

Abstract
Vesicular stomatitis virus (VSV) has been extensively utilized as a viral vector system for the induction of protective immune responses against a variety of pathogens. We constructed recombinant VSVs specifying either the Indiana or Chandipura virus G glycoprotein and expressing the West Nile virus (WNV) envelope (E) glycoprotein. Mice were intranasally vaccinated using a prime (Indiana)-boost (Chandipura) immunization approach and challenged with the virulent WNV-LSU-AR01. Ninety-percent (9 of 10) of the vaccinated mice survived as compared to 10% of the mock-vaccinated mice after WNV lethal challenge. Histopathological examination of brain tissues revealed neuronal necrosis in mock-vaccinated mice but not in vaccinated mice, and vaccinated, but not mock-vaccinated mice developed a strong neutralizing antibody response against WNV. Extensive immunological analysis using polychromatic flow cytometry staining revealed that vaccinated, but not mock-vaccinated mice developed robust cellular immune responses as evidenced by up-regulation of CD4+ CD154+ IFNγ+ T cells in vaccinated, but not mock-vaccinated mice. Similarly, vaccinated mice developed robust E-glycoprotein-specific CD8+ T cell immune responses as evidenced by the presence of a high percentage of CD8+ CD62Llow IFNγ+ cells. In addition, a sizeable population of CD8+ CD69+ cells was detected indicating E-specific activation of mature T cells and CD4+ CD25+ CD127low T regulatory (T reg) cells were down-regulated. These results suggest that VSV-vectored vaccines administered intranasally can efficiently induce protective humoral and cellular immune responses against WNV infections.
Keywords: West Nile virus, Vesicular stomatitis virus, Mucosal immunization, Viral vector, Humoral and cellular immunity
1. Introduction
1.1. West Nile virus (WNV)
West Nile virus (WNV) was first isolated more than 70 years ago from a febrile patient in the West Nile province of Uganda [1]. WNV is a positive-sense RNA virus belonging to genus Flavivirus in the Falviviridae family [2]. The lipid-bilayer membrane of the nascent virus contains 180 molecules of the envelope (E) and premembrane (preM) proteins organized into 60 asymmetric trimeric spikes of preM-E heterodimers [3], [4]. The E glycoprotein is the major antigenic determinant and is involved in virus binding and fusion [5]. WNV spread rapidly in North America after its initial introduction in New York [6]. WNV was transmitted via mosquito vectors and caused substantial morbidity and mortality in birds, horses and other animals including humans. Humans constitute a dead-end host because the virus does not efficiently replicate in humans. WNV can be transmitted by the intrauterine route [7], through breast milk [8], [9], blood transfusion [10], [11], [12], bone-marrow transplant [13], organ transplantation [14], [15] and through kidney dialysis [16], [17].
The human incubation period for West Nile is 2–14 days [18]. WNV-infected persons may exhibit a variety of clinical symptoms including fever, headache, muscle weakness, fatigue, nausea, vomiting, gastrointestinal manifestations, lymphadenopathy and non-pruritic maculopapular skin rash [19], [20], [21]. Additional non-neurological clinical manifestations include rhabdomyolysis [22], [23], pancreatitis [24], hepatitis [25], myositis, orchitis [26], chorioretinitis [27] and cardiac dysrhythmias [28]. Typically, less than 1% of patients suffer from West Nile neuroinvasive disease (WND) including West Nile meningitis (WNM), encephalitis (WNE) and acute flaccid paralysis (poliomyelitis-like syndrome, WNP) [29]. Among WND cases, an estimated 55–60% of the patients had WNE resulting in an estimated 20% case fatality. Additionally, 10–50% of mortalities in humans could be attributed to WNP [29].
1.2. WNV vaccines
The absence of effective treatment against WNV infection has encouraged vaccine development. A variety of different approaches have been employed to produce WNV vaccines including inactivated virus, subunit and DNA-based vaccines. Most of these vaccines appeared to be highly immunogenic, and in some cases protected against WNV-infection in experimental animals [30]. Recently, recombinant viruses expressing WNV antigens have been shown to induce strong immune responses and protection against WNV challenge in animals. Specifically, a recombinant live canarypox-vectored vaccine expressing the preM protein and the E glycoprotein induced strong immune responses in horses and cats [31], [32], [33], [34], that appeared to be partially protective [35]. Other viral-vectored vaccines that elicited protective immune responses in mice include a lentivirus vector based vaccine (TRIP/sEWNV) [36], and a measles virus-vectored vaccine [37]. Recombinant yellow fever virus (YFV) has also been used to express WNV preM and E proteins based on the extensive safety record of the YFV attenuated vaccine [38], [39]. A YFV recombinant vaccine (ChimeriVax™) has shown good immune responses in hamster, mice, non-human primates and humans [40], [41], [42]. A Phase II clinical trial with ChimeriVax™-WNV is currently underway [43].
1.3. Vesicular stomatitis-vectored vaccines
VSV is an enveloped, negative strand RNA virus belonging to the Rhabdoviridae family. Natural VSV infections of humans are rare causing at most mild flu-like illness [44]. VSV infectious viruses can be efficiently recovered by a reverse genetic approach that utilizes multiple plasmids expressing VSV genes. This methodology has enabled the rapid construction of recombinant VSV viruses expressing a variety of viral and bacterial antigens for vaccine purposes including influenza virus, bovine diarrhea virus, cotton-tail papillomavirus, human immunodeficiency virus, simian immunodeficiency virus, respiratory syncytial virus, hepatitis C, measles virus, Ebola virus, Lassa fever virus, Marburg virus, severe acute respiratory syndrome virus (SARS), and herpes simplex type-2 virus [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [63]. Recombinant VSVs have been also constructed and tested as vaccines for bacterial pathogens including Mycobacterium tuberculosis and Yersinia pestis [64], [65]. VSV-vectored vaccines have been administered via intranasal, intramuscular and subcutaneous routes and have been shown to elicit robust mucosal and systemic humoral and cellular immune responses [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [63], [66], [67], [68].
We constructed recombinant VSVs expressing the WNV E glycoprotein. A prime-boost approach was employed utilizing two different recombinant VSVs expressing either the Indiana or the Chandipura G glycoproteins for priming and boosting immunizations, respectively. Intranasal immunization of mice conferred high protection against lethal challenge with the virulent WNV strain WNV-LSU-AR01 [69]. Neuronal necrosis was observed in mock-vaccinated but not in vaccinated mice. These results suggest that VSV recombinant vaccines expressing the WNV E glycoprotein may be efficacious intranasal vaccines for animal and human use.
2. Materials and methods
2.1. Cells and plasmids
Baby hamster kidney cells (BHK-21) were obtained from the American Tissue Culture Collection (ATCC). These cells were grown using Dulbecco's modified minimal essential media (DMEM) supplemented with 10% fetal bovine serum (FBS) and appropriate amounts of antibiotics. The West Nile virus envelope (E) gene was obtained by first producing a cDNA of the E gene from the WNV-LSU-AR01 strain, and subsequently cloning this gene into the pcDNA3.1 plasmid (Invitrogen, Inc.) after PCR amplification. The E gene was further amplified by PCR from this plasmid using primers that introduced unique NotI and BamHI sites at the 5′ and 3′ using 5′ WNE-FLAG-Not-I (5′-GACGACGCGGCCGC ATGTTTAACTGCCTTGGAA TGAGC-3′) and 3′ WNE-FLAG-BamHI (5′-GCAGCAGGATCCAGCGTGCACGTTCACGG AGAGG-3′) primers. NotI and BamHI sites are italicized. The fragment was then cloned into plasmid p3XFLAG-CMV-14 (Sigma) placing the FLAG epitope coding sequence downstream and in-frame with the E glycoprotein sequence. All recombinant plasmids were confirmed by restriction endonuclease digestion and DNA sequencing.
2.2. Transient expression of the WNV E gene
BHK-21 cells were transfected with the WNV E-3XFLAG plasmid using Lipofectamine 2000 (Invitrogen) as suggested by the manufacturer. E glycoprotein was detected at 48 h post-transfection using anti-FLAG (Sigma) and anti-West Nile rabbit polyclonal antibody (Abcam). For immunofluorescence assay (IFA), cells were washed twice with phosphate buffered saline (PBS) and fixed with ice-cold methanol. Cells were then washed with PBS and wells were blocked with 2% BSA and 5% goat serum in TBS (Tris-buffered saline) for 1 h. Mouse anti-FLAG antibodies (Sigma) in blocking buffer and rabbit anti-WNV antibodies were added to respective wells at a 1:500 dilution and incubated for 1 h at room temperature. Cells were then washed six times with TBS and the secondary antibody Alexa Fluor® 488 goat anti-mouse IgG and goat anti-rabbit IgG (Invitrogen) were added to the respective wells at the same dilution. Cells were incubated in dark for 1 h. Finally, cells were washed six times with TBS and observed under a fluorescence microscope.
2.3. Construction of recombinant VSVs expressing the WNV E gene
Plasmid clones that efficiently expressed the WNV E gene were used as the template for PCR amplification of the gene, while at the same time introducing unique XhoI and NheI sites at the 5′ and 3′ ends of the gene fragment using 5′-XN2-Xho-I (5′-CCGCGGCTCGAG ATGTTT AACTGCCTTGGAATGAGC-3′) and 3′-XN2-Nhe-I (5′-GACGACGCTAGCGGATCACTAC TTGTCATCGTC-3′) primers. XhoI and NheI restriction sites are italicized. This DNA fragment was cloned into the pVSV-XN2-IN and pVSV-XN2-CH transfer vectors. Cells were infected with recombinant vaccinia virus expressing T7 polymerase (vTF7-3) at a multiplicity of infection (MOI) of 10 for 1 h. Subsequently, BHK 21 cells were co-transfected with pBS-N, pBS-P, pBS-L and pVSV-XN2 containing the WNV E gene and recombinant virus was recovered as described in detail previously [70], [71]. Control viruses having no exogenous inserted genes were also produced using pBS-N, pBS-P, pBS-L and the pVSV-XN2 (empty vector). Anti-FLAG and anti-WNV-E antibodies were used to detect expression of the E glycoprotein by IFA in VSV-infected BHK cells. Viral isolates expressing high amounts of the WNV E glycoprotein were selected through multiple rounds of plaque purification. Viral titers were determined and stocks were stored at −80 °C for vaccination studies.
2.4. Vaccination study
All animal studies were carried out after the appropriate approvals were obtained from the LSU Institutional Animal Care and Use Committee (IACUC) and BSL3 Biosafety Committee. Four groups of ten 4-week-old female Balb/c mice (Harlan, IN, USA) were used in this study. Each individual mouse was identified with an ear tag (National Band and Tag Company, KY, USA). Group I (vaccine group): These animals were mildly anesthetized by inhalation of 2–3% isoflurane and 10 μl dose of vaccine containing 105 PFU of the vaccine (rVSV-IN-WNV E) was administered intranasally using a 10 μl pipette (5 μl per nostril). Animals were boosted with the rVSV-CH-WNV E at 21 days post-vaccination using the same technique. One mouse from this group was not included in the fluorescence-activated cell sorting (FACS) analysis due to sample preparation problems (n = 9). Group II (control for vaccine group): Control group animals were vaccinated in the same way as described above with the exception that they were inoculated with 10 μl of uninfected cell culture supernatant. These animals were boosted at 21 days post-vaccination with uninfected cell culture supernatant. Animals belonging to Groups I and II were humanely euthanized at 14 days post-boost. Spleens were collected in Eppendorf tubes containing RPMI and processed by flow cytometry for intracellular cytokines and cell-surface markers associate with memory T cells, regulatory T cells and cytotoxic T cells among others. For serology, animals were bled by the sub-mandibular route (cheek bleed) using Golden-Rod lancets (Medipoint, NY). Animals were bled on 21 days post-vaccination and 14 days post-boost. Blood was collected in Becton Dickinson microtainers with serum separators (Becton Dickinson).
2.5. Challenge studies: Group III (challenge group) and Group IV (challenge group control)
These 20 animals were treated exactly in the same way as Groups I and II until the boost stage. At 10 days post-boost, these animals were transported to the animal biosafety level-3 (ABSL-3) facility for acclimatization. Blood was collected at 14 days post-boost (before challenge). Animals were challenged intraperitonially with 105 PFU of WNV-LSU-AR01 and observed 2–3 times a day for 18 days. Animals showing severe neurological symptoms (like ataxia and hunching posture) were humanely euthanized and dead animals were surgically processed immediately (thoracic and abdominal cavities opened up and placed in 10% formalin jars) for pathological studies.
2.6. Plaque reduction neutralization test (PRNT90)
Serum samples were inactivated by incubation at 65 °C for 30 min. Serial two-fold dilutions of the serum were incubated with equal volumes of 50 PFU of WNV-LSU-AR01 at 37 °C for 1 h. Serum-virus mixtures were then added to Vero cell monolayers in 12-well plates in triplicates and the plates were incubated for another hour. Plates were then overlaid with Dulbecco's modified minimum essential media (DMEM) containing 1% methyl cellulose and 2% fetal bovine serum. Plates were incubated at 37 °C for 72 h and then fixed with 10% formalin in phosphate buffered saline (PBS). Plates were washed three times with PBS and stained with 0.01% crystal violet. Plaques were counted and the highest dilution of serum resulting in reduction of 90% of the plaques was noted.
2.7. Polychromatic flow cytometric staining and analysis
Mouse splenocytes were adjusted to 107 cells/ml. One-hundred microlitre aliquots of splenocyte suspension was incubated with appropriately diluted concentrations of antibodies for 30 min at room temperature. Cells were washed once with PBS and fixed with 1X BD stabilizing fixative buffer (BD Biosciences) in distilled water. Cells were kept protected from light at 4 °C and flow cytometric acquisition was completed within 24 h of staining. Polychromatic (7 parameters) flow cytometric acquisition was performed on a LSR II Becton Dickinson instrument having three lasers (488 nm blue laser, 633 nm red laser and 407 violet laser) by using FITC, PE-Texas red, APC, APC-Cy7 and Pacific Blue as the available fluorochrome parameters. Single-stained controls for each fluorochrome were used for setting flow cytometry compensation. Monoclonal antibodies including CD127 FITC (A7R34, eBioscience), CD62L PE-Texas Red (MEL-14, Invitrogen), CD25 APC (3C7, BD Biosciences), CD4 APC-Cy7 (GK1.5, BD Biosciences) and CD8a Pacific Blue (53-6.7, BD Biosciences) were used. At least 50,000 events were collected by gating on CD4+ T cells and those data were analyzed using FlowJo software (TreeStar Inc.) version 8.7.1.
To test CD4+ or CD8+ T lymphocytes subsets for IFNγ production, intracellular cytokine flow cytometry (CFC) assay was employed in response to each WNV peptide pool stimulation as described previously [72]. Briefly, processed splenocytes were resuspended at 1 × 106 cells/ml in complete RPMI-10 with 10% FCS, and stimulated with 2 different WNV peptide pools at a final concentration of 1 μg/ml of each peptide pool. Peptide pools (15–19mers with 10–11 amino acids overlap) derived from the WNV E glycoprotein were based on the WNV-NY99 E amino acid sequence (NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH). The 67 peptide array was divided to generate two peptide pools. Peptide pool 1 (pp1) was made of peptides 1–34 and peptide pool 2 (pp2) was composed of peptides 35–67. For positive controls, PMA (50 ng/ml, Sigma) and ionomycin (1 μg/ml, Sigma) were used. Negative controls had no antigen or mitogen stimulation. Brefeldin A (10 μg/ml, Sigma) was added to cultures after the first hour, in a 6-h incubation period. Following stimulation, cells were stained for cell-surface markers with directly conjugated mAbs to CD69 FITC (H1.2F3, BD Biosciences), CD62L PE-TR, CD4 APC-Cy7 and CD8a pacific blue for 30 min at room temperature and washed with dPBS/BSA wash buffer. Cells were then fixed and permeabilized by using Cytofix/Cytoperm (BD Biosciences), washed twice in Perm Buffer (BD Biosciences), and stained with intracellular mAbs. IFNγ PE (XMG1.2, BD Biosciences) and/or CD154 APC (MR1, eBiosciences) were added to cells and incubated at room temperature for 30 min. Single color and isotype-matched control antibodies were used to confirm staining specificity. After washing, cells were resuspended in 1% paraformaldehyde in PBS and stored in the dark at 4 °C. Data were acquired within 24 h of staining using a LSR II instrument (BD Immunocytometry System) and FACSDiva software (BD Immunocytometry System). For each sample, 50,000 events were collected by gating on CD4+ T cells. Data analysis was performed using FlowJo software. Gated CD4+ and CD8+ T cells were further analyzed for its cytokine production. Positive cytokine responses were determined based on the percentage of cytokine responses obtained above background responses (unstimulated medium control) in each experiment.
2.8. Histopathology
Tissues (brain, lung, liver, bilateral kidneys, heart, spleen, skull, and vertebra) were collected from the mice, euthanized or after dead, and fixed by immersion in 10% neutral buffered formalin. The skull and vertebra were decalcified in 10% formic acid for 3 days. All sampled tissues were routinely processed into paraffin, and 3–4 μm sections were cut for hematoxylin and eosin staining (H&E). H&E sections of the nasal olfactory epithelium and bulb in the skull and four sections of the spinal cord including two consecutive anterior cervico-thoracic and two consecutive lumbar-sacral posterior sections in the vertebrae were examined under the light microscope.
2.9. Statistical analysis
Graphical presentation and statistical analysis of the results were performed by two-tailed Student's paired t-test using the GraphPad Prism 4.0 (GraphPad Software Inc., SanDiego, CA). Expressions of CD62L, CD154, CD25 and CD127 between immunized and mock challenged mice were determined by non-parametric Mann–Whitney t-test. For all statistical analysis, results were considered significant if p < 0.05. Mouse survival analysis was done with GraphPad Prism 5.01 (GraphPad Software Inc., SanDiego, CA) using the Gehan-Breslow-Wilcoxin test.
3. Results
3.1. Cloning and transient expression of the WNV E glycoprotein
The WNV-LSU-AR01 strain was isolated from a dead blue jay (Cyanocitta cristata) in Louisiana in 2001. Recently, the entire genome of this strain was sequenced and phylogenetically compared to 75 full WNV genomes deposited in GeneBank [69]. The E gene was amplified from viral RNA using specific primers as described in Section 2 and cloned into plasmid p3XFLAG (Sigma) placing the entire open reading frame of WNV E in-frame with the 3XFLAG coding sequence resulting in the addition of the 3XFLAG amino acid sequence immediately after the last carboxyl terminal amino acid of the E glycoprotein. The p3XFLAG-E plasmid was transfected into baby hamster kidney cells (BHK-21) and E glycoprotein expression was detected at 48 h post-transfection using anti-FLAG monoclonal antibody. The anti-FLAG antibody detected E glycoprotein expression in 3XFLAG-E transfected BHK cells, while mock-transfected BHK cells failed to react with the anti-FLAG antibody.
3.2. Construction of recombinant vesicular stomatitis virus (VSV) expressing the WNV E glycoprotein
To construct recombinant VSVs expressing the E glycoprotein, the E gene was amplified with primers engineered to have unique XhoI and NheI restriction sites at the E 5′ and 3′ termini, respectively. The amplified E gene (with the 3XFLAG coding sequence) was cloned within the unique XhoI and NheI restriction sites of plasmids pVSV-XN2-IN and pVSV-XN2-CH containing the Indiana and Chandipura G glycoprotein gene, respectively (Fig. 1A). Recombinant VSV was recovered after co-transfection of pVSV-XN2-E with three other plasmids encoding the VSV polymerase subunits (P and L), and the nucleocapsid (N), purified by filtration and extensively plaque-purified. The appropriate insertion of the WNV gene within the VSV genomes was confirmed by direct DNA sequencing of viral RNA after RT-PCR amplification of specific cDNA regions. WNV E expression was readily detected by indirect immunofluorescence assay (IFA) using anti-FLAG monoclonal antibody in recombinant VSV-infected BHK cells, while WNV E was not detected in mock-infected BHK cells (Fig. 1B). Cell lysates from BHK-21 cells infected with recombinant VSVs expressing the WNV E glycoprotein were tested for E glycoprotein expression in western immunoblots. Anti-FLAG antibody readily detected major protein species with apparent molecular masses of approximately 53–55 kDa, respectively in agreement with previous reports (Fig. 1C) [33], [73].
Fig. 1.

Construction of rVSVs expressing the LSU-AR01 E glycoprotein. (A) The WNV-LSU-AR01 E-FLAG fusion gene was cloned into the unique XhoI and NheI sites in pVSV-XN2. (B) IFA showing rVSV expression of WNV envelope glycoprotein after infection of BHK-21 cells. Expression of the WNV E glycoprotein was assayed using anti-FLAG and anti-West Nile antibodies. (C) Expression of the WNV E protein was with an apparent molecular mass of 53–55 kDa on a western immunoblot using anti-FLAG antibodies. Lane 1 is cell control, lanes 2 and 3 are rVSV-IN-WNE and rVSV-CH-WNE respectively, lane 4 is the molecular mass ladder and lane 5 is cell lysate from VSV-infected BHK-21 cells (control).
3.3. Mouse immunization and challenge schedule
Four groups of 4-week-old Balb/c mice (Harlan, IN, USA) were used for the vaccine-challenge experiments. All four groups of mice were vaccinated by intranasal administration of 105 PFU of pVSV-XN2-IN-E recombinant virus at day 0 and boosted with pVSV-XN2-CH-E (105 PFU) 21 days post-vaccination (Fig. 2A). Mice in Groups I and II were processed for immunological analyses (see Section 2), while Groups III and IV were challenged with 105 PFU of WNV-LSU-AR01 administered intraperitoneally. Mice in the challenge groups were observed for 18 days for clinical signs including ruffled fur, ataxia, hunching posture, lethargy and mortality. VSV-E vaccinated and boosted animals exhibited 90% survival, while only 10% of the mock-vaccinated animals survived WNV-LSU-AR01 challenge (p = 0.004) (Fig. 2B). Vaccinated animals appeared to have mild clinical signs post-challenge including mild fur ruffling, but recovered quickly to a full healthy status. In contrast, mock-vaccinated animals exhibited severe clinical signs post-challenge including high degree of fur ruffling, ataxia, lethargy and eventually death. Post-mortem histopathological examination revealed that none of the vaccinated mice showed any central nervous system (CNS) pathology as compared to mock-vaccinated animals, which exhibited severe neuronal necrosis and lymphoplasmacytic perivascular cuffing (Fig. 3 ). The single mouse in the vaccinated group that died at 12 days post-challenge had suppurative rhinitis which may be suggestive of bacterial infection. Mild suppurative inflammation was also observed in the visceral pleura and subpleura of three mock-vaccinated mice that died before 11 days post-challenge (not shown). There were no significant histopathological abnormalities within other tissues examined. In a separate set of experiments mice were vaccinated via the intramuscular route and challenged with a different strain of WNV (WNV-NY99) 9 weeks post-boost. The vaccine efficaciously protected 70% of the vaccinated mice (not shown).
Fig. 2.
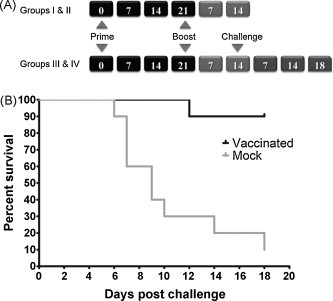
Vaccination and animal challenge schedule. (A) Schematic of the time-line followed for vaccination, boost-vaccination and challenge with WNV-AR01. (B) Kaplan-Meier Survival curves. Mice in challenge groups were challenged intraperitoneally with 105 PFU of WNV-LSU-AR01 14 days post-boost-vaccination and observed for 18 days. Ninety percent of the WNV vaccinated animals survived, while 90% of the mock-vaccinated animals died. A statistically significant difference was observed between the WNV and mock-vaccinated groups (p = 0.0004) using the Gehan-Breslow-Wilcoxin test.
Fig. 3.

Histopathology of cerebrum sections from mock-vaccinated and vaccinated mice after WNV challenge. (A) Mock-vaccinated group: Cerebral cortex showing large numbers of necrotic neurons (arrows), characterized by angular and shrunken cell bodies containing pyknotic nucleus and densely eosinophilic cytoplasm at 40× magnification. (B) WNV vaccinated group: Cerebral cortex showing normal neurons at 40× magnification. Cerebral cortex of mock-vaccinated (C) and WNV vaccinated (D) groups at 60× magnification. H&E stain, Bar = 50 μm (A and B) or 100 μm (C and D).
3.4. Induction of strong neutralizing antibody correlates with protection
The ability of mouse sera to neutralize WNV-LSU-AR01 strain was tested in a standard plaque reduction neutralization test (PRNT90). Vaccinated animals developed strong neutralizing antibody responses against the LSU-AR01 at 21 days after primary vaccination. Specifically, 9 of 10 mice developed PRNT90 titers of 1:32 and one mouse had a titer of 1:64. Neutralizing antibody titers increased at 14 days post-boost-vaccination. Specifically, 9 of 10 mice had a PRNT90 titer of 1:64, while the remaining mouse had a titer of 1:128.
CD154 expression in CD4+ T cells is intimately involved in the polyclonal activation of immature B cells [74]. Therefore, we compared the expression of CD154 in both vaccinated and mock-vaccinated mice after in vitro stimulation with PMA/ionomycin followed by FACS analysis (see Section 2). These experiments revealed the presence of a significantly higher population of CD4+CD154+IFNγ+ T cells in vaccinated mice compared to mock-vaccinated mice (mean value 1.73% versus 1.0% in vaccinated and mock-vaccinated mice respectively, p < 0.0001, Fig. 4A and C), as also indicated by the observed differences in their mean fluorescence intensities (Fig. 4B and D).
Fig. 4.

Correlates of T cell-mediated induction of humoral immune responses: (A) Representative contour plot showing increased percentage of CD4+CD154+IFNγ+ T cells in vaccinated mice compared to mock-vaccinated mice after 6 h of in vitro PMA/ION stimulation. (B) Histogram showing increased mean fluorescence intensity (MFI) of CD4+CD154+IFNγ+ T cells in a vaccinated mouse compared to a mock-vaccinated mouse. (C) Increased MFI in CD4+CD154+IFNγ+ T cells was observed in WNV mice compared to mock-vaccinated animals (p = 0.01). (D) The increased MFI percentage of CD4+CD154+IFNγ+ T cells suggests that activated CD4+ T cells stimulated B cells with the help of costimulatory signals inducing humoral immune responses. A statistically significant difference was observed in the percentage of CD4+CD154+IFNγ+ T cells between vaccinated and mock-vaccinated animals (p < 0.001).
3.5. Antigen-specific cellular immune responses
Antigen-specific cytokine responses were determined in all vaccinated and mock-vaccinated mice. Specifically, WNV-E specific T cell responses were measured using cytokine flow cytometry (CFC) to determine IFNγ responses. Overall, 7 of 9 vaccinated mice had detectable IFNγ responses (ranged from 0.07 to 0.80%) in splenic CD8+ T cells. CD4+ T cell positive IFNγ responses were absent in any of the vaccinated mice. Both peptide pools 1(E amino acids 291–554) and 2 (E amino acids 544–791) appeared to contain T cell epitopes, however, peptide pool 1 contained dominant T cell epitopes. One of the 9 mice developed antigen-specific IFNγ responses against both the WNV-E peptide pools. None of the mock-vaccinated mice had any detectable IFNγ responses above background levels.
3.6. Profiles of CD62L and CD69 expression
CD62L is a lymphocyte homing marker that is generally associated with extravasation of activated T cells to peripheral sites of inflammation. Generally, increased percentages of CD8+ T cells were present in the vaccinated mice compared to the mock-vaccinated mice (mean 18.3% and 15.3% for vaccinated and mock-vaccinated mice respectively, p = 0.01) (Fig. 5A). CD8+ T cell subsets in all vaccinated mice had lower CD62L expression compared to mock-vaccinated mice (p = 0.0003) (Fig. 5B). To further characterize the cells responsible for inducing cytokine responses, antigen-specific cytokine positive cells were determined. A significant population (0.73%) of the IFNγ positive cells was memory cells (CD8+ CD62L−) (Fig. 5C).
Fig. 5.

Induction of WNV E-specific CD8+ T cells: (A) A higher percentage of CD8+ T cells was present in vaccinated animals compared to mock-vaccinated controls (p = 0.01). (B) Down-regulation of CD62L expression in CD8+ T cells in vaccinated mice. A statistically significant difference was observed in the CD8+CD62L+ T cell populations (p = 0.0003) in the WNV vaccinated versus the mock-vaccinated animals. (C) Representative contour plots showing WNV E-specific CD8+ T cells. The majority of the IFNγ producing cells were CD8+CD62L− T cells (black circle) indicating the presence of activated effector T cells. (D) The percentage of CD8+CD69+ T cell population was increased (p = 0.012) in vaccinated mice versus mock-vaccinated mice indicating the presence of WNV E-specific stimulation of T cells.
CD69 is an early activation marker indicative of the presence of antigen-specific stimulation of mature T cells [75]. CD69 up-regulation of activated CD8+ T cells was detected in all the vaccinated mice following antigen stimulation compared to mock-vaccinated mice (mean 1.8% versus 0.8% in vaccinated and mock-vaccinated mice, p = 0.012) indicating E-specific stimulation of mature T cells in vaccinated animals (Fig. 5D).
3.7. Profile of T reg activation in vaccinated versus mock-vaccinated mice
Initial determination of CD4+T cell percentages in splenocytes revealed no significant differences between vaccinated and mock-vaccinated mice (Fig. 6A). CD127, the α-chain of the IL7 receptor, in combination with CD25, the α-chain of the IL2 receptor, were used to define the relative abundance of T reg cells within the population of conventional T cells [76]. Analysis of CD4+ CD25+CD127low cells revealed that vaccinated mice had a significantly lower population of these cells (mean 6.3%) in comparison to the mock-vaccinated mice (mean 7.3%) (p < 0.05) (Fig. 6B and C).
Fig. 6.

Role of regulatory T cells in vaccinated animals: (A) Percentage of CD4+ T cells in WNV vaccinated and mock-vaccinated animals. There was no statistically significant difference observed between these two groups. (B) Representative dot plots showing the gating strategy for T reg cells derived from spleenocytes. CD4+ T cells were first gated and plotted for CD25 and CD127. CD4+ CD25+CD127low T cells were defined as T regs. (C) Percentage of CD4+CD25+CD127low T reg cells. A statistically significant difference (p < 0.05) was observed between WNV-vaccinated and mock-vaccinated mice.
4. Discussion
VSV-vectored vaccines have shown exceptional promise for protecting animals and humans against different viral and bacterial pathogens. A VSV-vectored vaccine expressing the WNV-E glycoprotein was constructed and found to efficiently protect mice after intranasal administration against lethal WNV challenge. The salient features of this vaccine study are: (1) A prime-boost intranasal vaccination approach with recombinant VSVs expressing the WNV E glycoprotein produced robust CD8+IFNγ+ T cell responses; (2) This vaccine approach produced strong neutralizing titers against the WNV; (3) Vaccinated mice were protected against lethal challenge and they were free of neuronal necrosis, while unvaccinated mice exhibited severe neuronal necrosis and inflammation in the brain. These results suggest that a prime-boost VSV-vectored intranasal vaccine approach induces strong humoral and cellular immune responses that protect mice against WNV-induced neuronal necrosis.
Mucosal surfaces constitute the natural route of VSV infections. VSV is primarily a veterinary viral pathogen that infects cattle, horses, sheep and other animals. VSV infects animals via transmucosal and transcutaneous routes [77]. VSV may also be transmitted through sandflies, blackflies and mosquitoes [44], [78]. The VSV G glycoprotein is a potent immunogen and also serves important functions in virus-entry and virus-induced cell fusion [79]. Recombinant VSVs expressing a variety of viral and bacterial antigens have been constructed. Vaccine studies with these recombinant VSVs have showed that intranasal and intramuscular administration of the rVSVs were safe and efficient in inducing protective humoral and cellular immune responses against a variety of pathogens [78]. Of particular interest is the ability of the VSV-vector system to elicit strong humoral and cellular immune responses via the intranasal route [48], [49], [59], [60], [65], [80] that can be substantially easier to administer than intramuscularly injected vaccines. In these vaccine studies, although the “empty” VSV vector elicited robust humoral and cellular immune responses against VSV, these responses did not contribute to protection against a variety of pathogens indicating that specific immune responses against the expressed transgene were primarily responsible for protection [49], [58], [59], [60], [81], [82].
We constructed rVSVs that expressed the WNV-E glycoprotein and either the VSV Indiana G glycoprotein, or the Chandipura vesiculovirus G glycoprotein. This pair of rVSVs was used in a prime-boost-vaccination approach to maximize humoral immune responses against the WNV-E glycoprotein expressed by both viruses, while minimizing the anamnestic immune response against the VSV vector targeted predominantly against the G glycoprotein. This is largely accomplished because the Chandipura G and the VSV-Indiana G glycoproteins are approximately 60% different in their amino acid sequences [83]. Recombinant VSVs are known to non-specifically incorporate certain other viral and cellular glycoproteins into their virions without adversely affecting viral infectivity [70]. The insertion of the foreign E gene into the VSV genome did not adversely affect viral replication and infectivity, because rVSV containing the E gene replicated to similar titers with those of the VSV control virus that did not have a foreign gene inserted within their genomes (not shown). Moreover, rVSV-E isolates were stable, since multiple serial passages of virus stocks in BHK cells did not affect E glycoprotein expression and genomic stability (not shown). Although it is unclear whether the WNV E glycoprotein is inserted into VSV envelopes, these results suggested that rVSV-E were stable retaining wild-type levels of viral replication and infectivity. Recombinant VSV-E expressed WNV-E glycoprotein to high levels in BHK cells and the expressed E glycoprotein appeared to be fully glycosylated as evidenced by the apparent molecular mass of approximately 53–55 kDa in SDS-PAGE in agreement with published reports [33], [73].
Based on the known strong immune responses generated by VSV, especially when administered via the intranasal route, we devised an experimental vaccine protocol to vaccinate mice through the intranasal route using a prime-boost strategy. This prime-boost-vaccination approach resulted in 90% (9 of 10) of the mice surviving lethal challenge with the WNV-LSU-AR01 virulent strain. The single mouse from the vaccinated group of mice that died late in the experiment (12 days post-challenge) appeared to die from WNV-unrelated causes, since histopathological examination showed severe suppurative rhinitis but no histological abnormality in the brain. Therefore, the rVSV-E prime-boost intranasal vaccination protocol was highly efficacious in protecting mice against WNV infection. Similar results were obtained in a different experiment in which mice were vaccinated via the intramuscular route and challenged with WNV-NY99 strain instead of the LSU-AR01 9 weeks post-boost. In this experiment 70% of the mice survived indicating that intramuscular immunization may also provide protective immune responses against WNV infection.
Primary WNV infection is thought to result in local replication of the virus in peripheral organs and viremia that ultimately results in virus invading the CNS. WNV mortality is thought to be largely caused by replication of the virus in the CNS tissues of animals and the resultant immunopathological damage of CNS tissues. Accordingly, unvaccinated mice showed obvious clinical signs of neurological disease such as ataxia, hunching posture, lethargy and hindlimb paralysis. Histopathological examination of brain tissues showed neuronal necrosis, perivascular cuffing, and microgliosis. In contrast, only a few vaccinated mice developed mild clinical signs such as mild ruffled fur, but recovered quickly. Importantly, none of the vaccinated mice exhibited any neuronal necrosis.
The interaction of CD40 on B cells with CD154 (CD40L) on CD4+ T cells results in T cell mediated activation of B cells resulting in immunoglobulin class switching, somatic hypermutation and proliferation [84], [85], [86]. Accordingly, CD4+ CD154+ IFNγ+ T cells were up-regulated in vaccinated but not control mice indicating generation of T cell mediated B cell activation. The specificity of this response is not discernable, since it may be due to either or both VSV and WNV antigens. However, strong neutralizing antibody titers were also produced against WNV indicating the induction of E-specific humoral immune responses. This result is in agreement with previous reports showing that other VSV-vectored vaccines induced strong humoral immune responses against different VSV-expressed antigens. Specifically, recombinant VSVs expressing either the respiratory syncytial virus F glycoprotein [49], or rVSV expressing the severe acute respiratory syndrome (SARS) corona virus (SARS-CoV) produced high antibody titers against the F glycoprotein and SARC-CoV spike (S) glycoprotein, while strong immune responses against the VSV virus was noted [59].
The WNV E glycoprotein contains multiple predicted and experimentally verified cytotoxic T cell (CTL) epitopes [87], [88], [89], [90]. The availability of a library of overlapping peptides derived from the WNV E glycoprotein allowed the elucidation of antigen-specific cellular immune responses. Peptide pool 1 composed of the first 34 peptides averaging 12–18 amino acids each generated stronger cellular CD8+IFNγ+ T cell responses in in vitro proliferation assays, in comparison to peptide pool 2, which represented the carboxyl terminus-half of the WNV E glycoprotein. Peptide pool 1 contains the experimentally verified CTL epitope RSYCYLAT (E 347–354) while peptide pool 2 contains the CTL epitope IALTFLAV (E771–778), both of which have been shown to confer protection against lethal WNV-challenge in mice [87], [89]. In vitro stimulation of lymphocytes from vaccinated mice revealed the presence of antigen-specific IFNγ responses specifically in CD8+CD62Llow T cells. CD62L (L-selectin) mediates adhesion of resting lymphocytes to peripheral lymph nodes. Typically, high expression of CD62L (CD62Lhi) reveals entrapment of lymphocytes within lymph nodes, while low CD62L (CD62Llow) cell-surface expression (the result of T cell activation) is indicative of lymphocyte extravasation to sites of inflammation [91]. Splenocytes from vaccinated mice had significantly lower expression of the CD62L marker on E-specific IFNγ+ CD8+T cells revealing activation and extravasation of these cells to peripheral sites, potentially involved in killing virus-infected cells prior to transmission to the CNS. CD69 is an early activation marker that is absent in resting lymphocytes [75]. The up-regulation of the CD8+CD69+ E-specific T cell responses in vaccinated versus mock-vaccinated mice provides additional evidence for the stimulation of T cells. Accordingly, CD8+CD69+ E-specific population of T cells was up-regulated in vaccinated versus mock-vaccinated mice indicating the generation of activated memory CD8+ T cells. It is unclear whether the observed CD8+T cell memory responses confer long-term immunity against WNV infection. T regs are known to play important roles in down-regulation of anti-self immune responses [92], and to suppress proliferation and cytokine production of effector T cells [93]. Typically, during viral infections, up-regulation of humoral and cellular immune responses causes down-regulation of T reg activation. Typically, T regs express the FoxP3 and CD25 markers. The IL-7 receptor CD127 marker expression is inversely correlated to FoxP3 expression and CD127low CD25+ cells have been shown to be positive for FoxP3 [93], [94]. Consequently, the CD25+CD127low population was used to define T regs. As expected, there was a negative correlation between the relative population of T reg cells (CD4+CD25+CD127low) and antigen-specific CTL responses in the vaccinated mice. However, the specificity of this immune response cannot be discerned, since it most likely is caused by both VSV and E glycoprotein antigens.
A variety of experimental vaccine approaches have been reported to generate protective humoral and cellular immune responses against flaviviruses and specifically WNV. The relative role of humoral versus cellular immune responses has been extensively debated in the literature. Certain studies have suggested that a strong humoral immune response evidenced by the production of high titer anti-WNV titers is necessary and sufficient to protect mice from CNS infection, while other reports have argued that a cellular immune response characterized by a robust anti-WNV CD8+ T cell responses is necessary for protecting and clearing brain tissues from WNV [89], [95], [96]. One report has argued that CTL-immune responses may result in exacerbated immunopathology in brain and CNS tissues at infections with low WNV titers (103 PFU) [95]. In our experiments, 105 WNV PFU were inoculated intraperitoneally. Vaccinated mice had no evidence of neuronal necrosis suggesting the CD8+T cell responses conferred protection and virus clearance. It is probable that both humoral and cellular immune responses generated against the WNV E glycoprotein prevented the virus from entering CNS, potentially arresting the virus at peripheral sites. Alternatively, if some virus escaped peripheral immune surveillance, it is possible that CTLs cleared the virus from brain tissues before it could cause significant damage and resultant immunopathological manifestations.
In summary, the VSV-E-vectored vaccine appeared to elicit robust humoral and cellular immune responses that efficiently protected mice from WNV lethal challenge. Intranasal vaccination is second only to oral vaccination with regard to the relative ease of administration and patient compliance issues rendering this approach attractive for human use. Recently, single-cycle VSV-vectored vaccines have been shown to generate robust immune responses against a number of viral pathogens including HIV, Ebola, Marburg, Lassa, influenza, avian influenza, hepatitis C and RSV viruses [46], [47], [49], [54], [55], [56], [57], [58], [81], [97]. Based on these results, it is expected that single cycle VSV-WNV vaccines would be also efficacious. Additional improvements in attenuating VSV can be made by providing more than one viral protein in trans through complementing cells, as well as engineering additional mutations that are known to attenuate VSV.
Acknowledgements
This work was supported by NIH:NCRR 2P51RR000164 project. KGK and AI were supported by the Division of Biotechnology and Molecular Medicine (BIOMMED), LSU School of Veterinary Medicine. BP was supported by the FACS Immunology Core Laboratory of the Tulane National Primate Research Center (NIH:NCRR 2P51RR000164). The following reagent was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH: Peptide Array West Nile Virus Gene E, NR-435. We gratefully acknowledge Dr. John K. Rose (Yale University, School of Medicine) for providing us with the VSV reverse genetics systems and Ms. Li Huang and other BIOMMED staff for technical assistance.
References
- 1.Smithburn K.C., Hughes T.P., Burke A.W., Paul J.H. A neurotropic virus isolated from the blood of a native of Uganda. Am J Trop Med Hyg. 1940;20:471–492. [Google Scholar]
- 2.Lindenbach B.D., Thiel H.-J., Rice C.M. Flaviviridae: the viruses and their replication. In: Fields B.N., Knipe D.M., Howley P.M., editors. Fields’ virology. 5th Edition. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1101–1152. [Google Scholar]
- 3.Zhang Y., Corver J., Chipman P.R., Zhang W., Pletnev S.V., Sedlak D. Structures of immature flavivirus particles. Embo J. 2003;22(June (11)):2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y., Zhang W., Ogata S., Clements D., Strauss J.H., Baker T.S. Conformational changes of the flavivirus E glycoprotein. Structure. 2004;12(September (9)):1607–1618. doi: 10.1016/j.str.2004.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ledizet M., Kar K., Foellmer H.G., Bonafe N., Anthony K.G., Gould L.H. Antibodies targeting linear determinants of the envelope protein protect mice against West Nile virus. J Infect Dis. 2007;196(December (12)):1741–1748. doi: 10.1086/523654. [DOI] [PubMed] [Google Scholar]
- 6.Lanciotti R.S., Roehrig J.T., Deubel V., Smith J., Parker M., Steele K. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286(December (5448)):2333–2337. doi: 10.1126/science.286.5448.2333. [DOI] [PubMed] [Google Scholar]
- 7.CDC. Intrauterine West Nile virus infection--New York, 2002. MMWR Morb Mortal Wkly Rep 2002;51(December (50)):1135–6. [PubMed]
- 8.CDC. Possible West Nile virus transmission to an infant through breast-feeding--Michigan, 2002. MMWR Morb Mortal Wkly Rep 2002;51(October (39)):877–8. [PubMed]
- 9.Hayes E.B., O’Leary D.R. West Nile virus infection: a pediatric perspective. Pediatrics. 2004;113(May (5)):1375–1381. doi: 10.1542/peds.113.5.1375. [DOI] [PubMed] [Google Scholar]
- 10.CDC. Transfusion-associated transmission of West Nile virus--Arizona, 2004. MMWR Morb Mortal Wkly Rep 2004;53(September (36)):842–4. [PubMed]
- 11.CDC. Investigations of West Nile virus infections in recipients of blood transfusions. MMWR Morb Mortal Wkly Rep 2002;51(November (43)):973–4. [PubMed]
- 12.CDC. Detection of West Nile virus in blood donations--United States, 2003. MMWR Morb Mortal Wkly Rep 2003;52(August (32)):769–72. [PubMed]
- 13.Hiatt B., DesJardin L., Carter T., Gingrich R., Thompson C., de Magalhaes-Silverman M. A fatal case of West Nile virus infection in a bone marrow transplant recipient. Clin Infect Dis. 2003;37(November (9)):e129–e131. doi: 10.1086/378891. [DOI] [PubMed] [Google Scholar]
- 14.CDC. West Nile virus infection in organ donor and transplant recipients--Georgia and Florida, 2002. MMWR Morb Mortal Wkly Rep 2002;51(September (35)):790. [PubMed]
- 15.Iwamoto M., Jernigan D.B., Guasch A., Trepka M.J., Blackmore C.G., Hellinger W.C. Transmission of West Nile virus from an organ donor to four transplant recipients. N Engl J Med. 2003;348(May (22)):2196–2203. doi: 10.1056/NEJMoa022987. [DOI] [PubMed] [Google Scholar]
- 16.CDC. Possible dialysis-related west nile virus transmission--Georgia, 2003. MMWR Morb Mortal Wkly Rep 2004;53(August (32)):738–9. [PubMed]
- 17.Cairoli O. The West Nile Virus and the dialysis/transplant patient. Nephrol News Issues. 2005;19(November (12)):73–75. [PubMed] [Google Scholar]
- 18.Gea-Banacloche J., Johnson R.T., Bagic A., Butman J.A., Murray P.R., Agrawal A.G. West Nile virus: pathogenesis and therapeutic options. Ann Intern Med. 2004;140(April (7)):545–553. doi: 10.7326/0003-4819-140-7-200404060-00015. [DOI] [PubMed] [Google Scholar]
- 19.Del Giudice P., Schuffenecker I., Zeller H., Grelier M., Vandenbos F., Dellamonica P. Skin manifestations of West Nile virus infection. Dermatology. 2005;211(4):348–350. doi: 10.1159/000088506. [DOI] [PubMed] [Google Scholar]
- 20.Ferguson D.D., Gershman K., LeBailly A., Petersen L.R. Characteristics of the rash associated with West Nile virus fever. Clin Infect Dis. 2005;41(October (8)):1204–1207. doi: 10.1086/444506. [DOI] [PubMed] [Google Scholar]
- 21.Davis L.E., DeBiasi R., Goade D.E., Haaland K.Y., Harrington J.A., Harnar J.B. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60(September (3)):286–300. doi: 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- 22.Jeha L.E., Sila C.A., Lederman R.J., Prayson R.A., Isada C.M., Gordon S.M. West Nile virus infection: a new acute paralytic illness. Neurology. 2003;61(July (1)):55–59. doi: 10.1212/01.wnl.0000073617.08185.0a. [DOI] [PubMed] [Google Scholar]
- 23.Kulstad E.B., Wichter M.D. West Nile encephalitis presenting as a stroke. Ann Emerg Med. 2003;41(February (2)):283. doi: 10.1067/mem.2003.67. [DOI] [PubMed] [Google Scholar]
- 24.Perelman A., Stern J. Acute pancreatitis in West Nile fever. Am J Trop Med Hyg. 1974;23(November (6)):1150–1152. doi: 10.4269/ajtmh.1974.23.1150. [DOI] [PubMed] [Google Scholar]
- 25.Sampson B.A., Ambrosi C., Charlot A., Reiber K., Veress J.F., Armbrustmacher V. The pathology of human West Nile virus infection. Hum Pathol. 2000;31(May (5)):527–531. doi: 10.1053/hp.2000.8047. [DOI] [PubMed] [Google Scholar]
- 26.Smith R.D., Konoplev S., DeCourten-Myers G., Brown T. West Nile virus encephalitis with myositis and orchitis. Hum Pathol. 2004;35(February (2)):254–258. doi: 10.1016/j.humpath.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 27.Khairallah M., Ben Yahia S., Ladjimi A., Zeghidi H., Ben Romdhane F., Besbes L. Chorioretinal involvement in patients with West Nile virus infection. Ophthalmology. 2004;111(November (11)):2065–2070. doi: 10.1016/j.ophtha.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 28.Hayes E.B., Sejvar J.J., Zaki S.R., Lanciotti R.S., Bode A.V., Campbell G.L. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg Infect Dis. 2005;11(August (8)):1174–1179. doi: 10.3201/eid1108.050289b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sejvar J.J. The long-term outcomes of human West Nile virus infection. Clin Infect Dis. 2007;44(June (12)):1617–1624. doi: 10.1086/518281. [DOI] [PubMed] [Google Scholar]
- 30.Dauphin G., Zientara S. West Nile virus: recent trends in diagnosis and vaccine development. Vaccine. 2007;25(July (30)):5563–5576. doi: 10.1016/j.vaccine.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Grosenbaugh D.A., Backus C.S., Karaca K., Minke J.M., Nordgren R.M. The anamnestic serologic response to vaccination with a canarypox virus-vectored recombinant West Nile virus (WNV) vaccine in horses previously vaccinated with an inactivated WNV vaccine. Vet Ther. 2004;5(Winter (4)):251–257. [PubMed] [Google Scholar]
- 32.Siger L., Bowen R.A., Karaca K., Murray M.J., Gordy P.W., Loosmore S.M. Assessment of the efficacy of a single dose of a recombinant vaccine against West Nile virus in response to natural challenge with West Nile virus-infected mosquitoes in horses. Am J Vet Res. 2004;65(November (11)):1459–1462. doi: 10.2460/ajvr.2004.65.1459. [DOI] [PubMed] [Google Scholar]
- 33.Minke J.M., Siger L., Karaca K., Austgen L., Gordy P., Bowen R. Recombinant canarypoxvirus vaccine carrying the prM/E genes of West Nile virus protects horses against a West Nile virus-mosquito challenge. Arch Virol Suppl. 2004;(18):221–230. doi: 10.1007/978-3-7091-0572-6_20. [DOI] [PubMed] [Google Scholar]
- 34.Karaca K., Bowen R., Austgen L.E., Teehee M., Siger L., Grosenbaugh D. Recombinant canarypox vectored West Nile virus (WNV) vaccine protects dogs and cats against a mosquito WNV challenge. Vaccine. 2005;23(May (29)):3808–3813. doi: 10.1016/j.vaccine.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 35.Siger L., Bowen R., Karaca K., Murray M., Jagannatha S., Echols B. Evaluation of the efficacy provided by a Recombinant Canarypox-Vectored Equine West Nile Virus vaccine against an experimental West Nile Virus intrathecal challenge in horses. Vet Ther. 2006;7(Fall (3)):249–256. [PubMed] [Google Scholar]
- 36.Iglesias M.C., Frenkiel M.P., Mollier K., Souque P., Despres P., Charneau P. A single immunization with a minute dose of a lentiviral vector-based vaccine is highly effective at eliciting protective humoral immunity against West Nile virus. J Gene Med. 2006;8(March (3)):265–274. doi: 10.1002/jgm.837. [DOI] [PubMed] [Google Scholar]
- 37.Despres P., Combredet C., Frenkiel M.P., Lorin C., Brahic M., Tangy F. Live measles vaccine expressing the secreted form of the West Nile virus envelope glycoprotein protects against West Nile virus encephalitis. J Infect Dis. 2005;191(January (2)):207–214. doi: 10.1086/426824. [DOI] [PubMed] [Google Scholar]
- 38.Monath T.P. Yellow fever: an update. Lancet Infect Dis. 2001;1(August (1)):11–20. doi: 10.1016/S1473-3099(01)00016-0. [DOI] [PubMed] [Google Scholar]
- 39.Monath T.P. Prospects for development of a vaccine against the West Nile virus. Ann N Y Acad Sci. 2001;951(December):1–12. doi: 10.1111/j.1749-6632.2001.tb02680.x. [DOI] [PubMed] [Google Scholar]
- 40.Tesh R.B., Arroyo J., Travassos Da Rosa A.P., Guzman H., Xiao S.Y., Monath T.P. Efficacy of killed virus vaccine, live attenuated chimeric virus vaccine, and passive immunization for prevention of West Nile virus encephalitis in hamster model. Emerg Infect Dis. 2002;8(December (12)):1392–1397. doi: 10.3201/eid0812.020229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arroyo J., Miller C., Catalan J., Myers G.A., Ratterree M.S., Trent D.W. ChimeriVax-West Nile virus live-attenuated vaccine: preclinical evaluation of safety, immunogenicity, and efficacy. J Virol. 2004;78(November (22)):12497–12507. doi: 10.1128/JVI.78.22.12497-12507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monath T.P., Liu J., Kanesa-Thasan N., Myers G.A., Nichols R., Deary A. A live, attenuated recombinant West Nile virus vaccine. Proc Natl Acad Sci USA. 2006;103(April (17)):6694–6699. doi: 10.1073/pnas.0601932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall R.A., Khromykh A.A. ChimeriVax-West Nile vaccine. Curr Opin Mol Ther. 2007;9(October (5)):498–504. [PubMed] [Google Scholar]
- 44.Lichty B.D., Power A.T., Stojdl D.F., Bell J.C. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10(May (5)):210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Roberts A., Kretzschmar E., Perkins A.S., Forman J., Price R., Buonocore L. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J Virol. 1998;72(June (6)):4704–4711. doi: 10.1128/jvi.72.6.4704-4711.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts A., Buonocore L., Price R., Forman J., Rose J.K. Attenuated vesicular stomatitis viruses as vaccine vectors. J Virol. 1999;73(May (5)):3723–3732. doi: 10.1128/jvi.73.5.3723-3732.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartz J.A., Buonocore L., Roberts A., Suguitan A., Jr., Kobasa D., Kobinger G. Vesicular stomatitis virus vectors expressing avian influenza H5 HA induce cross-neutralizing antibodies and long-term protection. Virology. 2007;366(September (1)):166–173. doi: 10.1016/j.virol.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grigera P.R., Marzocca M.P., Capozzo A.V., Buonocore L., Donis R.O., Rose J.K. Presence of bovine viral diarrhea virus (BVDV) E2 glycoprotein in VSV recombinant particles and induction of neutralizing BVDV antibodies in mice. Virus Res. 2000;69(August (1)):3–15. doi: 10.1016/s0168-1702(00)00164-7. [DOI] [PubMed] [Google Scholar]
- 49.Kahn J.S., Roberts A., Weibel C., Buonocore L., Rose J.K. Replication-competent or attenuated, nonpropagating vesicular stomatitis viruses expressing respiratory syncytial virus (RSV) antigens protect mice against RSV challenge. J Virol. 2001;75(November (22)):11079–11087. doi: 10.1128/JVI.75.22.11079-11087.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brandsma J.L., Shlyankevich M., Buonocore L., Roberts A., Becker S.M., Rose J.K. Therapeutic efficacy of vesicular stomatitis virus-based E6 vaccination in rabbits. Vaccine. 2007;25(January (4)):751–762. doi: 10.1016/j.vaccine.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 51.Brandsma J.L., Shylankevich M., Su Y., Roberts A., Rose J.K., Zelterman D. Vesicular stomatitis virus-based therapeutic vaccination targeted to the E1, E2, E6, and E7 proteins of cottontail rabbit papillomavirus. J Virol. 2007;81(June (11)):5749–5758. doi: 10.1128/JVI.02835-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ezelle H.J., Markovic D., Barber G.N. Generation of hepatitis C virus-like particles by use of a recombinant vesicular stomatitis virus vector. J Virol. 2002;76(December (23)):12325–12334. doi: 10.1128/JVI.76.23.12325-12334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlereth B., Buonocore L., Tietz A., Meulen Vt V., Rose J.K., Niewiesk S. Successful mucosal immunization of cotton rats in the presence of measles virus-specific antibodies depends on degree of attenuation of vaccine vector and virus dose. J Gen Virol. 2003;84(August (Pt 8)):2145–2151. doi: 10.1099/vir.0.19050-0. [DOI] [PubMed] [Google Scholar]
- 54.Garbutt M., Liebscher R., Wahl-Jensen V., Jones S., Moller P., Wagner R. Properties of replication-competent vesicular stomatitis virus vectors expressing glycoproteins of filoviruses and arenaviruses. J Virol. 2004;78(May (10)):5458–5465. doi: 10.1128/JVI.78.10.5458-5465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones S.M., Feldmann H., Stroher U., Geisbert J.B., Fernando L., Grolla A. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med. 2005;11(July (7)):786–790. doi: 10.1038/nm1258. [DOI] [PubMed] [Google Scholar]
- 56.Daddario-DiCaprio K.M., Geisbert T.W., Geisbert J.B., Stroher U., Hensley L.E., Grolla A. Cross-protection against Marburg virus strains by using a live, attenuated recombinant vaccine. J Virol. 2006;80(October (19)):9659–9666. doi: 10.1128/JVI.00959-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daddario-DiCaprio K.M., Geisbert T.W., Stroher U., Geisbert J.B., Grolla A., Fritz E.A. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: an efficacy assessment. Lancet. 2006;367(April (9520)):1399–1404. doi: 10.1016/S0140-6736(06)68546-2. [DOI] [PubMed] [Google Scholar]
- 58.Geisbert T.W., Jones S., Fritz E.A., Shurtleff A.C., Geisbert J.B., Liebscher R. Development of a new vaccine for the prevention of Lassa fever. PLoS Med. 2005;2(June (6)):e183. doi: 10.1371/journal.pmed.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kapadia S.U., Rose J.K., Lamirande E., Vogel L., Subbarao K., Roberts A. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology. 2005;340(September (2)):174–182. doi: 10.1016/j.virol.2005.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Natuk R.J., Cooper D., Guo M., Calderon P., Wright K.J., Nasar F. Recombinant vesicular stomatitis virus vectors expressing herpes simplex virus type 2 gD elicit robust CD4+ Th1 immune responses and are protective in mouse and guinea pig models of vaginal challenge. J Virol. 2006;80(May (9)):4447–4457. doi: 10.1128/JVI.80.9.4447-4457.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haglund K., Forman J., Krausslich H.G., Rose J.K. Expression of human immunodeficiency virus type 1 Gag protein precursor and envelope proteins from a vesicular stomatitis virus recombinant: high-level production of virus-like particles containing HIV envelope. Virology. 2000;268(March (1)):112–121. doi: 10.1006/viro.1999.0120. [DOI] [PubMed] [Google Scholar]
- 62.Johnson J.E., Rodgers W., Rose J.K. A plasma membrane localization signal in the HIV-1 envelope cytoplasmic domain prevents localization at sites of vesicular stomatitis virus budding and incorporation into VSV virions. Virology. 1998;251(November (2)):244–252. doi: 10.1006/viro.1998.9429. [DOI] [PubMed] [Google Scholar]
- 63.Johnson J.E., Schnell M.J., Buonocore L., Rose J.K. Specific targeting to CD4+ cells of recombinant vesicular stomatitis viruses encoding human immunodeficiency virus envelope proteins. J Virol. 1997;71(July (7)):5060–5068. doi: 10.1128/jvi.71.7.5060-5068.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xing Z., Lichty B.D. Use of recombinant virus-vectored tuberculosis vaccines for respiratory mucosal immunization. Tuberculosis (Edinb) 2006;86(May–July (3–4)):211–217. doi: 10.1016/j.tube.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 65.Palin A., Chattopadhyay A., Park S., Delmas G., Suresh R., Senina S. An optimized vaccine vector based on recombinant vesicular stomatitis virus gives high-level, long-term protection against Yersinia pestis challenge. Vaccine. 2007;25(January (4)):741–750. doi: 10.1016/j.vaccine.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 66.Egan M.A., Chong S.Y., Rose N.F., Megati S., Lopez K.J., Schadeck E.B. Immunogenicity of attenuated vesicular stomatitis virus vectors expressing HIV type 1 Env and SIV Gag proteins: comparison of intranasal and intramuscular vaccination routes. AIDS Res Hum Retroviruses. 2004;20(September (9)):989–1004. doi: 10.1089/aid.2004.20.989. [DOI] [PubMed] [Google Scholar]
- 67.Ramsburg E., Rose N.F., Marx P.A., Mefford M., Nixon D.F., Moretto W.J. Highly effective control of an AIDS virus challenge in macaques by using vesicular stomatitis virus and modified vaccinia virus Ankara vaccine vectors in a single-boost protocol. J Virol. 2004;78(April (8)):3930–3940. doi: 10.1128/JVI.78.8.3930-3940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rose N.F., Marx P.A., Luckay A., Nixon D.F., Moretto W.J., Donahoe S.M. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell. 2001;106(September (5)):539–549. doi: 10.1016/s0092-8674(01)00482-2. [DOI] [PubMed] [Google Scholar]
- 69.Iyer AV, Boudreaux MJ, Wakamatsu N, Roy AF, Baghian A, Chouljenko VN, et al. The Louisiana West Nile virus strain LSU-AR01 isolated from a blue jay (Cyanocitta cristata) exhibits increased mouse neurovirulence in comparison to the prototypic New York-99 strain and is closely related to the Connecticut-99 mosquito isolate, Virus Genes, in press.
- 70.Schnell M.J., Buonocore L., Kretzschmar E., Johnson E., Rose J.K. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc Natl Acad Sci USA. 1996;93(October (21)):11359–11365. doi: 10.1073/pnas.93.21.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schnell M.J., Buonocore L., Whitt M.A., Rose J.K. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J Virol. 1996;70(April (4)):2318–2323. doi: 10.1128/jvi.70.4.2318-2323.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pahar B., Wang X., Dufour J., Lackner A.A., Veazey R.S., Virus-specific T cell responses in macaques acutely infected with SHIV(sf162p3) Virology. 2007;363(June (1)):36–47. doi: 10.1016/j.virol.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis B.S., Chang G.J., Cropp B., Roehrig J.T., Martin D.A., Mitchell C.J. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J Virol. 2001;75(May (9)):4040–4047. doi: 10.1128/JVI.75.9.4040-4047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brines R.D., Klaus G.G. Polyclonal activation of immature B cells by preactivated T cells: the role of IL-4 and CD40 ligand. Int Immunol. 1993;5(November (11)):1445–1450. doi: 10.1093/intimm/5.11.1445. [DOI] [PubMed] [Google Scholar]
- 75.Sancho D., Gomez M., Sanchez-Madrid F. CD69 is an immunoregulatory molecule induced following activation. Trends Immunol. 2005;26(March (3)):136–140. doi: 10.1016/j.it.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 76.Seddiki N., Santner-Nanan B., Martinson J., Zaunders J., Sasson S., Landay A. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203(July (7)):1693–1700. doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Office international des épizooties (Paris) O.I.E. Manual of diagnostic tests and vaccines for terrestrial animals 2004. fifth edition. OIE. Office international des épizooties; Paris: 2004. Vesicular stomatitis. pp. 129–35. [Google Scholar]
- 78.Clarke D.K., Cooper D., Egan M.A., Hendry R.M., Parks C.L., Udem S.A. Recombinant vesicular stomatitis virus as an HIV-1 vaccine vector. Springer seminars in immunopathology. 2006;28(November (3)):239–253. doi: 10.1007/s00281-006-0042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roche S., Rey F.A., Gaudin Y., Bressanelli S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science. 2007;315(February (5813)):843–848. doi: 10.1126/science.1135710. [DOI] [PubMed] [Google Scholar]
- 80.Reuter J.D., Vivas-Gonzalez B.E., Gomez D., Wilson J.H., Brandsma J.L., Greenstone H.L. Intranasal vaccination with a recombinant vesicular stomatitis virus expressing cottontail rabbit papillomavirus L1 protein provides complete protection against papillomavirus-induced disease. J Virol. 2002;76(September (17)):8900–8909. doi: 10.1128/JVI.76.17.8900-8909.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Publicover J., Ramsburg E., Rose J.K. A single-cycle vaccine vector based on vesicular stomatitis virus can induce immune responses comparable to those generated by a replication-competent vector. J Virol. 2005;79(November (21)):13231–13238. doi: 10.1128/JVI.79.21.13231-13238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cooper D., Wright K.J., Calderon P.C., Guo M., Nasar F., Johnson J.E. Attenuation of recombinant vesicular stomatitis virus-human immunodeficiency virus type 1 vaccine vectors by gene translocations and g gene truncation reduces neurovirulence and enhances immunogenicity in mice. J Virol. 2008;82(January (1)):207–219. doi: 10.1128/JVI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rose N.F., Roberts A., Buonocore L., Rose J.K. Glycoprotein exchange vectors based on vesicular stomatitis virus allow effective boosting and generation of neutralizing antibodies to a primary isolate of human immunodeficiency virus type 1. J Virol. 2000;74(December (23)):10903–10910. doi: 10.1128/jvi.74.23.10903-10910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grewal I.S., Foellmer H.G., Grewal K.D., Xu J., Hardardottir F., Baron J.L. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 1996;273(September (5283)):1864–1867. doi: 10.1126/science.273.5283.1864. [DOI] [PubMed] [Google Scholar]
- 85.Kawabe T., Naka T., Yoshida K., Tanaka T., Fujiwara H., Suematsu S. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1(June (3)):167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 86.O’Keefe G.M., Nguyen V.T., Benveniste E.N. Regulation and function of class II major histocompatibility complex, CD40, and B7 expression in macrophages and microglia: Implications in neurological diseases. J Neurovirol. 2002;8(December (6)):496–512. doi: 10.1080/13550280290100941. [DOI] [PubMed] [Google Scholar]
- 87.Brien J.D., Uhrlaub J.L., Nikolich-Zugich J. Protective capacity and epitope specificity of CD8(+) T cells responding to lethal West Nile virus infection. Eur J Immunol. 2007;37(July (7)):1855–1863. doi: 10.1002/eji.200737196. [DOI] [PubMed] [Google Scholar]
- 88.McMurtrey C.P., Lelic A., Piazza P., Chakrabarti A.K., Yablonsky E.J., Wahl A. Epitope discovery in West Nile virus infection: identification and immune recognition of viral epitopes. Proc Natl Acad Sci USA. 2008;105(February (8)):2981–2986. doi: 10.1073/pnas.0711874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Purtha W.E., Myers N., Mitaksov V., Sitati E., Connolly J., Fremont D.H. Antigen-specific cytotoxic T lymphocytes protect against lethal West Nile virus encephalitis. Eur J Immunol. 2007;37(July (7)):1845–1854. doi: 10.1002/eji.200737192. [DOI] [PubMed] [Google Scholar]
- 90.De Groot A.S., Saint-Aubin C., Bosma A., Sbai H., Rayner J., Martin W. Rapid determination of HLA B*07 ligands from the West Nile virus NY99 genome. Emerg Infect Dis. 2001;7(July–August (4)):706–713. doi: 10.3201/eid0704.010419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Waters W.R., Rahner T.E., Palmer M.V., Cheng D., Nonnecke B.J., Whipple D.L. Expression of L-Selectin (CD62L), CD44, and CD25 on activated bovine T cells. Infect Immun. 2003;71(January (1)):317–326. doi: 10.1128/IAI.71.1.317-326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang L., Zhao Y. The regulation of Foxp3 expression in regulatory CD4(+)CD25(+)T cells: multiple pathways on the road. J Cell Physiol. 2007;211(June (3)):590–597. doi: 10.1002/jcp.21001. [DOI] [PubMed] [Google Scholar]
- 93.Banham A.H. Cell-surface IL-7 receptor expression facilitates the purification of FOXP3(+) regulatory T cells. Trends Immunol. 2006;27(December (12)):541–544. doi: 10.1016/j.it.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 94.Liu W., Putnam A.L., Xu-Yu Z., Szot G.L., Lee M.R., Zhu S. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203(July (7)):1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y., Lobigs M., Lee E., Mullbacher A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J Virol. 2003;77(December (24)):13323–13334. doi: 10.1128/JVI.77.24.13323-13334.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shrestha B., Diamond M.S. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78(August (15)):8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Buonocore L., Blight K.J., Rice C.M., Rose J.K. Characterization of vesicular stomatitis virus recombinants that express and incorporate high levels of hepatitis C virus glycoproteins. J Virol. 2002;76(July (14)):6865–6872. doi: 10.1128/JVI.76.14.6865-6872.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]