

Abstract
Une lymphopénie est définie par un nombre de lymphocytes circulants inférieur à 1500/mm3 chez l’adulte et 4500/mm3 chez l’enfant avant huit mois. La lymphopénie peut être globale ou sélective, affectant une population lymphocytaire particulière. Le diagnostic étiologique doit tenir compte de l’âge, du contexte, des manifestations clinicobiologiques associées et des thérapeutiques reçues. Les lymphopénies de l’adulte peuvent être liées schématiquement à : (1) une insuffisance de production (carence en zinc, corticothérapie, déficits immunitaires primitifs…), (2) un excès de catabolisme (radiothérapie, chimiothérapie, traitements immunosuppresseurs, infection par le VIH ou lupus systémique, etc.), (3) une modification de la répartition des lymphocytes (infections virales, choc septique, brûlures étendues, hypersplénisme, granulomatoses, etc.), (4) les étiologies multifactorielles ou non identifiées (insuffisance rénale chronique, certaines hémopathies lymphoïdes, tumeur solide, causes ethniques, etc.). Chez l’enfant, à ces étiologies s’ajoutent d’autres déficits immunitaires primitifs d’expression sévère (défaut des précurseurs thymiques, déficit cytokinique, défaut de synthèse des récepteurs des lymphocytes B et T et défaut de la transduction du signal ou des interactions cellulaires). La lymphopénie CD4+ idiopathique de l’adulte est un diagnostic d’élimination. Cette affection rare se définit par une lymphopénie T CD4+ inférieure ou égale à 300/mm3 ou inférieure ou égale à 20 % des lymphocytes totaux, persistante en l’absence de diagnostic alternatif. Elle peut être asymptomatique, s’associer à des infections à germes opportunistes, ou se compliquer de symptômes auto-immuns (en particulier cytopénies) ainsi que de néoplasies. Le traitement, calqué sur la prise en charge des patients infectés par le VIH, peut nécessiter le recours à une immunothérapie spécifique dont le bénéfice clinique reste à évaluer.
Mots clés: Lymphopénie, Lymphopénie CD4+ idiopathique, Sarcoïdose, Lupus systémique, Infection virale
Abstract
Lymphocytopenia is defined by a lymphocyte count less than 1500/mm3 in adults and less than 4500/mm3 in children before the age of 8 months. Lymphocytopenia can be global or selectively affect a peculiar lymphocyte subpopulation. The patient's age, the context as well as the associated clinical manifestations and treatment prescribed must be taken into account in order to identify the etiology of lymphocytopenia. In adults, lymphocytopenia can be caused by: (1) insufficient thymic output (primary immune deficiencies, corticosteroid treatment, zinc deficiency, etc.), (2) increased lymphocyte catabolism (radiotherapy, chemotherapy, immunosuppressant, HIV infection, systemic lupus, etc.), (3) modified lymphocyte distribution (viral infections, septic shock, extensive burns, splenomegaly, granulomatosis, etc.), (4) multifactorial or unknown etiology (end-stage renal disease, lymphoid malignancies, solid tumor, ethnicity, etc.). In children, in addition to these etiologies, other immune deficiencies may be responsible for severe lymphocytopenia (thymocytes apoptosis, cytokine deficiencies, altered B-cell and T-cell receptor synthesis, signal transduction and cellular interactions deficiencies). Idiopathic CD4+ lymphocytopenia is a rare disorder. It is defined by a persisting lymphocyte CD4+ count less or equal to 300/mm3 or less or equal to 20% of total lymphocytes in the absence of alternative diagnosis. Clinical symptoms can be absent or include opportunistic infections, auto-immune manifestations, lymphoma or solid tumors. Treatment is similar to that of HIV-infected patients and sometimes relies on specific immunotherapy even though clinical benefit has not been evaluated.
Keywords: Lymphocytopenia, Idiopathic CD4+ lymphocytopenia, Sarcoidosis, Systemic lupus, Viral infection
1. Introduction
Les lymphocytes sont des cellules immunitaires dont on distingue trois principaux types : les lymphocytes B (LB) et les lymphocytes T (LT), acteurs de l’immunité adaptative, et les lymphocytes « natural killer » (NK) qui n’expriment pas de récepteur à l’antigène. Les LB sont générés dans divers tissus fœtaux et après la naissance dans la moelle osseuse, et les LT dans le thymus. Ces cellules qui ont des rôles très variés avec une spécialisation importante sont habituellement identifiées et classées selon l’expression de molécules de surface appelées cluster de différenciation (CD) et selon la nature du récepteur à l’antigène : récepteur B (B-cell receptor, BcR) pour les LB et récepteur T (T-cell receptor, TcR) αβ ou γδ pour les LT. La répartition sanguine des différentes populations lymphocytaires est détaillée dans le Tableau 1 . Produites en grand nombre (108 à 109 cellules par jour), ces cellules vont migrer vers les organes lymphoïdes secondaires (rate, ganglions, amygdales) et les autres tissus lymphoïdes périphériques, dont les tissus lymphoïdes associés aux muqueuses. Cette migration fait intervenir certaines chimiokines ainsi que différents récepteurs et molécules d’adhésion.
Tableau 1.
Répartition des sous-populations lymphocytaires chez l’adulte sain.
| Cellules | Valeur en % |
|---|---|
| Lymphocyte T (CD3+) | 75 ± 8 |
| Selon le récepteur | |
| Lymphocyte T αβ | 69 ± 6 |
| Lymphocyte T γδ | 5 ± 2 |
| Selon le co-récepteur | |
| Lymphocytes T CD3+CD4+ | 47 ± 8 |
| Lymphocytes T CD3+CD8+ | 26 ± 5 |
| Lymphocyte T CD3+CD4−CD8− | < 1 |
| Lymphocytes B (CD19+, CD20+) | 14 ± 2 |
| Lymphocytes NK (CD3−CD16+CD56+) | 12 ± 4 |
Chez l’adulte, le taux de lymphocytes dans le sang périphérique est compris entre 1500 et 4000/mm3, ce chiffre étant la résultante de la production centrale des lymphocytes, de leur migration tissulaire et de leur durée de vie. L’altération de l’un ou plusieurs de ces mécanismes peut être responsable d’une lymphopénie, situation fréquemment rencontrée en pratique clinique. La lymphopénie se définit par un nombre de lymphocytes circulants inférieur à 1500/mm3 chez l’adulte et 4500/mm3 chez l’enfant avant huit mois. Elle peut être globale ou porter sélectivement sur une population ou sous-population lymphocytaire et être alors sans retentissement sur le nombre de lymphocytes totaux. Peu de revues générales ont été consacrées à la démarche diagnostique à suivre dans ce contexte. Nous abordons ici dans un premier temps les déficits immunitaires primitifs (DIP) responsables d’une lymphopénie avant d’aborder les déficits immunitaires secondaires et la lymphopénie CD4+ idiopathique. Enfin nous proposons une démarche diagnostique pour l’exploration d’une lymphopénie chronique.
2. Les déficits immunitaires primitifs
2.1. Les syndromes d’immunodépression combinée sévère (DICS)
Les syndromes d’immunodépression combinée sévère (DICS) sont souvent révélés dès les premiers jours de vie par des infections bactériennes, virales et fongiques inhabituelles par le germe en cause, la localisation ou le caractère récidivant. Ils peuvent être responsables de lymphopénie. Souvent classés selon les populations cellulaires affectées (LT et/ou LB et/ou NK), ces déficits sont principalement diagnostiqués chez l’enfant mais des formes plus frustres peuvent être découvertes jusqu’à l’âge adulte. L’ensemble de ces déficits est rassemblé dans le Tableau 2 .
Tableau 2.
Causes innées des déficits immunitaires responsable d’une lymphopénie.
| Développement thymique | Syndrome de Di-Georges |
| FOXN-1 | |
| Précurseurs lymphoïdes | Déficit en ADA |
| Déficit en PNP | |
| Déficit en AK2 (dysgénésie réticulaire) | |
| Cytokines | Chaîne γ commune |
| Jak3 | |
| Chaîne α du récepteur à l’IL-7 | |
| Synthèse des TcR et BcR | RAG1/2 |
| NHEJ, Artemis, DNA-PKcs, cernunnos/XLF, DNA ligase IV | |
| Transduction du signal | CD3δ ou ɛ ou ζ |
| CD45 | |
| ZAP-70 | |
| STIM-1 et ORAI-1 | |
| Interactions cellulaires | TAP-1/2 |
| CMH classe II | |
| CD40-ligand ou CD40 | |
ADA : adénosine déaminase ; AK2 : adénylate kinase 2 ; BcR : récepteur des cellules B ; CD : cluster de différenciation ; CMH : complexe majeur d’histocompatibilité ; DNA-PKcs : DNA-Protein kinase catalytic subunit ; FOXN1 : Forkhead box protein N1 ; Jak : Janus associated kinase ; IL : interleukine ; NHEJ : Jonction terminale non homologue ; ORAI-1 : Calcium release-activated calcium channel protein 1 ; PNP : purine nucléoside phosphorylase ; RAG : gènes associés à la recombinaison ; STIM1 : molécule d’interaction stromale 1 ; TAP : transporteurs associés à l’apprêtement de l’antigène ; TcR : récepteur des cellules T ; ZAP : protéine associée à la chaîne zêta.
2.1.1. Les anomalies de développement thymique
Plusieurs syndromes peuvent s’accompagner de lymphopénie. Ainsi, le syndrome de Di-George est caractérisé par une anomalie de développement des troisième et quatrième arcs branchiaux conduisant à des anomalies cardiaques, une quasi-absence de développement du thymus ou des parathyroïdes et une dysmorphie faciale discrète mais caractéristique. Au cours de cette affection, on constate une lymphopénie profonde portant sur les LT avec parfois expansion des LB.
Par ailleurs, l’absence de développement thymique, des troubles marqués des phanères et l’absence de réponse proliférative lymphocytaire se voient au cours des mutations de Forkhead box protein N1 (FOXN1) [1].
2.1.2. Les anomalies du métabolisme des précurseurs lymphoïdes
Deux anomalies du métabolisme des purines ont été mises en évidence conduisant à l’accumulation de déchets toxiques : le déficit en adénosine déaminase (ADA), responsable de 15 % des DICS, et le déficit en purine nucléoside phosphorylase (PNP) plus rare. Le tableau du déficit en ADA dépend de l’activité enzymatique résiduelle : il peut être paucisymptomatique et de révélation tardive mais, en cas de déficit sévère, il est responsable d’un DICS, comme dans le déficit en PNP. Aux symptômes liés à l’immunodépression, s’ajoutent des signes neurologiques et des stigmates d’auto-immunité (déficit en PNP) ou des anomalies squelettiques pseudorachitiques (déficit en ADA) [2]. La lymphopénie intéresse les LT et les LB dans le déficit en ADA, alors qu’elle épargne les LB dans le déficit en PNP.
La dysgénésie réticulaire est une cause rare de DICS rapidement mortelle en l’absence de greffe de moelle, responsable de surdité sensorielle, d’un défaut des lignées myéloïdes et lymphoïdes et de développement thymique. La mutation du gène de l’adénylate kinase, enzyme mitochondriale, a été récemment identifiée [3], [4].
2.1.3. Les anomalies de la synthèse du TcR et du BcR
Ces déficits sont principalement liés à des anomalies de la recombinaison des segments variable (V), diversité (D) et jonction (J) codant pour les régions variables des immunoglobulines ou des chaînes V, D et J du TcR, étape indispensable à la genèse d’un répertoire diversifié des TcR et BcR. La défaillance de ce mécanisme commun entraîne un défaut de maturation des LB et des LT à l’origine d’une lymphopénie alors que les lymphocytes NK sont matures et fonctionnels. L’alymphocytose autosomique récessive est liée à des mutations non sens des gènes « recombination activating genes » (RAG) 1/2. D’autres anomalies de la recombinaison peuvent être liées à des mutations des enzymes « non-homologous end joining » (NHEJ), « DNA-Protein kinase catalytic subunit » (DNA-PKcs), « Artemis », « DNA ligase IV » (LIG4) ou « XRCC4-like factor » (XLF)/Cernunnos.
Les variants hypomorphes des déficits en RAG1/2 et en Artemis, certains déficits en LIG4 ou en ADA peuvent être à l’origine d’un syndrome d’Omenn. Celui-ci associe érythrodermie, alopécie, hépatosplénomégalie, polyadénopathie, manifestations autoimmunes, hyper-IgE et expansion oligloclonale des LT [5].
2.1.4. Les déficits de la réponse cytokinique
Pour établir une réponse immune, trois signaux différents sont nécessaires pour activer efficacement un lymphocyte :
-
•
la fixation de l’antigène par le TcR ou le BcR ;
-
•
un signal apporté par les molécules de co-stimulation ;
-
•
le troisième signal provenant des cytokines et des récepteurs.
Un déficit de ce troisième signal a été mis en évidence au cours de certains DICS : le déficit en chaîne γc, commune aux récepteurs à l’interleukine (IL)-2, -4, -7, -9, -15, -21 est à l’origine d’un DICS lié à l’X [6]. Exceptionnellement, il s’agit d’un déficit en chaîne α du récepteur à l’IL-7 [7] ou en Jak-3 impliquée dans la signalisation de nombreux récepteurs, dont des récepteurs aux cytokines [8].
2.1.5. Autres causes de DICS
Un défaut d’expression de l’une des chaînes δ, γ et ɛ constitutives du complexe CD3, peut être responsable de DICS [9]. Un déficit dans le motif kinasique du CD45 a également été décrit chez un nouveau-né [10]. Un déficit au niveau de « zeta-chain-associated protein kinase 70 » (ZAP-70) a été mis en évidence chez de nombreux patients qui présentaient un déficit sélectif en LT CD8+ avec des LT CD4+ anergiques [11]. Enfin, un défaut des flux calciques, nécessaires à l’activation lymphocytaire, peut être responsable de DICS. Ainsi, des mutations de « stromal interaction molecule 1 » (STIM1) [12] ou ORAI1 [13] ont été rapportées.
Un déficit de la présentation antigénique et de l’expression du CMH de classe I sur les LT peut être lié à une mutation du gène « transporter associated with antigen presentation » (TAP) [14]. Le tableau associe de manière variable infections, arthrite, vascularite cutanée ou des voies aériennes supérieures. La présentation clinique se rapproche parfois de la granulomatose avec polyangéite (GPA) (anciennement granulomatose de Wegener) et peut en constituer un diagnostic différentiel. En revanche, un déficit d’expression du CMH de classe II entraîne un défaut d’activation des LT avec une lymphopénie T CD4+ isolée [15].
Le syndrome de Wiskott-Aldrish, pathologie liée à un déficit en Wiskott-Aldrish Syndrome Protein (WASP) située sur le chromosome X, associe un purpura thrombopénique (microplaquettes), un eczéma, des manifestations auto-immunes et des infections répétées à pyogènes. La protéine WASP interagit avec l’actine et son déficit se traduit par une désorganisation du cytosquelette prédominant dans les lymphocytes et les plaquettes.
2.2. DIP avec atteinte humorale prédominante
Les déficits isolés en LB ne sont théoriquement pas responsables de lymphopénie du fait de la faible proportion des LB dans le sang circulant. Néanmoins, un déficit en LB (associé à une hypogammaglobulinémie) doit faire rechercher avant tout une agammaglobulinémie de Bruton, un syndrome de Good [16] ou un déficit immunitaire commun variable (DICV).
L’agammaglobulinémie liée à l’X est liée à un déficit de la Bruton tyrosine kinase (Btk). Cette mutation entraîne un défaut de maturation des LB expliquant l’absence de LB circulants et une altération du développement de tissus lymphoïdes secondaires. Les agammaglobulinémies autosomiques représentent moins de 20 % des cas et se caractérisent par un défaut d’expression ou de fonction du récepteur pré-B [17].
Le DICV est le plus fréquent des DIP symptomatiques de l’adulte. Il s’agit d’un syndrome hétérogène caractérisé par une hypogammaglobulinémie (ou un déficit portant sur au moins deux classes d’Ig) et une altération de la réponse anticorps. Au plan clinique s’associent de manière variable des infections, des manifestations auto-immunes, une granulomatose, des manifestations digestives (atrophie villositaire, maladie inflammatoire intestinale…) et une hyperplasie lymphoïde bénigne. Un lymphome peut compliquer l’évolution de ce syndrome.
Enfin, le syndrome de Good se définit par une hypogammaglobulinémie associée à un thymome, d’expression plus sévère que le DICV et de pronostic péjoratif. Une alymphocytose B est pratiquement constante et une lymphopénie T est retrouvée chez 15 % des patients, contrastant avec une infiltration de nombreux tissus par des LT [16].
3. Les déficits immunitaires secondaires
Les déficits immunitaires secondaires sont plus fréquents que les DIP et peuvent s’expliquer par trois grands types de mécanisme : un défaut de production, une modification de la répartition des lymphocytes et un excès de catabolisme.
3.1. Défaut de production
La plus importante en fréquence est la carence en zinc dont l’étiologie principale est la malnutrition. Cependant, une carence en zinc se retrouve également lors des pathologies rénales ou gastro-intestinales ou de l’intoxication alcoolique chronique et plus rarement du glucagonome. En plus de la lymphopénie, les formes les plus sévères se compliquent d’un défaut de réponse des lymphocytes aux mitogènes. Les autres anomalies mises en évidence sont une hypogammaglobulinémie, un déficit en protéines du complément et une réponse moindre à une stimulation antigénique [18]. Chez des souris subissant un régime carencé en zinc, le thymus semble être l’organe le plus sensible avec une apoptose élevée des précurseurs intrathymiques qui pourrait être la résultante de l’augmentation des glucocorticoïdes circulants alors que la lymphopoïèse et l’érythropoïèse médullaire semblent moins affectées [19]. Des carences en vitamine A, en vitamine C ou en vitamine B9 (folates) pourraient également participer à la lymphopénie ou au défaut de prolifération lymphoïde après stimulation.
3.2. Excès de catabolisme
Un certain nombre de causes est à l’origine d’un excès de catabolisme des lymphocytes, parmi lesquelles les traitements médicamenteux (corticothérapie, chimiothérapie, traitements immunosuppresseurs), la radiothérapie, les infections virales en particulier par le virus de l’immunodéficience humaine (VIH) et le LES.
Les médicaments peuvent induire une lymphopénie, soit par un effet toxique direct sur les lymphocytes : lymphopénie B spécifique au cours d’un traitement par rituximab, lymphopénie T secondaire à des anticorps anti-CD3 ou au sérum anti-lymphocytaire ou lymphopénie globale suite à un traitement par alemtuzumab (anti-CD52). Par ailleurs, certaines thérapeutiques entraînent une lymphopénie par des mécanismes toxiques non spécifiques. C’est le cas en particulier au cours des traitements par chimiothérapie ou immunosuppresseurs. Ainsi, la fludarabine, la cladribine et les autres analogues des purines ont un effet lymphopéniant prolongé intéressant principalement le compartiment LT-CD4+ qui peut persister plusieurs mois voire années après l’arrêt des traitements. La lymphopénie survenant cinq jours après une chimiothérapie pourrait être utile pour prédire la survenue d’une neutropénie [20].
L’infection par le VIH conduit à une lymphopénie se majorant progressivement liée à la destruction progressive des LT principalement de phénotype CD4+. De nombreux scenarii physiopathologiques ont été proposés. Celui qui domine actuellement fait état d’un cercle vicieux entre l’hyperactivation des LT résiduels et l’excès d’apoptose de ces derniers qui aggrave la lymphopénie. De nombreux autres virus sont responsables de lymphopénie (cytomégalovirus [CMV], virus de la rougeole, grippe, coronavirus, etc.).
Enfin, au cours du LES, on peut retrouver une lymphopénie qui en constitue un des critères diagnostiques. Les mécanismes sont encore mal compris mais il existe un excès d’apoptose ainsi que des auto-anticorps anti-lymphocytaires d’isotype IgG et IgM. La présence d’anticorps froids cytotoxiques dirigés contre les lymphocytes d’isotype IgM a été rapportée depuis longtemps et leur présence pourrait être corrélée à l’activité de la maladie [21]. Par ailleurs, des anticorps chauds d’isotype IgG dirigés contre des LT activés ont également été rapportés [22].
3.3. Redistribution
Au cours des granulomatoses, une redistribution des lymphocytes avec diminution de la lymphocytose sanguine est constatée alors qu’il existe une infiltration tissulaire de LT (image dite en miroir).
Ainsi, au cours de la sarcoïdose, la lymphopénie est un marqueur d’activité de même que l’absence de réaction aux tests antigéniques cutanés [23]. L’étude phénotypique des lymphocytes au cours de la sarcoïdose montre des stigmates d’activation tant au niveau sanguin qu’au niveau tissulaire (liquide de lavage broncho-alvéolaire). La redistribution lymphocytaire peut être responsable d’une lymphopénie sanguine parfois importante qui peut favoriser la survenue de rares infections opportunistes [24].
Au cours de la GPA (anciennement dénommée granulomatose de Wegener), la sévérité de la lymphopénie au diagnostic ou au cours des trois premiers mois de la prise en charge était plus prononcée chez les patients ayant développé une pneumopathie à Pneumocystis jirovecii au cours du suivi [25].
Enfin, au cours de la maladie de Crohn, la lymphopénie, présente chez 30 % des patients avant une chirurgie, peut servir de marqueur d’activité ainsi que de critère pronostique. Au plan histologique, elle est plus fréquemment associée à la présence de granulomes [26].
3.4. Pertes excessives
Au cours des entéropathies exsudatives ou du chylothorax, la perte excessive de lymphocytes peut s’accompagner d’une lymphopénie. Cependant, le mécanisme de la lymphopénie n’est pas toujours univoque : ainsi, au cours des ectasies lymphatiques digestives primitives (telle que la maladie de Waldmann), les pertes digestives sont également responsables de carences alimentaires multiples.
3.5. Étiologies multifactorielles
De nombreuses infections virales peuvent s’accompagner d’une lymphopénie initiale et celle-ci peut en constituer un élément diagnostique [27]. Elle est également présente à la phase initiale du choc septique [28], lors d’infections bactériennes en particulier à mycobactéries [29] mais aussi de diverses pneumopathies (Streptococus pneumoniae, Legionella, etc.) ou lors de brûlures étendues. Cette lymphopénie est souvent multifactorielle. Au cours de la rougeole, il a été montré que l’hémagglutinine virale induisait l’apoptose des cellules mononucléées sanguines [30]. Mais, cette lymphopénie peut aussi être liée à une infection directe des lymphocytes, à une élévation des taux de cortisol plasmatique ou encore à une redistribution. Elle n’a de valeur pronostique que dans quelques situations particulières. Au cours de l’infection à CMV chez le patient allogreffé, la baisse des lymphocytes dans les 14 jours suivants le diagnostic est associée à un pronostic péjoratif [31]. Par ailleurs, au cours de l’infection VIH, le taux de LT-CD4+ mais également la lymphocytose totale sont pronostiques [32].
Les hypercorticismes primaires ou l’administration de corticoïdes entraînent une lymphopénie prédominant sur les LT et en particulier sur les LT-CD4+. Celle-ci s’explique par une redistribution des lymphocytes circulants [33] et par une apoptose des thymocytes et des lymphocytes [34]. Par ailleurs, toutes les situations de stress aigu peuvent se compliquer de lymphopénie via l’augmentation des taux circulants de cortisol.
Des facteurs génétiques pourraient également intervenir dans la lymphopénie. Ainsi, les Éthiopiens ont une déplétion portant principalement sur les LT-CD4+ en dehors de toute infection VIH.
3.6. Mécanismes inconnus
Au cours de l’insuffisance rénale chronique, et en particulier chez les patients dialysés, il existe souvent une lymphopénie [35]. Elle est liée à une hyperactivation des lymphocytes qui expriment des niveaux plus élevés de CD57 et/ou de CD95 (Fas) et ont une sensibilité accrue à l’apoptose.
La lymphopénie initiale est fréquente au cours des lymphomes. Elle a une valeur pronostique au cours des lymphomes de Hodgkin [36] mais également des lymphomes diffus à grandes cellules et des lymphomes folliculaires. Elle peut être masquée par l’existence de cellules malignes circulantes.
Enfin, au cours des cancers solides, la lymphopénie peut également servir de marqueur pronostique [37].
Dans une étude portant sur 115 patients ayant un syndrome de Gougerot-Sjögren (SGS) primitif, Kirtava et al. ont mis en évidence une lymphopénie inférieure à 300/mm3 chez 5,2 % des malades au moment du diagnostic [38]. La présence d’une lymphopénie serait par ailleurs prédictive de la survenue d’un lymphome au cours de l’évolution [39]. Des auto-anticorps anti-CD4 ont été rapportés chez 12,6 % des patients présentant un SGS mais ils ne sont pas corrélés à la lymphopénie [40].
La lymphopénie CD4 idiopathique (LCI) est une cause rare de lymphopénie.
4. Lymphopénie CD4 idiopathique
4.1. Définition
La LCI a été rapportée en 1992 pour la première fois, puis, dans une série d’observations par le Center for Disease Control (CDC) [41] du fait de l’accumulation d’observations d’infections opportunistes chez des patients non infectés par le VIH. La définition retenue est de moins de 300 LT CD4+/mm3 ou moins de 20 % de LT CD4+ à deux reprises espacées d’au moins six mois. Il s’agit d’un diagnostic d’élimination qui nécessite d’exclure une étiologie virale (VIH), auto-immune ou inflammatoire ou un déficit immunitaire primitif ou acquis.
4.2. Épidémiologie
Bien qu’il soit difficile de donner une prévalence de la LCI avec précision, dans une première étude s’intéressant aux femmes sexuellement actives non porteuses du VIH, 2,4 % ont des LT CD4+ inférieurs à 350 et 1,6 % ont moins de 300 LT CD4+/mm3 [42]. Cependant, lors d’un contrôle, celle-ci était corrigée pour toutes les patientes testées. De même, chez 2030 donneurs de sang, une lymphopénie est mise en évidence chez 0,25 % des sujets. Ceux-ci présentent dans les jours suivants des symptômes de type infectieux et le contrôle effectué ultérieurement est normal [43].
4.3. Physiopathologie
Plusieurs études ont été menées sur la physiopathologie de la LCI. Il s’agit en fait d’un syndrome de présentation clinique hétérogène et les mécanismes physiopathologiques sont sans doute très divers.
Le phénotypage des LT circulants met en évidence un déficit de LT CD4+ qui peut être associé à une lymphopénie T CD8+ qui est inconstante et de mauvais pronostic [44]. Les lymphocytes circulants expriment Fas (CD95) à un niveau élevé à l’origine d’une apoptose spontanée et induite accrue [45]. Des auto-anticorps (anti-LT CD4+) ont également été rapportés chez un patient [46]. L’étude du répertoire des LT (séquençage du TcR) chez trois patients montrait une restriction du répertoire dont la signification est indéterminée [47]. Enfin, une diminution de l’activité de p56lck a été rapportée chez un patient. Elle pourrait être responsable d’un défaut d’activation et de prolifération des LT [48].
Il existe un déficit en cellules CD34+CD38−DR+ médullaires, population qui inclut des précurseurs lymphoïdes [49]. Une élévation possiblement compensatrice de l’IL-7 sérique a été rapportée associée à une augmentation des LB transitionnels CD10+CD27− [50].
Plus récemment, un défaut d’expression membranaire de CXCR-4 sur les LT a été mis en évidence chez six patients atteints de LCI. Ce récepteur et son ligand CCL-12 sont impliqués dans la différenciation lymphocytaire T thymique et dans la domiciliation des LT vers les organes lymphoïdes secondaires. Après traitement de ces LT in vitro par l’IL-2, le défaut d’expression membranaire de CXCR-4 était corrigé chez cinq des six patients étudiés [51].
Enfin, chez un patient présentant une lymphopénie modérée et isolée, Kuijpers et al. ont mis en évidence une mutation hypomorphe de Rag [52].
4.4. Manifestations cliniques/évolution
Les manifestations sont principalement en rapport avec un déficit de l’immunité cellulaire responsable d’événements évoquant dans certains cas un sida. Ainsi, ont été rapportées des mycobactérioses [53], des cryptococcoses [54], des histoplasmoses, des pneumocystoses [55], des toxoplasmoses [56], des cas de leuco-encéphalopathies multifocales progressives (LEMP), des infections récidivantes à virus varicelle-zona (VZV) ou à papillomavirus humain (HPV) ou encore des cas de candidoses disséminées. Cette immunodépression peut se compliquer de la survenue de tumeurs solides (cancer du col utérin associé à HPV), de lymphoproliférations bénignes ou de lymphomes malins [57].
Enfin, des signes latents ou patents d’auto-immunité sont également fréquemment rapportés : cytopénies auto-immunes, LES, vitiligo, thyroïdite auto-immune… [44]. Cependant, dans les observations, la lymphopénie est parfois secondaire aux manifestations auto-immunes ce qui pose des questions nosologiques.
L’évolution de la LCI reste mal connue en l’absence de cohortes de suivi importantes. Il semble que cette lymphopénie soit habituellement persistante et stable, même si des corrections spontanées restent possibles [44]. Au plan clinique, la survenue d’infections opportunistes n’est pas prévisible.
4.5. Traitement
Le traitement s’inspire des principes de prise en charge des sujets VIH+. Une prophylaxie par triméthoprime/sulfaméthoxasole (Bactrim®) est proposée chez les patients qui ont moins de 200 LT CD4+/mm3. En cas de prophylaxie secondaire suite à une cryptococcose, un traitement par fluconazole (Triflucan®) ou le voriconazole (Vfend®) est préconisé.
De nombreux traitements ont été proposés sans qu’aucun n’ait été validé par des études prospectives randomisées. Toujours par analogie avec des essais réalisés chez les patients VIH ayant une lymphopénie profonde, de nombreux patients ont été traités par de l’IL-2 avec une efficacité inconstante sur la lymphocytose et une efficacité clinique difficile à affirmer [58]. Cependant, la correction partielle de l’expression membranaire de CXCR4 sur les LT au cours du traitement par IL-2 soutient son utilisation au cours de la LCI [51]. Plus récemment, l’IL-7 a été proposée et une étude de phase I – phase II est en cours aux États-Unis dans le traitement de la LCI (NCT00839436 ; IL-7 [CYT107] Treatment of Idiopathic CD4 Lymphocytopenia : Expansion of CD4 T Cells [ICICLE]).
Par ailleurs, l’interféron gamma a été proposé dans la prise en charge de la méningite à cryptocoque ou des mycobactéries atypiques [59] avec une efficacité inconstante [60]. Enfin, l’intérêt du cidofovir au cours de la LEMP a été évoqué.
5. Conduite à tenir devant une lymphopénie
Le diagnostic étiologique d’une lymphopénie doit avant tout s’appuyer sur son taux, son ancienneté, l’existence d’autres cytopénies associées et la présence de signes cliniques d’accompagnement. Si la lymphopénie est isolée et modérée (supérieure à 1200/mm3 chez l’adulte), cela incite à un simple contrôle à distance.
Si la lymphopénie s’intègre dans un contexte particulier, on recherchera des arguments tant cliniques que biologiques en faveur d’une infection aiguë (responsable d’une lymphopénie transitoire) ou chronique (tuberculose, infection par le VIH ou par HTLV). Certaines maladies systémiques (SGS, LES, sarcoïdose) s’accompagnent le plus souvent d’une hypergammaglobulinémie polyclonale qu’il convient de rechercher. On réalisera également un dosage de la calcémie, de la phosphorémie, de l’enzyme de conversion de l’angiotensine, des anticorps anti-ADN natifs et des anticorps antinucléaires. On s’assurera également de l’absence d’adénopathie superficielle ou d’organomégalie qui orienterait vers une lymphoprolifération et s’assurer de l’absence d’argument pour une néoplasie profonde. Enfin, il faudra évaluer l’état nutritionnel du patient (clinique, albuminémie et dosage du zinc plasmatique) et éliminer une insuffisance rénale chronique.
Des explorations de base et un arbre décisionnel sont rassemblés sur la Fig. 1 .
Fig. 1.
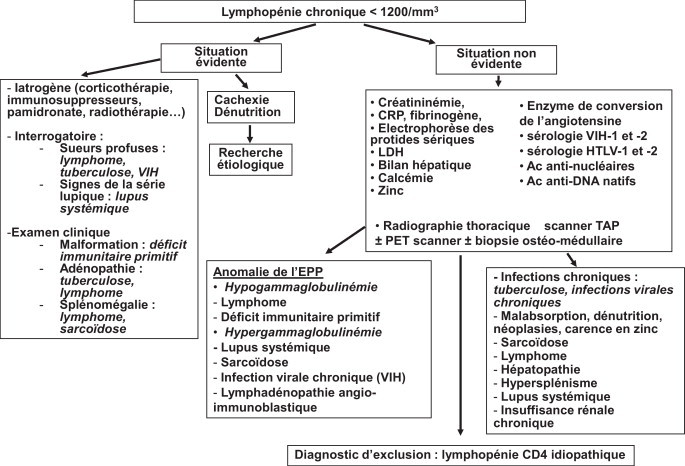
Conduite à tenir devant une lymphopénie chronique de l’adulte.
6. Conclusion
Les mécanismes et les étiologies pouvant être à l’origine d’une lymphopénie sont multiples et intriqués. Le plus souvent, celle-ci s’intègre dans un contexte clinique particulier et ne nécessite pas d’exploration complémentaire. Dans le cas contraire, il ne faut pas la négliger et il faut savoir rechercher une étiologie carentielle, infectieuse, dysimmunitaire ou néoplasique. Après des explorations bien conduites, le diagnostic de lymphopénie CD4+ idiopathique doit être envisagé afin d’adapter le suivi de ces patients.
Déclaration d’intérêts
L. Mouthon est consultant pour le laboratoire Cytheris.
Références
- 1.Pignata C., Fiore M., Guzzetta V., Castaldo A., Sebastio G., Porta F. Congenital Alopecia and nail dystrophy associated with severe functional T-cell immunodeficiency in two sibs. Am J Med Genet. 1996;65:167–170. doi: 10.1002/(SICI)1096-8628(19961016)65:2<167::AID-AJMG17>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 2.Camici M., Micheli V., Ipata P.L., Tozzi M.G. Pediatric neurological syndromes and inborn errors of purine metabolism. Neurochem Int. 2009;56:367–378. doi: 10.1016/j.neuint.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Lagresle-Peyrou C., Six E.M., Picard C., Rieux-Laucat F., Michel V., Ditadi A. Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet. 2009;41:106–111. doi: 10.1038/ng.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pannicke U., Honig M., Hess I., Friesen C., Holzmann K., Rump E.M. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet. 2009;41:101–105. doi: 10.1038/ng.265. [DOI] [PubMed] [Google Scholar]
- 5.Ege M., Ma Y., Manfras B., Kalwak K., Lu H., Lieber M.R. Omenn syndrome due to Artemis mutations. Blood. 2005;105:4179–4186. doi: 10.1182/blood-2004-12-4861. [DOI] [PubMed] [Google Scholar]
- 6.Macchi P., Villa A., Giliani S., Sacco M.G., Frattini A., Porta F. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995;377:65–68. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 7.Puel A., Ziegler S.F., Buckley R.H., Leonard W.J. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet. 1998;20:394–397. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- 8.Russell S.M., Tayebi N., Nakajima H., Riedy M.C., Roberts J.L., Aman M.J. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270:797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 9.Fischer A., de Saint Basile G., Le Deist F. CD3 deficiencies. Curr Opin Allergy Clin Immunol. 2005;5:491–495. doi: 10.1097/01.all.0000191886.12645.79. [DOI] [PubMed] [Google Scholar]
- 10.Kung C., Pingel J.T., Heikinheimo M., Klemola T., Varkila K., Yoo L.I. Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nat Med. 2000;6:343–345. doi: 10.1038/73208. [DOI] [PubMed] [Google Scholar]
- 11.Roifman C.M., Hummel D., Martinez-Valdez H., Thorner P., Doherty P.J., Pan S. Depletion of CD8+ cells in human thymic medulla results in selective immune deficiency. J Exp Med. 1989;170:2177–2182. doi: 10.1084/jem.170.6.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Picard C., McCarl C.A., Papolos A., Khalil S., Luthy K., Hivroz C. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–1980. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S.H., Tanasa B. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 14.de la Salle H., Hanau D., Fricker D., Urlacher A., Kelly A., Salamero J. Homozygous human TAP peptide transporter mutation in HLA class I deficiency. Science. 1994;265:237–241. doi: 10.1126/science.7517574. [DOI] [PubMed] [Google Scholar]
- 15.Nekrep N., Fontes J.D., Geyer M., Peterlin B.M. When the lymphocyte loses its clothes. Immunity. 2003;18:453–457. doi: 10.1016/s1074-7613(03)00086-4. [DOI] [PubMed] [Google Scholar]
- 16.Kelesidis T., Yang O. Good's syndrome remains a mystery after 55 years: a systematic review of the scientific evidence. Clin Immunol. 2010;135:347–363. doi: 10.1016/j.clim.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Z.Y., Zhao X.D., Jiang L.P., Liu E.M., Wang M., Yu J. Clinical characteristics and molecular analysis of 21 Chinese children with congenital agammaglobulinemia. Scand J Immunol. 2010;72:454–459. doi: 10.1111/j.1365-3083.2010.02457.x. [DOI] [PubMed] [Google Scholar]
- 18.Chandra R.K. Immunocompetence in undernutrition. J Pediatr. 1972;81:1194–1200. doi: 10.1016/s0022-3476(72)80262-2. [DOI] [PubMed] [Google Scholar]
- 19.King L.E., Osati-Ashtiani F., Fraker P.J. Apoptosis plays a distinct role in the loss of precursor lymphocytes during zinc deficiency in mice. J Nutr. 2002;132:974–979. doi: 10.1093/jn/132.5.974. [DOI] [PubMed] [Google Scholar]
- 20.Choi C.W., Sung H.J., Park K.H., Yoon S.Y., Kim S.J., Oh S.C. Early lymphopenia as a risk factor for chemotherapy-induced febrile neutropenia. Am J Hematol. 2003;73:263–266. doi: 10.1002/ajh.10363. [DOI] [PubMed] [Google Scholar]
- 21.Winfield J.B., Winchester R.J., Kunkel H.G. Association of cold-reactive antilymphocyte antibodies with lymphopenia in systemic lupus erythematosus. Arthritis Rheum. 1975;18:587–594. doi: 10.1002/art.1780180609. [DOI] [PubMed] [Google Scholar]
- 22.Litvin D.A., Cohen P.L., Winfield J.B. Characterization of warm-reactive IgG anti-lymphocyte antibodies in systemic lupus erythematosus. Relative specificity for mitogen-activated T cells and their soluble products. J Immunol. 1983;130:181–186. [PubMed] [Google Scholar]
- 23.Morell F., Levy G., Orriols R., Ferrer J., De Gracia J., Sampol G. Delayed cutaneous hypersensitivity tests and lymphopenia as activity markers in sarcoidosis. Chest. 2002;121:1239–1244. doi: 10.1378/chest.121.4.1239. [DOI] [PubMed] [Google Scholar]
- 24.Girard N., Cottin V., Hot A., Etienne-Mastroianni B., Chidiac C., Cordier J.F. Opportunistic infections and sarcoidosis. Rev Mal Respir. 2004;21:1083–1090. doi: 10.1016/s0761-8425(04)71582-x. [DOI] [PubMed] [Google Scholar]
- 25.Godeau B., Mainardi J.L., Roudot-Thoraval F., Hachulla E., Guillevin L., Huong Du L.T. Factors associated with Pneumocystis carinii pneumonia in Wegener's granulomatosis. Ann Rheum Dis. 1995;54:991–994. doi: 10.1136/ard.54.12.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heimann T.M., Bolnick K., Aufses A.H., Jr. Prognostic significance of severe preoperative lymphopenia in patients with Crohn's disease. Ann Surg. 1986;203:132–135. doi: 10.1097/00000658-198602000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cunha B.A., Pherez F.M., Schoch P. Diagnostic importance of relative lymphopenia as a marker of swine influenza (H1N1) in adults. Clin Infect Dis. 2009;49:1454–1456. doi: 10.1086/644496. [DOI] [PubMed] [Google Scholar]
- 28.Venet F., Davin F., Guignant C., Larue A., Cazalis M.A., Darbon R. Early assessment of leukocyte alterations at diagnosis of septic shock. Shock. 2010;34:358–363. doi: 10.1097/SHK.0b013e3181dc0977. [DOI] [PubMed] [Google Scholar]
- 29.Uppal S.S., Tewari S.C., Verma S., Dhot P.S. Comparison of CD4 and CD8 lymphocyte counts in HIV-negative pulmonary TB patients with those in normal blood donors and the effect of antitubercular treatment: hospital-based flow cytometric study. Cytometry B Clin Cytom. 2004;61:20–26. doi: 10.1002/cyto.b.20018. [DOI] [PubMed] [Google Scholar]
- 30.Iwasa T., Suga S., Qi L., Komada Y. Apoptosis of human peripheral blood mononuclear cells by wild-type measles virus infection is induced by interaction of hemagglutinin protein and cellular receptor, SLAM via caspase-dependent pathway. Microbiol Immunol. 2010;54:405–416. doi: 10.1111/j.1348-0421.2010.00231.x. [DOI] [PubMed] [Google Scholar]
- 31.Fries B.C., Khaira D., Pepe M.S., Torok-Storb B. Declining lymphocyte counts following cytomegalovirus (CMV) infection are associated with fatal CMV disease in bone marrow transplant patients. Exp Hematol. 1993;21:1387–1392. [PubMed] [Google Scholar]
- 32.Mofenson L.M., Harris D.R., Moye J., Bethel J., Korelitz J., Read J.S. Alternatives to HIV-1 RNA concentration and CD4 count to predict mortality in HIV-1-infected children in resource-poor settings. Lancet. 2003;362:1625–2167. doi: 10.1016/s0140-6736(03)14825-8. [DOI] [PubMed] [Google Scholar]
- 33.Fauci A.S. Mechanisms of corticosteroid action on lymphocyte subpopulations. I. Redistribution of circulating T and b lymphocytes to the bone marrow. Immunology. 1975;28:669–680. [PMC free article] [PubMed] [Google Scholar]
- 34.Herold M.J., McPherson K.G., Reichardt H.M. Glucocorticoids in T cell apoptosis and function. Cell Mol Life Sci. 2006;63:60–72. doi: 10.1007/s00018-005-5390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Litjens N.H., van Druningen C.J., Betjes M.G. Progressive loss of renal function is associated with activation and depletion of naive T lymphocytes. Clin Immunol. 2006;118:83–91. doi: 10.1016/j.clim.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Bierman P.J., Lynch J.C., Bociek R.G., Whalen V.L., Kessinger A., Vose J.M. The International Prognostic Factors Project score for advanced Hodgkin's disease is useful for predicting outcome of autologous hematopoietic stem cell transplantation. Ann Oncol. 2002;13:1370–1377. doi: 10.1093/annonc/mdf228. [DOI] [PubMed] [Google Scholar]
- 37.Ray-Coquard I., Cropet C., Van Glabbeke M., Sebban C., Le Cesne A., Judson I. Lymphopenia as a prognostic factor for overall survival in advanced carcinomas, sarcomas, and lymphomas. Cancer Res. 2009;69:5383–5391. doi: 10.1158/0008-5472.CAN-08-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirtava Z., Blomberg J., Bredberg A., Henriksson G., Jacobsson L., Manthorpe R. CD4+ T-lymphocytopenia without HIV infection: increased prevalence among patients with primary Sjogren's syndrome. Clin Exp Rheumatol. 1995;13:609–616. [PubMed] [Google Scholar]
- 39.Baimpa E., Dahabreh I.J., Voulgarelis M., Moutsopoulos H.M. Hematologic manifestations and predictors of lymphoma development in primary Sjogren syndrome: clinical and pathophysiologic aspects. Medicine (Baltimore) 2009;88:284–293. doi: 10.1097/MD.0b013e3181b76ab5. [DOI] [PubMed] [Google Scholar]
- 40.Henriksson G., Manthorpe R., Bredberg A. Antibodies to CD4 in primary Sjogren's syndrome. Rheumatology (Oxford) 2000;39:142–147. doi: 10.1093/rheumatology/39.2.142. [DOI] [PubMed] [Google Scholar]
- 41.Smith D.K., Neal J.J., Holmberg S.D. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. An investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med. 1993;328:373–379. doi: 10.1056/NEJM199302113280601. [DOI] [PubMed] [Google Scholar]
- 42.DeHovitz J.A., Feldman J., Landesman S. Idiopathic CD4+ T-lymphocytopenia. N Engl J Med. 1993;329:1045–1046. doi: 10.1056/NEJM199309303291418. [DOI] [PubMed] [Google Scholar]
- 43.Busch M.P., Valinsky J.E., Paglieroni T., Prince H.E., Crutcher G.J., Gjerset G.F. Screening of blood donors for idiopathic CD4+ T-lymphocytopenia. Transfusion. 1994;34:192–197. doi: 10.1046/j.1537-2995.1994.34394196614.x. [DOI] [PubMed] [Google Scholar]
- 44.Zonios D.I., Falloon J., Bennett J.E., Shaw P.A., Chaitt D., Baseler M.W. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112:287–294. doi: 10.1182/blood-2007-12-127878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laurence J., Mitra D., Steiner M., Lynch D.H., Siegal F.P., Staiano-Coico L. Apoptotic depletion of CD4+ T cells in idiopathic CD4+ T lymphocytopenia. J Clin Invest. 1996;97:672–680. doi: 10.1172/JCI118464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salit R.B., Hankey K.G., Yi R., Rapoport A.P., Mann D.L. Detection of CD4(+) T-cell antibodies in a patient with idiopathic CD4 T lymphocytopenia and cryptococcal meningitis. Br J Haematol. 2007;139:133–137. doi: 10.1111/j.1365-2141.2007.06781.x. [DOI] [PubMed] [Google Scholar]
- 47.Signorini S., Pirovano S., Fiorentini S., Stellini R., Bianchi V., Albertini A. Restriction of T-cell receptor repertoires in idiopathic CD4+ lymphocytopenia. Br J Haematol. 2000;110:434–437. doi: 10.1046/j.1365-2141.2000.02166.x. [DOI] [PubMed] [Google Scholar]
- 48.Hubert P., Bergeron F., Ferreira V., Seligmann M., Oksenhendler E., Debre P. Defective p56Lck activity in T cells from an adult patient with idiopathic CD4+ lymphocytopenia. Int Immunol. 2000;12:449–457. doi: 10.1093/intimm/12.4.449. [DOI] [PubMed] [Google Scholar]
- 49.Isgro A., Sirianni M.C., Gramiccioni C., Mezzaroma I., Fantauzzi A., Aiuti F. Idiopathic CD4+ lymphocytopenia may be due to decreased bone marrow clonogenic capability. Int Arch Allergy Immunol. 2005;136:379–384. doi: 10.1159/000084258. [DOI] [PubMed] [Google Scholar]
- 50.Malaspina A., Moir S., Chaitt D.G., Rehm C.A., Kottilil S., Falloon J. Idiopathic CD4+ T lymphocytopenia is associated with increases in immature/transitional B cells and serum levels of IL-7. Blood. 2007;109:2086–2088. doi: 10.1182/blood-2006-06-031385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scott-Algara D., Balabanian K., Chakrabarti L.A., Mouthon L., Dromer F., Didier C. Idiopathic CD4+ T-cell lymphocytopenia is associated with impaired membrane expression of the chemokine receptor CXCR4. Blood. 2010;115:3708–3717. doi: 10.1182/blood-2009-02-202796. [DOI] [PubMed] [Google Scholar]
- 52.Kuijpers T.W., Ijspeert H., van Leeuwen E.M., Jansen M.H., Hazenberg M.D., Weijer K.C. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood. 2011;117:5892–5896. doi: 10.1182/blood-2011-01-329052. [DOI] [PubMed] [Google Scholar]
- 53.Zaharatos G.J., Behr M.A., Libman M.D. Profound T-lymphocytopenia and cryptococcemia in a human immunodeficiency virus-seronegative patient with disseminated tuberculosis. Clin Infect Dis. 2001;33:E125–E128. doi: 10.1086/324086. [DOI] [PubMed] [Google Scholar]
- 54.Zonios D.I., Falloon J., Huang C.Y., Chaitt D., Bennett J.E. Cryptococcosis and idiopathic CD4 lymphocytopenia. Medicine (Baltimore) 2007;86:78–92. doi: 10.1097/md.0b013e31803b52f5. [DOI] [PubMed] [Google Scholar]
- 55.Matsuyama W., Tsurukawa T., Iwami F., Wakimoto J., Mizoguchi A., Kawabata M. Two cases of idiopathic CD4+ T-lymphocytopenia in elderly patients. Intern Med. 1998;37:891–895. doi: 10.2169/internalmedicine.37.891. [DOI] [PubMed] [Google Scholar]
- 56.Plonquet A., Bassez G., Authier F.J., Dray J.M., Farcet J.P., Gherardi R.K. Toxoplasmic myositis as a presenting manifestation of idiopathic CD4 lymphocytopenia. Muscle Nerve. 2003;27:761–765. doi: 10.1002/mus.10376. [DOI] [PubMed] [Google Scholar]
- 57.Ben Rejeb A., Ebdelli N., Bouali M.R., Goucha A., Bougrine F., Khediri F. Primary digestive tract Kaposi sarcoma with idiopathic CD4+ lymphocytopenia, HIV negative, HHV8 positive. Gastroenterol Clin Biol. 2001;25:707–710. [PubMed] [Google Scholar]
- 58.Trojan T., Collins R., Khan D.A. Safety and efficacy of treatment using interleukin-2 in a patient with idiopathic CD4(+) lymphopenia and Mycobacterium avium-intracellulare. Clin Exp Immunol. 2009;156:440–445. doi: 10.1111/j.1365-2249.2009.03910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cunningham-Rundles C., Murray H.W., Smith J.P. Treatment of idiopathic CD4 T lymphocytopenia with IL-2. Clin Exp Immunol. 1999;116:322–325. doi: 10.1046/j.1365-2249.1999.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sternfeld T., Nigg A., Belohradsky B.H., Bogner J.R. Treatment of relapsing Mycobacterium avium infection with interferon-gamma and interleukin-2 in an HIV-negative patient with low CD4 syndrome. Int J Infect Dis. 2010;14(Suppl. 3):e198–e201. doi: 10.1016/j.ijid.2009.08.004. [DOI] [PubMed] [Google Scholar]