

Abstract

A phenotypic whole cell high-throughput screen against the asexual blood and liver stages of the malaria parasite identified a benzimidazole chemical series. Among the hits were the antiemetic benzimidazole drug Lerisetron 1 (IC50 NF54 = 0.81 μM) and its methyl-substituted analogue 2 (IC50 NF54 = 0.098 μM). A medicinal chemistry hit to lead effort led to the identification of chloro-substituted analogue 3 with high potency against the drug-sensitive NF54 (IC50 NF54 = 0.062 μM) and multidrug-resistant K1 (IC50 K1 = 0.054 μM) strains of the human malaria parasite Plasmodium falciparum. Compounds 2 and 3 gratifyingly showed in vivo efficacy in both Plasmodium berghei and P. falciparum mouse models of malaria. Cardiotoxicity risk as expressed in strong inhibition of the human ether-a-go-go-related gene (hERG) potassium channel was identified as a major liability to address. This led to the synthesis and biological assessment of around 60 analogues from which several compounds with improved antiplasmodial potency, relative to the lead compound 3, were identified.
Introduction
Malaria, caused by the Plasmodium parasite genus and transmitted to humans by the bite of Plasmodium-infected female Anopheles mosquitoes, remains a life-threatening disease.1 Among the five species of Plasmodium parasites (Plasmodium falciparum (Pf), Plasmodium vivax, Plasmodium ovale, Plasmodium malariae, and Plasmodium knowlesi) that cause malaria in humans, P. falciparum is the most serious and often leads to death.2 A milder form of malaria is initiated in humans by P. vivax, P. ovale, and P. malariae and less frequently by P. knowlesi.3 According to the 2018 World Malaria Report, globally, approximately 219 million malaria cases and about 435 000 deaths were reported in 2017.4 On the basis of the figures published by the Institute of Health Metrics and Evaluation (IHME) and World Health Organization (WHO), 90% of reported deaths caused by malaria occur in sub-Saharan Africa.5 Worldwide, children under 5 years of age are the most vulnerable age group and accounted for 61% of the total malaria deaths in 2017.4,5 Malaria can be prevented by the use of indoor insecticide sprays and mosquito nets and avoiding the accumulation of stagnant water in the house or treatment using improved antimalarial medications.
Among the various antimalarial medications used for the treatment of malaria, artemisinin-based combination therapies (ACTs) have been found to be the most effective, especially against P. falciparum and have allowed millions of patients to be cured over the last few decades.6 Unfortunately, the emerging resistance to all existing antimalarials has become a recurring challenge for the goal of malaria eradication.7 In 2008, delayed parasite clearance by ACTs, which hinted at the emergence of resistance, were reported in patients from the eastern Thai-Cambodian border.8 Hence, there is a critical and urgent need to develop novel and affordable antimalarial therapeutic agents to tackle this rising problem.
Compounds possessing a benzimidazole core possess a broad spectrum of biological activities,9 including antimalarial activity10 (Figure 1). This scaffold is present in astemizole (brand name Hismanal), a second-generation antihistamine drug and antimalarial lead that was withdrawn from the market in most countries because of rare but potentially fatal side effects, such as QTc interval prolongation and related arrhythmias due to human ether-a-go-go related gene (hERG) channel blockade.11 Nor-astemizole is an active metabolite of astemizole with supposedly lower cardiac risks.12 Lerisetron, a related benzimidazole derivative, is an effective antagonist of the 5-HT3 receptor and was used in clinical trials as a highly potent antiemetic drug.13 Very few reports are available in the literature regarding efforts toward improving the off-target activity (hERG) of Astemizole and Lerisetron derivatives.14
Figure 1.

Pharmacologically active molecules containing the benzimidazole structure.
Herein, we disclose the synthesis, structure–activity relationship (SAR), and biological assessment of a series of benzimidazole derivatives based on the lead compound 3. The in vivo pharmacokinetic (PK) and efficacy studies on compound 3 are also described.
SAR Strategy
-
(1)
A broad range of cyclic and acyclic amines as the Eastern Substituent were introduced. To mitigate the hERG liability, nonbasic substituents or amines where the basicity of the amine is modulated, as well as bulky substituents or linear side chains plus carbon-linked ring systems were introduced.
-
(2)
Benzimidazole core replacements exemplified by incorporation of a variety of substituted aryl or heteroaryl rings instead of the Cl-phenyl group as the Western Substituent, were pursued.
-
(3)
In the Southern Substituent, the effect of benzyl group replacement using different carbon linkers or replacement of the phenyl moiety with heteroaryl or saturated systems was investigated.
The overall goal of the initial investigation was to identify an early lead compound suitable for a lead optimization campaign by addressing identified liabilities. In this regard, we aimed to mitigate the hERG liability of the series (ideally >10 μM, or at least a 100-fold safety index over asexual blood stage antiplasmodium activity) while retaining the excellent druglike properties and maintaining or improving Pf potency.
Chemistry
The 1,2,5-trisubstituted benzimidazoles were synthesized using a literature protocol, which leads to the final target compounds in five consecutive steps as shown in Scheme 1.
Scheme 1. General Synthetic Approach to the Synthesis of 2-Amino Benzimidazole Derivatives.

Reagents and conditions: (a) Et3N, acetonitrile (ACN), 50 °C, 16 h (56–97%) or K2CO3, dimethylformamide (DMF), 80 °C, 4–18 h (25–98%) or K2CO3, dimethyl sulfoxide (DMSO), 120 °C, 24 h, (93%); (b) Pt/C, H2 balloon, RT, MeOH, 8 h to 3 days (88–97%) or Fe powder, sat. aqu. NH4Cl, EtOH, 90 °C, 6–18 h (69–97%) or NH2NH2·H2O, MeOH, 80 °C, 2 h (61%); (c) triphosgene, dichloromethane (DCM), 25 °C, 16 h (68–94%); (d) POCl3, HCl, 150 °C, 4–24 h, (44–51%) or POCl3, PCl5, 110 °C, 1 h (45%); (e) CH(OMe)3, HCOOH, 100 °C 1–2 h (30–85%) or CH(OEt)3, para-toluene sulfonic acid (PTSA), tetrahydrofuran (THF), reflux, 2 h (88%); (f) lithium diisopropylamide (LDA), Cl3C–CCl3, THF, −78 °C, 4–5 h (33–96%) or LDA, CBr4, THF, −78 °C, 3 4 h (41%); (g) R3R4NH, Et3N, t-BuOH, 120 °C, 6 h to 18 days (9–91%).
The first step involves a nucleophilic aromatic substitution reaction (SNAr) between suitable amines, o-halo-nitro compounds 6, and an additional base [K2CO3 or Et3N] at elevated temperatures to obtain an amino-nitro intermediate 7,15 followed by a reduction step [Pt/C and H216 or iron powder and NH4Cl17 or hydrazine hydrate18] to afford diamines 8. The cyclization of 8 to the 1,3-dihydro-2H-benzo[d]imidazol-2-one 9 was carried out with triphosgene19 or with trimethyl orthoformate/HCOOH or triethyl orthoformate/PTSA to access benzimidazole intermediate 11.20 Transformation to the 2-halogen derivatives was performed on 9 using POCl3 and HCl or PCl5 to yield 10.21 Unfortunately, this reaction did not work well for some substrates. Alternatively, deprotonation of the benzimidazole 11 using lithium diisopropylamide (LDA) followed by subsequent halogenation with Cl3C–CCl3 or CBr4 yielded 12.22 The desired halogenated derivatives 12 were cleanly isolated and in high yield albeit initially an undesired side product 13 was observed when more than 1 equiv of LDA was used. Interestingly, double deprotonation/halogenation opened the door to cleanly attach additional halogen atoms to the benzene ring in position 4. These derivatives were further used for the attachment of aromatic rings via Pd-mediated coupling with boronic acids.23 The halogenated compounds 10, 12, and 13 were subjected to amination reactions using a broad range of amines in t-BuOH and triethylamine to access the corresponding final products 14–15 and 16–42 in good yields.
Furthermore, compound 43 were synthesized following a sodium triacetoxyborohydride-mediated reductive amination protocol (Scheme 2),24 which was carried out by the treatment of the amine 33 and a variety of aldehydes. The corresponding products (43) were obtained in generally low yields (16–49%).
Scheme 2. General Synthetic Route for the 2-Amino Benzimidazole Analogues (43).

Reaction and conditions: (a) R5-CHO, Na(OAc)3BH, AcOH, DCM, 20–27 °C, 2–24 h (16–49%).
Carbon-linked benzimidazole derivatives were synthesized as depicted in Scheme 3. The amide coupling between 44 and 8 was accomplished via the treatment with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) and diisopropyl ethyl amine (DIPEA) to yield amides 45.25 Ring closure and Boc-deprotection of 45 were accomplished in acetic acid when heated to 100 °C in a sealed tube. The resulting C-linked benzimidazoles 46 were isolated in low to good yields.26
Scheme 3. General Synthetic Route for the C-Linked Benzimidazoles 46.

Reagents and conditions: (a) HATU, DIPEA, DMF, 35 °C, 16 h (39–94%); (b) AcOH, 100 °C, 6 h (28–71%).
2-Amino benzimidazole derivatives 49 and 52 were synthesized by a Suzuki coupling reaction23 between the bromo compounds 47 or 50 with the appropriate aryl boronic acids, followed by Boc-deprotection using 4 N HCl in 1,4-dioxane in low to moderate yields (Scheme 4i,ii).
Scheme 4. General Synthetic Route for the Synthesis of 5-Substituted (49) and 4,5-Disubstituted 2-Amino Benzimidazoles (52).

Reaction and conditions: (a) R10-B(OH)2, Cs2CO3, Pd(dppf)Cl2, 1,4-dioxane/H2O (4:1), 95 °C, 4–24 h (45-97%); (b) 4 N HCl in 1,4-dioxane, 2–4 h, 20–27 °C (15–76%); for 49ea; R10 = 4-Py; EtOAc, neutralized with sat. NaHCO3 solution, 20 °C, 3 h (50%).
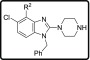
The incorporation of a nitrogen atom into the phenyl ring was carried out by selecting an appropriate commercially available chloropyridine isomer 53a and 53b (Scheme 5). Using an identical protocol carried out as mentioned earlier in the Scheme 1, the reaction between 53 with benzylamine yielded the desired amino-nitro products 54. These compounds (54) were subsequently reduced via catalytic hydrogenation with platinum on carbon and hydrogen to furnish diamino derivatives followed by the cyclization with trimethyl orthoformate and formic acid at elevated temperatures to access imidazopyridines 55 in low to good yields in a one-pot reaction. Deprotonation of Imidazopyridines 55 was realized with in situ generated LDA followed by treatment with hexachloroethane to afford chloro-imidazopyridines 56 in 91 and 37% yields, respectively. The chloro compounds 56, piperazine, and Et3N in t-BuOH were heated to 120 °C in a pressure tube to yield the desired products 57.
Scheme 5. Synthetic Route for the Synthesis of Imidazopyridines (57).

Reagents and conditions: (a) (i) Bn-NH2, K2CO3, DMF, 80 °C, 3–4 h, quantitative; (b) (i) Pt/C, H2 balloon, 20 °C, MeOH, 2–3 h; (ii) CH(OMe)3, HCOOH, 100 °C, 1–2 h (30–82%); (c) LDA, Cl3C–CCl3, THF, −78 °C, 4 h (37–91%); (d) piperazine, Et3N, t-BuOH, 120 °C, 18 h (25–80%).
Results and Discussion
In Vitro Antiplasmodium Activity, Solubility, and Cytotoxicity
All of the synthesized compounds were evaluated for in vitro antiplasmodium activity against the drug-sensitive strain (NF54) of P. falciparum. Compounds with PfNF54 <0.400 μM were also evaluated against a multidrug-resistant strain (K1) using chloroquine and artesunate as the reference drugs. Selected compounds were also assessed for cytotoxicity in a Chinese Hamster Ovary (CHO) cell line using emetine as a reference drug. Aqueous solubility was determined at pH 2.0 and 6.5. Additionally, selected compounds were further tested for their activity against the hERG channel and in vitro microsomal metabolic stability was assessed as a percentage of the test compound remaining after 30 min.
SAR of the Eastern Substituent
On the basis of the literature precedence, hERG inhibition liabilities can be drastically reduced by the removal of polar/basic moieties (e.g., reduction of the basicity of amines),27 formation of zwitterions (e.g., introduction of −COOH groups),28 disrupting the π–π interaction (e.g., replacing electron-donating groups with electron-withdrawing groups and introducing steric hindrance),29 and by discrete structural modifications (e.g., imparting rigidity).30 On the basis of the aforementioned strategies, we began exploring the Eastern Substituent by replacing the basic piperazine moiety with nonbasic ones, or replacing it with a variety of basic cyclic/bicyclic amines or introducing open-chain amines (2-, 3- or 4-carbon linkers) or adding acidic groups. For the investigation of the Eastern Substituent, we left the Western Substituent (phenyl with 5-chloro substituent) and the Southern Substituent (benzyl) unchanged, to have matched pairs for comparison.
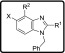
A broad range of 2-amino benzimidazoles were synthesized and evaluated for in vitro Pf activity, solubility, CHO cytotoxicity for comparison with 3 (Table 1), and for activity on the hERG channel. The morpholine derivative 16a displayed a noteworthy drop-in potency with an IC50 value of >1 μM (NF54) compared with 3. The piperazine derivatives 17 and 18 containing a nonbasic nitrogen also showed a substantial loss of antiplasmodium activity, demonstrating the importance of a basic nitrogen in the Eastern side chain. Compound 20, with a zwitterionic character, was inactive, whereas the related nitrile derivative 19 retained reduced activity, presumably due to the reduced basicity of the piperazine nitrogen. In a publication from the University of Helsinki and GlaxoSmithKline (GSK) Tres Cantos,31 a compound similar to 21 but without the Southern Substituent benzyl group was reported as being active; however, our derivative 21 proved to be completely inactive. Examples 22 and 24 without a benzyl group were resynthesized since they had been reported as active (185 and 66 nM); however, in our assays, these compounds were completely inactive. The follow-on derivatives containing a Bn-group (23 and 25) were also completely inactive in our assays. We also resynthesized the literature compound 26,32 which was at least weakly active in our assays despite having no basic nitrogen in the Eastern substituent. We did not pursue this approach any further, since the compounds without a basic nitrogen did not show improved hERG activity profiles (see the discussion later on and the data in Table 7). It is clear from Table 1 that incorporating a nonbasic Eastern substituent led to a considerable drop in the in vitro Pf activity for the above-mentioned compounds, demonstrating the importance of a basic nitrogen for antiplasmodium activity.
Table 1. Eastern Substituents: In Vitro Antiplasmodium Activity, Solubility, and Cytotoxicity Data of the Compounds with Reduced Basicity.
![]()

Mean from n values of ≥2 independent experiments with sensitive (NF54) and multidrug-resistant (K1) strains of P. falciparum.
Aqueous solubility using high-performance liquid chromatography (HPLC)-diode array detector (DAD)-mass spectrometry (MS).
CHO = Chinese hamster ovarian cell line.
Selectivity index (SI) = [IC50 (CHO)/IC50 (PfNF54)].
Data from Singh et al.33 ND = not determined.
Table 7. hERG Inhibition and Microsomal Stability Data of the Active Compounds.

Mean from n values of ≥2 independent experiments with multidrug-resistant (K1) and sensitive (NF54) strains of P. falciparum.
hERG determined at Novartis Institute for Tropical Diseases (NITD).
Human, mouse, and rat liver microsomes.
Obtained from NOVARTIS screening library and Pf activity determined at NITD. ND = not determined.
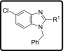
When compounds with a basic nitrogen in the Eastern substituent are compared against each other (Table 2), additional methyl groups on the ring were detrimental to activity (27) but connecting the Eastern substituent via a carbon linker results in the retention or improvement of potency (46a). A potency improvement was seen with the piperidine-4-amine side chain (28) where the basic nitrogen was moved slightly away from the benzimidazole core, leading to a 3-fold boost in potency. Consistent with this observation was the 2-fold shift in activity for the bicyclic Eastern substituent in 29 both for NF54 and K1.
Table 2. Eastern Substituents—In Vitro Antiplasmodium Activity, Solubility, and Cytotoxicity Data of the Compounds Retaining a Basic Nitrogen.

Mean from n values of ≥2 independent experiments with multidrug-resistant (K1) and sensitive (NF54) strains of P. falciparum.
Aqueous solubility using HPLC-DAD-MS.
CHO = Chinese hamster ovarian cell line.
Selectivity index (SI) = [IC50 (CHO)/IC50 (PfNF54)]. ND = not determined.
We next investigated the effect of open-chain analogues on antiplasmodium activity. Opening the piperazine ring to the ethylenediamine derivative 33 led to a 4-fold drop-in potency. However, we did see an improvement relating to the hERG activity of the compound, which will be discussed later. Potency dropped even further in the case of the related butanediamine (30) and propanediamine (32) derivatives. Methylation of the nitrogen (34) or dimethylation (35) resulted in nearly equipotent derivatives. Incorporation of two benzyl groups (43a) on the terminal nitrogen proved to be detrimental to activity. However, the compound with a single benzyl group on the terminal nitrogen (43b) displayed reasonably good activity, which was comparable to that of 3. Replacement of the phenyl group of 43b with a pyridyl or CF3-phenyl (43c and 43d) was not successful. When the terminal nitrogen was incorporated into a ring system (36–38), compounds were found to be less active. Overall, acyclic derivatives on the eastern end were less potent than their cyclic counterparts. Selected, potent compounds (IC50 NF54 < 0.400 μM) were screened against a multidrug-resistant strain (K1) to determine if these compounds are still effective against a resistant strain. All derivatives tested were also active against K1 within a twofold window, except for 38 (4-fold difference). In general, most of the analogues showed good solubility across both acidic (pH = 2) and neutral (pH = 6.5) conditions, with few exceptions. This active set of compounds (NF54 <0.400 μM) was also screened in a cytotoxicity assay using Chinese hamster ovarian (CHO) cells. The most potent compounds had a good safety window (SI = CHO CC50/NF54 IC50 > 500), except for compound 43b (SI < 35-fold). Overall, in vitro cytotoxicity was in general not a concern for the series.
SAR of the Western Substituent
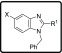
For the investigation of the Western substituent, Eastern substituents that gave active compounds based on the known derivatives (piperazine, piperidine, and diaminopropane) were chosen. The Southern substituent (benzyl) was left unchanged to have matched pairs for comparison (Table 3).
Table 3. In Vitro Antiplasmodium Activity, Solubility, and Cytotoxicity Data of the Western Substituent with Different Substituents in 5-Position.

Mean from n values of ≥2 independent experiments with multidrug-resistant (K1) and sensitive (NF54) strains of P. falciparum.
Aqueous solubility using HPLC-DAD-MS.
CHO = Chinese hamster ovarian cell line.
Selectivity index (SI) = [IC50 (CHO)/IC50 (PfNF54)].
Obtained from NOVARTIS screening library and Pf activity determined at NITD. ND = not determined.
In a series of compounds where the Eastern substituent is piperazinyl, the switch from a 5-methyl substituent (2) to 5-Cl (3) or 5-Br (16b) led to a ∼2-fold improvement in potency, whereas a 5-CF3 substituent (14c) led to a ∼3-fold loss of potency and a reduced safety index. As mentioned earlier, introducing steric hindrance on the core is a hERG mitigation strategy. Aromatic/heteroaromatic substituents were introduced in the 5-position (49a–d and 49ea), via coupling of the bromo derivative with the respective boronic acid derivatives. The consequence was a 25-fold or greater loss in potency. In a series of compounds where the Eastern substituent is piperidinyl, the 5-Cl (46a) and 5-Br (46b) derivatives picked up some potency in comparison to the piperazine derivatives in the NF54 assay but were equally potent in the K1 assay. There was a ∼4-fold drop-in activity for the corresponding 5-CF3 (46c) derivative, whereas an ester (46d) or sulfone (46e) substituent in the 5-position led to inactive compounds. In a series of compounds wherein the Eastern substituent is diaminopropyl, this trend was confirmed where the chloro and bromo derivatives are equipotent but a 5–6-fold drop-in potency was observed for the corresponding 5-CF3 substituent while there was more than a 10-fold drop-in potency for the unsubstituted derivative.
As mentioned earlier, introducing steric hindrance is a known hERG mitigation strategy. We accordingly synthesized and evaluated double halogenation products of the Western substituent. Several substituents were introduced in the 4-position of the benzimidazole scaffold while keeping the piperazinyl as the Eastern substituent (Table 4). Compound 42a with an additional chlorine atom at the 4-position showed equal or slightly improved Pf activity against both NF54 and K1 strains in comparison to the 4-unsubstituted derivative 3, whereas the 4-Br derivative 42k was equipotent or slightly less potent. Unfortunately, both derivatives were ∼4-fold more cytotoxic than the 4-unsubstituted compound 3 and the SI index dropped below 500 as a consequence. Introducing larger aromatic substituents in compounds 52a and 52b was not tolerated.
Table 4. In Vitro Antiplasmodium Activity, Solubility, and Cytotoxicity Data of the Disubstituted Western Substituent.

Mean from n values of ≥2 independent experiments with multidrug-resistant (K1) and sensitive (NF54) strains of P. falciparum.
Aqueous solubility using HPLC-DAD-MS.
CHO = Chinese hamster ovarian cell line.
Selectivity index (SI) = [IC50 (CHO)/IC50 (PfNF54)]. ND = not determined.
On the basis of literature precedence,34 we reasoned that incorporation of a nitrogen atom into the Western substituent might result in improved Pf potency and reduce hERG activity. Hence, a couple of changes to the core were investigated by incorporating an additional nitrogen (Table 5). Unfortunately, these modifications (57a and 57b) led to inactive compounds.
Table 5. In Vitro Antiplasmodium Activity of the Modified Cores Containing Additional Nitrogen Atoms.

Mean from n values of ≥2 independent experiments with multidrug-resistant (K1) and sensitive (NF54) strains of P. falciparum.
obtained from NOVARTIS screening library and Pf activity determined at NITD. ND = not determined.
SAR of the Southern Substituent
For the investigation of the Southern substituent, the piperazinyl moiety was fixed as the Eastern substituent on the basis that compounds with this moiety showed activity. The Western substituent was left unchanged, or we used the corresponding unsubstituted derivative, to have matched pairs for comparison (Table 6). Removal of the 1-carbon spacer in the N-benzyl group (16f) or introducing an additional spacer (16g) led to inactive compounds. Replacing the phenyl group with a hydrogen (60), cyclohexyl (14h), or 2-/4-pyridyl (59 and 14i) groups also resulted in inactive compounds. The combination of a one-carbon spacer and a phenyl ring (3) was found to be optimal for maintaining the Pf activity as the other N-replacements were not tolerated.
Table 6. SAR of the Southern Substituent.
![]()

Mean from n values of ≥2 independent experiments with multidrug-resistant (K1) and sensitive (NF54) strains of P. falciparum.
Obtained from NOVARTIS screening library and Pf activity determined at NITD. ND = not determined.
In Vitro, hERG Channel Activity, and Metabolic Stability
Selected compounds with good antiplasmodium activity were further evaluated for hERG channel inhibition (Table 7). Unfortunately, only one compound (33) showed some marginal improvement. The nonbasic compounds 18 and 19 showed only marginal improvement in the hERG liability but substantial loss in antiplasmodium activity. The only compound with a better hERG profile was the one containing an ethylenediamine side chain (33), leading to a still unsatisfactory 10-fold safety window.
The microsomal metabolic stability of the active analogues was evaluated in human, mouse, and rat liver microsomes35 and was found to be generally high for most compounds (except 19 and 27) across the three species.
Pharmacokinetic Studies in Rats
On the basis of the good in vitro antiplasmodium activity and solubility data, in vivo pharmacokinetics (PK) studies in rats were initiated for compound 3, with the goal to assess the exposure and oral bioavailability in rodent species. Following intravenous administration at 1 mg/kg dose, compound 3 displayed high total systemic clearance (>100% hepatic blood flow) and high volume of distribution (>5 L/kg), resulting in a moderate half-life (1.98 h) (Table 8). Following oral administration at 5 mg/kg dose, compound 3 showed rapid absorption, reasonable exposure, and moderate oral bioavailability (42%).
Table 8. Pharmacokinetic Parameters for Compound 3 Following Intravenous Dosing at 1 mg/kg and Oral Dosing at 5 mg/kg in Ratsa.
| intravenous
PK parameters |
oral
PK parameters |
|||||
|---|---|---|---|---|---|---|
| Vss (L/kg) | CL (mL/(min kg)) | T1/2 (h) | Cmax (μM) | Tmax (h) | AUC (μM h) | F (%) |
| 17.69 | 144 | 1.98 | 0.80 | 0.08 | 1.5 | 42 |
Vss = volume of distribution at steady state; CL = total systemic clearance; T1/2 = half-life, Cmax = maximum concentration of drug in plasma; Tmax = time to maximum concentration of drug in plasma; AUC = area under the curve; F = oral bioavailability; compounds were formulated in 30% N-methyl pyrrolidone/70% poly(ethylene glycol) (PEG)200 for intravenous administration and 0.5% methyl cellulose/0.5% Tween 80 in water for oral administration.
In Vivo Efficacy Studies
On the basis of the potent in vitro activity and promising in vitro physicochemical properties (Table 2 and 9), the frontrunner compounds were further profiled in vivo for efficacy in a Plasmodium berghei and P. falciparum mouse model. Compounds 2 and 3 were evaluated in the P. berghei mouse model (Table 10). One day post infection, P. berghei infected mice were treated orally with a single dose of 100 mg/kg of the respective compound. The key readouts were the percentage of parasitemia reduction on day 3 post infection compared with untreated mice and mouse survival prolongation (Figure 3). Both compounds showed good parasitemia reduction (>90%) with 6–7 days of survival. Among this, compound 3 displayed better in vivo activity (99%) presumably due to its better Cmax and exposure reached in these animals. Therefore, compound 3 was evaluated in the P. falciparum severe combined immunodeficiency (SCID) mouse model (Table 10 and Figure 3). On day 3 post infection, mice were dosed orally, once a day for 4 days at a dose of 25 mg/kg. 3 reduced 99.9% of the parasitemia in the Pf SCID mouse model at day 7 of the study. Overall, this compound displayed good in vivo potency and induced clearance of asexual parasites from peripheral blood comparable to chloroquine (dosed 4 × 50 mg/kg) in the P. falciparum mouse model.
Table 9. In Vitro Physicochemical Properties.
| parameters | 1 | 2 | 3 |
|---|---|---|---|
| structure | Lerisetron (R = H) | (R = Me) | (R = Cl) |
| molecular weighta | 292.4 | 306.4 | 326.8 |
| PSAa | 35.56 | 35.56 | 35.56 |
| cLog Pa | 3.3 | 3.8 | 4.1 |
| solubility (mM), (pH 6.8) | >1 | >0.1a | 0.77 |
| PAMPA (%FA) | 99 | 100 | 99 |
| mouseb/ratc/humanb | 29/–/26.6 | 36.7/172/43.5 | 45.3/79/<25 |
| mPPBd (%) | 93.7 | 97.8 |
In silico predictions; PSA = polar surface area; PAMPA = parallel artificial membrane permeability assay (fraction absorbed).
In vitro metabolic stability in liver microsomes (μL/(min mg)).
In vitro metabolic stability in hepatocytes (μL/min per 106 cells).
mPPB = mouse plasma protein binding.
Table 10. In Vivo Efficacy of Selected Analogues on Plasmodium Mouse Malaria Modelsa.
| parameters | 2 |
3 |
|
|---|---|---|---|
| malaria model | P. berghei | P. berghei | P. falciparum |
| dose (mg/kg) | 1 × 100 | 1 × 100 | 4 × 25 |
| activity (%) | 94 | 99 | >99.9 |
| survival (days) | 6 | 7 | |
| Cmax (μM) | 13.1 | 20.6 | 5.2 |
| AUC (μM h) | 165.4 | 377.1 | 86.0 |
Activity = average parasitemia reduction compared with untreated controls; survival = average lifespan after infection (6–7 days for untreated control mice); Cmax = maximum concentration of drug in plasma; AUC = area under the curve; compounds were formulated in 0.5% methyl cellulose/0.5% Tween 80 in water for oral administration.
Figure 3.

P. falciparum SCID mouse efficacy study for compound 3 compared with chloroquine (CQ).
Figure 2.

Exploration of SAR studies based on lead compound 3.
Conclusions and Discussion
A novel series of 1,2,5-trisubstituted benzimidazoles was identified from phenotypic whole cell high-throughput screening of the Novartis chemical library. A broad range of structurally diverse benzimidazoles were synthesized and assessed for in vitro antiplasmodium activity. SAR analysis indicated that several of the synthesized compounds displayed promising activity (IC50 NF54 <0.1 μM). As expected, addition of a functional group (H → Me → Cl) increases lipophilicity and plasma protein binding (Table 9). Overall, these compounds displayed good solubility, permeability, and metabolic stability in mouse and human species. However, in vitro clearance was moderate to high in rat and that translated to high in vivo total systemic clearance (Table 8). Additionally, the frontrunner compound displayed good parasitemia reduction in two malaria mouse models (Table 10 and Figure 3); however, survival rates as determined in the P. berghei model were poor and equated to the vehicle-treated animals. Further lead optimization needs to be carried out to address the unresolved hERG liability of the series, as well as the high plasma clearance and short half-life of the compounds (Scheme 6).
Scheme 6. Overview of the SAR Trends of the Series.

Experimental Section
All commercially available chemicals were purchased from either Sigma-Aldrich or Combi-Blocks. All solvents were dried by appropriate techniques. Unless otherwise stated, all solvents used were anhydrous. 1H NMR spectra were recorded on a Brucker Spectrometer at 300 or 400 MHz. Analytical thin-layer chromatography (TLC) was performed on aluminum-backed silica-gel 60 F254 (70–230 mesh) plates. Flash column chromatography was performed with Merck silica-gel 60 (70–230 mesh). Chemical shifts (δ) are given in ppm downfield from trimethylsilane (TMS) as the internal standard. Coupling constants, J, are recorded in hertz (Hz). Purity was determined by HPLC, and all compounds were confirmed to have >95% purity. The data that is not shown below is supplied in the Supporting Information.
Aqueous Solubility
Water solubility was analyzed using a miniaturized shake flask method. Ten millimolar stock solutions of each of the compounds were used to prepare calibration standards (10–220 μM) in DMSO. The same 10 mM stock solutions were accurately dispensed in duplicate into 96-well plates, and the DMSO, dried down (MiVac GeneVac, 90 min, 37 °C). Thereafter, the samples were reconstituted (200 μM) in an aqueous solution and shaken (20 h, 25 °C). The solutions were analyzed by means of HPLC-DAD (Agilent 1200 Rapid Resolution HPLC with a diode array detector). Best fit calibration curves were constructed using the calibration standards, which were used to determine the aqueous samples’ solubility.36
Metabolic Stability Assay
The metabolic stability assay was performed in duplicate in a 96-well microtiter plate. The test compounds (0.1 μM) were incubated (37 °C) in mouse, rat, and pooled human liver microsomes (final protein concentration of 0.4 mg/mL; XenoTech, Lenexa, KS) suspended in 0.1 M phosphate buffer (pH 7.4) for predetermined time points, in the presence and absence of the cofactor-reduced nicotinamide adenine dinucleotide phosphate (NADPH) (1 mM). The reactions were quenched by the addition of ice cold acetonitrile containing an internal standard (carbamazepine, 0.0236 μg/mL). The samples were centrifuged, and the supernatant was analyzed by means of liquid chromatography–tandem mass spectrometry (LC–MS/MS) (Agilent Rapid Resolution HPLC, AB SCIEX 4500 MS). The relative loss of the parent compound over time was monitored and plots were prepared for each compound of Ln% remaining versus time to determine the first-order rate constant for compound depletion. This was used to calculate the degradation half-life and in vitro intrinsic clearance value and subsequently to predict the in vivo intrinsic clearance and in vivo hepatic extraction ratio (EH). Metabolite searches were not conducted during the metabolic stability assay.35
In Vitro Antiplasmodium Assay
All parasite strains were acquired from MR4 (Malaria Research and Reference Reagent Resource Center, Manassas, VA). Compounds were tested using parasite lactate dehydrogenase assay as a marker for parasite survival.37 Briefly, the respective stock solutions of CQ diphosphate and test compounds were prepared to 2 mg/mL in distilled water (for CQ) and 100% DMSO for test compounds and then stored at −20 °C, and further dilutions were prepared on the day of the experiment. The cultures were synchronized in the ring stage as described previously using 15 mL of 5% (w/v) d-sorbitol in water.38 Synchronous cultures of PfNF54 (CQS) and Pf K1 (CQR) in the late trophozoite stage were prepared to 2% parasitemia and 2% hematocrit. Compounds were tested at starting concentrations of 10 000 ng/mL (1000 ng/mL for CQ), which were then serially diluted twofold in complete medium to give 10 concentrations with a final volume of 200 μL in each well. Parasites were incubated in the presence of the compounds at 37 °C in a specialized atmosphere of 4% CO2 and 3% O2 in nitrogen for 48 h. Following incubation, 100 μL of MalStat reagent and 15 μL of resuspended culture were combined, followed by addition of 25 μL of nitro blue tetrazolium chloride (NBT). The plates were kept in the dark for about 10 min to fully develop, and absorbance was measured at 620 nm on a microplate reader. Raw data were exported to Microsoft Excel for dose-response analysis.
Cytotoxicity Assay
Compounds were screened for in vitro cytotoxicity against Chinese hamster ovarian (CHO) mammalian cell lines, using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The reference standard, emetine, was prepared to 2 mg/mL in distilled water while stock solutions of test compounds were prepared to 20 mg/mL in 100% DMSO with the highest concentration of solvent to which the cells were exposed having no measurable effect on the cell viability. The initial concentration of the compounds and control was 100 μg/mL, which was serially diluted in complete medium with 10-fold dilutions to give six concentrations, the lowest being 0.001 μg/mL. Plates were incubated for 48 h with 100 μL of drug and 100 μL of cell suspension in each well and developed afterward by adding 25 μL of sterile MTT (Thermo Fisher Scientific) to each well, followed by 4 h of incubation in the dark. The plates were then centrifuged; the medium was aspirated, and 100 μL of DMSO was added to dissolve crystals before reading the absorbance at 540 nm. Data were analyzed, and the sigmoidal dose-response was derived using GraphPad Prism v 4.0 software (La Jolla). All experiments were performed for at least three independent biological repeats, each with technical triplicates.
In Vivo Efficacy Studies
P. berghei Mouse Model
In vivo antimalarial activity was assessed for groups of five female NMRI mice (20–22 g) intravenously infected on day 0 with P. berghei GFP ANKA malaria strain (2 × 107 parasitized erythrocytes, donation from AP Waters and CJ Janse, Leiden University). Untreated control mice die typically between day 6 and day 7 post infection. Experimental compounds were formulated (in 0.5% methyl cellulose/0.5% Tween 80) and administered orally in a volume of 10 mL/kg as a single dose (24 h postinfection) of 100 mg/kg. Parasitaemia was determined 72 h post infection using standard flow cytometry techniques. Activity was calculated as the difference between the mean percent parasitaemia for the control and treated groups expressed as a percent relative to the control group. The survival time in days was also recorded up to 30 days after infection. A compound was considered curative if the animal survived to day 30 after infection with no detectable parasites by slide reading.
P. falciparum SCID Mouse Model
Compound efficacy was assessed in the murine P. falciparum SCID model essentially as described.39 Briefly, compound 3, formulated in 7% Tween 80, 3% ethanol, was administered to a cohort of age-matched female immunodeficient NOD-scid IL-2Rγnull mice (The Jackson Laboratory, Bar Harbor, ME) previously engrafted with human erythrocytes (generously provided by the Blood Bank in Zürich, Switzerland). The mice were intravenously infected with 2 × 107P. falciparum Pf3D70087/N9-infected erythrocytes (day 0).40 On day 3 after infection, mice were randomly allocated to treatments that were administered once a day for 4 days (n = 2 mice) by oral gavage at 25 mL/kg. Parasitemia was measured by microscopy. Chimerism was monitored by flow cytometry using anti-murine erythrocyte TER119 monoclonal antibody (Pharmingen, San Diego, CA) and SYTO-16 and then analyzed by flow cytometry in serial 2 μL blood samples.
In vivo studies conducted at the Swiss TPH, Basel were approved by the veterinary authorities of the Canton Basel-Stadt (permit no. 1731 and 2303) based on Swiss cantonal (Verordnung Veterinäramt Basel-Stadt) and national regulations (the Swiss animal protection law, Tierschutzgesetz).
General Procedure 1 (GP1) for the Synthesis of 3, 14c, 14h, 14i, 15, 16a, 16b, 16f, 16g, 17–41, 42a, and 42k (Scheme 1); (Synthesis of Starting Material; See the Supporting Information)
Step 7: General Procedure for Synthesis of 16a, 16b, 16g, and 41
A mixture of appropriate 2-Cl-benzimidazole (1 equiv), corresponding amine (10–12 equiv), and Et3N (10 equiv) in tert-butanol (0.15 M) was heated in a pressure tube at a temperature of 120 °C for 6 h to 18 days. Then, the reaction mixture was allowed to cool to room temperature and saturated aqueous NaHCO3 solution (40 mL) and EtOAc (50 mL) were added. The organic phase was separated and the aqueous phase was added with an excess of EtOAc (2 × 50 mL). The organic phases were combined and dried over anhydrous Na2SO4 and concentrated in vacuo to afford a crude residue. The crude was purified by flash column chromatography using ISCO Teledyne RediSep Rf column eluting DCM/MeOH (0.5 M NH3 solution) or hexane/EtOAc to get pure products.
4-(1-Benzyl-5-chloro-1H benzo[d]imidazol-2-yl)morpholine (16a)
Flash column chromatography using hexane/EtOAc (60/40–40/60) followed by crystallization in ipropylether/DCM. (80 mg, 18% yield), White solid. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.67 (d, J = 3.0 Hz, 1H), 7.42–7.33 (m, 3H), 7.17–7.13 (m, 2H), 7.10 (dd, J = 9.0, 3.0 Hz, 1H), 6.94 (d, J = 9.0 Hz, 1H), 5.25 (s, 2H), 3.84–3.81 (m, 4H), 3.34–3.31 (m, 4H); HPLC purity 99%, retention time (Rt) = 4.815 min; method used (general positive); LC–MS (atmospheric pressure chemical ionization (APCI)) m/z = found 328.1 (M+) and 330.1 (M + 2)+ on a positive mode M 280 nm [calcd for C18H19ClN3O m/z = 328.1217 (M + H)+].
1-Benzyl-5-bromo-2-(piperazin-1-yl)-1H-benzo[d]imidazole (16b)
Flash column chromatography using DCM/MeOH (2% Et3N) (0–30% gradient). (100 mg, 57% yield), off white solid. 1H NMR (300 MHz, CD3OD) δ (ppm) 7.65 (dd, J = 1.9, 0.5 Hz, 1H), 7.39–7.27 (m, 3H), 7.24 (dd, J = 8.5, 1.9 Hz, 1H), 7.18–7.15 (m, 2H), 7.07 (dd, J = 8.5, 0.5 Hz, 1H), 5.34 (s, 2H), 3.28–3.24 (m, 4H), 2.99–2.96 (m, 4H); HPLC purity 99%, retention time (Rt) = 3.644 min; method used (general positive); LC–MS (APCI) m/z = found 371.1 (M+) and 373.1 (M + 2)+ on a positive mode M 280 nm [calcd for C18H20BrN4m/z = 371.0871 (M + H)+].
5-Chloro-1-phenethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole Hydrochloride (16g)
Flash column chromatography using DCM/MeOH/Et3N (94/4/2–92/6/2) yielded an oil. The material was dissolved in MeOH/EtOAc, and 2 M HCl in diethyl ether (2 mL) was added and concentrated in vacuo to dryness. Crystallization from EtOAc/MeOH provided 229 mg (72% yield) as a white solid. 1H NMR (300 MHz, CD3OD) δ (ppm) 7.73 (d, J = 8.8 Hz, 1H), 7.65 (d, J = 2.0 Hz, 1H), 7.53 (dd, J = 8.8, 2.0 Hz, 1H), 7.33–7.25 (m, 3H), 7.03–6.97 (m, 2H), 4.62 (t, J = 6.4 Hz, 2H), 3.64–3.60 (m, 4H), 3.39–3.34 (m, 4H), 3.20 (t, J = 6.4 Hz, 2H); HPLC purity 99%, retention time (Rt) = 4.040 min; method used (general positive); LC–MS (APCI) m/z = found 341.2 (M+) and 343.2 (M + 2)+ on a positive mode M 280 nm [calcd for C19H23Cl2N4m/z = 377.1300 (M + H)+].
N1-(1-Benzyl-5-bromo-1H-benzo[d]imidazol-2-yl)propane-1,3-diamine (41)
Flash column chromatography using DCM/MeOH (0.5M NH3 solution) (0–25% gradient). (52 mg, 31% yield), off white solid. 1H NMR (400 MHz, CD3OD) δ (ppm) 7.45 (dd, J = 1.8, 0.4 Hz, 1H), 7.36–7.25 (m, 3H), 7.16–7.12 (m, 2H), 7.09 (dd, J = 8.3, 1.8 Hz, 1H), 6.99 (dd, J = 8.3, 0.4 Hz, 1H), 5.25 (s, 2H), 3.56 (t, J = 6.7 Hz, 2H), 2.85 (t, J = 7.0 Hz, 2H), 1.91 (quin, J = 6.8 Hz, 2H); HPLC purity 99%, retention time (Rt) = 3.513 min; method used (general positive); LC–MS (APCI) m/z = found 359.1 (M+) and 361.1 (M + 2)+ on a positive mode M 280 nm [calcd for C17H20BrN4m/z = 359.0871 (M + H)+].
General Procedure 2 (GP2) for the Reductive Amination Reaction (Scheme 2) (43a–d)
To a mixture of N1-(1-benzyl-5-chloro-1H-benzo[d]imidazol-2-yl)ethane-1,2-diamine (33) (1 equiv), benzaldehyde (1.05 equiv), and acetic acid (2.0 equiv) in dichloroethane (DCE) (0.027 M) was added sodium triacetoxyborohydride (2.25 equiv) and stirring continued for 2–24 h at RT. TLC and LC–MS indicated the complete consumption of the starting material and showed a mass peak for monobenzylated (major) and in some cases, a dibenzylated (minor) product. Then, the reaction was quenched by the addition of NaOH (1 N) solution (30 mL) and extracted with EtOAc (2 × 50 mL). The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by flash chromatography using ISCO Teledyne and elution using DCM/MeOH (0.5 M NH3 solution) (0–10%) gradient to afford product. The purified oily/gummy compounds were dissolved in MeOH (5 mL), and a 2.0 M solution of HCl/ether (4 mL) was added and concentrate in vacuo to dryness. Et2O (6 mL) was added and stirred at room temperature of 25 °C for 1 h; the precipitate (ppt) was filtered and dried to afford a pure salt.
N1-Benzyl-N2-(1-benzyl-5-chloro-1H-benzo[d]imidazol-2-yl)ethane-1,2-diamine Hydrochloride (43b)
Flash column chromatography using DCM/MeOH (0.5 M NH3 solution) (0–10% gradient). (44 mg, 44% yield), yellow solid. 1H NMR (300 MHz, CD3OD) δ (ppm) 7.62–7.59 (m, 2H), 7.56–7.55 (m, 1H), 7.50–7.46 (m, 3H), 7.42–7.30 (m, 7H), 5.53 (s, 2H), 4.34 (s, 2H), 3.98 (t, J = 6.1 Hz, 2H), 3.51 (t, J = 6.2 Hz, 2H); HPLC purity 99%, retention time (Rt) = 0.911 min; method used (general positive); LC–MS (APCI) m/z = found 391.2 (M+) and 393.2 (M + 2)+ on a positive mode M 280 nm [calcd for C23H25Cl2N4m/z = 427.1456 (M + H)+].
Note: Dibenzylated (4 mg, 3.57%) product was also obtained.
N1-(1-Benzyl-5-chloro-1H-benzo[d]imidazol-2-yl)-N2-(pyridin-4-ylmethyl)ethane-1,2-diamine Hydrochloride (43c)
Flash column chromatography using DCM/MeOH (0.5 M NH3 solution) (0–5% gradient). (40 mg, 49% yield), yellow solid. 1H NMR (300 MHz, CD3OD) δ (ppm) 8.95–8.92 (m, 2H), 8.27 (d, J = 6.5 Hz, 2H), 7.57–7.56 (m, 1H), 7.42–7.33 (m, 7H), 5.57 (s, 2H), 4.66 (s, 2H), 4.06 (t, J = 5.9 Hz, 2H), 3.64 (t, J = 5.9 Hz, 2H); HPLC purity 99%, retention time (Rt) = 1.054 min; method used (general positive); LC–MS (APCI) m/z = found 392.2 (M+) and 394.2 (M + 2)+ on a positive mode M 280 nm [calcd for C22H24Cl2N5m/z = 428.1049 (M + H)+].
Note: Dibenzylated product was not observed.
General Procedure 3 (GP3) for the Synthesis of C-Linked Compounds (46a–e) (Scheme 3)
Step 1: General Procedure for Amide Coupling (45a)
Synthesis of tert-Butyl 4-((2-(benzylamino)-5-chlorophenyl)carbamoyl)piperidine-1-carboxylate (45a)
To a solution of the N1-benzyl-4-chlorobenzene-1,2-diamine (8a) (400 mg, 1.719 mmol) in DMF (8.0 mL) was added 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (394 mg, 1.719 mmol), HATU (784 mg, 2.063 mmol), and DIPEA (0.751 mL, 4.30 mmol). The reaction mixture was stirred at 35 °C. After 16 h, TLC indicated that the reaction was completed. H2O (30 mL) and EtOAc (60 mL) were added, the organic phase separated, and the aqueous phase extracted with a further portion of EtOAc (2 × 20 mL). The organic phases were combined and washed sequentially with saturated aqueous NaHCO3 (30 mL) and brine (30 mL), dried over anhydrous Na2SO4, and concentrated in vacuo to afford a crude residue of 45a (450 mg, 39% yield) as a brown oil, which was used directly to the next step without any further purification. TLC system Rf = 0.25 [hexane/EtOAc (4/6)]. HPLC purity 67%, retention time (Rt) = 4.756 min; method used (general negative); LC–MS (APCI) m/z = found 442.2 (M – H)− on a negative mode M 280 nm [calcd for C24H29ClN3O3m/z = 442.1897 (M – H)−].
Step 2: General Procedure for Cyclization and Deprotection of Boc Group (46a)
Synthesis of 1-Benzyl-5-chloro-2-(piperidin-4-yl)-1H-benzo[d]imidazole (46a)
A suspension of the crude tert-butyl 4-((2-(benzylamino)-5-chlorophenyl)carbamoyl)piperidine-1-carboxylate 45a (1.0 equiv) in acetic acid (60 equiv) was stirred and heated in a sealed tube at a temperature at 100 °C. After 6 h, TLC indicated that the reaction is completed. LC–MS indicated a corresponding mass ion peak. The reaction mixture was allowed to cool to 25 °C and concentrated in vacuo to afford a crude residue. EtOAc (50 mL) was added, and aqueous NaHCO3 solution (50 mL) was saturated. The organic phase was separated, and the aqueous phase, extracted, with a further portion of EtOAc (2 × 30 mL). The organic phases were combined, dried over anhydrous Na2SO4, and concentrated in vacuo to afford a residue. Purification by flash chromatography was carried out using ISCO Teledyne on a 12 g RediSep Rf column, and elution was carried out using DCM/MeOH (0.5 M NH3 solution) (0–25% gradient) to afford 46a (64 mg, 28% yield) as an off white solid. 1H NMR (300 MHz, CD3OD) δ (ppm) 7.67 (dd, J = 1.9, 0.5 Hz, 1H), 7.45 (dd, J = 8.7, 0.6 Hz, 1H), 7.40–7.27 (m, 4H), 7.15–7.09 (m, 2H), 5.61 (s, 2H), 3.54–3.47 (m, 2H), 3.45–3.40 (m, 1H), 3.18–3.05 (m, 2H), 2.15–1.98 (m, 4H); HPLC purity 98%, retention time (Rt) = 0.434 min; method used (general positive); LC–MS (APCI) m/z = found 326.2 (M+)+ and 328.2 (M + 2)+ on a positive mode M 280 nm [calcd for C19H21ClN3m/z = 326.1424 (M + H)+].
General Procedure 4 (GP4) for the Synthesis of 49a–e, 52a, and 52b (Scheme 4)
Step 1: General Procedure for Suzuki Cross-Coupling
Bromo compounds 47/50 (1.0 equiv) and appropriate aryl boronic acids (1.3 equiv) were taken in a pressure vial in 1,4-dioxane (0.1 M) and water (0.318 M) (5:1) followed by the addition of cesium carbonate (3.0 equiv) and [1,1′-bis(diphenylphosphino)-ferrocene]dichloropalladium(II) (0.1 equiv) and heated to 95 °C until TLC of the reaction indicated complete consumption of the starting material (approximately 4–21 h). The reaction mixture was cooled to room temperature and a saturated solution of NaHCO3 (30 mL) was added and extracted with EtOAc (2 × 40 mL). The combined organic layer was dried over anhydrous sodium sulfate and evaporated under reduced pressure. The crude residue was purified by flash chromatography using eluting hexane/EtOAc or DCM/MeOH gradient to afford the desired Suzuki cross-coupled product in moderate yields, which were used directly in the next step.
tert-Butyl 4-(1-Benzyl-5-(1H-pyrazol-3-yl)-1H-benzo[d]imidazol-2-yl)piperazine-1-carboxylate (48a)
Flash column chromatography using hexane/EtOAc (0–100% gradient). (66 mg, 45% yield), off white solid. HPLC purity 99%, retention time (Rt) = 1.226 min; method used (general positive); LC–MS (APCI) m/z = found 459.2 (M + H)+ on a positive mode M 280 nm [calcd for C26H31N6O2m/z = 459.25 (M + H)+].
Step 2: General Procedure for Boc-Deprotection
A Boc-protected compound (1 equiv) in 4 N HCl in dioxane (4 mL) was stirred for 2–20 h. TLC and LC–MS of the reaction indicated the complete consumption of the starting material. The reaction solvent was evaporated under reduced pressure and diethyl ether (5 mL) was added. Then, the ppt was filtered off and washed with an excess of diethyl ether (5 mL) to afford the desired hydrochloride salt in low to moderate yields.
1-Benzyl-2-(piperazin-1-yl)-5-(1H-pyrazol-3-yl)-1H-benzo[d]imidazole Hydrochloride (49a)
(35 mg, 67% yield), yellow solid. 1H NMR (300 MHz, CD3OD) δ (ppm) 8.08 (dd, J = 1.6, 0.6 Hz, 1H), 7.90–7.86 (m, 2H), 7.51–7.35 (m, 6H), 6.88 (d, J = 2.5 Hz, 1H), 5.63 (s, 2H), 3.90–3.87 (m, 4H), 3.50–3.46 (m, 4H); HPLC purity 99%, retention time (Rt) = 2.688 min; method used (general positive); LC–MS (APCI) m/z = found 359.1 (M + H)+ on a positive mode M 280 nm [calcd for C21H24ClN6m/z = 395.1751 (M + H)+].
Acknowledgments
Thierry Diagana, Fumiaki Yokokawa, and NITD in vivo PK and BA team members are acknowledged for their input. The in vitro (parasitology and absorption, distribution, metabolism, and excretion (ADME)) team at H3D, Nesia Barnes, Warren Olifant, Sumaya Salie, and Virgil Verghoog are acknowledged. The Novartis Research Foundation, University of Cape Town, South African Medical Research Council, and South African Research Chairs Initiative of the Department of Science and Technology, administered through the South African National Research Foundation are gratefully acknowledged for support (K.C.).
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- hERG
human ether-a-go-go related gene
- ACN
acetonitrile
- DCM
dichloromethane
- DCE
dichloroethane
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- THF
tetrahydrofuran
- LDA
lithium diisopropylamide
- PTSA
para-toluene sulfonic acid
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- DIPEA
diisopropyl ethyl amine
- CHO cells
chinese hamster ovarian cells
- EH
extraction ratio
- SAR
structure–activity relationships
- CQ
chloroquine
- PK
pharmacokinetics
- NMR
nuclear magnetic resonance
- TMS
trimethylsilane
- RT
room temperature
- NITD
Novartis Institute for Tropical Diseases
- GSK
GlaxoSmithKline
- WHO
World Health Organization
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00327.
Experimental section with additional details of the synthesis and characterization of selected compounds, references and 1H NMR spectra of the synthesized compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- White N. J.; Pukrittayakamee S.; Hien T. T.; Faiz M. A.; Mokuolu O. A.; Dondrop A. M. Malaria. Lancet 2014, 383, 723–735. 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- a Orth H.; Jensen B. O.; Holtfreter M. C.; Kocheril S. J.; Mallach S.; McKenzie C.; Muller-Stover I.; Henrich B.; Imwong M.; White N. J.; Haussinger D.; Richter J. Plasmodium knowlesi Infection Imported to Germany, January 2013. Euro Surveill. 2013, 20603 10.2807/1560-7917.es2013.18.40.20603. [DOI] [PubMed] [Google Scholar]; b de Koning-Ward T. F.; Dixon M. W. A.; Tilley L.; Gilson P. R. Plasmodium Species: Master Renovators of Their Host Cells. Nat. Rev. Microbiol. 2016, 14, 494–507. 10.1038/nrmicro.2016.79. [DOI] [PubMed] [Google Scholar]
- a Burrows J. N.; Chibale K.; Wells T. N. C. The State of the Art in Anti-malarial Drug Discovery and Development. Curr. Top. Med. Chem. 2011, 11, 1226–1254. 10.2174/156802611795429194. [DOI] [PubMed] [Google Scholar]; b Chin W.; Contacos P. G.; Coatney G. R.; Kimball H. R. A Naturally Acquired Quotidian-Type Malaria in Man Transferable to Monkeys. Science 1965, 149, 865 10.1126/science.149.3686.865. [DOI] [PubMed] [Google Scholar]; c Vythilingam I.; NoorAzian Y. M.; Huat T. C.; Ida Jiram A.; Yusri Y. M.; Azahari A. H.; NorParnia I.; NoorRain A.; LokmanHakim S. Plasmodium knowlesi in Humans, Macaques and Mosquitoes in Peninsular Malaysia. Parasites Vectors 2008, 1, 26 10.1186/1756-3305-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (WHO). World Malaria Report, 2018. http://www.who.int/malaria/publications/world-malaria-report-2018/en/ (accessed Sept 30, 2019).
- https://ourworldindata.org/malaria.
- Wells T. N. C.; Alonso P. L.; Gutteridge G. E. New Medicines to Improve Control and Contribute to the Eradication of Malaria. Nat. Rev. Drug Discovery 2009, 8, 879–891. 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- a Biamonte M. A.; Wanner J.; Le Roch K. G. Recent Advances in Malaria Drug Discovery. Bioorg. Med. Chem. Lett. 2013, 23, 2829–2843. 10.1016/j.bmcl.2013.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sinha S.; Medhi B.; Sehgal R. Challenges of Drug-Resistant Malaria. Parasite 2014, 21, 61 10.1051/parasite/2014059. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Antony H. A.; Parija S. C. Antimalarial Drug Resistance: An Overview. Trop. Parasitol. 2016, 1, 30–41. 10.4103/2229-5070.175081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Noedl H.; Se Y.; Schaecher K.; Smith B. L.; Socheat D.; Fukuda M. M. Evidence of Artemisinin-resistant Malaria in Western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]; b Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P. J.; Lindegardh N.; Socheat D.; White N. J. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2009, 361, 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ariey F.; Witkowski B.; Amaratunga C.; Beghain J.; Langlois A. C.; Khim N.; Kim S.; Duru V.; Bouchier C.; Ma L.; Lom P.; Leang R.; Duong S.; Sreng S.; Suon S.; Chuor C. M.; Bout D. M.; Menard S.; Rogers W. O.; Genton B.; Fandeur T.; Miotto O.; Ringwald P.; Le Bras J.; Berry A.; Barale J. C.; Fairhurst R. M.; Benoit-Vical F.; Mercereau-Puijalon O.; Menard D. A. Molecular Marker of Artemisinin-Resistant Plasmodium falciparum Malaria. Nature 2014, 505, 50–55. 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bansal Y.; Silakari O. The Therapeutic Journey of Benzimidazoles; A Review. Bioorg. Med. Chem. 2012, 20, 6208–6236. 10.1016/j.bmc.2012.09.013. [DOI] [PubMed] [Google Scholar]; b Keri R. S.; Hiremathad A.; Budagumpi S.; Nagaraja B. M. Comprehensive Review in Current Developments of Benzimidazole-Based Medicinal Chemistry. Chem. Biol. Drug Des. 2015, 86, 19–65. 10.1111/cbdd.12462. [DOI] [PubMed] [Google Scholar]; c Ates-Alagoz Z. Antimicrobial Activities of 1-H-Benzimidazole-Based Molecules. Curr. Top. Med. Chem. 2016, 16, 2953–2962. 10.2174/1568026616666160506130226. [DOI] [PubMed] [Google Scholar]; d Prajapat P.; Kumawat M.; Talesara G. L.; Kalal P.; Agarwal S.; Kapoor C. S. Benzimidazole Scaffold as a Versatile Biophore in Drug Discovery: A Review. Chem. Biol. Interface 2018, 8, 1–10. [Google Scholar]
- a Musonda C. C.; Whitlock G. A.; Witty M. J.; Brun R.; Kaiser M. Chloroquine-Astemizole Hybrids with Potent in vitro and in vivo Antiplasmodial Activity. Bioorg. Med. Chem. Lett. 2009, 19, 481–484. 10.1016/j.bmcl.2008.11.047. [DOI] [PubMed] [Google Scholar]; b Camacho J.; Barazarte A.; Gamboa N.; Rodrigues J.; Rojas R.; Vaisberg A.; Gilman R.; Charris J. Synthesis and Biological Evaluation of Benzimidazole-5-Carbohydrazide Derivatives as Anti-malarial, Cytotoxic and Anti-tubercular Agents. Bioorg. Med. Chem. 2011, 19, 2023–2029. 10.1016/j.bmc.2011.01.050. [DOI] [PubMed] [Google Scholar]; c Saify Z. S.; Azim M. K.; Ahmad W.; Nisa M.; Goldberg D. E.; Hussain S. A.; Akhtar S.; Akram A.; Arayne a.; Oksman A.; Khan I. A. New Benzimidazole Derivatives as Antiplasmodial Agents and Plasmepsin Inhibitor: Synthesis and Analysis of Structure-Activity Relationship. Bioorg. Med. Chem. Lett. 2012, 22, 1282–1286. 10.1016/j.bmcl.2011.10.018. [DOI] [PubMed] [Google Scholar]; d Worachartcheewan A.; Nantasenamat C.; Isarankura-Na-Ayudhya C.; Prachaysittikul V. QSAR Study of Amidino Bis-benzimidazole Derivatives as Potent Anti-malarial Agents Against Plasmodium falciparum. Chem. Pap. 2013, 67, 1462–1473. 10.2478/s11696-013-0398-5. [DOI] [Google Scholar]; e Kumar M.; Okombo J.; Mambwe D.; Taylor D.; Lawrence N.; Reader J.; van der Watt M.; Fontinha D.; Sanches-Vaz M.; Bezuidenhout B. C.; Lauterbach S. B.; Liebenberg D.; Birkholtz L.-M.; Coetzer T. L.; Prudencio M.; Egan T. J.; Wittlin S.; Chibale K. Multistage Antiplasmodium Activity of Astemizole Analogues and Inhibition of Hemozoin Formation as a Contributor to Their Mode of Action. ACS Infect. Dis. 2019, 5, 303–315. 10.1021/acsinfecdis.8b00272. [DOI] [PubMed] [Google Scholar]
- a Zhou Z.; Vorperian V. R.; Gong Q.; Zhang S.; January C. T. Block of HERG Potassium Channels by the Antihistamine Astemizole and Its Metabolites Desmethylastemizole and Norastemizole. J. Cardiovasc. Electrophysiol. 1999, 10, 836–843. 10.1111/j.1540-8167.1999.tb00264.x. [DOI] [PubMed] [Google Scholar]; b Chong C. R.; Chen X.; Shi L.; Liu J. O.; Sullivan D. J. A Clinical Drug Library Screen Identifies Astemizole as an Antimalarial Agent. Nat. Chem. Biol. 2006, 2, 415–416. 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]; c Tian J.; Vandermosten L.; Peigneur S.; Moreels L.; Rozenski J.; Tytgat J.; Herdewijn P.; Van den Steen P. E.; De Jonghe S. Astemizole Analogues with Reduced hERG inhibition as Potent Antimalarial Compounds. Bioorg. Med. Chem. 2017, 25, 6332–6344. 10.1016/j.bmc.2017.10.004. [DOI] [PubMed] [Google Scholar]
- Bachmann K. A. Norastemizole Sepracor. Curr. Opin. Invest. Drugs 2000, 1, 219–226. [PubMed] [Google Scholar]
- a Orjales A.; Mosquera R.; Labeaga L.; Rodes R. New 2-Piperazinylbenzimidazole Derivatives as 5-HT3 Antagonists. Synthesis and Pharmacological Evaluation. J. Med. Chem. 1997, 40, 586–593. 10.1021/jm960442e. [DOI] [PubMed] [Google Scholar]; b Gomez-de-Segura I. A.; Grande A. G.; De Miguel E. Antiemetic Effects of Lerisetron in Radiation-Induced Emesis in the Dog. Acta Oncol. 1998, 37, 759–763. 10.1080/028418698430160. [DOI] [PubMed] [Google Scholar]; c Cooper M.; Sologuren A.; Valiente R.; Smith J. Effects of Lerisetron, a New 5-HT3 Receptor Antagonist, on Ipecacuanha-Induced Emesis in Healthy Volunteers. Arzneimittelforschung 2011, 52, 689–694. 10.1055/s-0031-1299952. [DOI] [PubMed] [Google Scholar]
- a Huckel R. Lerisetron. FAES. Curr. Opin. Invest. Drugs 2003, 4, 874–877. [PubMed] [Google Scholar]; b Sann H.Use of a 5-HT3 Receptor Antagonist for the Manufacture of a Medicament for the Treatment of Non-Digestive Tract Derived Abdominal Disorders Associated with Pain. WO2005/089732A1, Sept 29, 2005.; c Chabbert C.; Venail F.. Serotonin 5-HT3 Receptor Antagonists for the Use in the Treatment of Lesional Vestibular Disorders. WO2010/133663A1, Nov 25, 2010.; d Waldmann H.; Triola G.; Wittinghofer A.; Bastiaens P.; Vartak N.; Papke B.; Zimmermann G.; Shehab I.; Schultz-Fademrecht C.; Koch U.. Benzimidazoles for the Treatment of Cancer. WO2014/027053A1, Feb 20, 2014.
- a Sheppard G.; Wang G.; Palazzo F.; Bell R.; Mantel R.; Wang J.; Hubbard R.; Kawai M.; Erickson S.; Bamaung N.. Protein Kinase Inhibitors. WO2007/079164A2, July 12, 2007.; b Mjalli A.; Grella B.; Subramanian G.; Arimilli M. N.; Gopalaswamy R.; Andrews R. C.; Davis S.; Guo X.; Zhu J.. Benzimidazole Derivatives, Compositions, and Methods of Use of as Aurora Kinase Inhibitors. WO2007/095124A2, August 23, 2007.
- Palmer B. D.; Smaill J. B.; Boyd M.; Boschelli D. H.; Doherty A. M.; Hamby J. M.; Khatana S. S.; Kramer J. B.; Kraker A. J.; Panek R. L.; Lu G. H.; Dahring T. K.; Thomas Winters R.; Hollis Showalter H. D.; Denny W. A. Structure-Activity Relationships for 1-phenylbenzimidazoles as Selective ATP Site Inhibitors of the Platelet-Derived Growth Factor Receptor. J. Med. Chem. 1998, 41, 5457–5465. 10.1021/jm9804681. [DOI] [PubMed] [Google Scholar]
- Neuthe K.; Popeney C. S.; Bialecka K.; Hinsch A.; Sokolowski A.; Veurmann W.; Haag R. Simple NIR Complexes and their Applicability in Dye-Sensitized Solar Cells. Polyhedron 2014, 81, 583–587. 10.1016/j.poly.2014.07.015. [DOI] [Google Scholar]
- Huang-Minlon The reaction of Hydrazine Hydrate on Nitro-Compounds and a New Route to Synthetic Oestrogens. J. Am. Chem. Soc. 1948, 70, 2802–2805. 10.1021/ja01188a048. [DOI] [PubMed] [Google Scholar]
- Gerhard S.; Ulrich I.; Rifat P.; Gary A. P.. Method for Treating Neoplasia by Exposure to N,N-Substituted Benzimidazol-2-ones. US6420410B1, July 16, 2002.
- Hamaguchi W.; Masuda N.; Isomura M.; Miyamoto S.; Kikuchi S.; Amano Y.; Honbou K.; Mihara T.; Watanabe T. Design and Synthesis of Novel Benzimidazole Derivatives as Phosphodiesterase 10A Inhibitors with Reduced CYP1A2 Inhibition. Bioorg. Med. Chem. 2013, 21, 7612–7623. 10.1016/j.bmc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- Oh S.; Kim S.; Kong S.; Yang G.; Lee N.; Han D.; Goo J.; Siqueira-Neto J. L.; Freitas-Junior L. H.; Song R. Synthesis and Biological Evaluation of 2,3-dihydroimidazolo[1,2-a] Benzimidazole Derivatives Against Leishmania donovani and Trypanosoma cruzi. Eur. J. Med. Chem. 2014, 84, 395–403. 10.1016/j.ejmech.2014.07.038. [DOI] [PubMed] [Google Scholar]
- Yamada A.; Nakata K.; Shiina I. Acylative Kinetic Resolution of Racemic Aromatic β-Hydroxy Esters Catalyzed by Chiral Nucleophilic N-(1-arylethyl)benzoguanidines. Tetrahedron: Asymmetry 2017, 28, 516–521. 10.1016/j.tetasy.2017.03.005. [DOI] [Google Scholar]
- Miyaura N.; Suzuki A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. 10.1021/cr00039a007. [DOI] [Google Scholar]
- Zhu A.; Zhan W.; Liang Z.; Yoon Y.; Yang H.; Grossniklaus H. E.; Xu J.; Rojas M.; Lockwood M.; Snyder J. P.; Liotta D. C.; Shim H. Dipyrimidine Amines: A Novel Class of Chemokine Receptor Type 4 Antagonists with High Specificity. J. Med. Chem. 2010, 53, 8556–8568. 10.1021/jm100786g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeur E.; Bradley M. Amide Bond Formation: Beyond the Myth of Coupling Reagents. Chem. Soc. Rev. 2009, 38, 606–631. 10.1039/B701677H. [DOI] [PubMed] [Google Scholar]
- Nchinda A. T.; Le Manach C.; Paquet T.; Cabrera D. G.; Wicht K. J.; Brunschwig C.; Njoroge M.; Abay E.; Taylor D.; Lawrence N.; Wittlin S.; Jimenez-Diaz M.-B.; Martinez M. S.; Ferrer S.; Angulo-Barturen I.; Lafuente-Monasterio M. J.; Duffy J.; Burrows J.; Street L. J.; Chibale K. Identification of Fast-Acting 2,6-Disubstituted Imidazopyridines that are Efficacious in the in Vivo Humanized Plasmodium falciparum NODscidIL2Rγnull Mouse Model of Malaria. J. Med. Chem. 2018, 61, 4213–4227. 10.1021/acs.jmedchem.8b00382. [DOI] [PubMed] [Google Scholar]
- a Fraley M. E.; Garbaccio R. M.; Arrington K. L.; Hoffman W. F.; Tasber E. S.; Coleman P. J.; Buser C. A.; Walsh E. S.; Hamilton K.; Fernandes C.; Schaber M. D.; Lobell R. B.; Tao W.; South V. J.; Yan Y.; Kuo L. C.; Prueksaritanont T.; Shu C.; Hartman G. D. Kinesin Spindle Protein (KSP) Inhibitors. Part 2: The Design, Synthesis, and Characterization of 2,4-Diaryl-2,5-dihydropyrrole Inhibitors of the Mitotic Kinesin KSP. Bioorg. Med. Chem. Lett. 2006, 16, 1775–1779. 10.1016/j.bmcl.2006.01.030. [DOI] [PubMed] [Google Scholar]; b Garbaccio R. M.; Fraley M. E.; Tasber E. S.; Olson C. M.; Hoffman W. F.; Arrington K. L.; Torrent M.; Buser C. A.; Walsh E. S.; Hamilton K.; Schaber M. D.; Fernandes C.; Lobell R. B.; Tao W.; South V. J.; Yan Y.; Kuo L. C.; Prueksaritanont T.; Hartman G. D. Kinesin Spindle Protein (KSP) Inhibitors. Part 2: Synthesis and Evaluation of Phenolic 2,4-Diaryl-2,5-dihydropyrroles with Reduced hERG Binding and Employment of a Phosphate Prodrug Strategy for Aqueous Solubility. Bioorg. Med. Chem. Lett. 2006, 16, 1780–1783. 10.1016/j.bmcl.2005.12.094. [DOI] [PubMed] [Google Scholar]
- a Thomson C. G.; Carlson E.; Chicchi G. G.; Kulagowski J. J.; Kurtz M. M.; Swain C. J.; Tsao K.-L. C.; Wheeldon A. Synthesis and Structure-Activity Relationship of 8-azabicyclo[3.2.1]octane Benzylamine NK1 Antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 811–814. 10.1016/j.bmcl.2005.11.026. [DOI] [PubMed] [Google Scholar]; b Xu J.; Wei L.; Mathvink R.; He J.; Park Y.-J.; He H.; Leiting B.; Lyons K. A.; Marsilio F.; Patel R. A.; Wu J. K.; Thornberry N. A.; Weber A. E. Discovery of Potent, and Selective Phenylalanine Based Dipeptidyl Peptidase IV Inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 2533–2536. 10.1016/j.bmcl.2005.03.055. [DOI] [PubMed] [Google Scholar]
- a Rowley M.; Hallett D. J.; Goodacre S.; Moyes C.; Crawforth J.; Sparey T. J.; Patel S.; Marwood R.; Patel S.; Thomas S.; Hitzel L.; O’Connor D.; Szeto N.; Castro J. L.; Hutson P. H.; MacLeod A. M. 3-(4-Fluoropiperidin-3-yl)2-phenylindoles as high Affinity, Selective, and Orally Bioavailable h5-HT2A Receptor Antagonists. J. Med. Chem. 2001, 44, 1603–1614. 10.1021/jm0004998. [DOI] [PubMed] [Google Scholar]; b Bilodeau M. T.; Balitza A. E.; Koester T. J.; Manley P. J.; Rodman L. D.; Buser-Doepner C.; Coll K. E.; Fernandes C.; Gibbs J. B.; Heimbrook D. C.; Huckle W. R.; Kohl N.; Lynch J. J.; Mao X.; McFall R. C.; McLoughlin D.; Miller-Stein C. M.; Rickert K. W.; Sepp-Lorenzino L.; Shipman J. M.; Subramanian R.; Thomas K. A.; Wong B. K.; Yu S.; Hartman G. D. Potent N-(1,3-Thiazol-2-yl)pyrdin-2-amine Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitors with Excellent Pharmacokinetics and Low Affinity for the hERG Ion Channel. J. Med. Chem. 2004, 47, 6363–6372. 10.1021/jm049697f. [DOI] [PubMed] [Google Scholar]
- a Aronov A. M. Predictive in Silico Modelling for hERG Channel Blockers. Drug Discovery Today 2005, 10, 149–155. 10.1016/S1359-6446(04)03278-7. [DOI] [PubMed] [Google Scholar]; b Jamieson C.; Moir E. M.; Ranakovic Z.; Wishart G. Medicinal Chemistry of hERG Optimizations: Highlights and Hang-Ups. J. Med. Chem. 2006, 49, 5029–5046. 10.1021/jm060379l. [DOI] [PubMed] [Google Scholar]
- Keurulainen L.; Vahermo M.; Puente-Felipe M.; Sandoval-Izquierdo E.; Crespo-Fernandez B.; Guijarro-Lopez L.; Huertas-Valentin L.; de las Heras-Duena L.; Leino T. O.; Siiskonen A.; Ballell-Pages L.; Sanz L. M.; Castaneda-Casado P.; Jimenez-Diaz M. B.; Martinez-Martinez M. S.; Viera S.; Kiuru P.; Calderon F.; Yli-Kauhaluoma J. A Developability-Focused Optimization Approach Allows Identification of in Vivo Fast-Acting Antimalarials N-[3-[(Benzimidazol-2-yl)amino]propyl]amides. J. Med. Chem. 2015, 58, 4573–4580. 10.1021/acs.jmedchem.5b00114. [DOI] [PubMed] [Google Scholar]
- Magistrado P. A.; Corey V. C.; Lukens A. K.; LaMonte G.; Sasaki E.; Meister S.; Wree M.; Winzeler E.; Wirth D. F. Plasmodium falciparum Cyclic Amine Resistance Locus (PfCARL), A Resistance Mechanism for Two Distinct Compound Classes. ACS Infect. Dis. 2016, 2, 816–826. 10.1021/acsinfecdis.6b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K.; Okombo J.; Brunschwig C.; Ndubi F.; Barnard L.; Wilkinson C.; Njogu P. M.; Njoroge M.; Laing L.; Machado M.; Prudencio M.; Reader J.; Botha M.; Nondaba S.; Birkholtz L.-M.; Lauterbach S.; Churchyard A.; Coetzer T. L.; Burrows J. N.; Yeates C.; Denti P.; Wiesner L.; Egan T. J.; Wittlin S.; Chibale K. Antimalarial Pyrido[1,2-a]benzimidazoles: Lead Optimization, Parasite Life Cycle Stage Profile, Mechanistic Evaluation, Killing Kinetics, and in Vivo Oral Efficacy in a Mouse Model. J. Med. Chem. 2017, 60, 1432–1448. 10.1021/acs.jmedchem.6b01641. [DOI] [PubMed] [Google Scholar]
- Pennington L. D.; Moustakas D. T. The Necessary Nitrogen Atom: A Versatile High-Impact Design Element for Multiparameter Optimization. J. Med. Chem. 2017, 60, 3552–3579. 10.1021/acs.jmedchem.6b01807. [DOI] [PubMed] [Google Scholar]
- Obach R. S. Prediction of Human Clearance of Twenty–Nine Drugs from Hepatic-Microsomal Intrinsic Clearance Data: An Examination of in Vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos. 1997, 27, 1350–1359. [PubMed] [Google Scholar]
- Hill P. A.; Young R. J. Getting Physical in Drug Discovery a Contemporary Perspective on Solubility and Hydrophobicity. Drug Discovery Today 2010, 15, 648–655. 10.1016/j.drudis.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Makler M. T.; Ries J. M.; Williams J. A.; Bancroft J. E.; Piper R. C.; Gibbins B. L.; Hinrichs D. J. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 1993, 48, 739–741. 10.4269/ajtmh.1993.48.739. [DOI] [PubMed] [Google Scholar]
- Lambros C.; Vanderberg J. P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- Jimenez-Díaz M. B.; Mulet T.; Viera S.; Gomez V.; Garuti H.; Ibanez J.; Alvarez-Doval A.; et al. Improved Murine Model of Malaria Using Plasmodium falciparum Competent Strains and Non-Myelodepleted NOD-scid IL2Rgammanull Mice Engrafted with Human Erythrocytes. Antimicrob. Agents Chemother. 2009, 53, 4533–4536. 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo-Barturen I.; Jiménez-Díaz M. B.; Mulet T.; Rullas J.; Herreros E.; Ferrer S.; Jiménez E.; Mendoza A.; Regadera J.; Rosenthal P. J.; Bathurst I.; Pompliano D. L.; Gómez de las Heras F.; Gargallo-Viola D. A Murine Model of Falciparum-Malaria by in Vivo Selection of Competent Strains in non-Myelodepleted Mice Engrafted with Human Erythrocytes. PLoS One 2008, 3, e2252 10.1371/journal.pone.0002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.