

Abstract
The underlying molecular mechanisms of cisplatin resistance in non-small cell lung cancer (NSCLC) are unclear. In this study, a novel differential methylation region located in the upstream regulatory region of the forkhead box F1 (FOXF1) gene was identified. The abnormal hypomethylation of FOXF1 increased the expression of FOXF1, and the high expression of FOXF1 promoted cell proliferation and inhibited cell apoptosis induced by cisplatin, which resulted in cisplatin resistance in NSCLC cells. In addition, FOXF1 promoted the expression of stem cell markers and self-renewal capability, indicating that FOXF1 regulated cisplatin resistance by promoting cancer stem cell properties in NSCLC cells. Moreover, a strong association was observed between FOXF1 upregulation and the presence of platinum-based chemotherapy resistance in patients with NSCLC. On the whole, the findings of this study indicate the regulatory mechanisms of cisplatin resistance by FOXF1 in NSCLC, and suggest that FOXF1 may be used as a prognostic biomarker of platinum-based chemotherapy resistance in NSCLC.
Keywords: non-small cell lung cancer, cisplatin resistance, forkhead box F1, DNA methylation, cancer stem cell
Introduction
Lung cancer is the leading cause of cancer-related mortality, and the major histological type of lung cancer is classified as non-small cell lung cancer (NSCLC) (occurring in 75% of cases), of which adenocarcinoma (40%) and squamous cell carcinoma (35%) are the two most common subtypes (1,2). At present, although surgery is the most effective treatment method for NSCLC, approximately 80% of patients with NSCLC require chemotherapy, as they are diagnosed at an advanced stage and are unable to undergo surgery. However, in clinical practice, >90% of patients with NSCLC receiving chemotherapy will eventually become resistant, which leads to treatment failure; thus, the 5-year survival rate is <20% (3). Therefore, the identification of the molecular mechanisms responsible for resistance to chemotherapy and a method with which to overcome drug resistance in NSCLC are urgently required.
In recent years, cancer stem cells have been considered to be the source of tumor heterogeneity and tumor expansion, and are the source of resistance to tumor chemotherapy in numerous types of cancer, including NSCLC (4,5). Cancer stem cells refer to a very small part of the tumor tissue and are also known as side population cells (6). Similar to normal stem cells, cancer stem cells have stem cell-like properties, such as differentiation potential, self-renewal capability, dormancy capability, DNA repair capability and drug efflux capability during tumor development and play important roles in tumor progression (7,8). Moreover, cancer stem cells cannot be eliminated completely by drug treatment and result in resistance to tumor chemotherapy, although the underlying basis of cancer stem cells in resistance to chemotherapy remains elusive (9,10). Therefore, cancer stem cells are considered to drive chemotherapeutic resistance in NSCLC, and the investigation into the regulatory mechanisms of cancer stem cell properties is essential for NSCLC therapy.
In the present study, DNA methylation microarray was used to detect the DNA methylation profiles of cisplatin-resistant A549/DDP cells and cisplatin-sensitive A549 cells, and it was found that the DNA methylation levels of forkhead box F1 (FOXF1) decreased significantly in the A549/DDP cells compared with the A549 cells. FOXF1 is a member of the forkhead box (FOX) transcription factor family, which is involved in various complex cellular processes and significantly implicated in cancer (11,12). Herein, it was found that the hypomethylation of FOXF1 increased the expression of FOXF1 and that the overexpression of FOXF1 enhanced the resistance of NSCLC cells to cisplatin by promoting cancer stem cell properties, such as self-renewal capability. In addition, clinical analysis found that high expression levels of FOXF1 in NSCLC tissues were associated with platinum-based chemotherapeutic resistance. These results not only shed light onto the mechanisms of cancer stem cells in chemotherapy-resistant NSCLC, but may also lead to the discovery of a biomarker that may be used for the identification of patients with NSCLC who are resistant to chemotherapy.
Materials and methods
Cell culture and transfections
A549/DDP (cisplatin-resistant A549) cells were established using increasing concentrations of cisplatin. Briefly, A549 cells (ATCC) in logarithmic growth were treated with 0.5 µmol/l of cisplatin. After 48 h, cisplatin was withdrawn and the cells were cultured without cisplatin until they recovered. The same treatment was performed again, and when the cells were considered to be resistant to cisplatin (cells can grow normally at the current cisplatin concentration), the concentration was gradually increased up to a final concentration of 3 µmol/l. When the induced cells had survived in 3 µmol/l of cisplatin for approximately 2 months with normal activity, the cells were confirmed to be cisplatin-resistant and named A549/DDP (the IC50 increased from 0.49 to 4.12 µmol/l) (data not shown). The A549/DDP cells were cultured with 2 µmol/l cisplatin.
NSCLC epithelial cell lines (A549, A549/DDP and H1299) and the normal lung epithelial cell line (16HBE) (ATCC) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37˚C in a humidified atmosphere with 5% CO2. The expression plasmid of FOXF1 was constructed by inserting cDNA into the pCDNA3.1 plasmid (Invitrogen; Thermo Fisher Scientific), and the shRNAs of FOXF1 was designed for lentivirus production (Shanghai GeneChem Co.). The plasmids were transfected into the cells using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific), and shRNA lentiviral vectors were transfected into the cells by the lentivirus. After 72 h, stable transfected cell lines were established for analysis.
Tissue sample collection
Primary NSCLC tissues were collected from the Cancer Center of Guangzhou Medical University (Guangzhou, China) with informed consent and Institutional Review Board (IRB) permission. A total of 70 patients with NSCLC were recruited into this study. All of the following criteria were met: Patients with primary NSCLC; a histological diagnosis of NSCLC with at least one measurable lesion; a TNM clinical stage of III to IV; first-line chemotherapy with platinum-based chemotherapy every 3 weeks for a maximum of 4 cycles. According to the Response Evaluation Criteria in Solid Tumors (RECIST), tissue samples were divided into 2 groups according to the patient's response assessed by medical image analysis and the detection of serum tumor markers after 4 cycles of platinum-based chemotherapy: Response or partial response, at least a 30% decrease in the sum of diameters of target lesions from pre-chemotherapy levels, taking as a reference the baseline sum diameters; stable or progressive disease, a decrease of <30% or an increase from pre-chemotherapy levels, taking as a reference the baseline sum diameters. Patients with chemotherapy response or partial response were considered to be chemotherapy-sensitive (R, responder), whereas patients with stable or progressive disease were grouped together and considered to be chemotherapy-resistant (NR, non-responder). The patient characteristics are presented in Table SI. Fresh NSCLC tissues were obtained by aspiration biopsy and immediately snap-frozen in liquid nitrogen and stored at -80˚C until use. All clinical and biological data on these samples were available.
All patients provided written informed consent, and the collection of NSCLC tissues for research purposes was approved by the relevant human research ethics committees of the Affiliated Cancer Hospital and Institute of Guangzhou Medical University.
DNA methylation assays
According to the manufacturer's instructions, total DNA was extracted from cells and tissue samples using the Genomic DNA Purification kit (Promega) and were then bisulfite-modified using the EpiTect Bisulfite kit (Qiagen).
Genome-wide methylation analysis was performed using the validated Illumina Infinium HumanMethylation 450K BeadChip. The methylation score of each CpG is represented as a β-value.
The detection of FOXF1 methylation was achieved by the quantitative measurement of methylated (C)/unmethyl-ated (T) allele peak ratios by pyrosequencing analysis. The assay was designed to detect FOXF1 CpG site methylation at chromosome 16 at position 86542659-86542696 (position 1, 6 CpG sites) and 86542770-86542808 (position 2, 6 CpG sites). Sequence accessions matched the UCSC Genome Browser Human assembly (GRCh37/hg19). The sequence reaction and detection were performed by pyrosequencing following the manufacturer's protocol (PyroMark Q96 Plate, Qiagen). The results are reported as a percentage of the methylated (C) allele over the background of un-methylated (T) allele (Methylation level = mC/(mC + umT) ×100%). The primers for FOXF1 pyrosequencing are listed in Table SII.
The DNA methyltransferase inhibitor 5-aza-2′-deoxycyti-dine (5-Aza-dC) (Sigma) was used to block DNA methylation. The cells were treated with 5-Aza-dC at 10 µM for 3 days. Drugs and culture medium were refreshed every day during treatment.
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA in cells was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific). Total RNA (1 µg) was used for cDNA synthesis using a Reverse Transcription kit (Takara), and the cDNA was then used for quantitative polymerase chain reaction (qPCR) using SYBR-Green Real-time PCR Master Mix (Toyobo). RT-qPCR was performed using the ABI ViiA™7Dx Real-Time PCR System (Life Technologies; Thermo Fisher Scientific) (thermocycling conditions: 50˚C, 2 min; 95˚C, 10 min; (95˚C, 15 sec; 60˚C, 1 min) 40 cycles). The expression levels of mRNA were normalized to GAPDH (method of quantification, 2-ΔΔCq) (13). The primers used for RT-qPCR are listed in Table SII.
Western blot analysis
The cells were harvested and lysed using RIPA buffer for 30 min at 4˚C. A total of 50 µg heat-denatured proteins were loaded onto a 15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) system, and then transferred to a polyvinylidene difluoride membrane for western blot analysis. After blocking non-specific binding sites with 5% (wt/vol) non-fat milk, 0.1% (vol/vol) Tween-20 diluted in Tris (pH 7.8)-buffered saline, rabbit polyclonal anti-FOXF1 (ab23194, 1:400 dilution), anti- aldehyde dehydrogenase 1 (ALDH1; ab52492, 1:500 dilution), anti-octamer-binding transcription factor 4 (OCT4; ab18976, 1:500 dilution) and anti-GAPDH (ab37168, 1:1,000 dilution) (all from Abcam) primary antibodies were added followed by incubation for 2 h at 37˚C. Subsequently, horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (1:2,000 dilution, A0208, Beyotime) was added and incubated for 2 h at 25˚C. The bound antibodies were detected using the ECL Plus Western Blotting Detection system (GE Healthcare). Actin was used as an internal control.
Cell immunofluorescence staining
NSCLC cells were stained using the standard immunofluorescence (IF) protocol. Briefly, NSCLC cells were pre-fixed with 4% paraformaldehyde and permeabilized with PBS containing 0.2% Triton X-100 and 5% BSA, and the cells were stained with direct anti-FOXF1 (ab23194, 1:200 dilution), anti-ALDH1 (ab52492, 1:200 dilution) and anti-OCT4 (ab18976, 1:200 dilution) antibodies (all from Abcam) overnight at 4˚C. The cells were then washed with PBS 3 times, and an appropriate secondary antibody (1:400 dilution, goat anti-rabbit-Texas Red, ab6719, Abcam) was added followed by incubation for 1 h at 25˚C. Finally, the cells were stained with DAPI and images were visualized using a fluorescence microscope (Leica).
Xenograft tumors in nude mice
For the xenograft model assay, 1x107 of A549/FOXF1-plasmid and A549/control-plasmid cells were subcutaneously injected into 4-week-old BALB/c athymic nude mice, respectively [8 mice (weight, 19.0, 18.0, 18.2, 18.5, 17.0, 16.3, 19.3 and 20.3 g), temperature, 18-22˚C; humidity, 50-60%, free access to food and water]. Each cell line was injected subcutaneously into the armpit of the right forelimb, for a total of 8 injections (4 A549/FOXF1-plasmid cells and 4 A549/control-plasmid cells). The longest diameter 'L' and the shortest diameter 'W' of tumors were measured 5 times in 30 days. The tumor volume was calculated using the following formula: Tumor volume (mm3) = π/6 xLxWxW. After 30 days, all experimental mice were sacrificed simultaneously and tumor weights were measured.
Animal experiments were approved by the Animal Ethics Committee of Guangzhou Medical University (2018-114). The experimental mice were anesthetized with an intraperitoneal injection of 0.7% pentobarbital sodium (70 mg/kg). The experimental mice were euthanized with carbon dioxide (flow rate, 20% of the chamber volume per minute) and subsequently also subjected to cervical dislocation.
Cell cytotoxicity assays
NSCLC cells were incubated with various concentrations of cisplatin (for the A549 and H1299 cells: 0, 0.5, 1, 1.5, 2, 2.5, 3, 3.5 and 4 µmol/l; for the A549/DDP cells: 0, 1, 2, 3, 4, 5, 6, 7 and 8 µmol/l). After 48 h, cisplatin-induced cytotoxicity was determined using the CCK8 kit (Beyotime) and represented as IC50 (µmol/l).
Colony-formation assay
NSCLC cells were treated with cisplatin (2 µmol/l) for 24 h, and the cells were then plated in 6-well plates (1,000 cells per well) and allowed to form colonies over 7 days. Cells were stained with Giemsa (Sigma-Aldrich, cells were pre-fixed with 4% paraformaldehyde and incubated for 15 min at 25˚C) and counted using ImageJ software (NIH, 1.52r).
Cell apoptosis assay
NSCLC cells were treated with cisplatin (2 µmol/l) for 24 h. Cell apoptosis was then determined using the Annexin V-FITC Apoptosis Detection kit (Beyotime) and by flow cytometry (Guava easyCyte HT, Millipore).
Sphere formation assay
NSCLC cells were plated in DMEM F12 serum-free medium reconstituted with 20 ng/ml of epidermal growth factor (EGF), 20 ng/ml of basic fibroblast growth factor (bFGF), 2% B27 and 1% methylcellulose (all from Sigma-Aldrich) (5,000 cells per well in a 6-well plate). After 4-7 days, microsphere-like structures were visible, and images of the microspheres were captured using a microscope (Leica).
Statistical analysis
All values are expressed as the means ± standard deviation (SD) from at least 3 separate experiments. The Student's unpaired t-test, ANOVA and receiver operating characteristic (ROC) curves were performed using SPSS 21.0 statistical software (IBM). Multiple comparisons between the groups was performed using the Bonferroni method. A two-tailed P-value was used in all analyses, and differences were considered statistically significant if the P-value was <0.05 (P<0.05).
Results
FOXF1 is epigenetically activated in cisplatin-resistant A549/DDP cells
Firstly, a DNA methylation microarray was used (Illumina Infinium 450K BeadChip) to detect the DNA methylation profiles (β-values) of the cisplatin-resistant A549/DDP cells and cisplatin-sensitive A549 cells. The results revealed that the 32 probes of FOXF1 were defined as 1,500 bp upstream region 1 of the transcription start site (TSS1500 region 1, 10 probes), 1,500 bp upstream region 2 of the transcription start site (TSS1500 region 2, 6 probes), First Exon (1stExon, 5 probes), gene body (3 probes) and 3′ untranslated region (3′UTR, 8 probes), and the differential region (fold change <0.5, P<0.01) were mapped to the TSS1500 region 1 of the FOXF1 gene and the locations of these 10 probes concentrated on a CpG island (Fig. 1A and B, and Table SIII). Moreover, the methylation status of the FOXF1 CpG island was determined in NSCLC epithelial cell lines (A549, A549/DDP and H1299) and a normal lung epithelial cell line (16HBE) using pyrosequencing analysis. It was confirmed that the DNA methylation levels of the FOXF1 CpG island were decreased in the A549/DDP cells compared with the A549, H1299 and 16HBE cells (Figs. 1A and C and S1, and Table SIV). Using western blot analysis and RT-qPCR, the mRNA and protein expression of levels FOXF1 were found to be higher in the A549/DDP compared with the A549, H1299 and 16HBE cells (Fig. 1D). According to the observation that the hypomethylation of the CpG island of FOXF1 facilitated the expression of FOXF1, it was also found that demethylation using the DNA methyltransferase inhibitor, 5-Aza-dC, promoted FOXF1 expression in the A549, 16HBE and H1299 cells (Fig. 1E). Therefore, these results indicate that cisplatin-induced DNA hypomethylation of FOXF1 can epigenetically activate the expression of FOXF1.
Figure 1.

Discovery of differential methylation region around FOXF1 gene upstream regulatory region in cisplatin-resistant A549/DDP cells compared with cisplatin-sensitive A549 cells. (A) The location of Illumina Infinium HumanMethylation 450k BeadChip FOXF1 probes and pyrosequencing primers. (B) Average β-values of probes mapping to different regions of the FOXF1 gene in A549/DDP cells and A549 cells. (C) Statistical analysis of the percentage of methylated CpGs in the FOXF1 gene. (D) FOXF1 protein levels detected by western blot analysis (upper panel) and FOXF1 mRNA levels detected by RT-qPCR (lower panel) in 1 normal lung cell lines (16HBE cells) and 3 NSCLC cell lines (A549, A549/DDP and H1299 cells). (E) FOXF1 protein levels detected by western blot analysis (upper panel) and FOXF1 mRNA levels detected by RT-qPCR (lower panel) in 16HBE, A549, A549/DDP and H1299 cells following treatment with the demethylating agent, 5-Aza-dC. n=3, *P<0.05 [(B) A549 compared to A549/DDP; (C) A549/DDP compared to 16HBE, A549 and H1299; (D) A549/DDP compared to 16HBE, A549 and H1299; (E) 5-Aza-dC compared to Control]. 5-Aza-dC, 5-aza-2′-deoxycytidine; FOXF1, forkhead box F1; NSCLC, non-small cell lung cancer.
FOXF1 regulates cisplatin resistance and is associated with platinum-based chemotherapeutic resistance in NSCLC
In order to investigate the regulatory functions of FOXF1 in cisplatin resistance, A549 and H1299 cells were established with the stable overexpression of FOXF1 (FOXF1 plasmids) and A549/DDP cells were established with the stable knockdown of FOXF1 (FOXF1 shRNAs) (Fig. 2A). In A549 and H1299 cells, the overexpression of FOXF1 protected the A549 and H1299 cells from cisplatin (IC50 increased from 0.51 to 1.33 µmol/l for the A549 cells and from 0.43 to 1.15 µmol/l for the H1299 cells); in the A549/DDP cells, the knockdown of FOXF1 restored sensitivity to cisplatin (IC50 decreased from 4.23 to 2.77 µmol/l) (Fig. 2B). The results of the colony-formation assay demonstrated that the overexpression of FOXF1 increased the proliferation of the A549 and H1299 cells treated with cisplatin, and the knockdown of FOXF1 decreased the proliferation of the A549/DDP cells treated with cisplatin (Fig. 2C). Moreover, the overexpression of FOXF1 decreased the cisplatin-induced apoptosis of A549 and H1299 cells, and the knockdown of FOXF1 increased the cisplatin-induced apoptosis of A549/DDP cells (Fig. 2D). In addition, the overexpression of FOXF1 decreased the cisplatin-induced apoptosis of 16HBE cells (Fig. S2). Thus, these findings suggest that FOXF1 plays important roles in the regulation of cisplatin resistance in NSCLC by promoting cell proliferation and inhibiting cell apoptosis.
Figure 2.


FOXF1 regulates cisplatin resistance by promoting cell proliferation and inhibiting cell apoptosis. (A) FOXF1 expression levels were increased in A549 and H1299 cells following transfection of FOXF1 plasmids and were decreased in A549/DDP cells following transfection of FOXF1 shRNAs. (B) Overexpression of FOXF1 increased resistance to cisplatin in A549 and H1299 cells (DDP treatment), and the knockdown of FOXF1 decreased resistance to cisplatin in A549/DDP cells (DDP treatment). (C) Overexpression of FOXF1 increased the colony-formation capability of A549 and H1299 cells (DDP treatment), and the knockdown of FOXF1 decreased the colony-formation capability in A549/DDP cells (DDP treatment) (clone formation rate = clones/inoculated amount of cells ×100%). (D) Overexpression of FOXF1 decreased the cisplatin-induced apoptosis of A549 and H1299 cells (DDP treatment), and the knockdown of FOXF1 increased the cisplatin-induced apoptosis of A549/DDP cells (DDP treatment). The late apoptotic cells are shown in the bar graph (n=3); *P<0.05 (FOXF1 plasmids compared to control plasmids, FOXF1 shRNAs compared to control shRNAs). FOXF1, forkhead box F1.
FOXF1 has been found to be associated with cisplatin resistance in NSCLC cells; therefore, the authors wished to determine whether the expression levels of FOXF1 may be associated with the outcome of chemotherapy in patients with NSCLC. A total of 35 platinum-based chemotherapy-resistant NSCLC tissues (NR tissues) and 35 platinum-based chemotherapy-sensitive NSCLC tissues (R tissues) were collected, and the expression levels of FOXF1 were measured in these samples using RT-qPCR. The results revealed that the expression levels of FOXF1 were significantly higher in the NR tissues compared with the R tissues (Fig. 3A). To determine the potential of FOXF1 in predicting the clinical outcome of platinum-based chemotherapy, ROC analysis of the expression levels of FOXF1 was performed and the area under the curve (AUC) of the ROC was calculated to assess the sensitivity and specificity of the prediction. The AUC value of the FOXF1 expression level was 0.66 (P<0.05) (Fig. 3B). On the whole, these findings suggested that FOXF1 was significantly associated with platinum-based chemotherapeutic resistance in patients with NSCLC and may thus be used as a potential biomarker for predicting the clinical outcome of platinum-based chemotherapy in NSCLC.
Figure 3.
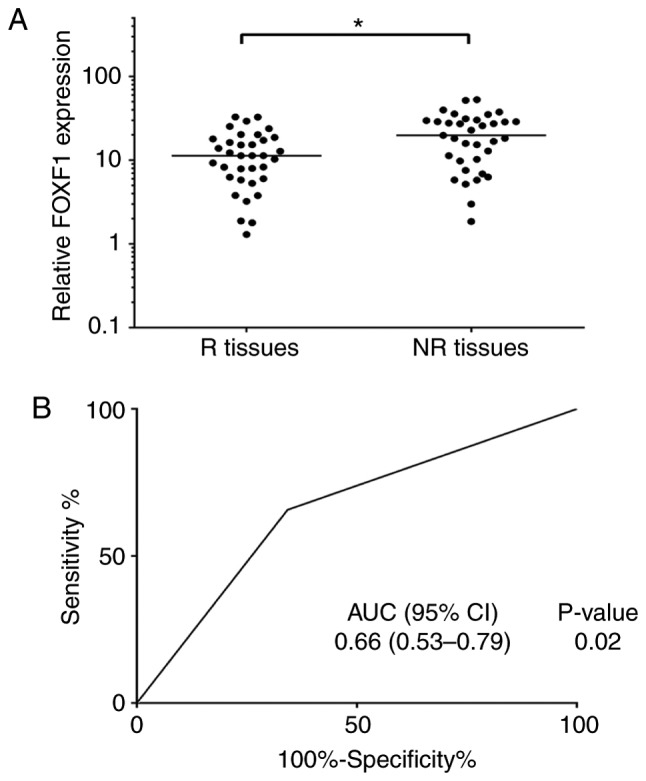
Expression levels of FOXF1 are associated with platinum-based chemotherapy response. (A) Scatter plots of expression of FOXF1 in responders and non-responders. Relative FOXF1 expression represents the ratio of FOXF1 expression in cancer tissue to FOXF1 expression in adjacent tissue [R, responder (response or partial response); NR, non-responder (stable or progressive disease)]. (B) ROC analyses assessing the association of FOXF1 expression level and platinum-based chemotherapy response (NR or R), where FOXF1 expression level was dichotomized and its categories represented by the score of 0 or 1 as follows: Score 1 (high risk) = FOXF1 level ≥ median; score 0 (low risk) = FOXF1 level < median. FOXF1, forkhead box F1.
FOXF1 induces the development of cancer stem cell properties in NSCLC cells
The stem cell markers, ALDH1 and OCT4, have been successfully used to isolate cancer stem cells from NSCLC cell lines and tissues, and it is widely accepted that NSCLC cancer stem cells are contained exclusively in the high ALDH1 and OCT4 expression cell compartment (14,15). Therefore, this study investigated the influence of the overex-pression and knockdown of FOXF1 on the expression of the stem cell markers, ALDH1 and OCT4. Using western blot analysis and RT-qPCR, ALDH1 and OCT4 were found to be highly expressed in the A549/DDP cells compared with the A549 and H1299 cells. The overexpression of FOXF1 resulted in an increase in ALDH1 and OCT4 expression in the A549 and H1299 cells, and the knockdown of FOXF1 resulted in a decrease in ALDH1 and OCT4 expression in the A549/DDP cells (Fig. 4A). Moreover, the regulatory effects of FOXF1 on ALDH1 and OCT4 expression were confirmed by immunofluorescence staining (Fig. 4B), and it was also found that the overexpression of FOXF1 increased the expression of ALDH1 and OCT4 in the 16HBE cells (Fig. S3).
Figure 4.

FOXF1 promotes the expression of stem cell markers. (A) Protein and mRNA expression levels of ALDH1 and OCT4 were increased in A549 and H1299 cells following transfection with FOXF1 plasmids and decreased in A549/DDP cells following transfection with FOXF1 shRNAs. (B) Immunofluorescence detection of ALDH1 and OCT4 in A549 and H1299 cells following transfection with FOXF1 plasmids and A549/DDP cells following transfection with FOXF1 shRNAs. (n=3); *P<0.05 (FOXF1 plasmids compared to control plasmids, FOXF1 shRNAs compared to control shRNAs). FOXF1, forkhead box F1; ALDH1, aldehyde dehydrogenase 1; OCT4, octamer-binding transcription factor 4.
The effect of FOXF1 on the self-renewal capacity of the cells was then examined using sphere formation assay. The results revealed that the number and size of spheres formed by the A549/DDP cells were greater than those formed by the A549 and H1299 cells. The overexpression of FOXF1 resulted in an increase in the number and size of spheres formed by the A549 and H1299 cells, and the knockdown of FOXF1 resulted in a decrease in the number and size of spheres formed by the A549/DDP cells (Fig. 5A). These data suggested that FOXF1 promoted the self-renewal ability of the NSCLC cells. Moreover, according to the results of the xenograft model assay, the overexpression of FOXF1 resulted in a significant increase in tumor volumes and weights compared with control-plasmid, indicating that FOXF1 promoted tumorigenesis (Fig. 5B). Taken together, these data demonstrate that the cisplatin-induced upregulation of FOXF1 expression is generally a critical determinant of stem cell-like properties, which results in cisplatin resistance in NSCLC cells.
Figure 5.

FOXF1 promotes the self-renewal capability and tumorigenesis capacity. (A) Overexpression of FOXF1 increased the self-renewal capability of A549 and H1299 cells, and knockdown of FOXF1 decreased self-renewal capability in A549/DDP cells. (n=3); *P<0.05 (FOXF1 plasmids compared to control plasmids, FOXF1 shRNAs compared to control shRNAs) (B) Overexpression of FOXF1 in A549 cells increased the volumes and weights of xenograft tumors. (n=4); *P<0.05 (FOXF1 plasmids compared to control plasmids). FOXF1, forkhead box F1.
Discussion
Resistance to chemotherapy is the main cause of chemotherapy failure and disease recurrence, and is a major obstacle in the treatment of a number of types of cancer, including NSCLC (16,17). The mechanisms responsible for resistance to chemotherapy are complex, and are related to several factors, such as drug uptake, cell cycle, DNA repair and others; the molecular mechanisms responsible for chemotherapeutic resistance have not yet been fully elucidated (18,19). Recently, it was found that the abnormal DNA methylation of the genome plays an important role in the chemotherapy resistance of cancers, including NSCLC (20,21). It was found that platinum-based chemotherapy can influence DNA methylation modification in cancer (22). In the present study, it was found that cisplatin influenced the genomic DNA methylation levels in A549/DDP cells, and it was confirmed that the DNA methylation levels of the FOXF1 gene upstream regulatory region were significantly decreased. FOXF1 is located in 16q24.1, and the upstream regulatory region of FOXF1 has a CpG island which can inhibit the transcription of FOXF1 by DNA methylation modification (23,24). It was also found that the hypomethylation of FOXF1 can increase the mRNA and protein expression levels of FOXF1 in NSCLC cells. FOXF1 has been found to be abnormally expressed in cancers, which is closely related to tumor occurrence and progression (25-27). Gialmanidis et al found that FOXF1 expression was elevated in NSCLC tissue samples and was associated with lymph node metastasis (28). Saito et al found that FOXF1 was involved in the regulation of the tumor-promoting properties of lung cancer-associated fibroblasts (29). In this study, it was found that the overexpression of FOXF1 promoted cisplatin resistance in NSCLC by increasing cell proliferation and inhibiting cell apoptosis. In addition, the expression levels of FOXF1 in were determined in NSCLC samples, and it was found that the expression levels of FOXF1 were higher in platinum-based chemotherapy-resistant NSCLC tissues compared with platinum-based chemotherapy-sensitive NSCLC tissues; the expression levels of FOXF1 were associated with the platinum-based chemotherapeutic response in patients with NSCLC. Therefore, these results indicate that cisplatin can promote the expression of FOXF1, which results in cisplatin resistance, by the hypomethylation of the FOXF1 gene upstream regulatory region.
There is strong evidence to indicate that abnormalities in DNA methylation can influence the dedifferentiation of cancer cells, which promotes stem cell properties in cancer cells, which then form cancer stem cells (30,31). In the course of cancer treatment, cancer stem cells have the characteristics of dormancy, strong DNA repair and self-renewal. As a result, they are 'difficult to kill' and become the source of chemotherapy resistance and cancer recurrence (32-35). Cancer stem cells have also been found in NSCLC and are closely related to chemotherapeutic resistance in NSCLC (36). Lopez-Ayllon et al found that cancer stem cells isolated from NSCLC cells were resistant to cisplatin (37). Bora-Singhal et al found that Gli1 promoted the self-renewal of NSCLC cancer stem cells by regulating Sox2, and promoted drug resistance (38). FOXF1 is one of the important transcription factors involved in the regulation of lung tissue differentiation and development, and is mainly involved in the regulation of the correct development of airway smooth muscle and cartilage (39). Wei et al found that abnormalities in FOXF1 can influence the dedifferentiation of cancer cells which promotes stem cell properties in cancer cells, which then form cancer stem cells (40). In this study, it was found that the overexpression of FOXF1 increased the expression of the stem cell markers, ALDH1 and OCT4, and promoted the self-renewal capacity and tumorigenesis capacity of NSCLC cells, suggesting that the abnormal high expression of FOXF1 induced by cisplatin can promote the stem cell-like properties of NSCLC cells. Thus, the ability of FOXF1 to initiate cisplatin resistance is dependent in turn on their ability to promote the cancer stem cell properties of NSCLC cells. Moreover, it was also found that the overexpression of FOXF1 increased the expression of stem cell markers and promoted cisplatin resistance in 16HBE normal human bronchial epithelial cells. As FOXF1 is involved in the regulation of normal lung tissue differentiation, the abnormal expression of FOXF1 can also promote the stem cell-like properties of normal lung epithelial cells and may be related to the occurrence of NSCLC.
According to the results of this study, we suggest that cisplatin can promote the transcription of FOXF1 by hypo-methylation of the upstream regulatory region of FOXF1 gene, and FOXF1 further promotes cancer stem cell properties which ultimately result in cisplatin resistance in NSCLC. Furthermore, the understanding of the molecular mechanisms of cisplatin resistance regulated by FOXF1 may also provide biomarkers and therapeutic targets for NSCLC chemotherapy.
Supplementary Data
Acknowledgements
Not applicable
Funding
This study was supported by grants from the National Natural Science Foundation of China (81501969, 81572258), Guangdong Basic and Applied Basic Research Foundation (2019A1515011092, 2019A1515010026) and Guangzhou key medical discipline construction project fund.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
JZ and JY conceived and designed the experiments. JZ, JY, and SZ performed the experiments and assembled the data. XX, WF, LD and ZJ obtained the tumors and tissues with clinical information where available. JY and BD performed the statistical analysis. JZ and JY analyzed the data and drafted the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
NSCLC tissues were collected from the Cancer Center of Guangzhou Medical University (Guangzhou, China) with written informed consent and permission from the Institutional Review Board. All patients provided written informed consent. The study protocol was approved by the ethics committee of Cancer Center of Guangzhou Medical University (approval no (2014) 100). Animal experiments were approved by the animal ethics committees of the Guangzhou Medical University (2018-114).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Lemjabbar-Alaoui H, Hassan OU, Yang YW, Buchanan P. Lung cancer: Biology and treatment options. Biochim Biophys Acta. 2015;1856:189–210. doi: 10.1016/j.bbcan.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 5.Adorno-Cruz V, Kibria G, Liu X, Doherty M, Junk DJ, Guan D, Hubert C, Venere M, Mulkearns-Hubert E, Sinyuk M, et al. Cancer stem cells: Targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015;75:924–929. doi: 10.1158/0008-5472.CAN-14-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–38. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 7.Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D, Huang S. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun. 2013;4:2467. doi: 10.1038/ncomms3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Y, Guo R, Wei J, Zhou Y, Ji W, Liu J, Zhi X, Zhang J. Effects of PI3K inhibitor NVP-BKM120 on overcoming drug resistance and eliminating cancer stem cells in human breast cancer cells. Cell Death Dis. 2015;6:e2020. doi: 10.1038/cddis.2015.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells-what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacDonagh L, Gray SG, Breen E, Cuffe S, Finn SP, O'Byrne KJ, Barr MP. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016;372:147–156. doi: 10.1016/j.canlet.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Bach DH, Long NP, Luu TT, Anh NH, Kwon SW, Lee SK. The dominant role of forkhead box proteins in cancer. Int J Mol Sci. 2018;19:E3279. doi: 10.3390/ijms19103279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laissue P. The forkhead-box family of transcription factors: Key molecular players in colorectal cancer pathogenesis. Mol Cancer. 2019;18:5. doi: 10.1186/s12943-019-0938-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 14.Sterlacci W, Savic S, Fiegl M, Obermann E, Tzankov A. Putative stem cell markers in non-small-cell lung cancer: A clini-copathologic characterization. J Thorac Oncol. 2014;9:41–49. doi: 10.1097/JTO.0000000000000021. [DOI] [PubMed] [Google Scholar]
- 15.Curtarelli RB, Gonçalves JM, Dos Santos LGP, Savi MG, Nör JE, Mezzomo LAM, Rodríguez Cordeiro MM. Expression of cancer stem cell biomarkers in human head and neck carcinomas: A systematic review. Stem Cell Rev. 2018;14:769–784. doi: 10.1007/s12015-018-9839-4. [DOI] [PubMed] [Google Scholar]
- 16.Kartal-Yandim M, Adan-Gokbulut A, Baran Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit Rev Biotechnol. 2016;36:716–726. doi: 10.3109/07388551.2015.1015957. [DOI] [PubMed] [Google Scholar]
- 17.Kunjachan S, Rychlik B, Storm G, Kiessling F, Lammers T. Multidrug resistance: Physiological principles and nanomedical solutions. Adv Drug Deliv Rev. 2013;65:1852–1865. doi: 10.1016/j.addr.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 19.Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist Updat. 2016;26:1–9. doi: 10.1016/j.drup.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Grasse S, Lienhard M, Frese S, Kerick M, Steinbach A, Grimm C, Hussong M, Rolff J, Becker M, Dreher F, et al. Epigenomic profiling of non-small cell lung cancer xenografts uncover LRP12 DNA methylation as predictive biomarker for carboplatin resistance. Genome Med. 2018;10:55. doi: 10.1186/s13073-018-0562-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stone A, Zotenko E, Locke WJ, Korbie D, Millar EK, Pidsley R, Stirzaker C, Graham P, Trau M, Musgrove EA, et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat Commun. 2015;6:7758. doi: 10.1038/ncomms8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flanagan JM, Wilson A, Koo C, Masrour N, Gallon J, Loomis E, Flower K, Wilhelm-Benartzi C, Hergovich A, Cunnea P, et al. Platinum-based chemotherapy induces methylation changes in blood DNA associated with overall survival in patients with ovarian cancer. Clin Cancer Res. 2017;23:2213–2222. doi: 10.1158/1078-0432.CCR-16-1754. [DOI] [PubMed] [Google Scholar]
- 23.Dharmadhikari AV, Szafranski P, Kalinichenko VV, Stankiewicz P. Genomic and epigenetic complexity of the FOXF1 locus in 16q24.1: Implications for development and disease. Curr Genomics. 2015;16:107–116. doi: 10.2174/1389202916666150122223252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo PK, Lee JS, Liang X, Han L, Mori T, Fackler MJ, Sadik H, Argani P, Pandita TK, Sukumar S. Epigenetic inactivation of the potential tumor suppressor gene FOXF1 in breast cancer. Cancer Res. 2010;70:6047–6058. doi: 10.1158/0008-5472.CAN-10-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fulford L, Milewski D, Ustiyan V, Ravishankar N, Cai Y, Le T, Masineni S, Kasper S, Aronow B, Kalinichenko VV, Kalin TV. The transcription factor FOXF1 promotes prostate cancer by stimulating the mitogen-activated protein kinase ERK5. Sci Signal. 2016;9:ra48. doi: 10.1126/scisignal.aad5582. [DOI] [PubMed] [Google Scholar]
- 26.Milewski D, Pradhan A, Wang X, Cai Y, Le T, Turpin B, Kalinichenko VV, Kalin TV. FoxF1 and FoxF2 transcription factors synergistically promote rhabdomyosarcoma carcinogenesis by repressing transcription of p21Cip1 CDK inhibitor. Oncogene. 2017;36:850–862. doi: 10.1038/onc.2016.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran L, Chen Y, Sher J, Wong EWP, Murphy D, Zhang JQ, Li D, Deniz K, Sirota I, Cao Z, et al. FOXF1 defines the core-regulatory circuitry in gastrointestinal stromal tumor. Cancer Discov. 2018;8:234–251. doi: 10.1158/2159-8290.CD-17-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gialmanidis IP, Bravou V, Petrou I, Kourea H, Mathioudakis A, Lilis I, Papadaki H. Expression of Bmi1, FoxF1, Nanog, and γ-catenin in relation to hedgehog signaling pathway in human non-small-cell lung cancer. Lung. 2013;191:511–521. doi: 10.1007/s00408-013-9490-4. [DOI] [PubMed] [Google Scholar]
- 29.Saito RA, Micke P, Paulsson J, Augsten M, Peña C, Jönsson P, Botling J, Edlund K, Johansson L, Carlsson P, et al. Forkhead box F1 regulates tumor-promoting properties of cancer-associated fibroblasts in lung cancer. Cancer Res. 2010;70:2644–2654. doi: 10.1158/0008-5472.CAN-09-3644. [DOI] [PubMed] [Google Scholar]
- 30.Toh TB, Lim JJ, Chow EK. Epigenetics in cancer stem cells. Mol Cancer. 2017;16:29. doi: 10.1186/s12943-017-0596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wickström M, Dyberg C, Milosevic J, Einvik C, Calero R, Sveinbjörnsson B, Sandén E, Darabi A, Siesjö P, Kool M, et al. Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemore-sistance. Nat Commun. 2015;6:8904. doi: 10.1038/ncomms9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carnero A, Garcia-Mayea Y, Mir C, Lorente J, Rubio IT, LLeonart ME. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat Rev. 2016;49:25–36. doi: 10.1016/j.ctrv.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 34.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: An evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 36.Suresh R, Ali S, Ahmad A, Philip PA, Sarkar FH. The role of cancer stem cells in recurrent and drug-resistant lung cancer. Adv Exp Med Biol. 2016;890:57–74. doi: 10.1007/978-3-319-24932-2_4. [DOI] [PubMed] [Google Scholar]
- 37.Lopez-Ayllon BD, Moncho-Amor V, Abarrategi A, Ibañez de Cáceres I, Castro-Carpeño J, Belda-Iniesta C, Perona R, Sastre L. Cancer stem cells and cisplatin-resistant cells isolated from non-small-lung cancer cell lines constitute related cell populations. Cancer Med. 2014;3:1099–1111. doi: 10.1002/cam4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bora-Singhal N, Perumal D, Nguyen J, Chellappan S. Gli1-mediated regulation of Sox2 facilitates self-renewal of stem-like cells and confers resistance to EGFR inhibitors in non-small cell lung cancer. Neoplasia. 2015;17:538–551. doi: 10.1016/j.neo.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahlapuu M, Enerbäck S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001;128:2397–2406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- 40.Wei HJ, Nickoloff JA, Chen WH, Liu HY, Lo WC, Chang YT, Yang PC, Wu CW, Williams DF, Gelovani JG, Deng WP. FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget. 2014;5:9514–9529. doi: 10.18632/oncotarget.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.