

Abstract
Malaria has been teasing human populations from a long time. Presently, several classes of antimalarial drugs are available in market, but the issues of toxicity, lower efficacy and the resistance by malarial parasites have decreased their overall therapeutic indices. Thus, the search for new promising antimalarials continues, however, the battle against malaria is far from over. Ferroquine is a derivative of chloroquine with antimalarial properties. It is the most successful of the chloroquine derivatives. Not only ferroquine, but also its derivatives have shown promising potential as antimalarials of clinical interest. Presently, much research is dedicated to the development of ferroquine derivatives as safe alternatives to antimalarial chemotherapy. The present article describes the structural, chemical and biological features of ferroquine. Several classes of ferroquine derivatives including hydroxyferroquines, trioxaferroquines, chloroquine-bridged ferrocenophanes, thiosemicarbazone derivatives, ferrocene dual conjugates, 4-N-substituted derivatives, and others have been discussed. Besides, the mechanism of action of ferroquine has been discussed. A careful observation has been made into pharmacologically significant ferroquine derivatives with better or equal therapeutic effects to that of chloroquine and ferroquine. A brief discussion of the toxicities of ferroquine derivatives has been made. Finally, efforts have been made to discuss the current challenges and future perspectives of ferroquine-based antimalarial drug development.
Keywords: Ferroquine, Ferroquine derivatives, Hydroxyferroquines, Chloroquine-bridged ferrocenophanes, Ferrocene dual conjugates, Future perspectives
Graphical abstract

Highlights
-
•
Structural, chemical and biological features of ferroquine have been discussed.
-
•
Several classes of ferroquine derivatives have been reviewed.
-
•
Mechanism of action of ferroquine has been described.
-
•
Challenges in ferroquine-based antimalarial drug development have been highlighted.
-
•
Perspectives in ferroquine-based antimalarial drug development have been outlined.
1. Introduction
Malaria is a mosquito-borne disease of human beings caused by parasitic protozoans of the Plasmodium genus [1]. Parasitic infections are recognized as one of the major causes of deaths in the third world countries. Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, and Plasmodium malariae are the four notorious species of the protozoal parasites causing malaria in humans. P. falciparum and P. vivax account for more than 95% of the malarial infections worldwide [2]. Among the various species of Plasmodium, P. falciparum is the most lethal one causing the most virulent form of human malaria. Annually, P. falciparum is known to cause 200–300 million infections and 1–3 million deaths [3]. Generally, malaria is associated with symptoms such as fatigue, fever, headaches and vomiting. In severe cases it may cause yellow skin, seizures, coma or even death [4]. Malaria has been reported to infect human populations for over 50,000 years [5]. The first evidence of Plasmodium was reported in a fossilized Culex mosquito in a piece of amber, almost 30 million years old [6].
Presently, several drugs such as chloroquine (CQ), amodiaquine, pyrimethamine, proguanil, mefloquine, atovaquone, primaquine, etc. (Fig. 1 ) are available in market for the treatment of malaria. The main target of most of the antimalarial drugs is the erythrocytic stage of malarial infection. From the last two decades, the resistance of P. falciparum strains to CQ, and the emergence of antifolate combination of sulfadoxine/pyrimethamine led to artemisinin combination therapy (ACT) [7]. ACT is now a worldwide treatment of uncomplicated malaria [8]. However, some recent fears of the onset of artemisinin resistance in Cambodia [9] stimulated the development of new antidotes against malaria.
Fig. 1.

Some of the market available antimalarial drugs.
In 1994, ferroquine [FQ, (SSR97193)] was designed by Biot and co-workers at the University of Lille. Later on, it was successfully synthesized by incorporating a ferrocene unit into the basic skeleton of CQ (Fig. 2 ) [10]. FQ was found to be remarkably effective against CQ-resistant P. falciparum [11] with no observable immunotoxic effects in naıve and infected young rats [12]. It acts on haematin and causes the inhibition of hemozoin formation [13]. The interesting antimalarial properties of FQ stimulated the extensive development of its analogues with the hope of increased efficacy, lower side effects and the ability to overcome resistance by malarial parasites. Presently, several classes of FQ derivatives and analogues have been prepared and tested for antimalarial properties, and interesting results have been obtained in several instances.
Fig. 2.

Schematic representation of the synthesis of ferroquine by Biot and co-workers.
2. Review background
Literature updates indicate an increasing interest in the development of antimalarial drugs from the last couple of decades. A through and exhaustive search of literature through SciFinder and PubMed showed that FQ and its derivatives are being extensively investigated as antimalarial agents. More than hundred research papers have appeared on antimalarial properties of FQ and its derivatives with interesting results. An analysis of the number of publications on “FQ and its derivatives as antimalarial agents” from 2000 to 15 indicated a steadily growing interest in the research on this topic. The period from 2000 to 15 was divided into three intervals viz. 2001–04, 2005–09 and 2010–15, and a graph was plotted as shown in Fig. 3 . It is clear from this figure that the number of research reports has increased considerably from 10 during 2000–04 through 23 during 2005–09 to 37 during 2010–15. A further look into the literature indicated that some review papers have appeared on FQ as an antimalarial agent [11], [14], [15], [16], [17], [18], [19], [20]. Besides, a book chapter by Biot et al. [21] has appeared on antimalarial properties of FQ. However, no relevant review has appeared on this topic after 2011. Therefore, an updated review on the recent advances in this field is largely missing. Thus, it was thought worthwhile to address all the recent advances in the development of FQ and its derivatives as antimalarials of clinical interest. The present review highlights the epidemiology of malaria as a global concern, and the need for the development of better FQ derivatives. Besides, chemical and biological features of FQ have been discussed. In addition, mechanism of action of FQ against malarial parasites has been described. A careful observation into pharmacologically significant FQ derivatives with better or equal therapeutic effects to that of CQ and FQ has been presented. In addition, issues of the toxicity of ferroquine derivatives have been outlined. Finally, current challenges and future perspectives of FQ-based antimalarial drug development have been discussed. This review is an updated reference material that will be of significant help to the researchers in antimalarial drug development.
Fig. 3.

A graphic representation of the increasing interest in the research on ferroquine and its derivatives as antimalarial agents from 2000–04 through 2005–09 to 2010–15.
3. Malaria: a clinical overview
Most often, malaria is characterized by symptoms of fever. The clinical appearances of malaria are attributed to erythrocytic phase of the parasitic development [22]. The initial malarial symptoms following prepatent period constitute the primary attack. As the disease progresses, there is relapse of symptoms at regular intervals of 48–72 h, which are called short term relapses. Besides, long term relapses after 20–60 days or more have also been reported. The malarial infections by P. vivax and P. ovale involve relapses due to reactivation of hypnozoites in liver. In falciparum and malariae infections, relapses are referred to as recrudescences. The recrudescences are thought to be due to persistent blood infections. During prepatent period, the infected individuals may not have any characteristic symptoms. At the end of erythrocytic schizogony phase, RBCs are ruptured by mature schizonts releasing merozoites into blood, and thus infecting more RBCs. The growing parasite gradually consumes and degrades intracellular proteins, more specifically haemoglobin, which results in malarial pigment formation and haemolysis of the infected RBCs. The transport properties of RBC membranes are altered, and they become more spherical and less deformable. The ruptured RBCs release several factors and toxins (red cell membrane lipid, glycosyl phosphatidyl inositol anchor of a parasite membrane protein), which directly initiate the release of cytokines such as tumour necrosis factor (TNF) and interleukin-1 from macrophages, leading to chills and high-grade fever. This occurs once in 48/72 h, corresponding to the erythrocytic cycle [23]. In P. falciparum malaria, infected RBCs develop sticky protrusions on cell membranes, which help them in adhesion and clumping. Besides, the RBCs may also stick to the walls of minute capillaries, venules and arterioles and block blood flow. These processes may lead to damage of vital organs including kidneys, brain, liver, lungs and gastrointestinal tract. The most serious complication of falciparum malaria is the cerebral malaria, wherein the blockage of the cerebral microcirculation by the parasitized RBCs seems as the basic underlying defect. However, there is no complete obstruction to blood flow as the survivors do not have any permanent neurological deficit. The P. vivax and P. ovale infections are generally benign with rare complications of significant morbidity and mortality. Some of the sporozoites of P. vivax and P. ovale hibernate in liver, and the hibernating hypnozoites reactivate leading to relapses of the clinical illness once in 2–3 months.
4. Epidemiology of malaria
Malaria is a global epidemic disease, which has been known in China, Mesopotamia and Egypt since 2700, 2000 and 1570 BC, respectively [24]. The prominence of malarial infections grew due to advancements in agriculture and migration of human populations to colonize new lands [25]. Presently, malaria is one of the major causes of human miseries due to increasing morbidity and mortality, and halt of intellectual and economic growth [26]. Malaria affects approximately 225 million people annually with 780,000 deaths, and most of these deaths occur in children below 5 years age [27]. Malarial epidemiology reports of 2009, indicated approximately 243 million malarial cases and more than 800,000 deaths, which makes malaria as one of the most important human diseases of parasitic origin. Out of the five Plasmodium species that infect human beings, P. falciparum is the most severe cause of deaths [28]. Approximately, 1.2 billion people are at high risk out of 3.4 billion people; who get exposure to the malarial infections every year, globally. World Health Organization (WHO) indicated more than 207 million people with symptomatic malaria in addition to 627,000 deaths in 2012 [29]. Most of the deaths occurred in children in Africa, with one child dying every minute [30]. Overall, malaria is an endemic disease that affects all the populations of the world.
5. Emergence of ferroquine: an exciting ferrocene conjugate
The strategic incorporation of ferrocenyl moieties into the structures of well-known antimalarial agents was first tried during the mid-1990s, and still yet these structural designs and their investigations are continuing. It was very much surprising to obtain disappointing results with the ferrocenyl incorporated mefloquine, quinine [31], artemisinin [32], [33] and atovaquone [34]. The antimalarial activities of these newly grafted molecules were either inferior or equal to the parent drug molecules. This failure of the ferrocenyl derivatives of mefloquine and quinine was attributed to their instability in acidic conditions [15].
In the late end of the 20th century, ferrocenyl compounds of fluoroquinolones were obtained via esterification of the carboxylic group of ciprofloxacin along with the addition of a ferrocenyl moiety [35]. The compounds produced by this approach were considerably more active than ciprofloxacin. Esterification was assumed to enhance activity along with the complementary efficacy enhancement due to the insertion of the ferrocene core. It was the derivatization of CQ that finally gave birth to potentially effective antimalarial agents. Several grafting experiments indicated that the simple addition product of basic CQ and ferrocene carboxylic acid formed via a salt bridge displayed poor antimalarial activity probably due to the antagonism between the two compounds [36]. In addition, condensation of ferrocene at C-3 (Fig. 4 ) or on the endocyclic nitrogen also decreased activity with respect to CQ [37]. The monoferrocenyl derivatives wherein the ferrocenyl group was linked to the terminal nitrogen along with a modulation in the lateral chain size also could not resulted in activity enhancement as compared to CQ [38]. Besides, these molecules demonstrated a high risk of cross-resistance with CQ. Particularly, inconsistent results were obtained with diferrocenyl conjugates probably because of their instability under the experimental conditions [38]. Surprisingly, it was ferrocene's insertion into the lateral chain of CQ that resulted in the formation of compounds with high activity against both CQ-susceptible and CQ-resistant strains of P. falciparum. The activity was independent of the substitution of the terminal nitrogen [10], [38], [39], [40]. One of the main determinants of the antimalarial activity of these compounds towards CQ-resistant parasites was the position of the ferrocene moiety within the lateral chain. FQ was the most active compound out of this class [41]. Additional studies were carried out to find out a molecule with better activity and a more reasonable cost for industrial production. However, the efforts failed to find out a possible competitor for FQ [38]. Further in vitro and in vivo experiments explored FQ as a potential antimalarial agent, which led to its entry into preclinical phase I development at Sanofi-Aventis [42]. However, in response to the WHO's regulations for reducing the emergence of parasitic resistance, FQ was used in combination therapy with artesunate. Subsequent development of FQ continued with its entry into clinical phase II studies in adult humans in late 2007 [43].
Fig. 4.

Energy minimized model of CQ created through MOPAC programme of Chem 3D Ultra. The encircled atoms indicate the target sites which were chemically modified to obtain new derivatives.
5.1. Chemical biological features
FQ is known as the first organometallic drug in which a ferrocenyl group is covalently skirted by a 4-aminoquinoline and a basic alkylamine [Fig. 5 (a)]. It bears planar chirality due to the presence of 1,2-unsymmetrically substituted ferrocene moiety [Fig. 5(b)]. For biological experiments, FQ is often used as a racemic mixture. There were no dramatic differences in the activities of the pure enantiomers of FQ on human or rodent parasites. Almost equal cytotoxicity has been observed for the two enantiomers. These facts have allowed the development of the racemic mixture of FQ as an antimalarial drug [44].
Fig. 5.

Chemistry of ferroquine; (a): Chemical structure of ferroquine indicating the ferrocene, basic alkylamine and the 4-aminoquinoline moieties, (b): R- and S-enantiomers of ferroquine.
A strong intramolecular hydrogen bond of length 2.17 Å between the amine N(11) and the amine N(24) (Fig. 6 ) has been documented to stabilize the structure of neutral FQ in solid-state [13] and solutions of organic solvents such as chloroform [45]. In comparison to the more flexible lateral side chain of CQ, the intramolecular hydrogen bond is stronger in FQ. Several studies have been carried out to demonstrate the effect of this intramolecular hydrogen bond on the antimalarial activity of FQ. Biot et al. [46] replaced the hydrogen atom on the aniline N(11) of FQ by a methyl group to produce FQ-Me. It was observed that FQ-Me exhibited similar physico-chemical properties as that of FQ, and also demonstrated ability to inhibit β-haematin formation. However, FQ-Me was significantly less active against CQ-susceptible and CQ-resistant strains. Thus, the hydrogen bonding interaction in the lateral side chain of FQ was suggested as contributing to its antimalarial activity. This interaction helps to maintain a particular geometric identity of the molecule. It was further proposed that the part of the molecule modified by isosterism (by the insertion of the ferrocenyl moiety instead of the alkyl chain) did not interact with the receptor. However, it properly orients the other functionalities of the molecule. Besides, the ferrocenic moiety tangles with lipidic structures and enables the drug to drip the transport system involved in resistance. It further helps in concentrating the drug at the proper site for the inhibition of hemozoin formation.
Fig. 6.
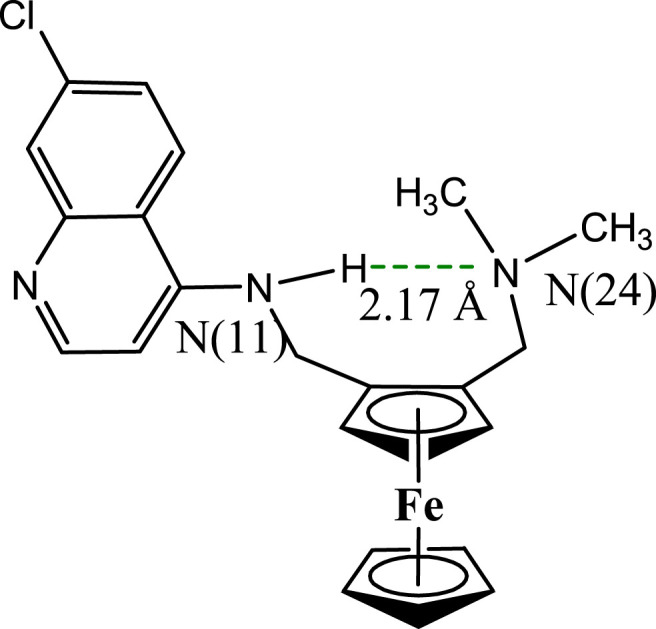
Depiction of a 2.17 Å long hydrogen bond between hydrogen of N(11) and N(24) in FQ.
FQ shares several properties with the parent antimalarial, CQ. CQ acts by accumulating in the digestive vacuoles of parasite and consequently prevents the crystallization of toxic heme into hemozoin. This results in membrane damage and the death of parasite [47], [48]. CQ complexes with haematin in solution, and inhibits β-haematin formation. Similar to CQ, FQ by nature is a weak base with ability to accumulate in the digestive vacuoles of parasite. However, the pKa of FQ is lower as compared to CQ, and therefore, it might be affecting this property CQ [13]. Interestingly, FQ also forms complexes with haematin in addition to the inhibition of haematin crystallization into β-haematin (IC50 0.8 versus 1.9 for CQ) [13], [17]. Moreover, FQ has been reported to produce significant amounts of hydroxyl radicals, in the digestive vacuoles of malarial parasites (acidic pH and presence of H2O2). The hydroxy free radicals are known to damage the parasite [45].
5.2. Ferroquine as an antimalarial agent
Rational drug development based on the molecular structure of chloroquine fostered the development of several ferrocene based molecules. However, among all the ferrocenyl chloroquine derivatives, FQ was the most active derivative in both in vitro and in vivo investigations. It was from the early days of its development considered as a lead compound. One of the most interesting properties of FQ is its ability to overcome the CQ-resistance problem. This property is enough for controlling P. falciparum, which is the principal causative agent of malaria. FQ has been tested in vitro on 16 different laboratory P. falciparum strains [40], [39], [46], [49] and eight sets of field isolates (total 441) collected from Gabon, Senegal, Cambodia and Thailand. In several tests on field isolates from different geographic regions of the world, a minor cross response with CQ was observed. However, the observed cross response was weak and thought as not having a clinical importance for a cross-resistance between the drugs. Moreover, there were no correlations between CQ and FQ responses on using standardized initial parasitaemia during the assays [50], [51], [52], [53], [54], [55]. In the in vivo investigations against Plasmodium vinckei, no antagonistic effects were observed between FQ and artesunate, and the survival time of the infected mice was increased on treatment with the combination of these two drugs [56]. Long et al. performed in vivo investigations on different murine Plasmodium species and observed that the curative dose of FQ (10 mg/kg/d for 4 days) was same irrespective of the susceptibility of the strains to CQ and the administration strategy, which demonstrated a good availability and the powerful activity of the drug. The studies of the effects on naive animal responses and Plasmodium infected hosts indicated two advantages of FQ over CQ. The first one was the rapid efficacy against blood parasites and the decrease of CD4+CD25+ T-cells, which are involved in the expression of susceptibility to experimental malaria infection [57]. Thus, a decrease in the number of these cells would be very helpful for the expression of protective immunity. The second advantage was the potential to maintain the proliferation ability of spleen cells in reply to different mitogens [12]. Thus, on account of the absence of any noticeable immuno-toxicity in rats along with in vitro efficacy against CQ-resistant strains, FQ was suggested as an effective alternative treatment for P. falciparum. Interestingly, the results in both in vitro tests on field isolates and in vivo investigations on rodent models revealed that the potential resistance to FQ was not dependent on a gene polymorphism that is involved in CQ resistance. This was basically observed in Cambodian isolates [54], [55], and further extended to fifteen P. falciparum laboratory strains, with four genes currently involved in drug resistance (pfcrt, pfmdr1, pfmrp, pfhne1) [58]. Additionally, pressure experiments were conducted to obtain FQ-resistant P. falciparum or rodent malaria parasites [15], [59], and it was found that fit-cost of FQ resistance was extremely high. The persisting parasites exhibited a reduced vitality, and the resistance that was observed in rodent malarial parasites was not genetically integrated [15].
5.3. Mechanism of action of ferroquine
It is a well-known fact that the basicity and lipophilicity of FQ are considerably different from CQ [13]. However, the lipophilicity of FQ approaches to that of CQ (log D = −0.77 and −1.2, respectively) when the former is protonated at the food vacuole pH of 5.2. Marked differences in lipophilicity can be observed at pH 7.4 (log D = 2.95 and 0.85 for FQ and CQ, respectively). Additionally, FQ has lower pKa values (pKa1 = 8.19 and pKa2 = 6.99) than CQ (pKa1 = 10.03 and pKa2 = 7.94, respectively). The differential pKa values determine the relative concentrations of the micro species at vacuolar pH such that the neutral and monoprotonated FQ micro species are 10-folds more concentrated at vacuolar pH than those of CQ. These micro species are known to interact with haematin and enable its conversion into hemozoin [60]. Some reports indicated that FQ is 100-folds more lipophilic than CQ at cytosolic pH. At around pH 5, FQ concentrates 50-folds more than CQ [46]. In accordance with the pH affected behaviour of FQ, it was hypothesized that FQ might target the lipid site of hemozoin formation more proficiently [61].
The molecular structure of FQ contains a strong intramolecular hydrogen bond between the anilino N(11) and the tertiary amino N(24) groups (Fig. 6). Results from several NMR experiments have shown that the structure of FQ in solutions (with low dielectric constants such as the lipid environment) is the same as in the solid phase [60]. The role of hydrogen bond on the antimalarial activity of FQ remained a subject of debate for some time. The flip/flop hydrogen bond between the open conformation of charged FQ and the folded conformation of the uncharged FQ should facilitate its transport from water to the hydrophobic membranes. To further justify the importance of hydrogen bond in the molecular structure of FQ, Bio et al. [46] documented that an FQ analogue with a methyl group in place of a hydrogen atom on the anilino N(11) had reduced antimalarial activities against both susceptible and resistant strains of CQ; despite having almost unaffected physicochemical properties to that of FQ. FQ is known to assume a notable conformation wherein the side chain stands in planarity with the quinoline ring, and the bulky ferrocenyl moiety is projected outside. In the molecular structure of FQ, the flexibility of the side chain is reduced due to rigid ferrocene core which leads to a thermodynamic increase in entropy. The increased entropy of FQ has been assumed as one of the main reasons for clustering in the lipids of the membranes [62]. FQ enables favourable interaction owing to the hydrophobic collapse with the lipid structures of the membranes, and the quinoline ring is exposed to water. Similar to CQ, FQ ensures complex formation with haematin in aqueous solutions (log K = 4.95 ± 0.05) in addition to strongly inhibiting β-haematin formation than CQ. Another reason for the high antimalarial activity of FQ in comparison to organic drugs might be the preferential localization of FQ at the lipid–water interface. FQ might help in the prevention of the conversion of haematin into hemozoin by the maintenance of toxic haematin in the aqueous environment, which further justifies the steady activity of FQ despite the resistance level of the strains.
Under the oxidizing conditions of the digestive vacuole of malarial parasites, FQ undergoes a reversible one-electron redox reaction [45] that results into the formation of the ferriquinium salt along with the generation of hydroxyl radicals as shown below.
| Fe(II) + H2O2 → FQ(III) + HO– + HO° |
This free radical generation in the micromolar range does not affect the stability of FQ. Moreover, the clustering of FQ close to the membrane of digestive vacuole causes the generation of ROS and lipid peroxidation. Both the mechanisms viz. inhibition of hemozoin formation and ROS generation eventually lead to death of the malarial parasites.
Researchers are continuously making efforts to further explore the mechanism of FQ as an antimalarial agent against different malarial parasitic strains both in vitro and in vivo. Recently, Dubar et al. [63] documented subcellular mapping of the generation of HO° in P. falciparum RBCs treated with FQ by using a ratiometric fluorescent probe. The oxidative damage was found to be consistent with the damage of the membrane of digestive vacuole leading to death of the parasites. Metallocenic moiety was proved as an important contributor to the overall mechanism of action of FQ. Besides, FQ was supposed to play a crucial role in the inhibition of merozoites reinvasion. Other studies by the same research group [64] related the breakdown of the parasite digestive vacuole membrane to FQ's ability of the generation of hydroxyl radicals.
6. Ferroquine derivatives as antimalarial agents
There was a huge interest in the development of antimalarial drugs based on FQ template after the initial success of FQ as an antimalarial agent. Several classes of structurally diverse molecules have been designed and developed in a hope to ensure resistance free death of malarial parasites. Some of the important classes of FQ derivatives are discussed in the following sub-sections.
6.1. Hydroxyferroquine derivatives
After the revelation of the strong antimalarial effects of FQ on CQ-resistant clones and field isolates, the design and development of hydroxyferroquine derivatives was thought to evolve new molecules with potential activity against P. falciparum and other strains. Biot et al. [40] synthesized three FQ derivatives (Fig. 7 ; 1–3), which closely mimicked hydroxychloroquine. Although the compounds 1–3 were less active than FQ, they displayed the inhibition of in vitro growth of P. falciparum quite higher than CQ. The IC50 values of 1–3 against 3D7 and W2 strains of P. falciparum in comparison to CQ and FQ are given in Table 1 . Interestingly, these compounds also exhibited antiviral effects with selectivity towards severe acute respiratory syndrome coronavirus (SARS-CoV infection). From the results in this research report, it may be suggested that these compounds are interesting alternatives to FQ in antimalarial therapy, and would be highly beneficent in geographical regions where malaria and viral infections co-exist.
Fig. 7.

Chemical structures of the hydroxyferroquine derivatives (1–3).
Table 1.
In vitro activities of 1–3 and 4–7 against P. falciparum strains (3D7 and W2) and (FcB1 and FcM2), respectively with respect to CQ and FQ. The IC50 values in  indicate compounds with better activities than CQ.
indicate compounds with better activities than CQ.

6.2. Trioxaferroquines
Trioxaquines are a class of hybrid molecules with dual modes of action. This class of compounds contains two covalently linked pharmacophores viz. a 1,2,4-trioxane and a 4-aminoquinoline. Such hybrid molecules are thought as possible alternatives to the recently growing resistance to artemisinin [65], [66]. Interestingly, the first generation trioxaquines were highly active against CQ-resistant strains of P. falciparum [67], [68]. Due to the success of hybrid molecules, their fame for the treatment of malaria or other neglected tropical diseases is becoming widespread [69], [70]. FQ may also be seen as a hybrid molecule wherein a ferrocenyl moiety has been inserted within the side chain of CQ [10], [17], [36]. Bellot et al. [71] reported a series of trioxaferroquines (Fig. 8 ; 4–7) as potential antimalarial agents against two CQ-resistant strains, FcB1 and FcM29 (Table 1). Chemically, the reported trioxaferroquines are hybrid molecules with 1,2,4-trioxane moieties covalently linked to FQ. Trioxaferroquines 4–7 displayed IC50 values in the range 16–43 nM. It may be realized that the covalent linking of quinoline and trioxaferrocene via ferrocene has significantly boosted the activities of both the fragments despite the inactivity of CQ towards these strains (Table 1). Also, the essentiality of the 4-aminoquinoline moiety of trioxaferroquines for their antimalarial activities in the case of CQ-resistant strains was highlighted. Compound 4 was the best trioxaferroquine (IC50 = 20 and 17 nM against FcB1 and FcM29, respectively). Thus, it was further investigated in vivo in P. vinckei petteri (2 × 107 parasitized erythrocytes) infected mice. The untreated mice had a survival time of 8 days. On a four day period, the mice were daily given oral dosages at 10 mg/kg/d and 25 mg/kg/d. The results indicated that parasitaemia was below detectable levels on day four even for mice treated at 10 mg/kg/day, which indicated a prompt clearance of the parasites. 0/5 and 2/5 were the numbers of mice treated at 30 days time period for treatments at 10 and 25 mg/kg/d, respectively. The mean survival time of mice treated at 25 mg/kg/d was ca. 18 days, with recurrence between 17 and 21 days. Therefore, the trioxaferroquines have high activity and are quite able to clear parasitaemia below detectable levels in mice treated at 10 mg/kg. However, it's quite important to note that a curative effect with no recurrence was not achieved at doses below 25 mg/kg.
Fig. 8.

Chemical structures of trioxaferroquines (4–7). The 1,2,4-trioxane moieties are covalently linked to ferroquine.
A critical evaluation of this report indicates that trioxaferroquines are a novel class of antiparasitic agents with profound in vivo and in vitro efficacy. However, there is need to lower the dosage below which a complete cure can be obtained. For that purpose, new molecules based on these templates are in need to be developed and investigated in future.
6.3. Chloroquine-bridged ferrocenophane derivatives
Salas et al. [72] documented five disubstituted ferrocene CQ derivatives wherein the terminal nitrogen atoms of the CQ derivatives bridged the two cyclopentadienyl rings of ferrocene (Fig. 9 : 18–22). The derivatives were compared to the corresponding monosubstituted ferrocene CQ analogues (Fig. 9: 13–17) and the corresponding organic fragments (Fig. 9: 8–12). All compounds were active against different strains of P. falciparum including CQ-sensitive D10, CQ-resistant Dd2 and CQ-resistant K1 (Table 2 ). It was observed that the bridged disubstituted ferrocenyl compounds 18–22 overcame resistance and were more active against the resistant strains (except compound 22) as compared to the sensitive ones. The branching of the methylene spacer had a profound effect on the activity as the derivatives lost activity with increase in branching. Besides, the presence of an intramolecular H-bond was not determinant for antiplasmodial activity. All the compounds were found to associate with haematin, however, this association was not an important force for the in vitro activity. The balance between lipophilicity and hydrophilicity of the compounds determined their activities. Addition of ferrocenyl unit largely increased the lipophilicity, which in past that has been related to improved transport of the drugs through membranes [13]. The calculated partition coefficient (log P) values between 4.5–5.0 and topological polar surfaces area (tPSA) of ∼26.0 Å2 gave the best balance to the compounds. The compact size, conformation, and the balance between lipophilicity and hydrophilicity in the bridged derivatives enabled them to escape the mechanisms of resistance by the pathogens. Thus, it may be supposed that the compounds 18–22 would benefit due to their slightly more hydrophilic nature and electronegative area available in addition to having an intramolecular hydrogen-bonding interaction that could allow the flip-flop mechanism. This flip-flop mechanism has been assumed to aid in the accumulation of FQ in crossing membranes.
Fig. 9.

Chemical structures of the disubstituted ferrocene CQ derivatives 18–22 (the terminal nitrogen atoms of the CQ derivatives bridged the two cyclopentadienyl rings of ferrocene), 13–17 (monosubstituted ferrocene CQ analogues) and 8–12 (the corresponding organic fragments).
Table 2.
In vitro antiplasmodial activities of 8–22 against CQ-sensitive P. falciparum strain (D10) and CQ-resistant strains (Dd2 and K1), respectively. The IC50 values have been taken in reference to the values of CQ and FQ. The IC50 values in  and indicate compounds with activities higher than FQ and CQ, respectively. On the other hand, the values in
and indicate compounds with activities higher than FQ and CQ, respectively. On the other hand, the values in  indicate compounds with activities higher than both CQ and FQ.
indicate compounds with activities higher than both CQ and FQ.

The analysis of the parameters that determine antiplasmodial activity in combination with the in silico calculations of these compounds may help in the design of more FQ analogues with adjusted lipophilicity/hydrophilicity and intramolecular hydrogen bonding. This might result in the development of more potent analogues than FQ.
6.4. Thiosemicarbazone derivatives
Thiosemicarbazones (TSCs) have exhibited potent antimalarial activities in the past [73]. The mechanism of the antimalarial activities of TSCs has been attributed to their reactive oxygen radicals production due to intrinsic metal (e.g. iron) coordinating properties [74]. Despite of disappointing activities of TSCs in Plasmodium berghei infected mice [75], there has been a renewed interest in TSCs. Several new lead compounds have been developed that kill certain protozoal parasites by the inhibition of cysteine proteases in addition to other novel targets [76], [77]. Biot et al. [78] reported the antimalarial activities of chimeras of TSCs and FQ (Fig. 10 ; 23–32) against 3D7, W2, FCR3 and BreI strains with origins from Africa, Indochina, Gambia and Brazil, respectively. The contributions of each molecular fragment including the ferrocenic derivatives 23, 24 and 28, 29 (without the 4-aminoquinoline moiety) and the organic compounds 26 & 27 and 31 & 32 were analysed. It was observed that the compounds 24–27 and 28–32 showed higher activity than the ferrocenyl TSCs 23 and 28, and demonstrated IC50 values in the low micromolar range (Table 3 ). Interestingly, the incorporation of the amino side chains in compounds 24 and 29 with respect to the compounds 23 and 28 led to a slight increase in the antimalarial activity. This indicated the incorporation of the basic amino group added potency by facilitating transport into the acidic food vacuole of the parasite. However, their low inhibitions against falcipain-2 suggested that some other inhibitory mechanisms may also be involved [79]. In accordance with the expectations; the chimeras of TSCs and FQ analogues 25 and 30 showed the best activity against the different strains of P. falciparum. Besides, the rigid metallocenic compounds 25 and 30, which were derived from FQ displayed better falcipain-2 inhibition as compared to the corresponding flexible alkyl analogues 26, 27, 31 and 32. It was interesting to note that only compound 26 showed the typical food vacuole abnormality associated with inhibitors of falcipain-2. All these investigations indicated that there may be two different modes of action; one targeting falcipain-2 and another acting independently. However, it was speculated that these compounds preferentially accumulate in the food vacuole, and therefore, showed better parasite than enzyme inhibition.
Fig. 10.

Chemical structures of the chimeras of thiosemicarbazones and FQ (23–32).
Table 3.
In vitro activities of 23–32 against P. falciparum strains (3D7, W2, FCR3 and BreI) with respect to CQ and FQ. Activities of 23–32 are in μM units whereas those of CQ and FQ are in nM units.
| Compounds | Plasmodium strains (IC50 values) |
|||
|---|---|---|---|---|
| 3D7 | W2 | FCR3 | BReI | |
| 23 | 30.2 ± 6.1 | 95.8 ± 20.4 | 74.0 ± 15.4 | 46.0 ± 14.4 |
| 24 | 33.0 ± 6.4 | 28.3 ± 9.9 | 31.2 ± 5.8 | 34.0 ± 8.1 |
| 25 | 0.2 ± 0.1 | 0.8 ± 0.2 | 0.2 ± 0.1 | 1.0 ± 0.4 |
| 26 | 0.3 ± 0.2 | 0.3 ± 0.1 | 0.6 ± 0.1 | 0.3 ± 0.1 |
| 27 | 0.1 ± 0.0 | 0.2 ± 0.1 | 0.8 ± 0.2 | 0.6 ± 0.2 |
| 28 | 64.2 ± 12.8 | 68.2 ± 10.6 | 30.2 ± 5.6 | 33.0 ± 8.5 |
| 29 | 20.7 ± 6.3 | 21.8 ± 7.9 | 22.2 ± 5.9 | 17.0 ± 3.5 |
| 30 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.8 ± 0.2 | 0.8 ± 0.2 |
| 31 | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.1 |
| 32 | 0.6 ± 0.1 | 2.0 ± 0.4 | 0.5 ± 0.2 | 0.7 ± 0.1 |
| CQ | 23.9 ± 4.6 | 540.0 ± 87.5 | 645.0 ± 63.1 | 396.2 ± 54.2 |
| FQ | 3.5 ± 0.5 | 7.1 ± 1.8 | 1.8 ± 0.3 | 3.0 ± 1.0 |
6.5. Ferrocene dual conjugates
Drug combination therapies have brought improvement in the treatment of malaria [80]. A careful antimalarial drug combination can cause delay in the selection of resistant mutants [81], [82]. However, there can be some complications with the simple drug combination therapies owing to the different pharmacokinetics of the combined drugs. Thus, the binding of active pharmacophores via covalent linkers seems a good alternative strategy. The binding of two active molecular fragments increases the bioavailability of the dual molecule produced and enables the merging of active molecules with independent modes of action that prevents the emergence of resistance [83]. Dual prodrug strategies have been previously explored for the treatment of malaria [84], [85], [86], [87], [88], [89], [90], [91], [92]. The linking of two active components targeting the thiol network and the heme detoxification pathway of malarial parasites provided both in vitro and in vivo proof of concept at the molecular level [84]. The covalent linker is known to play crucial roles as the cleavage of the linkage needs to occur under the specific conditions in the target cells.
Biot et al. [38] documented the antimalarial activity of two series of 4-aminoquinolines (Fig. 11 ; A and B) against HB3, Dd2 and W2. The reported compounds were structurally related to FQ. Overall, antimalarial screening, physicochemical parameters (partition coefficients) and the β-haematin inhibition properties indicated that the ferrocene moiety has to be covalently flanked by a 4-aminoquinoline and an alkylamine. Regardless of the lower antimalarial activity of the drug conjugates in comparison to FQ, unique modes of action of FQ and ferrocenyl analogues were observed. Chavain and co-workers [93] reported two series of ferrocenic antimalarial dual molecules wherein FQ analogue was conjugated with a glutathione reductase inhibitor via a cleavable amide bond (Fig. 12 ; A and B); for targeting two important pathways in malarial parasites (NF54 and K1 clones of P. falciparum). The dual molecules based on FQ analogues (64–69) were the most active in vitro against NF54 and K1 clones of P. falciparum with IC50 values in the nanomolar range (42.6–104.2 nM and 26.7–58.8 nM, respectively). However, they were slightly less active than the parent FQ analogues 58–63, which was assumed to be due to the cleavage and oxidative metabolism of the amide bond and the side chain of the FQ derivatives within the digestive vacuole.
Fig. 11.

Chemical structures of the two series (A and B) of 4-aminoquinolines.
Fig. 12.

Chemical structures of ferrocenic antimalarial dual molecules.
A critical analysis of these research reports indicated failure in achieving higher antimalarial activity of the dual conjugates; but satisfying evidences of unique mechanisms of action for FQ and ferrocenyl analogues were obtained.
6.6. 4-N-substituted analogues
Development of new and more potent FQ derivatives/analogues has been a continuing goal of researchers after the exploration of the antimalarial properties of FQ. Moreover, the thirst for the development of new derivatives is also important in that the effects of different functional groups and other chemical moieties can be worked for developing a rationale into FQ-based antimalarial drug development. Biot et al. [46] developed FQ derivatives (Fig. 13 : 88–92) by chemical transformations at the nitrogen atom placed at the 4-position of the quinoline ring to study the influence of their basic and lipophilic features on their antimalarial activities and inhibition of β-haematin formation. All the derivatives were screened against twelve strains of P. falciparum including 3D7, D6, 8425, Voll, L1, PA, Bres, FCR3, W2, K2, K14 and FCM29. The compounds 88–92 were more active than CQ against several CQ-resistant strains (Table 4 ). The developed compounds also showed better lipophilicity than both FQ and CQ at cytosolic and vacuolar pH indicating no simple relationship between activity, drug accumulation and inhibition of β-haematin formation. The increase in lipophilicity of the FQ derivatives was attributed to the increase in length of the side chain. Overall, the results were indicative of the fact that ferrocene moiety needs to be covalently flanked by a 4-aminoquinoline and an alkylamine in the molecular structures of the derivatives. Thus, antimalarial activity was suggested on account of the subtle combination of membrane penetration, localization (heme-targeting) and inhibition of hemozoin formation properties.
Fig. 13.

Chemical structures of 4-N-substituted FQ derivatives.
Table 4.
In vitro activities (IC50 values in nM) of 88–92 against the different strains of P. falciparum. The IC50 values in indicate compounds with activities higher than CQ.

6.7. Miscellaneous derivatives
Several efforts have been made to establish the role of ferrocenyl moiety in the antiplasmodial activity of FQ. In this direction, Blackie and co-workers [94] reported some structural analogues of FQ wherein the ferrocenyl moiety was replaced by the corresponding ruthenium-based moiety (Fig. 14 : Ruthenoquine, 93 and 94). Besides, some FQ analogues with different aromatic substituents (Fig. 14: 95–101) were also reported. It was interesting to observe that the ruthenoquine and its analogues demonstrated comparable potency to FQ (Table 5 ) against both the sensitive (D10) and resistant (K1) strains of P. falciparum; suggesting that the ferrocenyl fragment simply served as a hydrophobic spacer group.
Fig. 14.

Chemical structures of some structural analogues of FQ wherein the ferrocenyl moiety was replaced by the corresponding ruthenium-based moiety (Ruthenoquine, 93 and 94). Chemical structures of some FQ analogues with different aromatic substituents (95–101) are also presented.
Table 5.
A comparison of the in vitro antiplasmodial activities of ruthenoquine and its analogues with some FQ analogues against CQ-sensitive (D10 and 3D7) and chloroquine-resistant (K1) strains of P. falciparum. The IC50 values in indicate compounds with activities higher than FQ.

Organosilicon moieties are being incorporated into drug molecules in order to increase their lipophilicity and biological activity in addition to reducing toxicity for the enhancement of drug therapeutic values [95], [96], [97], [98]. Li et al. [99] documented the bioavailability features and in vitro antiplasmodial activities of a carbosilane congener of FQ and its heterometallic complexes (Fig. 15 : 102–107) against the chloroquine-sensitive (NF54) and chloroquine-resistant (Dd2) strains of P. falciparum. The silicon-containing congener was prepared by the incorporation of an organosilicon motif in the lateral side chain of FQ. The bioavailability studies indicated moderate solubility for all the compounds in PBS (phosphate buffered saline) buffer at physiological pH and room temperature. Compounds 105 and 106 were the least soluble (turbidimetric solubility = 1–5 μM). The compound 104 (turbidimetric solubility = 20–40 μM) had the best solubility properties. The bioavailability data suggested that the synthesized ferroquine derivatives were suitable for in vitro screening. All the compounds displayed high activity against the NF54 and Dd2 strains of P. falciparum (Table 6 ) with IC50 values in the range of 4.96 ± 0.76–30.73 ± 2.70 and 34.33 ± 3.37–77.19 ± 12.46 nM, respectively. However, complex 105 was the most active compound with IC50 values of 4.86 and 35.91 nM, respectively against NF54 and the Dd2 strains. Additionally, the metal complexes were able to inhibit the formation of synthetic hemozoin.
Fig. 15.

Chemical structure of the carbosilane congener of FQ (102) and its heterometallic complexes (103–107).
Table 6.
In vitro antiplasmodial activities of 102–107 against NF54 and Dd2 strains of P. falciparum. The IC50 values in and indicate compounds with activities higher than FQ and CQ, respectively. On the other hand, the values in indicate compounds with activities higher than both CQ and FQ.

A critical analysis of this research report shows that none of the derivatives had superior antimalarial activity in comparison to FQ against Dd2. However, meaningful structure–activity relationships, beneficial effects, and any evidences of cross resistance could be defined only after the compounds reported herein are screened against other sensitive and resistant strains of the malarial parasite. These results demonstrate the benefits of the incorporation of organosilicon moieties for the activity enhancement of existing and newly developed drugs.
Heterobimetallic complexes are bifunctional agents with two or more active metal centres capable of showing interactions with biological targets [100], [101]. Present scenario witnesses a growing interest in the design of heterometallic complexes as agents for the treatment of different diseases [102], [103]. The two different biologically active metals incorporated in the same molecule usually improve activity due to different interactions of the two different metals with multiple biological targets or by the improved physicochemical properties of the resulting heterometallic complex. Blackie et al. [104] in a quest to explore whether two metal centres in the same molecule lead to additive, synergistic or antagonistic effects towards antimalarial activity; reported the antimalarial properties of rhodium- and gold-based heterobimetallic complexes (Fig. 16 : 107–116) of FQ and its analogues against D10 (CQ-sensitive) and K1 (CQ-resistant) stains of P. falciparum. The authors observed that the coordination of gold or rhodium increased the efficacy of CQ particularly in the CQ-resistant K1 strain. Ferrocenyl ligands were more active than chloroquine-based gold or rhodium complexes against the resistant strains. Overall, it was observed that the complexation of the second metal brought little effect to the overall activity of the compounds. Basically, there appears an antagonistic effect on the overall efficacy by the complexation of the second metal. However, it needs to be understood that irrespective of the fact that gold and rhodium heterobimetallic complexes were unable to demonstrate additive or synergistic behaviour, the possibility of synergism with other heterobimetallic systems cannot be ruled out. Besides, these complexes can be screened against some other CQ-resistant and susceptible stains of P. falciparum before any strong conclusions can be derived.
Fig. 16.

Chemical structures of rhodium- and gold-based heterobimetallic complexes (108–116) of FQ and its analogues.
7. Pharmacologically significant systems
The discussion in this article explored several FQ derivatives with different structural modifications possessing promising antimalarial properties. The basic purpose of rational drug design using FQ template or its analogues was to obtain new synthetic derivatives or analogues with better activity, lower toxicity and other therapeutic benefits in comparison to the standard antimalarials like CQ and FQ. Of course, several classes of FQ derivatives have demonstrated better antimalarial activities than both CQ and FQ.
Hydroxyferroquine derivatives (1–3) demonstrated better activities than CQ against W2 strain of P. falciparum. Out of these three FQ derivatives, compounds 2 and 3 displayed 4- and 6-folds higher activity than CQ. However, compound 1 was only slightly more active than CQ. The four trioxaferroquines (4–7) were several folds more active than CQ against both FcB1 and FcM2 strains of P. falciparum. However, all the three hydroxyferroquine derivatives (1–3) and the four trioxaferroquines (4–7) displayed lower activities than FQ. The ferrocenophane analogues of ferroquine (11, 12, 13, 15, 16, 18, and 20) were more active than CQ against CQ-resistant Dd2 strains of P. falciparum. Besides, the ferrocenophane derivatives 11, 12, 14–17, 19 and 21 displayed higher activity than FQ against CQ-resistant K1 strains. The best compound 12 showed better activity than both CQ and FQ against CQ-sensitive D10. The 4-N-substituted analogues (88–92) were several folds more active than CQ against Voll, L1, PA, Bres, PCR3, W2, K2, K14 and FCM strains of P. falciparum. However, none of the derivatives was more active than FQ. Interestingly, the ruthenium-based analogue of FQ (ruthenoquine) was more active than FQ against CQ-resistant K1 strain of P. falciparum, and the other ruthenoquine analogue 94 was more active than FQ against CQ-sensitive D10 strain of P. falciparum. The FQ derivative 98 showed better activity than CQ against both CQ-sensitive 3D7 and CQ-resistant K1 strains whereas the derivatives 95 and 99 were more active than CQ against CQ-resistant K1 and CQ-sensitive 3D7 strains, respectively. Appreciable in vitro antiplasmodial activity was demonstrated by the carbosilane congener of FQ and its heterometallic complexes (102–107). The compounds 102–104 and 106 and 107 exhibited better activity than FQ against NF54 strain of P. falciparum. On the other hand, activity higher than CQ was displayed by 102–107 against Dd2 strain of P. falciparum. However, the best FQ derivative among all the congeners was 105, which was slightly more active than CQ against NF54 strain, and approximately 10-folds more active than FQ against the same plasmodium strain.
Overall, the discussion is this section indicates a positive move towards the discovery of efficient antimalarial agents using FQ as a base compound. A bright future can be seen for the different classes of FQ derivatives and analogues with promising activities against several geographically diverse strains of P. falciparum. A keen observation of the in vitro antimalarial properties of the FQ derivatives indicated that several derivatives demonstrated activities better than CQ and also FQ (in some cases) against the different sensitive and resistant strains of P. falciparum. Most of the active compounds demonstrated better activities than CQ but were considerably less active than FQ. Therefore, it is easy to envisage that the development of derivatives with better activity than FQ (and also CQ) is an achievement as far as the rational drug development based on FQ is concerned. The compounds 1–3 with activities better than CQ against W2 strain of P. falciparum can serve as templates for future development of efficient antimalarials against W2 strain of P. falciparum. Similarly, the compounds 4–7 may be used for the development of potential antimalarials against FcB1 and FcM2 strains of P. falciparum. Future development must include the in vivo testing of the compounds. The compounds 11, 12, 13, 15, 16, 18, and 20 deserve further evaluation in in vivo experiments for the treatment of malarial infections due to CQ-resistant Dd2 strains of P. falciparum. The ferrocenophane derivatives 11, 12, 14–17, 19 and 21 represent some interesting examples for future investigations against CQ-resistant K1 strains; as all these compounds demonstrated better activities than FQ. Developing 4-N-substituted FQ analogues 88–92 with several folds higher activities than CQ against Voll, L1, PA, Bres, PCR3, W2, K2, K14 and FCM strains indicated that these compounds have potential for progressing as future antimalarials against all these P. falciparum strains. Therefore, it will be of huge benefit to develop these compounds and their new generations as possible agents for the treatment of malaria. Ruthenoquine and compound 94 may be supposed to have a bright future for the development of future generations of drugs with a hope to treat the infections due to CQ-resistant K1 and D10 strains. Carbosilane congener of FQ and its heterometallic complexes also demonstrated exciting in vitro antimalarial activities against NF54 and Dd2 strains of P. falciparum. Some of the compounds in this class demonstrated better activities than FQ and CQ against the two pathogenic strains. However, it must be mentioned here that the best congener was 105 which was about 10-folds more active than FQ against the NF54 strain. Thus, carbosilane congeners find a special place in the development of new generations of antimalarial antidotes using FQ as a template. Such compounds may be tested in vivo for the possible effects against several strains of P. falciparum so that new efficient antimalarials may be developed.
In nutshell, several FQ derivatives and analogues demonstrated promising in vitro activities and deserve attention for further investigation in in vivo animal models. Besides, some exciting templates can be chosen for further development of new efficient derivatives and analogues.
8. Toxicity issues of ferroquine derivatives
Toxicity is one of the serious issues of importance that is often looked keenly while developing antimalarial drugs. Drug toxicity is acceptable when the drugs are less harmful than the disease. The main toxicity associated with the use of CQ is severe itching of the skin, often an adverse effect in African patients. Combination of CQ with proguanil has good tolerability but this combination is usually associated with mouth ulcers and gastrointestinal problems. The treatment with quinine has the side effects of hypoglycaemia. Halofantrine has been established as unsuitable for widespread use in humans due to its potential cardiotoxicity [105]. Interestingly, the famous FQ has been shown to respond negatively to the Ames and FETAX (Frog Embryo Teratogenesis Assay Xenopus) tests. Besides, it also showed negative results in the micronucleus in both in vitro and in vivo assays. However, it was found to be weakly mutagenic and genotoxic in similar types of experiments [106].
The hydroxyferroquines (1–3) were investigated for their toxic effects against Vero (kidney epithelial cells) cell cultures. The minimum toxic concentrations of the compounds against Vero cells were 40, 40 and 200 μM, respectively. FQ showed a minimum toxic concentration of 40 μM for the same cell culture. It can be seen that the compounds 1 and 2 have similar toxic effects to that of FQ whereas the compound 3 is considerably of less toxicity. The toxicity investigations indicated that compound 4 can be viewed as a promising alternative to FQ [40]. The FQ derivatives 8–22 reported by Salas et al. [72] were investigated for toxic effects against MCF-10A (normal breast epithelial cells) cells. The compounds 8,9, 11 and 12 had lower toxicity towards MCF-10A cells (IC50 = 74.7 ± 3.7–107.0 ± 2.1 μM) as compared to CQ and FQ, which displayed toxicities with IC50 values of 69.7 ± 4.8 and 26.5 ± 2.0 μM, respectively against the same cell line. Compound 9 was more toxic than CQ and less toxic than FQ as evident from its IC50 value of 47.9 ± 1.4 μM. However, the derivatives 13–22 were considerably more toxic than both FQ and CQ towards MCF-10A cells. The results were indicative of the fact that incorporation of the ferrocenyl moiety increased the toxicity, which has been previously seen in other ferrocenyl chloroquine compounds [107], [108], [109]. However, more detailed studies are needed for the determination of the toxicity of such compounds to comment on their real candidature as future drugs. Interestingly, the ruthenoquine (toxicity 50% dosage = 11.55 ± 0.06 μM) was demonstrated to observe a lower toxicity than FQ (toxicity 50% dosage = 1.06 ± 0.01 μM) towards LLC-MK2 (monkey kidney cells) cells with selective indices of 13.3 and 8.2, respectively [110].
It is clear from the discussion in this section that some FQ derivatives exhibit toxic effects towards normal cells. Some of the derivatives are mildly toxic while some exhibit pronounced toxicities in comparison to CQ and FQ. However, there are limited studies available on this subject and more investigations are needed to further comment on this issue. Besides, there is need to carry out the toxicity studies of FQ derivatives both in vitro and in vivo to visualize the potential of such compounds in the antimalarial chemotherapy.
9. Current challenges and future perspectives
There have been enormous advances in almost every sphere of science and technology during the last couple of decades. Despite these advances, the treatment of malaria is still far from over. No doubt, malaria has been a threat to human beings all over the world and a big challenge to our society. The applications of the presently available antimalarial drugs are limited due to their side effects, other drug issues, and the onset of resistance in several malarial parasites. The development of novel antimalarials is associated with many impediments and the main problem lies in the fact that no general guideline is available that could direct the synthesis of new active antimalarials with the least or no possibilities of the onset of resistance. Thus, which other antimalarial molecules are active and should be investigated in near future is still a mystery. Till now, there have been significant advances in the understanding of the molecular aetiology of malaria, but ideal therapeutic modalities are still missing. In view of these facts, it is very urgent to speed up the development of new antimalarials.
The therapeutic potential of the diversity of molecules (both ligands and metal complexes) can be fully harnessed for the design of novel and efficient antimalarials. So, it would be beneficent to explore other FQ derivatives with diverse molecular features and topologies as antimalarials. Besides, targeting and activation strategies may be helpful in the development of future generations of antimalarials with ability to overcome the disadvantages of FQ and its derivatives. The FQ derivatives should be obtained with no or reduced side effects, broad spectrum of activity, and no onset of drug resistance. Modern theoretical methods such as Density Functional Theory, Molecular Operating Environment and techniques such as high resolution electrospray mass spectrometry and multinuclear polarization transfer NMR (nuclear magnetic resonance) spectroscopy are greatly helpful and can surely improve our understanding of the chemical and biochemical reactivity of drugs, which might help in establishing some meaningful structure–activity relationships. Therefore, chemical studies of antimalarials under physiologically-relevant conditions (e.g. biological screening conditions) becomes very important in drug development rationales.
Nanotechnology can be faithful as far as several important issues of conventional antimalarial chemotherapy are concerned. Besides, nanotechnology is expected to help in developing a new generation of effective antimalarial therapies with potential to overcome the biological, biophysical and biomedical barriers that the body imposes against a chemotherapeutic intervention. The drug molecules trapped within nanostructured frameworks are protected from degradation in the blood system, which allows their safe delivery to target sites. Nanodrugs exhibit no or the least side effects towards normal cells with the potential to cross cancer cell barriers easily. Besides, these drugs can evade unproductive properties of conventional antimalarials. Thus, it would be of considerable interest if antimalarials are trapped into nano cages, which might avoid their poor bioavailability and ensure selective, specific and fast action of the trapped drugs. Nano identities of FQ and its derivatives may be expected to be effective replacements of their conventional counterparts.
A few reports indicated synergistic effects in several malarial cases by the use of new antimalarials in combination with the established drugs. Therefore, combination therapies need to be explored in order to get novel drug combinations in near future. Besides, it is very important to establish dosage ratios of the combined drugs in a particular formulation. In the present scenario, software and simulation programmes can be used to estimate drug therapeutic efficiency even before their actual syntheses. Thus, simulated drugs with good bioavailability, maximum solubility, remarkably less side effects and higher efficiency need to be designed and developed. Malarial infections generally involve multiple and complex biochemical pathways. Therefore, a successful treatment depends on pharmaceutical intervention at multiple pathways and sometimes with a combination of different drugs. Hence, we envisage the development of FQ-based designed multiple ligands, which may act at multiple biological targets and be quite helpful in the eradication of malaria.
In view of the discussion in this section and the understanding of the molecular mechanism of inhibition of malarial sporozoites by FQ and its derivatives, we foresee the design and development of new FQ analogues along with the development of nano counterparts of FQ and its derivatives. It will be really great to see how they work. Medicinal chemists need to develop safe and effective FQ derivatives (either in nano regime or the normal size range) for the treatment of malaria. Briefly, the future of FQ-based scafolds is quite bright in the treatment of malaria.
10. Conclusion
The inefficiency of quinine and its derivatives against malarial parasites and particularly the resistant strains of P. falciparum stimulated the development of other structurally relevant molecules for the treatment of malaria. Of course, FQ showed potential as a candidate for the treatment of malaria, however, the quest for the development of effective antimalarials still continues. Recently, much research is being carried out all over the world for the development of FQ derivatives as antimalarial agents and promising results have been obtained. Some of the FQ derivatives displayed several folds higher activities in comparison to CQ. However, research still needs to be carried out for exploring new molecules based on FQ scafolds as antimalarial agents. The FQ derivatives and analogues that have been reported with better activities than CQ and FQ against a plethora of malarial parasites need to be investigated for their toxic effects both in vitro and in vivo. This may help us to realize their real potential as antimalarial agents of clinical interest. Malaria greatly affects the economic status of particularly under developed nations and, therefore, needs to be eradicated as soon as possible. In this direction, the exploration of the antimalarial potentials of FQ derivatives along with their mechanisms of action is a ray of hope. Definitely, the future of FQ derivatives is quite bright since this is a step in the right direction towards the eradication of malaria.
Conflict of interest
There are no conflicts of interest to declare.
Plasmodium falciparum strains
3D7, 8425, BreI, Bres, D10, D6, Dd2, FcB1, FcM29, FCR3, HB3, K14, K2, L1, PA, Voll, K1 and NF54 are the different strains of Plasmodium falciparum collected from diverse geographical regions of the world.
Acknowledgements
Waseem A. Wani acknowledges the Research Management Centre of Universiti Teknologi Malaysia for post-doctoral research fellowship. Besides, Ehtesham Jameel thanks the University Department of Chemistry, B. R. Ambedkar Bihar University, Muzaffarpur, Bihar for all the help needed while writing this article.
Contributor Information
Waseem A. Wani, Email: waseemorg@gmail.com, waseem@ibd.utm.my.
Lee Ting Hun, Email: lee@ibd.utm.my.
References
- 1.Wotodjo A.N., Trape J.F., Richard V., Doucouré S., Diagne N., Tall A., Ndiath O., Faye N., Gaudart J., Rogier C., Sokhna C. No difference in the incidence of malaria in human-landing mosquito catch collectors and non-collectors in a Senegalese village with endemic malaria. PLoS One. 2015;10:e0126187. doi: 10.1371/journal.pone.0126187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vangapandu S., Jain M., Kaur K., Patil P., Patel S.R., Jain R. Recent advances in antimalarial drug development. Med. Res. Rev. 2007;27:65–107. doi: 10.1002/med.20062. [DOI] [PubMed] [Google Scholar]
- 3.Volkman S.K., Barry A.E., Lyons E.J., Nielsen K.M., Thomas S.M., Choi M., Thakore S.S., Day K.P., Wirth D.F., Hartl D.L. Recent origin of Plasmodium falciparum from a single progenitor. Science. 2001;293:482–484. doi: 10.1126/science.1059878. [DOI] [PubMed] [Google Scholar]
- 4.Caraballo H., King K. Emergency department management of mosquito-borne illness: malaria, dengue, and west nile virus. Emerg. Med. Pract. 2014;16:1–23. [PubMed] [Google Scholar]
- 5.Urban B.C., Ing R., Stevenson M.M. Early interactions between blood-stage plasmodium parasites and the immune system. Curr. Microbiol. Immunol. 2005;297:25–70. doi: 10.1007/3-540-29967-x_2. [DOI] [PubMed] [Google Scholar]
- 6.Poinar G., Jr. Plasmodium dominicana n. sp. (Plasmodiidae: Haemospororida) from tertiary Dominican amber. Syst. Parasitol. 2005;61:47–52. doi: 10.1007/s11230-004-6354-6. [DOI] [PubMed] [Google Scholar]
- 7.Beeson J.G., Boeuf P., Fowkes F.J. Maximizing antimalarial efficacy and the importance of dosing strategies. BMC Med. 2015;13:110. doi: 10.1186/s12916-015-0349-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eastman R.T., Fidlock D.A. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat. Rev. Microbiol. 2009;7:864–874. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noedl H., Se Y., Kurt S., Smith B.L., Socheat D., Fukuda M.M. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 10.Biot C., Glorian G., Maciejewski L.A., Brocard J.S., Domarle O., Blampain G., Millet P., Georges A.J., Abessolo H., Dive D., Lebibi J. Synthesis and antimalarial activity in vitro and in vivo of a new ferrocene-chloroquine analogue. J. Med. Chem. 1997;40:3715–3718. doi: 10.1021/jm970401y. [DOI] [PubMed] [Google Scholar]
- 11.Biot C. Ferroquine: a new weapon in the fight against malaria. Curr. Med. Chem. Anti-infect. Agents. 2004;3:135–147. [Google Scholar]
- 12.Pierrot C., Lafitte S., Dive D., Fraisse L., Brocard J., Khalife J. Analysis of immune response patterns in naive and Plasmodium berghei-infected young rats following a ferroquine treatment. Int. J. Parasitol. 2005;35:1601–1610. doi: 10.1016/j.ijpara.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Biot C., Taramelli D., Forfar-Bares I., Maciejewski L.A., Boyce M., Nowogrocki G., Brocard J.S., Basilico N., Olliaro P., Egan T.J. Insights into the mechanism of action of ferroquine. Relationship between physicochemical properties and antiplasmodial activity. Mol. Pharm. 2005;2:185–193. doi: 10.1021/mp0500061. [DOI] [PubMed] [Google Scholar]
- 14.Pradines B., Orlandi-Pradines E., Henry M., Bogreau H., Fusai T., Mosnier J., Baret E., Durand C., Bouchiba H., Penhoat K., Rogier C. Metallocenes and malaria: a new therapeutic approach. Ann. Pharm. Fr. 2005;63:284–294. doi: 10.1016/s0003-4509(05)82293-x. [DOI] [PubMed] [Google Scholar]
- 15.Dive D., Biot C. Ferrocene conjugates of chloroquine and other antimalarials: the development of ferroquine, a new antimalarial. ChemMedChem. 2008;3:383–391. doi: 10.1002/cmdc.200700127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blackie M.A., Chibale K. Metallocene antimalarials: the continuing quest. Met. Based Drugs. 2008;2008:495123. doi: 10.1155/2008/495123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubar F., Khalife J., Brocard J., Dive D., Biot C. Ferroquine, an ingenious antimalarial drug: thoughts on the mechanism of action. Molecules. 2008;13:2900–2907. doi: 10.3390/molecules13112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biot C., Nosten F., Fraisse L., Ter-Minassian D., Khalife J., Dive D. The antimalarial ferroquine: from bench to clinic. Parasite. 2011;18:207–214. doi: 10.1051/parasite/2011183207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biot C., Dive D. A new hope against malaria: the contribution of organometallic chemistry. Actual. Chim. 2011;353–354:93–96. [Google Scholar]
- 20.Sekhon B.S., Bimal N. Transition metal-based anti-malarials. J. Pharm. Educ. Res. 2012;3:52–63. [Google Scholar]
- 21.Biot C., Pradines B., Dive D. Ferroquine: a concealed weapon. In: Becker K., Becker K., editors. Apicomplexan Parasites: Molecular Approaches toward Targeted Drug Development. WILEY-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2011. pp. 397–411. [Google Scholar]
- 22.Brown G.V., Beck H.P., Molyneux M., Marsh K. Molecular approaches to epidemiology and clinical aspects of malaria. Parasitol. Today. 2000;16:448–451. doi: 10.1016/s0169-4758(00)01793-2. [DOI] [PubMed] [Google Scholar]
- 23.Clark I.A., Schofield L. Pathogenesis of malaria. Parasitol. Today. 2000;16:451–454. doi: 10.1016/s0169-4758(00)01757-9. [DOI] [PubMed] [Google Scholar]
- 24.Cox F.E. History of the discovery of the malaria parasites and their vectors. Parasit. Vectors. 2010;3 doi: 10.1186/1756-3305-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harper K., Armelagos G. The changing disease-scape in the third epidemiological transition. Int. J. Environ. Res. Publ. Health. 2010;7:675–697. doi: 10.3390/ijerph7020675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta V., Mittal M., Sharma V. Epidemiology of malaria in Amritsar District of India. Oman Med. J. 2014;29:142–145. doi: 10.5001/omj.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burrows J.N., Leroy D., Lotharius J., Waterson D. Challenges in antimalarial drug discovery. Future Med. Chem. 2011;3:1401–1412. doi: 10.4155/fmc.11.91. [DOI] [PubMed] [Google Scholar]
- 28.Stratton L., O'neill M.S., Kruk M.E., Bell M.L. The persistent problem of malaria: addressing the fundamental causes of a global killer. Soc. Sci. Med. 2008;67:854–862. doi: 10.1016/j.socscimed.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 29.WHO . 2013. World Malaria Report.http://www.who.int/malaria/world_malaria_report_2013/en/ Geneva, Switzerland. (last accessed 14.05.15.) [Google Scholar]
- 30.WHO, 2013. http://www.who.int/mediacentre/factsheets/fs094/en/, (last accessed 14.05.15.).
- 31.Biot C., Delhaes L., Maciejewski L.A., Mortuaire M., Camus D., Dive D., Brocard J.S. Synthetic ferrocenic mefloquine and quinine analogues as potential antimalarials. Eur. J. Med. Chem. 2000;35:707–714. doi: 10.1016/s0223-5234(00)00178-1. [DOI] [PubMed] [Google Scholar]
- 32.Paitayatat S., Tarnchompoo B., Thebtaranonth Y., Yuthavong Y. Correlation of antimalarial activity of artemisinin derivatives with binding affinity with ferroprotoporphyrin IX. J. Med. Chem. 1997;40:633–638. doi: 10.1021/jm960767v. [DOI] [PubMed] [Google Scholar]
- 33.Delhaes L., Biot C., Berry L., Maciejewski L.A., Camus D., Brocard J.S., Dive D. Novel ferrocenic artemisinin derivatives: synthesis, in vitro antimalarial activity and affinity of binding with ferroprotoporphyrin IX. Bioorg. Med. Chem. 2000;8:2739–2745. doi: 10.1016/s0968-0896(00)00206-6. [DOI] [PubMed] [Google Scholar]
- 34.Baramee A., Coppin A., Mortuaire M., Pelinski L., Tomavo S., Brocard J. Synthesis and in vitro activities of ferrocenic aminohydroxynaphthoquinones against Toxoplasma gondii and Plasmodium falciparum. Bioorg. Med. Chem. 2006;14:1294–1302. doi: 10.1016/j.bmc.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 35.Dubar F., Anquetin G., Pradines B., Dive D., Khalife J., Biot C. Enhancement of the antimalarial activity of ciprofloxacin using a double prodrug/bioorganometallic approach. J. Med. Chem. 2009;52:7954–7957. doi: 10.1021/jm901357n. [DOI] [PubMed] [Google Scholar]
- 36.Domarle O., Blampain G., Agnaniet H., Nzadiyabi T., Lebibi J., Brocard J., Maciejewski L., Biot C., Georges A.J., Millet P. In vitro antimalarial activity of a new organometallic analog, ferrocene chloroquine. Antimicrob. Agents Chemother. 1998;42:540–544. doi: 10.1128/aac.42.3.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biot C., Delhaes L., Abessolo H., Domarle O., Maciejewski L.A., Mortuaire M., Del-court P., Deloron P., Camus D., Dive D., Brocard J.S. Novel metallocenic compounds as antimalarials agents. Study of the position of ferrocene in chloroquine. J. Organomet. Chem. 1999;589:59–65. [Google Scholar]
- 38.Biot C., Daher W., Jarry C., Ndiaye C.H., Pelinski L., Khalife J., Fraisse L., Pelinski L., Jerry C., Brocard J., Khalife J., Forfar-Bares I., Dive D. Probing the role of the covalent linkage of ferrocene into a chloroquine template. J. Med. Chem. 2006;49:4707–4714. doi: 10.1021/jm060259d. [DOI] [PubMed] [Google Scholar]
- 39.Delhaes L., Abessolo H., Biot C., Berry L., Delron P., Karbwang J., Mortuaire M., Maciejewski L.A. Ferrochloroquine, a ferrocenyl analogue of chloroquine, retains a potent activity against resistant Plasmodium falciparum in vitro and P. vinckei in vivo. Parasitol. Res. 2001;87:239–244. doi: 10.1007/s004360000317. [DOI] [PubMed] [Google Scholar]
- 40.Biot C., Daher W., Chavain N., Fandeur T., Khalife J., Dive D., De Clercq E. Design and synthesis of hydroxyferroquine derivatives with antimalarial and antiviral activities. J. Med. Chem. 2006;49:2845–2849. doi: 10.1021/jm0601856. [DOI] [PubMed] [Google Scholar]
- 41.J. Brocard, J. Lebibi, L. Maciejewski, Antimalarial Organometallic Iron Complexes, Patent International, 1996, WO1996035698 A1.
- 42.A Comparative Safety and Activity with Ferroquine Associated with Artesunate versus Amodiaquine Associated with Artesunate in African Patients with Uncomplicated Malaria, http://clinicaltrials.gov/ct2/show/NCT00563914, (last accessed 14.05.15.).
- 43.http://clinicaltrials.gov/ct2/show/NCT00988507, (last accessed 20.04.15.).
- 44.Delhaes L., Biot C., Berry L., Delcourt P., Maciejewski L.A., Camus D., Brocard J.S., Dive D. Synthesis of ferroquine enantiomers: first investigation of effects of metallocenic chirality upon antimalarial activity and cytotoxicity. ChemBioChem. 2002;3:418–423. doi: 10.1002/1439-7633(20020503)3:5<418::AID-CBIC418>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 45.Chavain N., Vezin H., Dive D., Touati N., Paul J.F., Buisine E., Biot C. Investigation of the redox behavior of the new antimalarial, ferroquine. Mol. Pharm. 2008;5:710–716. doi: 10.1021/mp800007x. [DOI] [PubMed] [Google Scholar]
- 46.Biot C., Chavain N., Dubar F., Pradines B., Brocard J., Forfar I., Dive D. Structure activity relationships of 4-N-substituted ferroquine analogues. Time to re-evaluate the mechanism of action of ferroquine. J. Organomet. Chem. 2009;694:845–854. [Google Scholar]
- 47.Yayon A., Cabantchik Z.I., Ginsburg H. Susceptibility of human malaria parasites to chloroquine is pH dependent. Proc. Natl. Acad. Sci. 1985;82:2784–2787. doi: 10.1073/pnas.82.9.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorn A., Vippagunta S.R., Matile H., Jacquet C., Vennerstrom J.L., Ridley R.G. An assessment of drug haematin binding as a mechanism for inhibition of haematin polymerization by quinoline antimalarials. Biochem. Pharmacol. 1998;55:727–736. doi: 10.1016/s0006-2952(97)00510-8. [DOI] [PubMed] [Google Scholar]
- 49.Beagley P., Blackie M.A.L., Chibale K., Clarkson C., Meijboom R., Moss J.R., Smith P.J., Su H. Synthesis and antiplasmodial activity in vitro of new ferrocene chloroquine-analogues. Dalton Trans. 2003:3046–3051. [Google Scholar]
- 50.Atteke C., Ndong J.M., Aubouy A., Maciejewski L., Brocard J., Lebibi J., Deloron P. In vitro susceptibility to a new antimalarial organometallic analogue, ferroquine, of Plasmodium falciparum isolates from the Haut-Ogooué region of Gabon. J. Antimicrob. Chemother. 2003;51:1021–1024. doi: 10.1093/jac/dkg161. [DOI] [PubMed] [Google Scholar]
- 51.Pradines B., Tall A., Rogier C., Spiegel A., Mosnier J., Marrama L., Fusai T., Millet P., Panconi E., Trape J.F., Parzy D. In vitro activities of ferrochloroquine against 55 Senegalese isolates of Plasmodium falciparum in comparison with those of standard antimalarial drugs. Trop. Med. Int. Health. 2002;7:265–270. doi: 10.1046/j.1365-3156.2002.00848.x. [DOI] [PubMed] [Google Scholar]
- 52.Pradines B., Fusai T., Daries W., Laloge V., Rogier C., Millet P., Panconi E., Kombila M., Parzy D. Ferrocene-chloroquine analogues as antimalarial agents: in vitro activity of ferrochloroquine against 103 Gabonese isolates of Plasmodium falciparum. J. Antimicrob. Chemother. 2001;48:179–184. doi: 10.1093/jac/48.2.179. [DOI] [PubMed] [Google Scholar]
- 53.Chim P., Lim P., Sem R., Nhem S., Maciejewski L., Fandeur T. The in-vitro antimalarial activity of ferrochloroquine, measured against Cambodian isolates of Plasmodium falciparum. Ann. Trop. Med. Parasitol. 2004;98:419–424. doi: 10.1179/000349804225003361. [DOI] [PubMed] [Google Scholar]
- 54.Kreidenweiss A., Kremsner P.G., Dietz K., Mordmüller B. In vitro activity of ferroquine (SAR97193) is independent of chloroquine resistance in Plasmodium falciparum. Am. J. Trop. Med. Hyg. 2006;75:1178–1181. [PubMed] [Google Scholar]
- 55.Barends M., Jaidee A., Khaohirun N., Singhasivanon P., Nosten F. In vitro activity of ferroquine (SSR 97193) against Plasmodium falciparum isolates from the Thai-Burmese border. Malar. J. 2007;6:81. doi: 10.1186/1475-2875-6-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.L. Fraisse, D. Ter-Minassian, International Application Association between Ferroquine and an Artemisinine Derivative for Treating Malaria, 2012, US20120258945 A1.
- 57.Long T.T., Nakazawa S., Onizuka S., Huaman M.C., Kanbara H. Influence of CD4+CD25+ T cells on Plasmodium berghei NK65 infection in BALB/c mice. Int. J. Parasitol. 2003;33:175–183. doi: 10.1016/s0020-7519(02)00261-8. [DOI] [PubMed] [Google Scholar]
- 58.Henry M., Briolant S., Fontaine A., Mosnier J., Baret E., Amalvict R., Fusaï T., Fraisse L., Rogier C., Pradines B. In vitro activity of ferroquine is independent of polymorphisms in transport proteins genes implicated in quinoline resistance in plasmodium falciparum. Antimicrob. Agents Chemother. 2008;52:2755–2759. doi: 10.1128/AAC.00060-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daher W., Biot C., Fandeur T., Jouin H., Pelinski L., Viscogliosi E., Fraisse L., Pradines B., Brocard J., Khalife J., Dive D. Assessment of P. falciparum resistance to ferroquine in field isolates and in W2 strain under pressure. Malar. J. 2006;5:11. doi: 10.1186/1475-2875-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Villiers K.A., Marques H.M., Egan T.J. The crystal structure of halofantrineferriprotoporphyrin IX and the mechanism of action of arylmethanol antimalarials. J. Inorg. Biochem. 2008;102:1660–1667. doi: 10.1016/j.jinorgbio.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 61.Pisciotta J.M., Sullivan D. Hemozoin: oil versus water. Parasitol. Int. 2008;57:89–96. doi: 10.1016/j.parint.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Helms V. Attraction within the membrane. Forces behind transmembrane protein folding and supramolecular complex assembly. EMBO Rep. 2002;3:1133–1138. doi: 10.1093/embo-reports/kvf245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dubar F., Slomianny C., Khalife J., Dive D., Kalamou H., Gurardel Y., Grellier P., Biot C. The ferroquine antimalarial conundrum: redox activation and reinvasion inhibition. Angew. Chem. Int. Ed. 2013;52:7690–7693. doi: 10.1002/anie.201303690. [DOI] [PubMed] [Google Scholar]
- 64.Dubar F., Egan T.J., Pradines B., Kuter D., Ncokazi K.K., Forge D., Paul J.F., Pierrot C., Kalamou H., Khalife J., Buisine E., Rogier C., Vezin H., Forfar I., Slomianny C., Trivelli X., Kapishnikov S., Leiserowitz L., Dive D., Biot C. The antimalarial ferroquine: role of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem. Biol. 2011;6:275–287. doi: 10.1021/cb100322v. [DOI] [PubMed] [Google Scholar]
- 65.Dondorp A.M., Nosten F., Yi P., Das D., Phyo A.P., Tarning J., Lwin K.M., Ariey F., Hanpithakpong W., Lee S.J., Ringwald P., Silamut K., Imwong M., Chotivanich K., Lim P., Herdman T., An S.S., Yeung S., Singhasivanon P., Day N.P.J., Lindegardh N., Socheat D., White N.J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taylor S.M., Juliano J.J., Meshnick S.R. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009;361 doi: 10.1056/NEJMc091737. 1807–1807. [DOI] [PubMed] [Google Scholar]
- 67.Dechy-Cabaret O., Benoit-Vical F., Robert A., Meunier B. Preparation and antimalarial activities of “trioxaquines”, new modular molecules with a trioxane squeleton linked to a 4-aminoquinoline. ChemBioChem. 2000;4:281–283. doi: 10.1002/1439-7633(20001117)1:4<281::AID-CBIC281>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 68.Robert A., Dechy-Cabaret O., Cazelles J., Meunier B. From mechanistic studies on artemisinin derivatives to new modular antimalarial drugs. Acc. Chem. Res. 2002;35:167–174. doi: 10.1021/ar990164o. [DOI] [PubMed] [Google Scholar]
- 69.Walsh J.J., Bell A. Hybrid drugs for malaria. Curr. Pharm. Des. 2009;15:2970–2985. doi: 10.2174/138161209789058183. [DOI] [PubMed] [Google Scholar]
- 70.Cavalli A., Bolognesi M.L. Neglected tropical diseases: multitarget-directed ligands in the search for novel lead candidates against Trypanosomia and Leishmania. J. Med. Chem. 2009;52:7339–7359. doi: 10.1021/jm9004835. [DOI] [PubMed] [Google Scholar]
- 71.Bellot F., Coslédan F., Vendier L., Brocard J., Meunier B., Robert A. Trioxaferroquines as new hybrid antimalarial drugs. J. Med. Chem. 2010;53:4103–4109. doi: 10.1021/jm100117e. [DOI] [PubMed] [Google Scholar]
- 72.Salas P.F., Herrmann C., Cawthray J.F., Nimphius C., Kenkel A., Chen J., de Kock C., Smith P.J., Patrick B.O., Adam M.J., Orvig C. Structural characteristics of chloroquine-bridged ferrocenophane analogues of ferroquine may obviate malaria drug resistance mechanisms. J. Med. Chem. 2013;56:1596–1613. doi: 10.1021/jm301422h. [DOI] [PubMed] [Google Scholar]
- 73.Scovill J.P., Klayman D.L., Lambros C., Childs G.E., Notsch J.D.J. 2-Acetylpyridine thiosemicarbazones. 9. Derivatives of 2-acetylpyridine 1-oxide as potential antimalarial agents. Med. Chem. 1984;27:87–91. doi: 10.1021/jm00367a019. [DOI] [PubMed] [Google Scholar]
- 74.Ames J.R., Ryan M.D., Klayman D.L., Kovacic P.J. Charge transfer and oxy radicals in antimalarial action. Quinones, dapsone metabolites, metal complexes, imunium ions, and peroxides. Free Radic. Biol. Med. 1985;1:353–361. doi: 10.1016/0748-5514(85)90147-3. [DOI] [PubMed] [Google Scholar]
- 75.Klayman D.L., Acton N., Scovill J.P. 2-Acetylpyridine thiosemicarbazones. 12. Derivatives of 3-acetylisoquinoline as potential antimalarial agents. Arzneimittelforschung. 1986;36:10–13. doi: 10.1002/chin.198619236. [DOI] [PubMed] [Google Scholar]
- 76.Greenbaum D.C., Mackey Z., Hansell E., Doyle P., Gut J., Caffrey C.R., Lehrman J., Rosenthal P.J., McKerrow J.H., Chibale K. Synthesis and structure-activity relationships of Parasiticidal thiosemicarbazone cysteine protease inhibitors against plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J. Med. Chem. 2004;47:3212–3219. doi: 10.1021/jm030549j. [DOI] [PubMed] [Google Scholar]
- 77.Chipeleme A., Gut J., Rosenthal P.J., Chibale K. Synthesis and biological evaluation of phenolic Mannich bases of benzaldehyde and (thio)semicarbazone derivatives against the cysteine protease falcipain-2 and a chloroquine resistant strain of Plasmodium falciparum. Bioorg. Med. Chem. 2007;15:273–282. doi: 10.1016/j.bmc.2006.09.055. [DOI] [PubMed] [Google Scholar]
- 78.Biot C., Pradines B., Sergeant M.H., Gut J., Rosenthal P.J., Chibale K. Design, synthesis, and antimalarial activity of structural chimeras of thiosemicarbazone and ferroquine analogues. Bioorg. Med. Chem. Lett. 2007;17:6434–6438. doi: 10.1016/j.bmcl.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 79.Chiyanzu I., Clarkson C., Smith P.J., Lehman J., Gut J., Rosenthal P.J., Chibale K. Design, synthesis and anti-plasmodial evaluation in vitro of new 4-aminoquinoline isatin derivatives. Bioorg. Med. Chem. 2005;13:3249–3261. doi: 10.1016/j.bmc.2005.02.037. [DOI] [PubMed] [Google Scholar]
- 80.Peters W. The chemotherapy of rodent malaria. V. Dynamics of drug resistance. I. Methods for studying the acquisition and loss of resistance to chloroquine by Plasmodiun berghei. Ann. Trop. Med. Parasitol. 1968;62:277–287. [PubMed] [Google Scholar]
- 81.Peters W. The chemotherapy of rodent malaria. XIV. The action of some sulphonamides alone or with folic reductase inhibitors against malaria vectors and parasites. 4. The response of normal and drug-resistant strains of Plasmodium berghei. Ann. Trop. Med. Parasitol. 1971;65:123–129. [PubMed] [Google Scholar]
- 82.Peters W., Robinson B.L. The chemotherapy of rodent malaria XXXV. Further studies on the retardation of drug resistance by the use of a triple combination of mefloquine, pyrimethamine and sulfadoxine in mice infected with P. berghei and ‘P. berghei NS’. Ann. Trop. Med. Parasitol. 1987;78:459–466. doi: 10.1080/00034983.1984.11811850. [DOI] [PubMed] [Google Scholar]
- 83.Meunier B. Hybrid molecules with a dual mode of action: dream or reality. Acc. Chem. Res. 2008;41:69–77. doi: 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]
- 84.Charvet E.D., Delarue S., Biot C., Schwobel B., Boehme C.C., Mussigbrodt A., Maes L., Sergheraert C., Grellier P., Schirmer R.H., Becker K. A prodrug form of a plasmodium falciparum glutathione reductase inhibitor conjugated with a 4-anilinoquinoline. J. Med. Chem. 2001;44:4268–4276. doi: 10.1021/jm010268g. [DOI] [PubMed] [Google Scholar]
- 85.Cabaret O.D., Vical F.B., Loup C., Robert A., Gornitzka H., Bonhoure A., Vial H., Magnaval J.F., Séguéla J.P., Meunier B. Synthesis and antimalarial activity of trioxaquine derivatives. Chem. Eur. J. 2004;10:1625–1636. doi: 10.1002/chem.200305576. [DOI] [PubMed] [Google Scholar]
- 86.Romeo S., Dell'Agli M., Parapini S., Rizzi L., Galli G., Mondani M., Sparatore A., Taramelli D., Bosisio E. Plasmepsin II inhibition and antiplasmodial activity of primaquine-statine ‘double-drugs’. Bioorg. Med. Chem. Lett. 2004;14:2931–2934. doi: 10.1016/j.bmcl.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 87.Salom-Roig X.J., Hamzé A., Calas M., Vial H.J. Dual molecules as new antimalarials. Comb. Chem. High. Throughput Screen. 2005;8:49–62. doi: 10.2174/1386207053328219. [DOI] [PubMed] [Google Scholar]
- 88.Grellepois F., Grellier P., Bonnet-Delpon D., Bégué J.P. Design, synthesis and antimalarial activity of trifluoromethylartemisinin-mefloquine dual molecules. ChemBioChem. 2005;6:648–652. doi: 10.1002/cbic.200400347. [DOI] [PubMed] [Google Scholar]
- 89.Burgess S.J., Selzer A., Kelly J.X., Smilkstein M.J., Riscoe M.K., Peyton D.H. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J. Med. Chem. 2006;49:5623–5625. doi: 10.1021/jm060399n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Friebolin W., Jannack B., Wenzel N., Furrer J., Oeser T., Sanchez C.P., Lanzer M., Yardley V., Becker K., Davioud-Charvet E. Antimalarial dual drugs based on potent inhibitors of glutathione reductase from Plasmodium falciparum. J. Med. Chem. 2008;51:1260–1277. doi: 10.1021/jm7009292. [DOI] [PubMed] [Google Scholar]
- 91.Solaja B.A., Opsenica D., Smith K.S., Milhous W.K., Terzić N., Opsenica I., Burnett J.C., Nuss J., Gussio R., Bavari S. Novel 4-aminoquinolines active against chloroquine-resistant and sensitive P. falciparum strains that also inhibit botulinum serotype A. J. Med. Chem. 2008;51:4388–4391. doi: 10.1021/jm800737y. [DOI] [PubMed] [Google Scholar]
- 92.Kelly J.X., Smilkstein M.J., Brun R., Wittlin S., Cooper R.A., Lane K.D., Janowsky A., Johnson R.A., Dodean R.A., Winter R. Discovery of dual function acridones as a new antimalarial chemotype. Nature. 2009;459:270–273. doi: 10.1038/nature07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chavain N., Charvet E.D., Trivelli X., Mbeki L., Rottmann M., Brun R., Biot C. Antimalarial activities of ferroquine conjugates with either glutathione reductase inhibitors or glutathione depletors via a hydrolyzable amide linker. Bioorg. Med. Chem. 2009:8048–8059. doi: 10.1016/j.bmc.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 94.Blackie M.A.L., Beagley P., Croft S.L., Kendrick H., Moss J.R., Chibale K. Metallocene-based antimalarials: an exploration into the influence of the ferrocenyl moiety on in vitro antimalarial activity in chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum. Bioorg. Med. Chem. 2007;15:6510–6516. doi: 10.1016/j.bmc.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 95.Li Y., de Kock C., Smith P.J., Guzgay H., Hendricks D.T., Naran K., Mizrahi V., Warner D.F., Chibale K., Smith G.S. Synthesis, characterization, and pharmacological evaluation of silicon-containing aminoquinoline organometallic complexes as antiplasmodial, antitumor, and antimycobacterial agents. Organometallics. 2013;32:141–150. [Google Scholar]
- 96.Englebienne P., Van Hoonacker A., Herst C.V. The place of the bioisosteric sila-substitution in drug design. Drug Des. Rev. Online. 2005;2:467–483. [Google Scholar]
- 97.Chen C.A., Sieburth S.M., Glekas A., Hewitt G.W., Trainor G.L., Viitanen S.E., Garber S.S., Cordova B., Jeffry S., Klabe R.M. Drug design with a new transition state analog of the hydrated carbonyl: silicon-based inhibitors of the HIV protease. Chem. Biol. 2001;8:1161–1166. doi: 10.1016/s1074-5521(01)00079-5. [DOI] [PubMed] [Google Scholar]
- 98.Tacke R., Heinrich T., Bertermann R.D., Burschka C., Hamacher A., Kassack M.U. Sila haloperidol: a silicon analogue of the dopamine (D2) receptor antagonist haloperidol. Organometallics. 2004;23:4468–4477. [Google Scholar]
- 99.Li Y., de Kock C., Smith P.J., Chibale K., Smith G.S. Synthesis and evaluation of a carbosilane congener of ferroquine and its corresponding half-sandwich ruthenium and rhodium complexes for antiplasmodial and β-hematin inhibition activity. Organometallics. 2014;33:4345–4348. [Google Scholar]
- 100.de Hoog P., Pitie M., Amadei G., Gamez P., Meunier B., Kiss R., Reedjik J. DNA cleavage and binding selectivity of a heterodinuclear Pt–Cu(3-Clip-Phen) complex. J. Biol. Inorg. Chem. 2008;13:575–586. doi: 10.1007/s00775-008-0346-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dong X., Wang X., Lin M., Sun H., Yang X., Guo Z. Promotive effect of the platinum moiety on the DNA cleavage activity of copper-based artificial nucleases. Inorg. Chem. 2010;49:2541–2549. doi: 10.1021/ic100001x. [DOI] [PubMed] [Google Scholar]
- 102.Donzello M.P., Viola E., Ercolani C., Fu Z., Futur D., Kadish K.M. Tetra-2,3-pyrazinoporphyrazines with externally appended pyridine rings. New heteropentanuclear complexes carrying four exocyclic cis-platin-like functionalities as potential bimodal (PDT/cis-platin) anticancer agents. Inorg. Chem. 2012;51:12548–12559. doi: 10.1021/ic301989a. [DOI] [PubMed] [Google Scholar]
- 103.Pantoja J.F.G., Stern M., Jarzecki A.A., Royo E., Escajeda E.R., Ramirez A.V., Aguilera R.J., Contel M. Titanocene-phosphine derivatives as precursors to cytotoxic heterometallic TiAu2 and TiM (M = Pd, Pt) compounds. Studies of their interactions with DNA. Inorg. Chem. 2011;50:11099–11110. doi: 10.1021/ic201647h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Blackie M.A.L., Beagley P., Chibale K., Clarkson C., Moss J.R., Smith P.J. Synthesis and antimalarial activity in vitro of new heterobimetallic complexes: Rh and Au derivatives of chloroquine and a series of ferrocenyl-4-amino-7-chloroquinolines. J. Organomet. Chem. 2003;688:144–152. [Google Scholar]
- 105.Taylor W.R., White N.J. Antimalarial drug toxicity: a review. Drug Saf. 2004;27:25–61. doi: 10.2165/00002018-200427010-00003. [DOI] [PubMed] [Google Scholar]
- 106.Chatterjee T., Muhkopadhyay A., Khan K.A., Giri A.K. Comparative mutagenic and genotoxic effects of three antimalarial drugs, chloroquine, primaquine and amodiaquine. Mutagenesis. 1998;13:619–624. doi: 10.1093/mutage/13.6.619. [DOI] [PubMed] [Google Scholar]
- 107.Herrmann C., Salas P.F., Patrick B.O., de Kock C., Smith P.J., Adam M.J., Orvig C. 1,2-Disubstituted ferrocenyl carbohydrate chloroquine conjugates as potential antimalarial agents. Dalton Trans. 2012;41:6431–6442. doi: 10.1039/c2dt12050j. [DOI] [PubMed] [Google Scholar]
- 108.Herrmann C., Salas P.F., Cawthray J.F., de Kock C., Patrick B.O., Smith P.J., Adam M.J., Orvig C. 1,1′-Disubstituted ferrocenyl carbohydrate chloroquine conjugates as potential antimalarials. Organometallics. 2012;31:5736–5747. doi: 10.1039/c2dt12050j. [DOI] [PubMed] [Google Scholar]
- 109.Herrmann C., Salas P.F., Patrick B.O., de Kock C., Smith P.J., Adam M.J., Orvig C. Modular synthesis of 1,2- and 1,1′-disubstituted ferrocenyl carbohydrate chloroquine and mefloquine conjugates as potential antimalarial agents. Organometallics. 2012;31:5748–5759. doi: 10.1039/c2dt12050j. [DOI] [PubMed] [Google Scholar]
- 110.Pomel S., Dubar F., Forge D., Loiseau P.M., Biot C. New heterocyclic compounds: synthesis and antitrypanosomal properties. Bioorg. Med. Chem. 2015 doi: 10.1016/j.bmc.2015.03.029. [DOI] [PubMed] [Google Scholar]