

Abstract
In one-pot strategy, diazotization of methyl anthranilate 5 followed by addition of amino acid ester hydrochloride, we have prepared methyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanoates 6a–c. Starting with hydrazides 7a,b, N-alkyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanamides 9–10(a–h) and methyl-2-(2-(4-oxobenzotriazin-3(4H)-yl)alkanamido)alkanoates 11–12(a–e) were prepared via azide coupling. Hydrazones 13–15 were prepared via condensation of hydrazides 7a,b with 4-methoxybenzaldehyde, 4-dimethylaminobenzaldehyde, and/or arabinose. Molecular docking was done for synthesized compounds using MOE 2008-10 software. The compounds 9a, 12a, 12c, 13a, 13b, and 14b have the most pronounced strong binding affinities toward the target E. coli Fab-H receptor, whereas compounds 3, 11e, 12e, and 13a have the most pronounced strong binding affinities toward the target vitamin D receptor. The in vitro antibacterial activities of the highest binding affinity docked compounds were tested against E. coli, Staphylococcus aureus, and Salmonella spp. Majority of the tested compounds showed effective positive results against E. coli, while they were almost inactive against Staphylococcus aureus and Salmonella spp. The in vitro cytotoxic activities of the highest binding affinity-docked compounds were tested against the human liver carcinoma cell line (HepG2). Some compounds showed potent cytotoxic activity with low IC50 values, especially for 3 (6.525 μM) and 13a (10.97 μM) than that for standard drug doxorubicin (2.06 μM).
Introduction
Cancer is one of the major health challenges for all nations. Chemotherapy is one of the most effective targets used to heal malignancy. The main disadvantages of cancer chemotherapy are the dangerous side effects associated with it such as vomiting and spinal depression in addition to a lack of selectivity of drugs against the tumor cellular substance in the tumor compared to normal cellular substances. Hence, the search for anti-cancer drugs is the latest task that never ends. Benzotriazinones are key interactions at protein–protein interfaces that constitute important targets for small molecule inhibition because of their specific arrangements and biological importance. Therefore, one of the most corner stone principles in our research group is based on searching for new anticancer drugs.1,2 Benzotriazine and its derivatives possess a diverse range of biological activities of pharmacological activities including antimicrobial,3 anti-inflammatory,4 anti-depressant,5 anti-ulcer,6 anti-diarrhoeal,7 anaesthetics,8 and anti-cancer.9 Some current commercial benzotriazinone anticancer drugs such as α-hydroxylatedbenzotriazinone10 (A), N-arylbenzotriazinones11 (B), tirapazamine (1,2,4-benzotriazin-3-amine,1,4-dioxide)12 (C), and 4-[(4-oxobenzotriazin-3(4H)-yl)methyl]benzoic acid13 (D) show the possibilities to reduce recombinant human cancer cell growth in a culture.
One of the most important benzotriazin-4(3H)-one derivatives is 3-hydroxy-1,2,3-benzotriazin-4(3H)-one, which is one as a versatile reagent employed for peptide synthesis (Figure 1).14
Figure 1.

Some current commercial benzotriazinone anticancer drugs.
Due to the importance of vitamin D, which has been produced by plants and animals, we used the vitamin D receptor in our docking investigation. VDR is found in almost all cells and tissues of higher-order animals, further emphasizing the importance of the receptor. Evidence for the existence of VDR was first provided in 1969 by Haussler and Norman,15 and since then a substantial amount of data on the structure and function of VDR has been accomplished.
In the present work, we aimed to synthesize a series of compounds containing benzotriazinone moieties on the basis of structure modification of methyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanoates 6a–c for biological evaluation as antimicrobial and anticancer agents.
Results and Discussion
Chemistry
Early attempts for preparing 6a–c as the target molecules for structure modification of benzotriazinone were achieved from isatoic anhydride 1. Isatoic anhydride 1 was reacted with ammonia in the presence of ammonium carbonate to give anthranilamide 2, which subsequently diazotized using sodium nitrite and HCl solution at 0 °C to give benzotriazinone 3. Similarly, 3 was also formed by diazotization of anthranilhydrazide 4 in a moderate yield. Benzotriazinone 3 was reacted with methyl chloroacetate in DMF in the presence of potassium carbonate at 100 °C to give the chemoselective N-alkylated ester, methyl-2-(4-oxobenzotriazin-3(4H)-yl)acetate 6a as the only product on the basis of chemoselective reactivity of heterocyclic amides toward electrophiles, Scheme 1.16−18 The multistep reactions mentioned gave 6a in an overall high yield from available isatoic anhydride, but the alkyl halides needed to prepare other methyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanoates 6b–c were not available.
Scheme 1. Multistep Method for Preparation of Methyl 2-(4-Oxobenzotriazin-3(4H)-yl)acetate 6a.

Diazotization of methyl anthranilate 5 using sodium nitrite and HCl solution at 0 °C followed by addition of amino acid ester hydrochloride in the presence of triethyl amine in a one-pot strategy affording methyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanoates 6a–c in excellent yields, Scheme 2. This method has the advantage of simple work up, one pot, and the availability of reagents.
Scheme 2. Direct Preparation of Methyl 2-(4-Oxobenzotriazin-3(4H)-yl)alkanoates 6a–c and Corresponding Hydrazides 7a,b.

The structure assignment of methyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanoates 6a–c is based on full characterization, including 1H and 13C NMR spectroscopies. Thus, the 1H NMR spectrum of methyl-3-(4-oxobenzotriazin-3(4H)-yl)propanoate 6b revealed signals at δ 4.73–4.68, 3.65, and 2.95–2.89 ppm for OCH3, NCH2, and CH2CO groups, respectively. The 13C NMR spectrum of 6b showed signals at δ 171.0, 155.3, 51.9, 45.5, and 33.0 ppm for 2C=O groups, OCH3, NCH2, and CH2CO groups, respectively.
Methyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanoates 6a–c are excellent precursors for structure modification of the benzotriazinone ring system via the azide coupling method by an attachment of either amines or amino acid through a peptide bond. Thus, the esters 6a,b were reacted with hydrazine hydrate in ethanol under reflux condition for 6 h afforded the corresponding hydrazides 7a,b, Scheme 2. Hydrazides 7a,b were reacted with NaNO2 and HCl in water at 0 °C for 1 h to afford the corresponding azides 8a,b and were extracted with ethyl acetate. The in situ generated azide 8a,b solution was successively added to primary amines iso-propyl, n-butyl, tert-butyl, n-decyl, cyclohexyl, and benzyl amines and secondary amines piperidine and morpholine to give N-alkyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanamides 9–10(a–h) in good yields, Scheme 3.
Scheme 3. Azide Coupling Method for the Preparation of N-alkyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanamides 9–10(a–h).

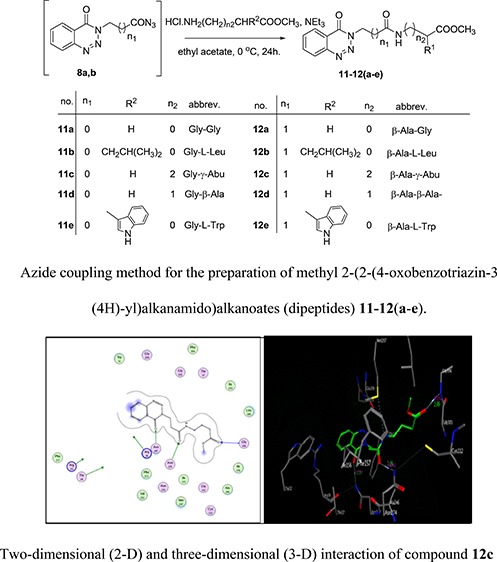
Next, the in situ generated azide 8a and 8b solution was simultaneously added to amino acid methyl ester hydrochloride glycine, l-leucine γ-aminobutyric acid, β-alanine, and l-tryptophane to give methyl-2-(2-(4-oxobenzotriazin-3(4H)-yl)alkanamido)alkanoates (dipeptides) 11–12(a–e) in good yields via the azide coupling method, Scheme 4.
Scheme 4. Azide Coupling Method for the Preparation of Methyl 2-(2-(4-Oxobenzotriazin-3(4H)-yl)alkanamido)alkanoates (Dipeptides) 11–12(a–e).

The structure assignments of N-alkyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanamides 9–10(a–h) and methyl-2-(2-(4-oxobenzotriazin-3(4H)-yl)alkanamido)alkanoates (dipeptides) 11–12(a–e) are based on full characterization, including 1H and 13C NMR spectroscopies. Thus, the 1H NMR spectrum of methyl-2-(2-(4-oxobenzotriazin-3(4H)-yl)acetamido)acetate 11a showed signals at δ 5.22, 4.14, and 3.77 ppm corresponding to NCH2, NHCH2, and OCH3, respectively. The 13C NMR spectrum of 11a showed signals at 170.1, 166.3, 155.9, 52.6, 52.5, and 41.4 ppm for 3C=O groups, OCH3, NCH2, and NHCH2 groups, respectively.
Also, a number of hydrazones 13–14(a,b) were prepared by condensation of hydrazides 7a,b with some aldehydes, namely, 4-methoxybenzaldehyde and 4-dimethylaminobenzaldehyde, in ethanol under reflux condition for 6 h and gave 13–14(a,b) in very good yields, Scheme 5.
Scheme 5. Condensation of 2-(4-Oxobenzotriazin-3(4H)-yl)alkanoic Acid Hydrazides 7a and 7b with Aldehydes.

Finally, the 3-(4-oxo-4H-benzo[d][1,2,3]triazin-3-yl)-propionic acid hydrazide 7b was condensed with arabinose in ethanol under reflux condition for 6 h and gave 15, Scheme 6.
Scheme 6. Condensation of 3-(4-Oxo-4H-benzo[d][1,2,3]triazin-3-yl)-propionic Acid Hydrazide7b with Arabinose.

Biological Activity
Molecular Docking Methodology
Bioinformatics including molecular modeling studies are very valuable at the present time in the field of drug discovery, saving money, and effort needed for the screening of new compounds by guiding and confining the investigation to possible target/targets. The use of docking simulation studies in our work is quite important to help in predicting the possible mode of the action and structure activity relationship of the active derivatives and guiding the research future directions in the compound optimization and biochemical enzyme assay for the possible target enzymes. Key interactions at protein–protein interfaces constitute important targets for small molecule inhibition because of their specific arrangements and biological importance.19
All the molecular modeling studies were carried out on Intel Core i3 CPU, 2.40 GHZ processor and 3 GB memory with a Windows 7 operating system using Molecular Operating Environment (MOE 2008-10 Chemical Computing Group, Canada) as the computational software. Anti-bacterial activities of the synthesized benzotriazinone derivatives were investigated through correlation with E. coli Fab-H inhibitory activities, and the anti-cancer activities of compounds were screened through detection of their abilities to act as the vitamin D receptor. The crystal structure of the E. coli FabH-CoA complex (PDB code: 1HNJ) and the crystal structure of the nuclear receptor for vitamin D bound to its natural ligand (PDB code: 1DB1) were obtained from the freely accessible protein data bank. The docking studies were performed after the verification process, which was performed by redocking of the cocrystallized ligand into the active site using the default settings. The synthesized compounds were docked within the active sites of the crystallized structures using the MOE dock tool in MOE, performed with the default values. Different conformers for each compound are imported by systematic conformational of the MOE and saved in an mdb data base file to be docked into the active site of the receptor. Each complex was analyzed for the interaction, and 2D and 3D images were taken by using the MOE visualizing tool.
The results were evaluated based on binding affinity calculation together with cluster size determination and visually through the possible interaction with key residues at the active site.
Anti-bacterial Activity
Molecular Modeling Study
As shown in Figure 2, compound 12a formed two hydrogen bonds as the sidechain acceptor with amino acid residues Arg 36 and Arg 249. However, the amino acid residues Thr 28, Arg 151, and Asn 247 were not oriented directly to form complete hydrogen bonds with the ligand structure. Moreover, there are 16 ligand exposures, which indicate good binding affinity of the compound with the receptor under study.
Figure 2.

Two-dimensional (2D) and three-dimensional (3D) interactions of compound 12a.
As shown in Figure 3, compound 12c formed two hydrogen bonds as the side-chain acceptor with amino acid residues Asn274 and Asn247 in addition to one hydrogen bond as a back-bone acceptor with the amino acid residue Gly 306. However, the amino acid residues Thr 28, Arg 36, and Arg 151 were not oriented directly to form complete hydrogen bonds with the ligand structure. Moreover, there are 10 ligand exposures, which indicate good binding affinity of the compound with the receptor under study.
Figure 3.

Two-dimensional (2D) and three-dimensional (3D) interactions of compound 12c.
As shown in Figure 4, compound 13b formed two hydrogen bonds as the sidechain acceptor with amino acid residue Asn274. The amino acid residues Asn 247, Arg 36, and Thr 28 were not oriented directly to form complete hydrogen bonds with the ligand structure. Moreover, there are four ligand exposures, which indicate good binding affinity of the compound with the receptor under study.
Figure 4.

Two-dimensional (2D) and three-dimensional (3D) interactions of compound 13b.
In Vitro Anti-bacterial Activity
The in vitro antibacterial activities of the highest binding affinity-docked compounds 3, 7b, 9a, 10f, 11a, 12a, 12b, 12c, 13a, 13b, and 14b were tested against E. coli, Staphylococcus aureus, and Salmonella spp. Majority of tested compounds gave effective positive results against E. coli with an inhibitory zone of about 1.0 cm, while they were inactive against Staphylococcus aureus and Salmonella spp.
Anti-cancer Activity
Molecular Modeling Study
As shown in Figure 5, compound 3 formed two hydrogen bonds as a side-chain acceptor with amino acid residues His 305 and His 379. However, the amino acid residues Tyr 143, Ser 237, Arg 274, and Ser 278 were not oriented directly to form complete hydrogen bonds with the ligand structure. Moreover, there are three ligand exposures, which indicate good binding affinity of the compound with the receptor under study.
Figure 5.

Two-dimensional (2D) and three-dimensional (3D) interactions of compound 3.
As shown in Figure 6, compound 11e formed two hydrogen bonds as a side-chain acceptor with amino acid residues Ser 237 and Arg 274 in addition to one hydrogen bond as the arene–cation interaction with the amino acid residue Arg 274. Another hydrogen bond formed with the amino acid residue Tyr 143 as the arene–arene interaction. However, the amino acid residues Tyr 143, Ser 237, Ser 278, His 305, and His 397 were not oriented directly to form complete hydrogen bonds with the ligand structure. Moreover, there are four ligand exposures, which indicate good binding affinity of the compound with the receptor under study.
Figure 6.

Two-dimensional (2D) and three-dimensional (3D) interactions of compound 11e.
As shown in Figure 7, compound 13a formed two hydrogen bonds with the amino acid residue Tyr 143 one of them as a side-chain acceptor and the other one as the arene–arene interaction. In addition, two hydrogen bonds as a side-chain acceptor with the amino acid residue Ser 237. It also formed one hydrogen bond as the arene–arene interaction with the amino acid residue Trp 286. However, the amino acid residues Arg 274, Ser 278, His 305, and His 397 were not oriented directly to form complete hydrogen bonds with the ligand structure. Moreover, there are five ligand exposures, which indicate good binding affinity of the compound with the receptor under study.
Figure 7.

Two-dimensional (2D) and three-dimensional (3D) interactions of compound 13a.
In Vitro Anti-cancer Activity
Potential cytotoxicity of the newly synthesized compounds was tested against the human liver carcinoma cell line (HepG2) using the method of Hansen et al.20 The in vitro anticancer screening was done by planting a tissue unit in Vacsera, Cairo A. R. Egypt.
This work was performed by a modification (Hansen et al., 1989)20 of the tetrazolium salt (MTT) method, Mosmann, 1983.21 Preferably, cells should be plated in triplicate wells. Relative cell proliferation/viability was measured when treated cells are compared with untreated cells.
The in vitro cytotoxic activities of the highest binding affinity docked compounds were tested against human liver carcinoma (HepG2) cancer cell lines. Many compounds showed potent cytotoxic activities with low IC50 values, especially for 3 (6.525 μM) and 13a (10.97 μM), than that for standard drug doxorubicin (2.06 μM).
On regarding IC50 values, It is clear that the tryptophan amino acid fragments enhanced the benzotriazinone anticancer activities. Moreover, the hydrazone 13a derived from methoxy benzaldehyde in the case of glycine was more active than that of one derived from β-alanine.
Conclusions
A series of 36 compounds were synthesized based on structure modification of the model benzotriazinone 3 as a potent HepG2 liver carcinoma inhibitor. Different benzotriazinone derivatives were synthesized via different chemistry protocols to obtain 2-(4-oxobenzotriazin-3(4H)-yl)alkanoates methyl esters, 2-(4-oxobenzotriazin-3(4H)-yl)alkanamide derivatives, and 2-(2-(4-oxobenzotriazin-3(4H)-yl)alkanamido)alkanoates methyl esters. The hydrazones of some hydrazides were obtained by reactions with some aldehydes and/or arabinose. Applying of Molecular docking on synthesized compounds leads us to choose the most promising derivatives for further biological studies. Some compounds possess strong binding affinity toward the target E.coli Fab-H receptor. Moreover, some compounds had strong binding affinity toward the target vitamin D receptor. The in vitro antibacterial activities of the highest binding affinity docked compounds gave effective positive results against E. coli. On the other hand, the in vitro cytotoxic activities were tested against the human liver carcinoma cell line (HepG2), and IC50 values of compounds 3, 11e, 12e, and 13a were 6.525, 12.4019, 59.85, and 10.97ug/mL respectively.
Experimental Section
General Procedure
Thin-layer chromatography (TLC) was carried out on silica gel 60 F254 aluminum sheets (E. Merck, layer thickness 0.2 mm) in the following solvent systems, S1: petroleum ether/ethyl acetate (2:1) and S2: petroleum ether/ethyl acetate (1:1). The spots on thin layer plates were detected by the UV lamp. Melting points were determined on a Buchi 510 melting-point apparatus, and the values are uncorrected. Elemental analyses were performed on a Flash EA-1112 instrument at the Microanalytical Laboratory, Faculty of Science, Suez Canal University, Ismailia, Egypt. 1H NMR spectra were measured on a Bruker spectrometer operating at 300 and 400 MHz, respectively, at nuclear magnetic resonance laboratory, kafr-El-shiekh University, Egypt. The mass spectra were measured with a KRATOS analytical compact; on MALDI-MS, the spectrometer used 2,5-dihydroxy benzoic acid (DHB) as a matrix.
Synthesis of Benzo[d][1,2,3]triazin-4(3H)-one (3)22
Method A: to anthranilamide (2) (13.6 g, 0.1 mol) in an ice bath, 11.3 g (0.198 mol) of NaNO2 and 50 mL of 8 M HCl were added and the mixture was stirred for 1 h at 0 °C. The solution was warmed to room temperature and neutralized by dropwise addition of 1 N NaOH. The formed precipitate was filtered off, washed with water, dried, and crystallized from methanol.
Method B: aqueous solution of sodium nitrite (3.9 g) was added to an ice-cold solution of anthranilohydrazide (4) (7.5 g) (0.05 mol) in of 2 N hydrochloric acid (15 mL) and distilled water (15 mL). The mixture was stirred for 5 min with subsequent neutralization by sodium carbonate, and the resulting white precipitate was filtered, washed with cold water, dried, and crystallized from methanol. White crystals (Method A, 88%), (Method B, 65%) Rf = 0.70 (S2), mp: 211–214 °C. 1H-NMR spectrum (300.0 MHz, CDCl3); δ ppm, (J, Hz): 11.92 (1H, bs, NH); 8.32 (1H, d, J = 7.6 Hz, Ar–H); 8.16 (1H, d, J = 8.0 Hz, Ar–H); 7.96 (1H, t, J = 8.0 Hz, Ar–H); 7.80 (1H, t, J = 7.2 Hz, Ar–H). MS (MALDI, positive mode, matrix DHB): m/z = 170 (M + Na)+. Anal. calcd for C7H5N3O (147.1) C, 57.14; H, 3.43; N, 28.56. found C, 57.30; H, 3.37; N, 28.46.
Synthesis of Methyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanoates (6a–c)
Method A: to an ice-cold solution of methyl anthranilate 5 (15.1 g, 0.1 mol) in diluted HCl contained in a 250 mL beaker, sodium nitrite (6.8 g, 0.116 mol) solution in water was added and the resulting diazonium solution was stirred at 0 °C for 5 min. Then, ammonium hydroxide was added dropwise until the solution becomes just alkaline. Amino acid ester hydrochlorides, glycine, β-alanine, and l-leucine (0.01 mol) were added dropwise, and the mixture was stirred for 5 min with subsequent neutralization by sodium carbonate. A precipitate out was filtered off and washed with ice-cold ethanol. Product 6a–c was crystallized from ethanol.
Method B: a solution of benzo[d][1,2,3]triazin-4(3H)-one (3) (0.15 g, 1.0 mmol) in DMF (30 mL), K2CO3 (2.0 mmol), and methyl chloroacetate (0.12 mL, 1.0 mmol) was mixed and heated at 100 °C for 12 h while the reaction was monitored via TLC. The reaction mixture was then cooled, and an ice/water mixture was added. A precipitate out was filtered off, washed with ice-cold ethanol, and crystallized from ethanol.
Methyl-2-(4-oxo(3H)-1,2,3-benzotriazin-3-yl)acetate (6a)
A yellow solid, (Method A, 69%), mp: 113–114 °C (Method B, 83%), (Lit. mp: 114–116 °C).23
Methyl-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanoate (6b)
Oil (Method A, 87%), Rf = 0.75 (S2) 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 8.28–8.23 (1H, m, Ar–H); 8.08–8.04 (1H, m, Ar–H); 7.91–7.86 (1H, m, Ar–H); 7.76–7.66 (1H, m, Ar–H); 4.73–4.68 (2H, m, NCH2); 3.65 (3H, s, OCH3); 2.95–2.89 (2H, m, CH2CO). 13C-NMR (75 MHz, CDCl3), δ ppm, 171.0; 155.3(2CO); 144.1; 134.8; 132.4; 128.3; 125.0; 119.6 (Ar–C); 51.9 (OCH3); 45.5 (NCH2); 33.0 (CH2CO). MS (MALDI, positive mode, matrix DHB): m/z = 256 (M + Na)+. Anal. calcd for C11H11N3O3 (233.2) C, 56.65; H, 4.75; N, 18.02. found C, 56.40; H, 4.62; N, 17.89.
Methyl-4-methyl-2-(4-oxo-4H-benzo[d][1,2,3]triazin-3-yl)pentanoate (6c)
White crystals (Method A, 84%), Rf = 0.32 (S2), mp: 197–200 °C. 1H NMR (300.0 MHz, DMSO-d6), δ ppm, (J, Hz): 8.38–6.64 (4H, m, Ar–H); 5.82–5.74 (1H, m, NCHCO); 4.46–4.41 (2H, m, CH2); 3.97 (3H, s, OCH3); 2.53–1.65 (1H, m, CH); 0.97 (6H, d, J = 7.0 Hz, 2CH3). MS (MALDI, positive mode, matrix DHB): m/z = 298 (M + Na)+. Anal. calcd for C14H17N3O3 (275.3) C, 61.08; H, 6.22; N, 15.26. found C, 61.17; H, 6.13; N, 15.21.
General Procedure for Syntheses of 2-(4-Oxo(3H)-1,2,3-benzotriazin-3-yl)alkanoic Acid Hydrazides 7a,b
To a solution of esters 6a,b (10.0 mmol) in ethanol (30 mL), hydrazine hydrate (3 mL, 48 mmol) was added. The reaction mixture was refluxed for 6 h; after cooling to room temperature, the precipitated hydrazide was filtered off, washed with water, and followed by recrystallization from aqueous ethanol.
2-(4-Oxo(3H)-1,2,3-benzotriazin-3-yl) Acetic Acid Hydrazide (7a)
A white solid (85%), mp: 142–144 °C. Lit mp: 146–147 °C.22
3-(4-Oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanoic Acid Hydrazide (7b)
Yellow crystals (80%), Rf = 0.38 (S2), mp: 165–168 °C. 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 9.11 (1H, s, NH); 8.27 (1H, d, J = 8.0 Hz, Ar–H); 8.21 (1H, d, J = 8.0 Hz, Ar–H); 8.12 (1H, t, J = 8.0 Hz, Ar–H); 7.96 (1H, t, J = 8.0 Hz, Ar–H); 4.61 (2H, t, J = 6.0 Hz, NCH2); 4.19 (2H, bs, NH2); 2.67 (2H, t, J = 6.0 Hz, CH2). 13C NMR (75 MHz, CDCl3), δ ppm, 169.2; 155.1 (2CO); 144.1; 135.8; 133.3; 128.4; 125.0; 119.7 (Ar–C); 46.5 (NCH2); 32.9 (CH2CO). MS (MALDI, positive mode, matrix DHB): m/z = 256 (M + Na)+. Anal. calcd for C10H11N5O2 (233.2) C, 51.50; H, 4.75; N, 30.03. found C, 51.65; H, 4.68; N, 30.10.
Syntheses of N-alkyl-2-(4-oxobenzotriazin-3(4H)-yl)alkanamides (9–10(a–h))
To a cold solution (− 5 °C) of hydrazides 7a,b (8.0 mmol) in acetic acid (60 mL), hydrochloric acid (5 N, 30 mL) was added portionwise under stirring a cold solution (0 °C) of sodium nitrite (0.7 g, 10.0 mmol) in water (30 mL). After stirring at the same temperature for 30 min, the in situ generated azide 8a,b was extracted with cold ethyl acetate and washed successively with cold water, 5% NaHCO3, and water. After drying over anhydrous sodium sulfate, the azide 8a,b was used without further purification in the next step. Amines (9.0 mmol) were added to the previously prepared cold dried solution of the azide 8a,b. Afterward, the mixture was kept for 12 h in the refrigerator and then at room temperature for another 12 h. The reaction mixture was washed with 0.1 N HCl, water, 5% NaHCO3, and water then dried over anhydrous sodium sulfate, the solvent was evaporated in vacuum and the residue was crystallized from ethyl acetate-petroleum ether to give products 9–10(a–h).
N-Isopropyl-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamide (9a)
White crystals (80%), Rf = 0.50 (S2), mp: 210–214 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.37 (1H, d, J = 8.0 Hz, Ar–H); 8.19 (1H, d, J = 8.0 Hz, Ar–H); 7.98 (1H, t, J = 7.6 Hz, Ar–H); 7.83 (1H, t, J = 7.6 Hz, Ar–H); 6.02 (1H, bs, NH); 5.07 (2H, s, NCH2CO); 4.15–4.10 (1H, m, CH); 1.20 (6H, d, J = 6.0 Hz, 2CH3). MS (MALDI, positive mode, matrix DHB): m/z = 269 (M + Na)+. Anal. calcd for C12H14N4O2 (246.3) C, 58.53; H, 5.73; N, 22.75. found C, 58.42; H, 5.89; N, 22.57.
N-Butyl-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamide (9b)
White crystals (83%), Rf = 0.38 (S2), mp: 192–194 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.28 (1H, d, J = 7.8 Hz, Ar–H); 8.12 (1H, d, J = 8.0 Hz, Ar–H); 7.91 (1H, t, J = 7.8 Hz, Ar–H); 7.76 (1H, t, J = 7.8 Hz, Ar–H); 6.06 (1H, s, NH); 5.02 (2H, s, NCH2CO); 3.25–3.20 (2H, m, CH2); 1.47–1.39 (2H, m, CH2); 1.31–1.22 (2H, m, CH2); 0.85 (3H, t, J = 7.2 Hz, CH3). (MALDI, positive mode, matrix DHB): m/z = 283 (M + Na)+. Anal. calcd for C13H16N4O2 (260.3) C, 59.99; H, 6.20; N, 21.52. found C, 60.22; H, 6.35; N, 21.48.
N-(tert-Butyl)-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamide (9c)
White crystals (83%), Rf = 0.45 (S1), mp: 216–220 °C. 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 8.39 (1H, d, J = 8.0 Hz, Ar–H); 8.21 (1H, d, J = 8.0 Hz, Ar–H); 8.01 (1H, t, J = 8.0 Hz, Ar–H); 7.85 (1H, t, J = 6.0 Hz, Ar–H); 5.78 (1H, bs, NH); 5.01 (2H, s, NCH2); 1.39 (9H, s, 3CH3). 13C NMR (75 MHz, CDCl3), δ ppm, 164.8; 155.8 (2CO); 144.4; 135.1; 132.5; 128.5; 125.2; 119.8 (Ar–C); 53.3 (C); 52.0 (NCH2); 28.8 (3CH3). (MALDI, positive mode, matrix DHB): m/z = 283 (M + Na)+. Anal. calcd for C13H16N4O2 (260.3) C, 59.99; H, 6.20; N, 21.52. found C, 59.89; H, 6.40; N, 21.48.
N-Dodecyl-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamide (9d)
White crystals (82%), Rf = 0.50 (S2), mp: 115–117 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.30 (1H, d, J = 7.6 Hz, Ar–H); 8.14 (1H, d, J = 8.0 Hz, Ar–H); 7.93 (1H, t, J = 7.6 Hz, Ar–H); 7.76 (1H, t, J = 7.6 Hz, Ar–H); 6.25 (1H, bs, NH); 5.03 (2H, s, NCH2CO); 3.88–3.06 (2H, m, NHCH2); 1.86 (2H, s, CH2); 1.57–1.52 (16H, m, 8CH2); 1.45–1.05 (2H, m, CH2); 0.83 (3H, t, J = 7.2 Hz, CH3). (MALDI, positive mode, matrix DHB): m/z = 395 (M + Na)+. Anal. calcd for C21H32N4O2 (372.5) C, 67.71; H, 8.66; N, 15.04. found C, 67.85; H, 8.60; N, 14.90.
N-Cyclohexyl-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamide (9e)
White crystals (85%), Rf = 0.36 (S2), mp: 240–242 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.30 (1H, d, J = 7.6 Hz, Ar–H); 8.13 (1H, d, J = 8.2 Hz, Ar–H); 7.92 (1H, t, J = 8.0 Hz, Ar–H); 7.77 (1H, t, J = 7.6 Hz, Ar–H); 5.81 (1H, bs, NH); 5.00 (2H, s, NCH2CO); 3.78–3.74 (1H, m, CH); 1.89–1.05 (10H, m, 5CH2). (MALDI, positive mode, matrix DHB): m/z = 309 (M + Na)+. Anal. calcd for C15H18N4O2 (286.3) C, 62.92; H, 6.34; N, 19.57. found C, 63.06; H, 6.25; N, 19.45.
N-Benzyl-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamide (9f)
White crystals (77%), Rf = 0.37 (S2), mp: 187–190 °C. 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 8.36 (1H, d, J = 8.0 Hz, Ar–H); 8.20 (1H, d, J = 8.0 Hz, Ar–H); 8.00 (1H, t, J = 8.0 Hz, Ar–H); 7.85 (1H, t, J = 8.0 Hz, Ar–H); 7.37–7.30 (5H, m, Ar–H); 6.54 (1H, bs, NH); 5.14 (2H, s, NCH2); 4.51 (2H, d, J = 6.0 Hz, HNCH2). 13C NMR (75 MHz, CDCl3), δ ppm, 166.0; 162.0 (2CO); 144.3; 137.6; 135.2; 132.7; 130.8; 128.7 (Ar–C); 128.6; 128.3; 127.8; 127.6; 127.0; 126.8 (Ar–C); 52.8 (NCH2); 43.9 (NCH2). (MALDI, positive mode, matrix DHB): m/z = 317 (M + Na)+. Anal. calcd for C16H14N4O2 (294.3) C, 65.30; H, 4.79; N, 19.04. found C, 65.42; H, 4.65; N, 18.87.
3-(2-Oxo-2-(piperidin-1-yl)ethyl)benzo[d][1,2,3]triazin-4(3H)-one (9g)
White crystals (65%), Rf = 0.45 (S2), mp:140–142 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.33–8.31 (1H, m, Ar–H); 8.15 (1H, d, J = 8.0 Hz, Ar–H); 7.95–7.90 (1H, m, Ar–H); 7.89–7.75 (1H, m, Ar–H); 5.28 (2H, s, NCH2CO); 3.58–3.51 (4H, m, 2NCH2); 1.68–1.54 (4H, m, 2CH2); 1.58 (2H, m,CH2). (MALDI, positive mode, matrix DHB): m/z = 295 (M + Na)+. Anal. calcd for C14H16N4O2 (272.3) C, 61.75; H, 5.92; N, 20.58. found C, 61.71; H, 5.80; N, 20.68.
3-(2-Morpholino-2-oxoethyl)benzo[d][1,2,3]triazin-4(3H)-one (9h)
White crystals (68%), Rf = 0.38 (S2), mp: 170–172 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.36 (1H, d, J = 8.0 Hz, Ar–H); 8.19 (1H, d, J = 8.0 Hz, Ar–H); 7.98 (1H, t, J = 7.6 Hz, Ar–H); 7.83 (1H, t, J = 8.0 Hz, Ar–H); 4.21 (2H, s, NCH2CO); 3.82–3.61 (4H, m, 2OCH2); 3.00 (4H, t, J = 4.8 Hz, 2NCH2). (MALDI, positive mode, matrix DHB): m/z = 297 (M + Na)+. Anal. calcd for C13H14N4O3 (274.3) C, 56.93; H, 5.14; N, 20.43. found C, 56.85; H, 5.04; N, 20.46.
N-Isopropyl-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamide (10a)
White crystals (82%), Rf = 0.30 (S2), mp: 175–178 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.32 (1H, d, J = 8.0 Hz, Ar–H); 8.13 (1H, d, J = 8.0 Hz, Ar–H); 7.95–7.91 (1H, m, Ar–H); 7.81(1H, t, J = 7.6 Hz, Ar–H); 5.85 (1H, bs, NH); 4.79 (2H, t, J = 7.2 Hz, NCH2); 4.11–4.02 (1H, m, NHCH); 2.83 (2H, t, J = 7.2 Hz, CH2CO); 1.12 (6H, d, J = 6.4 Hz, 2CH3). (MALDI, positive mode, matrix DHB): m/z = 283 (M + Na)+. Anal. calcd for C13H16N4O2 (260.3) C, 59.99; H, 6.20; N, 21.52. found C, 60.14; H, 6.14; N, 21.48.
N-Butyl-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamide (10b)
White crystals (85%), Rf = 0.40 (S2), mp: 140–142 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.04 (1H, d, J = 7.6 Hz, Ar–H); 8.13 (1H, d, J = 8.0 Hz, Ar–H); 7.95 (1H, t, J = 7.6 Hz, Ar–H); 7.80 (1H, t, J = 7.6 Hz, Ar–H); 6.09 (1H, bs, NH); 4.79 (2H, t, J = 7.2 Hz, NCH2); 3.36–3.14 (2H, m, NHCH2); 2.87 (2H, t, J = 7.2 Hz, CH2CO); 1.47–1.40 (2H, m, CH2); 1.32–1.23 (2H, m, CH2); 0.88(3H, t, J = 7.2 Hz, CH3). (MALDI, positive mode, matrix DHB): m/z = 297 (M + Na)+. Anal. calcd for C14H18N4O2 (274.3) C, 61.30; H, 6.61; N, 20.42. found C, 61.45; H, 6.55; N, 20.34.
N-(tert-Butyl)-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamide (10c)
White crystals (83%), Rf = 0.38 (S2), mp: 145–1 5°C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.04 (1H, d, J = 8.0 Hz, Ar–H); 7.86 (1H, d, J = 8.0 Hz, Ar–H); 7.68(1H, t, J = 7.6 Hz, Ar–H); 7.58(1H, t, J = 7.6 Hz, Ar–H); 5.51(1H, bs, NH); 4.50 (2H, t, J = 7.2 Hz, NCH2); 2.52 (2H, t, J = 7.6 Hz, CH2CO); 1.05 (9H, s, 3CH3). (MALDI, positive mode, matrix DHB): m/z = 297 (M + Na)+. Anal. calcd for C14H18N4O2 (274.3) C, 61.30; H, 6.61; N, 20.42. found C, 61.17; H, 6.70; N, 20.50.
N-Dodecyl-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamide (10d)
White crystals (88%), Rf = 0.38 (S2), mp: 115–118 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.36 (1H, d, J = 7.8 Hz, Ar–H); 8.17 (1H, d, J = 8.0 Hz, Ar–H); 7.98 (1H, t, J = 7.8 Hz, Ar–H); 7.84 (1H, t, J = 7.8 Hz, Ar–H); 5.87 (1H, bs, NH); 4.82 (2H, t, J = 7.2 Hz, NCH2); 3.27–3.22 (2H, m, NHCH2); 2.88 (2H, t, J = 7.2 Hz, CH2CO); 1.48 (2H, t, J = 6.8 Hz, CH2); 1.33–1.27 (18H, m, 9CH2); 0.89 (3H, t, J = 7.2 Hz, CH3) (MALDI, positive mode, matrix DHB): m/z = 409 (M + Na)+. Anal. calcd for C22H34N4O2 (386.5) C, 68.36; H, 8.87; N, 14.49. found C, 68.40; H, 8.81; N, 14.52.
N-Cyclohexyl-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamide (10e)
White crystals (83%), Rf = 0.25 (S2), mp: 138–140 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.39 (1H, d, J = 7.8 Hz, Ar–H); 8.20 (1H, d, J = 8.0 Hz, Ar–H); 8.00 (1H, t, J = 8 Hz, Ar–H); 7.86 (1H, t, J = 8 Hz, Ar–H); 5.47 (1H, bs, NH); 4.83 (2H, t, J = 6.8 Hz, NCH2); 2.86 (2H, t, J = 6.8 Hz, CH2CO); 2.02–1.07 (11H, m, CH, 5CH2). (MALDI, positive mode, matrix DHB): m/z = 323 (M + Na)+. Anal. calcd for C16H20N4O2 (300.4) C, 63.98; H, 6.71; N, 18.65. found C, 64.09; H, 6.63; N, 18.59.
N-Benzyl-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamide (10f)
White crystals (77%), Rf = 0.35 (S2), mp: 145–150 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.28 (1H, d, J = 8.0 Hz, Ar–H); 8.13 (1H, d, J = 8.0 Hz, Ar–H); 7.96–7.92 (1H, m, Ar–H); 7.80 (1H, t, J = 7.6 Hz, Ar–H); 7.34–7.22 (5H, m, Ar–H); 6.37 (1H, s, NH); 4.82 (2H, t, J = 7.2 Hz, NCH2); 4.44 (2H, d, J = 5.6 Hz, NHCH2); 2.92 (2H, t, J = 7.2 Hz, CH2CO). (MALDI, positive mode, matrix DHB): m/z = 331 (M + Na)+. Anal. calcd for C17H16N4O2 (308.3) C, 66.22; H, 5.23; N, 18.17. found C, 66.02; H, 5.30; N, 18.10.
3-(3-Oxo-3-(piperidin-1-yl)propyl)benzo[d][1,2,3]triazin-4(3H)-one (10g)
White crystals (75%), Rf = 0.38 (S2), mp: 128–130 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.33 (1H, d, J = 8.0 Hz, Ar–H); 8.13 (1H, d, J = 8.0 Hz, Ar–H); 7.95 (1H, t, J = 7.8 Hz, Ar–H); 7.80 (1H, t, J = 8.0 Hz, Ar–H); 4.80 (2H, t, J = 7.6 Hz, NCH2); 3.56–3.27 (4H, m, 2NCH2); 2.99 (2H, t, J = 7.6 Hz, CH2CO); 1.63–1.54 (6H, m, 3CH2). (MALDI, positive mode, matrix DHB): m/z = 309 (M + Na)+. Anal. calcd for C15H18N4O2 (286.3) C, 62.92; H, 6.34; N, 19.57. found C, 62.82; H, 6.40; N, 19.47.
3-(3-Morpholino-3-oxopropyl)benzo[d][1,2,3]triazin-4(3H)-one (10h)
White crystals (67%), Rf = 0.38 (S2), mp: 117–120 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.33–8.08 (1H, m, Ar–H); 8.14–8.11 (1H, m, Ar–H); 7.93–7.59 (1H, m, Ar–H); 7.53–7.48(1H, m, Ar–H); 4.76–4.72 (2H, m, NCH2); 3.62–3.56 (4H, m, 2OCH2); 3.44–3.39 (4H, m, 2NCH2); 2.94–2.90 (2H, m, CH2CO). (MALDI, positive mode, matrix DHB): m/z = 311 (M + Na)+. Anal. calcd for C14H16N4O3 (288.3) C, 58.32; H, 5.59; N, 19.43. found C, 58.23; H, 5.65; N, 19.31.
General Procedure for Dipeptides Methyl 2-(2-(4-Oxobenzotriazin-3(4H)-yl)alkanamido)alkanoates (Dipeptides) (11–12(a–e))
To a cold solution (−5 °C) of hydrazides (7a,b) (1.6 mmol) in acetic acid (12 mL), hydrochloric acid (5 N, 6 mL) and water (50 mL) were added portionwise under stirring a cold solution (0 °C) of sodium nitrite (0.14 g, 2.0 mmol) in water (6 mL). After stirring at the same temperature for 30 min, the azides 8a,b were extracted with cold ethyl acetate and washed successively with cold water, 5% NaHCO3, and water. After drying over anhydrous sodium sulfate, azides were used directly without further purification in the next step. Amino acid methyl ester hydrochlorides (1.8 mmol) were stirred in ethyl acetate (50 mL) with triethyl amine (0.2 mL) at 0 °C for 20 min. The formed triethyl amine hydrochloride was filtered off, and the filtrate was added to the previously prepared cold-dried solution of the azide. Afterward, the mixture was kept for 12 h in the refrigerator and then at room temperature for another 12 h. The reaction mixture was washed with 0.1 N HCl, water, 5% NaHCO3, and water then dried over anhydrous sodium sulfate. The solvent was evaporated in vacuum, and the residue was crystallized from ethyl acetate-petroleum ether to give the appropriate products 11–12(a–-e).
Methyl-2-(2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamido)acetate (11a)
White crystals (77%), Rf = 0.20 (S2), mp: 120–124 °C. 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 8.38 (1H, d, J = 8.0 Hz, Ar–H); 8.21 (1H, d, J = 8.0 Hz, Ar–H); 8.0 (1H, t, J = 8.2 Hz, Ar–H); 7.85 (1H, t, J = 8.2 Hz, Ar–H); 6.79 (1H, bs, NH); 5.21 (2H, s, NCH2); 4.14 (2H, d, J = 6.0 Hz, CH2CO); 3.77 (3H, s, OCH3). 13C NMR (75 MHz, CDCl3), δ ppm, 170.1; 166.3; 155.9 (3CO); 144.3; 135.2; 132.7; 128.6; 123.2; 119.7 (Ar–C); 52.6 (NCH2); 52.5 (OCH3); 41.4 (NCH2CO) (MALDI, positive mode, matrix DHB): m/z = 299 (M + Na)+. Anal. calcd for C12H12N4O4 (276.3) C, 52.17; H, 4.38; N, 20.28. found C, 52.03; H, 4.30; N, 20.34.
Methyl-4-methyl-2-[2-(4-oxo-4H-benzo[d][1,2,3]triazin-3-yl)-acetylamino]pentanoate (11b)
White crystals (72%), Rf = 0.44 (S2), mp: 131–132 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.28 (1H, d, J = 8.0 Hz, Ar–H); 8.07 (1H, d, J = 8.0 Hz, Ar–H); 7.99 (1H, t, J = 7.8 Hz, Ar–H); 7.79 (1H, t, J = 7.8 Hz, Ar–H); 6.26 (1H, bs, NH); 5.12 (2H, s, NCH2); 4.64–4.59 (1H, m, CH); 3.71 (3H, s, OCH3); 1.89 (1H, m, CH); 1.60–1.49 (2H, m, CH2); 0.92 (6H, t, J = 6.0 Hz, 2CH3). (MALDI, positive mode, matrix DHB): m/z = 355 (M + Na)+. Anal. calcd for C16H20N4O4 (332.4) C, 57.82; H, 6.07; N, 16.86. found C, 57.78; H, 6.11; N, 16.98.
Methyl-4-[2-(4-Oxo-4H-benzo[d][1,2,3]triazin-3-yl)-acetylamino]butanoate (11c)
White crystals (81%), Rf = 0.23 (S2), mp: 102–104 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.37 (1H, d, J = 7.8 Hz, Ar–H); 8.20 (1H, d, J = 7.8 Hz, Ar–H); 7.98 (1H, t, J = 7.8 Hz, Ar–H); 7.88 (1H, t, J = 7.8 Hz, Ar–H); 6.12 (1H, bs, NH); 5.07 (2H, s, NCH2); 3.70 (3H, s, OCH3); 3.33–3.31 (2H, m, NHCH2); 2.34 (2H, t, J = 7.2 Hz, CH2CO); 1.85–1.80 (2H, m, CH2). (MALDI, positive mode, matrix DHB): m/z = 327 (M + Na)+. Anal. calcd for C14H16N4O4 (304.3) C, 55.26; H, 5.30; N, 18.41. found C, 55.32; H, 5.37; N, 18.33.
Methyl-3-(2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamido)propanoate (11d)
White crystals (77%), Rf = 0.35 (S2), mp: 285–290 °C. 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 8.39 (1H, d, J = 8.0 Hz, Ar–H); 8.22 (1H, d, J = 8.0 Hz, Ar–H); 8.01 (1H, m, Ar–H); 7.86 (1H, m, Ar–H); 5.87 (1H, bs, NH); 5.12 (2H, s, NCH2); 3.70 (3H, s, OCH3); 3.64–3.5 (2H, m, NHCH2); 2.62 (2H, t, J = 6.8 Hz, CH2CO). (MALDI, positive mode, matrix DHB): m/z = 313 (M + Na)+. Anal. calcd for C13H14N4O4 (290.3) C, 53.79; H, 4.86; N, 19.30. found C, 53.60; H, 4.72; N, 19.21.
Methyl-3-(1H-indol-2-yl)-2-(2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetamido)propanoate (11e)
White crystals (78%), Rf = 0.23 (S2), mp: 202–204 °C. 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 10.93 (1H, s, NH); 8.90 (1H, d, J = 8.0 Hz, Ar–H); 8.28–7.94 (3H, m, Ar–H); 7.54–7.02 (4H, m, Ar–H); 7.00 (1H, bs, NH); 6.7 (1H, s, ArH); 5.11 (2H, s, NCH2); 4.67 (1H, t, J = 7.0 Hz, CH); 3.61 (3H, s, OCH3); 3.38–3.15 (2H, m, CH2). 13C NMR (75 MHz, CDCl3), δ ppm, 172.4; 166.7; 155.3 (3CO); 144.3; 136.6; 136.0; 135.0; 132.6; 129.3; 128.5; 127.6; 125.0; 124.3; 121.5; 119.0; 111.9; 109.6 (Ar–C); 53.9 (CHCO); 52.4 (NCH2); 51.9 (OCH3); 27.7(CHCH2). (MALDI, positive mode, matrix DHB): m/z = 428 (M + Na)+. Anal. calcd for C21H19N5O4 (405.4) C, 62.22; H, 4.72; N, 17.27. found C, 62.10; H, 4.78; N, 17.18.
Methyl-2-(3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamido)acetate (12a)
White crystals (58%), Rf = 0.11(S2), mp: 220–224 °C. 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 8.26 (1H, d, J = 8.0 Hz, Ar–H); 8.18 (1H, d, J = 8.0 Hz, Ar–H); 8.10 (1H, t, J = 8.0 Hz, Ar–H); 7.94 (1H, t, J = 8.0 Hz, Ar–H); 6.54 (1H, bs, NH); 4.38 (2H, d, J = 6.8 Hz, NHCH2CO); 4.07–4.00 (2H, m, NCH2); 3.31 (3H, s, OCH3); 1.21 (2H, t, J = 7.0 Hz, CH2CO). 13C NMR (75 MHz, CDCl3), δ ppm, 170.8; 158.4, 155.4 (3CO); 144.2; 136.6; 133.1; 128.3; 125.0; 119.9 (Ar–C); 60.2 (OCH3); 50.33 (NCH2); 38.3 (NHCH2); 21.2 (CH2CO). (MALDI, positive mode, matrix DHB): m/z = 313 (M + Na)+. Anal. calcd for C13H14N4O4 (290.3) C, 53.79; H, 4.86; N, 19.30. found C, 53.84; H, 4.81; N, 19.41.
Methyl-4-methyl-2-(3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamido)pentanoate(12b)
White crystals (75%), Rf = 0.49 (S2), mp: 138–140 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.32 (1H, d, J = 8.0 Hz, Ar–H); 8.15 (1H, d, J = 8.0 Hz, Ar–H); 7.97 (1H, t, J = 7.6 Hz, Ar–H); 7.82 (1H, t, J = 7.6 Hz, Ar–H); 6.35 (1H, d, J = 7.2 Hz, NH); 4.85–4.74 (2H, m, NCH2); 4.67–4.58 (1H, m, NHCH); 3.69 (3H, s, OCH3); 2.95 (2H, d, J = 6.8 Hz, CH2CO); 1.88 (1H, m, CH); 1.62–1.48 (2H, m, CH2); 0.89 (6H, t, J = 6.6 Hz, 2CH3). (MALDI, positive mode, matrix DHB): m/z = 369 (M + Na)+. Anal. calcd for C17H22N4O4 (346.4) C, 58.95; H, 6.40; N, 16.17. found C, 58.79; H, 6.28; N, 16.18.
Methyl-4-(3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamido)butanoate (12c)
White crystals (86%), Rf = 0.25 (S2), mp: 111–115 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.38 (1H, d, J = 7.6 Hz, Ar–H); 8.19 (1H, d, J = 7.6 Hz, Ar–H); 8.00 (1H, t, J = 7.6 Hz, Ar–H); 7.85 (1H, t, J = 7.6 Hz, Ar–H); 6.07 (1H, bs, NH); 4.82 (2H, t, J = 7.2 Hz, NCH2); 3.68 (3H, s, OCH3); 3.35–3.30 (2H, m, NHCH2); 2.89 (2H, t, J = 7.2 Hz, CH2CO); 2.39 (2H, t, J = 7.2 Hz, CH2CO); 1.88–1.81 (2H, m, CH2). (MALDI, positive mode, matrix DHB): m/z = 341 (M + Na)+. Anal. calcd for C15H18N4O4 (318.3) C, 56.60; H, 5.70; N, 17.60. found C, 56.46; H, 5.77; N, 17.61.
Methyl-3-[3-(4-oxo-4H-benzo[d][1,2,3]triazin-3-yl)-propionylamino]propanoate (12d)
White crystals (74%), Rf = 0.34 (S2), mp: 285–290 °C. 1H NMR (300.0 MHz, CDCl3), δ ppm, (J, Hz): 8.37 (1H, d, J = 8.0 Hz, Ar–H); 8.26 (1H, d, J = 8.0 Hz, Ar–H); 7.98 (1H, m, Ar–H); 7.86 (1H, m, Ar–H); 6.05 (1H, bs, NH); 3.69 (3H, s, OCH3); 3.62–3.56 (2H, m, NHCH2); 3.25–3.22 (2H, m, NCH2); 2.55 (2H, t, J = 6.0 Hz, CH2CO); 2.38 (2H, t, J = 6.6 Hz, CH2CO). (MALDI, positive mode, matrix DHB): m/z = 327 (M + Na)+. Anal. calcd for C14H16N4O4 (304.3) C, 55.26; H, 5.30; N, 18.41. found C, 55.37; H, 5.23; N, 18.52.
Methyl-2-(1H-indol-3-yl)-2-(3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanamido)acetate (12e)
White crystals (83.3%), Rf = 0.26 (S2), mp: 176–178 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.25 (1H, d, J = 8.0 Hz, Ar–H); 8.07 (1H, d, J = 8.0 Hz, Ar–H); 7.88 (1H, t, J = 7.6 Hz, Ar–H); 7.73 (1H, t, J = 7.6 Hz, Ar–H); 7.69–7.42 (1H, m, Ar–H); 7.29–7.24 (1H, m, Ar–H); 7.10–7.06 (1H, m, Ar–H); 7.03–7.00 (1H, m, Ar–H); 6.92 (1H, s, NH); 6.20 (1H, d, J = 7.6 Hz, NH); 4.91–4.88 (1H, m, CH); 4.69 (2H, d, J = 7.2 Hz, NCH2); 3.59 (3H, s, OCH3); 3.25 (1H, t, J = 8.8 Hz, NHCH); 2.77 (2H, t, J = 7.2 Hz, CH2CO). (MALDI, positive mode, matrix DHB): m/z = 428 (M + Na)+. Anal. calcd for C21H19N5O4 (405.4) C, 62.22; H, 4.72; N, 17.27. found C, 62.20; H, 4.81; N, 17.42.
Synthesis of N′-(4-Arylidene)-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)alkanoic Acid Hydrazide (13–14(a,b))
A mixture of hydrazides 7a,b (1.0 mmol) and carbonyl compound (1.0 mmol) was refluxed in ethanol (20 mL) for 6 h. After cooling to room temperature, the resulting solid was filtered off, washed with cold ethanol, and recrystallized from aqueous ethanol gave proper products 13–14(a,b).
N′-(4-Methoxybenzylidene)-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetohydrazide (13a)
White crystals (80%), Rf = 0.43 (S2), mp: 258–260 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 9.82 (1H, s, CH=); 8.80 (1H, d, J = 8 Hz, Ar); 8.33–6.87 (8H, m, NH & 2Ar–H); 5.64 (2H, s, NCH2CO); 3.79 (3H, s, OCH3). (MALDI, positive mode, matrix DHB): m/z = 360 (M + Na)+. Anal. calcd for C17H15N5O3 (337.3) C, 60.53; H, 4.48; N, 20.76. found C, 60.63; H, 4.36; N, 20.80.
N′-(4-(Dimethylamino)benzylidene)-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetohydrazide (13b)
White crystals (82%), Rf = 0.40 (S2), mp: 226–229 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.99 (1H, s, CH); 8.71 (1H, d, J = 8 Hz, Ar); 8.33–6.62 (8H, m, NH and 2Ar–H); 5.63 (2H, s, NCH2CO); 2.97 (6H, s, 2NCH3). (MALDI, positive mode, matrix DHB): m/z = 373 (M + Na)+. Anal. calcd for C18H18N6O2 (350.4) C, 61.70; H, 5.18; N, 23.99. found C, 61.73; H, 5.27; N, 24.07.
N′-(4-Methoxybenzylidene)-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanehydrazide (14a)
White crystals (87%), Rf = 0.75 (S2), mp: 215–217 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 9.82 (1H,s, CH=); 8.97 (1H, bs, NH); 8.26–8.01 (1H, m, Ar–H); 7.99–7.78 (1H, m, Ar–H); 7.67–7.64 (1H, m, Ar–H); 7.54 (1H, m, Ar–H); 7.41–7.38 (1H, m, Ar–H); 6.95–9.93 (1H, m, Ar–H); 6.85–6.83 (1H, m, Ar–H); 6.78–6.76 (1H, m, Ar–H); 4.85 (2H, t, J = 6.0 Hz, NCH2); 3.76 (3H, s, OCH3); 3.34 (2H, t, J = 7.2 Hz, CH2CO). (MALDI, positive mode, matrix DHB): m/z = 374 (M + Na)+. Anal. calcd for C18H17N5O3 (351.4) C, 61.53; H, 4.88; N, 19.93. found C, 61.48; H, 4.97; N, 19.89.
N′-(4-(Dimethylamino)benzylidene)-3-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)propanehydrazide (14b)
White crystals (81%), Rf = 0.75 (S2), mp: 228–230 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 8.89 (1H, bs, NH); 8.30 (1H, s, N=CH); 8.27–8.09 (1H, m, Ar–H); 8.02–7.89 (1H, m, Ar–H); 7.83–7.79 (1H, m, Ar–H); 7.76–7.75 (1H, m, Ar–H); 7.69–7.62 (1H, m, Ar–H); 7.53 (1H, m, Ar–H); 7.48–7.46 (1H, m, Ar–H); 7.45–7.39 (1H, m, Ar–H); 4.85 (2H, t, J = 7.2 Hz, NCH2); 3.34 (2H, t, J = 7.2 Hz, CH2CO); 2.97 (6H, s, 2NCH3). (MALDI, positive mode, matrix DHB): m/z = 387 (M + Na)+. Anal. calcd for C19H20N6O2 (364.4) C, 62.62; H, 5.53; N, 23.06. found C, 62.57; H, 5.42; N, 23.10.
Synthesis of 3-(4-Oxo-4H-benzo[d][1,2,3]triazin-3-yl)-propionic Acid (1,2,3,4,5-Pentahydroxy-pentylidene)-hydrazide (15)
A mixture of hydrazide 7b (1.0 mmol) and arabinose (1.0 mmol) was refluxed in ethanol (20 mL) for 6 h. After cooling to room temperature, the resulting solid was filtered, washed with cold ethanol, and recrystallized from aqueous ethanol. White crystals (87.5%), Rf = 0.30 (S2), mp: 302–304 °C. 1H NMR (400.0 MHz, CDCl3), δ ppm, (J, Hz): 11.10 (1H, s, NH); 8.21–7.40 (4H, m, Ar–H); 5.43–4.24 (4H, m, 2CH2); 3.61–2.52 (10H, m, arabinose). (MALDI, positive mode, matrix DHB): m/z = 404 (M + Na)+. Anal. calcd for C15H19N5O7 (381.3) C, 47.24; H, 5.02; N, 18.37. found C, 47.17; H, 5.12; N, 18.27.
Experimental Part of Biology
Experimental Part for Antimicrobial Studies
The concentration was 200 ppm in DMF.24 Samples (20 μL each) were analyzed using “Disc diffusion method”1″ against the following indicator strains:
-
a.
E. coli: “Escherichia coli”
-
b.
Staph: “Staphylococcus aureus (NCMB6571) (laboratory culture)”
-
c.
Salmonella spp. typhimurium
As shown in the Table 2, most of compounds more effective against E. coli in compared with staph and Salmonella spp. Most of compounds are in comparison with the starting material 3, which gave a negative result exposed a good activity against bacteria like; compounds 9a, 11a, 12a, 12b, 12c, 13a, 13b, and 14b. This data confirmed the identity between the experimental data and molecular docking data. Moreover, clear increasing for activity of the starting material benzotriazinone compound against bacteria gave a positive impression.
Table 2. Anti-bacterial Activity of the Compounds against E. coli, Staph, and Salmonella sppa.
| sample | E. coli | Staph | Salmonella spp. |
|---|---|---|---|
| control | –ve | –ve | –ve |
| (3) | –ve | –ve | –ve |
| (7b) | –ve | –ve | 1.0 |
| (9a) | 0.9 | –ve | –ve |
| (10f) | 0.7 | –ve | –ve |
| (11a) | 1.0 | –ve | 0.9 |
| (12a) | 1.1 | –ve | –ve |
| (12b) | 1.0 | –ve | –ve |
| (12c) | 1.1 | –ve | –ve |
| (13a) | 1.1 | –ve | –ve |
| (13b) | 1.1 | –ve | –ve |
| (14b) | 1.1 | –ve | –ve |
Results are presented as diameter of inhibitory zone in centimeter (cm).
Experimental Part of Anti-cancer Activity (HEPG −2 Cell Line)
In Vitro Anti-cancer Activity
Potential cytotoxicity of the newly synthesized compounds was tested against the human liver carcinoma cell line (HepG2) using the method of Hansen et al.19 The in vitro anticancer screening was done by planting a tissue unit in Vacsera, Cairo A. R. Egypt.
This assay was performed by a modification (Hansen et al., 1989) of the tetrazolium salt (MTT) method (Mosmann, 1983).20 Preferably, cells should be planted in triplicate wells. Relative cell proliferation/viability was measured when treated cells are compared with untreated cells.
-
1.
To each well of a 96-well flat-bottomed plate, add 25 μL of MTT (5 mg/mL stock solution).
-
2.
Incubate 2 h at 37o C.
-
3.
Add 100 μL of extraction buffer.
-
4.
Incubate overnight at 37° C.
-
5.
Read OD570.
-
6.
Include negative controls of no cells and extraction buffer as the blank.
-
7.
Note: it is not necessary to remove medium prior to addition of reagents or to mix reagents in the well.
Reagent Preparation
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylterazolium bromide; Sigma cat. no. M2128) is dissolved at a concentration of 5 mg/mL in sterile PBS at room temperature. The filter sterilized and stored in a dark container at 4 °C. Prepare fresh each month, extraction buffer 20% (W.V) was dissolved at 37 °C in a solution of 50% DMF (N,N-dimethyl formamide; Fluka cat. no. 40250) and 50% SDW. Adjust pH to 4.7 by adding 2.5% of 80% acetic acid and 2.5% 1 N HCl.
This is a colorimetric assay that measures the reduction of yellow 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) by mitochondrial succinate dehydrogenase. The MTT enters the cells and passes into the mitochondria where it is reduced to an insoluble, colored (dark purple) form azan product. The cells are then solubilized with an organic solvent (e.g., isopropanol), and the released, solubilized formazan reagent is measured spectrophotometrically. Since reduction of MTT can only occur in metabolically active cells, the level of activity is a measure of the viability of the cells.
Materials
MTT (5 mg/mL in PBS), filter and keep dark, prepare freshly acidic isopropanol (0.1 N HCl in absolute isopropanol) 96-well plate (flat bottom).
Procedure
-
1.
Plate cells (104–106 cells) in 200 mL of PBS in 96-well (flat bottom).
-
2.
Add 20 mL of MTT solution, mix well.
-
3.
Incubate for 4 h in 37 °C in dark.
-
4.
Remove the aliquot for analysis, add 200 mL acidic isopropanol, and mix well.
-
5.
Incubate additional 1 h in dark at 37 °C.
-
6.
Read plate in the ELISA reader, measure OD in 570 nm (the background wavelength is 630 nm).
As shown in the Table 1, the compound 3 shown an IC50 of 6.525 μg/mL whereas the derivatives 11e shown an IC50 of 12.40 μg/mL, 13a shown an IC50 of 10.97 μg/mL, and compound 12e shown an IC50 of 59.85 μg/mL. In comparing with the Doxorubicin standard IC50 of 2.06 μg/mL, We found that the starting material benzotriazinone 3 was the highest active compound having an IC50 of 6.525 μg/mL; this result showed that the starting material compound 3 need more attention as an anti-cancer drugs. We hope that these potential anticancer compounds could offer an effective treatment that is less likely to cause resistance and recurrence of cancer and less toxic to normal tissues than available chemotherapeutic agents.
Table 1. IC50 Values for Compounds 3, 11e, 12e, and 13a Tested on HEPG-2 Cell Line.
| compound | HEPG-2 IC50 (μg/mL) |
|---|---|
| stand doxorubicin | 2.06 |
| 3 | 6.525 |
| 11e | 12.4019 |
| 12e | 59.85 |
| 13a | 10.97 |
Acknowledgments
We would like to thank the Science & Technology Development Fund in Egypt STDF Project ID: 22909 for funding this research proposal.
The authors declare no competing financial interest.
References
- El-Rayes S.; Gomaa M. S.; Abouelmagd A.; Fathalla W.; Ali I. A. I. Synthesis and antiproliferative assay of triazolyl-2,2-dimethyl-3-phenylpropanoates as potential HDAC inhibitors. RSC Adv. 2019, 9, 13896. 10.1039/C9RA01277J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Rayes S. M.; Aboelmagd A.; Gomaa M. S.; Ali I. A. I.; Fathalla W.; Pottoo F. H.; Khan F. A. Convenient synthesis and antiproliferative activity of methyl 2-[3-(3-phenyl-quinoxalin-2-ylsulfanyl)propanamido]alkanoates and N-Alkyl 3-((3-phenylquinoxalin-2-yl)sulfanyl) propanamides. ACS Omega 2019, 4, 18555–18566. 10.1021/acsomega.9b02320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammaiah G.; Sarangapani M. Synthesis and Characterization of Biologically Significant 3-(2-Oxo-1,2,dihydroindol-3-ylideneamino)-3H-benzo[d][1,2,3] triazin-4-ones. Asian J. Chem. 2008, 20, 282. [Google Scholar]
- Daidone G.; Plescia S.; Raffa D.; Bajardi M. L.; Matera M.; Caruso A.; Leone M. G. Researches on anti-inflammatory agents. Studies on some new 3-(pyrazol-5yl)-1,2,3-benzotriazin-4-(3H)-ones and quinazolin −4 (3H)–ones. Farmaco 1990, 45, 391. [PubMed] [Google Scholar]
- Fiorino F.; Severino B.; De Angelis F.; Perissutti E.; Frecentese F.; Massarelli P.; Bruni G.; Collavoli E.; Santagada V.; Caliendo G. Synthesis and in-vitro pharmacological evaluation of new 5-HT1A receptor ligands containing a benzotriazinone nucleus. Arch. Pharm. 2008, 341, 20. 10.1002/ardp.200700151. [DOI] [PubMed] [Google Scholar]
- Takai H.; Obase H.; Nakamizo N.; Teranishi M.; Kubo K.; Shuto K.; Hashimoto T. Synthesis and pharmacological evaluation of piperidine derivatives with various heterocyclic rings at the 4-position. Chem. Pharm. Bull. 1985, 33, 1104. 10.1248/cpb.33.1104. [DOI] [PubMed] [Google Scholar]
- Fiorino F.; Caliendo G.; Perissutti E.; Severino B.; Frecentese F.; Preziosi B.; Izzo A. A.; Capasso R.; Santagada V. Synthesis by microwave irradiation and anti-diarrhoeal activity of benzotriazinone and saccharine derivatives. Arch. Pharm. 2005, 338, 548. 10.1002/ardp.200500134. [DOI] [PubMed] [Google Scholar]
- Caliendo G.; Fiorino F.; Grieco P.; Perissutti E.; Santagada V.; Meli R.; Raso G. M.; Zanesco A.; De Nucci G. Preparation and local anaesthetic activity of benzotriazinone and benzoyltriazole derivatives. Eur. J. Med. Chem. 1999, 34, 1043. 10.1016/S0223-5234(99)00126-9. [DOI] [Google Scholar]
- Raffa D.; Daidone G.; Maggio B.; Schillaci D.; Plescia F. Synthesis and antiproliferative activity of novel 3-(indazol-3-yl)-quinazolin-4(3H)-one and 3-(indazol-3-yl)-benzotriazin-4(3H)-one derivatives. Arch. Pharm. 1999, 332, 317.. [DOI] [PubMed] [Google Scholar]
- Jollimore J. V.; Vaughan K.; Hooper D. L. 1-Aryl-3-(carbamoylmethyl)triazenes: Synthesis, Spectroscopic Analysis and Cyclization to New 1,2,3-Benzotriazines. J. Org. Chem. 1996, 61, 210. 10.1021/jo951279i. [DOI] [Google Scholar]
- Witty D. R.; Bateson J.; Hervieu G. J.; Al-Barazanji K.; Jeffrey P.; Hamprecht D.; Haynes A.; Johnson C. N.; Muir A. I.; O’Hanlon P. J.; Stemp G.; Stevens A. J.; Thewlis K.; Winborn K. Y. Discovery of potent and stable conformationally constrained analogues of the MCH R1 antagonist SB-568849. Bioorg. Med. Chem. Lett. 2006, 16, 4872. 10.1016/j.bmcl.2006.06.061. [DOI] [PubMed] [Google Scholar]
- Kelson A. B.; McNamara J. P.; Pandey A.; Ryan K. J.; Dorie M. J.; McAfee P. A.; Menke D. R.; Brown J. M.; Tracy M. 1,2,4-Benzotriazine 1,4-dioxides. An important class of hypoxic cytotoxins with antitumor activity. Anti-Cancer Drug Des. 1998, 13, 575. [PubMed] [Google Scholar]
- Vaisburg A.; Paquin I.; Bernstein N.; Frechette S.; Gaudette F.; Leit S.; Moradei O.; Raeppel S.; Zhou N.; Bouchain G.; Woo S. H.; Jin Z.; Gillespie J.; Wang J.; Fournel M.; Yan P. T.; Trachy-Bourget M.-C.; Robert M.-F.; Lu A.; Yuk J.; Rahil J.; Macleod A. R.; Besterman J. M.; Li Z.; Delorme D. N-(2-Amino-phenyl)-4-(heteroarylmethyl)-benzamides as new histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6729. 10.1016/j.bmcl.2007.10.050. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Suzuki K.; Singh S. K. N-Acylation in combinatorial chemistry. ARKIVOC 2004, 1, 12. 10.3998/ark.5550190.0005.102. [DOI] [Google Scholar]
- David F.; Roman J. S.; Donna MP. Diet and Cancer 2005. pp 53–63.
- El Fekki I.; Ali I. A. I.; Fathalla W.; Alsheikh A. A.; El Tamney S. Synthesis of methyl [3-alkyl-2-(2,4-dioxo-3,4-dihydro-2H-quinazolin-1-yl)-acetamido]alkanoate. ARKIVOC 2017, 2017, 104. [Google Scholar]
- Megahed M.; Fathalla W.; Alsheikh A. A. Synthesis and Antimicrobial Activity of Methyl 2-(2-(2-Arylquinazolin-4-yl)oxy)Acetylamino Alkanoates. J. Heterocyclic Chem. 2018, 55, 2799. 10.1002/jhet.3347. [DOI] [Google Scholar]
- El Rayes S. M. Convenient synthesis of some methyl-N-[2-(3-oxo-6-p-tolyl-2,3,4,5-tetrahydropyridazin-2-yl)-acetylamino]amino acid esters. ARKIVOC 2008, 2008, 243–254. 10.3998/ark.5550190.0009.g23. [DOI] [Google Scholar]
- Fletcher S.; Hamilton A. D. Targeting protein–protein interactions by rational design: mimicry of protein surfaces. J. R. Soc., Interface 2006, 3, 215. 10.1098/rsif.2006.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M. B.; Nielsen S. E.; Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 1989, 119, 203–210. 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Gupta R.; Sirohi R.; Kishore D. A Novel approach to the synthesis of 4H tetrazolo[1,5-a][1,4]benzodiazepine-6-ones from1,2,3-benztriazin-4-(3H)-ones. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2006, 45, 2147. [Google Scholar]
- Van Heyningen E. 1, 2, 3-Benzotriazines. J. Am. Chem. Soc. 1955, 77, 6562–6564. 10.1021/ja01629a042. [DOI] [Google Scholar]
- Arnold L. D.; Julian D.; Ronald M.; Atlas G.C.; Charles H.; Sherman W. H. D.; Richard W.; Wu J.. Manual of industrial microbiology and biotechnology; 2nd Ed., American society for microbiology: Washington DC, United States, ISBN10 1555811280, 1999. [Google Scholar]