

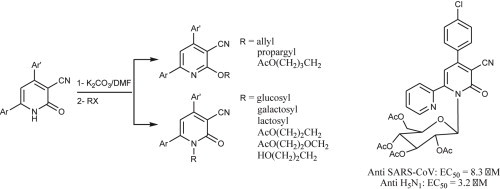
Abstract
Drug resistance and emergence of new pathogens highlight the need for developing new therapeutic agents. We focused on 2-oxonicotinonitrile (2-ONN) as derivative of the natural product 2-pyridinone.1 Herein, we describe the synthesis of 2-ONNs bearing two aryl groups, which we coupled with organohalides, including three glycosyl bromides, to prepare the nucleoside analogues. Coupling occurred mostly at the 2-ONN ring nitrogen to give the aimed targets, and in a few cases, it happened at the 2-oxo position giving O-alkylation products. Free 2-ONNs and their acetylated nucleosides were tested against a number of viruses. The nucleoside analogue 2aAc showed good anti SARS-CoV and anti influenza A (H5N1) activities. Additionally, 7b had good activity against Gram positive bacterium, Bacillis subtilis.
Keywords: 2-Oxonicotinonitrile, 2-Pyridinone, Nucleoside analogues, Glycosylation, Antimicrobial, Antiviral
Graphical abstract
Highlights
-
•
2-Oxonicotinonitriles with two aryl groups were coupled with several organohalides.
-
•
The coupling occurred mostly at the ring nitrogen.
-
•
In a few cases, the coupling occurred at the 2-oxo position.
-
•
Antimicrobial and antiviral activities were tested for acetylated and free compounds.
-
•
One nucleoside analogue showed good anti SARS and anti influenza activity.
1. Introduction
Drug resistance in microbial and viral infections has become a marked problem causing the failure of many treatments. The constant search for new drugs to be used alone or in combination with existing market drugs has become a necessity. In continuation of our efforts in finding such drug candidates, we have focused on the investigation of 2-oxonicotinonitrile (2-ONN) derivatives containing two aryl groups at the four- and six- positions of the ring. The 2-ONN ring contains the biologically important, naturally occurring 2-pyridinone motif [1], [2]. This motif has been found in compounds that are human immune deficiency virus 1 (HIV-1) nonnucleoside reverse transcriptase inhibitors [3], [4], [5], anti-herpes simplex virus 1 (HSV-1) agents [6], hepatitis C virus (HCV) NS5B polymerase inhibitors [7], and anti-hepatitis B virus (HBV) agents [8]. In addition to their antiviral activity, 2-pyridinone derivatives are known to possess activity against wild type [9], [10] and resistant [11] bacterial strains. The derivatives have also been shown to be antifungal [12] and antiprotozoal agents [13], [14].
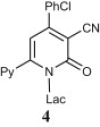
Herein, we discuss the synthesis of three 2-ONN diaryl derivatives (1a–c, Fig. 1 ) for antiviral and antimicrobial activity testing. For enhancing their potency as antiviral agents, our 2-ONN compounds were coupled with different halo-sugars and halo-alkyl derivatives, to make the corresponding nucleoside and acyclic nucleoside analogues. Nucleoside analogues are well known to exhibit potent antiviral activity [15].
Fig. 1.
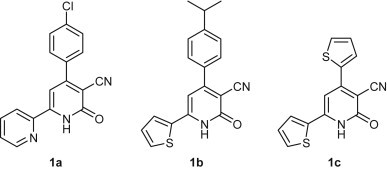
Structures of three synthesized 2-ONN derivatives.
2. Results and discussion
2.1. Chemistry
2-ONN derivatives 1a–c (Fig. 1) were synthesized by one-pot condensation [16], [17] of ethyl cyanoacetate with the corresponding aromatic ketone and aromatic aldehyde, in presence of ammonium acetate in refluxing ethanol (see Scheme 1 ). The 2-ONN ring was attached to 2-pyridyl (Py) and p-chlorophenyl (PhCl) groups in 1a, to 2-thienyl (Th) and 4-isopropylphenyl (PhPr) groups in 1b, and to two Th groups in 1c. Compounds 1a [18] and 1c [19] were reported to be prepared through their respective chalcones in two steps from the aldehyde and ketone. However, the one-pot method that we adapted afforded a shorter route for their synthesis. Compounds 1a–c were obtained as the amido tautomer as suggested by the presence of bands in the IR spectrum characteristic for NH and C O of the amide functional group. This was also emphasized by the presence of an 1H NMR peak at a chemical shift higher than 12 ppm, corresponding to the amide NH. Obtaining the amido tautomer rather than the imido was explained by a recent study showing that the amido tautomer was more stable and hence predominant [20].
Scheme 1.
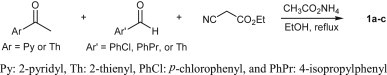
Preparation of 2-ONN derivatives 1a–c.
Derivatives 1a–c were coupled with different organohalides in presence of K2CO3 in N,N-dimethylformamide (DMF) in order to give the aimed nucleoside analogues (Table 1 ). A range of these coupling agents were O-acetylated (Table 1, entries 1–6), in which case the coupling reaction was followed by deacetylation with triethylamine (TEA) in H2O/MeOH. Products before and after deacetylation were separated by crystallization from ethanol for biological evaluation. Structures of these separated coupling products were confirmed by IR, NMR, and elemental analyses. Table 1 shows the structure of the final products after deacetylation and lists the yields for the coupling reaction itself.
Table 1.
Coupling of 2-ONN derivatives 1a–c with different organohalides.
| Entry | Deriv.a | Coupling agent | Producta,b | % Yieldc |
|---|---|---|---|---|
| 1 | 1a–c |  |
 |
2a: 66 |
| 2b: 58 | ||||
| 2c: 62 | ||||
| 2 | 1a–c | 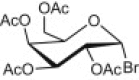 |
 |
3a: 87 |
| 3b: 70 | ||||
| 3c: 51 | ||||
| 3 | 1a | 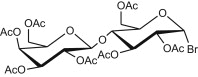 |
 |
66 |
| 4 | 1a | 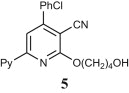 |
80 | |
| 5 | 1b,c | 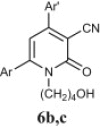 |
6b: 57 | |
| 6c: 53 | ||||
| 6 | 1a–c |
 d d
|
7a: 76 | |
| 7b: 62 | ||||
| 7cAc: 45 | ||||
| 7 | 1a–c | 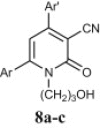 |
8a: 74 | |
| 8b: 88 | ||||
| 8c: 82 | ||||
| 8 | 1a | 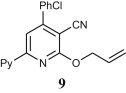 |
82 | |
| 9 | 1a | 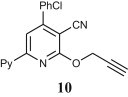 |
73 |
a: Ar = Py; Ar′ = PhCl, b: Ar = Th; Ar′ = PhPr, c: Ar = Ar′ = Th.
Glc: glucosyl, Gal: galactosyl, Lac: lactosyl.
Coupling yield before deprotection.
Coupling product was not deacetylated for 7cAc.
Entries 1–3 of Table 1 show the synthesis of seven 2-ONN based nucleoside analogues. Compounds 1a–c were coupled with both peracetylated α-D-glucopyranosyl and α-D-galactopyranosyl bromides leading to the formation of the corresponding N-glycosides 2a–c (entry 1) and 3a–c (entry 2), respectively, after deacetylation. Similarly, 1a was coupled with peracetylated α-D-lactosyl bromide (entry 3) to give the respective N-lactoside (4) after deacetylation. In all these seven cases, the acetylated N-β-glycosides were obtained in yields ranging from 51% to 87%, indicating that the coupling reaction occurred mainly at the nitrogen of the 2-ONN ring. Formation of the N-glycoside was confirmed by an IR band in the range of 1640–1660 cm‾1 corresponding to an amide C O functional group for each compound. Those resulting nucleosides had β-configuration as indicated by the 1H NMR coupling constant of the anomeric proton (J > 7.5 Hz) due to diaxial coupling with H-2′.
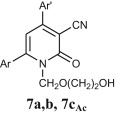
Preparation of acyclic nucleoside analogues were attempted by coupling our 2-ONNs with five different substituted alkyl halides. Coupling with 3-hydroxypropyl chloride led to the N- alkylation, as desired, to give 8a–c in good yields (entry 7). The 2-ONN ring of 1a–c reacted with 2-bromomethoxyethyl acetate at the nitrogen atom as well to give the N- alkylation products 7a and 7b after deprotection and the unprotected 7c Ac (entry 6). However, when 1a–c were coupled with 4-bromobutyl acetate, results varied. 1b and 1c underwent N- alkylation to give 6b and 6c as the final and major products, respectively (entry 5). On the other hand, 1a underwent coupling at the 2-oxo position with the same reagent and reaction conditions in a good yield to give, after deprotection, the O-alkylation product 5 (entry 4). This was judged by the absence of the IR band corresponding to the amide C O functional group. The coupling at the 2-oxo position of 1a was also seen with allyl bromide and propargyl bromide to give the O-alkylation products 9 (entry 8) and 10 (entry 9), respectively.
Based on the above results, only 1a among the three 2-ONN compounds under study was glycosylated at the 2-oxo position with a few simple organohalides. In all other cases coupling occurred mainly at the ring N atom. In fact, these results were in contrast to the ones we observed in our previous work [21], [22], where we have examined the coupling of the 2-ONN derivative 1d (Fig. 2 ) with 10 different glycosyl and substituted alkyl halides under the same reaction conditions. In all cases, alkylation occurred at the 2-oxo position of the 2-ONN ring of 1d and the corresponding O-glycosylation and O-alkylation products were separated in yields ranging from 48% to 87%. Compounds 1a–c differ from 1d by changing one of the aryl groups attached to the 2-ONN ring, while retaining the other one. Other published work showed that a very close 2-ONN structure (1e, Fig. 2) is also glycosylated at the 2-oxo position under the same conditions [23]. Furthermore, the similar 2-ONN compound 1f (Fig. 2) was also reported to couple with ethyl bromoacetate at the 2-oxo position under similar reaction condition to the one used in our work [24].
Fig. 2.
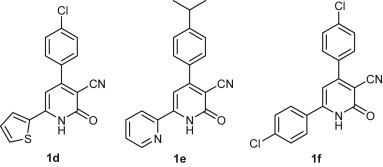
Structures of 2-ONNs in the literature.
Interestingly, reported results [25] showed that a compound different from 1d, only in that the positions of the PhCl and Th groups relative to the 2-ONN ring are interchanged, undergoes N-glucosylation under similar reaction conditions to ours. Keeping these contradicting results in mind, one can envision that regioselectivity of the reaction depends on the size of both the coupling agent and the substituents on the 2-ONN ring. Additionally, the type and relative positions of the aryl substituents on the ring induce an electronic effect that contributes to the relative electron density at the ring N and 2-oxo positions, which in turn controls the regioselectivity. It is to be noted that careful chromatographic separation of products of such reactions show that both N- and O-glycosylation/alkylation can occur, with one of them as the major product [26].
2.2. Biology
2.2.1. Evaluation of antimicrobial activities
Our new 2-ONN derivatives were screened in vitro for antibacterial activity against Bacillis subtilis and Escherichia coli, representing Gram positive and Gram negative bacteria, respectively, and antifungal activity against the fungi Aspergillus niger and Aspergillus virdi-nutans. Zone of inhibition screening tests based on well diffusion method were used to examine both the antibacterial and antifungal activities. Table 2 shows the diameter of inhibition zone in mm for the 2-ONN compounds against the selected bacteria and fungi. The results were compared to Ampicillin as a reference antibacterial drug and Dermatin as a reference antifungal drug to evaluate the potency of the tested compounds. This antimicrobial study was conducted for both the final deprotected form and O-acetylated form of the 2-ONN derivatives. As mentioned above, O-acetylated forms were originally obtained as synthetic intermediates. The latter were more lipophilic and could have different biological properties than the deacetylated counterparts. Derivatives in Table 2 that were variants of 2-ONN compounds from Table 1 by acetyl protection were denoted by the addition of Ac to the number of each compound. Since the tested samples were prepared as N,N-dimethylformamide (DMF) solutions, wells containing only the microorganism and DMF were used as a negative control. DMF resulted in no inhibition zone.
Table 2.
Antibacterial and antifungal inhibitory activities of 2-ONN derivatives.
| 2-ONN derivative (250 μg/disc) | Diameter of inhibition zone (mm) |
|||
|---|---|---|---|---|
| Bacillus subtilis | Escherichia coli | Asperigillus niger | Asperigillus virdi-nutans | |
| 2bAc | 1 | 0 | 7 | 7 |
| 2cAc | 2 | 0 | 8 | 6 |
| 2b | 10 | 0 | 7 | 8 |
| 2c | 2 | 0 | 8 | 6 |
| 3aAc | 1 | 0 | 7 | 5 |
| 3bAc | 1 | 0 | 9 | 6 |
| 3a | 2 | 0 | 7 | 7 |
| 3b | 3 | 5 | 8 | 6 |
| 3c | 4 | 0 | 8 | 8 |
| 5Ac | 3 | 0 | 9 | 7 |
| 5 | 2 | 0 | 7 | 5 |
| 7cAc | 10 | 0 | 8 | 7 |
| 7b | 23 | 0 | 7 | 6 |
| 8a | 3 | 0 | 9 | 9 |
| 9 | 1 | 0 | 7 | 7 |
| DMF | 0 | 0 | 0 | 0 |
| Ampicillin | 20 | 18 | – | – |
| Dermatin | – | – | 22 | 31 |
Among the tested compounds, 7b showed the highest antibacterial activity (23 mm) against B. subtilis, which was even higher than the standard drug Ampicillin (20 mm). However, it had no activity against the Gram negative bacteria E. coli. In fact, all the tested compounds except 3b showed no activity against E. coli. 3b had small antibacterial activity of 5 mm compared to that of Ampicillin (18 mm). Compounds 2b and 7c Ac showed moderate antibacterial activity against B. subtilis. Compounds 2b Ac, 2c Ac, 3a Ac, 3b Ac, 2c, 3a, 3b, 3c, 5 Ac, 5, 8a and 9 showed lower antibacterial activity against Gram positive bacteria compared to Ampicillin. These data proved that there was no obvious trend suggesting that increasing lipophilicity by addition of acetyl groups in the 2-ONN structure had an effect on the antibacterial activity. Antifungal zone of inhibition studies showed that all the tested compounds had moderate antifungal activity against Asperigillus niger and Asperigillus virdi-nutans compared to the standard drug Dermatin.
2.2.2. In vitro antiviral screening
A panel of different viral species was used to evaluate the in vitro antiviral activity of some of the 2-ONN compounds in this report. The panel consisted of HCV, HBV, HSV-1, HSV-2, severe acute respiratory syndrome coronavirus (SARS-CoV), West Nile virus (WNV), influenza A virus (serotypes H3N2, H5N1 and H1N1) and influenza B virus. The acetylated 2-ONN glucosides 2a Ac –c Ac, and the acetylated galactoside of 1c (3c Ac), which we designed as nucleoside analogues, in addition to uncoupled 2-ONN derivatives 1a–c were selected for this study. The compounds were evaluated for their antiviral activity in comparison to known drugs for each virus as controls. Antiviral activity was given as 50% effective concentration (EC50), which is the concentration of the compound in μM required to inhibit 50% of viral infection. Cytotoxic concentration (CC50) was determined as the concentration of the compound in μM at which 50% of cells become dead. Selectivity index (SI) was calculated as the ratio of CC50 to IC50 values, and was used along with the EC50 values to evaluate the bioactivity of 2-ONN derivatives.
Anti HCV and HBV activities were assessed using RNA replicon and DNA virion control assays, respectively. Anti HBV activity was compared to that of Lamivudine. Anti HSV-1 and HSV-2 as two additional representatives of DNA virus families were compared to acyclovir as the control drug. Similarly, anti HCV activity of the compounds was compared to that of human interferon-alpha (HIA) and 2′C-methylcytidine (2′C-MC). As summarized in Table 3 results showed that among the tested compounds, 1c showed the best antiviral activity against HSV-1 and HSV-2 (EC50 > 12 μM), while 2b Ac, 2c Ac and 3c Ac were moderately active against HSV-1 and HSV-2 with an EC50 of 67.2, >60, and >60 μM, respectively. However, the SI values are much lower than reference drugs. All tested compounds were inactive against HCV and HBV as the EC50 values are higher than 100 μM. Anti SARS-CoV and WNV activities were investigated in comparison with M128533 and infergen, respectively as control drugs. Results were obtained using both visual and neutral red tests. Among the tested compounds, only 2a Ac was slightly active by visual, but not neutral red test for SARS-CoV.
Table 3.
Antiviral activity of compounds 1a–c, 2a–cAc, and 3cAc.
| 2-ONN derivative | HCV EC50a | HBV EC50a | HSV-1 |
HSV-2 |
SARS-CoV |
WNV EC50a | |||
|---|---|---|---|---|---|---|---|---|---|
| EC50a | SI | EC50a | SI | EC50a | SI | ||||
| 1a | >100 | >100 | >300 | 1 | >300 | 1 | >0.32 | ND | >2.2 |
| 1b | >100 | >100 | >300 | 1 | >300 | 1 | >26 | ND | >2.2 |
| 1c | >100 | >100 | >12 | <4.7 | >12 | <4.8 | >100 | ND | >4.6 |
| 2aAc | >100 | >100 | >300 | 1 | >300 | 1 | 8.3 | 4.3 | >5.1 |
| 2bAc | >100 | >100 | 67.2 | >4.4 | 60.3 | >4.9 | >100 | ND | >4.7 |
| 2cAc | >100 | >100 | >60 | <4 | >60 | <4 | >32 | ND | >3 |
| 3cAc | >100 | >100 | >60 | <2.4 | >60 | <2.4 | 10 | 1.9 | >3.6 |
| HIA | 2 | – | – | – | – | – | – | – | – |
| 2′C-MC | 1.6 | – | – | – | – | – | – | – | – |
| Lamivudine | – | 0.035 | – | – | – | – | – | – | – |
| Acyclovir | – | – | 3.3 | >30.3 | 9.6 | >10.4 | – | – | – |
| M128533 | – | – | – | – | – | – | 0.59 | >170 | – |
| Infergen | – | – | – | – | – | – | – | – | 0.00004 |
EC50 is measured in μM.
Anti influenza activity was screened using ribavirin as the control drug. Cytopathic effect and cytotoxicity were used as control assays and the resulting data were reported in Table 4 . The screened compounds against influenza B virus showed that all tested compounds were not active. On the other hand, 2a Ac was slightly active against H5N1 influenza A virus. The 2-ONN derivatives were moderately active against H1N1 serotype.
Table 4.
Anti influaza activity of 1a–c, 2a–cAc, and 3cAc.
| 2-ONN derivative | Type-B |
Type-A |
||||||
|---|---|---|---|---|---|---|---|---|
| H3N2 |
H5N1 |
H1N1 |
||||||
| EC50a | SI | EC50a | SI | EC50a | SI | EC50a | SI | |
| 1a | >36 | ND | >2.6 | 0 | >100 | >0 | 26 | >3.8 |
| 1b | 46 | >2.2 | 22 | >4.5 | 32 | >3.1 | ND | – |
| 1c | >100 | ND | 16 | >4.2 | >100 | >0 | 32 | >3.1 |
| 2aAc | >3.2 | ND | 24 | >4.2 | 3.2 | 1.3 | ND | – |
| 2bAc | 40 | >2.2 | 33 | >3 | 32 | 1 | ND | – |
| 2cAc | 32 | 1 | 40 | >2.5 | >100 | >0 | ND | – |
| 3cAc | >12 | ND | >32 | ND | >19 | ND | >28 | ND |
| Ribavirin | 2.4 | >130 | 2 | >160 | 4.2 | >76 | 10 | >32 |
EC50 is measured in μM.
3. Conclusion
A series of 2-oxonicotinonitrile based analogues of nucleosides and acyclic nucleosides were prepared by coupling 2-ONNs with bromosugars and other organohalides. Some attempts of the coupling resulted in reaction at the 2-oxo position instead of the 2-ONN ring nitrogen. Seemingly, a combination of steric and electronic factors had an effect on the regioselectivity of the coupling reaction. Changing the coupling reaction conditions, such as the type of the base used, could lead to different selectivity [24]. Among the synthesized 2-ONN derivatives and their nucleosides, 7b showed good activity against the Gram positive bacterium B. subtilis, and 2a Ac was active against SARS-CoV and influenza A H5N1 virus. The 2-ONN compounds showed moderate antifungal activity.
4. Experimental results and protocols
4.1. Materials, instruments and general considerations
Starting materials and reagents were purchased from Aldrich and had a minimum purity of 97%. DMF was dried by distillation from CaCl2. 4-Bromobutyl acetate [27] and 2-bromomethoxyethyl acetate [28] were prepared according to literature from tetrahydrofuran and 1,3-dioxane, respectively. Other solvents and reagents were purchased from commercial sources and used as received. Reaction progress was monitored by TLC analysis using aluminium-backed plates pre-coated with Merck silica gel 60 F254. Spots separated on the TLC plates were visualized by UV light and/or charring with a solution of 10% H2SO4 in EtOH. Column chromatography was carried out on Whatman™ 60–120 mesh silica. Melting points were measured using an Electrothermal IA 9100 apparatus. IR spectra (KBr disc) were recorded on either a Pye Unicam Sp-3-300 or a Shimadzu FTIR 8101 PC infrared spectrophotometer. The 1H and 13C NMR spectra were recorded on JEOL-JNM-LA 300 MHz spectrometer. Chemical shifts (δ) were reported in ppm and were referenced to tetramethylsilane as a standard (0.00 ppm). Chemical shifts for 13C NMR were referenced relative to DMSO (39.50 ppm). Elemental microanalysis was done on a Perkin Elmer 240 analyzer. Analyses indicated by the symbols of the elements were within ±0.4% of the theoretical values.
4.2. Chemistry
4.2.1. General procedure for the preparation of 2-ONN 1a–c
A mixture of the aromatic ketone (0.01 mol), the aromatic aldehyde (0.01 mol), ethyl cyanoacetate (1.13 g, 0.01 mol), and ammonium acetate (6.16 g, 0.08 mol) in ethanol (30 mL) was refluxed for 12 h. After cooling, the precipitate was filtered off, washed with ethanol, and dried. Recrystallization from DMF/Ethanol (1:10 v:v) afforded the 2-ONN derivatives 1a-c.
4.2.1.1. 4-(4-Chlorophenyl)-2-oxo-6-(2-pyridyl)nicotinonitrile (1a)
This compound was prepared from 2-acetylpyridine (1.21 g) and 4-chlorobenzaldehyde (2.4 g) according to the general procedure described above, and was obtained as a green powder (1.55 g, 50.4% yield), mp 318–320 °C (lit [18]. mp 209–214 °C). IR (KBr, cm‾1): 3294 (NH), 2217 (C N), 1655 (C O, amide). 1H NMR (300 MHz, DMSO-d 6): δ 7.32 (s, 1H, Ar–H, 2-ONN ring), 7.59 (t, 1H, J = 5.1 Hz, Ar–H, Py), 7.68 (d, 2H, J = 8.7 Hz, Ar–H), 7.78 (d, 2H, J = 8.7 Hz, Ar–H), 8.04 (t, 1H, J = 7.5 Hz, Ar–H, Py), 8.30 (d, 1H, J = 8.1 Hz, Ar–H, Py), 8.77 (d, 1H, J = 4.8 Hz, Ar–H, Py), 12.38 (br, 1H, NH). Anal. Calcd for C17H10ClN3O (307.73): C, 66.35; H, 3.28; N, 13.65. Found: C, 66.34; H, 3.28; N, 13.66.
4.2.1.2. 4-(4-Isopropylphenyl)-2-oxo-6-(2-thienyl)nicotinonitrile (1b)
This compound was prepared from 2-acetylthiophene (1.26 g) and 4-isopropylbenzaldehyde (1.48 g) according to the general procedure described above, and was obtained as a yellow powder (1.2 g, 38% yield), mp 310–311 °C. IR (KBr, cm‾1): 3278 (NH), 2210 (C N), 1636 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.24 (d, 6H, J = 6.9 Hz, CH 3(i-Pr)), 2.98 (m, 1H, CH (i-Pr)), 7.23 (s, 1H, 2-ONN ring), 7.29 (t, 1H, J = 3.9 Hz, Th), 7.43 (d, 2H, J = 8.1 Hz, Ar–H), 7.64 (d, 2H, J = 8.1 Hz, Ar–H), 7.87 (d, 1H, J = 5.1 Hz, Th), 8.06 (d, 1H, J = 4.8 Hz, Th), 12.74 (br, 1H, NH). Anal. Calcd for C19H16N2OS (320.41): C, 71.22; H, 5.03; N, 8.74. Found: C, 71.23; H, 5.04; N, 8.73.
4.2.1.3. 2-Oxo-4,6-di(2-thienyl)nicotinonitrile (1c)
This compound was prepared from 2-acetylthiophene (1.26 g) and thiophene-2-carbaldehyde (1.12 g) according to the general procedure described above, and was obtained as a yellow powder (1.2 g, 43% yield), mp > 300 °C (lit [19]. mp 318 °C dec.) IR (KBr, cm‾1): 3273 (NH), 2208 (C N), 1636 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 7.22 (s, 1H, 2-ONN ring), 7.26 (t, 1H, J = 3.9 Hz, Th), 7.32 (t, 1H, J = 3.9 Hz, Th), 7.88 (d, 1H, J = 5.1 Hz, Th), 7.98 (m, 2H, Th), 8.07 (d, 1H, J = 4.8 Hz, Th), 12.74 (br, 1H, NH). Anal. Calcd for C14H8N2OS2 (284.36): C, 59.13; H, 2.84; N, 9.85. Found: C, 59.12; H, 2.85; N, 9.86.
4.2.2. General procedure for glycosylation and alkylation
The 2-ONN derivatives 1a–c (0.010 mol) were stirred with potassium carbonate (1.38 g, 0.010 mol) in dry DMF (15 mL) for 1 h, followed by the addition of the appropriate peracetylated glycosyl bromide (0.011 mol) or alkyl halide (0.010 mol) in portions. Except for allyl and propargyl derivatives, the reaction mixture was stirred at room temperature overnight, filtered to remove insoluble materials, and then the solvent was evaporated under reduced pressure. For allyl and propargyl derivatives, the reaction mixture was refluxed for 5 h after stirring overnight; and after cooling it was filtered, and the filtrate was poured into ice-water to give the crude product as a precipitate, which in turn was filtered off and dried. The crude product obtained in all cases was purified by recrystallization from 95% ethanol.
4.2.3. General procedure for deacetylation
TEA (1 mL) was added to a solution of the acetylated compound (0.01 mol) in MeOH (20 mL) and 3 drops of water. The mixture was stirred overnight at room temperature, and then the solvent was evaporated under reduced pressure. The residue was repeatedly co-evaporated with MeOH until TEA was removed, and then purified by crystallization from 95% ethanol.
4.2.4. Compounds prepared by glycosylation/alkylation followed by deacetylation
4.2.4.1. 4-(4-Chlorophenyl)-1-(β-D-glucopyranosyl)-2-oxo-6-(2-pyridyl)nicotinonitrile (2a)
Compound 1a (3 g) was reacted with peracetylated glucosyl bromide (3.2 g) according to the general procedure described for glycosylation, to give a yellow powder (4.2 g, 66% yield), mp 220–222 °C. IR (KBr, cm‾1): 2225 (C N), 1747 (C O, acetoxy), 1662 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.78, 1.94, 1.99 and 2.03 (4s, 12H, 4CH 3CO), 4.08 (dd, 1H, J 5′,6′ = 4.67, J 6′,6″ = 12.22 Hz, H-6′), 4.22 (dd, 1H, J 5′,6″ = 4.9, J 6′,6″ = 12.2 Hz, H-6″), 4.39 (m, 1H, H-5′), 5.07 (t, 1H, J 3′,4′ = 9.4 Hz, H-4′), 5.23 (t, 1H, J 1′,2′ = 8.2, J 2′,3′ = 9.6 Hz, H-2′), 5.63 (t, 1H, J 2′,3′ = 9.6, J 3′,4′ = 9.3 Hz, H-3′), 6.72 (d, 1H, J 1′,2′ = 8.1 Hz, H-1′), 7.32 (s, 1H, Py), 7.59 (t, 1H, J = 5.1 Hz, Py), 7.68 (d, 2H, J = 8.7 Hz, Ar–H), 7.78 (d, 2H, J = 8.7 Hz, Ar–H), 8.05 (t, 1H, J = 7.5 Hz, Py), 8.25 (d, 1H, J = 8.1 Hz, Py) 8.64 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C31H28ClN3O10 (638.02): C, 58.36; H, 4.42; N, 6.59. Found: C, 58.34; H, 4.43; N, 6.58. This powder (6 g) was deprotected according to the general procedure described for deacetylation, to give 2a as a grey powder (3.4 g, 88% yield), mp 300–302 °C. IR (KBr, cm‾1): 3394 (broad, 4OH), 2223 (C N), 1660 (C O, amide). 1H NMR (300 MHz, DMSO-d6/D2O): δ 3.24–3.63 (m, 6H, H-6′, H-6″, H-5′, H-4′, H-3′, and H-2′), 6.13 (d, 1H, J 1′,2′ = 8.4 Hz, H-1′), 7.23 (s, 1H, 2-ONN ring), 7.43 (t, 1H, J = 5.1 Hz, Py), 7.60 (d, 2H, J = 8.7 Hz, Ar–H), 7.65 (d, 2H, J = 8.7 Hz, Ar–H), 7.95 (t, 1H, J = 7.5 Hz, Py), 8.23 (d, 1H, J = 8.1 Hz, Py), 8.68 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C23H20ClN3O6 (469.87): C, 58.79; H, 4.29; N. 8.94. Found: C, 58.78; H, 4.29; N, 8.93.
4.2.4.2. 1-(β-D-Glucopyranosyl)-4-(4-isopropylphenyl)-2-oxo-6-(2-thienyl)nicotinonitrile (2b)
Compound 1b (3.2 g) was reacted with peracetylated glucosyl bromide (4.1 g) according to the general procedure described for glycosylation to give a yellow powder (3.78 g, 58% yield), mp 225–227 °C. IR (KBr, cm‾1): 2215 (C N), 1744 (C O, acetoxy), 1643 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.24 (d, 6H, J = 6.9 Hz, CH 3 (i-Pr)), 1.78, 1.85, 1.98 and 2.03 (4s, 12H, 4CH 3CO), 2.98 (m, 1H, CH (i-Pr)), 4.09 (dd, 1H, J 5′,6′ = 4.7, J 6′,6″ = 12.2 Hz, H-6′), 4.33 (dd, 1H, J 5′,6″ = 4.9, J 6′,6″ = 12.2 Hz, H-6″), 4.33 (m, 1H, H-5′), 5.03 (t, 1H, J 3′,4′ = 9.4, H-4′), 5.20 (t, 1H, J 1′,2′ = 8.2, J 2′,3′ = 9.6 Hz, H-2′), 5.62 (t, 1H, J 2′,3′ = 9.6, J 3′,4′ = 9.0 Hz, H-3′), 6.42 (d, 1H, J = 8.7 Hz, H-1′), 7.23 (t, 1H, J = 3.9 Hz, Th), 7.42 (d, 2H, J = 8.1 Hz, Ar–H) 7.56 (s, 1H, 2-ONN ring), 7.62 (d, 2H, J = 8.1 Hz, Ar–H), 7.84 (d, 1H, J = 5.1 Hz, Th), 8.03 (d, 1H, J = 4.8 Hz, Th). 13C NMR (75 MHz, DMSO-d6): δ 20.12 (CH3(i-Pr)), 20.25, 20.26, 20.28 and 20.30 (4CH3CO), 34.2 (CH(i-Pr)), 61.5 (C-6′), 68.0 (C-4′), 68.8 (C-3′), 71.4 (C-2′), 72.2 (C-5′), 93.8 (C-1′), 115.2 (C N), 118.2, 128.5, 129.8, 130.2, 130.8, 131.2, 132.2, 132.8, 134.5, 135.2, 142.2, 151.2 (Ar–C), 161.5, 168.0, 169.4, 170.2 and 170.6 (5 C O). Anal. Calcd for C33H34N2O10S (650.70): C, 60.91; H, 5.27; N, 4.31. Found: C, 60.92; H, 5.27; N, 4.30. This powder (6.6 g) was deprotected according to the general procedure described for deacetylation to give 2b as a yellow powder (4.58 g, 96% yield), mp 298–300 °C. IR (KBr, cm‾1): 3391 (broad, 4OH), 2215 (C N), 1644 (C O, amide). 1H NMR (300 MHz, CDCl3/D2O): δ 1.26 (d, 6H, J = 6.9 Hz, CH 3 (i-Pr)), 2.88 (m, 1H, CH (i-Pr)), 3.14 (m, 3H, H-3′, H-6′ and H-6″), 3.89 (m, 3H, H-2′, H-4′ and H-5′), 6.18 (d, 1H, J 1′,2′ = 8.4 Hz, H-1′), 7.27 (t, 1H, J = 3.9 Hz, Th), 7.38 (d, 2H, J = 8.1 Hz, Ar–H), 7.51 (d, 2H, J = 8.1 Hz, Ar–H), 7.62 (s, 1H, 2-ONN ring), 7.64 (d, 1H, J = 5.1 Hz, Th), 8.21 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C25H26N2O6S (482.55): C, 62.23; H, 5.43; N, 5.81. Found: C, 62.24; H, 5.44; N, 5.80.
4.2.4.3. 1-(β-D-Glucopyranosyl)-2-oxo-4,6-di(2-thienyl)nicotinonitrile (2c)
Compound 1c (2.8 g) was reacted with peracetylated glucosyl bromide (4.1 g) according to the general procedure described for glycosylation to give a green powder (3.78 g, 62% yield), mp 225–226 °C. IR (KBr, cm‾1): 2213 (C N), 1748 (C O, acetoxy), 1639 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.84, 1.98, 1.99 and 2.02 (4s, 12H, 4CH 3CO), 4.05 (dd, 1H, J 5′,6′ = 4.6, J 6′,6″ = 12.2 Hz, H-6′), 4.17 (dd, 1H, J 5′,6″ = 4.9, J 6′,6″ = 12.2 Hz, H-6″), 4.35 (m, 1H, H-5′), 5.03 (t, 1H, J 3′,4′ = 9.4 Hz, H-4′), 5.23 (t, 1H, J 1′,2′ = 8.4 Hz, H-2′), 5.65 (t, 1H, J 2′,3′ = 9.0, J 3′,4′ = 9.6 Hz, H-3′), 6.43 (d, 1H, J 1′,2′ = 8.7 Hz, H-1′), 7.25 (t, 1H, J = 3.9 Hz, Th), 7.31 (t. 1H, J = 3.9 Hz, Th), 7.52 (s, 1H, 2-ONN ring), 7.87 (d, 1H, J = 5.2 Hz, Th), 7.98 (m, 2H, Th), 8.05 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C28H26N2O10S2 (614.64): C, 54.71; H, 4.26; N, 4.56. Found: C, 54.72; H, 4.25; N, 4.55. This powder (6.1 g) was deprotected according to the general procedure described for deacetylation to give 2c as a green powder (4.41 g, 91.4% yield), mp 300–302 °C. IR (KBr, cm‾1): 3360 (broad, 4OH), 2216 (C N), 1640 (C O, amide). 1H NMR (300 MHz, DMSO-d6/D2O): δ 3.36–3.40 (m, 6H, H-6′, H-6″, H-5′, H-4′, H-3′ and H-2′), 5.98 (d, 1H, J 1′,2′ = 8.4 Hz, H-1′), 7.23 (t, 1H, J = 3.9 Hz, Th), 7.29 (d, 1H, J = 5.2 Hz, Th), 7.65 (s, 1H, 2-ONN ring), 7.82 (d, 1H, J = 5.2 Hz, Th), 7.94 (m, 2H, Th), 7.98 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C20H18N2O6S2 (446.50): C, 53.80; H, 4.06; N, 6.27. Found: C, 53.81; H, 4.08; N, 6.26.
4.2.4.4. 4-(4-Chlorophenyl)-1-(β-D-galactopyranosyl)-2-oxo-6-(2-pyridyl)nicotinonitrile (3a)
Compound 1a (3.07 g) was reacted with peracetylated galactosyl bromide (4.1 g) according to the general procedure described for glycosylation a pale yellow powder (5.59 g, 87% yield), mp 184–186 °C. IR (KBr, cm‾1): 2229 (C N), 1753 (C O, acetoxy), 1650 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.63, 1.84, 1.87 and 2.05 (4s, 12H, 4CH 3CO), 3.87 (dd, 1H, J 5′,6′ = 6.0, J 6′,6″ = 11.1 Hz, H-6′), 4.01 (dd, 1H, J 5′,6′ = 6.3, J 6′,6″ = 11.1 Hz, H-6″), 4.56 (m, 1H, H-5′), 5.23 (t, 1H, J 3′,2′ = 10.2, J 3′,4′ = 2.8 Hz, H-3′), 5.38 (t, 1H, J 2′,1′ = 8.4, J 2′,3′ = 10.3 Hz, H-2′), 5.43 (t, 1H, J 4′,3′ = 2.8, J 4′,5′ = 2.7 Hz, H-4′), 6.51 (d, 1H, J 1′,2′ = 8.4 Hz, H-1′), 7.33 (s,1H, 2-ONN ring), 7.43 (t, 1H, J = 5.1 Hz, Py), 7.53 (d, 2H, J = 8.7 Hz, Ar–H), 7.64 (d, 2H, J = 8.7 Hz, Ar–H), 7.90 (t, 1H, J = 7.5 Hz, Py), 8.12 (d, 1H, J = 8.1 Hz, Py), 8.45 (d, 1H, J = 4.8 Hz, Py). 13C NMR (75 MHz, DMSO-d6): δ 20.78, 20.84, 20.89 and 20.91 (4CH3CO), 62.1 (C-6′), 68.06 (C-4′), 68.15 (C-2′), 70.66 (C-3′), 71.72 (C-5′), 94.64 (C-1′), 114.4, 115.8 (C N), 122.6, 126.3, 129.4, 130.6, 130.9, 134.8, 135.8, 138.1, 150.2, 153.1, 156.2, 156.4 (Ar–C), 162.3, 169.4, 170.0, 170.3 and 170.5 (5 C O). Anal. Calcd for C31H28ClN3O10 (638.02): C, 58.36; H, 4.42; N, 6.59. Found: C, 58.35; H, 4.43; N, 6.58. This powder (6.3 g) was deprotected according to the general procedure described for deacetylation to give 3a as a yellow powder (4 g, 91% yield), mp 230–232 °C. IR (KBr, cm‾1): 3416 (broad, 4OH), 2220 (C N), 1659 (C O, amide). 1H NMR (300 MHz, DMSO-d6/D2O): δ 3.55 (m, 3H, H-3′, H-6′ and H-6″), 3.83 (m, 3H, H-2′, H-4′ and H-5′), 6.09 (d, 1H, J 1′,2′ = 7.8 Hz, H-1′), 7.23 (s, 1H, 2-ONN ring), 7.53 (t, 1H, J = 5.1 Hz, Py), 7.62 (d, 2H, J = 8.7 Hz, Ar–H), 7.71 (d, 2H, J = 8.7 Hz, Ar–H), 7.99 (t, 1H, J = 7.5 Hz, Py), 8.41 (d, 1H, J = 8.1 Hz, Py), 8.70 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C23H20ClN3O6 (469.87): C, 58.79; H, 4.29; N, 8.94. Found: C, 58.79; H, 4.28; N, 8.93.
4.2.4.5. 1-(β-D-Galactopyranosyl)-4-(4-isopropylphenyl)-2-oxo-6-(2-thienyl)nicotinonitrile (3b)
Compound 1b (3.2 g) was reacted with peracetylated galactosyl bromide (4.1 g) according to the general procedure described for glycosylation to give a yellow powder (4.58 g, 70% yield), mp 160–161 °C. IR (KBr, cm‾1): 2218 (C N), 1754 (C O, acetoxy), 1644 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.23 (d, 6H, J = 6.9 Hz, CH 3 (i-Pr)), 1.63, 1.84, 1.88 and 2.04 (4s, 12H, 4CH 3CO), 2.88 (m, 1H, CH (i-Pr)), 3.87 (dd, 1H, J 5′,6′ = 6.0, J 6′,6″ = 11.1 Hz, H-6′), 4.01 (dd, 1H, J 5′,6′ = 6.3, J 6′,6″ = 11.1 Hz, H-6″), 4.43 (m, 1H, H-5′), 5.19 (t, 1H, J 3′,2′ = 10.2, J 3′,4′ = 2.8 Hz, H-3′), 5.26 (t, 1H, J 2′,1′ = 8.4, J 2′,3′ = 10.3 Hz, H-2′), 5.44 (t, 1H, J 4′,3′ = 2.80, J 4′,5′ = 2.78 Hz, H-4′), 6.26 (d, 1H, J 1′,2′ = 8.4 Hz, H-1′), 7.30 (t, 1H, J = 3.9 Hz, Th), 7.48 (d, 2H, J = 8.1 Hz, Ar–H) 7.50 (s, 1H, 2-ONN ring), 7.54 (d, 2H, J = 8.1 Hz, Ar–H), 7.76 (d, 1H, J = 5.1 Hz, Th), 8.01 (d, 1H, J = 4.8 Hz, Th). 13C NMR (75 MHz, DMSO-d6): δ 20.12 (CH3 (i-Pr)), 20.84, 20.85, 20.87 and 20.94 (4CH3CO), 33.82 (CH (i-Pr)), 61.9 (C-6′), 68.03 (C-4′), 68.24 (C-2′), 70.60 (C-3′), 71.81 (C-5′), 94.7 (C-1′), 114.2, 115.6 (C N), 127.3, 128.7, 129.1, 129.6, 132.1, 133.4, 142.5, 151.3, 153.1, 154.2, 157.1 (Ar–C), 162.2, 169.4, 169.9, 170.3 and 170.5 (5 C O). Anal. Calcd for C33H34N2O10S (650.70): C, 60.91; H, 5.27; N, 4.31. Found: C, 60.90; H, 5.28; N, 4.32. This powder (6.5 g) was deprotected according to the general procedure described for deacetylation to give 3b as a yellow powder (1.56 g, 85.5% yield), mp 220–222 °C. IR (KBr, cm‾1): 3418 (broad, 4OH), 2216 (C N), 1642 (C O, amide). 1H NMR (300 MHz, DMSO-d6/D2O): δ 1.21 (d, 6H, J = 6.9 Hz, CH 3 (i-Pr)), 2.93 (m, 1H, CH (i-Pr)), 3.34–3.48 (m, 3H, H-3′, H-6′ and H-6″), 3.52–3.61 (m, 3H, H-2′, H-4′ and H-5′), 5.92 (d, 1H, J 1′,2′ = 8.1 Hz, H-1′), 7.19 (t, 1H, J = 3.9 Hz, Th), 7.41 (d, 2H, J = 8.1 Hz, Ar–H), 7.62 (d, 2H, J = 8.1 Hz, Ar–H), 7.66 (s, 1H, 2-ONN ring), 7.73 (d, 1H, J = 5.1 Hz, Th), 7.99 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C25H26N2O6S (482.55): C, 62.23; H, 5.43; N, 5.81. Found: C, 62.24; H, 5.43; N, 5.82.
4.2.4.6. 1-(β-D-Galactopyranosyl)-2-oxo-4,6-di(2-thienyl)-nicotinonitrile (3c)
Compound 1c (2.8 g) was reacted with peracetylated galactosyl bromide (4.1 g) according to the general procedure described for glycosylation to give a colourless solid (3.05 g, 51% yield), mp 210–212 °C. IR (KBr, cm‾1): 2221 (C N), 1745 (C O, acetoxy), 1634 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.69, 1.82., 1.88 and 2.04 (4s, 12H, 4CH 3CO), 3.91 (dd, 1H, J 5′,6′ = 6.0, J 6′,6″ = 10.1 Hz, H-6′), 3.99 (dd, 1H, J 5′,6′ = 6.3, J 6′,6″ = 10.1 Hz, H-6″), 4.44 (m, 1H, H-5′), 5.21 (t, 1H, J 3′,2′ = 10.23, J 3′,4′ = 2.8 Hz, H-3′), 5.28 (t, 1H, J 2′,1′ = 8.4, J 2′,3′ = 10.3 Hz, H-2′), 5.43 (t, 1H, J 4′,3′ = 2.8, J 4′,5′ = 2.78 Hz, H-4′), 6.23 (d, 1H, J 1′,2′ = 8.4 Hz, H-1′), 7.20 (s, 1H, 2-ONN ring), 7.24 (t, 1H, J = 3.9 Hz, Th), 7.29 (t, 1H, J = 5.2 Hz, Th), 7.82 (d, 1H, J = 5.2 Hz, Th), 7.86 (m, 2H, Th), 8.05 (d, 1H, J = 4.8 Hz, Th). 13C NMR (75 MHz, DMSO-d6): δ 20.84, 20.85, 20.88 and 20.93 (4CH3CO), 61.9 (C-6′), 68.06 (C-4′), 68.23 (C-2′), 70.59 (C-3′), 71.82 (C-5′), 94.79 (C-1′), 112.5, 115.6 (C N), 129.1, 129.6, 129.7, 131.1, 131.7, 132.3, 132.5, 136.6, 142.3, 148.7 and 153.2 (Ar–C), 162.7, 169.4, 169.9, 170.3 and 170.5 (5 C O). Anal. Calcd for C28H26N2O10S2 (614.64): C, 54.71; H, 4.26; N, 4.56. Found: C, 54.72; H, 4.26; N, 4.57. This solid (6.14 g) was deprotected according to the general procedure described for deacetylation to give 3c as a yellow powder (3.96 g, 89% yield), mp 245–246 °C. IR (KBr, cm‾1): 3420 (broad, 4OH), 2218 (C N), 1645 (C O, amide). 1H NMR (300 MHz, DMSO-d6/D2O): δ 3.47–3.78 (m, 6H, H-6′, H-6″, H-5′, H-4′, H-3′ and H-2′), 5.95 (d, 1H, J 1′,2′ = 8.4 Hz, H-1′), 7.23 (t, 1H, J = 3.9 Hz, Th), 7.31 (d, 1H, J = 5.2 Hz, Th), 7.56 (s, 1H, 2-ONN ring), 7.81 (d, 1H, J = 5.1 Hz, Th), 7.93 (m, 2H, Th), 8.07 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C20H18N2O6S2 (446.50): C, 53.80; H, 4.06; N, 6.27. Found: C, 53.82; H, 4.04; N, 6.26.
4.2.4.7. 4-(4-Chlorophenyl)-1-[O4′-(β-D-galactopyranosyl)-β-D-glucopyranosyl]-2-oxo-6-(2-pyridyl)nicotinonitrile (4)
Compound 1a (3.07 g) was reacted with peracetylated O 4-(β-D-galactopyranosyl)-β-D-glucopyranosyl bromide (7.2 g) according to the general procedure described for glycosylation to give a yellow powder (5.2 g, 66% yield), mp 220–221 °C. IR (KBr, cm‾1): 2223 (C N), 1747 (C O, acetoxy), 1662 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.77, 1.86, 1.87, 1.89, 1.90, 1.98 and 2.05 (7s, 21H, 7CH 3CO), 3.87 - 4.01 (m, 3H, H-2′b, H-6′a and H-6′b), 4.14 (dd, 1H, J 6″a,6′a = 11.4, J 6″a,5′a = 5.6 Hz, H-6″a), 4.36 (m, 1H, H-5′b), 4.68 (dd, 1H, J 6″b,5′b = 6.3 Hz, H-6″b), 4.87 (m, 1H, H-5′a), 4.95 (dd, 1H, J 1′b,2′b = 7.6 Hz, H-1′b), 5.04 (dd, 1H, J 4′b,3′b = 3.1, J 4′b,5′b = 3.8 Hz, H-4′b), 5.21 (dd, 1H, J 2′a,1′a = 8.7, J 2′a,3′a = 8.4 Hz, H-2′a), 5.27 (dd, 1H, J 4′a,3′a = 9.1, J 4′a,5′a = 9.9 Hz, H-4′a), 5.30 (d, 1H, J 3′b,4′b = 3.1 Hz, H-3′b), 5.38 (dd, 1H, J 3′a,2′a = 8.4, J 3′a,4′a = 9.1 Hz, H-3′a), 6.54 (d, 1H, J 1′a,2′a = 8.7 Hz, H-1′a), 7.23 (s, 1H, 2-ONN ring), 7.42 (t, 1H, J = 5.1 Hz, Py), 7.60 (d, 2H, J = 8.7 Hz, Ar–H), 7.66 (d, 2H, J = 8.7 Hz, Ar–H), 7.93 (t, 1H, J = 7.5 Hz, Py), 8.49 (d, 1H, J = 8.1 Hz, Py), 8.61 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C43H44ClN3O18 (926.27): C, 55.76; H, 4.79; N, 4.54. Found: C, 55.77; H, 4.78; N, 4.55. This powder (9.2 g) was deprotected according to the general procedure described for deacetylation to give 4 as a yellow powder (4.8 g, 75% yield), mp 275–277 °C. IR (KBr, cm‾1): 3410 (broad, 7OH), 2220 (C N), 1655 (C O, amide). 1H NMR (300 MHz, DMSO-d6/D2O): δ 3.40–3.76 (4m, 12H, H-2′b, H-3′b, H-4′b, H-5′b, H-6′b, H-6″b, H-2′a, H-3′a, H-4′a, H-5′a, H-6′a and H-6″a), 5.81 (d, 1H, J 1′b,2′b = 7.8 Hz, H-1′b), 5.92 (d, 1H, J 1′a,2′a = 8.9 Hz, H-1′a), 7.24 (s, 1H, 2-ONN ring), 7.43 (t, 1H, J = 5.1 Hz, Py), 7.62 (d, 2H, J = 8.7 Hz, Ar–H), 7.68 (d, 2H, J = 8.7 Hz, Ar–H), 7.92 (t, 1H, J = 7.5 Hz, Py), 8.48 (d, 1H, J = 8.1 Hz, Py), 8.60 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C29H30ClN3O11 (632.01): C, 55.11; H, 4.78; N, 6.65. Found: C, 55.12; H, 4.77; N, 6.65.
4.2.4.8. 4-(4-Chlorolphenyl)-2-(4-hydroxybutyloxy)-6-(2-pyridyl)nicotinonitrile (5)
Compound 1a (3.07 g) was reacted with 4-bromobutyl acetate (1.95 g) according to the general procedure described for alkylation to give a green powder (3.4 g, 80% yield), mp 130–132 °C. IR (KBr, cm‾1): 2220 (C N), 1739 (C O, acetoxy). 1H NMR (300 MHz, DMSO-d6): δ 1.83 (m, 2H, CH 2), 1.99 (s, 3H, CH 3CO), 2.94 (m, 2H, CH 2),, 4.08 (t, 2H, J = 5.7 Hz, CH 2), 4.64 (t, 2H, J = 6.3 Hz, CH 2), 7.43 (t, 1H, J = 5.1 Hz, Py), 7.65 (d, 2H, J = 8.7 Hz, Ar–H), 7.78 (d, 2H, J = 8.7 Hz, Ar–H), 8.10 (s, 1H, 2-ONN ring), 8.12 (t, 1H, J = 7.5 Hz, Py), 8.43 (d, 1H, J = 8.1 Hz, Py), 8.60 (d, 1H, J = 4.8 Hz, Py). 13C NMR (75 MHz, DMSO-d6): δ 20.65 (CH3CO), 24.75 (CH2), 24.87 (CH2), 63.42 (CH2), 66.85 (CH2), 93.85, 113.4, 114.8 (C N), 121.6, 125.4, 128.9, 130.1, 134.4, 135.0, 137.5, 149.5, 152.9, 154.9, 156.0, 163.9 and 170.2 (Ar–C, C N and C O). Anal. Calcd for C23H20ClN3O3 (421.88): C, 65.48; H, 4.78; N, 9.96. Found: C, 65.47; H, 4.79; N, 9.98. This powder (4.21 g) was deprotected according to the general procedure described for deacetylation to give 5 as a yellow powder (2.6 g, 86% yield), mp 145–146 °C. IR (KBr, cm‾1): 3418 (broad, OH), 2221 (C N). 1H NMR (300 MHz, DMSO-d6): δ 1.63 (m, 2H, CH 2), 1.87 (m, 2H, CH 2), 3.28 (t, 1H, J = 4.5 Hz, OH), 3.48 (t, 2H, J = 5.7 Hz, CH 2), 4.62 (t, 2H, J = 6.3 Hz, CH 2), 7.53 (t, 1H, J = 5.1 Hz, Py), 7.68 (d, 2H, J = 8.7 Hz, Ar–H), 7.77 (d, 2H, J = 8.7 Hz, Ar–H), 8.0 (t, 1H, J = 7.5 Hz, Py), 8.12 (s, 1H, 2-ONN ring), 8.43 (d, 1H, J = 8.1 Hz, Py), 8.74 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C21H18ClN3O2 (379.84): C, 66.40; H, 4.78; N, 11.06. Found: C, 66.41; H, 4.78; N, 11.08.
4.2.4.9. 1-(4-Hydroxybutyl)-4-(4-isopropylphenyl)-2-oxo-6-(2-thienyl)nicotinonitrile (6b)
Compound 1b (3.2 g) was reacted with 4-bromobutyl acetate (1.95 g) according to the general procedure for alkylation to give a yellow powder (2.55 g, 57% yield), mp 240–242 °C. IR (KBr, cm‾1): 2221 (C N), 1731 (C O, acetoxy), 1643 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.25 (d, 6H, J = 6.9 Hz, CH 3 (i-Pr)), 1.84 (m, 2H, CH 2), 1.99 (s, 3H, CH 3CO), 2.94 (m, 2H, CH 2), 3.10 (m, 1H, CH (i-Pr)), 4.09 (t, 2H, J = 5.7 Hz, CH 2), 4.53 (t, 2H, J = 6.3 Hz, CH 2), 7.21 (t, 1H, J = 3.9 Hz, Th), 7.43 (d, 2H, J = 8.1 Hz, Ar–H), 7.62 (d, 2H, J = 8.1 Hz, Ar–H), 7.76 (s, 1H, 2-ONN ring), 7.81 (d, 1H, J = 5.1 Hz Hz, Th), 8.09 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C25H26N2O3S (434.55): C, 69.10; H, 6.03; N, 6.45. Found: C, 69.11; H, 6.02; N, 6.45. This powder (4.34 g) was deprotected according to the general procedure described for deacetylation to give 6b as a yellow powder (3.5 g, 92% yield), mp 260–262 °C. IR (KBr, cm‾1): 3450 (OH), 2218 (C N), 1643 (C O, amide). Anal. Calcd for C23H24N2O2S (392.51): C, 70.38; H, 6.16; N, 7.14. Found: C, 70.37; H, 6.17; N, 7.13.
4.2.4.10. 1-(4-Hydroxybutyl)-2-oxo-4,6-di(2-thienyl)nicotinonitrile (6c)
Compound 1c (2.8 g) was reacted with 4-bromobutyl acetate (1.95 g) according to the general procedure described for alkylation to give a yellow powder (2.1 g, 53% yield), mp 210–212 °C. IR (KBr, cm‾1): 2212 (C N), 1731 (C O, acetoxy), 1644 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.76 (m, 2H, CH 2), 1.84 (m, 2H, CH 2), 1.99 (s, 3H, CH 3CO), 4.06 (t, 2H, J = 5.7 Hz, CH 2), 4.53 (t, 2H, J = 6.3 Hz, CH 2), 7.23 (t, 1H, J = 3.9 Hz, Th), 7.31 (d, 1H, J = 5.2 Hz, Th), 7.82 (d, 1H, J = 5.2 Hz, Th), 7.85 (s, 1H, 2-ONN ring), 7.97 (m, 2H, Th), 8.14 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C20H18N2O3S2 (398.50): C, 60.28; H, 4.55; N, 7.03. Found: C, 60.27; H, 4.56; N, 7.02. This powder (4 g) was reacted according to the general procedure described for deacetylation to give 6c as a yellow powder (2 g, 86% yield), mp 260–262 °C. IR (KBr, cm‾1): 3450 (OH), 2212 (C N), 1638 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.68 (m, 2H, CH 2), 1.85 (m, 2H, CH 2), 3.45 (t, 1H, J = 4.48 Hz, OH), 3.55 (t, 2H, J = 5.7 Hz, CH 2), 4.50 (t, 2H, J = 6.3 Hz, CH 2), 7.16 (t, 1H, J = 3.9 Hz, Th), 7.26 (d, 1H, J = 5.2 Hz, Th), 7.68 (d, 1H, J = 5.2 Hz, Th), 7.85 (m, 2H, Th), 7.90 (s, 1H, 2-ONN ring), 7.96 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C18H16N2O2S2 (356.46): C, 60.65; H, 4.52; N, 7.86. Found: C, 60.65; H, 4.53; N, 7.85.
4.2.4.11. 4-(4-Chlorophenyl)-1-[(2-hydroxyethoxy)methyl]-2-oxo-6-(2-pyridyl)nicotinonitrile (7a)
Compound 1a (3.07 g) was reacted with 2-bromomethoxyethyl acetate (1.97 g) according to the general procedure described for alkylation to give a yellow powder (3.2 g, 76% yield), mp 220–222 °C. IR (KBr, cm‾1): 2221 (C N), 1734 (C O, acetoxy), 1661 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 2.02 (s, 3H, CH 3CO), 3.38 (t, 2H, J = 5.4 Hz, CH 2), 4.43 (t, 2H, J = 5.4 Hz, CH 2), 4.72 (s, 2H, NCH 2O), 7.52 (t, 1H, J = 5.1 Hz, Py), 7.63 (d, 2H, J = 8.7 Hz, Ar–H), 7.74 (d, 2H, J = 8.7 Hz, Ar–H), 8.02 (t, 1H, J = 7.5 Hz, Py), 8.12 (s, 1H, 2-ONN ring), 8.48 (d, 1H, J = 8.1 Hz, Py), 8.72 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C22H18ClN3O4 (423.85): C, 62.34; H, 4.28; N, 9.91. Found: C, 62.33; H, 4.29; N, 9.92. This powder (4.2 g) was deprotected according to the general procedure described for deacetylation to give 7a a yellow powder (1.2 g, 86% yield), mp 260–262 °C. IR (KBr, cm‾1): 3426 (OH), 2219 (C N), 1659 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 3.40 (t, 2H, J = 5.3 Hz, CH 2), 3.83 (t, 2H, J = 5.0 Hz, CH 2), 4.63 (t, 1H, J = 5.3 Hz, OH), 5.98 (s, 2H, NCH 2O), 7.55 (t, 1H, J = 5.1 Hz, Py), 7.61 (d, 2H, J = 8.7 Hz, Ar–H), 7.69 (d, 2H, J = 8.7 Hz, Ar–H), 7.98 (t, 1H, J = 7.5 Hz, Py), 8.05 (s, 1H, 2-ONN ring), 8.40 (d, 1H, J = 8.1 Hz, Py), 8.72 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C20H16ClN3O3 (381.81): C, 62.91; H, 4.22; N, 11.01. Found: C, 62.92; H, 4.23; N, 11.00.
4.2.4.12. 1-[(2-Hydroxyethoxy)methyl]-4-(4-isopropylphenyl)-2-oxo-6-(2-thienyl)nicotinonitrile (7b)
Compound 1b (3.2 g) was reacted with 2-bromomethoxyethyl acetate (1.97 g) according to the general procedure described for alkylation to give a yellow powder (3.3 g, 62% yield), mp 180–182 °C. IR (KBr, cm‾1): 2218 (C N), 1735 (C O, acetoxy), 1643 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.24 (d, 6H, J = 6.9 Hz, CH 3 (i-Pr)), 2.02 (s, 3H, CH 3CO), 2.97 (m, 1H, CH (i-Pr)), 3.34 (t, 2H, J = 5.4 Hz, OCH 2 (c)), 4.45 (t, 2H, J = 5.4 Hz, OCH 2 (d)), 4.72 (s, 2H, NCH 2O (a)), 7.22 (t, 1H, J = 3.9 Hz, Th), 7.43 (d, 2H, J = 8.1 Hz, Ar–H), 7.62 (d, 2H, J = 8.1 Hz, Ar–H), 7.79 (s, 1H, 2-ONN ring), 7.82 (d, 1H, J = 5.1, Hz, Th), 8.11 (d, 1H, J = 4.8 Hz, Th). 13C NMR (75 MHz, DMSO-d6): δ 20.6 (CH3CO), 23.6 ( CH3 (i-Pr)), 33.3 (CH (i-Pr)), 61.9 (CH2O (d)), 65.1 (OCH2 (c)), 91.2 (NCH2O), 112.2, 115.2 (C N), 126.7, 128.2, 128.6, 129.0, 131.2, 133.0, 133.4, 142.4, 150.6, 152.5, 156.2 (Ar–C) and 163.6, 170.3 (2C = O). Anal. Calcd for C24H24N2O4S (436.92): C, 66.03; H, 5.54; N, 6.42. Found: C, 66.02; H, 5.53; N, 6.44. This powder (4.3 g) was deprotected according to the general procedure described for deacetylation to give 7b as a yellow powder (2.78 g, 85% yield), mp 200–201 °C. IR (KBr, cm‾1): 3428 (OH), 2221 (C N), 1662 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.23 (d, 6H, J = 6.9 Hz, CH 3 (i-Pr)), 2.99 (m, 1H, CH (i-Pr)), 3.57 (t, 2H, J = 5.3 Hz, CH 2), 3.80 (t, 2H, J = 5.0 Hz, CH 2), 4.52 (t, 1H, J = 5.3 Hz, OH), 5.40 (s, 2H, NCH 2O), 7.23 (t, 1H, J = 3.9 Hz, Th), 7.45 (d, 2H, J = 8.1 Hz, Ar–H), 7.63 (d, 2H, J = 8.1 Hz, Ar–H), 7.65 (s, 1H, 2-ONN ring), 7.80 (d, 1H, J = 5.1 Hz, Th), 7.85 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C22H22N2O3S (394.49): C, 66.98; H, 5.62; N, 7.10. Found: C, 66.99; H, 5.62; N, 7.11.
4.2.5. Compounds prepared by alkylation without deacetylation
4.2.5.1. 1-[(2-Acetoxyethoxy)methyl]-2-oxo-4,6-di(2-thienyl)nicotinonitrile (7cAc)
This compound was prepared from 1c (2.8 g) and 2-bromomethoxyethyl acetate (1.97 g) according to the general procedure described above for alkylation and was obtained as a yellow powder (1.8 g, 45% yield), mp 160–161 °C. IR (KBr, cm‾1): 2214 (C N), 1732 (C O, acetoxy), 1643 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 2.06 (s, 3H, CH 3CO), 3.38 (t, 2H, J = 5.4 Hz, CH 2), 4.44 (t, 2H, J = 5.4 Hz, CH 2), 4.72 (s, 2H, NCH 2O (a)), 7.23 (t, 1H, J = 3.9 Hz, Th), 7.33 (d, 1H, J = 5.2 Hz, Th), 7.79 (m, 2H, Th), 7.82 (s, 1H, 2-ONN ring), 7.83 (d, 1H, J = 5.2 Hz, Th), 8.15 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C19H16N2O4S2 (400.47): C, 56.98; H, 4.03; N, 7.00. Found: C, 56.99; H, 4.04; N, 7.02.
4.2.5.2. 4-(4-Chlorolphenyl)-1-(3-hydroxypropyl)-2-oxo-6-(2-pyridyl)nicotinonitrile (8a)
This compound was prepared from 1a (3.07 g) and 3-hydroxypropyl chloride (0.94 g) according to the general procedure described above, and was obtained as a yellow powder (2.7 g, 74% yield), mp 174–176 °C. IR (KBr, cm‾1): 3328 (OH), 2221 (C N), 1652 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.95 (m, 2H, CH 2), 3.31 (t, 2H, J = 5.3 Hz Hz, CH 2), 3.62 (t, 2H, J = 5.0 Hz, CH 2), 4.69 (t, 1H, J = 5.3 Hz, OH), 7.33 (t, 1H, J = 5.1 Hz, Py), 7.53 (d, 2H, J = 8.7 Hz, Ar–H), 7.69 (d, 2H, J = 8.7 Hz, Ar–H), 8.01 (t, 1H, J = 7.5 Hz, Py), 8.12 (s, 1H, 2-ONN ring), 8.44 (d, 1H, J = 8.1 Hz, Py), 8.75 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C20H16ClN3O2 (365.81): C, 65.67; H, 4.41; N, 11.49. Found: C, 65.68; H, 4.40; N, 11.48.
4.2.5.3. 1-(3-Hydroxypropyl)-4-(4-isopropylphenyl)-2-oxo-6-(2-thienyl)nicotinonitrile (8b)
This compound was prepared from 1b (3.2 g) and 3-hydroxypropyl chloride (0.94 g) according to the general procedure described for alkylation and was obtained as a yellow powder (3.3 g, 88% yield), mp 180–182 °C. IR (KBr, cm‾1): 3384 (OH), 2219 (C N), 1660 (C O, amide). Anal. Calcd for C22H22N2O2S (378.49): C, 69.81; H, 5.86; N, 7.40. Found: C, 69.82; H, 5.85; N, 7.42.
4.2.5.4. 3-Cyano-1-(3-hydroxypropyl)-4,6-di(2-thienyl)-2-pyridone (8c)
This compound was prepared from 1c (2.8 g) and 3-hydroxypropyl chloride (0.94 g) according to the general procedure described for alkylation and was obtained as a yellow powder (2.8 g, 82% yield), mp 180–181 °C. IR (KBr, cm‾1): 3460 (OH), 2217 (C N), 1643 (C O, amide). 1H NMR (300 MHz, DMSO-d6): δ 1.93 (m, 2H, CH 2), 3.33 (t, 2H, J = 5.3 Hz, CH 2), 3.63 (t, 2H, J = 5.0 Hz, CH 2), 4.56 (t, 1H, J = 5.3 Hz, OH), 7.23 (t, 1H, J = 3.9 Hz, Th), 7.32 (d, 1H, J = 5.2 Hz, Th), 7.63 (d, 1H, J = 5.2 Hz, Th), 7.83 (s, 1H, 2-ONN ring), 7.97 (m, 2H, Th), 8.14 (d, 1H, J = 4.8 Hz, Th). Anal. Calcd for C17H14N2O2S2 (342.44): C, 59.63; H, 4.12; N, 8.18. Found: C, 59.62; H, 4.13; N, 8.17.
4.2.5.5. 2-(Allyloxy)-4-(4-chlorolphenyl)-6-(2-pyridyl)nicotinonitrile (9)
This compound was prepared from 1a (2.36 g) and allyl bromide (0.93 g) according to the general procedure described for alkylation and was obtained as a yellow powder (2.19 g, 82% yield), mp 176–177 °C. IR (KBr, cm‾1): 2222 (C N). 1H NMR (300 MHz, DMSO-d6): δ 5.15 (d, 2H, J = 5.4 Hz, CH 2), 5.32 (d, 1H, J = 10.2 Hz), 5.56 (d, 1H, J = 17.1 Hz), 6.17 (m, 1H), 7.33 (t, 1H, J = 5.1 Hz, Py), 7.53 (d, 2H, J = 8.7 Hz, Ar–H), 7.75 (d, 2H, J = 8.7 Hz, Ar–H), 8.01 (t, 1H, J = 7.5 Hz, Py), 8.13 (s, 1H, 2-ONN ring), 8.43 (d, 1H, J = 8.1 Hz, Py), 8.74 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C20H14ClN3O (347.80): C, 69.07; H, 4.06; N, 12.08. Found: C, 69.08; H, 4.06; N, 12.09.
4.2.5.6. 4-(4-Chlorophenyl)-2-(prop-2-ynyloxy)-6-(2-pyridyl)nicotinonitrile (10)
This compound was prepared from 1a (3.05 g) and propargyl bromide (1.18 g) according to the general procedure described for alkylation and was obtained as a yellow powder (2.5 g, 73% yield), mp 180–182 °C. IR (KBr, cm‾1): 2224 (C N). 1H NMR (300 MHz, DMSO-d6): δ 3.68 (s, 1H, ≡CH), 5.30 (d, 2H, J = 7.2 Hz, CH 2), 7.32 (t, 1H, J = 5.1 Hz, Py), 7.54 (d, 2H, J = 8.7 Hz, Ar–H), 7.76 (d, 2H, J = 8.7 Hz, Ar–H), 8.02 (t, 1H, J = 7.5 Hz, Py), 8.13 (s, 1H, 2-ONN ring), 8.44 (d, 1H, J = 8.1 Hz, Py), 8.74 (d, 1H, J = 4.8 Hz, Py). Anal. Calcd for C20H12ClN3O (345.78): C, 69.47; H, 3.50; N, 12.15. Found: C, 69.46; H, 3.52; N, 12.16.
4.3. Biology
4.3.1. Antimicrobial screening
The 2-ONN compounds were screened against the specified microorganisms using the well diffusion method [29]. In this method, bacteria were grown on the nutrient agar and fungi were grown on the Czapek's-Dox agar medium slants. The spores were inoculated in sterilized media, then shaken to make their dispersion homogeneous, and then poured in plates and left to solidify. Solutions of the tested compounds in DMF were prepared in a concentration of 250 μg/mL. Wells of 1 cm diameter were punched into the agar medium and filled with 1 mL of the compound solution. The plates were left for 24 h in a refrigerator to allow diffusion. Next, the plates were incubated at 35 °C for 48 h, in case of antibacterial screening, and at 28 °C for 5–7 days, in case of antifungal screening. The diameter of inhibition zones was measured in mm at the end of an incubation period. Ampicillin and Dermatin were used as references for bacteria and fungi, respectively.
4.3.2. In vitro antiviral screening
In vitro activity screening against viruses was carried out by Health Specialist, Virology Branch, Division of Microbiology and Infection Diseases (DMID) at the National Institute of Allergy and Infection Diseases (NIAID) and the National Institute of Health (NIH), USA. The compounds were evaluated for their antiviral activity against HCV and HBV using RNA Hybridization (Replicon) and DNA Hybridization (virion) control assays. Cytopathic effect and neutral red control assays were used with the other viruses.
Acknowledgements
The authors of this article thank Mr. Ahmed Saleh of the Microbiology Department at Zagazig University for antimicrobial testing and the Division of Microbiology and Infection Diseases (DMID)/National Institute of Allergy and Infection Diseases (NIAID) and the National Institute of Health (NIH), USA for conducting the antiviral activity studies. This work was sponsored by Zagazig University, Egypt.
Footnotes
2-ONN: 2-oxonicotinonitrile.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2013.12.055.
Appendix A. Supplementary data
The following is the supplementary data related to this article.
References
- 1.Jessen H.J., Gademann K. 4-Hydroxy-2-pyridone alkaloids: structures and synthetic approaches. Nat. Prod. Rep. 2010;27:1168–1185. doi: 10.1039/b911516c. [DOI] [PubMed] [Google Scholar]
- 2.Vegi S.R., Boovanahalli S.K., Patro B., Mukkanti K. SPF32629A and SPF32629B: enantioselective synthesis, determination of absolute configuration, cytotoxicity and antibacterial evaluation. Eur. J. Med. Chem. 2011;46:1803–1812. doi: 10.1016/j.ejmech.2011.02.039. [DOI] [PubMed] [Google Scholar]
- 3.Gomez R., Jolly S., Williams T., Tucker T., Tynebor R., Vacca J., McGaughey G., Lai M.-T., Felock P., Munshi V., DeStefano D., Touch S., Miller M., Yan Y., Sanchez R., Liang Y., Paton B., Wan B.-L., Anthony N. Design and synthesis of pyridone inhibitors of non-nucleoside reverse transcriptase. Bioorg. Med. Chem. Lett. 2011;21:7344–7350. doi: 10.1016/j.bmcl.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 4.Medina-Franco J.L., Martínez-Mayorga K., Juárez-Gordiano C., Castillo R. Pyridin-2(1H)-ones: a promising class of HIV-1 non-nucleoside reverse transcriptase inhibitors. Chem. Med. Chem. 2007;2:1141–1147. doi: 10.1002/cmdc.200700054. [DOI] [PubMed] [Google Scholar]
- 5.Goldman M.E., Nunberg J.H., O'Brien J.A., Quintero J.C., Schleif W.A., Freund K.F., Gaul S.L., Saari W.S., Wai J.S., Hoffman J.M. Pyridinone derivatives: specific human immunodeficiency virus type 1 reverse transcriptase inhibitors with antiviral activity. Proc. Natl. Acad. Sci. 1991;88:6863–6867. doi: 10.1073/pnas.88.15.6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scala A., Cordaro M., Risitano F., Colao I., Venuti A., Sciortino M., Primerano P., Grassi G. Diastereoselective multicomponent synthesis and anti-HSV-1 evaluation of dihydrofuran-fused derivatives. Mol. Diversity. 2012;16:325–333. doi: 10.1007/s11030-012-9367-0. [DOI] [PubMed] [Google Scholar]
- 7.J. Li, A.S.-T. Lui, K.L. McCaleb, F.X. Talamas, Preparation of arylpyridones as Hepatitis C virus NS5B polymerase inhibitors useful as antivirals, WO2010049331A1 (2010).
- 8.Lv Z., Sheng C., Wang T., Zhang Y., Liu J., Feng J., Sun H., Zhong H., Niu C., Li K. Design, synthesis, and antihepatitis B virus activities of novel 2-pyridone derivatives. J. Med. Chem. 2010;53:660–668. doi: 10.1021/jm901237x. [DOI] [PubMed] [Google Scholar]
- 9.Li Q., Mitscher L.A., Shen L.L. The 2-pyridone antibacterial agents: bacterial topoisomerase inhibitors. Med. Res. Rev. 2000;20:231–293. doi: 10.1002/1098-1128(200007)20:4<231::aid-med1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 10.J.P.H. Bosselaers, D.L.J. Bylemans, T.M.J. Kempen, Biocidal compositions comprising thiol group modulating enzyme inhibitors and pyrion compounds, WO2010060948A2 (2010).
- 11.Chu D.T.W. Recent progress in novel macrolides, quinolones, and 2-pyridones to overcome bacterial resistance. Med. Res. Rev. 1999;19:497–520. doi: 10.1002/(sici)1098-1128(199911)19:6<497::aid-med3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 12.Jayasinghe L., Abbas H.K., Jacob M.R., Herath W.H.M.W., Nanayakkara N.P.D. N-Methyl-4-hydroxy-2-pyridinone analogues from Fusarium oxysporum. J. Nat. Prod. 2006;69:439–442. doi: 10.1021/np050487v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan X., Feng D., Qu Y., Zhang X., Wang J., Loiseau P.M., Andrei G., Snoeck R., Clercq E.D. Practical and efficient synthesis of pyrano[3,2-c]pyridone, pyrano[4,3-b]pyran and their hybrids with nucleoside as potential antiviral and antileishmanial agents. Bioorg. Med. Chem. Lett. 2010;20:809–813. doi: 10.1016/j.bmcl.2009.12.102. [DOI] [PubMed] [Google Scholar]
- 14.Kumarihamy M., Fronczek F.R., Ferreira D., Jacob M., Khan S.I., Nanayakkara N.P.D. Bioactive 1,4-dihydroxy-5-phenyl-2-pyridinone alkaloids from Septoria pistaciarum. J. Nat. Prod. 2010;73:1250–1253. doi: 10.1021/np1000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Clercq E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohamed S.F., Youssef M.M., Amr A.-G.E., Kotb E.R. Antimicrobial activities of some synthesized pyridines, oxazines and thiazoles from 3-aryl-1-(2-naphthyl)prop-2-en-1-ones. Sci. Pharm. 2008;76:279–303. [Google Scholar]
- 17.Abadi A.H., Abouel-Ella D.A., Lehmann J., Tinsley H.N., Gary B.D., Piazza G.A., Abdel-Fattah M.A.O. Discovery of colon tumor cell growth inhibitory agents through a combinatorial approach. Eur. J. Med. Chem. 2010;45:90–97. doi: 10.1016/j.ejmech.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rashad A., Sayed H., Shamroukh A., Awad H. Preparation of some fused pyridopyrimidine and pyridothienotriazine derivatives for biological evaluation. Phosphorus, Sulfur Silicon Relat. Elem. 2005;180:2767–2777. [Google Scholar]
- 19.El-Kerdawy M.M., El-Emam A.A. Synthesis and biological testing of some α-thienyl and α-furyl derivatives. J. Chem. Soc. Pak. 1987;9:285–293. [Google Scholar]
- 20.Davoodnia A., Attar P., Morsali A., Eshghi H., Tavakoli-Hoseini N., Khadem S. Experimental and theoretical studies on the tautomerism in 2-aminopyridines and 2(1H)-pyridinones: synthesis of 2-amino-4-aryl-3-cyano-6-(3,4-dimethoxyphenyl)pyridines and 4-aryl-3-cyano-6-(3,4-dimethoxyphenyl)-2(1H)-pyridinones. Bull. Korean Chem. Soc. 2011;32:1873–1878. [Google Scholar]
- 21.El-Sayed H.A., Moustafa A.H., Haikal A.E.-F.Z., Abu-El-Halawa R., El Ashry E.S.H. Synthesis, antitumor and antimicrobial activities of 4-(4-chlorophenyl)-3-cyano-2-(β-O-glycosyloxy)-6-(thien-2-yl)-nicotinonitrile. Eur. J. Med. Chem. 2011;46:2948–2954. doi: 10.1016/j.ejmech.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 22.Moustafa A.H., El-Sayed H.A., Haikal A.E.-F.Z., El Ashry E.S.H. Synthesis of acyclovir and HBG analogues having nicotinonitrile and its 2-methyloxy 1,2,3-triazole. Nucleosides, Nucleotides Nucleic Acids. 2011;30:340–352. doi: 10.1080/15257770.2011.582850. [DOI] [PubMed] [Google Scholar]
- 23.Saad H.A., Abdel-Hafez S.H. Synthesis and biological activity of some nucleoside analogs of 3-cyanopyridin-2-one. Curr. Org. Synth. 2012;9:413–426. [Google Scholar]
- 24.Rashad A.E., Shamroukh A.H., El-Hashash M.A., El-Farargy A.F., Yousif N.M., Salama M.A., Mostafa A., El-Shahat M. Synthesis and anti-avian influenza virus (H5N1) evaluation of some novel nicotinonitriles and their N-acylic nucleosides. J. Heterocycl. Chem. 2012;49:1130–1135. [Google Scholar]
- 25.Al-Neyadi S.S., Hassan A.H., Abdou I.M. Microwave-assisted synthesis of 2(1H)-pyridones and their glucosides as cell proliferation inhibitors. Nucleosides, Nucleotides Nucleic Acids. 2011;30:120–134. doi: 10.1080/15257770.2010.551646. [DOI] [PubMed] [Google Scholar]
- 26.Abdou I.M., Attia A.M., Strekowski L. Glucopyranosides derived from 6-aryl-5-cyano-2-(methylthio)pyrimidin-4(3H)ones. Nucleosides, Nucleotides Nucleic Acids. 2002;21:15. doi: 10.1081/NCN-120006527. [DOI] [PubMed] [Google Scholar]
- 27.G.P.M. Mommen, M.R.J. Hamzink, G. Zomer, J.A.P.J.M. De, H.D. Meiring, Protein organophosphate affinity tag for affinity chromatography and mass spectrometric analysis of proteomic samples, WO2012081978A1 (2012).
- 28.Robins M.J., Hatfield P.W. Nucleic acid related compounds. 37. Convenient and high-yield syntheses of N-[(2-hydroxyethoxy)methyl] heterocycles as “acyclic nucleoside” analogues. Can. J. Chem. 1982;60:547–553. [Google Scholar]
- 29.Mehmood Z., Ahmad I., Mohammad F., Ahmad S. Indian medicinal plants: a potential source of anticandidal drugs. Pharm. Biol. 1999;37:237–242. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.