

Abstract
Keratin 17 (KRT17) has been demonstrated to be a potential biological marker for the prediction of prognosis in particular types of cancer. The aim of the present study was to investigate the molecular mechanisms underlying the function of KRT17 in the pancreatic cancer (PAC) cell line PANC-1 and the potential of KRT17 as a therapeutic target for PAC. KRT17 expression levels were analyzed using quantitative PCR and compared with histological data using bioinformatics tools in PAC samples and three human PAC cell lines. Cell proliferation was determined using an MTT assay, in addition to cell cycle distribution and apoptosis analysis using flow cytometry, colony formation assay using Giemsa staining and cell motility analysis using a Transwell migration assay. Tumor growth was evaluated in vivo in nude mice. The expression levels of a number of signaling molecules were measured to establish the potential mechanism by which silencing KRT17 expression affected PAC PANC-1 cells. Increased levels of KRT17 expression were observed in human PAC compared with normal tissues, as well as in three human PAC cell lines (MIA PaCa-2, PANC-1 and KP-3 cells) compared with the H6c7 human immortal pancreatic duct epithelial cell line. High expression levels of KRT17 in PAC samples were associated with poor overall survival (P=0.036) and disease-free survival (P=0.017). Lentivirus-mediated KRT17 silencing inhibited cell proliferation, colony formation and migration, but promoted apoptosis and resulted in cell cycle arrest in the G0/G1 phase in PANC-1 cells. In addition, KRT17 knockdown inhibited in vivo tumor growth. KRT17 knockdown induced dysregulation of ERK1/2 and upregulation of the pro-apoptotic Bcl-2 protein Bad. In conclusion, the present study demonstrated that elevated KRT17 levels are positively associated with pancreatic cancer progression; KRT17 knockdown suppressed cell growth, colony formation, migration and tumor growth, and induced apoptosis and cell cycle arrest, affecting ERK1/2/Bad signaling. Therefore, the results of the present study suggested that KRT17 may be a potential target for the treatment of pancreatic cancer.
Keywords: pancreatic cancer, KRT17, small RNA interference, cell proliferation, apoptosis
Introduction
Over recent years, the understanding of the causes of pancreatic cancer (PAC) has improved, but this has not improved the efficacy of treatment for patients with PAC (1). Surgical resection, which is effective in alleviating clinical symptoms and results in improved long-term survival, is only suitable for early pancreatic ductal carcinoma (2). Although postoperative radiotherapy and chemotherapy reduce metastatic development, no notable improvement in patient survival has been observed (3). Therefore, PAC remains among the most aggressive types of cancer with a 1-year survival rate of ~19% in the USA, and a 5-year survival rate of only 5% worldwide according to epidemiological data from 2010 (3). Therefore, there remains a need to understand the pathogenesis of PAC, identify novel therapeutic targets and develop new therapeutic strategies (4).
Keratin belongs to the superfamily of intermediate filament proteins and can be categorized into two major classes: Type I acidic protein and type II basic protein (5,6). The two types of keratin provide structural support through the use of heterodimers to assemble 10-nm filaments, thus maintaining cell integrity (7). Keratin 17 (KRT17) is a type I keratin and comprises 432 amino acids (8,9). The KRT17 gene is located on chromosome 17q21.2 and is mainly expressed in epithelial appendages, such as hair follicles and sebaceous glands, but not on healthy skin (10). However, KRT17 expression can be induced under specific circumstances, such as injury (11), viral infection (12) and psoriasis (13,14). KRT17 is a multifunctional protein and is involved in the regulation of cellular processes, including proliferation, differentiation and inflammation (15). Increasing evidence has suggested that mutations in KRT17 are associated with pachyonychia congenita type 2 (16–18) and steatocystoma multiplex (19).
Studies have demonstrated that the expression levels of KRT17 are abnormally high in multiple types of cancer, such as cervical (20–22), breast (23,24), gastric (25) and oral (26–28) cancer and lung adenocarcinoma (29), and have recently been reported in pancreatic cancer (30). KRT17 is considered to be a diagnostic and prognostic marker for a variety of diseases, in particular PAC (12,20,21,31,32). However, the underlying mechanism of KRT17 function in PAC remains unknown.
In the present study, the role of KRT17 in PAC and the potential mechanisms underlying KRT17 function were explored, as KRT17 inhibition may be a novel target for PAC treatment.
Materials and methods
Human PAC samples
PAC tumor and adjacent non-tumor samples were obtained from 18 patients (10 male and 8 female patients) that underwent surgical resection at the Department of Hepatobiliary Surgery, the First Affiliated Hospital of Wannan Medical College (Wuhu, China) between November 2016 and December 2018. The Medical Ethics Committee of the First Affiliated Hospital of Wannan Medical College (Wuhu, China) approved the experimental protocols in the present study and written informed consent was obtained from the patients. All patients included in the present study had no history of chemoradiotherapy or other pancreatic diseases. The ages of patients at the time of diagnosis ranged between 46 and 74 years (mean, 56). All patients were characterized clinically by two pathologists in accordance with the criteria of the American Joint Committee on Cancer (33).
Bioinformatics analysis of KRT expression
KRT17 mRNA expression levels in PAC and non-tumor tissues were analyzed using the GEPIA website according to the operating instructions on the website (34). In addition, overall survival and disease-free survival of patients with PAC were evaluated using Log-rank test according to the default settings on the GEPIA website online. The expression of KRT17 protein in PAC and non-tumor tissues was also compared using The Human Protein Atlas website (35–37).
Cell culture
The human PAC cell lines MIA PaCa-2, PANC-1 and KP-3, the human immortalized pancreatic duct epithelial cell line H6c7 and 293T cells were obtained from Guangzhou Cellcook Cell Biotechnology, Ltd (http://www.cellcook.com). All cells were maintained in RPMI-1640 medium (Sangon Biotech Co., Ltd.) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin/streptomycin (Sangon Biotech Co., Ltd.) and 2% L-glutamine (Sangon Biotech Co., Ltd.) at 37°C in a 5% CO2 incubator.
Construction of the KRT17-shRNA lentivirus
Two short hairpin RNA (shRNA) sequences specifically targeting KRT17 (GeneBank ID: X62571.1; 5′-CAGTCGCGTTTGCGACTGG-3′ and 5′-CTTCCTTGCCTGATGACAA-3′) and a scrambled (Scr) shRNA sequence (5′-TTCTCCGAACGTGTCACGT-3′) for use as a negative control were designed and purchased from iGene Biotechnology Co., Ltd. The KRT17 or Scr shRNA (2 ng of each shRNA used) was cloned into the enhanced green fluorescent protein (eGFP) containing pReceiver-Lv193× lentivector (iGene Biotechnology Co., Ltd.) using FastDigest KpnI (cat. no. FD0524) and XhoI (cat. no. FD0694) restriction endonucleases (both Thermo Fisher Scientific, Inc.) to produce plasmids termed KRT17-shRNA-1, KRT17-shRNA-2 and Scr-shRNA. A lentivector kit (Lenti-Pac™; iGene Biotechnology Co., Ltd.) was used to produce KRT17 or scrambled shRNA. 293T cells were co-infected with KRT17-shRNA-1, KRT17-shRNA-2 or Scr-shRNA plasmids to produce KRT17-shRNA-1, KRT17-shRNA-2 or Scr-shRNA lentiviruses. On day 3 post-transfection, the lentivectors were collected and resuspended in PBS. PANC-1 cells were cultured in 6-well plates (5×104 cells/well) and infected with KRT17-shRNA-1, KRT17-shRNA-2 or Scr-shRNA lentivectors in PBS containing 5 µg/ml polybrene (Merck KGaA). Cells were filtered with 2 µg/ml puromycin (Sangon Biotech) until they stably expressed KRT17-shRNA or Scr-shRNA. The same Scr-shRNA sequences were used and performed simultaneously with the corresponding KRT17-shRNA-1 or KRT-shRNA-2 target sequences, but the two experiments were not performed at the same time.
Efficiency of KRT17-shRNA knockdown
Human 293T cells or pancreatic cancer cells were plated into 6-well plates (5×104 cells/well) and infected with KRT17-shRNA or Scr-shRNA lentiviruses. Cells were incubated at 37°C in a 5% CO2 atmosphere for 48 h (multiplicity of infection, 5), and a suspension of lentivectors (5 µl/well) was added to the RPMI-1640 medium. After infection for 3 days, eGFP expression in the cells was quantified using an inverted fluorescence microscope (BX63; Olympus Corporation) at ×200 magnification to ascertain infection efficiency. The evaluation formula of infection efficiency is: Number of cells expressing EGFP/total number of cells ×100%. Cells with >75% infection efficiency were collected for further analysis, as described below.
Reverse transcription-quantitative (RT-q)PCR
Total RNA from each PAC sample or cultured cells was extracted using TRIzol® reagent (Sangon Biotech) according to the manufacturer's instructions and quantified using a SpectraMax QuickDrop (Molecular Devices, LLC.). Reverse transcription was performed using a SYBR® Green Quantitative RT-PCR kit (cat. no. QR0100-1KT; Sigma-Aldrich; Merck KGaA) according to the manufacturer's instructions. First, 50 ng RNA template was mixed with 10 µl 2X SYBR Green quantitative RT-PCR buffer, 1.125 µl each primer (10 µmol/l) and 0.125 µl MMLV RT Enzyme, and ddH2O was added to a total volume of 25 µl. The mixture was incubated at 43°C for 30 min for first strand synthesis. PCR was conducted using the following thermocycling conditions: Denaturation at 94°C for 30 sec, followed by 40 cycles of 95°C for 5 sec, 55°C for 15 sec and 72°C for 10 sec. The primer sequences were designed and synthesized (iGene Biotech) as follows: KRT17 forward, 5′-AGGTGCGTACCATTGTGGAA-3′ and reverse, 5′-ATCAGGCAAGGAAGCATGGG-3′; GAPDH (internal control) forward, 5′-TGACTTCAACAGCGACACCCA-3′ and reverse, 5′-CACCCTGTTGCTGTAGCCAAA-3′. Other primer sequences are listed in Table I. Each sample was processed in triplicate. Data were analyzed using the 2−ΔΔCq method (38) and normalized against GAPDH.
Table I.
Primer sequences used for quantitative PCR.
| Gene | Primer sequence (5′-3′) |
|---|---|
| ERK1/2 | |
| Forward | TTACTGCGCTTCAGACATGAGA |
| Reverse | ATCTGTTTCCATGAGGTCCTGT |
| Akt | |
| Forward | CGGGCACATCAAGATAACGGAC |
| Reverse | TCGTTGTCCTCCAGCACCTCAG |
| Bad | |
| Forward | CCCAGAGTTTGAGCCGAGTG |
| Reverse | GCTGTGCTGCCCAGAGGTT |
| p38 MAPK | |
| Forward | GCTGAAGATTCTGGATTTTG |
| Reverse | GTTCTTCCAGTCAACAGCTC |
| SAPK/JNK | |
| Forward | CCGCTGTCCCTTTGTCTTG |
| Reverse | GCACATCTATTCTGTTCCATACTACC |
| CAPS3 | |
| Forward | CTGGAAAACCCAAACTTTTCATTA |
| Reverse | GCCAGGAAAAGTAACCAGGTGC |
| CAPS7 | |
| Forward | CGGTCCTCGTTTGTACCGTC |
| Reverse | CGCCCATACCTGTCACTTTATCA |
| GAPDH | |
| Forward | TGACTTCAACAGCGACACCCA |
| Reverse | CACCCTGTTGCTGTAGCCAAA |
Western blotting
Total protein from each sample was extracted using RIPA lysis buffer (Sigma-Aldrich; Merck KGaA). Protein quantitation was performed using a QuantiPro™ BCA Assay kit (Sigma-Aldrich; Merck KGaA). Equal amounts (~10 µg) of total protein from each sample were loaded onto a 15% gel, resolved using SDS-PAGE and subsequently transferred to Immobilon®-P PVDF membranes (EMD Millipore). The membranes were blocked with 1% bovine serum albumin (BSA; cat. no. SRE0096; Sigma-Aldrich; Merck KGaA) dissolved in TBS + 0.1% (v/v) Tween-20 (TBST) for 60 min at 37°C and incubated with the appropriate primary monoclonal antibody overnight at 4°C. The following mouse primary monoclonal antibodies were used: Mouse anti-KRT17 (1:1,000; cat. no. WH0003872M1), rabbit anti-Phospho-p38 MAPK (1:1,000; cat. no. 4511), rabbit anti-p38 MAPK (1:1,000; cat. no. 2387), rabbit anti-Phospho-Akt (1:1,000; cat. no. 4060), rabbit anti-Akt (1:1,000; cat. no. 4691), or mouse anti-GAPDH (1:5,000; cat. no. G8795) all from Sigma-Aldrich (Merck KGaA). After rinsing 3–5 times with TBST, horseradish peroxidase-conjugated goat anti-mouse IgG (H+L) antibody (1:10,000; cat. no. AP308P; Sigma-Aldrich; Merck KGaA) was added as the secondary antibody, incubated at 37°C for 1 h, and then quantified using an ECL substrate kit (cat. no. ab133406; Abcam). The densitometry values of the immune response bands were quantified using ImageJ software (version 1.50; National Institutes of Health).
MTT assay
Logarithmic phase cells (2,000 cells/well) transfected with lentiviral KRT17-shRNA-1, KRT17-shRNA-2 or Scr-shRNA were plated into the wells of 96-well plates and maintained at 37°C in a 5% CO2 incubator. Cell proliferation was measured each day for 5 days using a Cell Proliferation Kit I (Roche Diagnostics) in accordance with the manufacturer's instructions. Finally, the absorbance at a wavelength of 490/570 nm in each well of the plate was measured using a microplate reader (Norgen Biotek Corp.).
Detection of apoptosis and cell cycle
Apoptosis was quantified using an Annexin V-FITC Apoptosis Detection kit (Sigma-Aldrich; Merck KGaA) according to the manufacturer's protocol. After culturing for 4 days, PANC-1 cells that had been infected with lentiviral KRT17-shRNA-1, KRT17-shRNA-2 or Scr-shRNA were harvested and stained with a propidium iodide solution (Sigma-Aldrich; Merck KGaA) according to the manufacturer's instructions to achieve a final density of 1×106 cells/ml. KRT17-shRNA-1-, KRT17-shRNA-2- and Scr-shRNA-infected PANC-1 cells were added to the wells of a 6-well plate (1×106 cells/well) and fixed in 70% ethanol for 1 h on ice. A 5 µl aliquot of Annexin V-FITC was added to 100 µl cell suspension, mixed and incubated for 10 min at room temperature. A FACSCalibur™ flow cytometer (BD Biosciences) was used to measure the percentage of apoptotic cells according to the manufacturer's protocol.
Colony-formation assay
Logarithmic phase cells infected with lentiviral KRT17-shRNA-1, KRT17-shRNA-2 or Scr-shRNA were seeded into 6-well plates (800 cells/well) and maintained at 37°C in a 5% CO2 incubator for 14 days. After discarding the cultured RPMI-1640 medium, 4% paraformaldehyde (Sigma-Aldrich; Merck KGaA) was added to each plate (1 ml/well) and incubated for 60 min at room temperature to fix the cells. After removing the supernatant, cellular clones were stained with 1 ml/well staining solution from the Giemsa staining kit (Thermo Fisher Scientific, Inc.) for 1 min and examined the number of colonies containing >50 cells under a light microscope (magnification, ×200). Three wells per group were stained and examined as biological replicates.
Transwell migration assay
A Transwell migration assay was performed to evaluate cell migration. The wells were coated with 80 µl collagen I (Thermo Fisher Scientific, Inc.), and 50 µl serum-free RPMI-1640 medium (Thermo Fisher Scientific, Inc.) was added. A total of 220 µl cell suspension (1×105 cells/ml) transfected with either KRT17-shRNA or Scr-shRNA lentivector were then added to the upper wells, and RPMI-1640 media with 30% FBS (Sigma-Aldrich; Merck KGaA) was added to the lower wells. After culture at 37°C for 24 h, cells in the lower wells were fixed with a 4% formaldehyde solution (Sigma-Aldrich; Merck KGaA) for 15 min and stained with 1% crystal violet (Sigma-Aldrich; Merck KGaA) at room temperature for 30 min. Cells were counted using a light microscope and each experiment in each group was performed in triplicate.
Tumorigenesis analysis
A total of 20 nude mice (18-22 g) were purchased from the Institute of Comparative Medicine of Yangzhou University (Yangzhou, China). The animal experiments were approved by The First Affiliated Hospital of Wannan Medical College & Yijishan Hospital Animal Experimental Ethics Committee (Wuhu, China). All animals were kept in an SPF animal room at 37°C and 60–70% humidity, with a 12-h light/dark cycle and free access to food and water. Tumorigenesis was assessed using nude mice as previously described (39). PANC-1 cells (1×105 cells in 200 µl PBS per mouse) transfected with KRT17-shRNA-1 or Scr-shRNA lentivector were injected subcutaneously or below the armpit into female BALB/c mice (4-weeks old). Five weeks after the initial injection, the dimensions of the tumors were measured every three days and volumes were calculated using the following formula: Tumor volume=(π/6) × L × W × W, where L represented the longest diameter and W was the shortest diameter. At the time of sacrifice, the mean weight of each excised tumors was calculated.
Statistical analysis
Three separate experiments were conducted for each condition. Data are presented as means ± SD. Statistical tests were performed using GraphPad Prism software for Windows (version 8.0.1; GraphPad Software, Inc.). Overall survival (OS) or disease free survival (DFS) was performed by Log-rank test. Multiple group comparisons were performed using least-significant difference (LSD) as a post-hoc method after one-way analysis of variance, and comparisons between two groups in all figures were performed using student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
KRT17 expression in human pancreatic cancer samples
To investigate KRT17 expression in human PAC tissue and matched adjacent non-tumor (normal) tissue, 18 samples were randomly selected from a total of 18 human PAC specimens for qPCR. The results indicated that the average expression levels of KRT17 mRNA in PAC tissues were significantly elevated compared with normal tissues (P<0.001; Fig. 1A). Bioinformatics results from historical samples in The Human Protein Atlas also suggested that, compared with normal tissues, KRT17 mRNA levels in PAC tissues were higher (P<0.05; Fig. 1B), and the protein expression levels of KRT17 were higher in PAC compared with normal tissues based on the immunohistochemical staining results from The Human Protein Atlas website (Fig. 1C). In addition, bioinformatics analysis suggested that high expression of KRT17 in PAC tissues was negatively associated with overall survival (log-rank P=0.036; Fig. 1D) and disease-free survival (log-rank P=0.017; Fig. 1E).
Figure 1.

KRT17 expression levels in PAC tissue samples. (A) Quantitative PCR results demonstrated significantly increased expression levels of KRT17 mRNA in PAC tissues compared with adjacent normal tissues. n=18. (B) Bioinformatics analysis revealed that the expression levels of KRT17 were higher in T compared with N tissues. (C) Immunohistochemical staining from The Human Protein Atlas demonstrated higher KRT17 expression levels in PAC compared with normal samples. (D and E) Kaplan-Meier survival analyses from the GEPIA website showed that (D) overall survival and (E) disease-free survival of patients with high expression levels of KRT17 were significantly lower compared with those with low expression levels of KRT17 in PAC. *P<0.05, ***P<0.001. KRT17, Keratin 17; PAC, pancreatic cancer; T, tumor; N, normal; HR, hazard ratio.
Identification of lentivirus-mediated shRNA for human KRT17 silencing
To explore the functional role of KRT17 in PAC both in vitro and in vivo, the expression levels of KRT17 mRNA were initially examined in three human pancreatic cancer cell lines: MIA PaCa-2, PANC-1 and KP-3, in addition to its expression levels in the pancreatic duct epithelial cell line H6c7. The results demonstrated that KRT17 mRNA levels were elevated in the human PAC cell lines MIA PaCa-2, PANC-1 and KP-3 compared with H6c7 cells (Fig. 2A) and confirmed by western blotting (Fig. 2B). Subsequently, PANC-1 cells were selected for follow-up studies, and a lentivirus designed to silence KRT17 expression in PANC-1 cells was constructed for functional analysis. The results demonstrated that GFP expression levels in PANC-1 cells transfected with KRT17-shRNA-1-, KRT17-shRNA-2- or Scr-shRNA-lentivirus indicated high rates of infection efficiency, >90% after 48 h (Fig. 2C). qPCR results indicated that the silencing efficiency of transfection of KRT17-shRNA in PANC-1 cells was ~60–80% (Fig. 2D and E), further confirmed by western blot analysis (Fig. 2F and G).
Figure 2.
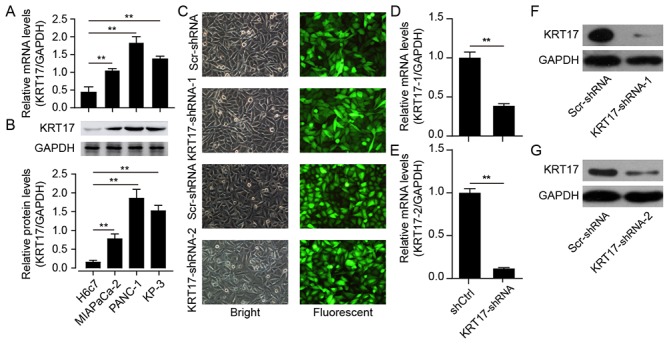
Efficient KRT17 silencing in human PANC-1 cells using KRT17-shRNA-mediated RNA interference. (A and B) Expression levels of KRT17 (A) mRNA and (B) protein in the PAC cell lines MIA PaCa-2, PANC-1 and KP-3 were significantly higher compared with the immortal ductal epithelial H6c7 cells. (C) Representative images of GFP expression in PANC-1 cells after transfection with Scr-shRNA, KRT17-shRNA-1 or KRT17-shRNA-2 lentiviruses. (D and E) KRT17 mRNA expression following transfection with KRT17-shRNA-1 or KRT17-shRNA-2 was downregulated compared with that in cells transfected with the Scr-shRNA lentivirus. GAPDH was used as the internal control. (F and G) Expression levels of KRT17 protein in PANC-1 cells following lentiviral transfection KRT17-shRNA-1 or KRT17-shRNA-2 were decreased compared with Scr-shRNA. **P<0.01. KRT17, Keratin 17; PAC, pancreatic cancer; Scr, scrambled; sh, short hairpin.
Knockdown of KRT17 significantly reduces cell proliferation of PANC-1 cells
To demonstrate the effect of KRT17-shRNA in vitro, PANC-1 cells were infected with lentiviral KRT17-shRNA-1, KRT17-shRNA-2 or Scr-shRNA. Cell viability was monitored every day for five days using an MTT assay. The results demonstrated that KRT17-shRNA significantly decreased cell viability 72 h post-transfection with KRT17-shRNA-1 or KRT17-shRNA-2 lentivirus and that cell viability decreased over the following two days compared with cells in the Scr-shRNA group (P<0.01; Fig. 3A and B). Taken together, these results suggested that KRT17 promoted pancreatic cancer cell progression via accelerated cell proliferation.
Figure 3.
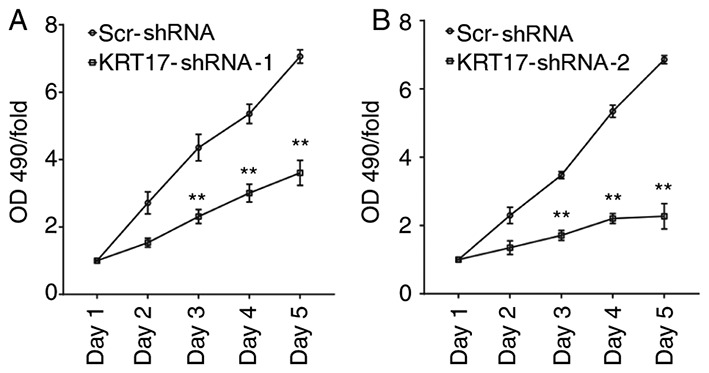
KRT17 knockdown inhibits cell proliferation in human PANC-1 cells. (A) Cell proliferation in human PANC-1 cells after transfection of lentiviral KRT17-shRNA-1 was relatively stable for 5 consecutive days compared with the Scr-shRNA group on corresponding days as determined by MTT assay. (B) Cell proliferation in human PANC-1 cells after transfection of lentiviral KRT17-shRNA-2 continued to decline for 5 consecutive days compared with the Scr-shRNA group on corresponding days. **P<0.01. KRT17, Keratin 17; Scr, scrambled; sh, short hairpin.
KRT17 knockdown induces cell cycle arrest in PANC-1 cells
As KRT17-shRNA significantly inhibited PANC-1 cell proliferation, the underlying molecular mechanism was investigated using flow cytometry to analyze the cell cycle. The results indicated that following transfection with the KRT17-shRNA-1 lentivirus, 65.58±2.02, 27.71±1.44 and 5.21±0.54% of cells were in the G0/G1, S and G2/M phase, respectively [Fig. 4A (upper panel) and B]. Following transfection with the Scr-shRNA lentivirus, 52.28±0.99, 44.56±3.05 and 5.27±0.95% of cells were in the G0/G1, S and G2/M phase, respectively (Fig. 4B). Additionally, following transfection with the KRT17-shRNA-2 lentivirus, 67.92±1.61, 9.52±0.93 and 22.8±1.08% of cells were in the G0/G1, S and G2/M phase, respectively [Fig. 4A (bottom panel) and C]. Following transfection with the Scr-shRNA lentivirus, 60.78±1.23, 18.77±1.63 and 21.21±1.63% of the cells transfected with KRT17-shRNA-2 lentivirus were in the G0/G1, S and G2/M phase, respectively (Fig. 4C). These results suggested that inhibition of KRT17 expression resulted in cell cycle arrest in the G0/G1 phase compared with the Scr-shRNA group cells (all P<0.01; Fig. 4).
Figure 4.

KRT17 silencing arrested the cell cycle at the G0/G1 phase. (A) Representative images of the cell cycle distribution in PANC-1 cells transfected with the KRT17-shRNA-1 or Scr-shRNA lentivirus. (B) Proportion of total cells at different phases of the cell cycle following transfection with KRT17-shRNA-1 or Scr-shRNA lentivirus. (C) Representative images of the cell cycle distribution in PANC-1 cells transfected with the KRT17-shRNA-2 or Scr-shRNA lentivirus. (D) Proportion of total cells at different phases of the cell cycle following transfection with KRT17-shRNA-2 or Scr-shRNA lentivirus. **P<0.01. KRT17, Keratin 17; Scr, scrambled; sh, short hairpin.
KRT17 silencing increases PANC-1 cell apoptosis
To analyze whether KRT17 silencing was associated with apoptosis in PANC-1 cells, infected PANC-1 cells were sorted using flow cytometry after treatment with FITC-conjugated Annexin V. Cells were infected with lentiviral KRT17-shRNA-1, KRT17-shRNA-2 or Scr-shRNA and sorted using flow cytometry after 5 days shown in a representative picture in Fig. 5A. As presented in Fig. 5B and C, 5.26±0.09 and 4.70±0.23% of cells infected with Scr-shRNA lentivirus were apoptotic, but in PANC-1 cells infected with KRT17-shRNA-1 or KRT17-shRNA-2 lentivirus, the proportion of apoptotic was increased to 12.75±0.90 and 16.79±1.00%, respectively, compared with the control cells (P<0.01). These results demonstrated that KRT17 knockdown increased apoptosis in human PANC-1 cells.
Figure 5.
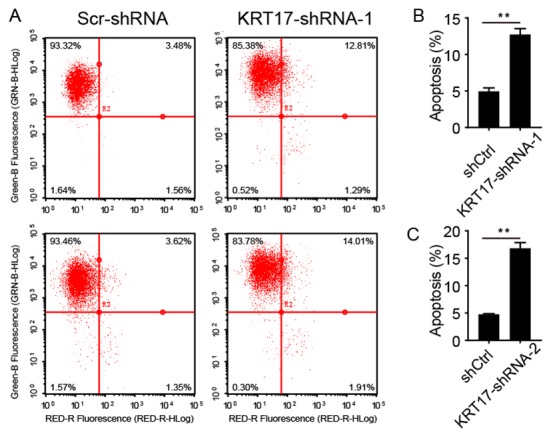
KRT17 knockdown increases apoptosis in human PANC-1 cells. (A) Representative fluorescence of PANC-1 cells infected with the Scr-shRNA, KRT17-shRNA-1 or KRT17-shRNA-2 lentivirus. (B) Proportion of apoptotic PANC-1 cells was increased following KRT17-shRNA-1 knockdown compared with the Scr-shRNA group. (C) Proportion of apoptotic PANC-1 cells following KRT17-shRNA-2 knockdown was elevated compared with the control cells. **P<0.01. KRT17, Keratin 17; sh, short hairpin.
Knockdown of KRT17-shRNA inhibits colony formation capability
The ability of PANC-1 cells to form colonies following treatment with KRT17-shRNA or Scr-shRNA lentivirus was explored. As presented in Fig. 6, KRT17 silencing significantly reduced the number of colonies (62±6 colonies/well in PANC-1 cells transfected with KRT17-shRNA-1; 37±4 colonies/well in PANC-1 cells transfected with KRT17-shRNA-2) compared with 164±8 and 157±18 colonies/well in PANC-1 cells in the Scr-shRNA groups (P<0.01).
Figure 6.

KRT17 knockdown inhibits colony formation in human PANC-1 cells. (A) Giemsa staining of PANC-1 clones in 6-well plates 14 days post-transfection with the Scr-shRNA or KRT17-shRNA-1 lentiviruses. (B) Mean number of PANC-1 colonies counted following KRT17-shRNA-1--mediated knockdown was decreased compared with the corresponding Scr-shRNA groups. (C) Giemsa staining of PANC-1 clones in 6-well plates 14 days post-transfection with the Scr-shRNA or KRT17-shRNA-2 lentiviruses. (D) Mean number of PANC-1 colonies counted following KRT17-shRNA-2-mediated knockdown was decreased compared with the corresponding Scr-shRNA groups. **P<0.01. KRT17, Keratin 17; sh, short hairpin; Scr, scrambled.
Knockdown of KRT17 inhibits the migration of PANC-1 cells
To investigate the effects of KRT17 knockdown on the migration of human PANC-1 cells, the migration capability of PANC-1 cells transfected with KRT17-shRNA-1 or Scr-shRNA lentivirus was measured using a Transwell assay. Compared with the Scr-shRNA group, the number of migratory cells in the KRT17-shRNA-1 group was significantly lower (P<0.01, Fig. 7A and B), suggesting that knockdown of KRT17 resulted in reduced PANC-1 cell migration. This finding further emphasized the function of KRT17 in the migration of PANC-1 cells.
Figure 7.
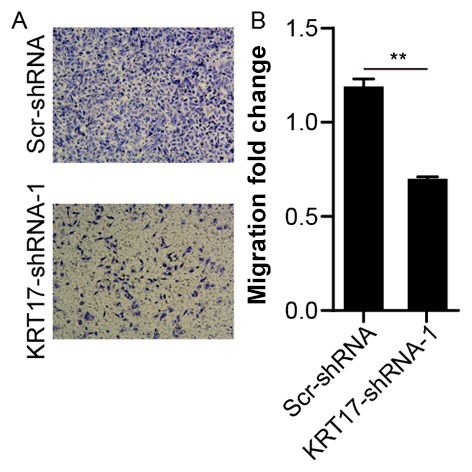
KRT17 knockdown inhibits the migration of human PANC-1 cells. (A) Representative images of migratory PANC-1 cells transfected with the KRT17-shRNA-1 lentivector. (B) The number of migrated PANC-1 cells transfected with the KRT17-shRNA-1 lentivector was significantly decreased compared with the Scr-shRNA group. **P<0.01. KRT17, Keratin 17; sh, short hairpin; Scr, scrambled.
KRT17 silencing suppresses tumor growth in vivo
To ascertain the role of KRT17-shRNA as a potential target for the treatment of PAC, BALB/c nude mice were subcutaneously injected with PANC-1 cells transfected with either KRT17-shRNA-1 or Scr-shRNA lentivector. The results demonstrated that, compared with the Scr-shRNA group, tumors grew more slowly in the KRT17-shRNA group (Fig. 8A). In addition, tumors from mice in the KRT17-shRNA group were also smaller compared with the Scr-shRNA-group (Fig. 8B and C). This result showed that KRT17 silencing inhibited PAC tumorigenesis in vivo. Taken together, these data indicate that targeting KRT17 with lentiviral-KRT17-shRNA could have an inhibitory effect in vivo on pancreatic cancers in which KRT17 is overexpressed.
Figure 8.
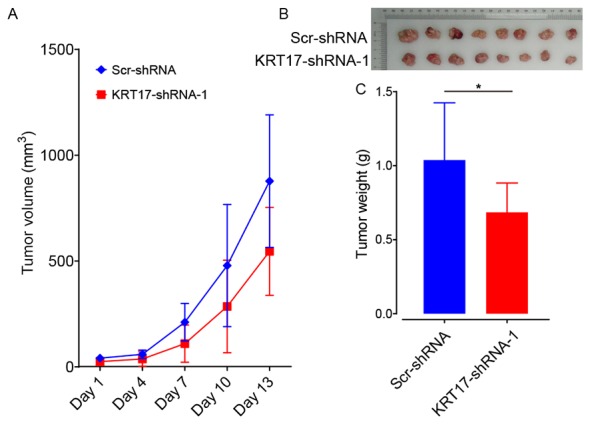
KRT17 silencing inhibits tumor growth in vivo. (A) Changes in tumor volume after injection of PANC-1 cells transfected with the KRT17-shRNA-1 or Scr-shRNA lentivector. (B) The diameter of PANC-1 tumors was lower in the KRT17-shRNA-1 group compared with the Scr-shRNA group 5 weeks after transplantation. (C) Mean tumor weight was reduced in the KRT17-shRNA-1 group compared with the Scr-shRNA group 5 weeks after cell injection. n=10. *P<0.05. KRT17, Keratin 17; sh, short hairpin; Scr, scrambled.
KRT17 silencing in PANC-1 cells affects ERK1/2/Bad signaling
To investigate the underlying signaling mechanisms of the effect of KRT17 silencing in PANC-1 cells, the expression levels of signaling molecules relevant to cell proliferation and apoptosis were analyzed using qPCR, including ERK1/2, Akt, Bad, p38 MAPK, SAPK/JNK, caspase-3 and caspase-7. The results demonstrated that ERK1/2 was significantly downregulated in KRT17-shRNA-treated PANC-1 cells compared with Scr-shRNA-treated cells (P<0.01; Fig. 9A). By contrast, Bad was significantly upregulated compared with the Scr-shRNA group (P<0.01). Western blot analysis further confirmed that ERK1/2 expression levels were decreased, whereas Bad expression levels were increased in the KRT17-shRNA group compared with the Scr-shRNA group. In addition, KRT17 knockdown exhibited no effects on the expression of p38 MAPK, Akt and phospho-Akt.
Figure 9.
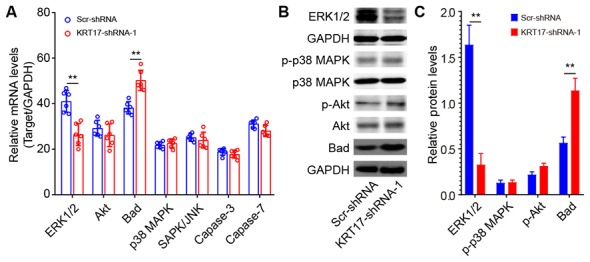
Effects of KRT17 knockdown on cell proliferation-related signaling molecules in PANC-1 cells. (A) Levels of proliferation-related signaling molecules were measured by quantitative PCR. Unpaired t-test. (B) The levels of pERK1/2, p-Akt, p-p38 MAPK and Bad expression following KRT17 knockdown. (C) Quantitative analysis of the grey value of (B). **P<0.01. Keratin 17; Scr, scrambled; p-, phosphor; sh, short hairpin; JNK, C-Jun N-terminal kinase 1.
Discussion
The results of the present study demonstrated that KRT17 expression levels were elevated in human PAC samples compared with normal tissues. Bioinformatics analysis also demonstrated high expression levels of KRT17 mRNA and protein in PAC samples compared with normal tissues. In addition, elevated expression levels of KRT17 were associated with the risk of PAC progression, including less favorable overall and disease-free survival.
Using qPCR and western blotting, the present study demonstrated that KRT17 expression levels were elevated in three PAC cell lines: MIA PaCa-2, PANC-1 and KP-3. Cell proliferation and colony formation of PANC-1 cells were significantly diminished in KRT17-shRNA-transfected cells, whereas the percentage of those cells that had undergone apoptosis was significantly higher compared with the proportion of Scr-shRNA-transfected cells. In addition, KRT17 silencing significantly reduced tumor growth in vivo. These data suggested that elevated KRT17 expression enhanced the development of PAC. Of note, the results of the present study revealed that KRT17 silencing arrested the cell cycle at the G0/G1 phase, suggesting that KRT17 augmented human PAC cell growth and colony formation by inhibiting the cell cycle. However, the underlying mechanism of the cell cycle arrest in KRT17-knockdown PANC-1 cells requires additional investigation in future studies.
KRT17 is primarily expressed in the epidermis and its appendages (40). Previous studies have demonstrated that abnormal expression levels of KRT17 are associated with cell proliferation and tumor development. For example, high expression levels of KRT17 promote cell proliferation and tumorigenesis in skin tumors (41), oral squamous cell carcinoma (22,26,42), cervical squamous cell carcinoma (20,21,43,44) and gastric cancer (25). However, reports on the functional role of KRT17 in pancreatic cancer are limited. A recent study has confirmed that high expression of KRT17 is positively associated with less favorable prognosis in patients with pancreatic cancer (45). However, the underlying mechanism of this has not been reported. The present study also revealed aberrantly increased expression levels of KRT17 in PAC samples, which indicated that KRT17 may serve a role in PAC tumorigenesis.
In the present study, the role of KRT17 in human PAC was analyzed using transfection of PANC-1 cells with KRT17-shRNA. The results demonstrated that KRT17 silencing suppressed cell proliferation and facilitated apoptosis in human PANC-1 cells. In addition, KRT17 knockdown decreased the migratory ability of PANC-1 cells. The results indicated that abnormally high expression levels of KRT17 were implicated in human PAC progression and may be associated with advanced tumor stage and distant metastasis of pancreatic cancer, as observed in the previous results of this study.
To explore the effects of KRT17 knockdown in human PANC-1 cells, the changes in the expression levels of a number of stress and apoptosis molecules were analyzed using qPCR and/or western blot analysis. The results demonstrated that ERK1/2 was significantly downregulated following lentiviral transfection-mediated KRT17 knockdown, and the expression levels of the Bad protein were significantly increased. Accumulating evidence suggests that ERK1/2 activation contributes to the epithelial-mesenchymal transition (EMT) in intrahepatic cholangiocarcinoma (46), although inhibition of the ERK1/2 pathway has failed to prevent transforming growth factor β-induced EMT and migration. The present study demonstrated that KRT17 knockdown decreased the level of ERK1/2 phosphorylation, suggesting that upregulation of KRT17 participated in EMT and migration in PAC tumorigenesis. In the majority of cancer types, ERK1/2 signaling promotes cell survival by inhibiting the function of pro-death proteins, including Bad (47). In the present study, Bad phosphorylation increased following KRT17 silencing using KRT17-shRNA lentivirus transfection. These results suggested that KRT17 may increase PAC tumorigenesis via ERK1/2/Bad signaling.
The present study demonstrated that KRT17 expression levels were elevated in PAC tissues and pancreatic cancer cell lines, MIA PaCa-2, PANC-1 and KP-3 cells. Lentivirus-mediated KRT17 knockdown inhibited cell proliferation and clone formation by arresting the cells in the G0/G1 phase and by accelerating apoptosis. The role of KRT17 may be to augment pancreatic cancer tumorigenesis via upregulation of ERK1/2 and downregulation of Bad expression. These results suggested that KRT17 may serve a role in PAC tumorigenesis and may be useful as a potential therapeutic target for the treatment of PAC.
Acknowledgements
Not applicable.
Funding
This study was supported by the Academic Funding Project for Top Talents of Discipline (Professional) in Anhui Province (grant no. gxbjZD17), Anhui Provincial Centralized Local Science and Technology Development Special Project (grant no. YDZX20183400004899) and Anhui Science and Technology Research Fund Project (grant no. 1501041156).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
GW and XW designed the study. PC, ZS and XF performed the cytology experiments. PC and SX conducted the animal experiments. XF, GW and JW analyzed the data. PC and XF wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by The First Affiliated Hospital of Wannan Medical College and Yijishan Hospital Medical Ethics Committee (Wuhu, China). Written informed consent was provided by all patients. Animal experiments were approved by The First Affiliated Hospital of Wannan Medical College & Yijishan Hospital Animal Experimental Ethics Committee (Wuhu, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Saad AM, Turk T, Al-Husseini MJ, Abdel-Rahman O. Trends in pancreatic adenocarcinoma incidence and mortality in the United States in the last four decades; a SEER-based study. BMC Cancer. 2018;18:688. doi: 10.1186/s12885-018-4610-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu D, Ansari D, Zhou Q, Sasor A, Said Hilmersson K, Andersson R. Galectin 4 is a biomarker for early recurrence and death after surgical resection for pancreatic ductal adenocarcinoma. Scand J Gastroenterol. 2019;54:95–100. doi: 10.1080/00365521.2018.1561937. [DOI] [PubMed] [Google Scholar]
- 3.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falasca M, Kim M, Casari I. Pancreatic cancer: Current research and future directions. Biochim Biophys Acta. 2016;1865:123–132. doi: 10.1016/j.bbcan.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 5.van den Hurk R, Dijkstra G, van Mil FN, Hulshof SC, van den Ingh TS. Distribution of the intermediate filament proteins vimentin, keratin, and desmin in the bovine ovary. Mol Reprod Dev. 1995;41:459–467. doi: 10.1002/mrd.1080410408. [DOI] [PubMed] [Google Scholar]
- 6.Somji S, Cao L, Mehus A, Zhou XD, Sens MA, Dunlevy JR, Garrett SH, Zheng Y, Larson JL, Sens DA. Comparison of expression patterns of keratin 6, 7, 16, 17, and 19 within multiple independent isolates of As(+3)- and Cd (+2)-induced bladder cancer: Keratin 6, 7, 16, 17, and 19 in bladder cancer. Cell Biol Toxicol. 2011;27:381–396. doi: 10.1007/s10565-010-9169-z. [DOI] [PubMed] [Google Scholar]
- 7.Jacob JT, Coulombe PA, Kwan R, Omary MB. Types I and II keratin intermediate filaments. Cold Spring Harb Perspect Biol. 2018;10(pii):a018275. doi: 10.1101/cshperspect.a018275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herrmann H, Aebi U. Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu Rev Biochem. 2004;73:749–789. doi: 10.1146/annurev.biochem.73.011303.073823. [DOI] [PubMed] [Google Scholar]
- 9.Herrmann H, Aebi U. Intermediate filaments: Structure and assembly. Cold Spring Harb Perspect Biol. 2016;8(pii):a018242. doi: 10.1101/cshperspect.a018242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurokawa I, Takahashi K, Moll I, Moll R. Expression of keratins in cutaneous epithelial tumors and related disorders-distribution and clinical significance. Exp Dermatol. 2011;20:217–228. doi: 10.1111/j.1600-0625.2009.01006.x. [DOI] [PubMed] [Google Scholar]
- 11.Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441:362–365. doi: 10.1038/nature04659. [DOI] [PubMed] [Google Scholar]
- 12.Proby CM, Churchill L, Purkis PE, Glover MT, Sexton CJ, Leigh IM. Keratin 17 expression as a marker for epithelial transformation in viral warts. Am J Pathol. 1993;143:1667–1678. [PMC free article] [PubMed] [Google Scholar]
- 13.Shi X, Jin L, Dang E, Chang T, Feng Z, Liu Y, Wang G. IL-17A upregulates keratin 17 expression in keratinocytes through STAT1- and STAT3-dependent mechanisms. J Invest Dermatol. 2011;131:2401–2408. doi: 10.1038/jid.2011.222. [DOI] [PubMed] [Google Scholar]
- 14.Zhang W, Dang E, Shi X, Jin L, Feng Z, Hu L, Wu Y, Wang G. The pro-inflammatory cytokine IL-22 up-regulates keratin 17 expression in keratinocytes via STAT3 and ERK1/2. PLoS One. 2012;7:e40797. doi: 10.1371/journal.pone.0040797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L, Zhang S, Wang G. Keratin 17 in disease pathogenesis: From cancer to dermatoses. J Pathol. 2019;247:158–165. doi: 10.1002/path.5178. [DOI] [PubMed] [Google Scholar]
- 16.Ghazawi FM, Hassani-Ardakani K, Henriques L, Jafarian F. Identification of a novel substitution mutation (R103C) in the rod domain of the keratin 17 gene associated with pachyonychia congenita type 2. Int J Dermatol. 2019;58:233–236. doi: 10.1111/ijd.14082. [DOI] [PubMed] [Google Scholar]
- 17.Agarwala M, Salphale P, Peter D, Wilson NJ, Pulimood S, Schwartz ME, Smith FJD. Keratin 17 mutations in four families from India with pachyonychia congenita. Indian J Dermatol. 2017;62:422–426. doi: 10.4103/ijd.IJD_321_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ofaiche J, Duchatelet S, Fraitag S, Nassif A, Nougue J, Hovnanian A. Familial pachyonychia congenita with steatocystoma multiplex and multiple abscesses of the scalp due to the p.Asn92Ser mutation in keratin 17. Br J Dermatol. 2014;171:1565–1567. doi: 10.1111/bjd.13123. [DOI] [PubMed] [Google Scholar]
- 19.Zang D, Zhou C, He M, Ma X, Zhang J. A novel mutation (p.Arg94Gly) of keratin 17 in a Chinese family with steatocystoma multiplex. Eur J Dermatol. 2011;21:142–144. doi: 10.1684/ejd.2011.1207. [DOI] [PubMed] [Google Scholar]
- 20.Mockler D, Escobar-Hoyos LF, Akalin A, Romeiser J, Shroyer AL, Shroyer KR. Keratin 17 is a prognostic biomarker in endocervical glandular neoplasia. Am J Clin Pathol. 2017;148:264–273. doi: 10.1093/ajcp/aqx077. [DOI] [PubMed] [Google Scholar]
- 21.Escobar-Hoyos LF, Yang J, Zhu J, Cavallo JA, Zhai H, Burke S, Koller A, Chen E, Shroyer KR. Keratin 17 in premalignant and malignant squamous lesions of the cervix: Proteomic discovery and immunohistochemical validation as a diagnostic and prognostic biomarker. Mod Pathol. 2014;27:621–630. doi: 10.1038/modpathol.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikami Y, Fujii S, Nagata K, Wada H, Hasegawa K, Abe M, Yoshimoto RU, Kawano S, Nakamura S, Kiyoshima T. GLI-mediated keratin 17 expression promotes tumor cell growth through the anti-apoptotic function in oral squamous cell carcinomas. J Cancer Res Clin Oncol. 2017;143:1381–1393. doi: 10.1007/s00432-017-2398-2. [DOI] [PubMed] [Google Scholar]
- 23.Kirjushkina MS, Tchipysheva TA, Ermilova VD, Guelstein VI. Breast tumor diagnosis in cytologic aspirates using monoclonal antibodies to keratin 8 and 17. Acta Cytol. 1997;41:307–312. doi: 10.1159/000332517. [DOI] [PubMed] [Google Scholar]
- 24.Merkin RD, Vanner EA, Romeiser JL, Shroyer ALW, Escobar-Hoyos LF, Li J, Powers RS, Burke S, Shroyer KR. Keratin 17 is overexpressed and predicts poor survival in estrogen receptor-negative/human epidermal growth factor receptor-2-negative breast cancer. Hum Pathol. 2017;62:23–32. doi: 10.1016/j.humpath.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Ide M, Kato T, Ogata K, Mochiki E, Kuwano H, Oyama T. Keratin 17 expression correlates with tumor progression and poor prognosis in gastric adenocarcinoma. Ann Surg Oncol. 2012;19:3506–3514. doi: 10.1245/s10434-012-2437-9. [DOI] [PubMed] [Google Scholar]
- 26.Khanom R, Nguyen CT, Kayamori K, Zhao X, Morita K, Miki Y, Katsube K, Yamaguchi A, Sakamoto K. Keratin 17 is induced in oral cancer and facilitates tumor growth. PLoS One. 2016;11:e0161163. doi: 10.1371/journal.pone.0161163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikami T, Maruyama S, Abe T, Kobayashi T, Yamazaki M, Funayama A, Shingaki S, Kobayashi T, Jun C, Saku T. Keratin 17 is co-expressed with 14-3-3 sigma in oral carcinoma in situ and squamous cell carcinoma and modulates cell proliferation and size but not cell migration. Virchows Arch. 2015;466:559–569. doi: 10.1007/s00428-015-1735-6. [DOI] [PubMed] [Google Scholar]
- 28.Regenbogen E, Mo M, Romeiser J, Shroyer ALW, Escobar-Hoyos LF, Burke S, Shroyer KR. Elevated expression of keratin 17 in oropharyngeal squamous cell carcinoma is associated with decreased survival. Head Neck. 2018;40:1788–1798. doi: 10.1002/hed.25164. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Liu L, Cao L, Wen Q. Keratin 17 promotes lung adenocarcinoma progression by enhancing cell proliferation and invasion. Med Sci Monit. 2018;24:4782–4790. doi: 10.12659/MSM.909350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takenami T, Maeda S, Karasawa H, Suzuki T, Furukawa T, Morikawa T, Takadate T, Hayashi H, Nakagawa K, Motoi F, et al. Novel biomarkers distinguishing pancreatic head Cancer from distal cholangiocarcinoma based on proteomic analysis. BMC Cancer. 2019;19:318. doi: 10.1186/s12885-019-5548-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Jong EM, van Vlijmen IM, van Erp PE, Ramaekers FC, Troyanovski SM, van de Kerkhof PC. Keratin 17: A useful marker in anti-psoriatic therapies. Arch Dermatol Res. 1991;283:480–482. doi: 10.1007/BF00371788. [DOI] [PubMed] [Google Scholar]
- 32.Babu S, Mockler DC, Roa-Pena L, Szygalowicz A, Kim NW, Jahanfard S, Gholami SS, Moffitt R, Fitzgerald JP, Escobar-Hoyos LF, Shroyer KR. Keratin 17 is a sensitive and specific biomarker of urothelial neoplasia. Mod Pathol. 2019;32:717–724. doi: 10.1038/s41379-018-0177-5. [DOI] [PubMed] [Google Scholar]
- 33.Karia PS, Morgan FC, Califano JA, Schmults CD. Comparison of tumor classifications for cutaneous squamous cell carcinoma of the head and neck in the 7th vs. 8th edition of the AJCC cancer staging manual. JAMA Dermatol. 2018;154:175–181. doi: 10.1001/jamadermatol.2017.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45((W1)):W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thul PJ, Lindskog C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018;27:233–244. doi: 10.1002/pro.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindskog C. The human protein atlas-an important resource for basic and clinical research. Expert Rev Proteomics. 2016;13:627–629. doi: 10.1080/14789450.2016.1199280. [DOI] [PubMed] [Google Scholar]
- 37.Ponten F, Jirstrom K, Uhlen M. The human protein atlas-a tool for pathology. J Pathol. 2008;216:387–393. doi: 10.1002/path.2440. [DOI] [PubMed] [Google Scholar]
- 38.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 39.Sun Q, Zhang Y, Su J, Li T, Jiang Y. Role of hydroxysteroid dehydrogenase-like 2 (HSDL2) in human ovarian cancer. Med Sci Monit. 2018;24:3997–4008. doi: 10.12659/MSM.909418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Troyanovsky SM, Leube RE, Franke WW. Characterization of the human gene encoding cytokeratin 17 and its expression pattern. Eur J Cell Biol. 1992;59:127–137. [PubMed] [Google Scholar]
- 41.Depianto D, Kerns ML, Dlugosz AA, Coulombe PA. Keratin 17 promotes epithelial proliferation and tumor growth by polarizing the immune response in skin. Nat Genet. 2010;42:910–914. doi: 10.1038/ng.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolokythas A, Schwartz JL, Pytynia KB, Panda S, Yao M, Homann B, Sroussi HY, Epstein JB, Gordon SC, Adami GR. Analysis of RNA from brush cytology detects changes in B2M, CYP1B1 and KRT17 levels with OSCC in tobacco users. Oral Oncol. 2011;47:532–536. doi: 10.1016/j.oraloncology.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 43.Escobar-Hoyos LF, Shah R, Roa-Pena L, Vanner EA, Najafian N, Banach A, Nielsen E, Al-Khalil R, Akalin A, Talmage D, Shroyer KR. Keratin-17 promotes p27KIP1 nuclear export and degradation and offers potential prognostic utility. Cancer Res. 2015;75:3650–3662. doi: 10.1158/0008-5472.CAN-15-0293. [DOI] [PubMed] [Google Scholar]
- 44.Hobbs RP, Batazzi AS, Han MC, Coulombe PA. Loss of keratin 17 induces tissue-specific cytokine polarization and cellular differentiation in HPV16-driven cervical tumorigenesis in vivo. Oncogene. 2016;35:5653–5662. doi: 10.1038/onc.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roa-Pena L, Leiton CV, Babu S, Pan CH, Vanner EA, Akalin A, Bandovic J, Moffitt RA, Shroyer KR, Escobar-Hoyos LF. Keratin 17 identifies the most lethal molecular subtype of pancreatic cancer. Sci Rep. 2019;9:11239. doi: 10.1038/s41598-019-47519-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sritananuwat P, Sueangoen N, Thummarati P, Islam K, Suthiphongchai T. Blocking ERK1/2 signaling impairs TGF-β1 tumor promoting function but enhances its tumor suppressing role in intrahepatic cholangiocarcinoma cells. Cancer Cell Int. 2017;17:85. doi: 10.1186/s12935-017-0454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017;284:4177–4195. doi: 10.1111/febs.14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.