

Abstract
Immunodeficiencies caused by infectious agents may result from disruption of normal host barriers or dysregulation of cellular immunity, the latter serving to promote survival of the infectious agent through immune evasion. Such infections may be followed by opportunistic infections with a variety of other microorganisms. Classic infectious causes of immunodeficiency in companion animals are the immunodeficiency retroviruses, including feline immunodeficiency virus and feline leukemia virus. Other important causes include canine distemper virus; canine parvovirus 2; feline infectious peritonitis virus; rickettsial organisms that infect leukocytes; Leishmania; and fungal pathogens, such as Cryptococcus. Considerable research effort has been invested in understanding the mechanisms of pathogen-induced immunosuppression, with the hope that effective therapies may be developed that reverse the immunodeficiencies developed and in turn assist the host to clear persistent or life-threatening infectious diseases.
Keywords: Feline immunodeficiency virus, Feline leukemia virus, Anaplasma phagocytophilum, Ehrlichia canis, Distemper virus, Parvovirus
The classic example of immunodeficiency caused by an infectious agent is the acquired immunodeficiency syndrome, caused by human immunodeficiency virus (HIV). Similarly, the best known pathogens of companion animals causing immunodeficiencies are the feline retroviruses feline immunodeficiency virus (FIV) and feline leukemia virus (FeLV). However, several other pathogens are capable of disrupting normal immune function. Many infectious agents disrupt host barriers to infection. This may result from the inflammatory response to a pathogen or direct damage by the microbe itself. Examples include disruption of the gastrointestinal mucosal barrier by canine parvovirus, destruction of nasal turbinates by Aspergillus fumigatus in canine sinonasal aspergillosis, or paralysis of the respiratory cilia by Bordetella bronchiseptica. Anaplasma phagocytophilum disables neutrophil function, ensuring its survival within a cell normally charged with antimicrobial substances. Viruses, such as canine distemper virus, cause lymphopenia; the outcome of infection depends on the balance between viral destruction of the immune system and the ability of the remaining immune defenses to eliminate the virus.
Disruption of immune function by infectious agents may serve to promote the infectious agent's survival through host immune evasion. Immunosuppression having the greatest impact clinically often occurs as a result of infection with organisms that are able to persist within the host. Ideally, a pathogen is able to adapt such that it can coexist with the host, without causing death of the host or severe illness, in a way that maximizes the pathogen's transmission efficiency.
The types of opportunistic infections that occur in patients that are immune compromised as a result of an underlying immunosuppressive infection depend upon the mechanisms of immunosuppression. Impairment of normal host barrier function or the function of granulocytes is generally associated with a broad spectrum of bacterial infections and sometimes infection with opportunistic fungi, such as Aspergillus spp Impairment of cell-mediated immunity (CMI) results in infections with opportunistic pathogens, such as Nocardia spp, Mycobacterium spp, Toxoplasma gondii, and a variety of fungal pathogens. Reactivation of dormant pathogens, such as feline herpesvirus, may also occur with depression of CMI.
The purpose of this article is to highlight some of the mechanisms by which persistent infectious microorganisms cause acquired immunodeficiency in companion animal species, and the consequences of the resulting disturbance in immune function.
Viral infections causing immunodeficiency
Canine Distemper Virus Infection
Canine distemper virus (CDV) causes canine distemper, a common disease of dogs worldwide that is associated with a high degree of morbidity and mortality. The virus also infects several other species, including foxes, raccoons, skunks, ferrets, and free-ranging and captive felids. Disease in dogs is most prevalent in regions where vaccination of young dogs against the disease is either not performed or is poorly timed, and epidemics continue to occur in shelter environments in developed countries.1
Canine distemper virus is a Morbillivirus related to measles virus and has been used to study the pathogenesis of measles virus infection. Morbilliviruses are enveloped RNA viruses that survive poorly in the environment. Based on genetic variation within the viral hemagglutinin (H) gene, a multitude of different strains of CDV exist that vary in their geographic distribution, cell tropism, and virulence. Although CDV infects a variety of different cell types, including epithelial, mesenchymal, neuroendocrine, and hematopoietic cells, the marked tropism of CDV for immune cells is critical in respect to its ability to cause immunosuppression. Viral components involved in CDV-induced immunodeficiency include the viral hemagglutinin; the V protein (a nonstructural phosphoprotein); and the nucleocapsid (N) protein.
Dogs are generally exposed to CDV through contact with infected oronasal secretions. The virus initially infects monocytes within lymphoid tissue in the upper respiratory tract and tonsils and is subsequently disseminated via the lymphatics and blood to the entire reticuloendothelial system. Direct viral destruction of a significant proportion of the lymphocyte population, and especially CD4+ T cells, occurs within the blood, tonsils, thymus, spleen, lymph nodes, bone marrow, mucosa-associated lymphoid tissue, and the hepatic Kupffer cells.1, 2, 3 This viral destruction is associated with an initial lymphopenia and transient fever that occurs a few days after infection. Subsequently, there is a second stage of cell-associated viremia, after which CDV infects cells of the lower respiratory; gastrointestinal tract; central nervous system; urinary tract; and red and white blood cells, including additional lymphoid cells.
Elimination of CDV by the host depends on humoral and CMI.1, 4 Because the virus is lymphocytolytic, the outcome of infection depends on the rate at which the host is able to remove the virus before the virus has sufficient time to cause severe immune system injury. Dogs mounting a partial immune response may undergo recovery from acute illness but fail to eliminate the virus completely, leading to a spectrum of more chronic disease manifestations that often involve the uvea, lymphoid organs, footpads, and especially the CNS. Opportunistic infections may also have the chance to develop in these dogs.
Dogs with canine distemper may develop profound lymphopenia and leucopenia. Lymphopenia results from generalized depletion of T and B cells in a variety of tissues (Fig. 1 ). CD4+ T cells are preferentially depleted during the acute phase, which is followed by CD8+ cell depletion.5, 6 Necrosis of hematopoietic cells within the bone marrow may result in leucopenia.7
Fig. 1.
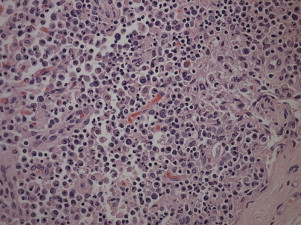
Severe cortical lymphoid necrosis in a mandibular lymph node from a 5-month-old female spayed German Shepherd cross that was euthanized as a result of canine distemper virus infection.
Infection of ferrets has been used as a model of CDV-induced immunosuppression.8 CDV infection of ferrets leads to dramatic reduction in cell-mediated immune function with markedly depressed lymphocyte proliferative activity, and to some extent delayed type hypersensitivity responses. The virus enters lymphocytes following binding of the viral H gene to the primary receptor for the virus, signaling lymphocyte activation molecule (CD150, SLAM). The expression of SLAM appears to be upregulated in response to CDV infection.9 SLAM is also expressed on antigen-presenting cells, such as dendritic cells and activated monocytes, and infection of these cells, which may predominate in the chronic phase of infection, has been hypothesized to be associated with impaired antigen presentation.1, 6 Infection of dendritic cells within the thymus may lead to impaired maturation and selection of T cells, with subsequent release of immature CD5- T cells, including cells that may have the potential for autoreactivity.6 Lymphocyte apoptosis also occurs independent of viral infection in canine distemper, although the mechanisms have not yet been elucidated.10 The presence of the viral V protein is essential to permit rapid replication of CDV in T cells and critical in CDV-mediated immunosuppression. This protein almost completely antagonizes alpha interferon, TNF-alpha, Il-6, gamma-interferon, and Il-2 in the acute phase of infection.3 Suppression of the cytokine response is associated with severe immunosuppression and a fatal outcome in ferrets. Finally, the N protein of Morbilliviruses may interfere with the immune response through the binding of the CD32 (Fc-gamma) receptor on B cells, resulting in impaired differentiation of B cells into plasma cells.11 Binding of this receptor on dendritic cells12 is associated with impairment of antigen presentation by dendritic cells and resulting disruption of T cell function.
The most common secondary infections in canine distemper are secondary bacterial infections that contribute to bronchopneumonia. Bordetella bronchiseptica is also a common co-pathogen in dogs with distemper. Dogs may be diagnosed with bordetellosis in the early stages of distemper, the underlying CDV infection being overlooked. Other opportunistic infections that have been identified in dogs with distemper include toxoplasmosis,13 salmonellosis,14 nocardiosis,15, 16 and generalized demodicosis (Sykes and colleagues, unpublished observations, 2006). In one study from Brazil, canine distemper was the most common underlying immunosuppressive disease predisposing to nocardiosis in dogs.16 Infection with Pneumocystis carinii was associated with CDV infection in a mink,17 and concurrent neosporosis and canine distemper was reported in a raccoon.18
Canine Parvovirus 2 and Feline Panleukopenia Virus Infection
Although parvoviruses do not cause chronic, persistent infections in dogs and cats, parvoviral replication creates the perfect storm for development of acute and severe opportunistic bacterial infections. The combination of leukopenia, disruption of the gastrointestinal barrier, and the immature immune system of the young animals that are most susceptible to these viruses is associated with the common development of sepsis, which is frequently the cause of death.
Canine parvovirus 2 (CPV-2) and feline panleukopenia virus (FPV) are small, nonenveloped DNA viruses. Since its emergence in 1978, CPV has subsequently mutated to CPV-2a; CPV-2b; and in the last decade, CPV-2c, which was first documented in Italy and has subsequently spread to dogs on every continent, with the exception of Australia. The CPV-2c strain appears to be particularly virulent and there has been some debate regarding the ability of current vaccines to protect against it and the ability of commercially available SNAP ELISA tests to detect the virus.19
CPV and FPV have tropism for rapidly dividing cells. As such, they exert an effect on the host that resembles the outcome of treatment with a chemotherapeutic drug. The virus binds and enters cells using the transferrin receptor.20 Cells preferentially involved are the crypt cells of the gastrointestinal tract, bone marrow, and lymphoid tissue. Leukopenia results from sequestration of neutrophils within damaged gastrointestinal tissue and is compounded by destruction of white cell precursors within the bone marrow. Damage to the gastrointestinal barrier can result in translocation of enteric bacteria. In the face of the massive immunosuppression that ensues as a result of virus-induced neutropenia and lymphopenia, the host fails to contain bacterial replication and bacteremia and sepsis ensue. Treatment of secondary infections with broad-spectrum parenteral antimicrobial drugs is critical to permit recovery of dogs and cats from parvoviral infection. Bacterial causes of sepsis reported in infected animals include Escherichia coli, Salmonella spp, and Clostridium difficile. Giardia infection also exacerbates illness.21 Immunosuppression may also contribute to replication of other co-infecting enteric viruses, such as enteric coronavirus, which in turn exacerbate the damage to the gastrointestinal mucosa. Similarly, CPV-induced immunosuppression potentiates the development of postvaccinal canine distemper encephalitis.22
The importance of secondary infections in the pathogenesis of parvovirus infections is highlighted by the fact that experimental infection of germfree cats is not associated with development of clinical illness, despite the associated reduction in white cell count.23
Feline Retroviral Infections
Feline leukemia virus and feline immunodeficiency virus are common causes of viral-induced immunodeficiency in cats, although the underlying mechanisms by which they exert immunodeficiency are still incompletely understood. Subtypes of FeLV and FIV are defined based on variations in the env gene sequence, which also influences their pathogenicity.
Feline Leukemia Virus Infection
There are four different subtypes of the gamma retrovirus FeLV: FeLV-A, FeLV-B, FeLV-C, and FeLV-T. Each subtype uses a different receptor to enter cells (Table 1 ).24, 25, 26, 27 All cats infected with FeLV-B, FeLV-C, and FeLV-T are co-infected with FeLV-A, with FeLV-A being the only type that is transmitted between animals. The other subtypes arise through recombination or point mutation within FeLV-A during the course of infection and influence the clinical expression of disease (see Table 1). FeLV-T, a T-cell tropic variant, is unique amongst gamma retroviruses in that it requires two host proteins to enter and infect cells.27 As a result of its T-cell tropism, FeLV-T infection may be particularly associated with immunodeficiency in cats.
Table 1.
Host cellular receptors involved in FeLV infection
| FeLV Subtype | Receptor | Receptor Function | Comments | References |
|---|---|---|---|---|
| FeLV-A | FeTHTR1 | Thiamine transporter protein | Present in all cats with FeLV; transmitted exogenously | Mendoza et al24 |
| FeLV-B | FePit1 or FePit2 | Inorganic phosphate transporter protein | Results from recombination between FeLV-A and feline endogenous FeLV-related retrovirus sequences; may accelerate development of lymphoma or enhance neuropathogenicity | Anderson et al25 |
| FeLV-C | FLVCR | Heme transporter protein | Arises from point mutations in FeLV-A env gene; associated with non-regenerative anemia | Keel et al26 |
| FeLV-T | FePit1 or FLVCR plus a soluble cofactor encoded by endogenous FeLV-related retrovirus sequence, usually FeLIX | Transporter protein (variable) | Arises from point mutations in FeLV-A env gene; associated with severe immunosuppression | Anderson27 |
Transmission of FeLV-A primarily occurs through prolonged, close contact with salivary secretions, although other routes of transmission, including through biting, can also occur. After an initial phase of viremia, FeLV replicates within rapidly dividing lymphoid, myeloid, and epithelial cells, such as those lining the intestinal crypts.28 As with distemper, when cellular destruction exceeds the ability of the host's immune system to suppress viral replication, persistent viremia and progressive FeLV-related disease results.
Clinical outcomes of FeLV infection include tumor development, especially lymphoma or leukemia; non-regenerative anemia; marrow failure, which in turn can result from myelophthisis, myelodysplasia, or myelofibrosis; neurologic manifestations, such as anisocoria; reproductive failure; gastrointestinal disease; and immunodeficiency. The development of opportunistic infections may result from marrow failure or cell-mediated immunodeficiency. The immunosuppressive properties of FeLV have been linked at least in part to the transmembrane viral envelope peptide, p15E.29 This viral protein inhibits T- and B-cell function, inhibits cytotoxic lymphocyte responses, alters monocyte morphology and distribution, and has been associated with impaired cytokine production and responsiveness.30, 31, 32 Kittens persistently infected with FeLV have impaired T-cell, and to a lesser extent, B-cell function.33, 34, 35, 36 Infected cats may develop lymphopenia, thymic atrophy, and depletion of lymphocytes within lymph node paracortical zones. CD4+ T-cell malfunction may contribute to a decreased humoral and cellular immune response in affected cats.37, 38 The response to vaccination may also be impaired. Neutrophil function is also impaired in cats that are FeLV-infected.39, 40, 41 Opportunistic infections documented in cats that are FeLV-infected include bacterial infections of the upper and lower urinary tract, hemoplasmosis, respiratory tract infections, feline infectious peritonitis (FIP), and chronic stomatitis, although there is little evidence in the literature to support an increased prevalence of these infections in cats with FeLV as opposed to cats not infected with FeLV. Some infections, such as cryptococcosis, appear to occur with the same frequency in cats that are FeLV positive as in cats that are FeLV negative, but may be more severe and refractory to therapy (Fig. 2 ).42
Fig. 2.
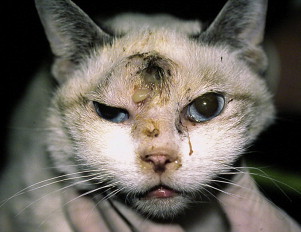
Siamese cat with FeLV infection and concurrent severe cryptococcal rhinosinusitis that was refractory to therapy with antifungal drugs.
Feline Immunodeficiency Virus Infection
FIV is a lentivirus that is primarily transmitted between cats by biting. FIV invades cells via the primary receptor CD134, which is expressed on feline CD4+ T lymphocytes; B lymphocytes; activated macrophages43, 44; and the secondary receptor CXCR4, a chemokine receptor.
The mechanisms of immunosuppression in FIV infection are complex, and despite more than 20 years of research on the subject, not completely understood. Paradoxically, immune suppression and immune hyperactivation have been documented in infected cats. A comprehensive review of the subject is beyond the scope of this article but has been recently published elsewhere.45
Central to FIV-induced immunosuppression is a progressive reduction in CD4+ T-cell numbers. The number of CD4+ T cells in peripheral blood declines shortly after infection, owing to initial viral replication within target activated CD4+ T cells and macrophages. After this acute phase of infection, numbers of CD4+ T cells rebound and viremia is suppressed (Fig. 3 ). Neutropenia can also occur during this phase46 and it has been suggested that this may result from neutrophil apoptosis.47 CD4+/CD25+ T regulator cells have recently been shown to be infected and activated during acute infection. When activated, these cells inhibit proliferation and induce apoptosis of other activated CD4+ or CD8+ T cells, which may also contribute to persistence of FIV and further immunosuppression.45, 48, 49 Evidence also points to altered dendritic cell function during acute FIV infection.50, 51 The impairment of T-cell function in acute FIV infection has been suggested to result from cytokine dysregulation, immunologic anergy, and increased apoptosis.45 In turn, this is associated with an inability to mount a primary immune response to opportunistic pathogens.
Fig. 3.
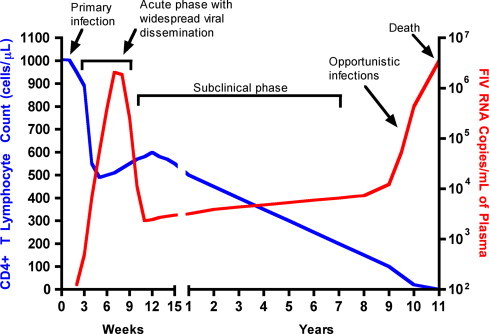
Graph depicting the pathogenesis of feline immunodeficiency virus infection. As the CD4+ count declines, production of antibody is limited and cats with advanced infection may test negative using antibody tests. Opportunistic infections ensue.
A prolonged asymptomatic period follows, sometimes lasting years or even the lifetime of the cat, which is associated with a gradual decline in CD4+ T-cell numbers; a reduction in the CD4+/CD8+ ratio; generalized lymphoid depletion; and in some cats, hyperglobulinemia, which results from B-cell hyperactivation. In addition to a decline in cell numbers, although activated, paradoxically, T cells develop a reduced ability to respond to antigenic stimulation. Altered lymphocyte expression of cell surface molecules, including CD4, cytokine receptors and major histocompatibility complex (MHC) II antigens, and continued alteration of dendritic cell function, also contribute to immunosuppression. Dysregulation of cytokine production occurs. Cats chronically infected with FIV fail to produce Il-2, Il-6, and Il-12 in response to T gondii infection, instead producing elevated levels of the antiinflammatory cytokine Il-10.45, 52
Ultimately these changes lead to opportunistic infections, most commonly bacterial infections of the mouth; chronic bacterial skin infections; persistent viral upper respiratory tract infections; mycobacterial infections; hemoplasmosis; toxoplasmosis; and parasitic infections, such as demodicosis and severe flea burdens.
Feline Coronavirus Infection
FIP virus infection is associated with a profound, virus-induced depletion of CD4+ and CD8+ cells and hypergammaglobulinemia, suggesting virus-induced dysregulation of the immune response.53 The mechanism of T cell depletion is not clear, because the virus does not infect lymphocytes, only monocytes and macrophages. Infection of antigen-presenting cells, specifically dendritic cells, by the virus has been hypothesized to cause T-cell apoptosis.53 Despite the profound T-cell deficiency that accompanies FIP, opportunistic infections are rarely reported, perhaps partly as a result of the rapidly fatal clinical course of disease.
Bacterial infections causing immunodeficiency
Perhaps the best examples of bacterial infections causing immunodeficiency are those of the tick-borne pathogens Ehrlichia canis and Anaplasma phagocytophilum, which are described later in this article. Bartonella spp. and hemotropic mycoplasmas (hemoplasmas) may also be capable of inducing chronic immunodeficiencies. Human infection with Bartonella bacilliformis infection may be immunosuppressive and many patients have succumbed with secondary bacterial infections, especially salmonellosis.54 Impaired leukocyte function, cyclic CD8+ lymphopenia, and diminished expression of adhesion molecules and MHC Class II molecules by CD8+ and B lymphocytes, respectively, were documented in one study of Bartonella vinsonii subspecies berkhoffii-infected dogs.55 Hemoplasma-induced immunosuppression is not a new phenomenon and has been recognized as a problem in experiments involving chronically infected laboratory rodents and in sheep chronically infected with Mycoplasma ovis.56, 57 The clinical importance of immunosuppression induced by Bartonella spp and hemoplasmas in cats and dogs requires further investigation.
Ehrlichia canis Infection
Ehrlichia canis is a gram negative intracellular bacteria that causes canine monocytic ehrlichiosis (CME), arguably the most important infectious disease of dogs exposed to ticks worldwide. The organism is transmitted by the brown dog tick, Rhipicephalus sanguineus. The organism infects monocytes, in which it forms morulae. In the United States, disease is diagnosed most frequently in dogs living in the southeastern and southwestern states, but because of chronic, subclinical infection, dogs can be transported to non-endemic regions and subsequently develop disease. Different strains of E canis exist but the degree by which these vary in virulence is poorly characterized.
The course of CME has been divided into acute, subclinical, and chronic phases, although in naturally infected dogs, these phases are often not readily distinguishable. Clinical signs of acute disease include depression, inappetence, fever, and weight loss. Ocular and nasal discharges, edema, hemorrhages, and neurologic signs may also occur. The organism replicates in reticuloendothelial cells with generalized lymphadenopathy and splenomegaly, and transient cytopenias, especially thrombocytopenia, may occur. After the acute phase, which may last up to 6 weeks, a subclinical phase may develop that lasts months to years. During this phase, the organism appears to evade host immune responses through antigenic variation. Ultimately, a small percentage of these infected dogs develop chronic CME. Chronic CME is characterized by signs that include lethargy, inappetence, fever, weight loss, bleeding tendencies, pallor, lymphadenopathy, splenomegaly, dyspnea, anterior uveitis, polyuria/polydipsia, muscle wasting, polyarthritis, and edema. Dogs with severe chronic ehrlichiosis may develop marrow failure, with aplastic pancytopenia. Severe disease may also be associated with a protein-losing nephropathy and development of neurologic signs. Some dogs have bone marrow plasmacytosis and peripheral granular lymphocytosis. Hyperglobulinemia is a frequent finding on the serum chemistry profile and usually results from a polyclonal gammopathy, although monoclonal gammopathies have also been reported.58 High antibody titers to E canis, occasionally exceeding 1:1,000,000, are also common.
The chronic phase may also be associated with development of secondary opportunistic infections. The precise underlying mechanism of the immunodeficiency that develops and how it relates to successful persistence of E canis has not been elucidated. Not all dogs that develop chronic infections are pancytopenic, so leukopenia alone does not explain the predisposition for opportunistic infection. Furthermore, the types of infections reported, such as viral papillomatosis; generalized demodicosis; protozoal infections, such as neosporosis and opportunistic mycoses, suggest a defect develops in CMI (Fig. 4 ).59 E canis infection has also been suggested to predispose dogs to development of canine leishmaniasis.60 Infection of a canine cell line with E canis resulted in suppression of MHC Class II expression.61 In one study, acute experimental infection with E canis was not associated with measurable suppression of CMI or humoral immune responses.62 Alterations in immune responses during chronic infection require further evaluation.
Fig. 4.
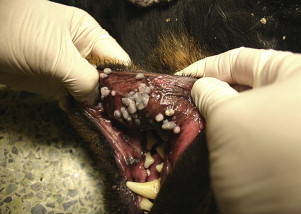
Viral papillomatosis in a male neutered Rottweiler cross with chronic canine monocytic ehrlichiosis
From Ettinger SJ, Feldman EC. Textbook of veterinary internal medicine. 7th edition. St. Louis (MO): Saunders; 2010. Figure 206-1; with permission.
Anaplasma Phagocytophilum Infection
Like E canis, Anaplasma phagocytophilum is an obligate, tick-transmitted intracellular bacteria that forms morulae within leukocytes. In contrast to E canis which infects monocytes, A phagocytophilum infects granulocytes, primarily the neutrophil,63 and causes granulocytic anaplasmosis, a disease of humans, dogs, horses, ruminants, and occasionally cats (Fig. 5 ). The vector ticks are generally those belonging to the Ixodes persulcatus complex, primarily I scapularis and I pacificus in the United States, and I ricinus in Europe. Numerous small wild mammals, deer, and possibly birds, act as reservoir hosts for the organism. Several genetic variants have been identified and there is increasing evidence of strain variation in host specificity and pathogenicity.
Fig. 5.
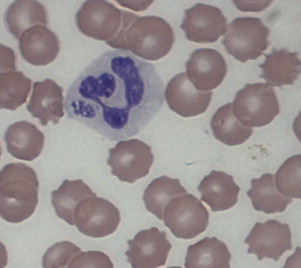
Morulae of Anaplasma phagocytophilum within a canine neutrophil
From Ettinger SJ, Feldman EC. Textbook of veterinary internal medicine. 7th edition. St. Louis (MO): Saunders; 2010. Figure 206-2; with permission.
Immunosuppression resulting from A phagocytophilum infection results primarily from impairment of neutrophil function by the bacteria. After inoculation into the host, A phagocytophilum attaches to sialylated ligands on the surface of neutrophils, after which it enters neutrophils via caveolae-mediated endocytosis, bypassing phagolysosomal pathways. A phagocytophilum then actively disables neutrophil bactericidal functions, in particular neutrophil superoxide production, thus promoting its own survival.64, 65 A phagocytophilum also reduces neutrophil mobility and phagocytosis,66 and reduces endothelial adherence and transmigration of neutrophils.67 By inhibiting neutrophil apoptosis, the organism is able to survive in a well-differentiated cell that normally has a very short lifespan. The impairment of neutrophil function and leukopenia that develop as a result of A phagocytophilum infection is occasionally associated with development of opportunistic infections in some humans and animals with granulocytic anaplasmosis. The best example of this is tick pyemia, which is a debilitating lameness and paralysis that develops in infected lambs in Europe, most commonly as a result of disseminated Staphylococcus aureus or Pasteurella spp infection. Infection with A phagocytophilum may influence the outcome of infection with Borrelia burgdorferi, which can be co-transmitted by Ixodes ticks, possibly as a result of impaired neutrophil function.68
Immunosuppression caused by protozoal and fungal pathogens
Leishmaniasis, caused by the protozoal parasite Leishmania infantum, is a chronic progressive disease transmitted by the sand fly. The mechanisms of immunosuppression induced by this organism are perhaps the best studied amongst protozoal parasites. The disease is most common in the Mediterranean basin and South America. The organism causes a systemic disease in dogs characterized by lymphadenopathy, crusting skin lesions, weight loss, anemia, ocular lesions, polyarthritis, and protein-losing nephropathy. The infection is often associated with other infections, especially ehrlichiosis and babesiosis, and occasionally with neoplastic disease, especially hematopoietic tumors.69 Leishmania infantum invades mononuclear phagocytes, evading the phagolysosome, and survives within them through inhibition of the respiratory burst, inhibition of macrophage function and apoptosis, and impairment of antigen presentation through inhibition of MHC Class I and MHC Class II molecule expression. The protozoan also appears to impair macrophage and neutrophil chemotaxis, and interferes with Il-12 transcription.70 The Leishmania spp surface protein gp63 is a key protein that mediates entry and survival within macrophages. It also allows the organism to resist complement and was recently shown to bind to and suppress the activity of NK cells.71
Several fungal pathogens are capable of causing immunosuppression, including Aspergillus spp, Candida spp, and Cryptococcus spp. Cryptococcus neoformans and Cryptococcus gattii are highly immunosuppressive fungal pathogens, although co-infections with other pathogens are rarely documented. Cryptococcal organisms possess several potent virulence factors that are capable of suppressing or orchestrating the immune response in favor of fungal growth and persistence. The cryptococcal capsular polysaccharide, glucuronoxylomannan, has attracted the most attention in this regard (Fig. 6 ). It effectively inhibits phagocytosis and interferes with migration of leukocytes from the bloodstream into tissues by causing them to shed selectin. It can also deplete complement and directly inhibits T-cell responses.72, 73 There is a shift from a Th1 to a Th2 immune response, the Th1 response being normally required for organism clearance. The cryptococcal urease enzyme was shown to promote accumulation of immature dendritic cells within the lung, and an associated shift in the immune response to a non-protective Th2-cytokine dominated response.71
Fig. 6.
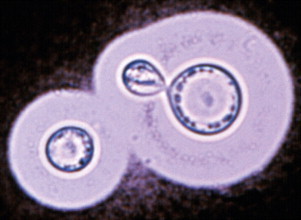
India ink preparation showing encapsulated yeasts of Cryptococcus spp within cerebrospinal fluid. Immunosuppressive properties of the organism have been associated with the glucuronoxylomannan capsule.
From Malik M, Krockenberger M, O'Brien CR, et al. Cryptococcosis. In: Greene CE. Infectious diseases of the dog and cat. 3rd edition. St. Louis (MO): Saunders/Elsevier; 2006. p. 584–98. Figure 61-6B; with permission.
Summary
This review highlights the mechanisms of immunosuppression in just a small subset of the huge variety of infectious agents that are capable of inducing immunosuppression to promote their own survival within the host. The degree of immunosuppression and the mechanisms by which immunodeficiency develops are highly variable and complex. Pathogen surface molecules and cellular receptor tropisms play an important role in determining the initial immune cells infected. Because of the cascading mechanisms involved in normal immune cell recruitment, cytokine and antibody production, pathogens frequently disrupt the function of immune cells that do not undergo direct infection. Considerable research effort has been invested in understanding the mechanisms of pathogen-induced immunosuppression, with the hope that effective therapies may be developed that reverse the immunodeficiencies developed and in turn assist the host to clear persistent or life-threatening infectious diseases.
Acknowledgments
The authors thank Dr Ellen E. Sparger for her review of the retroviral section of this article.
References
- 1.Beineke A., Puff C., Seehusen F. Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet Immunol Immunopathol. 2009;127(1–2):1–18. doi: 10.1016/j.vetimm.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 2.Von Messling V., Milosevic D., Cattaneo R. Tropism illuminated: lymphocyte-based pathways blazed by lethal morbillivirus through the host immune system. Proc Natl Acad Sci U S A. 2004;101(39):14216–14221. doi: 10.1073/pnas.0403597101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Von Messling V., Svitek N., Cattaneo R. Receptor (SLAM [CD150]) recognition and the V protein sustain swift lymphocyte-based invasion of mucosal tissue and lymphatic organs by morbillivirus. J Virol. 2006;80(12):6084–6092. doi: 10.1128/JVI.00357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appel M.J., Shek W.R., Summers B.A. Lymphocyte-mediated immune cytotoxicity in dogs infected with virulent canine distemper virus. Infect Immun. 1982;37(2):592–600. doi: 10.1128/iai.37.2.592-600.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwatsuki K., Okita M., Ochikubo F. Immunohistochemical analysis of the lymphoid organs of dogs naturally infected with canine distemper virus. J Comp Pathol. 1995;113(2):185–190. doi: 10.1016/s0021-9975(05)80033-7. [DOI] [PubMed] [Google Scholar]
- 6.Wünschmann A., Kremmer E., Baumgärtner W. Phenotypical characterization of T and B cell areas in lymphoid tissues of dogs with spontaneous distemper. Vet Immunol Immunopathol. 2000;73(1):83–98. doi: 10.1016/s0165-2427(99)00156-7. [DOI] [PubMed] [Google Scholar]
- 7.Baumgärtner W., Boyce R.W., Alldinger S. Metaphyseal bone lesions in young dogs with systemic canine distemper virus infection. Vet Microbiol. 1995;44(2–4):201–209. doi: 10.1016/0378-1135(95)00013-z. [DOI] [PubMed] [Google Scholar]
- 8.Kauffman C.A., Bergman A.G., O'Connor R.P. Distemper virus infection in ferrets: an animal model of measles-induced immunosuppression. Clin Exp Immunol. 1982;47(3):617–625. [PMC free article] [PubMed] [Google Scholar]
- 9.Wenzlow N., Plattet P., Wittek R. Immunohistochemical demonstration of the putative canine distemper virus receptor CD150 in dogs with and without distemper. Vet Pathol. 2007;44(6):943–948. doi: 10.1354/vp.44-6-943. [DOI] [PubMed] [Google Scholar]
- 10.Schobesberger M., Summerfield A., Doherr M.G. Canine distemper virus-induced depletion of uninfected lymphocytes is associated with apoptosis. Vet Immunol Immunopathol. 2005;104(1–2):33–44. doi: 10.1016/j.vetimm.2004.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kerdiles Y.M., Cherif B., Marie J.C. Immunomodulatory properties of morbillivirus nucleoproteins. Viral Immunol. 2006;19(2):324–334. doi: 10.1089/vim.2006.19.324. [DOI] [PubMed] [Google Scholar]
- 12.Schneider-Schaulies J., Schneider-Schaulies S. Receptor interactions, tropism, and mechanisms involved in morbillivirus-induced immunomodulation. Adv Virus Res. 2008;71:173–205. doi: 10.1016/S0065-3527(08)00004-3. [DOI] [PubMed] [Google Scholar]
- 13.Ehrensperger F., Pospischil A. [Spontaneous mixed infections with distemper virus and Toxoplasma in dogs]Dtsch Tierarztl Wochenschr. 1989;96(4):184–186. [in German] [PubMed] [Google Scholar]
- 14.Smith H.W., Buxton A. Incidence of salmonellae in feces of dogs suffering from distemper. Nature. 1950;166(4228):824. doi: 10.1038/166824a0. [DOI] [PubMed] [Google Scholar]
- 15.Fawi M.T., Tag el Din M.H., el-Sanousi S.M. Canine distemper as a predisposing factor for Nocardia asteroides infection in the dog. Vet Rec. 1971;88(13):326–328. doi: 10.1136/vr.88.13.326. [DOI] [PubMed] [Google Scholar]
- 16.Ribeiro M.G., Salerno T., Mattos-Guaraldi A.L. Nocardiosis: an overview and additional report of 28 cases in cattle and dogs. Rev Inst Med Trop Sao Paulo. 2008;50(3):177–185. doi: 10.1590/s0036-46652008005000004. [DOI] [PubMed] [Google Scholar]
- 17.Dyer N.W., Schamber G.J. Pneumocystosis associated with canine distemper virus infection in a mink. Can Vet J. 1999;40(8):577–578. [PMC free article] [PubMed] [Google Scholar]
- 18.Lemberger K.Y., Gondim L.F., Pessier A.P. Neospora caninum infection in a free-ranging raccoon (Procyon lotor) with concurrent canine distemper virus infection. J Parasitol. 2005;91(4):960–961. doi: 10.1645/GE-407R.1. [DOI] [PubMed] [Google Scholar]
- 19.Lamm C.G., Rezabek G.B. Parvovirus infection in domestic companion animals. Vet Clin North Am Small Anim Pract. 2008;38(4):837–850. doi: 10.1016/j.cvsm.2008.03.008. viii-ix. [DOI] [PubMed] [Google Scholar]
- 20.Hueffer K., Parrish C.R. Parvovirus host range, cell tropism and evolution. Curr Opin Microbiol. 2003;6(4):392–398. doi: 10.1016/s1369-5274(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 21.Pollock R.V. Experimental canine parvovirus infection in dogs. Cornell Vet. 1982;72(2):103–119. [PubMed] [Google Scholar]
- 22.Krakowka S., Olsen R.G., Axthelm M.K. Canine parvovirus infection potentiates canine distemper encephalitis attributable to modified live-virus vaccine. J Am Vet Med Assoc. 1982;180(2):137–139. [PubMed] [Google Scholar]
- 23.Carlson J.H., Scott F.W., Duncan J.R. Feline Panleukopenia. I. Pathogenesis in germfree and specific pathogen-free cats. Vet Pathol. 1977;14(1):79–88. doi: 10.1177/030098587701400110. [DOI] [PubMed] [Google Scholar]
- 24.Mendoza R., Anderson M.M., Overbaugh J. A putative thiamine transport protein is a receptor for feline leukemia virus subgroup A. J Virol. 2006;80(7):3378–3385. doi: 10.1128/JVI.80.7.3378-3385.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson M.M., Lauring A.S., Robertson S. Feline Pit2 functions as a receptor for subgroup B feline leukemia viruses. J Virol. 2001;75(22):10563–10572. doi: 10.1128/JVI.75.22.10563-10572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keel S.B., Doty R.T., Yang Z. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319(5864):825–828. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 27.Anderson M.M., Lauring A.S., Burns C.C. Identification of a cellular cofactor required for infection by feline leukemia virus. Science. 2000;287(5459):1828–1830. doi: 10.1126/science.287.5459.1828. [DOI] [PubMed] [Google Scholar]
- 28.Kipar A., Kremendahl J., Grant C.K. Expression of viral proteins in feline leukemia virus-associated enteritis. Vet Pathol. 2000;37(2):129–136. doi: 10.1354/vp.37-2-129. [DOI] [PubMed] [Google Scholar]
- 29.Good R.A., Ogasawara M., Liu W.T. Immunosuppressive actions of retroviruses. Lymphology. 1990;23(2):56–59. [PubMed] [Google Scholar]
- 30.Cianciolo G.J., Copeland T.D., Oroszlan S. Inhibition of lymphocyte proliferation by a synthetic peptide homologous to retroviral envelope proteins. Science. 1985;230(4724):453–455. doi: 10.1126/science.2996136. [DOI] [PubMed] [Google Scholar]
- 31.Mitani M., Cianciolo G.J., Snyderman R. Suppressive effect on polyclonal B-cell activation of a synthetic peptide homologous to a transmembrane component of oncogenic retroviruses. Proc Natl Acad Sci U S A. 1987;84(1):237–240. doi: 10.1073/pnas.84.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haraguchi S., Good R.A., Day-Good N.K. A potent immunosuppressive retroviral peptide: cytokine patterns and signaling pathways. Immunol Res. 2008;41(1):46–55. doi: 10.1007/s12026-007-0039-6. [DOI] [PubMed] [Google Scholar]
- 33.Cockerell G.L., Hoover E.A., Krakowka S. Lymphocyte mitogen reactivity and enumeration of circulating B- and T-cells during feline leukemia virus infection in the cat. J Natl Cancer Inst. 1976;57(5):1095–1099. doi: 10.1093/jnci/57.5.1095. [DOI] [PubMed] [Google Scholar]
- 34.Hebebrand L.C., Mathes L.E., Olsen R.G. Inhibition of concanavalin A stimulation of feline lymphocytes by inactivated feline leukemia virus. Cancer Res. 1977;37(12):4532–4533. [PubMed] [Google Scholar]
- 35.Mathes L.E., Olsen R.G., Hebebrand L.C. Abrogation of lymphocyte blastogenesis by a feline leukaemia virus protein. Nature. 1978;274(5672):687–689. doi: 10.1038/274687a0. [DOI] [PubMed] [Google Scholar]
- 36.Perryman L.E., Hoover E.A., Yohn D.S. Immunologic reactivity of the cat: immunosuppression in experimental feline leukemia. J Natl Cancer Inst. 1972;49(5):1357–1365. [PubMed] [Google Scholar]
- 37.Trainin Z., Wernicke D., Ungar-Waron H. Suppression of the humoral antibody response in natural retrovirus infections. Science. 1983;220(4599):858–859. doi: 10.1126/science.6302837. [DOI] [PubMed] [Google Scholar]
- 38.Wernicke D., Trainin Z., Ungar-Waron H. Humoral immune response of asymptomatic cats naturally infected with feline leukemia virus. J Virol. 1986;60(2):669–673. doi: 10.1128/jvi.60.2.669-673.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lafrado L.J., Olsen R.G. Demonstration of depressed polymorphonuclear leukocyte function in nonviremic FeLV-infected cats. Cancer Invest. 1986;4(4):297–300. doi: 10.3109/07357908609017509. [DOI] [PubMed] [Google Scholar]
- 40.Hoffmann-Jagielska M., Winnicka A., Jagielski D. Influence of naturally acquired feline leukemia virus (FeLV) infection on the phagocytic and respiratory burst activity of neutrophils and monocytes of peripheral blood. Pol J Vet Sci. 2005;8(2):93–97. [PubMed] [Google Scholar]
- 41.Wardini A.B., Guimarães-Costa A.B., Nascimento M.T. Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J Gen Virol. 2010;91(Pt 1):259–264. doi: 10.1099/vir.0.014613-0. [DOI] [PubMed] [Google Scholar]
- 42.Sykes JE, Malik R. Cryptococcosis. In: Greene CE, editor. Infectious diseases of the dog and cat. 4th edition. St Louis (MO): Saunders Elsevier, in press.
- 43.de Parseval A., Chatterji U., Sun P. Feline immunodeficiency virus targets activated CD4+ T cells by using CD134 as a binding receptor. Proc Natl Acad Sci U S A. 2004;101(35):13044–13049. doi: 10.1073/pnas.0404006101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimojima M., Miyazawa T., Ikeda Y. Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science. 2004;303(5661):1192–1195. doi: 10.1126/science.1092124. [DOI] [PubMed] [Google Scholar]
- 45.Tompkins M.B., Tompkins W.A. Lentivirus-induced immune dysregulation. Vet Immunol Immunopathol. 2008;123(1–2):45–55. doi: 10.1016/j.vetimm.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamamoto J.K., Sparger E., Ho E.W. Pathogenesis of experimentally induced feline immunodeficiency virus infection in cats. Am J Vet Res. 1988;49(8):1246–1258. [PubMed] [Google Scholar]
- 47.Sprague W.S., Terwee J.A., Vandewoude S. Temporal association of large granular lymphocytosis, neutropenia, proviral load, and FasL mRNA in cats with acute feline immunodeficiency virus infection. Vet Immunol Immunopathol. 2010;134(1–2):115–121. doi: 10.1016/j.vetimm.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mexas A.M., Fogle J.E., Tompkins W.A. CD4+CD25+ regulatory T cells are infected and activated during acute FIV infection. Vet Immunol Immunopathol. 2008;126(3–4):263–272. doi: 10.1016/j.vetimm.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vahlenkamp T.W., Tompkins M.B., Tompkins W.A. Feline immunodeficiency virus infection phenotypically and functionally activates immunosuppressive CD4+CD25+ T regulatory cells. J Immunol. 2004;172(8):4752–4761. doi: 10.4049/jimmunol.172.8.4752. [DOI] [PubMed] [Google Scholar]
- 50.Dean G.A., LaVoy A., Yearley J. Cytokine modulation of the innate immune response in feline immunodeficiency virus-infected cats. J Infect Dis. 2006;193(11):1520–1527. doi: 10.1086/503873. [DOI] [PubMed] [Google Scholar]
- 51.Lehman T.L., O'Halloran K.P., Hoover E.A. Utilizing the FIV model to understand dendritic cell dysfunction and the potential role of dendritic cell immunization in HIV infection. Vet Immunol Immunopathol. 2010;134(1–2):75–81. doi: 10.1016/j.vetimm.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levy J.K., Liang Y., Ritchey J.W. Failure of FIV-infected cats to control Toxoplasma gondii correlates with reduced IL2, IL6, and IL12 and elevated IL10 expression by lymph node T cells. Vet Immunol Immunopathol. 2004;98(1–2):101–111. doi: 10.1016/j.vetimm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 53.de Groot-Mijnes J.D., van Dun J.M., van der Most R.G. Natural history of a recurrent feline coronavirus infection and the role of cellular immunity in survival and disease. J Virol. 2005;79(2):1036–1044. doi: 10.1128/JVI.79.2.1036-1044.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weinman D. Human bartonella infection and African sleeping sickness. Bull N Y Acad Med. 1946;22(12):647–670. [PMC free article] [PubMed] [Google Scholar]
- 55.Pappalado B.L., Brown T., Gebhardt D. Cyclic CD8+ lymphopenia in dogs infected with Bartonella vinsonii subspecies berkhoffii. Vet Immunol Immunopathol. 2000;75(1–2):43–57. doi: 10.1016/s0165-2427(00)00182-3. [DOI] [PubMed] [Google Scholar]
- 56.Baker H.J., Cassell G.H., Lindsey J.R. Research complications due to Haemobartonella and Eperythrozoon infections in experimental animals. Am J Pathol. 1971;64(3):625–632. [PMC free article] [PubMed] [Google Scholar]
- 57.Philbey A.W., Barron R.C., Gounden A. Chronic eperythrozoonosis in an adult ewe. Vet Rec. 2006;158:662–664. doi: 10.1136/vr.158.19.662. [DOI] [PubMed] [Google Scholar]
- 58.Breitschwerdt E.B., Woody B.J., Zerbe C.A. Monoclonal gammopathy associated with naturally occurring canine ehrlichiosis. J Vet Intern Med. 1987;1(1):2–9. doi: 10.1111/j.1939-1676.1987.tb01980.x. [DOI] [PubMed] [Google Scholar]
- 59.Schroeder H., Jardine J.E., Davis V. Systemic phaeohyphomycosis caused by Xylohypha bantiana in a dog. J S Afr Vet Assoc. 1994;65(4):175–178. [PubMed] [Google Scholar]
- 60.Mekuzas Y., Gradoni L., Oliva G. Ehrlichia canis and Leishmania infantum co-infection: a 3-year longitudinal study in naturally exposed dogs. Clin Microbiol Infect. 2009 doi: 10.1111/j.1469-0691.2008.02150.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 61.Harrus S., Waner T., Friedmann-Morvinski D. Down-regulation of MHC class II receptors of DH82 cells, following infection with Ehrlichia canis. Vet Immunol Immunopathol. 2003;96(3–4):239–243. doi: 10.1016/j.vetimm.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 62.Hess P.R., English R.V., Hegarty B.C. Experimental Ehrlichia canis infection in the dog does not cause immunosuppression. Vet Immunol Immunopathol. 2006;109(1–2):117–125. doi: 10.1016/j.vetimm.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 63.Carrade D.D., Foley J.E., Borjesson D.L. Canine granulocytic anaplasmosis – a review. J Vet Intern Med. 2009;23(6):1129–1141. doi: 10.1111/j.1939-1676.2009.0384.x. [DOI] [PubMed] [Google Scholar]
- 64.Rikihisa Y. Ehrlichia subversion of host innate responses. Curr Opin Microbiol. 2006;9:95–101. doi: 10.1016/j.mib.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 65.Carlyon J.A., Fikrig E. Mechanism of evasion of neutrophil killing by Anaplasma phagocytophilum. Curr Opin Hematol. 2006;13:28–33. doi: 10.1097/01.moh.0000190109.00532.56. [DOI] [PubMed] [Google Scholar]
- 66.Garyu J.W., Choi K.S., Grab D.J. Defective phagocytosis in Anaplasma phagocytophilum infected neutrophils. Infect Immun. 2005;73:1187–1190. doi: 10.1128/IAI.73.2.1187-1190.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Choi K.S., Garyu J., Park J. Diminished adhesion of Anaplasma phagocytophilum-infected neutrophils to endothelial cells is associated with reduced expression of leukocyte surface selectin. Infect Immun. 2003;71:4586–4594. doi: 10.1128/IAI.71.8.4586-4594.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nyarko E., Grab D.J., Dumler J.S. Anaplasma phagocytophilum-infected neutrophils enhance transmigration of Borrelia burgdorferi across the human blood brain barrier in vitro. Int J Parasitol. 2006;36:601–605. doi: 10.1016/j.ijpara.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 69.Foglia Manzillo V., Pagano A., Guglielmino R. Extranodal gammadelta-T-cell lymphoma in a dog with leishmaniasis. Vet Clin Pathol. 2008;37(3):298–301. doi: 10.1111/j.1939-165X.2008.00048.x. [DOI] [PubMed] [Google Scholar]
- 70.Zambrano-Villa S., Rosales-Borjas D., Carrero J.C. How protozoan parasites evade the host immune response. Trends Parasitol. 2002;18(6):272–278. doi: 10.1016/s1471-4922(02)02289-4. [DOI] [PubMed] [Google Scholar]
- 71.Lieke T., Nylén S., Eidsmo L. Leishmania surface protein gp63 binds directly to human natural killer cells and inhibits proliferation. Clin Exp Immunol. 2008;153(2):221–230. doi: 10.1111/j.1365-2249.2008.03687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yauch L.E., Lam J.S., Levitz S.M. Direct inhibition of T-cell responses by the Cryptococcus capsular polysaccharide glucuronoxylomannan. PLoS Pathog. 2006;2(11):e120. doi: 10.1371/journal.ppat.0020120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Osterholzer J.J., Surana R., Milam J.E. Cryptococcal urease promotes the accumulation of immature dendritic cells and a non-protective T2 immune response within the lung. Am J Pathol. 2009;174(3):932–943. doi: 10.2353/ajpath.2009.080673. [DOI] [PMC free article] [PubMed] [Google Scholar]