signal Sequence Mediates the Targeting and Retention of the Human UDP-Glucuronosyltransferase 1A6 to the Endoplasmic Reticulum. J. Biol. Chem. 1999, 274, 31401–31409. 10.1074/jbc.274.44.31401. [DOI] [PubMed] [Google Scholar]
- Kirchmair J.; Markt P.; Distinto S.; Wolber G.; Langer T. Evaluation of the Performance of 3D Virtual Screening Protocols: RMSD Comparisons, Enrichment Assessments, and Decoy Selection—What Can We Learn from Earlier Mistakes?. J. Comput.-Aided Mol. Des. 2008, 22, 213–228. 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- Chen H.; Lyne P. D.; Giordanetto F.; Lovell T.; Li J. On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors. J. Chem. Inf. Model. 2006, 46, 401–415. 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- Erickson J. A.; Jalaie M.; Robertson D. H.; Lewis R. A.; Vieth M. Lessons in Molecular Recognition: The Effects of Ligand and Protein Flexibility on Molecular Docking Accuracy. J. Med. Chem. 2004, 47, 45–55. 10.1021/jm030209y. [DOI] [PubMed] [Google Scholar]
- Singh R.; Wu B.; Tang L.; Hu M. Uridine Diphosphate Glucuronosyltransferase Isoform-Dependent Regiospecificity of Glucuronidation of Flavonoids. J. Agric. Food Chem. 2011, 59, 7452–7464. 10.1021/jf1041454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma M. G.; van der Woude H.; Bogaards J.; Boeren S.; Vervoort J.; Cnubben N. H. P.; van Iersel M. L. P. S.; van Bladeren P. J.; Rietjens I. M. C. M. Regioselectivity of Phase II Metabolism of Luteolin and Quercetin by UDP-Glucuronosyl Transferases. Chem. Res. Toxicol. 2002, 15, 662–670. 10.1021/tx0101705. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy S.; Hao Q.; Al-Rohaimi A.; Hesse L. M.; von Moltke L. L.; Greenblatt D. J.; Court M. H. UDP Glucuronosyltransferase (UGT) 1A6 Pharmacogenetics: II. Functional Impact of the Three Most Common Nonsynonymous UGT1A6 Polymorphisms (S7A, T181A, and R184S). J. Pharmacol. Exp. Ther. 2005, 313, 1340–1346. 10.1124/jpet.104.081968. [DOI] [PubMed] [Google Scholar]
- Sigrell J. A.; Cameron A. D.; Mowbray S. L. Induced fit on sugar binding activates ribokinase. J. Mol. Biol. 1999, 290, 1009–1018. 10.1006/jmbi.1999.2938. [DOI] [PubMed] [Google Scholar]
- Ito S.; Fushinobu S.; Jeong J.-J.; Yoshioka I.; Koga S.; Shoun H.; Wakagi T. Crystal Structure of an ADP-Dependent Glucokinase from Pyrococcus Furiosus: Implications for a Sugar-Induced Conformational Change in ADP-Dependent Kinase. J. Mol. Biol. 2003, 331, 871–883. 10.1016/S0022-2836(03)00792-7. [DOI] [PubMed] [Google Scholar]
- Qasba P. K.; Ramakrishnan B.; Boeggeman E. Substrate-Induced Conformational Changes in Glycosyltransferases. Trends Biochem. Sci. 2005, 30, 53–62. 10.1016/j.tibs.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Osmani S. A.; Bak S.; Møller B. L. Substrate Specificity of Plant UDP-Dependent Glycosyltransferases Predicted from Crystal Structures and Homology Modeling. Phytochemistry 2009, 70, 325–347. 10.1016/j.phytochem.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Offen W.; Martinez-Fleites C.; Yang M.; Kiat-Lim E.; Davis B. G.; Tarling C. A.; Ford C. M.; Bowles D. J.; Davies G. J. Structure of a Flavonoid Glucosyltransferase Reveals the Basis for Plant Natural Product Modification. EMBO J. 2006, 25, 1396–1405. 10.1038/sj.emboj.7600970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H.; Yang T.; Whitaker B. D.; Shangguan L.; Fang J. Calcium/Calmodulin Alleviates Substrate Inhibition in a Strawberry UDP-Glucosyltransferase Involved in Fruit Anthocyanin Biosynthesis. BMC Plant Biol. 2016, 16, 197. 10.1186/s12870-016-0888-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riches Z.; Collier A. C. Posttranscriptional Regulation of Uridine Diphosphate Glucuronosyltransferases. Expert Opin. Drug Metab. Toxicol. 2015, 11, 949–965. 10.1517/17425255.2015.1028355. [DOI] [PubMed] [Google Scholar]
- Wu B.; Morrow J. K.; Singh R.; Zhang S.; Hu M. Three-Dimensional Quantitative Structure-Activity Relationship Studies on UGT1A9-Mediated 3-O-Glucuronidation of Natural Flavonols Using a Pharmacophore-Based Comparative Molecular Field Analysis Model. J. Pharmacol. Exp. Ther. 2011, 336, 403–413. 10.1124/jpet.110.175356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonk M. L.; Berdini V.; Hartshorn M. J.; Mooij W. T. M.; Murray C. W.; Taylor R. D.; Watson P. Virtual Screening Using Protein–Ligand Docking: Avoiding Artificial Enrichment. J. Chem. Inf. Comput. Sci. 2004, 44, 793–806. 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- He X.-Z.; Wang X.; Dixon R. A. Mutational Analysis of the Medicago Glycosyltransferase UGT71G1 Reveals Residues That Control Regioselectivity for (Iso)Flavonoid Glycosylation. J. Biol. Chem. 2006, 281, 34441–34447. 10.1074/jbc.M605767200. [DOI] [PubMed] [Google Scholar]
- Wetterhorn K. M.; Newmister S. A.; Caniza R. K.; Busman M.; McCormick S. P.; Berthiller F.; Adam G.; Rayment I. Crystal Structure of Os79 (Os04g0206600) from Oryza Sativa: A UDP-Glucosyltransferase Involved in the Detoxification of Deoxynivalenol. Biochemistry 2016, 55, 6175–6186. 10.1021/acs.biochem.6b00709. [DOI] [PubMed] [Google Scholar]
- Michlmayr H.; Varga E.; Malachová A.; Fruhmann P.; Piątkowska M.; Hametner C.; Šofrová J.; Jaunecker G.; Häubl G.; Lemmens M.; et al. UDP-Glucosyltransferases from Rice, Brachypodium, and Barley: Substrate Specificities and Synthesis of Type A and B Trichothecene-3-O-β-d-Glucosides. Toxins 2018, 10, 111. 10.3390/toxins10030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu N. K.; Kole L.; Basu M.; Chakraborty K.; Mitra P. S.; Owens I. S. The Major Chemical-Detoxifying System of UDP-Glucuronosyltransferases Requires Regulated Phosphorylation Supported by Protein Kinase C. J. Biol. Chem. 2008, 283, 23048–23061. 10.1074/jbc.M800032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volak L. P.; Court M. H. Role for Protein Kinase C Delta in the Functional Activity of Human UGT1A6: Implications for Drug-Drug Interactions between PKC Inhibitors and UGT1A6.. Xenobiotica 2010, 40, 306–318. 10.3109/00498251003596817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M.; Koga T.; Sakai H.; Yamanaka H.; Fujiwara R.; Yokoi T. N-Glycosylation Plays a Role in Protein Folding of Human UGT1A9. Biochem. Pharmacol. 2010, 79, 1165–1172. 10.1016/j.bcp.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Nagaoka K.; Hanioka N.; Ikushiro S.; Yamano S.; Narimatsu S. The Effects of N-Glycosylation on the Glucuronidation of Zidovudine and Morphine by UGT2B7 Expressed in HEK293 Cells. Drug Metab. Pharmacokinet. 2012, 27, 388–397. 10.2133/dmpk.DMPK-11-RG-135. [DOI] [PubMed] [Google Scholar]
- Lampe J. N. Advances in the Understanding of Protein-Protein Interactions in Drug Metabolizing Enzymes through the Use of Biophysical Techniques. Front. Pharmacol. 2017, 8, 521. 10.3389/fphar.2017.00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER: Fully Automated Protein Structure Prediction in CASP8. Proteins 2009, 77, 100–113. 10.1002/prot.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A.; Yang J.; Zhang Y. COFACTOR: An Accurate Comparative Algorithm for Structure-Based Protein Function Annotation. Nucleic Acids Res. 2012, 40, W471–W477. 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhang Y. I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 2015, 43, W174–W181. 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Zhang Y. LOMETS: A Local Meta-Threading-Server for Protein Structure Prediction. Nucleic Acids Res. 2007, 35, 3375–3382. 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Skolnick J. SPICKER: A Clustering Approach to Identify near-Native Protein Folds. J. Comput. Chem. 2004, 25, 865–871. 10.1002/jcc.20011. [DOI] [PubMed] [Google Scholar]
- Shen M.-y.; Sali A. Statistical Potential for Assessment and Prediction of Protein Structures. Protein Sci. 2006, 15, 2507–2524. 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L.; Mackerell A. D.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassov V. Z.; Yan L.; Flook P. K. The Dominant Role of Side-Chain Backbone Interactions in Structural Realization of Amino Acid Code. ChiRotor: A Side-Chain Prediction Algorithm Based on Side-Chain Backbone Interactions. Protein Sci. 2007, 16, 494–506. 10.1110/ps.062447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassov V. Z.; Flook P. K.; Yan L. LOOPER: A Molecular Mechanics-Based Algorithm for Protein Loop Prediction. Protein Eng., Des. Sel. 2008, 21, 91–100. 10.1093/protein/gzm083. [DOI] [PubMed] [Google Scholar]
- Colovos C.; Yeates T. O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.; Robertson D. H.; Brooks C. L.; Vieth M. Detailed Analysis of Grid-Based Molecular Docking: A Case Study of CDOCKER-A CHARMm-Based MD Docking Algorithm. J. Comput. Chem. 2003, 24, 1549–1562. 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.