

Abstract
We report the design, synthesis and efficacy of a new class of gel-like nano-carrier, or ‘nanogel’, prepared via templated electrostatic assembly of anionic hyaluronic acid (HA) polysaccharides with the cationic peptide amphiphile poly-L-lysine (PLL). Small molecules and proteins present during nanogel assembly become directly encapsulated within the carrier and are precisely released by tuning the nanogel HA:PLL ratio to control particle swelling. Remarkably, nanogels exhibit versatile and complimentary mechanisms of cargo delivery depending on the biologic context. For example, in mammalian cells, nanogels are rapidly internalized and escape the endosome to both deliver membrane-impermeable protein cargo into the cytoplasm and improve chemotherapeutic potency in drug resistant cancer cells. In bacteria, nanogels permeabilize microbial membranes to sensitize bacterial pathogens to the action of a loaded antibiotic. Thus, peptide nanogels represent a versatile, readily scalable and bio-responsive carrier capable of augmenting and enhancing the utility of a broad range of biomolecular cargoes.
Keywords: peptides, carbohydrates, bioassembly, nanotechnology, drug delivery
Background
Modern high throughput screening campaigns have identified numerous biochemical probes and therapeutic candidates with unprecedented specificity and potency. These agents, if successfully translated into the clinic, could transform strategies in precision medicine and lead to the design of highly selective drugs and diagnostics. Yet, many potentially efficacious molecules are abandoned due to poor solubility, low bioavailability, rapid systemic clearance, and off-target biodistribution to healthy tissues leading to dose-limiting adverse events.1 Even many clinically approved pharmaceutics must be formulated with toxic adjuvants and/or excipients that can compound the side effects of the active agent.2, 3
Incorporation of diagnostic or therapeutic cargo into bioresponsive nanomaterials, such as a polymer-4 or lipid-based nanoparticle,5 can address these challenges by improving the pharmacologic and therapeutic properties of loaded agents when parenterally administered. Chemical ligation or physical encapsulation of biomolecular cargo within the nano-carrier matrix leads to enhanced aqueous solubility, improved serum stability and affords preferential localization to diseased tissues through size-dependent passive targeting.6, 7 Considerable efforts are now being made to develop bioresponsive nano-scale vehicles to improve the transport of sensitive protein and nucleic-acid agents for genome editing, biotherapy and biosensing applications.8 While a number of ‘smart’ delivery systems have been designed to address this need, clinical and commercial translation of these platforms has remained elusive due to their significant chemical complexity, substantial cost to scale, and toxicity of the matrix constituents upon carrier degradation in physiologic environments.9–11
Herein, we report the development of a bioresponsive nano-carrier formed from readily available organic building blocks selected from the list of generally recognized as safe (GRAS) compounds by the U.S. Food and Drug Administration.12 A library of GRAS components were systematically screened under electrospray ionization conditions to identify combinations that form physically cross-linked nanomaterials in high yield and low cost. During these studies, we observed that spraying hyaluronic acid (HA) nanodroplets into a bath of ɛ-poly-L-lysine (PLL) templated the co-assembly of the polymers into electrostatically complexed ‘gel-like’ nanoparticles, herein referred to as a nanogel (Figure 1). This facile, aqueous approach eliminates the toxic co-solvents common in the synthesis of many synthetic nanoparticle scaffolds, and yields gram-quantities of nanogels in <1 hour and at low cost. Further, these biosourced nanomaterials exploit a number of physicochemical properties characteristic of nanogel carriers,13, 14 to produce a versatile and biocompatible delivery platform. For example, HA-PLL nanogels are defined by a non-covalently crosslinked network that enables facile encapsulation of small molecules or proteins under mild synthesis conditions conducive to sensitive cargos. The payload is then controllably released upon swelling of the hydrogel-like particle matrix in physiologic environments. A series of biophysical and cell-based assays demonstrates that this behavior allows peptide-templated nanogels to augment and enhance the utility of delivered cargoes, notably improving the potency of loaded antibiotics and chemotherapeutics, as well as successfully delivering otherwise membrane-impermeable proteins to the cytoplasm of treated cells. Hence, peptide-polysaccharide nanogels represent a novel and scalable new class of bioresponsive carriers that can improve the therapeutic and diagnostic utility of bioactive payloads challenged by poor cell permeability, low bioavailability and limited solubility.
Figure 1.
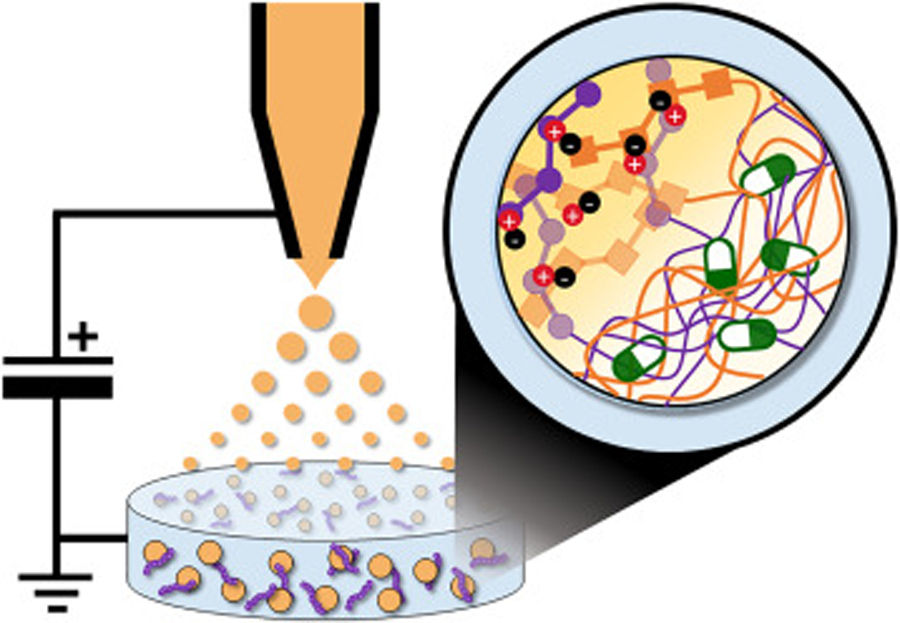
Production of peptide-polysaccharide nanogels by electrospray ionization. An aqueous solution of anionic hyaluronic acid (HA; orange) is infused through an electrically-charged capillary, causing it to spray as nano-scale droplets. Contact of HA nanodroplets with ɛ-poly-L-lysine (PLL; purple) in the bath solution leads to electrostatic assembly of nanogel particles. Therapeutic agents or biochemical sensors (green/white) present during nanogel assembly become physically entrapped within the particle network.
Methods
Materials:
Hyaluronic Acid (100kDa) was purchased from Lifecore Biomedical (Chaska, MN). 0.1% (w/v) poly-L-lysine (x=400) was purchased from Alamanda Polymers (Huntsville, AL). Vancomycin hydrochloride, MTT powder and o-nitrophenyl-β-D-galactopyranoside were purchased from Chem-Impex (Wood Dale, IL). Doxorubicin hydrochloride was purchased from Oakwood Chemical (Estill, SC). Formic acid, acetonitrile, Mueller-Hinton broth, Nunc Lab-Tek Chamber Slides and Sodium Chloride were purchased from Thermo Fisher Scientific (Waltham, MA). 300 kDa MWCO dialysis tubing was purchased from Spectrum (Rancho Dominguez, CA). RPMI 1640 culture medium was purchased from Lonza (Basel, Switzerland). Fetal Bovine Serum (FBS), L-glutamine (L-Gln), Trypsin, PBS and DMEM were purchased from Corning (Corning, NY). Gentamicin and Tris were purchased from VWR (Radnor, PA). HUVEC (ATCC PCS-100–010), Vascular Cell Basal Medium and the Endothelial Growth Cell Kit – VEGF were purchased from ATCC. EmbryoMax Ultrapure Water with 0.1% Gelatin and Triton X-100 were purchased from Sigma Aldrich (St. Louis, MO). Paraformaldehyde was purchased from Chem Cruz. InVivoMAb anti-human CD44 antibody (Hermes-1 clone) was purchased from BioXCell (West Lebanon, NH). Native E. coli beta Galactosidase (β-Gal) protein was purchased from Abcam (Cambridge, United Kingdom). Additional reagents and cell lines utilized in experiments were generous gifts. Green fluorescent protein and cancer cell lines were kindly provided by the laboratory of Dr. Joel P. Schneider (NCI, Frederick, MD). Pseudomonas aeruginosa, Escherichia coli, Acinetobacter baumannii, Salmonella enterica and Staphylococcus aureus bacterial strains were generous gifts from the laboratories of Dr. Zissis Chroneos (Penn State, College of Medicine, Hershey, PA), Dr. Pak Kin Wong (Penn State, Biomedical Engineering, University Park, PA) and Dr. Kenneth Keiler (Penn State, Biochemistry and Molecular Biology, University Park, PA).
Nanogel Synthesis and Formulation:
Nanogels were synthesized by diluting PLL (0.1% w/v, 3 mL) into sterile water (27 mL) to achieve a 30 mL bath solution of 0.01% w/v peptide. The solution was filtered through a 0.2 µm syringe filter into a glass petri dish with submerged stainless steel wire as a ground. The complimentary HA spray solution was prepared at various concentrations depending on desired N:P ratio. Here, dry polysaccharide (114.8 mg, 1 µmol for N:P of 10) was dissolved in sterile water (4 mL) at 37°C and filtered to a final volume of 3 mL. The HA solution was loaded into a 5 mL syringe and attached to a 0.5 inch 28G needle (Hamilton, Reno, NV) charged at 8 – 24 kV via a high voltage power supply (230–30R, Spellman, Hauppauge, NY). HA was infused through the charged capillary (0.1 mL/min) via a syringe pump. The particle solution was incubated at 37°C for 1 hour before centrifugation (10,000 × g, 30 minutes). Particles were washed with sterile DI water, frozen at −80°C in an isopropanol bath before lyophilization, and the dry powder stored in the freezer until use.
Particle Physiochemical Characterization:
Particle size and surface charge was measured via dynamic light scattering (DLS) and zeta potential measurements, respectively, using a Zetasizer Nano ZS (Malvern, United Kingdom). Dry particles (0.2 mg/mL) were suspended in DI water to prepare a stock solution, as well as to pre-equilibrate nanogels in an aqueous environment prior to their use. For size determination, nanogels (1 mL) were added to a clean polystyrene microcuvette. Three independent measurements were taken at a 175° scattering angle, with sample position and attenuation optimized by the instrument. Measurements were taken at 25°C with a 2 minute equilibration time. Phase analysis light scattering (PALS) assisted zeta potential measurements were performed by adding the solution of nanogels to a disposable folded capillary cell (Malvern, DTS1070). Three independent measurements were collected at 25°C, with three replicates per sample. To monitor particle swelling, nanogels (0.2 mg/mL) were suspended in 37°C DMEM media. DLS measurements were performed at regular intervals between 0 – 72 hours after particle resuspension (n = 3 for each time point).
Scanning electron microscopy was performed by air drying particles onto specimen stubs and sputter coating with iridium (Ir). Images were taken on a NanoSEM 630 (FEI, Hillsboro, OR) with a 5 keV landing energy.
Cargo Loading and Release from Nanogels:
To prepare loaded nanogels, the electrospray process was performed as previously described to formulate particles at an N:P of 10, unless specified otherwise. Loading was performed as described below for each of the tested cargos. Loaded nanogels were then collected via centrifugation (10,000 × g, 30 minutes) and washed with DI water.
For GFP-loaded particles, GFP was dissolved (3 mg) in the 30 mL PLL (0.01%) bath solution during the spray. Following centrifugation, the pellet was resuspended in DI water and passed through a 0.2 µm centrifugal filter. Loading was characterized by measuring fluorescence (λex = 470 nm, λem = 515 nm) of a 100 µL aliquot of particles in a 96 well plate on a microplate reader (Cytation 3, BioTek; Winooski, VT). A similar protocol was used for loading of the β-Gal enzyme, with minor modification. β-Gal was dissolved (15 mg) in the 30 mL PLL (0.01%) bath solution and particles filtered after electrosparying. Loading was characterized by measuring absorbance (λ = 280 nm) of particles in a quartz cuvette (path length = 1cm) on a UV/Vis Spectrophotometer (Cary 60, Agilent; Santa Clara, CA).
For doxorubicin-loaded particles, unloaded particles (0.5 mg) were suspended in DI water (0.25 mL). Doxorubicin hydrochloride (0.5 mg, 9×10−4 mmol; 1:1 NG:DOX mass ratio) was solubilized in DI water (0.25 mL) and stirred with the nanogels overnight. Loaded particles were centrifuged and washed three times before being passed through a 0.2 µm centrifugal filter. Loading was characterized by measuring fluorescence (λex = 480 nm, λem = 570 nm) of a 100 µL aliquot in a 96 well microplate reader.
For vancomycin-loaded particles, vancomycin hydrochloride (300 mg, 0.2 mmol) was dissolved in the HA spray solution. Following electrospray particle synthesis, loading was characterized by sonicating NGVAN in DI water to disrupt the particles, and then subjected to reverse-phase HPLC equipped with a silica gel C18(2) analytical column (5 μm bead, 200 x 4.6 mm; Phenomenex, Torrance, CA). HPLC solvents consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in 9:1 acetonitrile:water). A linear gradient of 0–100% solvent B over 25 minutes was employed and eluted compounds monitored at a 280 nm UV wavelength.
To assess release from cargo-loaded NGs, particles were dispersed in DI water (3 mL) and loaded into 300 kDa MWCO dialysis tubing. Samples were submerged in a bath of 37°C PBS (90 mL), and at predetermined time points aliquots collected from multiple locations throughout the bath and pooled (n ≥ 5). Quantification of cargo release was characterized using the same protocol as was employed to assess initial loading. Volume was adjusted to that of the initial bath to ensure concentration consistency.
Biomacromolecular Delivery:
A549 cells were cultured in RPMI-1640 supplemented with FBS (10% v/v), L-Gln (2 mм), and gentamicin (0.05 mg/mL). For microscopy studies, cells were seeded into a 4-well chamber slide at 10,000 cells/well and allowed to adhere overnight. Cells were treated for 24 or 72 hours with blank media (control), free GFP (1.5 µg/mL), or an equivalent concentrations of NGGFP. In separate experiments, competitive inhibition of particle uptake was studied by co-incubating NGGFP with A549 cells for 24 or 72 hours in the presence of excess HA (750 µg/mL) or an anti-CD44 monoclonal antibody (10 µg/mL, concentration based on similar experiments15). During the final hour of treatment, a 12 µL aliquot of transferrin-Texas Red (7.5 µg/mL final concentration) was added to each well to label endosomes. Cells were washed twice with PBS and fixed with paraformaldehyde in PBS (4%) for 20 minutes at 37°C. Following fixation, wells were washed with PBS and chamber walls were removed. A sterilized cover slip was mounted using ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific). Slides were cured at room temperature for 24 hours and stored at 4°C, before being mounted onto an Olympus FLUOVIEW 10i Confocal Microscope (Olympus, Tokyo, Japan) using 359nm, 489nm, and 595nm single photon lasers for DAPI, GFP, and Texas Red, respectively.
To evaluate delivery of β-Gal from nanogels, A549 cells were seeded in 24-well plates at 3×104 cells/well and allowed to adhere overnight. Cells were then treated for 24 hours with blank media (control), 10–100 µg/mL of free β-Gal protein, or equivalent concentrations of β-Gal loaded into the nanogel carrier (NGβ-GAL). Cells were washed extensively with PBS to remove any protein or particles that had non-specifically adsorbed to the cell surface, followed by addition of the chromogenic substrate o-nitrophenyl-β-D-galactopyranoside (ONPG) to reach a final concentration of 1mg/mL. After 1 hour of incubation the supernatant was transfered to a fresh plate and dye conversion measured via absorbance at 420 nm using a microplate reader.
Chemotherapeutic Toxicity:
A549 and NCI/ADR-RES were plated into a 96-well plate (2×103 cells/well) and incubated overnight to adhere. Cells were treated for 48 hours with 0.05 – 30 µg/mL of free DOX, or 0.005 – 3 µg/mL of equivalent drug loaded into nanogels. Cells treated with blank media or 20% DMSO served as negative and positive controls, respectively. After treatment cells were washed with blank media before addition of 100 µL MTT solution (0.5 mg/mL) and incubated for 2 hours. Supernatant was removed and formazan product solubilized by addition of 100 µL DMSO and absorbance read with a microplate reader at 540 nm. Percent viability was calculated with the following equation: (Absorbancetreatment – Absorbancenegative control)/(Absorbancepositive control – Absorbance negative control) X 100%. The resulting data was analyzed using nonlinear regression of semi log data as performed by GraphPad Prism.
Bacterial MIC Determination:
Pseudomonas aeruginosa, Escherichia coli, Acinetobacter baumannii, Salmonella enterica and Staphylococcus aureus were cultured in cation adjusted Mueller-Hinton broth (CAMHB) at 37°C in a shaking incubator (200 rpm), as advised by the Clinical and Laboratory Standards Institute (CLSI). Growth was monitored by performing optical density (OD) readings at 600 nm (OD600). Triplicate 2-fold serial dilutions of each treatment were made in CAMHB and added to a 96-well microtiter plate. Bacteria were diluted to an OD600 of 0.002, and added directly to the treated wells in a 1:1 volume ratio. The microtiter plates were incubated overnight (∼18 h) at 37°C, and bacterial growth was assessed by visual evaluation. The MIC was determined by observing the lowest concentration at which the compound prevented a significant increase in growth compared to the untreated control.
Biocompatibility Assays:
HUVEC (PCS-100–010) were cultured in supplemented Vascular Cell Basal Medium and grown in flasks coated for 15 minutes at 37°C with 0.1% gelatin. Cell suspensions were plated into a 96-well plate (5×103 cells/well) and allowed to adhere overnight. Cells were treated for 48 hours with blank nanogels dissolved in culture media at 0.2 – 40 µg/mL, before measuring viability via the MTT assay as described above.
Hemolysis studies were performed on fresh bovine red blood cells, a close surrogate to human erythrocytes.16 bRBCs were washed three times in hemolysis buffer (10mM Tris, 150mM NaCl, pH 7.4) via centrifugation (3,460 rpm, 10 minutes) at 4°C, and prepared to a working concentration of 0.25% v/v in hemolysis buffer. 75 µL of the bRBCs solution, and an equivalent volume of NG at 4 – 4000 µg/mL, were plated in a 96 well plate. Blank buffer and 1% Triton X-100 were included as negative and positive controls, respectively. After a 24 hour incubation at 37°C, plates were centrifuged (4,000 rpm, 10 minutes) at 4°C to pellet intact bRBCs. 100 µL of the supernatant was transferred to a clean 96 well plate and absorbance at 415 nm was measured to quantitative percentage hemolysis.
Results
Peptide Nanogel Synthesis and Characterization.
Exploiting bio-sourced and biodegradable GRAS compounds is an attractive strategy in the design and assembly of environmentally-sensitive, or ‘green’, engineered nanomaterials. These components are often inexpensive, readily available in bulk, and present a lower barrier to regulatory approval when compared to synthetic analogues. With this in mind, we began screening a library of commercially available GRAS compounds to identify suitable combinations that could prepare bioresponsive nanoparticles via electrospray ionization. Electrospraying is a procedure by which a high voltage is applied to a solution as it’s passed through a capillary tip.17 Coulombic repulsion within the ejected solution generates a fine nanodroplet mist that is collected in a bath solution containing a complimentary cross-linker. This facile synthesis method provides a convenient means to rapidly screen different permutations of oppositely charged molecules to assess their potential to form competent nanomaterials in bulk. During these studies we found that many of the electrosprayed GRAS mixtures generated surface films, fibrous amalgams or large amorphous aggregates. However, one particular combination – spraying a solution of HA into a bath of PLL – rapidly generated a monodisperse suspension of particles ~120 nm in diameter (Figure 2A), which remained colloidally stable in bulk solution (Figure 2B). Interestingly, the final size of the electrostatically complexed nanogels appeared to be insensitive to changes in the voltage applied to the metal capillary tip during electrospray synthesis (Figure S1, Supporting Information).
Figure 2.
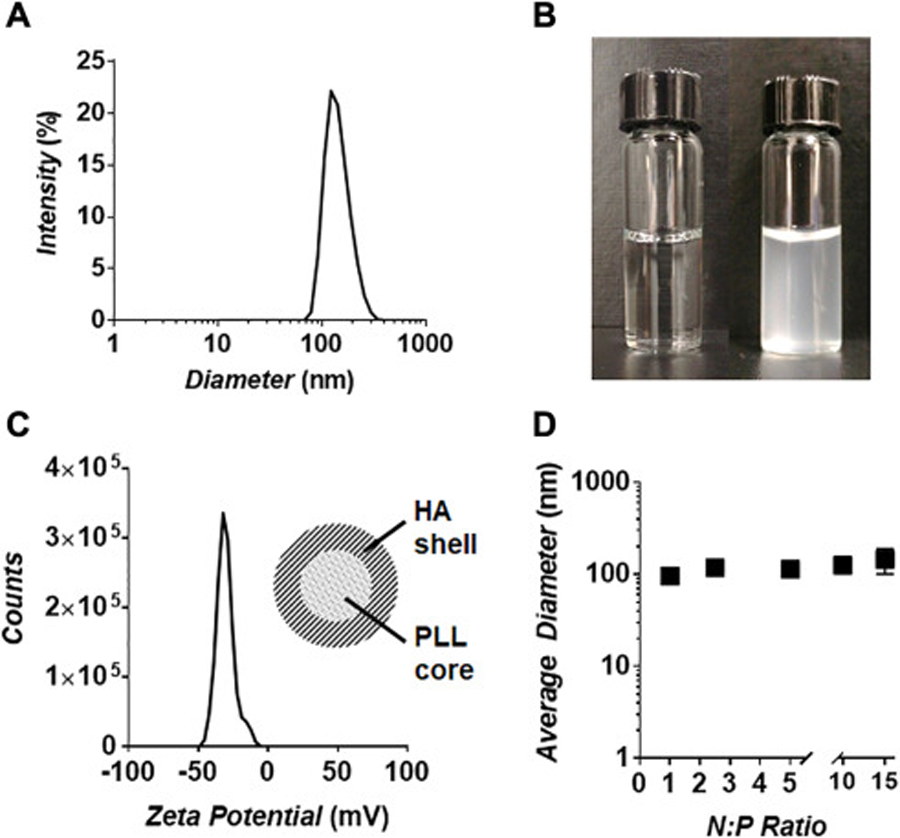
(A) Characterization of peptide nanogel size via dynamic light scattering (DLS). (B) Image of the electrospray bath solution before (left) and immediately after (right) nanogel synthesis. Rapid change in solution turbidity illustrates the high yield production of particles via this method, which remain colloidally stable. (C) Zeta potential analysis of purified nanogels. Negative surface charge suggests a core-shell particle architecture in which an anionic HA corona surrounds a cationic PLL core (inset). (D) Nanogel particle size at 1 – 15 N:P ratio utilized in their synthesis.
To better understand the relative distribution of the HA and PLL components within the nanogel matrix we employed zeta potential measurements to probe the particle’s surface composition. Figure 2C shows that nanogels possess a highly electronegative surface charge (−35 mV), indicating that the particles are likely comprised of an anionic HA shell that surrounds a PLL-rich core. This negative surface charge is contradictory to initial expectations, in which spraying HA into a bath of PLL was anticipated to yield nanogels with a PLL-rich exterior and, hence, a cationic surface charge. One possible explanation is that contact of the HA nanodroplets with PLL does initially produce a corona of electrostatically complexed peptide. However, the high-speed centrifugation employed for particle purification may compact the dense HA-PLL surface layer into the particle interior. Subsequent displacement of liquid HA from the core to the surface could allow complexation of the exuded carbohydrate around the newly formed PLL-rich compacted nucleus. In effect, the centrifugation step may cause an inversion of the two phases after initial assembly, leading to the core-shell architecture predicted from our zeta potential measurements (Figure 2C, inset).
Next, we tested how the density of electrostatic cross-links in the particle network impacts the size of assembled nanogels. This is achieved by changing the concentration of HA sprayed into a PLL bath of fixed concentration. This procedure tunes the stoichiometric ratio of negative (N; COO- of HA) to positive (P; NH3+ of PLL) groups available to assemble the particle matrix. We found that varying the N:P ratio from 1 to 15 formed particles of uniform size (Figure 2D; Figure S2, Supporting Information). Further, we observed a similar surface charge for particles prepared at an N:P of 5 and 10, suggesting that the core-shell architecture of nanogels is not influenced by the relative number of cross-links that comprise their network (Figure S3, Supporting Information). Attempts to form nanogels at N:P ratios <1 resulted in instable particles that could not be accurately measured by DLS or zeta potential analysis. Collectively, these results indicate that the cross-linking density of the nanogel network can be carefully tuned independent of particle size and, in turn, may allow for control over the swelling behavior of the particles in physiologic conditions.
In order to test this assertion, we suspended nanogels formulated with different N:P ratios in 37°C cell culture media, and measured the change in particle size as a function of time. Figure 3A shows that the rate of nanogel swelling is inversely related to the particle N:P ratio. Further, the time to nanogel failure, or the point at which the network can no longer sustain a competent gel, increased from 0.5 – 72 hours as the N:P ratio was raised from 1 to 15. Interestingly, nearly all of the tested nanogel compositions reached a maximum swollen diameter of ~300 nm, representing an approximate 2.5 – 3.0 fold increase in size, before complete decomposition (as indicated by disappearance of a coherent DLS signal). Conversely, nanogels stored in deionized water remained stable and showed no time-dependent change in size (Figure S4, Supporting Information). It is important to note that these studies employ nanogels that have been pre-equilibrated in deionized water before analysis. Hence, any change in particle size due to electrostatic repulsion of the HA-rich network has already taken place, and thus we can independently measure the effects of counterion influx on particle swelling.
Figure 3.

(A) Relative nanogel swelling in physiologic media at N:P ratios of 1 to 15. ‘X’ signifies disruption of particle integrity as indicated by loss of DLS signal. Note, N:P = 1 nanogels rapidly degrade between 0 and 0.5 hours. (B) Scanning electron micrograph of nanogel particles before (top) and after (bottom) 18 hours of swelling; (scale bar = 500 nm). (C) Linear change in nanogel failure time as a function of particle N:P ratio.
Scanning electron microscopy (SEM) performed on particles before and after swelling in buffer demonstrate that the nanogels possess a smooth surface topology that is maintained throughout their expansion without apparent fragmentation or particle agglomeration (Figure 3B). Together, these results suggest that disruption of the electrostatic interactions within the nanogel matrix by infiltrating salt ions leads to bulk disentanglement and subsequent expansion of the biopolymer network. The matrix continues to swell until a critical crosslinking density threshold is reached and particle integrity is lost. Importantly, we found that the time to nanogel failure is linearly dependent with the N:P ratio used during their synthesis (Figure 3C). This suggests that nanogel decomposition can be carefully controlled with temporal resolution to afford precise release of encapsulated cargo.
Loading and Release of Biomolecular Cargo from Nanogels
To demonstrate the utility of nanogels for drug delivery applications, we performed a series of release experiments following the encapsulation of three different molecular cargoes: the model biomacromolecule green fluorescent protein (GFP), the small molecule chemotherapeutic doxorubicin (DOX) and the antibiotic vancomycin (VAN). For these studies, we employed nanogels formed from an N:P of 10, as these particles could be readily prepared in bulk and showed a multi-day swelling profile amenable to sustained drug delivery (Figure 3A). Loading of the various cargoes into the nanogel carrier was performed using three different procedures that were adjusted for each of model payloads (Figure 4A). This included incorporation of the agent into either the sprayed HA solution (VAN) or the PLL bath (GFP), leading to its direct encapsulation during particle assembly. In the case of DOX, we found that incubating pre-assembled nanogels with the hydrophobic drug led to optimal loading. Attempts to include DOX in the spray or bath solutions led to amorphous precipitates during particle assembly. At any rate, these varied loading methods highlight the ability of the nanogel electrospray synthesis procedure to be readily adapted for encapsulation of a broad spectrum of cargoes with vastly different solubility and physiochemical properties. However, efficient cargo loading remains a challenge, and we were only able to achieve 4%, 0.3% and 0.1% loading of VAN, GFP and DOX, respectively. This indicates that further optimization of the loading conditions are necessary before these formulations can be scaled for clinical applications. Finally, DLS analysis confirmed that the loading of these varied agents into the nanogel carrier did not significantly impact their size (Figure S5, Supporting Information).
Figure 4.
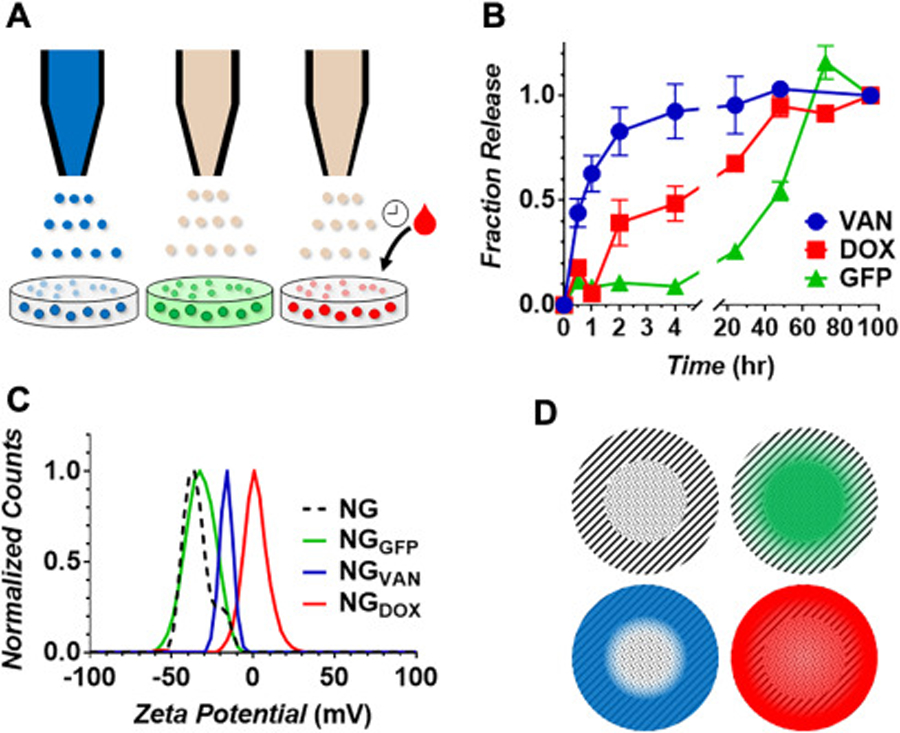
(A) Schematic showing the various loading methods utilized for the encapsulation of molecular cargo within nanogels. Vancomycin (VAN, blue) is suspended in the HA spray solution, while Green fluorescent protein (GFP, green) is present in the bath solution, leading to their encapsulation during nanogel assembly. Doxorubicin (DOX, red) is incubated with pre-formed nanogels leading to its adsorption within the particle amphiphilic matrix. (B) Fraction of cargo released as a function of time. (C) Zeta potential analysis of un-loaded nanogels (NG) or formulations encapsulating the various molecular cargoes. (D) Illustration of sub-particle localization of GFP (green), VAN (blue) or DOX (red) within the nanogel carrier.
Next, physiologic release of the encapsulated cargo was assessed by loading each nanogel formulation into dialysis tubing and suspending it in a release media of 37°C PBS. The concentration of loaded drug or GFP liberated to the dialysis media was then monitored as a function of time via UV-Vis or fluorescence spectroscopy. Results in Figure 4B show that VAN, a hydrophilic drug, is rapidly released from the nanogel carrier with first order kinetics, achieving 90% drug release within 4 hours. DOX, on the other hand, displays a zero-order release rate of ~2.5% per hour, leading to its complete release after 48 hours. Interestingly, GFP loaded formulations showed a very different release profile. The majority of encapsulated protein (>75%) was retained within the nanogel carrier over the first 24 hours, followed by a rapid release phase that occurred between 24 and 72 hours. Taken together with the nanogel swelling data (Figure 3A), this suggests that much of the loaded macromolecular protein remains entrapped within the carrier network as it swells, only achieving complete release upon particle disruption.
So far, our data shows that changes in the cross-linking density of the nanogel carrier, as well as physiochemical properties of the cargo, can be used to carefully control the release profile of loaded agents. In an effort to elucidate how the method of encapsulation impacts distribution of the cargo within the carrier, and thus influences its release, we performed zeta potential analysis on the loaded formulations (Figure 4C). Unloaded control nanogels (NG) are characterized by a surface zeta potential of −35.3 mV, which did not significantly change when GFP is encapsulated within the carrier (NGGFP). This suggests that the protein is largely sequestered within the PLL-rich core (see schematic representation in Figure 4D). Here, suspension of the negatively charged GFP protein, which possesses an isoelectric point of ~5.7,18 in the electrospray bath solution likely led to its initial complexation with the cationic PLL cross-linker before nanogel assembly. Conversely, as VAN is contained in the HA spray solution, we would anticipate that the drug is entrapped within the HA corona of the nanogel and thereby partially passivate the electronegative surface charge. This is corroborated by zeta measurements which show an increase in the surface potential of VAN-loaded nanogels to −15.4 mV. Finally, DOX loaded formulations showed a complete neutralization of particle surface charge as indicated by a zeta potential of 0.6 mV. This is likely a result of the loading method employed for DOX encapsulation, in which pre-formed nanogels are incubated with a saturated solution of the drug to drive it into the particle network. At these saturating concentrations it is likely that a fraction of the unloaded DOX molecules adsorb to the particle surface and thus neutralizes its exterior charge. It is worth noting that due to the small size of these nanogels (~120 nm) we were not able to employ more advanced analytical methods, such as Raman microscopy or TOF-SIMS, to better assess the distribution of loaded molecules within the particle network. Taken together, our data suggests that, in addition to nanogel N:P ratio and physiochemical properties of the loaded agents, the method of encapsulation may be utilized to control intra-particular localization and distribution of the cargo and, thus, potentially provide an additional degree of freedom to control release.
Intracellular Delivery of Protein Payloads from Nanogels
Based on the favorable protein and drug release profiles from the nanogels, we next assessed their potential to deliver bioactive proteins into cells through two independent in vitro experiments. The first involved incubating GFP-loaded nanogels with cancer cells to evaluate intracellular delivery of the protein cargo using fluorescent confocal microscopy (Figure 5). These studies reveal a remarkable capacity of nanogels to shuttle the otherwise membrane-impermeable GFP biomacromolecule into the cytoplasm of cells (Figure 5A), leading to a >11 fold enhancement in intracellular fluorescence compared to cells treated with free GFP (Figure 5B). To investigate potential mechanisms behind the preferential uptake of NGGFP, we performed two complementary competitive inhibition experiments in which cells were co-incubated with GFP-loaded particles in the presence of either an excess of free HA (750 µg/mL) or an anti-CD44 antibody. Previous studies have shown that HA functionalized nanoparticles bind to and are internalized by CD44-like cell adhesion receptors expressed on the surface of most mammalian cells.19, 20 Hence, our competitive inhibition experiments evaluate the contribution of HA within the nanogel matrix to the endocytic uptake of the particles into cells. Results in Figure 5, panels A and B, show that blocking CD44 receptors by co-incubating cells with an excess of free HA or via use of an anti-CD44 antibody substantially decreases the uptake of NGGFP particles, resulting in a 3–10 fold loss of intracellular fluorescence compared to cells treated with NGGFP without blocking (Figure 5B).
Figure 5.

(A) Merged confocal microscopy images of DAPI (blue, cell nuclei) and GFP (green) fluorescence after a 72 hour incubation of A549 lung carcinoma cells with free GFP, or GFP-loaded nanogels in the absence (NGGFP) or presence (NGGFP + mAb) of a monoclonal anti-CD44 blocking antibody (60x magnification; scale bar = 10µm). Fluorescent micrograph of cells co-incubated with NGGFP and an excess of free HA (NGGFP + HA) is shown in Figure S6 of the Supporting Information. (B) Quantitation of average GFP fluorescence per cell for each treatment condition (n=15; statistical comparison made relative to GFP control, with * indicating a p < 0.01). (C) Fluorescent confocal microscopy images of A549 cells treated with NGGFP at 24 and 72 hours. Samples are co-stained with texas-red labeled transferrin (TransferrinTR) to visualize endosomes. Individual fluorescence channels and merged images shown (60x magnification; scale bar = 10µm).
Collectively, these results suggest that nanogels are internalized into cells primarily through receptor-dependent endocytic mechanisms mediated by HA-CD44 binding. However, careful inspection of cells treated with only the NGGFP particles for 72 hours (Figure 5A, middle panel) reveals that a large fraction of the delivered GFP signal is diffusely localized throughout the cytoplasm. Taken together, our data suggests that nanogels are initially internalized into cells via endocytosis and subsequently escape the endosome to accumulate within the cytoplasm. To test this, we incubated cells with NGGFP particles for 24 or 72 hours and then co-stained the endosomes using fluorescently-labeled transferrin (Figure 5C). At early incubation times (24 hours), a significant fraction of the GFP fluorescence is detected both bound to the cell membrane, as well as intracellularly co-localized with the red endosomal marker (transferrin), yielding a yellow/orange signal. Conversely, at the 72 hour time point, no significant co-localization of GFP and transferrin fluorescence is observed. These results support our assertion that NGGFP particles are initially internalized by receptor-mediated endocytosis, followed later by endosomal escape to ultimately deliver loaded contents to the cell cytoplasm. Previous studies showing that high molecular weight PLL can induce endosomal leakage in a pH-dependent manner suggest similar permeabilization occurs when this amphiphile is released from our nanogel carrier during particle degradation in the acidic endosome.21, 22
In a second set of experiments, nanogels were loaded with β-galactosidase (β-GAL) before being incubated with live A549 cells. Similar to GFP-loaded formulations, no change in nanogel particle size (Figure S5, Supporting Information) or surface charge (Figure S7A, Supporting Information) was observed following β-GAL encapsulation. Importantly, compared to the extraordinarily stable and robust GFP, this homotetrameric enzyme represents a more sensitive model protein as it is only functional when all four sub-units are properly assembled. Following incubation, delivery of functional β-GAL into cells was quantified using the enzyme-specific ONPG chromogenic substrate. This assay not only tests the utility of nanogels to deliver the fragile enzyme, but it confirms that β-GAL remains bioactive after release as indicated by its ability to successfully convert the reporter substrate. Gratifyingly, as shown in Figure S7B of the Supporting Information, results of these delivery experiments reveal that β-GAL is more efficiently uptaken into A549 cells, and remains bioactive, when loaded into the nanogel carrier compared to controls treated with the free protein alone. These findings further support the utility of peptide-polysaccharide nanogels to deliver a broad range of sensitive biomacromolecular payloads.
Therapeutic Efficacy of Drug-Loaded Nanogels
Next, we evaluated the chemotherapeutic potency of DOX delivered from the nanogel carrier against drug-sensitive (A549) and multidrug resistant (NCI/ADR-RES) cancer cell lines. Free DOX demonstrates an IC50 of 0.4 µg/mL (0.7 µM) and 6.5 µg/mL (12.0 µM) towards A549 and NCI/ADR-RES, respectively. Gratifyingly, delivery of DOX via the nanogel carrier resulted in a marked increase in drug potency towards both cell lines (Figure 6). For example, treatment of A549 with NGDOX led to a >10-fold enhancement in drug cytotoxicity (IC50 = 0.03 µg/mL equivalent drug) when compared to samples treated with free DOX alone. Based on our previous experiments showing rapid uptake of nanogels into cancer cells (Figure 5), it is likely that this enhanced potency results from the carrier’s ability to preferentially shuttle loaded cargo into the cytoplasm and, thus, generates a high intracellular concentration of delivered DOX. Finally, control samples incubated with empty nanogels (NG) show the carrier itself is well tolerated by these cells.
Figure 6.
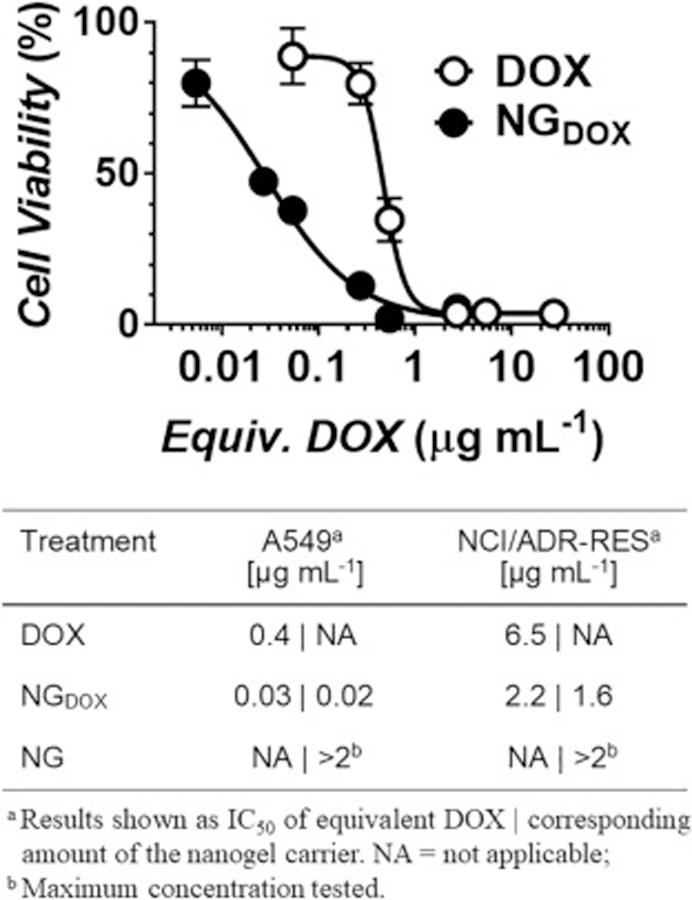
Representative toxicity profile of free DOX or DOX-loaded nanogels (NGDOX) against A549 lung carcinoma cells after 48 hours of incubation. GraphPad Prism software was used to fit cytotoxicity curves and, as shown in the table, calculate IC50 values for drug-sensitive A549 or multidrug resistant NCI/ADR-RES cancer cell lines.
Next, we evaluated the antibacterial activity of VAN-loaded nanogels against a panel of Gram-negative and Gram-positive bacterial pathogens (Table 1). It is important to note that clinical use of VAN is generally limited to Gram-positive infections, as most Gram-negative pathogens are innately resistant to the drug. This is due to the permeability barrier of the Gram-negative outer membrane, which prevents the large glycoprotein drug from diffusing into the cell wall and reaching its enzymatic target. Not surprisingly, then, weak activity of VAN was observed towards all four of the Gram-negative bacteria tested in our study, leading to inhibition of bacterial growth only at the highest concentration (144 µg/mL). As expected, the drug potently killed the control Gram-positive strain S. aureus (MIC = 4.5 µg/mL). Remarkably, when VAN is loaded into our nanogel carrier, we observed a significant increase in its potency towards both Gram-negative and Gram-positive bacteria. For instance, treatment of E. coli with NGVAN led to an equivalent drug MIC of 36 µg/mL, a 4-fold enhancement compared to the activity of free VAN. Likewise, NGVAN killed the Gram-positive S. aureus strain at a 15-times greater potency relative to bacteria treated with VAN alone.
Table 1.
Minimum inhibitory concentration (MIC) of free VAN or VAN-loaded nanogels (NGVAN) against a panel of Gram-negative (−) or Gram-positive (+) bacterial pathogens.
| Treatment |
P. aeruginosa (−)a [μgmL−1] |
A. baumannii (−)a [μgmL−1] |
S. enterica (−)a [μgmL−1] |
E. coli (−)a [μgmL−1] |
S. aureus (+)a [μgmL−1] |
|---|---|---|---|---|---|
| VAN | 144b | NA | 144b | NA | 144b | NA | 144b | NA | 4.5 | NA |
| NGVAN | 72 | 44 | 72 | 44 | 72 | 44 | 36 | 22 | 0.3 | 0.2 |
| NG | NA | >100b | NA | >100b | NA | >100b | NA | >100b | NA | >0.3b |
Results shown as MIC of equivalent VAN | corresponding amount of the nanogel carrier. NA = not applicable.
Maximum concentration tested.
The improvement in VAN efficacy when loaded into nanogels is likely due to complimentary bioactivity of the HA and PLL particle constituents. In the context of HA, evidence indicates that many bacterial pathogens utilize this polysaccharide, which is a ubiquitous component of the mammalian host extracellular matrix, as a carbon source during tissue invasion and replication.23, 24 Further, engagement of the cationic PLL side chains with negatively charged phospholipids displayed from the surface of bacteria induces a negative membrane curvature that ultimately permeabilizes the cell wall.25 Together, this suggests that HA in the nanogel matrix may preferentially recruit bacteria to the surface of nanogels, where subsequent disruption of the cell wall via PLL exposes permeabilized microbes to VAN as it is released from the carrier. Interestingly, we observed no toxic effects of un-loaded nanogels (NG) towards bacteria for all tested concentrations, indicating that PLL-mediated membrane permeabilization of the carrier is not independently sufficient to induce significant antimicrobial activity.
Nanogel Biocompatibility
Finally, the biocompatibility of our nanogels was assessed in healthy human endothelial cells and bovine red blood cells following a 24 hour incubation (Figure 7). No overt toxicity was observed towards human umbilical vein endothelial cells (HUVEC), as indicated by preservation of cell viability across a range of nanogel concentrations up to 40 µg/mL (Figure 7A). Similarly, particles were non-hemolytic towards bovine RBCs (Figure 7B, note 0 – 2% total hemolysis for all nanogel conditions) even when employed at concentrations orders of magnitude greater than what was required to achieve therapeutic responses in our drug delivery experiments (Figure 6 and Table 1). Collectively, these results demonstrate that peptide-polysaccharide nanogels are a highly biocompatible delivery platform with a low potential for systemic toxicity when parenterally administered.
Figure 7.
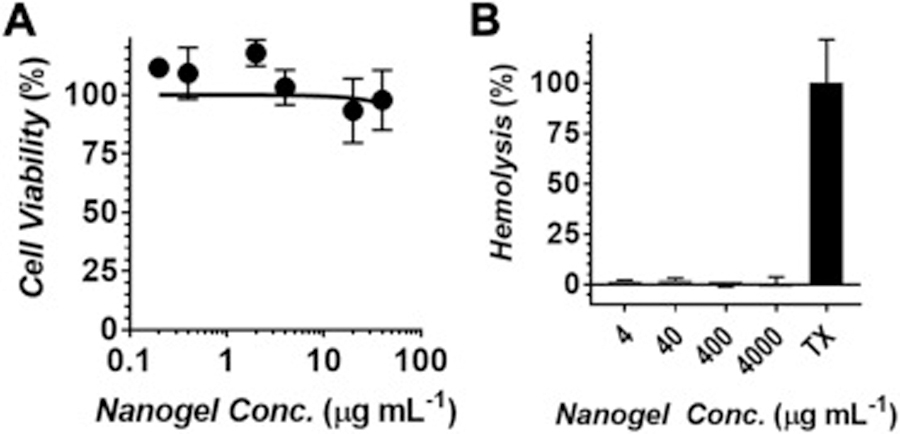
Biocompatability of nanogels following a 24 hour incubation with increasing concentrations of the empty nanogel carrier. (A) Viability of human umbilical vein endothelial cells (HUVEC). (B) Percentage of bovine red blood cell hemolysis. TX = Triton X-100 positive control.
Discussion
Enhancing the utility of therapeutic and diagnostic agents via nanoparticle-based delivery platforms requires nanomaterials that are chemically tractable, synthetically facile and are innately biocompatible. Here, we detail the synthesis and characterization of a new class of nanogel particle that can be rapidly prepared in high yield and purity via electrostatic complexation of complimentary charged HA and PLL biopolymers. Peptide-polysaccharide nanogels represent a unique class of bioresponsive nanoparticles with tunable swelling and release profiles. Remarkably, nanogels display broad efficacy in a range of delivery applications, including successful delivery of a membrane-impermeable protein into cells, improving the potency of loaded chemotherapeutics towards drug-sensitive and -resistant cancer cells, and sensitization of intractable bacterial pathogens to antibiotic.
In vitro studies suggest that nanogels augment the activity of delivered cargo through various complimentary mechanisms, dependent on the biologic context. In mammalian cells, binding of nanogel HA to CD44 adhesion receptors on the cell surface leads to direct intracellular uptake of the loaded cargo. Subsequent pH-dependent fusion of PLL delivered within the acidic endosome mediates endosomal escape of nanogels to the cytoplasm. Our studies show this mechanism allows nanogels to deliver otherwise membrane-impermeable protein cargo into the cytoplasm of cells, while enhancing the local intracellular concentration of loaded drug for potent therapeutic efficacy. In the context of bacteria, our studies support the potential for HA and PLL to recruit and permeabilize pathogens, respectively, leading to improved potency of antibiotics encapsulated within the nanogel carrier. Importantly, while these nano-scale materials are capable of potentially augmenting the activity of a variety of loaded biosensors and drugs, they are also inherently biocompatible, non-toxic and non-hemolytic. Thus, peptide-polysaccharide nanogels represent a potential theranostic platform with broad applications in drug delivery and biomedical imaging.
Supplementary Material
Acknowledgements
We thank the laboratory of Dr. Joel Schneider at the National Cancer Institute for providing cancer cell lines. We also thank the laboratories of Dr. Zissis Chroneos, Dr. Pak Kin Wong and Dr. Kenneth Keiler (Penn State, University Park, PA) for sharing the bacterial strains employed in these studies. Confocal microscopy was performed at the Penn State Microscopy and Cytometry Facility – University Park, PA. We also acknowledge the Penn State Materials Characterization Laboratory – University Park, PA for use of the dynamic light scattering instrumentation.
This work was supported by a Human Health and the Environment seed award provided to S. H. Medina by the Pennsylvania State University. A. W. Simonson was supported by funds from the Penn State Graduate Research Fellowship.
Footnotes
All authors declare no conflicts of interest.
Contributor Information
Andrew W. Simonson, Department of Biomedical Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, USA
Atip Lawanprasert, Department of Biomedical Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, USA.
Tyler D. P. Goralski, Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, Pennsylvania 16802, USA
Kenneth C. Keiler, Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, Pennsylvania 16802, USA
Scott H. Medina, Department of Biomedical Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, USA
References
- 1.Hodgson J, ADMET—turning chemicals into drugs. Nat. Biotechnol 2001;19:722–726 [DOI] [PubMed] [Google Scholar]
- 2.Grodowska K and Parczewski A, Organic solvents in the pharmaceutical industry. Acta. Pol. Pharm 2010;67:3–12 [PubMed] [Google Scholar]
- 3.Loftsson T and Brewster ME, Pharmaceutical applications of cyclodextrins: basic science and product development. J. Pharm. Pharmacol 2010;62:1607–1621 [DOI] [PubMed] [Google Scholar]
- 4.Kumari A, Yadav SK and Yadav SC, Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf., B 2010;75:1–18 [DOI] [PubMed] [Google Scholar]
- 5.Allen TM and Cullis PR, Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Delivery Rev 2013;65:36–48 [DOI] [PubMed] [Google Scholar]
- 6.Singh R and Lillard JW Jr, Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol 2009;86:215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torchilin V, Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Delivery Rev 2011;63:131–135 [DOI] [PubMed] [Google Scholar]
- 8.Tirelli N, (Bio) responsive nanoparticles. Curr. Opin. Colloid Interface Sci 2006;11:210–216 [Google Scholar]
- 9.Kwok KW, Dong W, Marinakos SM, Liu J, Chilkoti A, Wiesner MR, et al. , Silver nanoparticle toxicity is related to coating materials and disruption of sodium concentration regulation. Nanotoxicology 2016;10:1306–1317 [DOI] [PubMed] [Google Scholar]
- 10.Shin SW, Song IH and Um SH, Role of physicochemical properties in nanoparticle toxicity. Nanomaterials 2015;5:1351–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yildirimer L, Thanh NT, Loizidou M and Seifalian AM, Toxicology and clinical potential of nanoparticles. Nano Today. 2011;6:585–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Food U, Administration D, Food U and Administration D, Generally recognized as safe (GRAS). US Food and Drug Administration. 2015
- 13.Soni KS, Desale SS and Bronich TK, Nanogels: An overview of properties, biomedical applications and obstacles to clinical translation. J. Controlled Release. 2016;240:109–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neamtu I, Rusu AG, Diaconu A, Nita LE and Chiriac AP, Basic concepts and recent advances in nanogels as carriers for medical applications. Drug Delivery. 2017;24:539–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim Y and Kumar S, CD44-Mediated Adhesion to Hyaluronic Acid Contributes to Mechanosensing and Invasive Motility. Mol. Cancer Res 2014;12:1416–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanahan DJ, Watts RM and Pappajohn D, Some chemical characteristics of the lipids of human and bovine erythrocytes and plasma. J. Lipid Res 1960;1:421–432 [PubMed] [Google Scholar]
- 17.Xie J, Lim LK, Phua Y, Hua J and Wang C-H, Electrohydrodynamic atomization for biodegradable polymeric particle production. J. Colloid Interface Sci 2006;302:103–112 [DOI] [PubMed] [Google Scholar]
- 18.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins M, Appel R, et al. , 2005. The Proteomics Protocols Handbook, edited by Walker JM, Totowa: Humana Press, [Google Scholar]
- 19.Almalik A, Karimi S, Ouasti S, Donno R, Wandrey C, Day PJ, et al. , Hyaluronic acid (HA) presentation as a tool to modulate and control the receptor-mediated uptake of HA-coated nanoparticles. Biomaterials. 2013;34:5369–5380 [DOI] [PubMed] [Google Scholar]
- 20.Aruffo A, Stamenkovic I, Melnick M, Underhill CB and Seed B, CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313 [DOI] [PubMed] [Google Scholar]
- 21.Varkouhi AK, Scholte M, Storm G and Haisma HJ, Endosomal escape pathways for delivery of biologicals. J. Controlled Release. 2011;151:220–228 [DOI] [PubMed] [Google Scholar]
- 22.Wagner E, Plank C, Zatloukal K, Cotten M and Birnstiel ML, Influenza virus hemagglutinin HA-2 N-terminal fusogenic peptides augment gene transfer by transferrin-polylysine-DNA complexes: toward a synthetic virus-like gene-transfer vehicle. Proc. Natl. Acad. Sci 1992;89:7934–7938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirayama Y, Yoshimura M, Ozeki Y, Sugawara I, Udagawa T, Mizuno S, et al. , Mycobacteria exploit host hyaluronan for efficient extracellular replication. PLoS Pathog 2009;5:e1000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marion C, Stewart JM, Tazi MF, Burnaugh AM, Linke CM, Woodiga SA, et al. , Streptococcus pneumoniae can utilize multiple sources of hyaluronic acid for growth. Infect. Immun 2012;80:1390–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyldgaard M, Mygind T, Vad BS, Stenvang M, Otzen DE and Meyer RL, The antimicrobial mechanism of action of epsilon-poly-l-lysine. Appl. Environ. Microbiol 2014;80:7758–7770 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.