

Abstract
Acute lung injury (ALI) is characterized by tissue damage and inflammatory cytokine secretion; however, the therapeutic options available to treat ALI remain limited. Necrostatin-1 (Nec-1) has the ability to attenuate cell necroptosis in various inflammatory diseases. The present study evaluated the protective effects of Nec-1 on a mouse model of lipopolysaccharide-induced ALI. Histological alterations in the lungs were evaluated through hematoxylin and eosin staining, and the expression levels of cytokines in the bronchoalveolar lavage fluid and lung tissues were determined by ELISA. In addition, accumulated production of reactive oxygen species was determined by staining with DCFH-DA probes, western blotting and immunofluorescence. The results revealed that treatment with the necroptosis inhibitor, Nec-1, exerted significant protective effects on ALI-induced inflammation and necroptosis. The key proteins involved in necroptosis were markedly reduced, including receptor-interacting serine/threonine-protein kinase (RIP)1 and RIP3. Notably, antioxidant proteins were upregulated by Nec-1, which may attenuate oxidative stress. Furthermore, treatment with Nec-1 markedly suppressed necroptosis in the pulmonary alveoli RLE-6TN cell line. Taken together, these data revealed a novel association between ALI and necroptosis, and suggested that necroptosis inhibitors may be used as effective anti-inflammatory drugs to treat ALI.
Keywords: acute lung injury, necroptosis, reactive oxygen species, inflammation, necrostatin-1
Introduction
Acute lung injury (ALI) is a major cause of morbidity and mortality, which is characterized by strong pulmonary inflammation (1,2), leading to cell death and ultimately to respiratory failure. Lipopolysaccharide (LPS) derived from gram-negative bacteria is considered the principal pathogen-associated molecular cause of ALI (3). LPS is widely used to induce pulmonary inflammation in experimental models of ALI (4). As reported, acute lung damage is associated with initiation of an inflammatory response that leads to a series of pathological alterations in lung tissues, including changes in the ultrastructure of pulmonary alveoli (5), expression of inflammatory cytokines (6), cell damage (7) and accumulation of reactive oxygen species (ROS) (8). Although pharmacological therapies for ALI have been developed in recent decades, the mortality rate remains high.
The excessive inflammatory response associated with ALI causes cell damage, with the damaged cells being eliminated by apoptosis, which is a mechanism that protects cells from inflammation and necrosis (9). However, a high degree of apoptosis overwhelms the capacity of the immune system to clear dead cells, thus resulting in necrosis. Necrosis is a type of cell death morphologically characterized by cell swelling and cytomembrane rupture (10). Programmed necrosis, or necroptosis, is a regulated form of necrosis, which is induced by various initiators, particularly tumor necrosis factor α (TNF-α), which is a downstream product of LPS stimulation of Toll-like receptor 4 (TLR4) and the most extensive inducer of necroptosis (11). During the process of LPS-TLR4-TNF-α-induced necroptosis, receptor-interacting serine/threonine-protein kinase (RIP)1 interacts with RIP3 to induce its activation, thus leading to rupture of the cell membrane. RIP1-RIP3 forms a complex to promote necroptosis-induced tissue injury (12). There is a general consensus that necroptosis is a potent inducer of inflammation (13), whereas necrostatin-1 (Nec-1) is an inhibitor of RIP1 that has been confirmed to be a potent and specific allosteric inhibitor of necroptosis. Promising results have been obtained with Nec-1 in numerous experimental disease models, including ischemia-reperfusion injury (14,15), intestinal inflammation (16), acute pancreatitis (17) and neurodegenerative disease (18). Therefore, inhibition of necroptosis may be a promising strategy to improve the outcome of overloaded necroptosis-induced localized or severe and systemic inflammatory disorders.
Under physiological conditions, ROS are regulated by antioxidants (19), which can either be endogenously generated or externally supplemented. However, overproduction of ROS has been implicated in the development of various chronic and degenerative diseases, including cancer, and respiratory, neurodegenerative and digestive diseases (20). ROS are likely to be among the inducers of acute or chronic inflammation that lead to cellular damage, since antioxidants reduce activation of inflammatory pathways (21). The antioxidant defense system can be overwhelmed during sustained severe inflammation, as observed in acute lung inflammation, inflammatory bowel disease, neurodegenerative disorders, cardiovascular diseases and aging (22). Therefore, certain antioxidants are essential to regulate biochemical pathways that lead to the appropriate functioning of organs. Antioxidant supplementation has been reported to lessen endogenous antioxidant depletion and to alleviate oxidative damage in experimental research (20). Therefore, the present study evaluated the protective effects of Nec-1 on LPS-induced ALI.
Materials and methods
Animals
All experiments using male mice (age, 8–10 weeks; weight, 22–24 g). All animal procedures were approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University. A total of 30 healthy wild-type male C57B/6 mice (age, 6–8 weeks; weight, 18–22 g) purchased from the Animal Research Center of Wenzhou Medical University were maintained and acclimated in a specific pathogen-free environment for 2 weeks prior to experimentation, with free access to food and water at the Wenzhou Medical University under a controlled temperature (25°C) with 50% humidity and 12-h light/dark cycles.
Cells and cell culture
The rat alveolar epithelial RLE-6TN cell line, which was derived from alveolar type II cells isolated from a 56-day-old male F344 rat by airway perfusion with a pronase solution, was purchased from American Type Culture Collection. Cells were cultured in DMEM/F12 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.). The cell model of ALI was induced using TNF-α (40 µM; Sigma-Aldrich; Merck KGaA). The treatment group was co-treated with TNF-α and Nec-1 (50 µM; Enzo Life Sciences, Inc.) for 6 h at 37°C. The mock group was treated with saline for 6 h at 37°C. The cells were harvested at 6 h after initiation of the experiment for subsequent analyses.
Animal model and experimental setup
All animal procedures were approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University. Mice were fasted for 8 h prior to the experiment. Before induction of ALI, the mice were anesthetized by inhalation of sevoflurane and were transtracheally injected with either Nec-1 (5 mg/kg; Enzo Life Sciences, Inc.) or saline as a control. LPS-induced ALI was induced by transtracheal injection of LPS (Sigma-Aldrich; Merck KGaA) at a dose of 10 mg/kg 30 min after Nec-1 or saline treatment. Additionally, the mice of sham-operation were transtracheally injected with saline and treated with saline only. During the experiment, the mice were monitored every 30 min. All mice survived until sacrifice 6 h after initiation of the experiment. The mice were sacrificed by cervical dislocation under 3% sevoflurane inhalation, after which, the lungs were quickly removed. Lung samples were either placed in a buffered solution for histopathological analysis, or snap-frozen for protein isolation or for analyzing apoptosis by flow cytometry. Prior to tissue collection, death was confirmed after 1–2 min of cardiac and respiratory observation.
Histopathological examination of the lungs
The lung tissues were collected after 6 h of induction of ALI. The tissues were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 24 h at room temperature. Paraffin-embedded tissues from each mouse were sectioned at 5 µm and were then stained with hematoxylin for 5 min and eosin for 10 sec at room temperature. Each lung tissue section was observed in five fields of view using a light microscope (magnification, ×200).
Transmission electron microscopy (TEM)
Mouse lung tissues were minced into small pieces (1 mm3) and fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer for 4 h at 4°C. The tissues were post-fixed in 1% osmium tetroxide in 1% K4Fe(CH)6 for 1 h at 37°C, and dehydrated through graded concentrations of alcohol (50, 75, 90 and 100%; 30 min at room temperature per concentration), clarified in propylene oxide for 12 h at 45°C, and embedded in a mixture of Epon 812 and Durcupan for 3 days at 60°C (Sigma-Aldrich; Merck KGaA). The tissues were subsequently sectioned with an ultramicrotome (100 nm). The longitudinal sections were placed onto copper grids, stained with 7.7% uranyl acetate (50 µl) at room temperature for 30 min, and 2.6% lead nitrate (50 µl) at room temperature for 2 sec. Subsequently, the sections were visualized under a H-600 electron microscope (magnification, ×17000; Hitachi Ltd.).
Immunofluorescence analysis
Immunofluorescence was used to detect RIP1 and RIP3 expression. Cells were treated with TNF-α (40 µM) and Nec-1 (50 µM), washed with PBS, fixed with 4% paraformaldehyde for 15 min at 4°C, and were then permeabilized with 0.5% Triton X-100 for 10 min at room temperature. After blocking with 5% BSA (Beyotime Institute of Biotechnology) for 1 h at 37°C, the cells were incubated with either anti-RIP1 (cat. no. 3493; 1:200; Cell Signaling Technology, Inc.) or anti-RIP3 (cat. no. ab62344; 1:200; Abcam) antibodies overnight at 4°C. The slides were then incubated with Alexa Fluor® 488-conjugated goat anti-rabbit IgG antibody (cat. no. ab150077; 1:200; Abcam,) for 1 h at 37°C. The nuclei were then stained with DAPI for 5 min at room temperature. Images were obtained under a fluorescence microscope.
Protein extraction and western blotting
Snap-frozen lung tissues or cells were homogenized and resuspended in lysis buffer (cat. no. P0013B; Beyotime Institute of Biotechnology) containing 1% phenylmethylsulfonyl fluoride (cat. no. ST506; Beyotime Institute of Biotechnology) for 30 min at 4°C. Subsequently, the lysate was centrifuged at 12,000 × g for 15 min at 4°C and the supernatant was collected. Protein concentrations were determined using a bicinchoninic acid (BCA) assay kit (cat. no. P0012; Beyotime Institute of Biotechnology) at 37°C. The proteins (50 µg/lane) were separated by SDS-PAGE on 10% gels and were then transferred to PVDF membranes (EMD Millipore). The transferred membranes were blocked with 5% skim milk for 1 h at room temperature. The membranes were incubated at 4°C overnight with the following primary antibodies: Anti-RIP1 (cat. no. 3493; Cell Signaling Technology, Inc.), anti-RIP3 (cat. no. ab62344; Abcam), anti-GAPDH (cat. no. 5174; Cell Signaling Technology, Inc.), anti-nuclear factor erythroid 2-related factor 2 (NRF2; cat. no. ab137550; Abcam) and anti-heme oxygenase 1 (HO-1; cat. no. ab13243; Abcam). All aforementioned primary antibodies were diluted to 1:1,000 with WB antibody diluent (cat. no. P0023A; Beyotime Institute of Biotechnology). Subsequently, the membranes were incubated with HRP-conjugated anti-rabbit IgG secondary antibody (cat. no. 7074; 1:3,000; Cell Signaling Technology, Inc.) at room temperature for 1 h. Protein bands were detected using the Immobilon ECL Ultra Western HRP substrate (EMD Millipore) and Image Lab software (version 3.0; Bio-Rad Laboratories Inc.), with GAPDH as the loading control.
RNA isolation and reverse transcription-quantitative PCR (RT-qPCR)
RNA was isolated from lung tissues stored in RNAlater or from the rat alveolar epithelial cell line using TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. After determining RNA concentration, 1 µg RNA was used for cDNA synthesis using the PrimeScript™ RT Reagent kit for RT-PCR (Takara Bio, Inc.), according to the manufacturer's protocol. qPCR analysis was performed on cDNA using the ABI7500 instrument (Applied Biosystems; Thermo Fisher Scientific, Inc.) with TB Green® Premix Ex Taq™ II kit (Takara Bio, Inc.), according to the manufacturer's protocol. The thermocycling conditions were as follows: Pre-conditioning at 95°C for 30 sec; annealing for 5 sec at 95°C and amplification for 15 sec at 60°C for 40 cycles; final extension for 10 sec at 60°C. The relative mRNA expression levels were normalized the internal reference gene GAPDH using the 2−ΔΔCq cycle threshold method (23). The primer sequences for qPCR are listed in Table I.
Table I.
Sequences of primers used for reverse transcription-quantitative PCR.
| A, Mouse | ||
|---|---|---|
| Sequence (5′→3′) | ||
| Gene | Forward | Reverse |
| GAPDH | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA |
| HO-1 | AGGTCCTGAAGAAGATTGC | TCTCCAGAGTGTTCATTCG |
| NRF-2 | AGCATCCTCTCCACTGAT | GGTCACAGCCTTCAATAGT |
| B, Rat | ||
| Sequence (5′→3′) | ||
| Gene | Forward | Reverse |
| GAPDH | GTGCAGTGCCAGCCTCGTC | GGCAGCACCAGTGGATGCAG |
| HO-1 | GGTGACAGAAGAGGCTAAG | TAGTATCTTGAACCAGGCTAG |
| NRF-2 | CCGAGTTACAGTGTCTTAATAC | TGGAGAGGATGCTGCTAA |
HO-1, heme oxygenase 1; NRF-2, nuclear factor erythroid 2-related factor 2.
Isolation of primary alveolar epithelial cells
The lungs of were rapidly removed, incubated with 1.0 mg/ml collagenase I (Thermo Fisher Scientific, Inc.) for 1 h at 37°C and minced into small pieces (1 mm3) on ice. The single lung cell suspensions were used for further ROS and apoptosis analyses.
Quantification of ROS levels
Intracellular ROS intensity was measured using the Reactive Oxygen Species Assay kit (cat. no. S0033; Beyotime Institute of Biotechnology), according to the manufacturer's protocol. Fluorescence was detected by flow cytometry using FACSAria (BD Biosciences) and data were analyzed using FlowJo software (version 10; FlowJo, LLC).
Analysis of late apoptosis
Single lung cell suspensions were immunolabeled with an apoptosis kit (cat. no. 640914; BioLegend, Inc.), according to the manufacturer's protocol. Flow cytometry was performed with a FACSAria flow cytometer after gating the living cells. Data were analyzed using FlowJo software (version 10; FlowJo, LLC).
Isolation of bronchoalveolar lavage fluid (BALF)
Mice were sacrificed and BALF samples were collected by slow infusion and extraction with 0.2 ml cold PBS 4 times. The procedure was repeated four times and 0.8 ml BALF per mouse was collected for subsequent research. The concentration of total proteins in BALF was estimated using a BCA protein assay kit (cat. no. P0010; Beyotime Institute of Biotechnology). The levels of SOD and MDA in BALF were measured using a commercial SOD assay kit (cat. no. A001-3-2; Nanjing Jiancheng Bioengineering Institute) and MDA activity kit (cat. no. A003-4-1; Naning Jiancheng Bioengineering Institute), according to the manufacturer's instructions.
Analysis of inflammatory cytokines
The expression levels of inflammatory cytokines, TNF-α (cat. no. F11630), interleukin (IL)-1β (cat. no. F10770) and IL-6 (cat. no. F10830), in BALF and lung tissue homogenates were quantified using ELISA kits, according to the manufacturer's protocol (Westang Technology Company; www.westang.com).
Statistical analyses
Data are presented as the mean ± standard error or standard deviation of at least three independent experiments. One-way ANOVA was performed followed by Tukey's multiple comparison test using GraphPad Prism software (version 5; GraphPad Software Inc.). P<0.05 was considered to indicate a statistically significant difference.
Results
Nec-1 protects against LPS-induced inflammation and lung injury in ALI
To investigate the role of Nec-1 in ALI, a mouse model of ALI was established by transtracheal injection of LPS (10 mg/kg). Nec-1 (5 mg/kg) was administered 1 h before LPS induction. The histopathological results revealed that Nec-1 reduced LPS-induced lung injury compared with in mice challenged with LPS only, although the damage was more severe than that in the sham-operated mice (Fig. 1A). Subsequently, pulmonary inflammatory cytokine levels were detected by ELISA. Compared with the high level of secretory total proteins (Fig. 1B), and inflammatory cytokines in the BALF (Fig. 1C) and lung tissues (Fig. 1D) of mice in the ALI group, Nec-1-treated mice exhibited lower levels of secretory total proteins and inflammatory cytokines, including TNF-α, IL-1β and IL-6 (Fig. 1B-D; Tables II and III). These findings indicated that inflammation and injury were alleviated in the lungs of Nec-1-treated mice.
Figure 1.

Nec-1 protects against LPS-induced inflammatory response and lung injury in a mouse model of ALI. (A) Histopathological analysis of the effects of Nec-1 on LPS-induced experimental ALI using hematoxylin and eosin staining (magnification, ×100 and ×200). The black arrow indicates areas of lung injury. (B) Effects of Nec-1 on LPS-induced experimental ALI were assessed by estimating the levels of total secretory proteins using a bicinchoninic acid kit. (C and D) Levels of secreted inflammatory cytokines isolated from (C) BALF and (D) lung tissues were estimated by ELISA, in order to assess the effects of Nec-1 on LPS-induced ALI. Data are shown as the mean ± SEM from at least three independent experiments (n=5 mice/group). *P<0.05, as indicated. ALI, acute lung injury; BALF, bronchoalveolar lavage fluid; IL, interleukin; LPS, lipopolysaccharide; Nec-1, necrostatin-1; TNF-α, tumor necrosis factor-α.
Table II.
Inflammatory cytokine levels in bronchoalveolar lavage fluid.
| Group | TNF-α (pg/ml) | IL-1β (pg/ml) | IL-6 (pg/ml) |
|---|---|---|---|
| Sham operation | 21.24±7.53 | 73.704±13.18 | 39.98±4.98 |
| LPS-ALI | 476.06±90.14a | 165.19±32.21a | 360.29±19.52a |
| Nec-1 + LPS-ALI | 150.17±49.83b | 83.06±23.47b | 182.27±38.64b |
P<0.05 vs. the Sham operation group
P<0.05 vs. the LPS-ALI group. ALI, acute lung injury; IL, interleukin; LPS, lipopolysaccharide; Nec-1, necrostatin-1; TNF-α, tumor necrosis factor-α.
Table III.
Inflammatory cytokines in lung tissues.
| Group | TNF-α (pg/ml) | IL-1β (pg/ml) | IL-6 (pg/ml) |
|---|---|---|---|
| Sham operation | 40.34±18.16 | 268.81±58.06 | 63.75±24.24 |
| LPS-ALI | 394.81±60.17a | 727.01±86.83a | 293.13±17.40a |
| Nec-1 + LPS-ALI | 187.46±56.25b | 552.68±29.06b | 161.67±39.77b |
P<0.05 vs. the Sham operation group
P<0.05 vs. the LPS-ALI group. ALI, acute lung injury; IL, interleukin; LPS, lipopolysaccharide; Nec-1, necrostatin-1; TNF-α, tumor necrosis factor-α.
Suppressive effects of Nec-1 on necroptosis in LPS-induced ALI
The anti-necroptotic effects of Nec-1 on LPS-induced mice were assessed by western blotting. As shown in Fig. 2A and B, the expression levels of RIP1 and RIP3 were significantly decreased in response to Nec-1 treatment compared with in the untreated ALI group following exposure to LPS. Since late apoptosis also reflects the level of necroptosis in ALI pathogenesis, flow cytometry was performed with Annexin V and propidium iodide (PI) double-stained primary lung cells. As shown in Fig. 2C and D, LPS exposure significantly increased the percentage of late apoptotic cells (right upper quadrant) compared with in the control group, and this was effectively attenuated in lungs pretreated with Nec-1. Furthermore, Nec-1 reduced LPS-induced alterations, including increased number of necrosomes, lamellar body abnormality, disappearance of microvilli, karyorrhexis, cellular swelling and cytomembrane rupture (Fig. 2E). Taken together, these observations strongly indicated that Nec-1 protects lungs from necroptosis during ALI in vivo.
Figure 2.

Suppressive effects of Nec-1 on necroptosis in a mouse model of LPS-treated ALI. (A) Western blotting of lysates from mouse lung tissues was used to estimate the expression levels of necroptosis-associated proteins (RIP1 and RIP3). (B) Semi-quantification of the protein expression levels of RIP1 and RIP3. Representative images of at least three independent experiments are shown. (C and D) Estimation of the degree of apoptosis in primary lung cells, as determined by flow cytometry after staining with Annexin V and PI. (E) Ultrastructure of alveolar epithelial cells in the indicated mouse models (green arrow, lamellar body; red arrow, microvilli). Data are presented as the mean ± SEM from at least three independent experiments (n=5 mice/group). *P<0.05, as indicated. ALI, acute lung injury; LPS, lipopolysaccharide; Nec-1, necrostatin-1; PI, propidium iodide; RIP, receptor-interacting serine/threonine-protein kinase.
Nec-1 attenuates ROS response to LPS-induced ALI in vivo
LPS elicits a robust ROS response and leads to the production of ROS. To determine whether Nec-1 mediated protection against oxidative stress dysfunction, through enhancing the expression of antioxidant-associated proteins that mediate ROS homeostasis, the expression levels of NRF2 and HO-1 were detected. The expression levels of these antioxidants were increased in ALI mice treated with Nec-1 compared with in those in the saline-treated control group and LPS-treated ALI group (Fig. 3A and B). Nec-1 also enhanced the induction of anti-ROS gene expression, including NRF2 and HO-1, in the lung tissue of mice. As shown in Table IV and Fig. 3D, increased superoxide dismutase 2 (SOD2) and decreased malondialdehyde (MDA) levels were observed in the Nec-1-treated ALI group. ROS intensity in primary lung cells was estimated using an ROS assay kit. Nec-1-treated mice exhibited hypoactive DCFH-DA mean fluorescence intensity (Fig. 3E), suggesting that internal ROS production was decreased in the Nec-1 + LPS-ALI group compared with the LPS-ALI group. These results suggested that Nec-1 may be a mediator of ROS to maintain intrinsic homeostasis.
Figure 3.

Nec-1 attenuates the ROS response to LPS-induced ALI in vivo. (A) Western blotting of lysates in the indicated mouse lung tissues was used to estimate the expression levels of ROS-associated proteins (NRF2 and HO-1). (B) Semi-quantification of the protein expression levels of NRF2 and HO-1. Representative images of at least three independent experiments are shown. (C) mRNA expression levels of NRF2 and HO-1 in lung tissues from sham-operated, LPS-ALI and Nec-1 + LPS-ALI mice were detected by reverse transcription-quantitative PCR and were normalized to GAPDH. (D) Levels of SOD2 and MDA in the lung tissue homogenates were detected using commercial kits. (E) Intracellular ROS production of the indicated lung cells was analyzed by DCFH-DA staining and flow cytometry. Data are presented as the mean ± SEM from at least three independent experiments (n=5 mice/group). *P<0.05, as indicated. ALI, acute lung injury; HO-1, heme oxygenase-1; LPS, lipopolysaccharide; MDA, malondialdehyde; Nec-1, necrostatin-1; NRF2, nuclear factor erythroid 2-related factor 2; SOD2, superoxide dismutase.
Table IV.
Concentration of SOD2 and MDA in lung tissues.
| Group | SOD2 (U/mg protein) | MDA (nmol/mg protein) |
|---|---|---|
| Sham operation | 281.87±24.1 | 1.38±0.13 |
| LPS-ALI | 133.68±24.8a | 4.01±0.07a |
| Nec-1 + LPS-ALI | 293.47±24.6b | 1.72±0.18b |
P<0.05 vs. the Sham operation group
P<0.05 vs. the LPS-ALI group. ALI, acute lung injury; LPS, lipopolysaccharide; MDA, malondialdehyde; Nec-1, necrostatin-1; SOD2, superoxide dismutase 2.
Nec-1 regulates necroptosis and ROS in TNF-α-stimulated RLE-6TN cells
Similar to the inhibitory effects of Nec-1 on necroptosis in vivo, Nec-1 also exerted protective effects in vitro. Cell damage was induced by TNF-α, and necroptosis was assessed with flow cytometry after staining with Annexin V and PI. Nec-1 significantly reduced the percentage of late apoptotic cells exposed to TNF-α (Fig. 4A and B). Western blot and immunofluorescence analyses also indicated that RIP1 and RIP3 expression was reduced in Nec-1-pretreated damaged RLE-6TN cells (Fig. 4C-F). Furthermore, NRF2 and HO-1 were upregulated to attenuate LPS-induced ROS dysfunction in the Nec-1-pretreated group (Fig. 5A and B). As expected, Nec-1 also enhanced induction of anti-ROS gene expression, including NRF2 and HO-1, in injured Nec-1-treated RLE-6TN cells (Fig. 5C). The intensity of the ROS response was also evaluated by DCFH-DA probes using flow cytometry. Nec-1 was able to downregulate the increased mean fluorescent intensity of ROS in TNF-α-stimulated RLE-6TN cells (Fig. 5D), suggesting that internal ROS production was decreased in the Nec-1 + TNF-α group compared with the LPS-TNF-α group.
Figure 4.

Nec-1 regulates necroptosis in TNF-α-stimulated RLE-6TN cells. (A) Representative flow cytometry plots and (B) quantification of the degree of apoptosis of alveolar epithelial RLE-6TN cells, as determined using flow cytometry after staining with Annexin V and PI. (C) Western blotting of lysates from the indicated alveolar epithelial RLE-6TN cells was used to detect the expression levels of necroptosis-associated proteins RIP1 and RIP3. (D) Semi-quantification of the protein expression levels of RIP1 and RIP3. Representative images of at least three independent experiments are shown. (E and F) Representative images of immunofluorescence analysis of RIP1 and RIP3 in the indicated cell models (magnification, ×100). Data are presented as the mean ± SD from at least three independent experiments. *P<0.05, as indicated. ALI, acute lung injury; Nec-1, necrostatin-1; PI, propidium iodide; RIP, receptor-interacting serine/threonine-protein kinase; TNF-α, tumor necrosis factor-α.
Figure 5.

Nec-1 regulates ROS in TNF-α-stimulated RLE-6TN cells. (A) Western blotting of lysates in the indicated alveolar epithelial RLE-6TN cells was used to detected the expression levels of ROS-associated proteins (NRF2 and HO-1). (B) Semi-quantification of the protein expression levels of NRF2 and HO-1. Representative images of at least three independent experiments are shown. (C) mRNA expression levels of NRF-2 and HO-1 in RLE-6TN cells from the mock, TNF-α-ALI and Nec-1 + TNF-α-ALI groups were detected by reverse transcription-quantitative PCR and were normalized to GAPDH. (D) Intracellular ROS generation was analyzed by DCFH-DA staining using flow cytometry. Data are presented as the mean ± SD from at least three independent experiments. *P<0.05, as indicated. ALI, acute lung injury; HO-1, heme oxygenase-1; Nec-1, necrostatin-1; NRF2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α.
Discussion
ALI is characterized by a life-threatening inflammatory response of the lungs to various insults (24). The early inflammatory phase of ALI is characterized by alveolar epithelial and endothelial barrier damage and dysfunction. Hemorrhage and protein-rich pulmonary edema are followed by the proliferative phase, which involves alveolar epithelial cell proliferation, interstitial fibrosis and air space obliteration (25). Subsequently, necrosis, fibrosis and emphysema are observed alongside the loss of normal lung structure, thus resulting in acute respiratory distress syndrome (26). Despite continuous improvements in the medical management of ALI, it remains associated with a high level of morbidity and mortality (1). Therefore, it is necessary to gain a deep insight into the mechanism underlying ALI, in order to develop a novel therapeutic strategy.
An appropriate balance among cell death, proliferation and differentiation is critical for maintaining lung homeostasis and retaining its vital role as an immunological barrier against pathogens (27). However, pathological processes, including bacterial infection, viral infection and autoimmune disorders, jeopardize these processes. TNF-α and its receptor (TNFR) are essential mediators of cell death and inflammation, which have important roles in the immune response of pulmonary alveolar cells (28). The LPS-TNF-α-TNFR axis signaling can result in cell apoptosis, which is a means for eliminating unhealthy cells and limiting the inflammatory cascade. Once the degree of apoptosis exceeds the elimination capacity of immune cells, necrosis occurs. Necroptosis is a form of cell necrosis (29); since its discovery more than two decades ago, it has been considered a combination of cellular death and functional response, and its study represents an area of ongoing research. RIP1 has been reported to shift the balance between cell survival, apoptosis and necroptosis upon TNF-α stimulation. It acts as a kinase in the ‘necrosome’ complex, triggering the process of RIP3-dependent necroptosis (30). In the present study, it was demonstrated that necroptosis was associated with the pathogenesis of ALI, and Nec-1 exerted marked therapeutic effects by attenuating pulmonary alveolar injury and dysfunction, thereby notably improving the pulmonary alveolar cellular function in ALI mice. This effect may be due to the inhibition of proteins associated with the execution of necroptosis and reduced inflammatory cytokine production. Notably, Nec-1 reduced the expression of necroptotic proteins, RIP1 and RIP3. In addition, microscopic analysis suggested that Nec-1 protected damaged alveolar epithelial cells against cell swelling, karyorrhexis and cytomembrane rupture. Therefore, the necroptotic inhibitor Nec-1 may be considered a candidate for the treatment of ALI.
It has previously been reported that ROS are essential for necroptosis-induced cellular injury (31). Cellular ROS are diverse hyper-reactive derivatives of oxygen; cellular antioxidants maintain ROS at physiological levels. Excessive accumulation of ROS during cellular stress can overwhelm the antioxidant system and, if not alleviated, this can result in oxidative stress, oxidative damage to cellular constituents and cell death. Elevated levels of ROS appear to be particularly important in cellular necroptosis (32). RIP3-dependent necroptosis can trigger inflammasome activation by inducing alterations in the redox state, intracellular ion concentrations and cellular metabolic status (33). RIP3 regulates the cell metabolic state and is involved in oxidative phosphorylation. Furthermore, activated RIP3 enhances energy metabolism, resulting in ROS accumulation (34). The NRF2/HO-1 signaling pathway serves a vital role in oxidative stress. Under homeostatic conditions, NRF2 is not functionally activated and exists in the cytoplasm; however, once cells are subjected to oxidative stress, NRF2 is translocated into the nucleus and combines with the antioxidant response element to trigger HO-1 expression, thereby regulating the expression of oxidant or antioxidant-related genes and enzymes, such as SOD and MDA, and finally exerting its antioxidant defense function (35). Therefore, strengthening the antioxidant process may be considered a potential strategy to reduce the level of cellular necroptosis, in order to reduce the extent of tissue damage. This study revealed that treatment with Nec-1 (a specific inhibitor of RIP1) attenuated LPS-induced ALI in mice and TNF-α-induced cellular ALI by generating anti-ROS proteins. Furthermore, blockade of ROS production by Nec-1 treatment significantly inhibited the extent of cell death. These results strongly suggested that increased ROS levels may be responsible for necroptosis-induced cell damage and death.
Increased cellular injury stimulates ROS generation, which contributes to the promotion of cell swelling (36). ROS also induce cellular stress to promote cell necroptosis. Reduced cell necroptosis results in inhibition of ROS and increased antioxidative capacity, which can limit the irreversible damage to the cells. Decreased ROS can also inhibit inflammatory signaling, and contribute to the attenuation of local and systematic inflammation (37). A physiological ROS level is essential to induce an anti-ROS response and maintain cellular homeostasis (37). Notably, the ROS pathway is a double-edged sword; it helps pulmonary alveolar cells survive protein synthesis-associated stress; however, when cellular injury is too severe, it leads to inflammation and changes that ultimately damage these cells (Fig. 6). Therefore, artificially strengthening antioxidant ability may be a promising therapeutic strategy.
Figure 6.
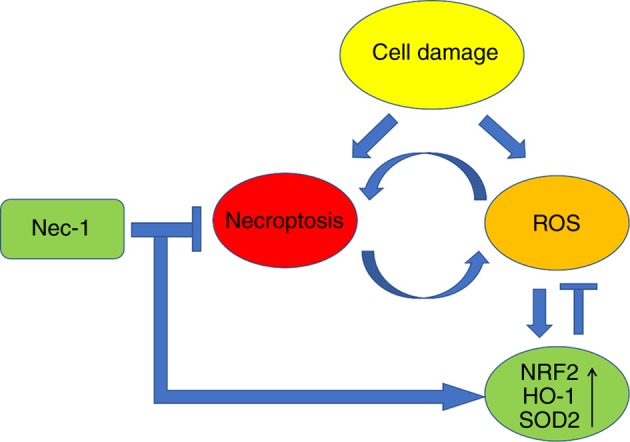
Schematic diagram of Nec-1-mediated protection from ALI-associated inflammatory injuries. Nec-1 triggered anti-ROS production by inhibiting necroptosis. Nec-1 also alleviated ALI symptoms, such as alveolar cell death, alveolar wall disorder, lung edema and the expression of inflammatory cytokines by directly suppressing necroptosis. HO-1, heme oxygenase-1; Nec-1, necrostatin-1; NRF2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; SOD2, superoxide dismutase 2.
The present study treated the experimental ALI models with Nec-1, which is a specific inhibitor of RIP1 and demonstrated that Nec-1 exerted marked anti-necroptosis and anti-inflammatory effects on LPS-stimulated mouse lung tissue in vivo and TNF-α-stimulated rat alveolar epithelial cells in vitro. Notably, inflammatory cytokines within BALF and lung tissue were decreased in the Nec-1-treated ALI mouse model. In addition, treatment with Nec-1 suppressed the expression of necroptosis-associated proteins, RIP1 and RIP3. Conversely, anti-ROS proteins were upregulated in the Nec-1-treated group compared with in the control and non-Nec-1-treated ALI groups. Positive anti-inflammatory and protective effects were also observed in the TNF-α-stimulated RLE-6TN cell line. These findings offer insights into the inhibitory effects of Nec-1 on the progression of ALI. It may be hypothesized that Nec-1 acts as a potential therapeutic agent for treating inflammatory lung diseases, such as ALI.
Several time windows for Nec-1 treatment to prevent ALI were analyzed and it was revealed that 6 h of pretreatment was required for effective prevention in the experimental ALI mouse model (data not shown). In addition, Nec-1 did not exert a promising therapeutic effect on ALI mice when administered post-ALI induction (data not shown). Typically, >6 h is required for Nec-1 to induce the production of anti-ROS proteins during ALI progression. Therefore, it is reasonable to propose that pretreatment with Nec-1 is required for the induction of NRF2 and HO-1, and to exert protective effects. Based on the current findings, Nec-1 may be a novel ALI therapeutic agent if used in patients with ALI at the early stage.
In conclusion, this study reported that necroptosis was effectively induced in rat alveolar epithelial cells and in a mouse model of LPS-induced ALI, whereas this necroptosis was blocked by Nec-1. Further investigations revealed increased expression of antioxidants (NRF2, HO-1 and SOD2) in response to Nec-1. HO-1 is an anti-ROS gene that is activated upon cell injury. Nec-1 actively enhanced the expression of HO-1 to attenuate the ROS response to LPS-induced ALI; therefore, decreased ROS production may be considered the most important inhibitory mechanism of necroptosis. These results may be beneficial to understand the detailed mechanisms of ALI caused by bacterial infections and could provide novel prospects for further molecular targeted therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Wenzhou Science and Technology Bureau (grant no. Y20170144).
Availability of data and materials
All data used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
BL and LL conceived and designed the experiments. BL, ZJ, XC, LZ, CW, BC and YT performed the experiments and analyzed the data. LL wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All animal procedures were approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: A clinical review. Lancet. 2007;369:1553–1564. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Zou C, Li J, Xiong S, Chen Y, Wu Q, Li Q, Weathington NM, Han S, Snavely C, Chen BB, Mallampalli RK. Mortality factor 4 like 1 protein mediates epithelial cell death in a mouse model of pneumonia. Sci Transl Med. 2015;7:311ra171. doi: 10.1126/scitranslmed.aac7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coon TA, McKelvey AC, Lear T, Rajbhandari S, Dunn SR, Connelly W, Zhao JY, Han S, Liu Y, Weathington NM, et al. The proinflammatory role of HECTD2 in innate immunity and experimental lung injury. Sci Transl Med. 2015;7:295ra109. doi: 10.1126/scitranslmed.aab3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayala P, Vivar R, Montalva R, Olmos P, Meneses M, Borzone GR. Elastin degradation products in acute lung injury induced by gastric contents aspiration. Respir Res. 2018;19:165. doi: 10.1186/s12931-018-0873-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo S, Jiang K, Wu H, Yang C, Yang Y, Yang J, Zhao G, Deng G. Magnoflorine ameliorates lipopolysaccharide-induced acute lung injury via suppressing NF-κB and MAPK activation. Front Pharmacol. 2018;9:982. doi: 10.3389/fphar.2018.00982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morales-Ortiz J, Deal V, Reyes F, Maldonado-Martínez G, Ledesma N, Staback F, Croft C, Pacheco A, Ortiz-Zuazaga H, Yost CC, et al. TLT-1 is a prognostic indicator in ALI/ARDS and prevents tissue damage in the lungs in a mouse model. Blood. 2018;132:2495–2505. doi: 10.1182/blood-2018-03-841593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Yu Y, Gorshkov B, Haigh S, Bordan Z, Weintraub D, Rudic RD, Chakraborty T, Barman SA, Verin AD, et al. Hsp70 suppresses mitochondrial reactive oxygen species and preserves pulmonary Microvascular barrier integrity following exposure to bacterial toxins. Front Immunol. 2018;9:1309. doi: 10.3389/fimmu.2018.01309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorman AM, Healy SJ, Jäger R, Samali A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol Ther. 2012;134:306–316. doi: 10.1016/j.pharmthera.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Dhuriya YK, Sharma D. Necroptosis: A regulated inflammatory mode of cell death. J Neuroinflammation. 2018;15:199. doi: 10.1186/s12974-018-1235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He S, Wang X. RIP kinases as modulators of inflammation and immunity. Nat Immunol. 2018;19:912–922. doi: 10.1038/s41590-018-0188-x. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Zhang J, Yan R, Tian J, Zhang Y, Zhang J, Chen M, Cui Q, Zhao L, Hu R, et al. Receptor-interacting protein kinase 3 promotes platelet activation and thrombosis. Proc Natl Acad Sci USA. 2017;114:2964–2969. doi: 10.1073/pnas.1610963114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 14.Pavlosky A, Lau A, Su Y, Lian D, Huang X, Yin Z, Haig A, Jevnikar AM, Zhang ZX. RIPK3-mediated necroptosis regulates cardiac allograft rejection. Am J Transplant. 2014;14:1778–1790. doi: 10.1111/ajt.12779. [DOI] [PubMed] [Google Scholar]
- 15.Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, Zhang ZX, Jevnikar AM. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant. 2013;13:2805–2818. doi: 10.1111/ajt.12447. [DOI] [PubMed] [Google Scholar]
- 16.Liu ZY, Wu B, Guo YS, Zhou YH, Fu ZG, Xu BQ, Li JH, Jing L, Jiang JL, Tang J, Chen ZN. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am J Cancer Res. 2015;5:3174–3185. [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikseresht S, Khodagholi F, Nategh M, Dargahi L. RIP1 inhibition rescues from LPS-induced RIP3-mediated programmed cell death, distributed energy metabolism and spatial memory impairment. J Mol Neurosci. 2015;57:219–230. doi: 10.1007/s12031-015-0609-3. [DOI] [PubMed] [Google Scholar]
- 19.Hwang S, Kim JK. Effects of NADPH oxidase inhibitors and mitochondria-targeted antioxidants on amyloid β1-42-induced neuronal deaths in mouse mixed cortical cultures. Chonnam Med J. 2018;54:159–166. doi: 10.4068/cmj.2018.54.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Ren Z, Zhang J, Chuang CC, Kandaswamy E, Zhou T, Zuo L. Role of ROS and nutritional antioxidants in human diseases. Front Physiol. 2018;9:477. doi: 10.3389/fphys.2018.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang C, Chen S, Zhang T, Li D, Huang Z, Huang J, Qin Y, Chen B, Cheng G, Ma F, Zhou M. TLR3 ligand PolyI:C prevents acute pancreatitis through the interferon-β/Interferon-α/β receptor signaling pathway in a Caerulein-induced pancreatitis mouse model. Front Immunol. 2019;10:980. doi: 10.3389/fimmu.2019.00980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han J, Kim YS, Lim MY, Kim HY, Kong S, Kang M, Choo YW, Jun JH, Ryu S, Jeong HY, et al. Dual roles of graphene oxide to attenuate inflammation and elicit timely polarization of macrophage phenotypes for cardiac repair. ACS Nano. 2018;12:1959–1977. doi: 10.1021/acsnano.7b09107. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O'Kane CM, Krasnodembskaya AD. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am J Respir Crit Care Med. 2017;196:1275–1286. doi: 10.1164/rccm.201701-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sousse LE, Herndon DN, Andersen CR, Zovath A, Finnerty CC, Mlcak RP, Cox RA, Traber DL, Hawkins HK. Pulmonary histopathologic abnormalities and predictor variables in autopsies of burned pediatric patients. Burns. 2015;41:519–527. doi: 10.1016/j.burns.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malaviya R, Sunil VR, Venosa A, Verissimo VL, Cervelli JA, Vayas KN, Hall L, Laskin JD, Laskin DL. Attenuation of nitrogen mustard-induced pulmonary injury and fibrosis by anti-tumor necrosis factor-α antibody. Toxicol Sci. 2015;148:71–88. doi: 10.1093/toxsci/kfv161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McConnell AM, Yao C, Yeckes AR, Wang Y, Selvaggio AS, Tang J, Kirsch DG, Stripp BR. p53 regulates progenitor cell quiescence and differentiation in the airway. Cell Rep. 2016;17:2173–2182. doi: 10.1016/j.celrep.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Malaviya R, Laskin JD, Laskin DL. Anti-TNFα therapy in inflammatory lung diseases. Pharmacol Ther. 2017;180:90–98. doi: 10.1016/j.pharmthera.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 30.Raju S, Whalen DM, Mengistu M, Swanson C, Quinn JG, Taylor SS, Webster JD, Newton K, Shaw AS. Kinase domain dimerization drives RIPK3-dependent necroptosis. Sci Signal. 2018;11(pii):eaar2188. doi: 10.1126/scisignal.aar2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen C, Wang C, Han S, Wang Z, Dong Z, Zhao X, Wang P, Zhu H, Sun X, Ma X, et al. Aldehyde dehydrogenase 2 deficiency negates chronic low-to-moderate alcohol consumption-induced cardioprotecion possibly via ROS-dependent apoptosis and RIP1/RIP3/MLKL-mediated necroptosis. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1912–1918. doi: 10.1016/j.bbadis.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 32.Chauhan AK, Min KJ, Kwon TK. RIP1-dependent reactive oxygen species production executes artesunate-induced cell death in renal carcinoma Caki cells. Mol Cell Biochem. 2017;435:15–24. doi: 10.1007/s11010-017-3052-7. [DOI] [PubMed] [Google Scholar]
- 33.Shan B, Pan H, Najafov A, Yuan J. Necroptosis in development and diseases. Genes Dev. 2018;32:327–340. doi: 10.1101/gad.312561.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 35.Xu C, Qiao L, Ma L, Guo Y, Dou X, Yan S, Zhang B, Roman A. Biogenic selenium nanoparticles synthesized by Lactobacillus casei ATCC 393 alleviate intestinal epithelial barrier dysfunction caused by oxidative stress via Nrf2 signaling-mediated mitochondrial pathway. Int J Nanomedicine. 2019;14:4491–4502. doi: 10.2147/IJN.S199193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Q, Lv H, Wen Z, Ci X, Peng L. Isoliquiritigenin activates nuclear factor erythroid-2 related factor 2 to suppress the NOD-like receptor protein 3 inflammasome and inhibits the NF-κB pathway in macrophages and in acute lung injury. Front Immunol. 2017;8:1518. doi: 10.3389/fimmu.2017.01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lien CF, Lee WS, Wang IC, Chen TI, Chen TL, Yang KT. Intermittent hypoxia-generated ROS contributes to intracellular zinc regulation that limits ischemia/reperfusion injury in adult rat cardiomyocyte. J Mol Cell Cardiol. 2018;118:122–132. doi: 10.1016/j.yjmcc.2018.03.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used and/or analyzed during the present study are available from the corresponding author on reasonable request.