

Abstract
When the central nervous system (CNS) is under viral attack, defensive antiviral responses must necessarily arise from the CNS itself to rapidly and efficiently curb infections with minimal collateral damage to the sensitive, specialized and non-regenerating neural tissue. This presents a unique challenge because an intact blood–brain barrier (BBB) and lack of proper lymphatic drainage keeps the CNS virtually outside the radar of circulating immune cells that are at constant vigilance for antigens in peripheral tissues. Limited antigen presentation skills of CNS cells in comparison to peripheral tissues is because of a total lack of dendritic cells and feeble expression of major histocompatibility complex (MHC) proteins in neurons and glia. However, research over the past two decades has identified immune effector mechanisms intrinsic to the CNS for immediate tackling, attenuating and clearing of viral infections, with assistance pouring in from peripheral circulation in the form of neutralizing antibodies and cytotoxic T cells at a later stage. Specialized CNS cells, microglia and astrocytes, were regarded as sole sentinels of the brain for containing a viral onslaught but neurons held little recognition as a potential candidate for protecting itself from the proliferation and pathogenesis of neurotropic viruses. Accumulating evidence however indicates that extracellular insult causes neurons to express immune factors characteristic of lymphoid tissues. This article aims to comprehensively analyze current research on this conditional alteration in the protein expression repertoire of neurons and the role it plays in CNS innate immune response to counter viral infections.
Abbreviations: BBB, blood–brain barrier; BDV, borna disease virus; CCL, chemokine (C-C motif) ligand; CD, cluster of differentiation; CNS, central nervous system; CMV, cytomegalovirus; CTL, cytotoxic T lymphocytes; CXCL, chemokine (C-X-C motif) ligand; DEN, dengue fever virus; EBV, Epstein-Barr virus; EMCV, encephalomyocarditis virus; HCMV, human cytomegalovirus; HIV, human immunodeficiency virus; HSV, Herpes simplex virus; HTLV 1, human T-lymphotropic virus 1; IFN, interferon; IL, interleukin; IRF, interferon regulatory factors; ISGs, interferon stimulated genes; JEV, Japanese encephalitis virus; LMV, lymphocytic choriomeningitis virus; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MHC, major histocompatibility complex; MHV, mouse hepatitis virus; MMPs, matrix metalloproteinases; NFκB, nuclear factor κB; NO, nitric oxide; PAMP, pathogen-associated molecular patterns; PI3-K, phosphatidyl inositol-3 kinase; PKR, dsRNA-dependent protein kinase; PRR, pattern recognition receptors; RABV, Rabies virus; RIG, retinoic acid-inducible gene; RLR, RIG-like receptors; SINV, Sindbis virus; SIV, Simian immunodeficiency virus; SLE, St Louis encephalitis virus; SSPE, subacute sclerosing panencephalitis; STAT, signal transducers and activator of transcription; TCR, T cell receptor; TIMP, tissue inhibitor of MMPs; TLR, Toll-like receptor; TMEV, Theiler's murine encephalomyelitis virus; TNF-α, tumor necrosis factor alpha; VSV, vescicular stomatitis virus; WNV, West Nile virus; YFV, yellow fever virus
Keywords: Neurotropic virus, Major histocompatibility complex (MHC), Lymphocytes, Toll-like receptors, Cytokines, Chemokines, Interferons
1. Introduction
There are about 1011 neurons in the human brain comprising the major cell type involved in the vital motor and cognitive functions in the body (Williams and Herrup, 1988). Neurotropic viruses have been characterized by their propensity for infecting central nervous system (CNS) cells. The co-evolution of such viruses with the mammalian nervous system has witnessed a constant dueling struggle for survival. Interactions between the immune system and the central nervous system constitute the most complex and interactive regulatory network in mammals. Several features of the CNS cells have evolved unique defensive mechanisms to keep infections at bay, some of which have ironically left neurotropic viruses at a selective advantage for sustaining their life-cycle within the very same cells. While the blood–brain barrier (BBB) is an efficient mechanical check-point for filtering out blood-borne infections from entering the CNS, at the same time, it also cordons off the key cellular and molecular components which are active in immune surveillance in the peripheral organs; for instance “professional” antigen presenting cells called dendritic cells roving in the circulation are not allowed entry into the CNS. The major histocompatibility complex (MHC) class I molecules, important for antigen presentation to cytotoxic T lymphocytes (CTLs) are expressed on most nucleated cells outside the CNS in an uninfected state. Until recently constitutive expression of these MHC proteins in normal neuronal cell cultures or brain slices (Lampson, 1995) were not detected. This apparent absence of MHC on CNS cells was looked upon as a benefit to the sensitive organ system as a robust immune attack on non-replicating virus-infected neurons may cause much greater injury than the infection itself; a fatal risk that such a vital organ system cannot afford to take under any circumstance. Thus deficient antigen presentation to infiltrating T cells along with the lack of a conventional lymphatic drainage system in the CNS are key factors responsible for keeping the CNS in an immunologically resting state, a unique harbor conducive to viral persistence. Additionally, interactions between neurons and the glial cells as well as constitutive neurotrophins and transforming growth factor-β secretion, might help in maintaining this immunological quiescence (Dorries, 2001, Neumann et al., 1998). However, some activated/memory CD4+ and CD8+ T cells patrol the CNS at random even in the absence of pathogenic signals, and these either flow away into the peripheral circulation or die in the brain in the absence of antigen recognition (Ransohoff et al., 2003). Thus, vertebrate antiviral mechanisms in the CNS are not constitutively active.
Sophisticated modes of intracellular transport in the physiologically complex neural tissue are favorable for viral sustenance and replication. Neurons, with their extensive cellular processes and interconnections, allow a natural freeway for disseminating viral particles and spread of infection. Understandably, CNS immune response to a viral infection must necessarily be a rapid, stringently controlled process that effectively curtails viral spread with minimal bystander damage to specialized, non-regenerating neural tissue, and preventing an autoimmune response. Extensive studies have focused on the active participation of glial cells (Farina et al., 2007, Hanisch and Kettenmann, 2007) in resisting viral attack while neurons have been interpreted to play more passive secondary roles in viral immune resistance. Recent research however indicates neurons to employ more direct mechanisms to fight a viral attack by hosting and regulating innate and adaptive immune responses in the CNS (Biber et al., 2007, Levite, 2008). Thus the long-held idea of the neuron being a mute victim to a viral infection seems no longer tenable. The main focus of this article is to delineate hitherto reported fundamental mechanisms of antiviral responses elicited by neurons.
Neurotropic viruses causing acute infection in humans include Japanese encephalitis virus (JEV) (Ghosh and Basu, 2009), West Nile virus (WNV), Venezuelan equine, and California encephalitis viruses, dengue fever virus (DEN), yellow fever virus (YFV), St Louis encephalitis virus (SLE), Murray Valley encephalitis virus (MVE), Tick-borne encephalitis (Ghosh and Basu, 2008) and Kunjin virus, polio, coxsackievirus, echovirus, paramyxoviruses (causing mumps and measles), influenza, and rabies viruses as well as members of the family Herpesviridae such as herpes simplex, varicella-zoster, cytomegalo and Epstein-Barr viruses (Griffin, 2003). Viruses causing latent infection include herpes simplex and varicella-zoster viruses and those causing slow virus infection include JC polyomaviruses, and retroviruses such as human T-lymphotropic virus 1(HTLV-1) and human immunodeficiency virus (HIV) (Fazakerley and Walker, 2003, Lepoutre et al., 2009, Petito et al., 1999). In case of HIV, the transactivator viral protein, Tat, that is implicated in neuronal death responsible for neurological deficits, has been shown to differ in their neurotoxic properties depending upon the clades in which the virus belong (Mishra et al., 2008). Many viruses that result in chronic infections in the rodent CNS have proven useful models for examining various mechanisms in participation and regulation of immune processes in the brain. Neurons are main targets in the murine CNS for many kinds of viruses, including JEV, Sindbis virus (SINV), WNV, vesicular stomatitis virus and lymphocytic choriomeningitis virus. Viruses causing chronic infection along with myelin loss include two well studied RNA virus models—Theiler's murine encephalomyelitis virus (TMEV), belonging to the non-enveloped Picornaviridae family (Brahic and Roussarie, 2009), and mouse hepatitis virus (MHV), belonging to the enveloped Coronaviridae family (Bergmann et al., 2006). Table 1 shows a list of major neurotropic viruses infecting humans.
Table 1.
Major neurotropic viruses causing human infections.
| Virus Name | Family | Genus | Genome |
|---|---|---|---|
| Human immunodeficiency virus | Retroviridae | Lentivirus | ss (+) RNA-RT |
| Human T-lymphotropic virus-I | Deltaretrovirus | ||
| Japanese encephalitis virus | Flaviviridae | Flavivirus | ss (+) RNA |
| West Nile virus | |||
| Kunjin virus | |||
| St. Louis Encephalitis | |||
| Murray Valley encephalitis virus | |||
| Dengue virus | |||
| Yellow fever virus | |||
| Tick borne encephalitis | |||
| Sindbis virus | Togaviridae | Alphavirus | |
| Polio virus | Picornaviridae | Enterovirus | |
| Coxsackie virus | |||
| Echovirus | |||
| Rabies virus | Rhabdoviridae | Lyssavirus | ss (-) RNA |
| Vesicular stomatitis virus | Vesiculovirus | ||
| Herpes simplex virus | Herpesviridae | Simplexvirus | ds DNA |
| Varicella zoster virus | Varicellovirus | ||
| Cytomegalovirus | Cytomegalovirus | ||
| Epstein-Barr virus | Lymphocryptovirus | ||
Neurotropic viruses that are commonly known to cause infections in humans are depicted in the table. These viruses have been shown to be associated with CNS inflammation and neurodegeneration in humans. Apart from the ones categorized in the table, there are some other neurotropic viruses that have been shown to infect other vertebrate animals. Similarly, these viruses may also infect non-human hosts.
Neurotropic viruses follow the general routes of virus entry into the body. Herpes virus and HIV may break mucous membranes of mouth and genital tracts, while Coxsackie, polio, entero- and echoviruses may take the gastrointestinal route. The bite of a vector organism – most often mosquitoes – can transmit arboviruses like Flavivirus, toga and bunya viruses transdermally into subcutaneous tissues. The bite of a rabid animal or an infected monkey can transmit rabies virus or herpes virus simiae from the skin into muscles transdermally. Infected blood or blood products is probably the only portal of entry for neurotropic viruses such as HIV, HTLV-I, HTLV-II, CMV, EBV and WNV to directly enter the blood. Respiratory tract infections such as mumps, measles, rubella and herpesviruses are known to take a nervous route to the brain. It is noteworthy here that these viruses reach the CNS from extraneural sites, thus virus-recognizing immune cells are first activated in regional peripheral lymph nodes (Savarin and Bergmann, 2008), thereby suggesting that adaptive immune responses are initiated against neurotropic viruses even before innate mechanisms are triggered in the CNS upon infection; a concept that has been verified even in experimental viral infections (Bergmann et al., 2006, Griffin, 2003).
CNS viral infections may manifest acute, latent or chronic pathology, depending on the resultant effect of a virtual tug-of-war between viral infectivity and host immune resistance. Viral strain and titre, route of infection/inoculation, age, genetic background of the individual infected (van den Pol, 2009) and extent of neuronal injury – due to virus-induced neuronal or glial cell death or secondary damage inflicted by immune mediators on infected cells – are factors deciding the neuroinvasiveness, tropism, viral clearance or latency. Modulation of innate immune response by virulent strains also contributes to the list of factors influencing disease prognosis. Maturity level of the infected neuron is a determining factor in virus replication and neuronal susceptibility to SINV-induced cell death which is found to be independent of the immune response (Burdeinick-Kerr and Griffin, 2005, Havert et al., 2000). In some cases, rise in viral titres causes alterations in integrity of BBB and microglial activation in the host, even in the absence of clear clinical signs of disease (Fabis et al., 2008). As immune processes take control of the viral infection, the clinical symptoms start appearing (Bergmann et al., 1999, Parra et al., 1999).
2. Neuronal sensing of viral antigens
The earliest stage of antiviral innate immune response is triggered by specific recognition of conserved structural moieties or pathogen-associated molecular patterns (PAMPs) on viral components by host pattern recognition receptors (PRRs). Over the past decade, extensive studies on the members of the membrane-bound Toll-like receptor family (TLRs) and cytosolic Retinoic acid-inducible gene (RIG)-like receptors (RLRs) have characterized these proteins to play a central role in specific sensing of viral RNA for initiating antiviral immune response in the host cell as well as evading host defense systems by employing anti-TLR mechanisms in viruses (Bowie and Haga, 2005). Further, certain viruses may employ TLR-mediated cellular activation early in the infection process such as host cell entry and replication (Rassa and Ross, 2003).
Downstream signaling through TLRs and RLRs activates interferon regulatory factors (IRFs) and nuclear factor κB (NFκB) which leads to the production of type 1 interferons (IFN) and pro-inflammatory cytokines which directly stimulate antiviral actions and modulate adaptive immune responses (Akira et al., 2006). TLRs have also been shown to form multimeric complexes which effectively broaden the spectrum of cognate pathogenic molecules that CNS cells can register. Microglia and astrocytes are the major cell types bearing TLRs and capable of mounting innate immune response against neurotropic viruses in the CNS (Bieback et al., 2002, Rassa et al., 2002). The potential role of neurons in mounting an innate immune response seemed plausible with the discovery of TLR expression in neurons. Double-stranded (ds) RNA is a conserved molecular pattern retained by many viruses, by virtue of which, it can be considered a viral PAMP. dsRNA-dependent protein kinase (PKR) can be considered as an intracellular pattern recognition receptor for poly(I:C), a synthetic analogue of viral dsRNA. However, mice with defective PKR expression can still respond to poly(I:C). This observation led Alexopoulou et al. (2001) to demonstrate that TLR3 carries out poly (I:C) induced activation of NFκB and type I interferon production; steps which mimic viral infection. Also, TLR3 knockout mice displayed significant resistance to poly (I:C)-induced shock in comparison to wild-type mice (Alexopoulou et al., 2001). However doubts remained regarding TLR3 working as a bonafide antiviral receptor inducing IFN production when challenged with a real viral infection. In 2004, Tabeta et al. had shown using Tlr3 knockout mice, that TLR3 mediates murine CMV-induced type I IFN production, and contributes to overall immune defense against the virus (Tabeta et al., 2004). Although TLR3 localization had shown cell type variance (Matsumoto et al., 2003), recent studies on NT2-N human post mitotic neurons have shown TLR-3 expression upon viral infection. These cells also expressed inflammatory cytokines (TNF-α, IL-6), chemokines (CCL-5 and CXCL-10), and antiviral molecules (2′5′OAS and IFN-β) after treatment with dsRNA–a by-product of viral infection and ligand of TLR-3. Similar results were also seen after treating NT2-N cells with recombinant IFN-β, wherein bacterial cell wall constituent lipopolysaccharide (LPS) treatment also lead to the increase in chemokines (CCL-5 and CXCL-10) and antiviral molecules (2′5′OAS and IFN-β) but caused no change in inflammatory cytokine (TNF-α, IL-6) levels (Lafon et al., 2006). More recent research has shown that primary human neurons and two human neuronal cell lines (NT2-N and CHP-212) express all 10 members of the human TLR family. To determine the functionality of TLRs these cells which were treated with poly-I:C or ssRNA showed increased expression of IFN-α/β and IFN-regulatory factors (IRFs) in time and dose dependent manner. For investigating the potential of Herpes simplex virus type1 (HSV-1) infection for TLR activation and subsequent intracellular IFN-α/β induction, it was shown that a remarkable increase in IFN-α mRNA and protein expression takes place with little effect on IFN-β mRNA and protein expression in NT-2N cells (Zhou et al., 2009). Analysis of viral growth kinetics and IFN production in WNV infected neuronal cultures from wild-type and TLR3−/− mice has revealed that in primary cortical neurons TLR-3 is not essential for regulating IFN gene induction but, nevertheless, exerts a subtle restriction of WNV replication (Daffis et al., 2008).
Gene expression analysis using microarray of the neuronal cell line NT2-N following rabies virus (RABV) and HSV-1 infection have been shown to result in a 24% and 4.9% upregulation of the respective 56 genes of immunity cluster. Genes showing greatest increase include those involved in sensing dsRNA, namely, PKR, RIG-1 and TLR-3, IFN-β, genes coding for chemokines CCL-5, CCL-3, CCL-4, CCL-20, CXCL-9, CXCL-10, CXCL-11 and CXCL-3, genes coding for the interferon regulatory factors IRF-7 and IRF-1, and genes coding for the activators of transcription STAT-1, STAT-2 and NF-κB. Upregulation was also found in the expression of genes of factors of complement pathway, genes for inflammatory cytokines IL-6, IL-1α, TNF-α and IL-15 (Prehaud et al., 2005). Chemokines induce cell migration, regulate immune cell trafficking and along with controlling a variety of other cell functions they are versatile messengers while interacting amongst a diversity of cell types (de Haas et al., 2007). CNS chemokine expression has been extensively discussed in microglia and astrocytes under both physiological and pathological conditions such as homeostasis, synaptic transmission, development, injury, and disease-associated neuroinflammation (Bertollini et al., 2006, Charo and Ransohoff, 2006, Ubogu et al., 2006). Recent studies have reported neuronal chemokine expression discussing the potential roles of neuronal chemokines in neuro-neuronal and neuro-glial interaction. Chemokine receptors can activate intracellular signal transducers like adenylcyclase, phospholipases, GTPases like Rho, Rac, and Cdc42 and major kinase pathways like mitogen-activated protein kinase (MAPK) and phosphatidyl inositol-3 kinase (PI3-K) (Balkwill, 1998, Mellado et al., 2001). Constitutive expression of neural CCL2 was also reported in a human neuronal cell line (Coughlan et al., 2000) and during human CNS development (Meng et al., 1999). Neuronal CCL5 and CXCL10 expression and release was reported to get induced after viral infection in vitro and in vivo (Klein et al., 2005, Sui et al., 2004).
Menager et al. (2009) have recently reported the formation of large (1–3 μm) spherical inclusions, composed of TLR3 and viral components in the perinuclear region of rabies virus infected neurons. These spherical aggregates now identified to be the previously reported Negri bodies (NBs) seen in rabies infection, were untraceable in TLR3 deficient mice along with heightened resistance and lowered virus multiplication and infection levels in the brain. This study exemplifies an intelligent mechanism for neurotropic viral survival by compartmentalized viral replication within the cell after sabotaging normal functioning of neural proteins (Menager et al., 2009). TLR2 has been found to mediate cellular responses in HCMV infection. In cells deficient in TLR2 or CD14 (a co-receptor for TLR2 and TLR4), UV-inactivated HCMV virions failed at NFkB activation and IL-6 and IL-8 induction (Compton et al., 2003). TLR7 and 8 have been reported as essential components for recognition of ssRNA viruses and ssRNA (Diebold et al., 2004, Heil et al., 2004, Lund et al., 2004). TLR3 and TLR9, that recognize DNA unmethylated at CpG motifs, are both reported to be involved in immune defense against mouse CMV infection (Tabeta et al., 2004).
Studies have discussed the critical importance of RLR signaling in determining the pathological outcome of vescicular stomatitis virus (VSV), JEV, and encephalomyocarditis virus (EMCV) (Kumar et al., 2006, Loo et al., 2008). Innate immune programs in the CNS can apparently mediate antiviral immunity independent of adaptive immunity. Induction of interferon regulatory factor (IRF)-3 target genes by RLR signaling has shown to directly control viral replication in infected cells (Anderson et al., 2008, Daffis et al., 2007, Hiscott, 2007) Transcription of RIG-I and melanoma differentiation associated gene 5 (MDA-5), the prototypical RLR proteins have been reported to get induced in WNV infection of primary cortical neurons (Daffis et al., 2007).
TLR3 and RLR signaling induces IFN-α/β in neurons and the latter activates a myriad interferon stimulated genes (ISGs) that act in concert to launch multi-leveled but specific antiviral programs like inducing antiviral genes, promoting antigen presentation, inhibiting viral replication and transcription and regulating lymphocyte function and survival (Borden et al., 2007, Ireland et al., 2008). In WNV infection a greater susceptibility of neurons in comparison to macrophages is considered to be due to undetectable expression levels of RIG-I/MDA-5 as well as ISG54 and ISG56, indicating an influential role of PRRs along with ISGs in determining viral permissiveness (Daffis et al., 2007).
Viral persistence in neurons can be explained by evolved immune evasive mechanisms adopted by viruses for antagonizing, suppressing or blocking adaptor and target proteins participating in IFN-α/β mediated antiviral pathways (Weber and Haller, 2007).
3. Innate immune responses induced after viral entry are mostly glia-mediated
Coronavirus infection of the rodent CNS has delivered novel insights into the immune regulation of acute and persistent viral infection at the cellular level and is a good model for studying chronic demyelinating diseases, such as multiple sclerosis (Bergmann et al., 2006). Bearing similarity with other models of viral-induced encephalitis, MHV infection induces a robust CNS inflammatory response comprising both the innate and adaptive immune components. CNS infection is initially presented by fast, active and coordinated expression of matrix metalloproteinases (MMPs), chemokines, a tissue inhibitor of MMPs (TIMP-1) and pro-inflammatory cytokines viz. IL-1α, Il-1β, TNF-α, IL-6 and IL-12 primarily in microglia and astrocytes (Bergmann et al., 2006, Mishra et al., 2007). In case of JEV infection also, such responses have been observed (Ghoshal et al., 2007, Mishra et al., 2007). Enhanced or sustained viral replication is associated with raised levels of TNF-α and IL-6 although TNF-α does not seem to play direct antiviral roles other than disrupting the BBB (Wang et al., 2004). In HIV-1 infection it has been observed that the viral Vpr protein deregulates expression of various important cytokines and inflammatory proteins in infected microglia (Rom et al., 2009). Detailed analysis in subsequent studies clearly indicates that both virus-infected and uninfected glial cells, predominantly astrocytes, provide these early inflammatory signals (Lane et al., 2000). Activated peripheral lymphocytes contribute in upregulating adhesion molecules and chemokine receptors, both facilitating the entry of circulating lymphocytes into the CNS. Together, these molecules facilitate BBB disruption and attract innate immune effectors, which further enhance the expression of inflammatory factors. Once the CNS integrity is compromised, MMP expression ushers tissue influx of inflammatory cells, activation of cytokine secretion and CNS damage (Yong et al., 2001). In neurotropic canine distemper virus infection, a morbillivirus related to human measles virus, induction of MMP-2 expression in astrocytes and MMP-9 in neurons was noted (Khuth et al., 2001).
Apoptotic neurons induce microglial cells to release neuroprotective molecules, such as anti-inflammatory cytokines and growth factors, while inhibiting synthesis of nitric oxide (NO) and pro-inflammatory cytokines (Minghetti et al., 2005). Cumulatively, these observations therefore indicate a molecular cross-talk that is apparent between glial cells and neurons such that stressed neurons under a viral attack can protect themselves from further damage by activated microglia.
4. Neuronal gain of adaptive immune function upon viral infection
There is profound interdependence in the varied mechanisms adopted by resident CNS and infiltrating cells to defend against viral attack (Savarin and Bergmann, 2008). The brain is considered a unique organ, with different and sometimes absent expression of the MHC-encoded cell surface molecules that normally bind and present antigenic peptides on the surfaces of peripheral cells for recognition (binding) by the antigen-specific T cell receptors. CD8+ CTLs mediate their antiviral effects by recognizing viral peptides presented by class I MHC molecules on the surfaces of infected target cells, whereas class II molecules present antigen to CD4 “helper” T cells. Engagement of the T cell receptor (TCR) with the peptide-loaded MHC leads to perforation of the target cell membrane, delivery of cytolytic granules and lysis of the infected cell. In most tissues, the loss of infected cells is accompanied by enhanced uninfected cell division to replenish the cell population. However, this may not be an optimal strategy for clearance of all viral infections in the CNS (Patterson et al., 2002a, Patterson et al., 2002b), lest vital neuronal functions get compromised. To limit immune cell entry into the CNS and restrict T cell-neuron interactions, multiple anatomic and biochemical barriers exist within the brain, including the BBB, limited lymphatic drainage and the paucity of class I MHC on resident brain cells. Nevertheless, activated T lymphocytes patrol the CNS parenchyma (Hickey et al., 1991) and are recruited even more into the brain after infection by many neurotropic viruses (Binder and Griffin, 2001). While non-specific T cell trafficking into the CNS may be antigen independent, the persistence of virus-specific T cells and their antiviral functions in the brain definitely depends on MHC restricted antigen recognition (Bergmann et al., 2006).
MHC expression in the CNS is actively neuron-regulated. Healthy, conducting neurons are known to robustly suppress MHC induction in the neuronal membrane as well as in adjoining glial cell surfaces, while disruption of normal electrical activity by pro-inflammatory signals switches on MHC induction (Wekerle, 2005). The classical MHC molecules (also referred to as HLA molecules in humans) play a vital role in the complex immunological dialogue that must occur between T cells and other cells of the CNS in the pathogenesis of acute viral encephalitis. MHC class I gene expression has been reported in a diverse population of healthy, uninjured neurons along with the expression of many MHC class I receptors and multiple mediators of downstream signal transduction (Boulanger and Shatz, 2004). Neurons also express non-classical MHC class Ib molecules, including H-2M (Loconto et al., 2003), Qa-1 (Huh et al., 2000) and HLA-G (Lafon et al., 2005, Maier et al., 1999). Neuronal expressions of these molecules have been found increased after exposure to both cytokines (Neumann et al., 1997) and viral infections (Irwin et al., 1999, Loconto et al., 2003, Redwine et al., 2001).
Human autopsy samples have shown an increased expression of both class I and II antigens in brain in cases of HIV-induced encephalitis (Kennedy and Gairns, 1992). The expression of MHC class I mRNA in neurons has also been shown to increase following rabies virus infection; a pathogenic strain of the virus induced a greater expression of MHC class I proteins than a non-virulent strain (Irwin et al., 1999). Lymphocytic choriomeningitis virus (LCMV) is known to establish lifelong, systemic persistence when introduced in utero or at birth. LCMV infection has been shown to significantly elevate MHC class I expression in the CNS as well as in peripheral tissues, that is dependent on IFN-α/β receptor as only genetic deletion of the receptors reduced MHC I to normal levels. This shows that a well adapted pathogen such as LCMV can also chronically stimulate the innate immune system and consequently alter the expression of antigen presenting machinery in an immunologically pecialized compartment like the CNS (Truong et al., 2009). Herpes simplex virus (HSV) has also been reported to cause upregulation of MHC class I on neurons, both in vitro as well as in vivo (Abendroth et al., 2000). In subacute sclerosing panencephalitis (SSPE), measles virus (MV) persists in neurons, resulting in a fatal chronic infection. MHC class I mRNA expression has been found to be upregulated in SSPE tissues studied, and the expression was definitively seen on neurons, though MV may not be directly involved in the induction of MHC class I on neurons; it is proposed that cytokines such as IFN-γ may play an important role. But, the paucity of neurons co-expressing MHC class I and MV antigens in SSPE and sub-acute measles encephalitis suggests that such cells are either rapidly cleared by CTLs, or, alternatively, lack of co-expression of MHC class I on MV infected neurons favors MV persistence in them by escaping CTL recognition (Gogate et al., 1996). JEV is known to infect neurons and neural progenitor cells. Recently we have shown that JEV infection leads to expression of MHC class I and costimulatory molecules CD40, CD80, and CD86 on neural progenitor cells that otherwise does not express them (Das et al., 2009). Hence, it can be said that following neurotropic viral infection, neural progenitor cells possibly behave as immunogenic cells and contribute to both the innate and adaptive immune axes. Fig. 1 is a schematic illustrating pathways involved in MHC expression and TLR3 activation in a virus infected neuron.
Fig. 1.
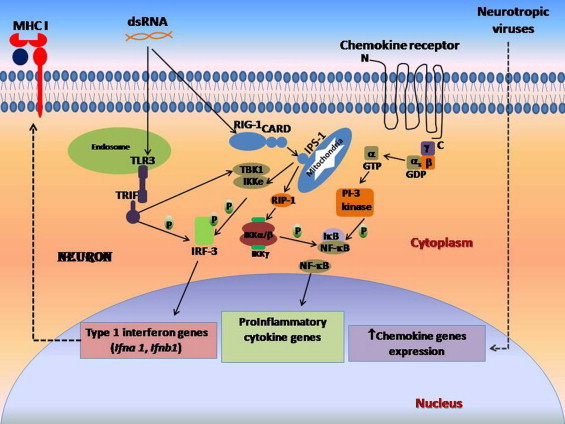
Viral infection causes major histocompatibility complex class I (MHC I) expression and production of type 1 interferons, cytokines and chemokines.
Double stranded RNA from viruses interacts with toll-like receptor (TLR3) located in endosomes compartment which in turn activates adapter molecule TIR-domain-containing adapter-inducing interferon-β (TRIF) leading to phosphorylation of Interferon regulatory factor 3 (IRF-3). TRIF, upon ligation, can also activate protein kinases tank binding kinase 1 (TBK-1) and I kappa B kinase complex (IKKe), that also leads to phosphorylation of IRF-3. IRF-3 activation can also occur via retinoid inducible gene 1 (RIG-1) dependent pathway. Phosphorylated IRF-3 induces expression of type 1 interferon genes that are believed to be associated with MHC class I expression on neurons. CARD of RIG-1 interacts with adapter molecule interferon-β promoter stimulator 1 (IPS-1) located on mitochondria, leading to activation of receptor interacting protein RIP-1 which in turn activate IKK α/β-IKK γ complex, that leads to phosphorylation of inhibitory kappa B (IκB) and subsequent release and activation of NFκB. NFκB induces expression of several pro-inflammatory cytokine genes in the neuron. Following neurotropic viral infection, it has been shown that there is increased expression G-protein coupled chemokine receptor expression on neurons. The GTP-activated stimulatory subunit (αs) of these receptors activates PI-3 kinase that lead to the phosphorylation of I kappa B and subsequent release of NFκB. Neurotropic viral infection has also been reported to cause upregulation of several chemokine genes in the neurons, through yet-to-be-defined pathways. Dashed denotes pathways that are not fully elucidated.
VSV causing acute infection of the CNS results in a rapid and sustained upregulation of MHC class I expression on microglia, whereas class II expression was markedly delayed (Steel et al., 2009) but whether it is also expressed on neurons is yet to be studied. Recently, it has been shown in a well-characterized simian immunodeficiency (SIV)/pigtailed macaque model, that expression of the MHC class I allele Mane-A*10 was significantly correlated with lower amounts of activated macrophages, SIV RNA, and neuronal dysfunction in the CNS than Mane-A*10 negative animals (Mankowski et al., 2008). It is noteworthy in this context that similar to viral proteins which antagonize IFN-α/β function, certain members of the herpesvirus and retrovirus families encode proteins which downregulate MHC antigen presentation for evading T cell function (Antoniou and Powis, 2008). Lack of MHC on neuronal surface, however, may not be the primary factor governing the partial CNS immune priviledge. Studies have reported some neurons overexpressing MHC class-I genes to be resistant to CTL-induced lysis following viral infection in vivo but get lysed effectively while grown in isolation in vitro (Rall et al., 1995). The involvement of some neural or glial factors intervening in CTL-neuron interactions has been suggested that determines neural viability in the face of a viral attack (Boulanger and Shatz, 2004).
5. IFN-γ mediates adaptive antiviral cross-talk between T cells and neurons
CD8+ T cells are the primary inhibitors of viral replication in neurons (Divito et al., 2006) and their function is dependent on IFN-γ expression which rises rapidly in acute inflammatory reactions caused by viral infection (Binder and Griffin, 2001). While virus-infected cells release IFN-α/β, activated NK cells and CD8+ and CD4+ cells produce IFN-γ (Rottenberg and Kristensson, 2002). Since the entry of these cells into the CNS is minimal under normal circumstances; IFN-γ is usually not detectable in the brain (Fabry et al., 1994). The role of IFN-γ in protecting the CNS and viral clearance has been described for many neurotropic viruses (Hausmann et al., 2005, Patterson et al., 2002a, Patterson et al., 2002b, Rodriguez et al., 2003, Shrestha et al., 2006). SINV-infected neurons have been shown to respond to IFN-γ by lowering virus replication through both viral protein and RNA synthesis interference (Burdeinick-Kerr and Griffin, 2005). Cell-type specific and collaborative functioning of IFN-γ and perforin released from CD8+ cells have been reported in the non-lethal MHV model (Bergmann et al., 2006). Direct neuronal response to IFN-γ occurs by the expression of many antiviral genes signaled through the Jak/Stat-1 pathway (Chesler and Reiss, 2002). For instance, the nitric oxide synthase gene, induced in neurons by IFN-γ, is important for suppression of VSV replication. Suppression of this gene aggravates SINV induced disease, increases mortality and may play a role in viral clearance (Chesler and Reiss, 2002, Tucker et al., 1996). It is noteworthy here that effective viral clearance mediated by IFN-γ differs among neuronal populations. Protection by IFN-γ in neuronal borna disease virus (BDV) infection was found to be complete in cerebellar cultures, while hippocampal neuron cultures were only partially protected (Freidl et al., 2004). Most likely neurons differ either in the expression of IFN-γ receptor subunits involved in antiviral intracellular signaling pathways or in their ability to collaborate with type I IFN functions, important for the inhibition of replication of other viruses (Pierce et al., 2005, Sainz et al., 2005, Vollstedt et al., 2004).
Previous reports have mainly discussed the beneficial effects of IFN-γ on the survival (Chang et al., 1990), growth (Erkman et al., 1989) and differentiation of neurons in vitro (Jonakait et al., 1994). IFN-γ also affords neural protection from destructive encephalitis during HSV-1 infection of the CNS by inhibiting virus-induced apoptosis. Levels of IFN-γ, released during acute and chronic infections and immunological reactions are sustained for a long time after viral clearance and during viral latency (Fan et al., 1993). But prolonged exposure of neurons to IFN-γ at concentrations similar to those detected after chronic viral infection in humans and mice has been correlated with major dendritic retraction (Li et al., 1999, Masliah et al., 1997, Rockstroh et al., 1998). IFN-γ therefore plausibly has a ‘dual-edged’ role in its antiviral responses: it promotes viral clearance while also contributing to the pathology by inducing dendritic retraction. It is interesting to note the coincidence of dendritic retraction (Park et al., 1996) with elevated IFN-γ expression (Kristensson et al., 1994, Lau and Yu, 2001) in many types of acute inflammatory reactions, including those triggered by trauma, stroke, and axotomy.
Virus-laden neurons may bypass or evade T cell antiviral functions by their inefficiency in upregulating MHC class-I molecules, their expression of antiapoptotic molecules and an intrinsic non-cytolytic mechanism to inhibit CTL and NK cell effector functions (Burdeinick-Kerr et al., 2007, Divito et al., 2006). Virus-specific T cells express NK cell inhibitory receptor NKG2A that can dimerize with CD94 to inhibit CTL and NK cell effector functions. In mice, CD94-NKG2A complex binds the ligand Qa-1, a non-classical MHC molecule homologous to human HLA-E. In a systemic HSV-1 infection model, spleen isolates of T cells upregulated CD94 NKG2A and still retained IFN-γ production capacity in response to peptide stimulation (Wojtasiak et al., 2004). In a corneal infection model, most granzyme B+-CD8+ effector T cells were found to express CD94-NKG2A and showed weakened cytotoxicity against Qa-1-expressing target cells. By blocking the Qa-1/CD94-NKG2A interaction using anti-Qa-1 mAb in vitro, this insufficiency was rectified. The study also showed that approximately 35% of neurons from latent virus-infected trigeminal ganglion neurons express Qa-1 (Suvas et al., 2006). In contrast to this observation, upregulation of the NKG2D ligands RAE-1, MULT1 and HS60 in MHV infection augments CD8+ T cell cytolysis without affecting IFN-γ production or demyelination (Walsh et al., 2008). These studies indicate the crucial role of IFN-γ in adopting separate effector mechanisms for controlling virus replication within a single target, the CNS.
6. Conclusion
Despite the exceedingly complex structural and functional makeup of the CNS, it seems to have a limited but effective arsenal for defending and containing neurotropic viral infections. Immune events in the CNS that have been traced till date are a net result of the combined actions of resident brain cells and the recruited peripheral leukocytes, making it overtly difficult to identify the immune component that originates with resident cell populations. Although most of the immune function associated with the brain in the face of a viral attack has been hitherto attributed primarily to events launched by microglia and astrocytes, accumulating evidence now suggests a less vigorous but no less important neural participation in the antiviral events. Research is however at its nascent stages in elucidating the antiviral immune mechanisms orchestrated by neurons in intraneural and extraneural sites. The complex communications between neuron, virus and the immune system is only beginning to be deciphered. Also, mechanisms directing differential responses of neuronal populations to immune mediators are only poorly understood. Many doubts and questions remain unresolved regarding the level at which environmental factors and innate immunity interacts in modulating the molecular cross-talk between neuronal and glial elements in the virus-infected CNS. The molecular mechanisms coupling detection of viruses to induction of IFN genes have only begun to be identified. Much research needs to be carried out to understand neuronal cell demise due to virus encoded proteins and further immune processes that follow this cell death. The viral or cellular mechanisms of induction of neuronal apoptosis are still incompletely understood. Cell type-specific actions of T cell mediated antiviral functions probably contribute to viral persistence. Neuronal response to immune effectors is site-specific. Even a single virus type can act unpredictably in infected individuals, infecting different regions of the brain to cause different symptoms, or resulting in CNS disease in only a small minority of infected individuals. Such diversity of consequences of viral infection makes it difficult to analyze the pathogenesis of the disease in a particular target organ, let alone tracking defense response tracking in individual cell types. With sophisticated technological advancements, however, we are optimistic that further understanding of the role of neurons in launching immunity within the CNS will be of immense help to develop precise therapeutics and designing vaccines for treatment and prophylaxis of the broad range of viral diseases in the CNS.
Acknowledgements
The work in the author's laboratory is funded by the grant from the Department of Biotechnology (Award#BT/PR/5799/MED/14/698/2005 and BT/PR8682/Med/14/1273/2007), and the Council of Scientific and Industrial Research ((27(0173)/07/EMR-II), Government of India. A.N. is a recipient of Junior Research Fellowship from Council of Scientific and Industrial Research. K.D. is a recipient of Research Associateship in Biotechnology and Life Sciences from the Department of Biotechnology, Government of India.
References
- Abendroth A., Simmons A., Efstathiou S., Pereira R.A. Infection with an H2 recombinant herpes simplex virus vector results in expression of MHC class I antigens on the surfaces of human neuroblastoma cells in vitro and mouse sensory neurons in vivo. J. Gen. Virol. 2000;81(Pt 10):2375–2383. doi: 10.1099/0022-1317-81-10-2375. [DOI] [PubMed] [Google Scholar]
- Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Anderson J.P., Mueller J.L., Misaghi A., Anderson S., Sivagnanam M., Kolodner R.D., Hoffman H.M. Initial description of the human NLRP3 promoter. Genes Immun. 2008;9(8):721–726. doi: 10.1038/gene.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou A.N., Powis S.J. Pathogen evasion strategies for the major histocompatibility complex class I assembly pathway. Immunology. 2008;124(1):1–12. doi: 10.1111/j.1365-2567.2008.02804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. The molecular and cellular biology of the chemokines. J. Viral Hepat. 1998;5(1):1–14. doi: 10.1046/j.1365-2893.1998.00081.x. [DOI] [PubMed] [Google Scholar]
- Bergmann C.C., Lane T.E., Stohlman S.A. Coronavirus infection of the central nervous system: host-virus stand-off. Nat. Rev. Microbiol. 2006;4(2):121–132. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann C.C., Altman J., Hinton D., Stohlman S. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the CNS. J. Immunol. 1999;163(6):3379–3387. [PubMed] [Google Scholar]
- Bertollini C., Ragozzino D., Gross C., Limatola C., Eusebi F. Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacology. 2006;51(4):816–821. doi: 10.1016/j.neuropharm.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Biber K., Neumann H., Inoue K., Boddeke H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30(11):596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Bieback K., Lien E., Klagge I.M., Avota E., Schneider-Schaulies J., Duprex W.P., Wagner H., Kirschning C.J., Ter Meulen V., Schneider-Schaulies S. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 2002;76(17):8729–8736. doi: 10.1128/JVI.76.17.8729-8736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder G.K., Griffin D.E. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science. 2001;293(5528):303–306. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- Borden E.C., Sen G.C., Uze G., Silverman R.H., Ransohoff R.M., Foster G.R., Stark G.R. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007;6(12):975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger L.M., Shatz C.J. Immune signaling in neural development, synaptic plasticity and disease. Nat. Rev. Neurosci. 2004;5(7):521–531. doi: 10.1038/nrn1428. [DOI] [PubMed] [Google Scholar]
- Bowie A.G., Haga I.R. The role of Toll-like receptors in the host response to viruses. Mol. Immunol. 2005;42(8):859–867. doi: 10.1016/j.molimm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Burdeinick-Kerr R., Griffin D.E. Gamma interferon-dependent, noncytolytic clearance of sindbis virus infection from neurons in vitro. J. Virol. 2005;79(9):5374–5385. doi: 10.1128/JVI.79.9.5374-5385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdeinick-Kerr R., Wind J., Griffin D.E. Synergistic roles of antibody and interferon in noncytolytic clearance of Sindbis virus from different regions of the central nervous system. J. Virol. 2007;81(11):5628–5636. doi: 10.1128/JVI.01152-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahic M., Roussarie J.P. Axon-Myelin interactions during a viral infection of the central nervous system. PLoS Pathog. 2009;5(9):e1000519. doi: 10.1371/journal.ppat.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.Y., Martin D.P., Johnson E.M., Jr. Interferon suppresses sympathetic neuronal cell death caused by nerve growth factor deprivation. J. Neurochem. 1990;55(2):436–445. doi: 10.1111/j.1471-4159.1990.tb04155.x. [DOI] [PubMed] [Google Scholar]
- Charo I.F., Ransohoff R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006;354(6):610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Chesler D.A., Reiss C.S. The role of IFN-gamma in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002;13(6):441–454. doi: 10.1016/s1359-6101(02)00044-8. [DOI] [PubMed] [Google Scholar]
- Compton T., Kurt-Jones E.A., Boehme K.W., Belko J., Latz E., Golenbock D.T., Finberg R.W. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 2003;77(8):4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan C.M., McManus C.M., Sharron M., Gao Z., Murphy D., Jaffer S., Choe W., Chen W., Hesselgesser J., Gaylord H. Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience. 2000;97(3):591–600. doi: 10.1016/s0306-4522(00)00024-5. [DOI] [PubMed] [Google Scholar]
- Daffis S., Samuel M.A., Keller B.C., Gale M., Jr., Diamond M.S. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 2007;3(7):e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S., Samuel M.A., Suthar M.S., Gale M., Jr., Diamond M.S. Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 2008;82(21):10349–10358. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S., Ghosh D., Basu A. Japanese encephalitis virus induce immuno-competency in neural stem/progenitor cells. PLoS One. 2009;4(12):e8134. doi: 10.1371/journal.pone.0008134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haas A.H., van Weering H.R., de Jong E.K., Boddeke H.W., Biber K.P. Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol. Neurobiol. 2007;36(2):137–151. doi: 10.1007/s12035-007-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold S.S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Divito S., Cherpes T.L., Hendricks R.L. A triple entente: virus, neurons, and CD8+ T cells maintain HSV-1 latency. Immunol. Res. 2006;36(1–3):119–126. doi: 10.1385/IR:36:1:119. [DOI] [PubMed] [Google Scholar]
- Dorries R. The role of T-cell-mediated mechanisms in virus infections of the nervous system. Curr. Top Microbiol. Immunol. 2001;253:219–245. doi: 10.1007/978-3-662-10356-2_11. [DOI] [PubMed] [Google Scholar]
- Erkman L., Wuarin L., Cadelli D., Kato A.C. Interferon induces astrocyte maturation causing an increase in cholinergic properties of cultured human spinal cord cells. Dev. Biol. 1989;132(2):375–388. doi: 10.1016/0012-1606(89)90234-0. [DOI] [PubMed] [Google Scholar]
- Fabis M.J., Phares T.W., Kean R.B., Koprowski H., Hooper D.C. Blood–brain barrier changes and cell invasion differ between therapeutic immune clearance of neurotrophic virus and CNS autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 2008;105(40):15511–15516. doi: 10.1073/pnas.0807656105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabry Z., Raine C.S., Hart M.N. Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol. Today. 1994;15(5):218–224. doi: 10.1016/0167-5699(94)90247-X. [DOI] [PubMed] [Google Scholar]
- Fan J., Bass H.Z., Fahey J.L. Elevated IFN-gamma and decreased IL-2 gene expression are associated with HIV infection. J. Immunol. 1993;151(9):5031–5040. [PubMed] [Google Scholar]
- Farina C., Aloisi F., Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28(3):138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Fazakerley J.K., Walker R. Virus demyelination. J. Neurovirol. 2003;9(2):148–164. doi: 10.1080/13550280390194046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freidl G., Hofer M., Auber B., Sauder C., Hausmann J., Staeheli P., Pagenstecher A. Borna disease virus multiplication in mouse organotypic slice cultures is site-specifically inhibited by gamma interferon but not by interleukin-12. J. Virol. 2004;78(3):1212–1218. doi: 10.1128/JVI.78.3.1212-1218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D., Basu A. Present perspectives on flaviviral chemotherapy. Drug Discov. Today. 2008;13(13–14):619–624. doi: 10.1016/j.drudis.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Ghosh D., Basu A. Japanese encephalitis—a pathological and clinical perspective. PLoS Negl. Trop. Dis. 2009;3(9):e437. doi: 10.1371/journal.pntd.0000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal A., Das S., Ghosh S., Mishra M.K., Sharma V., Koli P., Sen E., Basu A. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia. 2007;55(5):483–496. doi: 10.1002/glia.20474. [DOI] [PubMed] [Google Scholar]
- Gogate N., Swoveland P., Yamabe T., Verma L., Woyciechowska J., Tarnowska-Dziduszko E., Dymecki J., Dhib-Jalbut S. Major histocompatibility complex class I expression on neurons in subacute sclerosing panencephalitis and experimental subacute measles encephalitis. J. Neuropathol. Exp. Neurol. 1996;55(4):435–443. doi: 10.1097/00005072-199604000-00006. [DOI] [PubMed] [Google Scholar]
- Griffin D.E. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003;3(6):493–502. doi: 10.1038/nri1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch U.K., Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hausmann J., Pagenstecher A., Baur K., Richter K., Rziha H.J., Staeheli P. CD8 T cells require gamma interferon. J. Virol. 2005;79(21):13509–13518. doi: 10.1128/JVI.79.21.13509-13518.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havert M.B., Schofield B., Griffin D.E., Irani D.N. Activation of divergent neuronal cell death pathways in different target cell populations during neuroadapted sindbis virus infection of mice. J. Virol. 2000;74(11):5352–5356. doi: 10.1128/jvi.74.11.5352-5356.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F., Hemmi H., Hochrein H., Ampenberger F., Kirschning C., Akira S., Lipford G., Wagner H., Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Hickey W.F., Hsu B.L., Kimura H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991;28(2):254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 2007;282(21):15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- Huh G.S., Boulanger L.M., Du H., Riquelme P.A., Brotz T.M., Shatz C.J. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290(5499):2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland D.D., Stohlman S.A., Hinton D.R., Atkinson R., Bergmann C.C. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 2008;82(1):300–310. doi: 10.1128/JVI.01794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D.J., Wunner W.H., Ertl H.C., Jackson A.C. Basis of rabies virus neurovirulence in mice: expression of major histocompatibility complex class I and class II mRNAs. J. Neurovirol. 1999;5(5):485–494. doi: 10.3109/13550289909045377. [DOI] [PubMed] [Google Scholar]
- Jonakait G.M., Wei R., Sheng Z.L., Hart R.P., Ni L. Interferon-gamma promotes cholinergic differentiation of embryonic septal nuclei and adjacent basal forebrain. Neuron. 1994;12(5):1149–1159. doi: 10.1016/0896-6273(94)90322-0. [DOI] [PubMed] [Google Scholar]
- Kennedy P.G., Gairns J. Major histocompatibility complex (MHC) antigen expression in HIV encephalitis. Neuropathol. Appl. Neurobiol. 1992;18(5):515–522. doi: 10.1111/j.1365-2990.1992.tb00818.x. [DOI] [PubMed] [Google Scholar]
- Khuth S.T., Akaoka H., Pagenstecher A., Verlaeten O., Belin M.F., Giraudon P., Bernard A. Morbillivirus infection of the mouse central nervous system induces region-specific upregulation of MMPs and TIMPs correlated to inflammatory cytokine expression. J. Virol. 2001;75(17):8268–8282. doi: 10.1128/JVI.75.17.8268-8282.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R.S., Lin E., Zhang B., Luster A.D., Tollett J., Samuel M.A., Engle M., Diamond M.S. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005;79(17):11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensson K., Aldskogius M., Peng Z.C., Olsson T., Aldskogius H., Bentivoglio M. Co-induction of neuronal interferon-gamma and nitric oxide synthase in rat motor neurons after axotomy: a role in nerve repair or death? J. Neurocytol. 1994;23(8):453–459. doi: 10.1007/BF01184069. [DOI] [PubMed] [Google Scholar]
- Kumar H., Kawai T., Kato H., Sato S., Takahashi K., Coban C., Yamamoto M., Uematsu S., Ishii K.J., Takeuchi O. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006;203(7):1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon M., Megret F., Lafage M., Prehaud C. The innate immune facet of brain: human neurons express TLR-3 and sense viral dsRNA. J. Mol. Neurosci. 2006;29(3):185–194. doi: 10.1385/JMN:29:3:185. [DOI] [PubMed] [Google Scholar]
- Lafon M., Prehaud C., Megret F., Lafage M., Mouillot G., Roa M., Moreau P., Rouas-Freiss N., Carosella E.D. Modulation of HLA-G expression in human neural cells after neurotropic viral infections. J. Virol. 2005;79(24):15226–15237. doi: 10.1128/JVI.79.24.15226-15237.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson L.A. Interpreting MHC class I expression and class I/class II reciprocity in the CNS: reconciling divergent findings. Microsc. Res. Tech. 1995;32(4):267–285. doi: 10.1002/jemt.1070320402. [DOI] [PubMed] [Google Scholar]
- Lane T.E., Liu M.T., Chen B.P., Asensio V.C., Samawi R.M., Paoletti A.D., Campbell I.L., Kunkel S.L., Fox H.S., Buchmeier M.J. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J. Virol. 2000;74(3):1415–1424. doi: 10.1128/jvi.74.3.1415-1424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau L.T., Yu A.C. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J. Neurotrauma. 2001;18(3):351–359. doi: 10.1089/08977150151071035. [DOI] [PubMed] [Google Scholar]
- Lepoutre V., Jain P., Quann K., Wigdahl B., Khan Z.K. Role of resident CNS cell populations in HTLV-1-associated neuroinflammatory disease. Front. Biosci. 2009;14:1152–1168. doi: 10.2741/3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levite M. Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Curr. Opin. Pharmacol. 2008;8(4):460–471. doi: 10.1016/j.coph.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Li Q., Eiden L.E., Cavert W., Reinhart T.A., Rausch D.M., Murray E.A., Weihe E., Haase A.T. Increased expression of nitric oxide synthase and dendritic injury in simian immunodeficiency virus encephalitis. J. Hum. Virol. 1999;2(3):139–145. [PubMed] [Google Scholar]
- Loconto J., Papes F., Chang E., Stowers L., Jones E.P., Takada T., Kumanovics A., Fischer Lindahl K., Dulac C. Functional expression of murine V2R pheromone receptors involves selective association with the M10 and M1 families of MHC class Ib molecules. Cell. 2003;112(5):607–618. doi: 10.1016/s0092-8674(03)00153-3. [DOI] [PubMed] [Google Scholar]
- Loo Y.M., Fornek J., Crochet N., Bajwa G., Perwitasari O., Martinez-Sobrido L., Akira S., Gill M.A., Garcia-Sastre A., Katze M.G. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008;82(1):335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J.M., Alexopoulou L., Sato A., Karow M., Adams N.C., Gale N.W., Iwasaki A., Flavell R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U.S.A. 2004;101(15):5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier S., Geraghty D.E., Weiss E.H. Expression and regulation of HLA-G in human glioma cell lines. Transplant. Proc. 1999;31(4):1849–1853. doi: 10.1016/s0041-1345(99)00186-4. [DOI] [PubMed] [Google Scholar]
- Mankowski J.L., Queen S.E., Fernandez C.S., Tarwater P.M., Karper J.M., Adams R.J., Kent S.J. Natural host genetic resistance to lentiviral CNS disease: a neuroprotective MHC class I allele in SIV-infected macaques. PLoS One. 2008;3(11):e3603. doi: 10.1371/journal.pone.0003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E., Heaton R.K., Marcotte T.D., Ellis R.J., Wiley C.A., Mallory M., Achim C.L., McCutchan J.A., Nelson J.A., Atkinson J.H. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann. Neurol. 1997;42(6):963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- Matsumoto M., Funami K., Tanabe M., Oshiumi H., Shingai M., Seto Y., Yamamoto A., Seya T. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 2003;171(6):3154–3162. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- Mellado M., Rodriguez-Frade J.M., Vila-Coro A.J., Fernandez S., Martin de Ana A., Jones D.R., Toran J.L., Martinez A.C. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001;20(10):2497–2507. doi: 10.1093/emboj/20.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menager P., Roux P., Megret F., Bourgeois J.P., Le Sourd A.M., Danckaert A., Lafage M., Prehaud C., Lafon M. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri Bodies. PLoS Pathog. 2009;5(2):e1000315. doi: 10.1371/journal.ppat.1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng S.Z., Oka A., Takashima S. Developmental expression of monocyte chemoattractant protein-1 in the human cerebellum and brainstem. Brain Dev. 1999;21(1):30–35. doi: 10.1016/s0387-7604(98)00065-5. [DOI] [PubMed] [Google Scholar]
- Minghetti L., Ajmone-Cat M.A., De Berardinis M.A., De Simone R. Microglial activation in chronic neurodegenerative diseases: roles of apoptotic neurons and chronic stimulation. Brain Res. Brain Res. Rev. 2005;48(2):251–256. doi: 10.1016/j.brainresrev.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Mishra M., Vetrivel S., Siddappa N.B., Ranga U., Seth P. Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: significance of dicysteine C30C31 motif. Ann. Neurol. 2008;63(3):366–376. doi: 10.1002/ana.21292. [DOI] [PubMed] [Google Scholar]
- Mishra M.K., Koli P., Bhowmick S., Basu A. Neuroprotection conferred by astrocytes is insufficient to protect animals from succumbing to Japanese encephalitis. Neurochem. Int. 2007;50(5):764–773. doi: 10.1016/j.neuint.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Neumann H., Misgeld T., Matsumuro K., Wekerle H. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc. Natl. Acad. Sci. U.S.A. 1998;95(10):5779–5784. doi: 10.1073/pnas.95.10.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H., Schmidt H., Cavalie A., Jenne D., Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J. Exp. Med. 1997;185(2):305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.S., Bateman M.C., Goldberg M.P. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol. Dis. 1996;3(3):215–227. doi: 10.1006/nbdi.1996.0022. [DOI] [PubMed] [Google Scholar]
- Parra B., Hinton D.R., Marten N.W., Bergmann C.C., Lin M.T., Yang C.S., Stohlman S.A. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J. Immunol. 1999;162(3):1641–1647. [PubMed] [Google Scholar]
- Patterson C.E., Lawrence D.M., Echols L.A., Rall G.F. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma interferon dependent. J. Virol. 2002;76(9):4497–4506. doi: 10.1128/JVI.76.9.4497-4506.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson C.E., Daley J.K., Rall G.F. Neuronal survival strategies in the face of RNA viral infection. J. Infect. Dis. 2002;186(Suppl. 2):S215–S219. doi: 10.1086/344265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petito C.K., Kerza-Kwiatecki A.P., Gendelman H.E., McCarthy M., Nath A., Podack E.R., Shapshak P., Wiley C.A. Review: neuronal injury in HIV infection. J. Neurovirol. 1999;5(4):327–341. doi: 10.3109/13550289909029474. [DOI] [PubMed] [Google Scholar]
- Pierce A.T., DeSalvo J., Foster T.P., Kosinski A., Weller S.K., Halford W.P. Beta interferon and gamma interferon synergize to block viral DNA and virion synthesis in herpes simplex virus-infected cells. J. Gen. Virol. 2005;86(Pt 9):2421–2432. doi: 10.1099/vir.0.80979-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehaud C., Megret F., Lafage M., Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 2005;79(20):12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall G.F., Mucke L., Oldstone M.B. Consequences of cytotoxic T lymphocyte interaction with major histocompatibility complex class I-expressing neurons in vivo. J. Exp. Med. 1995;182(5):1201–1212. doi: 10.1084/jem.182.5.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff R.M., Kivisakk P., Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003;3(7):569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- Rassa J.C., Meyers J.L., Zhang Y., Kudaravalli R., Ross S.R. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. U.S.A. 2002;99(4):2281–2286. doi: 10.1073/pnas.042355399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassa J.C., Ross S.R. Viruses and Toll-like receptors. Microbes Infect. 2003;5(11):961–968. doi: 10.1016/s1286-4579(03)00193-x. [DOI] [PubMed] [Google Scholar]
- Redwine J.M., Buchmeier M.J., Evans C.F. In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. Am. J. Pathol. 2001;159(4):1219–1224. doi: 10.1016/S0002-9440(10)62507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockstroh J.K., Kreuzer K.A., Sauerbruch T., Spengler U. Protein levels of interleukin-12 p70 holomer, its p40 chain and interferon-gamma during advancing HIV infection. J. Infect. 1998;37(3):282–286. doi: 10.1016/s0163-4453(98)92138-7. [DOI] [PubMed] [Google Scholar]
- Rodriguez M., Zoecklein L.J., Howe C.L., Pavelko K.D., Gamez J.D., Nakane S., Papke L.M. Gamma interferon is critical for neuronal viral clearance and protection in a susceptible mouse strain following early intracranial Theiler's murine encephalomyelitis virus infection. J. Virol. 2003;77(22):12252–12265. doi: 10.1128/JVI.77.22.12252-12265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rom I., Deshmane S.L., Mukerjee R., Khalili K., Amini S., Sawaya B.E. HIV-1 Vpr deregulates calcium secretion in neural cells. Brain Res. 2009;1275:81–86. doi: 10.1016/j.brainres.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg M., Kristensson K. Effects of interferon-gamma on neuronal infections. Viral Immunol. 2002;15(2):247–260. doi: 10.1089/08828240260066206. [DOI] [PubMed] [Google Scholar]
- Savarin C., Bergmann C.C. Neuroimmunology of central nervous system viral infections: the cells, molecules and mechanisms involved. Curr. Opin. Pharmacol. 2008;8(4):472–479. doi: 10.1016/j.coph.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainz B., Jr., LaMarca H.L., Garry R.F., Morris C.A. Synergistic inhibition of human cytomegalovirus replication by interferon-alpha/beta and interferon-gamma. Virol. J. 2005;2:14. doi: 10.1186/1743-422X-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B., Wang T., Samuel M.A., Whitby K., Craft J., Fikrig E., Diamond M.S. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J. Virol. 2006;80(11):5338–5348. doi: 10.1128/JVI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel C.D., Hahto S.M., Ciavarra R.P. Peripheral dendritic cells are essential for both the innate and adaptive antiviral immune responses in the central nervous system. Virology. 2009;387(1):117–126. doi: 10.1016/j.virol.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y., Potula R., Dhillon N., Pinson D., Li S., Nath A., Anderson C., Turchan J., Kolson D., Narayan O. Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am. J. Pathol. 2004;164(5):1557–1566. doi: 10.1016/S0002-9440(10)63714-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvas S., Azkur A.K., Rouse B.T. Qa-1b and CD94-NKG2a interaction regulate cytolytic activity of herpes simplex virus-specific memory CD8+ T cells in the latently infected trigeminal ganglia. J. Immunol. 2006;176(3):1703–1711. doi: 10.4049/jimmunol.176.3.1703. [DOI] [PubMed] [Google Scholar]
- Tabeta K., Georgel P., Janssen E., Du X., Hoebe K., Crozat K., Mudd S., Shamel L., Sovath S., Goode J. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. U.S.A. 2004;101(10):3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong P., Heydari S., Garidou L., McGavern D.B. Persistent viral infection elevates central nervous system MHC class I through chronic production of interferons. J. Immunol. 2009;183(6):3895–3905. doi: 10.4049/jimmunol.0803085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker P.C., Griffin D.E., Choi S., Bui N., Wesselingh N. Inhibition of nitric oxide synthesis increases mortality in Sindbis virus encephalitis. J. Virol. 1996;70(6):3972–3977. doi: 10.1128/jvi.70.6.3972-3977.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubogu E.E., Cossoy M.B., Ransohoff R.M. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol. Sci. 2006;27(1):48–55. doi: 10.1016/j.tips.2005.11.002. [DOI] [PubMed] [Google Scholar]
- van den Pol A.N. Viral infection leading to brain dysfunction: more prevalent than appreciated? Neuron. 2009;64(1):17–20. doi: 10.1016/j.neuron.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollstedt S., Arnold S., Schwerdel C., Franchini M., Alber G., Di Santo J.P., Ackermann M., Suter M. Interplay between alpha/beta and gamma interferons with B, T, and natural killer cells in the defense against herpes simplex virus type 1. J. Virol. 2004;78(8):3846–3850. doi: 10.1128/JVI.78.8.3846-3850.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh K.B., Lodoen M.B., Edwards R.A., Lanier L.L., Lane T.E. Evidence for differential roles for NKG2D receptor signaling in innate host defense against coronavirus-induced neurological and liver disease. J. Virol. 2008;82(6):3021–3030. doi: 10.1128/JVI.02032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Town T., Alexopoulou L., Anderson J.F., Fikrig E., Flavell R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004;10(12):1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Wojtasiak M., Jones C.M., Sullivan L.C., Winterhalter A.C., Carbone F.R., Brooks A.G. Persistent expression of CD94/NKG2 receptors by virus-specific CD8 T cells is initiated by TCR-mediated signals. Int. Immunol. 2004;16(9):1333–1341. doi: 10.1093/intimm/dxh136. [DOI] [PubMed] [Google Scholar]
- Weber F., Haller O. Viral suppression of the interferon system. Biochimie. 2007;89(6–7):836–842. doi: 10.1016/j.biochi.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wekerle H. Planting and pruning in the brain: MHC antigens involved in synaptic plasticity? Proc. Natl. Acad. Sci. U.S.A. 2005;102(1):3–4. doi: 10.1073/pnas.0408495101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.W., Herrup K. The control of neuron number. Annu. Rev. Neurosci. 1988;11:423–453. doi: 10.1146/annurev.ne.11.030188.002231. [DOI] [PubMed] [Google Scholar]
- Yong V.W., Power C., Forsyth P., Edwards D.R. Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2001;2(7):502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Ye L., Wan Q., Zhou L., Wang X., Li J., Hu S., Zhou D., Ho W. Activation of Toll-like receptors inhibits herpes simplex virus-1 infection of human neuronal cells. J. Neurosci. Res. 2009;87(13):2916–2925. doi: 10.1002/jnr.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]