

Abstract
We report the design and synthesis of a series of dipeptide-type inhibitors with novel P3 scaffolds that display potent inhibitory activity against SARS-CoV 3CLpro. A docking study involving binding between the dipeptidic lead compound 4 and 3CLpro suggested the modification of a structurally flexible P3 N-(3-methoxyphenyl)glycine with various rigid P3 moieties in 4. The modifications led to the identification of several potent derivatives, including 5c–k and 5n with the inhibitory activities (Ki or IC50) in the submicromolar to nanomolar range. Compound 5h, in particular, displayed the most potent inhibitory activity, with a Ki value of 0.006 μM. This potency was 65-fold higher than the potency of the lead compound 4 (Ki = 0.39 μM). In addition, the Ki value of 5h was in very good agreement with the binding affinity (16 nM) observed in isothermal titration calorimetry (ITC). A SAR study around the P3 group in the lead 4 led to the identification of a rigid indole-2-carbonyl unit as one of the best P3 moieties (5c). Further optimization showed that a methoxy substitution at the 4-position on the indole unit was highly favorable for enhancing the inhibitory potency.
Keywords: SARS, SARS-CoV 3CLpro, Dipeptide, Peptidomimetics, Cysteine protease inhibitors
Graphical abstract

Highlights
-
•
A series of low-molecular-weight dipeptide-type anti-SARS agents were synthesized.
-
•
Compounds have shown potent inhibitory activities.
-
•
Discovery of potent lead compound was achieved by introducing P3 indole-2-carbonyl unit.
-
•
Compounds 5h exhibited excellent inhibitory activity with K i value of 6.0 nM.
1. Introduction
Severe acute respiratory syndrome is a highly contagious and fatal respiratory disease that has infected more than 8000 individuals, 10% of which died within a few months of the emergence of the disease between November 2002 and early 2003 [1]. Extensive collaborative research cooperation between the leading experts and the World Health Organization (WHO) led to the rapid identification of a novel coronavirus (CoV) as the etiological agent underlying the pandemic SARS infection. The SARS outbreak was consequently successfully controlled during the beginning of 2003 [2], [3]. A reemergence of a SARS-CoV pandemic is still considered to be a potential risk, and new strains of SARS could potentially be more severe than the strains that contributed to the 2003 outbreak. Since 2003, two additional human coronavirus, NL63 and HKU1, have been identified in patients around the world. These new viruses have been characterized and were found to be significantly less lethal than SARS-CoV [4], [5], [6]. A new SARS-like virus, HCoV-EMC, was recently identified in at least two individuals, one of whom died [7]. The first case of a fatal respiratory illness similar to the deadly SARS was very recently confirmed in Britain [8]. The possibility of a future SARS-like pandemic remains, and no vaccines or antiviral agents have yet been developed to prevent or treat SARS-like infections.
SARS-CoV encodes a chymotrypsin-like protease (CLpro) that plays a pivotal role in the replication of the virus [9]. Unlike common serine proteases that contain a Ser-His-Asp catalytic triad in the active site, SARS-CoV 3CLpro contains a Cys–His catalytic dyad (Cys145 and His41) that is functionally analogous to the porcine transmissible gastroenteritis virus main protease (Cys145 and His41) and the human coronavirus 229E main protease (Cys145 and His41) [10]. Cys act as a nucleophile, whereas His functions as a general base [10], [11]. Since SARS-CoV 3CLpro plays an important role in the virus life cycle, it has been recognized as a viable target for anti-SARS drug development.
Our ongoing efforts have led to the development of several inhibitors with moderate to remarkable potency against SARS-CoV 3CLpro. As shown in Fig. 1 , a first tripeptidic lead compound (1) bearing an electrophilic ketone as a warhead moiety was developed from the natural peptide sequence after extensive structural modifications [12]. Regarding the mechanism of action, the electrophilic ketone of 1 can be reacted to the active site Cys-SH group resulting in the formation of hemithioketal intermediate between the enzyme and inhibitors to interfere the enzyme function, and hence it was predicted be a reversible inhibitor of SARS-CoV 3CLpro. A subsequent SAR study afforded promising inhibitors 2 and 3 with excellent inhibitory activities (K i) of 4.1 and 3.1 nM, respectively [13]. Recently, we reported a series of low molecular weight dipeptide-type inhibitors in which the P3 valine moiety was removed form the lead compound 2 (see Fig. 1). This study led to the identification of a compound 4 as a promising lead with an inhibitory activity (K i) of 0.39 μM (Fig. 2 ) [14]. From a structural point of view, the P3 N-arylglycyl moiety was recognized as crucial to the activity of 4, as the amine functionality of glycyl moiety formed a hydrogen bond with the backbone amino acid residue Glu166 of 3CLpro in the docking study.
Fig. 1.

Structures of potent tripeptidomimetics (1–3).
Fig. 2.

Docking pose of the lead compound 4 with the SARS-CoV 3CL protease active site. Only residues that are contacted with the ligand (4) are highlighted.
This study was extended, as reported here, by rigidifying the P3 N-arylglycyl unit in 4 in search of a more suitable motif with a favorable conformation would provide better interactions and increase the inhibitory activity toward SARS-CoV 3CLpro. Accordingly, we designed and synthesized a series of dipeptide-type inhibitors with novel P3 scaffolds and tested the inhibitory activities of these compounds against SARS-CoV 3CLpro. Several analogs were identified as exhibiting potent inhibitory activities relative to the lead compound 4. In particular, a compound bearing a P3 4-methoxyindole-2-carbonyl group (5h) exhibited excellent inhibitory activity, with a K i or IC50 value of 0.006 or 0.74 μM, respectively. Extensive molecular docking studies of some compounds were conducted to model the binding interactions of these inhibitors.
2. Results and discussion
2.1. Chemistry
The synthesis of the target compounds 5a–r was envisioned as the assembly of two key fragments: the peptidics 14 and the C-terminal benzothiazole derivative 18 (Scheme 1, Scheme 2). As shown in Scheme 1 , the peptidic intermediates 14a–r were synthesized via a coupling reaction between various carboxylic acids (8a–r) and leucine tert-butyl ester (12), followed by deprotection of tert-butyl group. The carboxylic acids, such as 5-oxopyrrolidine-2-carboxylic acid (8a), 1H-pyrrole-2-carboxylic acid (8b), 1H-indole-2-carboxylic acid (8c), 5-methoxy-1H-indole-2-carboxylic acid (8d), 5-hydroxy-1H-indole-2-carboxylic acid (8e), 5-chloro-1H-indole-2-carboxylic acid (8f), 6-methoxy-1H-indole-2-carboxylic acid (8g), 4-methoxy-1H-indole-2-carboxylic acid (8h), 4-hydroxy-1H-indole-2-carboxylic acid (8k), 1H-benzo[d]imidazole-2-carboxylic acid (8n), benzo[d]thiazole-2-carboxylic acid (8o), indoline-2-carboxylic acid (8p), benzofuran-2-carboxylic acid (8q), and 1H-indole-3-carboxylic acid (8r) were commercially available.
Scheme 1.

Synthetic outline for the preparation of 8l, 8m, 8i, 8j and 14 Reagents and conditions: i) BBr3 (1 M in DCM), rt, 2 h; ii) DEAD, PPh3, i-PrOH or i-BuOH, THF, rt, 30 min; iii) LiOH·H2O, THF, 50 °C, 12 h; iv) POCl3/DMF, 100 °C, 2 h or CH3COCl, anhydrous AlCl3, 1,2-dichloroethane, reflux, 2 h; v) Et3SiH, CF3COOH, rt, 2 h; vi) 4 M NaOH, ethanol, 60 °C, 2 h; vii) 8, EDC·HCl, HOBt·H2O, TEA, DMF, 0 °C-rt, 2 h; viii) TFA, CH2Cl2, 0 °C-rt, 1 h, and subsequent coupling reaction without further characterizations. Note: the substituents R and R1 were indicated in Table 1.
Scheme 2.

Synthetic outline for the preparation of 5a–r Reagents and conditions: i) HN(OMe)Me·HCl, EDC·HCl, HOBt·H2O, Et3N, DMF, 0 °C-rt, 2 h; ii) benzothiazole, n-BuLi, THF, −78 °C-rt, 3–5 h; iii) TFA, CH2Cl2, 0 °C-rt, 1 h; iv) 14, HBTU, DIPEA, DMF, 0 °C-rt, 2 h followed by HPLC purification. Note: the substituents R and R1 were indicated in Table 1.
The carboxylic acids 8i, 8j, 8l, and 8m were synthesized as shown in Scheme 1. Briefly, the commercially available methyl indole-2-carboxylate 6 underwent O-demethylation in the presence of boron tribromide (BBr3) to produce the corresponding 4-hydroxyindole ester 7 [15], [16], which was subsequently treated with various alcohols under Mitsunobu conditions using diethyl azodicarboxylate (DEAD) and triphenylphosphine (PPh3) in THF, followed by deprotection with lithium hydroxide·water (LiOH·H2O) to furnish the 4-isopropoxy (8i) and/or 4-isobutyloxy indole-2-carboxylic acid (8j), respectively [15], [17].
The commercially available 5-methoxyindole-2-caboxylic acid ethyl ester 9 was submitted to a Vilsmeier–Haack formylation in the presence of phosphoryloxy chloride (POCl3) in DMF to produce 10a [18], [19]. On the other hand, acylation of 9 with acetyl chloride in the presence of anhydrous aluminum chloride (AlCl3) afforded compound 10b [19], [20]. Both the indole-3-formaldehyde (10a) and the indole-3-acetyl (10b) derivatives were reduced in the presence of triethyl silylhydride (Et3SiH) [19] to furnish 11a and 11b, which were hydrolyzed to afford the corresponding indole-2-carboxylic acids 8l and 8m, respectively.
The synthesis of the peptidic fragments 13a–r was achieved from the leucine tert-butyl ester (12) in the presence of various carboxylic acids (8) via a 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) –1-hydroxybenzotriazole (HOBt)-mediated coupling method in the presence of triethylamine (TEA) in DMF. The N-protected amino acid tert-butyl esters (13) were subsequently deprotected with trifluoroacetic acid/water (10:1) over 1 h to afford the N-protected amino acids (14a–r), which were used directly in subsequent steps.
The synthesis of the other key intermediate 18 was achieved using a method reported previously [12], [13], [14]. Briefly, the optically pure l-glutamic acid ester 15 was converted to the γ-lactam-acid 16 [21], [22] by treatment with bromoacetonitrile, followed by reduction with PtO2 (5%), cyclization, and hydrolysis (Scheme 2 ). Further coupling of 16 to N,O-dimethylhydroxylamine via the EDC–HOBt method afforded the Weinreb amide 17 [21]. The Weinreb amide 17 was then coupled to benzothiazole in the presence of n-butyl lithium (n-BuLi) at −78 °C to furnish 18, which was then deprotected and subsequently coupled to the peptides 14 in the presence of O-benzotriazole- N,N,N′,N′-tetramethyluroniumhexafluoro phosphate (HBTU) and DIPEA in DMF to afford the title compounds 5a–r. All compounds were purified by reverse phase HPLC for the biological evaluation and characterized by 1H NMR, 13C NMR, and mass spectrometry. The purity of each compound exceeded 95%.
2.2. Biological assay
The K i or IC50 values of the synthesized compounds against SARS-CoV 3CLpro are listed in Table 1, Table 2 . The compounds were subjected to a fluorometric protease inhibitory assay using a procedure similar to that mentioned in earlier studies [22], [23]. Briefly, the kinetic parameters were determined at a constant substrate concentration, and the inhibitor concentrations were varied to assess the K i values [12], [13], [14]. The IC50 values were determined only for certain potent inhibitors, based on the apparent decrease in the substrate concentration (H-TSAVLQSGFRK-NH2) upon digestion by R188I SARS-CoV 3CLpro, as described previously [24], [25], [26]. The cleavage reaction was monitored by analytical HPLC, and the cleavage rates were calculated from the decrease in the substrate peak area. Table 1, Table 2 report the K i or IC50 values as the mean of 3 independent experiments.
Table 1.
SARS-CoV 3CLpro inhibitory activities (Ki) of 5a–r.
| Entry no. | Inhibitors | Ki (μM) | Entry no. | Inhibitors | Ki (μM) |
|---|---|---|---|---|---|
| 5a |  |
2.7 | 5j | 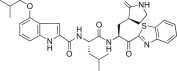 |
0.030 |
| 5b |  |
1.7 | 5k | 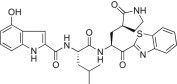 |
0.026 |
| 5c | 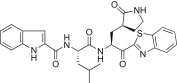 |
0.065 | 5l |  |
6.7 |
| 5d | 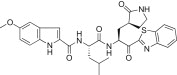 |
0.067 | 5m | 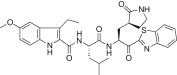 |
7.5 |
| 5e |  |
0.16 | 5n | 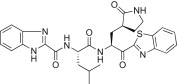 |
0.022 |
| 5f | 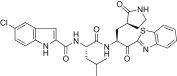 |
0.028 | 5o | 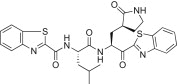 |
0.80 |
| 5g | 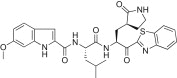 |
0.33 | 5p | 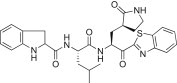 |
0.12 |
| 5h | 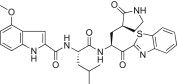 |
0.0063 | 5q |  |
14 |
| 5i | 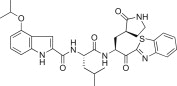 |
0.048 | 5r |  |
0.68 |
Table 2.
SARS-CoV 3CLpro inhibitory activities (IC50) of selected compounds.
| Entry no. | IC50 (μM) |
|---|---|
| 5c | 1.50 |
| 5d | 4.60 |
| 5f | 4.80 |
| 5h | 0.74 |
| 5j | 5.20 |
| 5k | 1.50 |
| 5n | 1.30 |
2.3. Structure–activity relationship study
In a previous study, we reported a series of low molecular weight dipeptide-type SARS-CoV 3CLpro inhibitors. Among them, compound 4 bearing P3 N-(3-methoxyphenyl)glycyl exhibited potent inhibitory activity with a K i value of 0.39 μM. In this study, we envisioned that substitutions at the P3 N-arylglycyl unit in 4 could improve the inhibitory potency against 3CLpro. Therefore, we designed and synthesized a series of analogs bearing a rigid P3 motif for evaluation against SARS-CoV 3CLpro. In a first attempt as shown in Table 1, the N-(3-methoxyphenyl)glycyl unit was replaced with the dl-pyroglutamyl (5a; K i = 2.70 μM) or the pyrrole-2-carbonyl (5b; K i = 1.70 μM) as a rigid P3 scaffold in 4. The inhibitory activities of the resulting compounds were dramatically reduced compared to the activity of 4 (K i = 0.39 μM). On the other hand, the introduction of an indole-2-carbonyl unit (5c; K i = 0.065 μM or IC50 = 1.50 μM) yielded a 40- or 25-fold higher inhibitory potency relative to 5a or 5b, respectively, and a 6-fold more potent activity relative to 4. These studies suggested that the rigid indole-2-carbonyl, which was introduced in place of the P3 moiety (N-(3-methoxyphenyl)glycyl) in the lead compound 4, displayed appreciable inhibitory activity. Thus, compound 5c served as a lead compound for further optimization steps.
The substituent effects at different positions on the P3 indole unit in 5c were investigated by introducing a wide variety of substituents. Initially, substituents were introduced at the 5-position of the indole unit in 5c, including 5-methoxy (5d; K i = 0.067 μM or IC50 = 4.60 μM), 5-hydroxyl (5e; K i = 0.160 μM), or 5-chloro (5f; K i = 0.028 μM or IC50 = 4.80 μM). This result suggested that the 5-chloro substituent on the indole unit (5f) exhibited good inhibitory potency relative to, respectively, lead compound 5c, 5-methoxy (5d) and 5-hydroxy (5e) derivatives. Next, a methoxy group was introduced at the 6-position on the indole unit, as shown in 5g (K i = 0.333 μM). The activity of 5g was lower than the activity of 5d; thus, the substitution at the 6-position on the P3 indole-2-carbonyl in 5c did not significantly improve the inhibitory activity. On the other hand, the methoxy substitution at the 4-position on the indole unit (5h; K i = 0.006 μM or IC50 = 0.74 μM) exhibited excellent inhibitory activity, with 10- or 55-fold increases in activity relative to, respectively, the 5-methoxy (5d) or 6-methoxy (5g) derivatives. This finding revealed that the methoxy substitution at the 4-position on the indole unit in 5c significantly improved the inhibitory activity.
The 4-methoxy group on the indole unit of 5h was examined by substitution with 4-isopropoxyl (5i; K i = 0.048 μM), 4-isobutyloxyl (5j; K i = 0.030 μM or IC50 = 5.20 μM), or 4-hydroxyl (5k; K i = 0.026 μM or IC50 = 1.30 μM) moieties; however, the inhibitory activities of 5i, 5j, or 5k were lower than the activity of the 4-methoxy derivative 5h. This study strongly suggested that the optimal methoxy group at the 4-position on the indole in 5c was more important than the isopropoxy, isobutyloxy, or hydroxyl groups.
We tested the introduction of substitutions at the 3-position on the indole unit by starting with the lead compound 5d. The 3-methyl (5l; K i = 6.71 μM) or 3-ethyl (5m; K i = 7.51 μM) groups were introduced first; however, the inhibitory activities of both 5l and 5m were severely reduced relative to the activity of 5d. These results suggested that substitutions at the 3-position on the indole unit were not a fruitful direction of study.
The P3 indole unit in 5c was next examined by varying the heterocycle by replacing it with benzimidazole, benzothiazole, benzofuran, or indoline scaffolds. The compounds bearing a benzimidazole (5n; K i = 0.022, IC50 = 1.30 μM) exhibited 3-fold potent inhibitory activity than the lead compound 5c; however, the other indoline (5p; K i = 0.120 μM) or benzothiazole (5o; K i = 0.800 μM) or a benzofuran (5q; K i = 14.1 μM), were lower than the activity of the lead compound 5c. Compound 5q, bearing a benzofuran unit, displayed a severely reduced inhibitory potency compared to the lead compound 5c. The observed low activity of 5q was attributed to the disruption of hydrogen bonding interactions at the P3 position. This result suggested that the hydrogen bonding properties of the amine moiety of the indole unit were very important for achieving effective inhibitory activities (see Fig. 4 ).
Fig. 4.

Molecular docking pose and binding interactions of compounds 5h, 5c, 5q and 5r (orange sticks) bound to SARS-CoV 3CLpro (PDB ID: 1WOF). Only the residues (yellow color), which are engaged in binding to the ligands (orange sticks), are highlighted. Dotted black lines represent the hydrogen bonding interaction. (A) Mode of interactions of 5h, particularly the P3-indole-2-carbonyl to the Glu166 and Gln189 of 3CLpro, which was also observed for compound 5c (B); (C) The hydrogen bond between Glu-166 and P3-benzofuran moiety was omitted in the case of 5q; (D) The indole-3-carbonyl of 5r looses their hydrogen bonding interactions to Glu166 and Gln189. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The position of the carbonyl substitution on the indole unit in 5c was examined next. The inhibitory activity of the derivative (5r; K i = 0.683 μM) bearing an indole-3-carbonyl was reduced relative to the activity of the compound bearing an indole-2-carbonyl (5c; K i = 0.065 μM). The substitution may have interrupted a hydrogen bonding interaction with the protease (see Fig. 4). These results strongly suggested that the 2-carbonyl substitution on the indole unit in 5c was important to the inhibitory potency.
2.4. Molecular docking study
The binding mode of the most potent compound, 5h was computationally modeled using a three-dimensional structure of SARS-CoV 3CLpro based on the reported crystal structure [27]. A procedure similar to the procedure described previously was used here [12], [13], [14]. The molecular docking of 5h (orange sticks) was examined, in comparison with a lead compound 4 (blue sticks) and a structurally similar tripeptidic ligand (light blue sticks), the docking structure of which was elucidated by X-ray crystallography (PDB ID. 1WOF, K i = 10.7 μM, Fig. 3) [27]. Several minimization processes were performed using the MMFF94X force field to model the solvation environment surrounding the inhibitor. A molecular simulation was subsequently performed. As shown in Fig. 3, the 5h moieties P1′–P2 interacted with the same region of the protease as did the lead compound 4 and the original ligand. The heterocycle unit mimicked the P3 valine of the reported substrate, and the phenyl ring in the indole unit partially occupied the S4-pocket. The rigid P3 moiety (4-methoxyindole unit) of 5h appeared to occupy a region of the active site with an appropriate volume and in a conformation that was more favorable than the conformation of the structurally flexible N-(3-methoxyphenyl)glycyl moiety in 4. Indeed, the fit of the rigid P3 moiety of 5h may have contributed to the 65-fold higher inhibitory potency of 5h as compared to 4. The detailed interactions between 5h and the active site are shown in Fig. 4A. Hydrogen bonds between the amine moiety (-NH) of the indole unit and the backbone amino acid residue Glu166 of 3CLpro were observed with a bond length of 2.673 Å.
Fig. 3.
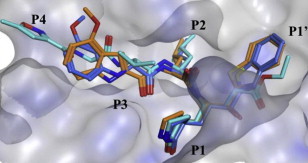
Molecular dynamics simulated pose of the compound 5h (orange stick) bound to SARS-CoV 3CLpro (PDB ID: 1WOF). (A) Overlapped view of 5h with an original vinyl ester (light blue stick) and the lead compound 4 (blue stick). The 5h moieties P1′–P2 interacted with the same region of the protease as the lead compound 4 and original ligand. The heterocycle unit mimics the P3-valine of the reported ligand and the phenyl ring in indole unit partially occupying the S4-pocket, respectively. The rigid P3-4-methoxyindole unit in 5h occupying with an appropriate volume and favorable conformation compared to the structurally flexible N-(3-methoxyphenyl)glycyl in 4. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Insight into the structure–activity relationships was sought by performing docking studies and analyzing the binding interactions of certain compounds that displayed notable variations in their inhibitory activities (Fig. 4B–D). The initial lead compound 5c, bearing a P3 indole-2-carbonyl unit, was docked (Fig. 4B), revealing hydrogen bonding interactions similar to those predicted for 5h (Fig. 4A); however, 5q, bearing a benzofuran-2-carbonyl P3 moiety, could not form a hydrogen bond interaction with the backbone amino acid residue Glu166 (Fig. 4C). The loss of hydrogen bond interactions could have reduced the inhibitory activity of compound 5q (K i = 14.1 μM) compared to the activity of compound 5c (K i = 0.065 μM). The compound 5r (K i = 0.683 μM), bearing an indole-3-carbonyl unit, engaged in two fewer hydrogen bonding interactions to the amino acid residues Glu166 and Gln189 of the protease relative to 5c, as shown in Fig. 4D. This study revealed that the optimal positioning of a carbonyl unit on the indole unit was very important for achieving a potent inhibitory activity.
2.5. Isothermal titration calorimetry study
In order to evaluate in-depth bending interaction between the most potent compound 5h and the SARS protease, the isothermal titration calorimetry study was demonstrated (Fig. 5 ). The titration was performed at 25 °C by injecting 10 μL aliquots of protease solution into the calorimetric cell (volume ∼ 1.4 mL) containing the inhibitor 5h at a concentration of 6 μM. The concentration of protease in the syringe was 109 μM. The heat evolved upon each injection of protease was obtained from the integral of the calorimetric signal. The heat associated with inhibitor binding was obtained by subtracting the heat of dilution from the heat of reaction. The individual heats were plotted against the molar ratio, and the enthalpy change (ΔH) and association constant (K a = 1/K d) were obtained by nonlinear regression of the data. The binding affinity of compound 5h was 16 nM which is in very good agreement with the K i value (0.006 μM).
Fig. 5.

Isothermal titration calorimetry of compound 5h.
3. Conclusion
We describe here the design, synthesis, and biological evaluation of a series of dipeptide-type inhibitors with novel P3 scaffolds against SARS-CoV 3CLpro. A docking study involving binding between the dipeptidic lead compound 4 and 3CLpro motivated the modification of a flexible P3 N-(3-methoxyphenyl)glycine in 4 to various structurally rigid moieties. This modification led to the identification of several potent derivatives, including 5c–k and 5n, which displayed inhibitory activities (K i or IC50) in the submicromolar to nanomolar range. Compounds 5c, 5f, 5h, 5k and 5n, in particular, exhibited the most potent inhibitory activities, with K i values of 0.065, 0.028, 0.006 0.026 and 0.022 μM, respectively. These compounds are attractive leads for a further development effort toward potent peptidomimetics with suitable pharmaceutical profiles. A SAR study around the P3 site in the lead compound 4 led to the identification of a rigid indole-2-carbonyl unit as one of the best P3 moieties (5c). Further optimization of 5c showed that an optimal methoxy substitution at the 4-position on the P3 indole unit enhanced the inhibitory activity significantly. The 2-carbonyl substitution on the P3 indole was also found to be important to the inhibitory potency against SARS-CoV 3CLpro.
4. Experimental section
4.1. Materials and methods
Reagents and solvents were purchased from Wako Pure Chemical Ind., Ltd. (Osaka, Japan) and Aldrich Chemical Co. Inc. (Milwaukee, WI) and were used without further purification. Analytical thin layer chromatography (TLC) was performed on Merck Silica Gel 60F254 pre-coated plates. Preparative HPLC was performed using a C18 reverse-phase column (19 × 100 mm; Sun-Fire Prep C18 OBD™, 5 μm) with a binary solvent system: a linear gradient of CH3CN in 0.1% aqueous TFA at a flow rate of 6 mL/min. Compounds were detected at 254 nm and 230 nm. All solvents used for HPLC were HPLC-grade. All other chemicals were of analytical grade or better. 1H and 13C NMR spectra were obtained using a JEOL 400 MHz spectrometer, a Varian Mercury 300 spectrometer (300 MHz), or a BRUKER AV600 spectrometer (600 MHz) with tetramethylsilane as an internal standard. Chemical shifts (δ) are expressed in ppm using solvent as an internal standard. The multiplicities of the resonance peaks are indicated as singlet (s), broad singlet (bs), doublet (d), triplet (t), quartet (q), and multiplet (m). The J values are given in Hz, and the relative number of protons was determined by integration. The solvent used for each spectrum is reported. High-resolution mass spectra (ESI or EI) were recorded on a micromass Q-Tof Ultima API or a JEOL JMS-GCmate BU-20 spectrometer. Mass spectra (ESI) were recorded on LCMS-2010EV (SHIMADZU).
4.2. Synthesis of methyl 4-hydroxy-1H -indole-2-carboxylate (7) [17]
To an ice-cold solution of methyl 4-methoxy-1H-indole-2-carboxylate 6 (0.850 g, 4.1 mmol) in DCM (10 mL) was added BBr3 (1.0 mL in DCM, 4.0 mmol). The solution was stirred for 1 h, and another equivalent (1.0 mL in DCM) of BBr3 was added. After stirring for another hour, the mixture was poured over crushed ice and the pH was adjusted to 7 by adding solid NaHCO3. The solution was extracted with DCM (60 mL), dried over Na2SO4, filtered, and evaporated under reduced pressure to give 7 [17].
4.3. Synthesis of 8a–r
The carboxylic acids 8a–h, 8k and 8n–r were commercially available.
4.3.1. Synthesis of 4-isopropoxy-1H-indole-2-carboxylic acid (8i) [16]
DEAD (0.227 mL, 2.9 mmol) was slowly added to a solution of 4-hydroxy-1H-indole-2-carboxylic acid methyl ester (7) (0.800 g, 4.0 mmol), triphenylphosphine (0.750 g, 2.9 mmol), and isopropanol (0.215 mL, 2.90 mmol) in 2 mL THF. Stirring was continued for 30 min, and the solvent was then evaporated. The crude mixture was purified by chromatography to give a pure methyl 4-isopropoxy-1H-indole-2-carboxylate, which was dissolved in 5 mL THF. A solution containing LiOH·H2O (2.94 mmol) in THF:H2O (10:1) was added, and the mixture was stirred overnight at 50 °C. The solvent was then evaporated, and the residue was partitioned between water and EtOAc. The water layer was acidified with 2 NHCl and extracted twice with EtOAc (50 mL × 2). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and evaporated to give 8i [16].
4.3.2. Synthesis of 4-isobutoxy-1H-indole-2-carboxylic acid (8j) [16]
Compound 8j was synthesized from 7 with isobutanol using methods similar to the method described for the preparation of 8i [16].
4.3.3. Synthesis of ethyl 3-formyl-5-methoxy-1H-indole-2-carboxylate (10a) [19]
A solution of ethyl 5-methoxy-1H-indole-2-carboxylate (9) (1.68 g, 7.6 mmol) in anhydrous N,N-dimethylformamide (3 mL) was added dropwise onto an ice-cooled solution of phosphorus oxychloride (1.73 mL, 15 mmol) in DMF (5 mL). The reaction mixture was heated at 110 °C for 2.5 h. After cooling, the reaction was quenched with ice water and made alkaline by the addition of a 2% sodium hydroxide solution. The precipitate was collected and purified by column chromatography to give 10a [19].
4.3.4. Synthesis of ethyl 3-acetyl-5-methoxy-1H-indole-2-carboxylate (10b) [20]
Ethyl 5-methoxy-1H-indole-2-carboxylate (9) (1.68 g, 7.6 mmol) was added to a mixture of acetyl chloride (0.55 mL, 7.6 mmol) and aluminum chloride (1.07 g, 7.6 mmol) in anhydrous 1,2-dichloroethane (10 mL). The reaction mixture was heated under reflux for 2.5 h. After cooling, the reaction mixture was poured over crushed ice and made acidic by the addition of 3 N HCl. The mixture was extracted with dichloromethane (20 mL × 2). The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by silica gel column chromatography to provide 10b [20].
4.3.5. Synthesis of ethyl 5-methoxy-3-methyl-1H-indole-2-carboxylate (11a)
A mixture of 10a (1.09 g, 4.40 mmol), triethylsilane (1.43 g, 16.0 mmol), and 2,2,2-trifluoroacetic acid (5.94 mL) was stirred at room temperature for 5 h. After quenching with a saturated solution of sodium carbonate, the mixture was extracted with ethyl acetate (20 mL × 2). The combined organic layers were washed with a saturated solution of sodium bicarbonate, brine, and then the solution was dried. After evaporating the solvent, the residue was purified by column chromatography to give ethyl 5-methoxy-3-methyl-1H-indole-2-carboxylate 11a. Yield 89% from 10a; yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.57 (br s, 1H), 7.29–7.25 (m, 1H merged with CDCl3), 7.03–6.98 (m, 2H), 4.41 (q, J = 7.2 Hz, 2H), 3.87 (s, 3H), 2.58 (s, 3H), 1.42 (t, J = 7.2 Hz, 3H). HRMS (ESI): m/z calcd for C13H16NO3 [M + H]+ 234.1130, found 234.1129.
4.3.6. Synthesis of ethyl 3-ethyl-5-methoxy-1H-indole-2-carboxylate (11b)
The compound 11b was synthesized from 10b using a procedure similar to that described for the preparation of 10a. Yellow solid; yield 65%; 1H NMR (400 MHz, CDCl3): δ 8.65 (br s, 1H), 7.27–7.20 (m, 1H merged with CDCl3), 7.06–7.04 (m, 1H), 7.00 (dd, J = 8.8, 2.4 Hz, 1H), 4.41 (q, J = 7.2 Hz, 2H), 3.87 (s, 3H), 3.08 (q, J = 7.5 Hz, 2H), 1.42 (t, J = 7.2 Hz, 3H), 1.29–1.23 (t, J = 7.5 Hz, 3H). HRMS (ESI): m/z calcd for C14H18NO3 [M + H]+ 248.1287, found 248.1285.
4.4. Synthesis of 5-methoxy-3-methyl-1H-indole-2-carboxylic acid (8l)
To a solution of the ethyl 5-methoxy-3-methyl-1H-indole-2-carboxylate 11a (0.100 g, 0.35 mmol) in THF (3 mL) at room temperature was added LiOH·H2O in water (0.102 g, 2.45 mmol). After 3 h stirring, the solvent was completely evaporated under reduced pressure, and the resulting residue was neutralized with 2 N HCl. The solution was extracted with EtOAc (20 mL × 2), dried over Na2SO4, filtered, and evaporated under reduced pressure to give the corresponding acids 8l. Yield 85%; brown solid; 1H NMR (400 MHz, CD3OD): δ 7.35–7.18 (m, 1H), 7.17–7.00 (m, 1H), 6.92–6.89 (m, 1H), 3.82–3.79 (m, 3H), 2.55 (s, 3H); HRMS (ESI): m/z calcd for C11H12NO3 [M + H]+ 206.0817, found 206.0818.
4.5. Synthesis of 3-ethyl-5-methoxy-1H-indole-2-carboxylic acid (8m)
The synthesis of 8m was achieved from 11b using a procedure similar to that described for the preparation of 8l. Yield 81%; brown solid; 1H NMR (400 MHz, CD3OD): δ 7.38–7.23 (m, 1H), 7.06 (s, 1H), 6.98 (d, J = 8.9 Hz, 1H), 3.88 (s, 3H), 3.11 (q, J = 7.4 Hz, 2H), 1.28 (t, J = 7.5 Hz, 3H); HRMS (ESI): m/z calcd for C12H14NO3 [M + H]+ 220.0974, found 220.0975.
4.6. General synthetic procedure for the preparation of 13a–r
To a solution of the commercially available l-leucine tert-butyl ester (12, 0.89 mmol) in DMF (15 mL) were added an appropriate carboxylic acid (8, 1.1 mmol), HOBt·H2O (1.1 mmol), and EDC·HCl (1.1 mmol). The resulting solution was cooled to 0 °C under ice bath conditions, and TEA was added dropwise. After 5 min, the ice bath was removed, and the mixture was allowed to stir for 2 h at ambient temperature. DMF was removed under high vacuum, and the resulting residue was dissolved in EtOAc (30 mL). The organic layer was washed with 5% citric acid (20 mL × 2), 5% NaHCO3 (20 mL × 2), and brine (20 mL). The solution was dried over Na2SO4, filtered, and evaporated under reduced pressure to give compound 13. The resulting crude compound was purified by silica gel column chromatography using hexane–EtOAc as eluents.
4.6.1. (2S)-tert-Butyl 4-methyl-2-(5-oxopyrrolidine-2-carboxamido)pentanoate (13a)
Yield 51% from 12 with 5-oxopyrrolidine-2-carboxylic acid (8a); white solid; 1H NMR (400 MHz, CDCl3): δ 4.58–4.44 (m, 1H), 4.21–4.16 (m, 1H), 2.60–2.48 (m, 1H), 2.47–2.38 (m, 1H), 2.37–2.25 (m, 1H), 2.24–2.12 (m, 1H), 1.65–1.51 (m, 3H), 1.45 (s, 9H), 0.95–0.83 (m, 6H); HRMS (ESI): m/z calcd for C15H27N2O4 [M + H]+ 299.1971, found 299.1973.
4.6.2. (S)-tert-Butyl 2-(1H-pyrrole-2-carboxamido)-4-methylpentanoate (13b)
Yield 63% from 12 with 1H-pyrrole-2-carboxylic acid (8b); white solid; 1H NMR (400 MHz, CDCl3): δ 6.94–6.89 (m, 1H), 6.65–6.62 (m, 1H), 6.35 (d, J = 8.3 Hz, 1H), 6.24–6.20 (m, 1H), 4.74–4.65 (m, 1H), 1.76–1.53 (m, 3H), 1.48 (s, 9H), 0.97 (t, J = 6.8 Hz, 6H); HRMS (ESI): m/z calcd for C15H25N2O3 [M + H]+ 281.1865, found 281.1862.
4.6.3. (S)-tert-Butyl 2-(1H-indole-2-carboxamido)-4-methylpentanoate (13c)
Yield 54% from 12 with 1H-indole-2-carboxylic acid (8c); yellow solid; 1H NMR (400 MHz, CDCl3): δ 9.10 (s, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.30–7.26 (m, 1H merged with CDCl3), 7.14 (t, J = 8.0 Hz, 1H), 6.92 (s, 1H), 6.60 (d, J = 8.4 Hz, 1H), 4.78–4.71 (m, 1H), 1.79–1.58 (m, 3H), 1.50 (s, 9H), 1.00 (t, J = 6.0 Hz, 6H); HRMS (ESI): m/z calcd for C19H27N2O3 [M + H]+ 331.2022, found 331.2012.
4.6.4. (S)-tert-Butyl 2-(5-methoxy-1H-indole-2-carboxamido)-4-methylpentanoate (13d)
Yield 61% from 12 with 5-methoxy-1H-indole-2-carboxylic acid (8d); fluorescent solid; 1H NMR (400 MHz, CDCl3): δ 9.33 (s, 1H), 7.49 (d, J = 8.7 Hz, 1H), 6.88–6.84 (m, 2H), 6.80 (dd, J = 8.8, 2.2 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 4.80–4.70 (m, 1H), 3.85 (s, 3H), 1.80–1.61 (m, 3H), 1.50 (s, 9H), 0.99 (t, J = 6.2 Hz, 6H); HRMS (ESI): m/z calcd for C20H29N2O4 [M + H]+ 361.2127, found 361.2116.
4.6.5. (S)-tert-Butyl 2-(5-hydroxy-1H-indole-2-carboxamido)-4-methylpentanoate (13e)
Yield 63% from 12 with 5-hydroxy-1H-indole-2-carboxylic acid (8e); yellow solid; 1H NMR (400 MHz, CD3OD): δ 7.97 (br s, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 8.8 Hz, 1H), 7.02 (s, 1H), 6.95 (d, J = 2.2 Hz, 1H), 6.81 (dd, J = 8.9, 2.4 Hz, 1H), 4.60–4.51 (m, 1H), 1.80–1.64 (m, 3H), 1.47 (s, 9H), 1.00 (d, J = 6.2 Hz, 3H), 0.96 (d, J = 6.3 Hz, 3H); HRMS (ESI): m/z calcd for C19H27N2O4 [M + H]+ 347.1971, found 347.1958.
4.6.6. (S)-tert-Butyl 2-(5-chloro-1H-indole-2-carboxamido)-4-methylpentanoate (13f)
Yield 72% from 12 with 5-chloro-1H-indole-2-carboxylic acid (8f); white solid; 1H NMR (400 MHz, CDCl3): δ 9.53 (s, 1H), 7.59 (s, 1H), 7.32 (d, J = 8.7 Hz, 1H), 7.22 (dd, J = 8.7, 1.9 Hz, 1H), 6.86–6.82 (m, 2H), 4.78–4.72 (m, 1H), 1.78–1.61 (m, 3H), 1.51 (s, 9H), 1.00 (t, J = 6.2 Hz, 6H); HRMS (ESI): m/z calcd for C19H26ClN2O3 [M + H]+ 365.1632, found 365.1631.
4.6.7. (S)-tert-Butyl 2-(6-methoxy-1H-indole-2-carboxamido)-4-methylpentanoate (13g)
Yield 67% from 12 with 6-methoxy-1H-indole-2-carboxylic acid (8g); white solid; 1H NMR (400 MHz, CDCl3): δ 9.14 (br s, 1H), 7.50 (d, J = 8.7 Hz, 1H), 6.85–6.82 (m, 2H), 6.81 (dd, J = 8.7, 2.3 Hz, 1H), 6.59 (d, J = 8.4 Hz, 1H), 4.78–4.71 (m, 1H), 3.86 (s, 3H), 1.80–1.62 (m, 3H), 1.49 (s, 9H), 0.99 (t, J = 6.3 Hz, 6H); HRMS (ESI): m/z calcd for C20H29N2O4 [M + H]+ 361.2127, found 361.2123.
4.6.8. (S)-tert-Butyl 2-(4-methoxy-1H-indole-2-carboxamido)-4-methylpentanoate (13h)
Yield 69% from 12 with 4-methoxy-1H-indole-2-carboxylic acid (8h); white solid; 1H NMR (400 MHz, CDCl3): δ 9.11 (br s, 1H), 7.20 (t, J = 8.0 Hz, 1H), 7.04–7.01 (m, 2H), 6.55 (br d, 1H), 6.51 (d, J = 8.0 Hz, 1H), 4.76–4.68 (m, 1H), 3.96 (s, 3H), 1.79–1.61 (m, 3H), 1.49 (s, 9H), 0.99 (t, J = 5.7 Hz, 6H); HRMS (ESI): m/z calcd for C20H29N2O4 [M + H]+ 361.2127, found 361.2125.
4.6.9. (S)-tert-Butyl 2-(4-isopropoxy-1H-indole-2-carboxamido)-4-methylpentanoate (13i)
Yield 72% from 12 with 4-isopropoxy-1H-indole-2-carboxylic acid (8i); viscous oil; 1H NMR (400 MHz, CDCl3): δ 8.98 (br s, 1H), 7.17 (t, J = 8.0 Hz, 1H), 7.05 (s, 1H), 6.98 (d, J = 8.2 Hz, 1H), 6.56–6.50 (m, 2H), 4.76–4.70 (m, 1H), 3.49 (s, 1H), 1.76–1.62 (m, 3H), 1.49 (s, 9H), 1.42–1.41 (m, 6H), 0.99 (t, J = 6.2 Hz, 6H); HRMS (ESI): m/z calcd for C22H33N2O4 [M + H]+ 389.2440 found 389.2447.
4.6.10. (S)-tert-Butyl 2-(4-isobutoxy-1H-indole-2-carboxamido)-4-methylpentanoate (13j)
Yield 70% from 12 with 4-isobutoxy-1H-indole-2-carboxylic acid (8j); yellow solid; 1H NMR (400 MHz, CDCl3): δ 9.09 (br s, 1H), 7.18 (t, J = 8.0 Hz, 1H), 7.06 (s, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.49 (d, J = 7.8 Hz, 1H), 4.77–4.73 (m, 1H), 3.87 (d, J = 6.5 Hz, 2H), 2.23–2.15 (m, 1H), 1.80–1.62 (m, 3H), 1.50 (s, 9H), 1.11 (s, 3H), 1.09 (s, 3H), 0.99 (t, J = 5.8 Hz, 6H); HRMS (ESI): m/z calcd for C23H35N2O4 [M + H]+ 403.2597, found 403.2613.
4.6.11. (S)-tert-Butyl 2-(4-hydroxy-1H-indole-2-carboxamido)-4-methylpentanoate (13k)
Yield 55% from 12 with 4-hydroxy-1H-indole-2-carboxylic acid (8k); yellow solid; 1H NMR (400 MHz, CDCl3): δ 9.06 (br s, 1H), 7.23 (s, 1H), 7.11 (t, J = 7.8 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 6.82 (d, J = 8.9 Hz, 1H), 6.50 (d, J = 7.5 Hz, 1H), 4.85–4.76 (m, 1H), 1.76–1.62 (m, 3H), 1.53 (s, 9H), 1.00–0.94 (m, 6H); HRMS (ESI): m/z calcd for C19H26N2O4Na [M + Na]+ 369.1790, found 369.1783.
4.6.12. (S)-tert-Butyl 2-(5-methoxy-3-methyl-1H-indole-2-carboxamido)-4-methylpentanoate (13l)
Yield 63% from 12 with 5-methoxy-3-methyl-1H-indole-2-carboxylic acid (8l); yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.83 (br s, 1H), 7.27–7.25 (m, 1H merged with CDCl3), 7.02–6.99 (m, 1H), 6.95 (dd, J = 8.9, 2.4 Hz, 1H), 6.54 (d, J = 8.0 Hz, 1H), 4.80–4.73 (m, 1H), 3.88 (s, 3H), 2.60 (s, 3H), 1.80–1.63 (m, 3H), 1.51 (s, 9H), 1.00 (t, J = 6.3 Hz, 6H); HRMS (ESI): m/z calcd for C21H30N2O4Na [M + Na]+ 397.2103 found 397.2097.
4.6.13. (S)-tert-Butyl 2-(3-ethyl-5-methoxy-1H-indole-2-carboxamido)-4-methylpentanoate (13m)
Yield 65% from 12 with 3-ethyl-5-methoxy-1H-indole-2-carboxylic acid (8m); yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.92 (br s, 1H), 7.29–7.25 (m, 1H merged with CDCl3), 7.02–7.00 (m, 1H), 6.96 (dd, J = 8.9, 2.5 Hz, 1H), 6.58 (d, J = 8.0 Hz, 1H), 4.80–4.74 (m, 1H), 3.87 (s, 3H), 3.08–2.97 (m, 2H), 1.80–1.64 (m, 3H), 1.50 (s, 9H), 1.38 (t, J = 7.7 Hz, 3H), 1.00 (t, J = 5.9 Hz, 6H); HRMS (ESI): m/z calcd for C22H32N2O4Na [M + Na]+ 411.2260, found 411.2260.
4.6.14. (S)-tert-Butyl 2-(1H-benzo[d]imidazole-2-carboxamido)-4-methylpentanoate (13n)
Yield 74% from 12 with 1H-benzo[d]imidazole-2-carboxylic acid (8n); white solid; 1H NMR (400 MHz, CDCl3): δ 11.3 (br s, 1H), 7.91 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.58 (d, J = 7.2 Hz, 1H), 7.40–7.30 (m, 2H), 4.80–4.72 (m, 1H), 1.82–1.64 (m, 3H), 1.49 (s, 9H), 1.02–0.98 (m, 6H); HRMS (ESI): m/z calcd for C18H26N3O3 [M + H]+ 332.1974, found 332.1980.
4.6.15. (S)-tert-Butyl 2-(benzo[d]thiazole-2-carboxamido)-4-methylpentanoate (13o)
Yield 45% from 12 with benzo[d]thiazole-2-carboxylic acid (8o); white viscous oil; 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 8.1 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 8.7 Hz, 1H), 7.55 (t, J = 7.1 Hz, 1H), 7.49 (t, J = 7.0 Hz, 1H), 4.78–4.71 (m, 1H), 1.80–1.66 (m, 3H), 1.50 (s, 9H), 1.02–0.98 (m, 6H); HRMS (ESI): m/z calcd for C18H25N3O3S [M + H]+ 349.1586, found 349.1596.
4.6.16. tert-Butyl 2-(indoline-2-carboxamido)-4-methylpentanoate (13p)
Yield 69% from 12 with indoline-2-carboxylic acid (8p); white solid; 1H NMR (400 MHz, CDCl3): δ 7.41 (br d, 1H), 7.31 (br d, 1H), 7.13–7.04 (m, 2H), 6.83–6.76 (m, 1H), 6.75–6.72 (m, 1H), 4.60–4.40 (m, 2H), 3.63–3.50 (m, 1H), 3.15–3.00 (m, 1H), 1.75–1.64 (m, 3H), 1.42 (s, 9H), 1.01–0.98 (m, 6H); HRMS (ESI): m/z calcd for C19H29N2O3 [M + H]+ 333.2178, found 333.2174.
4.6.17. (S)-tert-Butyl 2-(benzofuran-2-carboxamido)-4-methylpentanoate (13q)
Yield 81% from 12 with benzofuran-2-carboxylic acid (8q); white solid; 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.47 (s, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 7.04 (d, J = 8.5 Hz, 1H), 4.80–4.71 (m, 1H), 1.80–1.60 (m, 3H), 1.50 (s, 9H), 1.00 (t, J = 6.3 Hz, 6H); HRMS (ESI): m/z calcd for C19H26NO4 [M + H]+ 332.1862, found 332.1857.
4.6.18. (S)-tert-Butyl 2-(1H-indole-3-carboxamido)-4-methylpentanoate (13r)
Yield 87% from 12 from 1H-indole-3-carboxylic acid (8r); white solid; 1H NMR (400 MHz, CDCl3): δ 8.19 (br s, 1H), 7.57 (d, J = 7.5 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.26–7.12 (m, 3H), 5.98 (br d, 1H), 4.55–4.48 (m, 1H), 1.52–1.30 (m, 3H), 1.37 (s, 9H), 0.86–0.80 (m, 6H); HRMS (ESI): m/z calcd for C19H27N2O3 [M + H]+ 331.2022, found 331.2028.
4.7. General synthetic procedure for the preparation of 14a–r
To a solution of the corresponding tert-butyl ester 13 (2 mmol) in CH2Cl2 (2 mL) at 0 °C was added TFA/H2O (10:1, 3 mL). After 5 min stirring, the reaction mixture was allowed to stir at room temperature for 1 h. The solvent was completely evaporated under reduced pressure to give the corresponding acids 14, which were directly used in the subsequent step without further characterization.
4.8. Synthetic procedure for the preparation of tert-butyl ((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl) carbamate (18)
Compound 16 was prepared through sequential reactions from the intermediate 15, as reported previously [12], [13], [14].
To a solution containing the acid 16 (0.540 g, 1.8 mmol) in DMF (30 mL) were added EDC·HCl (0.418 g, 2.1 mmol), HOBt·H2O (0.334 g, 2.1 mmol), and N,O-dimethylhydroxylamine (0.213 g, 2.1 mmol) at room temperature. The solution was cooled to 0 °C, and TEA (0.304 mL, 2.1 mmol) was then added slowly. After 2 h, the DMF was evaporated, and the resulting residue was dissolved in ethyl acetate (100 mL). The organic phase was subsequently washed with 5% citric acid (20 mL × 2), 5% NaHCO3 (20 mL × 2), and brine (50 mL). The organic layer was then dried over Na2SO4 and concentrated under reduced pressure to yield the Weinreb amide derivative 17, which was purified by column chromatography (EtOAc/MeOH = 9.5:0.5).
To a solution of benzothiazole (10.0 mmol) in THF (20 mL) at −78 °C was added n-BuLi (2.0 M in THF, 1.67 mL) dropwise over 20 min. After 1 h stirring, the Weinreb amide 17 (0.640 g, 2.0 mmol) in THF (10 mL) was slowly added over 10 min, and the solution was stirred for 3 h. The reaction was quenched with sat. NH4Cl and allowed to stir at 0 °C for 20 min. The mixture was evaporated and dissolved in EtOAc (100 mL). This solution was washed with water (50 mL) and brine (40 mL), and then dried over Na2SO4. The organic layer was concentrated under reduced pressure, and the resulting residue was subjected to flash chromatography (EtOAc/MeOH = 9:1) to obtain the pure compound 18.
The characterization data for the compounds 17 and 18 are reported elsewhere [12], [13], [14].
4.8.1. Synthetic procedure for the preparation of N-((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-5-oxopyrrolidine-2-carboxamide (5a)
To a solution of 18 (0.200 g, 0.5 mmol) in CH2Cl2 (3 mL) at 0 °C was added TFA/H2O (10:1, 2 mL), and the solution was stirred for 1 h. After evaporating the solvent under reduced pressure, the corresponding deprotected lactam residue (0.100 g, 0.53 mmol) was coupled to the carboxylic acid 14a (0.136 g, 0.38 mmol) using the coupling agent HBTU (0.147 g, 0.38 mmol) in the presence of diisopropylethylamine (0.050 mL, 0.38 mmol) in DMF (3 mL) at 0 °C. After 5 min stirring, the ice bath was removed, and the solution was allowed to stir for 2 h under ambient conditions. The solvent was then evaporated under high vacuum, and the residue was dissolved in ethyl acetate (50 mL). The organic layer was washed with 5% citric acid (20 mL × 2), 5% NaHCO3 (20 mL × 2), and brine (25 mL). The solution was dried over Na2SO4, filtered, and evaporated under reduced pressure to give compound 5a. Yield 43%; white solid; 1H NMR (400 MHz, CD3OD): δ 8.20 (d, J = 8.4 Hz, 1H), 8.12 (d, J = 7.8 Hz, 1H), 7.68–7.55 (m, 2H), 5.75–5.65 (m, 1H), 4.50–4.40 (m, 1H), 4.30–4.20 (m, 1H), 3.40–3.28 (m, 2H merged with CD3OD), 2.80–2.60 (m, 1H), 2.59–2.18 (m, 5H), 2.17–2.01 (m, 2H), 2.00–1.75 (m, 1H), 1.74–1.40 (m, 3H), 1.00–0.86 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.5, 182.0, 181.6, 175.1, 165.5, 154.8, 138.4, 129.3, 128.5, 126.5, 123.7, 58.3, 55.7, 53.3, 41.7, 40.3, 39.5, 33.6, 30.0, 29.1, 26.8, 26.5, 25.9, 23.3, 21.9; HRMS (ESI): m/z calcd for C25H32N5O5S [M + H]+ 514.2124 found 514.2115.
The compounds 5b–r was synthesized from 18 with the 14b–r using a procedure similar to that described for the preparation of 5a.
4.8.2. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-1H-pyrrole-2-carboxamide (5b)
Yield 38%; white solid; 1H NMR (400 MHz, CD3OD): δ 8.14–8.05 (m, 2H), 7.62–7.54 (m, 2H), 6.95–6.85 (m, 2H), 6.18–6.15 (m, 2H), 5.72–5.65 (m, 1H), 4.70–4.42 (m, 1H), 3.36–3.10 (m, 2H merged with CD3OD), 2.80–2.56 (m, 1H), 2.54–2.40 (m, 1H), 2.39–2.20 (m, 1H), 2.19–1.99 (m, 2H), 1.98–1.60 (m, 3H), 1.00–0.85 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.5, 181.8, 175.6, 165.4, 163.5, 154.7, 138.3, 129.3, 128.4, 126.5, 126.4, 123.7, 123.3, 112.8, 110.3, 55.6, 53.1, 41.9, 41.6, 40.0, 34.0, 28.8, 26.0, 23.4, 22.1; HRMS (ESI): m/z calcd for C25H30N5O4S [M + H]+ 496. 2019 found 496. 2011.
4.8.3. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-1H-indole-2-carboxamide (5c)
Yield 51%; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.10–8.06 (m, 2H), 7.63–7.54 (m, 3H), 7.47–7.38 (m, 1H), 7.23–7.15 (m, 2H), 7.14–7.04 (m, 1H), 5.70–5.67 (m, 1H), 4.75–4.68 (m, 1H), 3.49–3.29 (m, 2H), 2.78–2.62 (m, 1H), 2.61–2.41 (m, 1H), 2.39–2.24 (m, 1H), 2.23–2.03 (m, 2H), 2.02–1.72 (m, 3H), 0.98–0.88 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 175.4, 165.4, 164.0, 154.7, 138.2, 131.6, 129.2, 128.9, 128.4, 126.4, 125.2, 123.6, 122.2, 121.2, 113.0, 105.3, 55.7, 53.4, 41.8, 41.5, 40.0, 33.9, 28.8, 26.0, 23.3, 22.1; HRMS (ESI): m/z calcd for C29H32N5O4S [M + H]+ 546.2175 found 546.2157.
4.8.4. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-5-methoxy-1H-indole-2-carboxamide (5d)
Yield 46%; light yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.10–8.06 (m, 1H), 8.00–7.96 (m, 1H), 7.56–7.48 (m, 2H), 7.07–7.00 (m, 2H), 6.96–6.92 (m, 2H), 5.82–5.68 (m, 1H), 4.80–4.75 (m, 1H), 3.85 (s, 3H), 3.44–3.26 (m, 2H), 2.70–2.42 (m, 2H), 2.41–2.14 (m, 1H), 2.13–1.90 (m, 2H), 1.89–1.64 (m, 3H), 0.98–0.88 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.5, 181.8, 175.4, 165.6, 164.0, 155.8, 154.7, 138.3, 133.8, 132.0, 129.2, 128.3, 127.1, 126.4, 123.6, 116.7, 113.9, 105.2, 103.2, 56.2, 55.7, 53.4, 41.5, 40.0, 33.9, 29.3, 28.8, 26.1, 23.4, 22.0; HRMS (ESI): m/z calcd for C30H34N5O5S [M + H]+ 576.2281 found 576.2294.
4.8.5. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-5-hydroxy-1H-indole-2-carboxamide (5e)
Yield 37%; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.08–8.04 (m, 2H), 7.57–7.50 (m, 2H), 7.30–7.23 (m, 1H), 7.02–6.93 (m, 2H), 6.81 (dd, J = 8.9, 2.3 Hz, 1H), 5.70–5.65 (m, 1H), 4.75–4.65 (m, 1H), 3.40–3.21 (m, 2H merged with CD3OD), 2.80–2.60 (m, 1H), 2.59–2.40 (m, 1H), 2.39–2.22 (m, 1H), 2.21–1.93 (m, 2H), 1.92–1.62 (m, 3H), 1.00–0.90 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 175.4, 165.6, 165.4, 164.0, 154.7, 152.3, 138.2, 133.5, 132.7, 129.6, 128.4, 128.3, 126.5, 123.6, 116.2, 113.6, 105.8, 104.6, 55.7, 53.4, 41.8, 41.6, 40.0, 34.0, 28.8, 26.1, 23.3, 22.1; HRMS (ESI): m/z calcd for C29H32N5O5S [M + H]+ 562.2124 found 562.2133.
4.8.6. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-5-chloro-1H-indole-2-carboxamide (5f)
Yield 41%; white solid; 1H NMR (400 MHz, CD3OD): δ 8.00–7.93 (m, 2H), 7.50–7.40 (m, 3H), 7.28 (t, J = 8.0 Hz, 1H), 7.06 (dd, J = 8.8, 2.0 Hz, 1H), 7.05–6.97 (m, 1H), 5.62–5.56 (m, 1H), 4.65–4.55 (m, 1H), 3.30–3.12 (m, 2H merged with CD3OD), 2.70–2.62 (m, 1H), 2.52–2.30 (m, 2H), 2.29–2.13 (m, 1H), 2.11–1.83 (m, 2H), 1.82–1.55 (m, 2H), 0.90–0.80 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.5, 181.8, 175.3, 165.6, 163.5, 154.7, 138.3, 136.6, 133.2, 129.8, 128.4, 128.3, 126.7, 125.3, 123.6, 121.9, 114.4, 104.7, 104.6, 55.6, 53.4, 41.8, 41.5, 40.0, 33.9, 28.8, 26.0, 23.3, 22.1; HRMS (ESI): m/z calcd for C29H31ClN5O4S [M + H]+ 580.1785 found 580.1795.
4.8.7. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-6-methoxy-1H-indole-2-carboxamide (5g)
Yield 38%; white solid; 1H NMR (400 MHz, CDCl3): δ 8.97 (br d, 1H), 8.72 (br d, 1H), 8.18–8.06 (m, 1H), 8.00–7.92 (m, 1H), 7.58–7.44 (m, 2H), 7.00–6.79 (m, 1H), 6.78–6.75 (m, 1H), 6.40–6.25 (m, 1H), 5.85–5.68 (m, 1H), 4.93–4.80 (m, 1H), 3.87 (s, 3H), 3.39–3.13 (m, 2H), 2.70–2.55 (m, 1H), 2.54–2.40 (m, 1H), 2.39–2.29 (m, 1H), 2.28–2.04 (m, 2H), 2.03–1.92 (m, 1H), 1.90–1.66 (m, 3H), 0.96–0.80 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 181.5, 175.6, 165.2, 164.1, 159.6, 155.9, 139.4, 138.9, 130.5, 129.2, 128.4, 126.4, 123.6 (2 carbons), 123.2, 112.7, 105.8, 94.8, 55.8, 55.7, 53.3, 41.8, 41.5, 40.0, 34.0, 28.8, 26.0, 23.3, 22.1; HRMS (ESI): m/z calcd for C30H34N5O5S [M + H]+ 576.2281 found 576.2268.
4.8.8. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-4-methoxy-1H-indole-2-carboxamide (5h)
Yield 45%; light yellow solid; 1H NMR (400 MHz, CDCl3): δ 9.65 (br s, 1H), 8.89 (br s, 1H), 8.57 (br s, 1H), 8.08–8.06 (m, 1H), 7.98–7.94 (m, 1H), 7.56–7.49 (m, 2H), 7.25–6.81 (m, 4H), 6.75–6.43 (m, 1H), 5.99–5.60 (m, 1H), 5.00–4.80 (m, 1H), 3.92 (s, 3H), 3.45–3.10 (m, 2H), 2.98–1.64 (m, 8H), 0.99–0.83 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 181.7, 175.3, 165.4, 164.0, 154.7, 152.8, 140.3, 138.2, 130.1, 129.2, 128.4, 126.49, 126.43, 123.6, 119.6, 104.8, 104.5, 103.7, 55.7, 53.4, 41.8, 41.5, 40.0, 34.0, 30.6, 28.8, 26.0, 23.3, 22.1; HRMS (ESI): m/z calcd for C30H34N5O5S [M + H]+ 576.2281 found 576.2264.
4.8.9. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-4-isopropoxy-1H-indole-2-carboxamide (5i)
Yield 49%; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.07–8.01 (m, 2H), 7.56–7.44 (m, 2H), 7.34–7.25 (m, 1H), 7.20–7.09 (m, 1H), 7.07–6.95 (m, 1H), 6.60–6.48 (m, 1H), 5.72–5.62 (m, 1H), 4.78–4.60 (m, 2H), 3.40–3.18 (m, 2H), 2.80–2.65 (m, 1H), 2.64–2.40 (m, 1H), 2.39–2.20 (m, 1H), 2.19–2.00 (m, 2H), 1.99–1.62 (m, 3H), 1.40–1.27 (m, 6H), 0.99–0.83 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 175.4, 165.4, 164.0, 154.6, 153.7, 140.1, 138.2, 130.2, 129.2, 128.3, 126.4, 126.2, 123.6, 121.3, 106.0, 103.36, 103.32, 71.1, 55.7, 53.4, 41.8, 41.5, 40.0, 34.0, 28.7, 26.0, 23.4, 22.5 (2 carbons), 22.1; HRMS (ESI): m/z calcd for C32H38N5O5S [M + H]+ 604.2594 found 604.2603.
4.8.10. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-4-isobutoxy-1H-indole-2-carboxamide (5j)
Yield 52%; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.04–8.00 (m, 2H), 7.52–7.44 (m, 2H), 7.35–7.20 (m, 1H), 7.10 (t, J = 8.0 Hz, 1H), 7.01–6.97 (m, 1H), 6.50–6.42 (m, 1H), 5.75–5.65 (m, 1H), 4.75–4.65 (m, 1H), 3.87–3.82 (m, 2H), 3.40–3.20 (m, 2H), 2.76–2.47 (m, 1H), 2.45–2.28 (m, 2H), 2.27–2.15 (m, 1H), 2.14–2.00 (m, 2H), 1.99–1.65 (m, 3H), 1.09–1.06 (m, 6H), 0.99–0.83 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 175.5, 165.4, 164.0, 155.1, 154.6, 139.8, 138.2, 130.1, 129.2, 128.3, 126.4, 126.3, 123.6, 120.3, 106.0, 103.1, 101.2, 75.3, 55.7, 53.4, 41.8, 41.5, 40.0, 34.1, 29.7, 28.7, 26.0, 23.4, 22.1, 19.7 (2 carbons); HRMS (ESI): m/z calcd for C33H40N5O5S [M + H]+ 618.2750 found 618.2767.
4.8.11. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-4-hydroxy-1H-indole-2-carboxamide (5k)
Yield 51%; white solid; 1H NMR (400 MHz, CD3OD): δ 8.00–7.95 (m, 2H), 7.50–7.38 (m, 2H), 7.24–7.12 (m, 1H), 6.97–6.91 (m, 1H), 6.85–6.78 (m, 1H), 6.31 (d, J = 7.6 Hz, 1H) 5.62–5.54 (m, 1H), 4.62–4.55 (m, 1H), 3.35–3.10 (m, 2H merged with CD3OD), 2.70–2.53 (m, 1H), 2.51–2.21 (m, 2H), 2.20–2.12 (m, 2H), 2.10–1.83 (m, 1H), 1.82–1.58 (m, 3H), 0.90–0.82 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 175.4, 165.4, 154.7, 152.8, 140.3, 138.2, 130.1, 129.2, 128.4, 126.5, 126.49, 126.43, 123.6, 119.6, 104.8, 104.5, 103.1, 55.7, 53.4, 41.8, 41.5, 40.0, 34.0, 28.8, 26.0, 23.3, 22.1; HRMS (ESI): m/z calcd for C29H32N5O5S [M + H]+ 562.2124, found 562.2122.
4.8.12. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-5-methoxy-3-methyl-1H-indole-2-carboxamide (5l)
Yield 46%; white solid; 1H NMR (400 MHz, CDCl3): δ 8.56 (br s, 1H), 7.90–7.84 (m, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.60–7.49 (m, 2H), 7.35–7.22 (m, 1H merged with CDCl3), 7.14–7.11 (m, 1H), 6.99–6.87 (m, 1H), 5.75–5.65 (m, 1H), 4.90–4.81 (m, 1H), 3.86 (s, 3H), 3.49–3.34 (m, 2H), 2.70–2.59 (m, 1H), 2.58–2.29 (m, 3H), 2.47 (s, 3H), 2.28–2.12 (m, 1H), 2.08–1.91 (m, 1H), 1.93–1.67 (m, 2H), 1.03–0.88 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.1, 181.2, 175.7, 165.4, 155.5, 152.8, 140.3, 138.2, 133.1, 129.2, 128.7, 126.4, 123.7, 122.7, 117.5, 116.9, 113.9, 101.5, 56.5, 56.0, 41.5, 40.2, 33.5, 29.4, 28.3, 25.5, 24.9, 23.4, 22.1, 18.7, 15.9; HRMS (ESI): m/z calcd for C31H36N5O5S [M + H]+ 590.2437 found 590.2443.
4.8.13. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-3-ethyl-5-methoxy-1H-indole-2-carboxamide (5m)
Yield 41%; white solid; 1H NMR (400 MHz, CDCl3): δ 8.00–7.97 (m, 1H), 7.84–7.74 (m, 1H), 7.55–7.47 (m, 2H), 7.00–6.85 (m, 2H), 6.70–6.52 (m, 1H), 5.91–5.68 (m, 1H), 4.90–4.85 (m, 1H), 3.86 (s, 3H), 3.49–3.38 (m, 2H), 3.00–2.73 (m, 2H), 2.72–2.58 (m, 1H), 2.57–2.38 (m, 1H), 2.37–2.01 (m, 4H), 2.00–1.68 (m, 2H), 1.40–1.21 (m, 3H), 1.02–0.88 (m, 6H); HRMS (ESI): m/z calcd for C32H38N5O5S [M + H]+ 604.2594 found 604.2607.
4.8.14. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-1H-benzo[d]imidazole-2-carboxamide (5n)
Yield 49%; white solid; 1H NMR (400 MHz, CD3OD): δ 8.15–8.07 (m, 2H), 7.70–7.68 (m, 2H), 7.57–7.53 (m, 2H), 7.43–7.38 (m, 2H), 5.73–5.68 (m, 1H), 4.73–4.70 (m, 1H), 3.40–3.28 (m, 2H merged with CD3OD), 2.80–2.66 (m, 1H), 2.65–2.35 (m, 2H), 2.34–2.20 (m, 1H), 2.19–1.95 (m, 2H), 1.93–1.70 (m, 2H), 1.00–0.96 (m, 6H); HRMS (ESI): m/z calcd for C28H31N6O4S [M + H]+ 547.2127, found 547.2127.
4.8.15. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)benzo[d]thiazole-2-carboxamide (5o)
Yield 43%; white solid; 1H NMR (400 MHz, CD3OD): δ 8.03–7.95 (m, 4H), 7.51–7.40 (m, 4H), 5.62–5.58 (m, 1H), 4.64–4.51 (m, 1H), 3.30–3.12 (m, 2H merged with CD3OD), 2.70–2.61 (m, 1H), 2.50–2.30 (m, 2H), 2.29–2.10 (m, 1H), 2.09–1.74 (m, 2H), 1.73–1.64 (m, 2H), 0.90–0.84 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.3, 181.8, 174.6, 165.4, 164.3, 161.7, 154.7, 154.4, 138.3, 138.1, 129.2, 128.4, 128.2, 128.1, 126.4, 125.5, 123.7, 123.6, 55.6, 53.7, 42.1, 41.5, 40.0, 33.8, 28.7, 26.0, 23.3, 22.1; HRMS (ESI): m/z calcd for C28H30N5O4S2 [M + H]+ 564.1739, found 564.1741.
4.8.16. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)indoline-2-carboxamide (5p)
Yield 43%; white solid; 1H NMR (400 MHz, CD3OD): δ 8.18–8.08 (m, 2H), 7.68–7.50 (m, 2H), 7.49–7.25 (m, 1H), 7.24–7.14 (m, 2H), 7.08–6.99 (m, 1H), 5.72–5.63 (m, 1H), 4.85–4.75 (m, 1H), 4.74–4.49 (m, 1H), 3.72–3.47 (m, 1H), 3.43–3.10 (m, 1H), 2.85–2.60 (m, 2H), 2.59–2.32 (m, 2H), 2.30–2.18 (m, 1H), 2.16–2.00 (m, 2H), 1.99–1.55 (m, 3H), 1.00–0.90 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 175.4, 165.4, 164.0, 154.7, 138.4, 138.2, 131.6, 129.2, 128.9, 128.4, 126.4, 125.2, 123.6, 122.8, 121.2, 113.0, 105.3, 55.7, 53.4, 41.8, 41.5, 40.0, 33.9, 28.8, 26.0, 23.3, 22.1; HRMS (ESI): m/z calcd for C29H34N5O4S [M + H]+ 548.2332 found 548.2328.
4.8.17. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)benzofuran-2-carboxamide (5q)
Yield 51%; white solid; 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 7.2 Hz, 1H), 7.70–7.64 (m, 1H), 7.61–7.53 (m, 2H), 7.51–7.39 (m, 1H), 7.38–7.26 (m, 3H), 7.24–7.16 (m, 2H), 5.72–5.56 (m, 1H), 4.83–4.79 (m, 1H), 3.49–3.30 (m, 2H), 2.70–2.55 (m, 1H), 2.54–2.22 (m, 2H), 2.21–2.00 (m, 2H), 1.99–1.70 (m, 3H), 1.04–0.99 (m, 6H); HRMS (ESI): m/z calcd for C29H31N4O5S [M + H]+ 547.2015 found 547.2019.
4.8.18. N-((S)-1-(((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-1H-indole-3-carboxamide (5r)
Yield 43%; light yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.20–8.06 (m, 1H), 7.98 (d, J = 7.2 Hz, 1H), 7.63–7.51 (m, 2H), 7.40–7.33 (m, 1H), 7.29–7.11 (m, 4H merged with CDCl3), 5.80–5.66 (m, 1H), 4.65–4.50 (m, 1H), 3.49–3.29 (m, 2H), 2.60–2.42 (m, 2H), 2.41–2.23 (m, 1H), 2.22–1.95 (m, 2H), 1.85–1.27 (m, 3H), 0.98–0.88 (m, 6H); 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 175.4, 165.4, 164.0, 154.7, 138.4, 138.2, 131.6, 129.2, 128.9, 128.4, 126.4, 125.2, 123.6, 122.8, 121.2, 113.0, 105.3, 55.7, 53.4, 41.8, 41.5, 40.0, 33.9, 28.8, 26.0, 23.3, 22.1; HRMS (ESI): m/z calcd for C29H31N5O4SNa [M + Na]+ 568.1994 found 568.1990.
5. Molecular docking study
The crystal structure of the SARS-CoV 3CLpro protease in complex with a substrate analog inhibitor (coded 1WOF) [27] was obtained from the Protein Data Bank (PDB; http://www.rcsb.org/pdb/home/home.do). Initially, a binding model of 5h with 3CLpro was simulated to form a basis for comparison with our previous lead 4 and substrate analogs, using molecular operating environment (MOE) software. Several minimization processes were performed using the MMFF94X force field to model the solvation environment surrounding the inhibitor. Structures having a relatively low binding free energy and a high number of cluster members were selected for the subsequent docking conformation optimization step. The minimized energies of 5h, obtained from the docking study, were −41.49 and −37.51 kcal/mol. The substrate analog was removed from the crystal structure and the docking studies were performed using 5c, 5q, and 5r according to a method similar to the method described for the 5h docking study.
6. Isothermal titration calorimetry
The binding of 5h was studied by isothermal titration calorimetry (ITC) using a VP-ITC microcalorimeter from MicroCal/GE Healthcare (Northampton, MA, USA). SARS-CoV 3CLpro and the inhibitor were dissolved in buffer composed of 10 mM sodium phosphate, pH 7.4, 10 mM NaCl, 1 mM TCEP, and 1 mM EDTA with 2% DMSO. The titration was performed at 25 °C by injecting 10 μL aliquots of protease solution into the calorimetric cell (volume ∼ 1.4 mL) containing the inhibitor at a concentration of 6 μM. The concentration of protease in the syringe was 109 μM. The heat evolved upon each injection of protease was obtained from the integral of the calorimetric signal. The heat associated with inhibitor binding was obtained by subtracting the heat of dilution from the heat of reaction. The individual heats were plotted against the molar ratio, and the enthalpy change (ΔH) and association constant (K a = 1/K d) were obtained by nonlinear regression of the data (see Fig. 4).
Acknowledgments
This research was supported by Grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, including a Grant-in-aid for Young Scientists (Tokubetsu Kenkyuin Shorei-hi) 23·01104 and a Grant-in-aid for Scientific Research 23659059. E.F. acknowledges support from the National Institutes of Health (Grants GM57144 and GM56550) and from the National Science Foundation (MCB-1157506).
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2013.07.037.
Appendix A. Supplementary data
References
- 1.He J.-F., Peng G.-W., Min J., Yu D.-W., Liang W.-L., Zhang S.-Y., Xu R.-H., Zheng H.-Y., Wu X.-W., Xu J., Wang Z.-H., Fang L., Zhang X., Li H., Yan X.-G., Lu J.-H., Hu Z.-H., Huang J.-C., Wan Z.-Y., Hou J.-L., Lin J.-Y., Song H.-D., Wang S.-Y., Zhou X.-J., Zhang G.-W., Gu B.-W., Zheng H.-J., Zhang X.-L., He M., Zheng K., Wang B.-F., Fu G., Wang X.-N., Chen S.-J., Chen Z., Hao P., Tang H., Ren S.-X., Zhong Y., Guo Z.-M., Liu Q., Miao Y.-G., Kong X.-Y., He W.-Z., Li Y.-X., Wu C.-I., Zhao G.-P., Chiu R.W.K., Chim S.S.C., Tong Y.-K., Chan P.K.S., Tam J.S., Lo Y.M.D. Science. 2004;303:1666–1669. [Google Scholar]
- 2.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.-E., Humphrey C.D., Shieh W.-J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.-Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Drosten C., Guenther S., Preiser W., Van Der Werf S., Brodt H.-R., Backer S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A.M., Berger A., Burguiére A.-M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.-C., Mueller S., Rickerts V., Stuermer M., Vieth S., Klenk H.-D., Osterhaus A.D.M.E., Schmitz H., Doerr H.W.N. N. Engl. J. Med. 2003;348:1967–1977. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 4.Pyrc K., Berkhout B., Van der Hoek L. J. Virol. 2007;81:3051–3057. doi: 10.1128/JVI.01466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fielding B.C. Future Microbiol. 2011;6:153–159. doi: 10.2217/fmb.10.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui L.-J., Zhang C., Zhang T., Lu R.-J., Xie Z.-D., Zhang L.-L., Liu C.-Y., Zhou W.-M., Ma X.-J., Tan W.-J. Adv. Virol. 2011 doi: 10.1155/2011/129134. No. 129134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 8.http://www.independent.co.uk/life-style/health-and-families/health-news/sarslike-virus-spreads-persontoperson-in-the-uk-8492750.html.
- 9.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chem M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Günther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 10.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 11.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L.M., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Sydnes M.O., Hayashi Y., Sharma V.K., Hamada T., Bacha U., Barrila J., Freire E., Kiso Y. Tetrahedron. 2006;62:8601–8609. doi: 10.1016/j.tet.2006.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Regnier T., Sarma D., Hidaka K., Bacha U., Freire E., Hayashi Y., Kiso Y. Bioorg. Med. Chem. Lett. 2009;19:2722–2727. doi: 10.1016/j.bmcl.2009.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konno S., Thanigaimalai P., Yamamoto T., Nakada K., Kakiuchi Y.R., Takayama K., Yamazaki Y., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Chen S.H., Freire E., Hayashi Y. Bioorg. Med. Chem. 2013;21:412–424. doi: 10.1016/j.bmc.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Chen S-E., Naser-Tavakolian A., Schön A., Freire E., Hayashi Y. Eur. J. Med. Chem. 2013;65:436–447. doi: 10.1016/j.ejmech.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R. Hersperger, P. Janser, E. Pfenninger, H.J. Wuethrich, W. Miltz, From PCT Int. Appl., 2005077932, 25 August 2005.
- 16.K.X. Chen, S. Venkatraman, F.G. Njoroge, S.B. Rosenblum, C.A. Lesburg, J.S. Duca, N.-Y. Shih, F. Velazquez, G.N. Anilkumar, Q. Zeng, J.A. Kozlowski, From PCT Int. Appl., WO 2008082484 A1 20080710, 2008.
- 17.Gray N.M., Dappen M.S., Cheng B.K., Cordi A.A., Biesterfeldt J.P., Hood W.F., Monahan J.B. J. Med. Chem. 1991;34:1283–1292. doi: 10.1021/jm00108a007. [DOI] [PubMed] [Google Scholar]
- 18.Lee J.H., So J.-H., Jeong J.H., Choi E.B., Lee Y.-R., Chang Y.-T., Kim C.-H., Bae M.A., Ahn J.H. Chem. Commun. 2011;47:7500–7502. doi: 10.1039/c1cc11253h. [DOI] [PubMed] [Google Scholar]
- 19.Piscitelli F., Ligresti A., La Regina G., Coluccia A., Morera L., Allarà M., Novellino E., Marzo V.D., Silvestri R. J. Med. Chem. 2012;55:5627–5631. doi: 10.1021/jm201485c. [DOI] [PubMed] [Google Scholar]
- 20.Bruel A., Logé C., de Tauzia M.-L., Ravache M., Le Guevel R., Guillouzo C., Lohier J.-F., de Oliveira Santos J.S., Lozach O., Meijer L., Ruchaud S., Bénédetti H., Robert J.-M. Eur. J. Med. Chem. 2012;57:225–233. doi: 10.1016/j.ejmech.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Tian Q., Nayyar N.K., Babu S., Chen L., Tao J., Lee S., Tibbetts A., Moran T., Liou J., Guo M., Kennedy T.P. Tetrahedron Lett. 2001;42:6807–6809. [Google Scholar]
- 22.Webber S.E., Okano K., Little T.L., Reich S.H., Xin Y., Fuhrman S.A., Matthews D.A., Love R.A., Hendrickson T.F., Patick A.K., Meador J.W., III, Ferre R.A., Brown E.L., Ford C.E., Binford S.L., Worland S.T. J. Med. Chem. 1998;41:2786–2805. doi: 10.1021/jm980071x. [DOI] [PubMed] [Google Scholar]
- 23.Bacha U., Barrila J., Gabelli B., Kiso Y., Amzel L.M., Freire E. Chem. Biol. Drug Des. 2008;72:34–39. doi: 10.1111/j.1747-0285.2008.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrila J., Bacha U., Freire E. Biochemistry. 2006;45:14908–14916. doi: 10.1021/bi0616302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akaji K., Konno H., Onozuka M., Makino A., Saito H., Nosaka K. Bioorg. Med. Chem. 2008;16:9400–9408. doi: 10.1016/j.bmc.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akaji K., Konno H., Mitsui H., Teruya K., Shimamoto Y., Hattori Y., Ozaki T., Kusunoki M., Sanjoh A. J. Med. Chem. 2011;54:7962–7973. doi: 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- 27.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. PLoS Biol. 2005;3:1742–1752. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.