

Abstract
Dihydropyrimidines are the most important heterocyclic ring systems which play an important role in the synthesis of DNA and RNA. Synthetically they were synthesized using Multi-component reactions like Biginelli reaction and Hantzschdihydropyridine. In the past decades, such Biginelli type dihydropyrimidones have received a considerable amount of attention due to the interesting pharmacological properties associated with this heterocyclic scaffold. In this review, we highlight recent developments in this area, with a focus on the DHPMs, recently developed as anti-inflammatory, anti-HIV, anti-tubercular, antifungal anticancer, antibacterial, antifilarial, antihyperglycemic, antihypertensive, analgesic, anti-convulsant, antioxidant, anti-TRPA1, anti-SARS, and anti-cancer activity and α1a binding affinity.
Keywords: Dihydropyrimidinones, Biginelli reaction, Hantzsch, Anticancer, Anti-HIV
Graphical abstract

Highlights
-
•
This review is focused on synthetic prospective of dihydropyrimidinones.
-
•
This review is also focused on medicinal prospective of dihydropyrimidinones.
-
•
It includes structure-activity relationship study of different activities.
1. Introduction
Heterocyclic chemistry is an important branch of organic chemistry accounting for nearly one-third of modern publications [1]. Heterocyclic compounds have vital role in our biological system. They are an integral part of many pharmacologically active molecules, natural products and nucleic acids. The base pair of DNA& RNA (guanine, cytosine, adenine and thymine) are also made up of heterocyclic compounds like purine, pyrimidine etc. Heterocyclic compound are also present in large variety of drug candidate like antitumor, antibiotic, anti-inflammatory, antidepressant, antimalarial, anti-HIV, antimicrobial, antibacterial, antifungal, antiviral, antidiabetic, herbicidal, fungicidal and insecticidal agents [2]. Some naturally occurring molecules are discovered and having a good biological activity against the many diseases e.g. quinine is used as an antimalarial drug, vinblastine and vincristine are also used as anticancer agent. In this review, our focus will be on the 3,4-dihydropyrimidine (DHPM) ring. Basically it is a selective review on dihydropyrimidinones. Literature of last two decades is incorporated in this review. The pyrimidine is the most important heterocyclic moiety. Pyrimidine derivatives have various therapeutic applications in medicinal chemistry. One anticipated reason for their activity is presence of a pyrimidine base in thymine, cytosine and uracil, which are essential building blocks of nucleic acids, DNA & RNA [3]. Number of chemical compounds consisting of pyrimidine as core nucleus were synthesized and evaluated for antihypertensive [4], anticancer [5], antimicrobial [6], antihyperglycemic [7], antiarrhythmic, anti-inflammatory [8], analgesic [9], antibacterial [10], anti-HIV [11] and antitubercular activity [12]. Due to the wide range of therapeutic properties scientist have attracted towards developing new dihydropyrimidine molecules. Recently in 2013, Dragovich et al. synthesized substituted 2-thio-6-oxo-1,6-dihydropyrimidines inhibitors of human lactate dehydrogenase [5]. The DHPMs are synthesized with the help of Multi-component reactions (MCR). MCR are special types of theoretically useful organic reactions in which three or more starting materials react to give a desired product [13]. Combine synthesis pathways generally show advantages over continuous or atypical approaches with respect to time, speed, yield and reproducibility. Among organic reactions, MCR with more than two starting materials are allowed to form a complex product. Therefore, they constitute a superior tool for diversity oriented and complexity-generating synthesis for drug discovery [14]. Commonly used multi component reaction for heterocyclic synthesis are Biginelli reaction and Hantzsch dihydropyridine synthesis [15] etc. Few pharmacologically active dihydropyrimidinones are shown in Fig. 1 .
Fig. 1.

Dihydropyrimidinone skeleton containing drugs.
Dihydroprymidinone nucleus is also found in marine natural alkaloids Batzelladine A and B which are known to inhibit the binding of HIV gp-120 to CD4 cells [16], [17]. Various other synthetic analogs such as monastrol [18], L-771,688 [19], SQ 32926 [20], have been developed (Fig. 2 ). Monastrol is the most important anticancer compound and has an ability to cross the cell membrane. It causes mitosis by reversible and specific inhibition of Eg5 myosin kinase. Various other analogs of monstraol such as oxo-monastrol, thio and 3,4-methylenedioxy derivatives of manostrol have been developed and tested against HT-29 colon cancer cell lines. 3,4-methylenedioxy analog was found to be 30 times more potent than monastrol [18].
Fig. 2.

DHPM containing various natural and synthetic analogs.
Various drug interactions and side effects have been reported for some of the drugs containing dihyropyrimidinone nucleus. Monastrol is well known as mitotic kinesin inhibitors [21] but it is found to have neurotoxicity as one of the major side effect. Interaction of aminophylline and topiramate has been studied carefully which revealed that aminophylline containing the dihyroprymidinone nucleus markedly attenuated the anticonvulsant potential of topiramate in the mouse maximal-electroshock-induced seizure model [22]. Seizure prolonging action of aminophylline has also been observed which is mainly attributed by blocking adenosine receptors [23]. However, seizure prolonging action was of aminophylline was antagonized by RO 15-1788, which is partial benzodiazepine agonist [24], [23]. Interaction of 5-flourouracil with misonidazole has revealed that the clearance of the former was significantly reduced. Misonidazole is a radiosensitiser of hypoxic cells which has been shown to enhance antitumor activity of several chemotherapy drugs [25].
2. Synthetic strategies
Research and development in the past years have effectively accomplished the purpose of introduction of various synthetic strategies. Numerous synthetic strategies have been outlined for the synthesis of DHPMs illustrated in Scheme 1 . In 1893, Pietro Bignelli's had reported the synthesis of dihydropyrimidine (monastrol) 4 by condensation of ethyl acetoacetate 3, 3-hydroxybenzaldehyde 1 and thiourea 2 under slightly acidic condition using concentrated hydrochloric acid as catalyst in appropriate solvent such as ethanol [26]. Matthews et al. treated β-keto ester 5 with aldehyde 1 and urea 2 for the synthesis of DHPMs 6 [27]. In 2004, Holla et al. published one pot synthesis of thiazolodihydropyrimidinones 8 by condensing benzaldehyde1 with 2,4-dichloro-5-fluoroacetophenones 7 and thiourea 2 in the presence of sodium hydroxide and ethanolic potassium hydroxide under the Claisen-Schmidt reaction conditions [28]. Yadlapalli et al. were synthesized DHPMs with the help of 1-(piperidin-1-yl)butane-1,3-dione 9, benzaldehyde 1, an excess of thiourea 2 in ethanol under mild acidic condition provided the expected DHPMs in good to excellent yield [29]. In 2010, Shaabni et al. described the synthesis of a new class of 3,4-dihydropyrimidine-2(1H) one derivatives in a one-pot process by a four-component condensation reaction of an aliphatic or aromatic amine 11, diketene 12, an aromatic aldehyde 1 and urea/thiourea 2 in the presence of p-toluenesulfonic acid (p-TsOH.H2O) as a catalyst in dichloromethane at ambient temperature. Since the number of possible combinations in four-component reactions is greater than the three-component reactions, the diversity of Biginelli reaction is more explored under four-component reaction strategy [30].
Scheme 1.

Various approaches for synthesis of 3,4-dihydropyrimidinones.
In 2000, Kappe had developed alternative synthetic strategy for the synthesis of DHPMs apart from the traditional Biginelli condensation. The enone 14 was condensed with protected urea or thiourea derivative 15 under the neutral conditions. Deprotection of intermediate with hydrochloric acid has led to the emergence of dihydropyrimidines 18 [31]. Sondhi et al. synthesized 2-thiopyrimidine derivatives with 3-isothiocyanatobutanal 17 with commericially available functionalized amines 16 in absolute methanol for the synthesis of 19. Kappe and Sondhi's reaction conditions for DHPMs synthesis are illustrated in Scheme 2 .
Scheme 2.

Kappe and Sondhi's strategies for synthesis of 3,4-dihydropyrimidine.
Various efficient methods on the synthesis of dihydropyrimidines have been developed in the past decade which is very well described in Scheme 1, Scheme 2. But still, various recent alternative approaches have been reported for the synthesis of this attractive molecule as described in Scheme 3 . Multi-component reactions have been very successful in generating complex molecules in a single and simple synthetic procedure [32], [33]. In 2005, Gong et al. reported the use of β-cyclodextrin-propyl sulfonic acid (2 mol %) as a catalyst in the synthesis of 3,4-dihydropyrimidinones 20 through a one-pot-multicomponent reaction of benzaldehydes 1, urea/thiourea 2 and ethyl acetoacetate 3 under solvent-free conditions at 80 °C [2]. Cyclodextrins are macrocyclic oligosaccharides, possessing hydrophobic cavities which bind to the substrate via non-covalent interactions. This property of cyclodextrin is utilized in various applications [33], [34]. Benazzouz et al. described a simple and efficient synthetic startegy for a new series of 4-aryl-6-methyl-5-(2-oxo-2Hchromene-3-carbonyl)-3,4-dihydropyrimidin-2(1H)-ones/thiones 22 or 23 [35]. The reaction was initiated using 3-(acetoacetyl) coumarin derivatives as key synthon 21, para-substituted benzaldehyde 1 and urea/thiourea 3 in the presence of sulfuric acid, acting as a catalyst, under refluxing acetonitrile.
Scheme 3.

Alternative synthetic approaches utilized for the synthesis of 3,4-dihydropyrmidin-2(1H)-ones.
In 2016, Saher et al. synthesized 3,4-dihydropyrimidones using Keggin and Dawson type polyoxometalates as an acid catalysts. The keggin type polyoxometalates have acidic and oxidizing properties which are mainly dependent upon the nature of components and composition that can be easily modified according to the reaction needs. The Bignelli reaction was carried out using two solvent EtOH (polar protic) and MeCN (polar aprotic) under reflux conditions. The condensation of ethyl/methyl acetoacetate 3, benzaldehyde 1, and urea 2 in the presence of H4SiMo12O4 as catalyst, using ethanol and MeCN as solvent for 1.5 h afforded 3,4-dihydropyrimidin-2(1H)-ones 20 in a moderate to good yields (52–82%) [36]. In 2015, Treptow et al. reported the fatty acid synthesis of 3,4-dihydropyrimidinones 20 by utilizing the bignelli multi-component reaction. The reaction was carried out by reacting aromatic aldehyde 1, urea or thiourea 2, β-ketoesters 3, in the presence of catalytic amount of InCl3 (10mol %) using acetonitrile as solvent, thereby affording DHPM-fatty acid synthesis in good to excellent yields [37]. Qiu et al. described cerium (III) trislaurylsulfonate (Ce(LS)3) as an efficient, stable catalyst which is a combination of lewis acid with surfactant. This catalyst is being ultilised to catalyse one-pot bignelli as well as solvent free esterification reactions. 3,4-dihyropyrimidinone were obtained in 83% yield by employing a reaction of benzaldehydes 1, ethylacetoacetate 3, and urea 2 in presence of catalytic amount of Ce(C12H25SO3)3, and one drop of conc. HCl in ethanol at 80 °C for 6 h as described in Scheme 3 [38].
In 2014, Kour et al., reported the use of green heterogeneous catalyst, SiO2-CuCl2, silica supported copper (II) chloride for the one-pot synthesis of DHPM under microwave and thermal conditions. The reaction was carried out with similar substrates, as benzaldehyde 1, urea 2, and ethylacetoacetate 3. A comparative study was undertaken using solvent-free conditions at 40 °C using diphenic acid under thermal conditions (condition A) and under microwave exposure at 80 °C in acetonitrile without the use of diphenic acid (condition B) as described in Scheme 3. Diphenic acid is used to provide polar conditions to the reaction and to make the carbonyl group of aldehyde more polar for the nucleophile-electrophile attack. It was found that reaction carried out in microwave radiation was fast, clean, and produce excellent yields. The structure-activity relationship study has also been conducted for both the conditions and it was found that in case of aromatic aldehyde substituted with electron-donating groups afforded 80–85% yield. While aromatic aldehydes substituted with electron-withdrawing group afforded 80–95% yield and heteroaromatic aldehydes end up with 90–95% yields. In case of aliphatic aldehyde, like formaldehyde produce a yield of 92% whereas acetaldehyde and butanal afforded the product in trace amounts [39].
Many synthetic approaches have been conducted under solvent-free conditions for the synthesis of 3,4-dihyrdopyrimidin-2(1H)-one using Bignelli reaction conditions as described in Scheme 4 . The nature of solvent play a crucial role in the reaction, as higher values of dielectric constant induces higher reaction yields. In 2011, Kumaran et al. reported an efficient synthesis of 3,4-dihydropyrimidinone 20 and corresponding thione with 98% yield using lanthanium oxide (La2O3) as catalyst from aromatic aldehydes 1, urea/thiourea 2, and β-ketoesters 3 without using any solvent under the effect of microwave radiation at 320 W for 20–60 s [40]. Slimi et al. synthesized 3,4-dihydropyrimidinone 20 or thione and their derivatives via bignelli reaction involving aromatic aldehydes 1, urea/thiourea 2, and β-ketoesters 3 in the presence of bismuth nitrate in acetonitrile or PPh3 without solvent at room temperature [41]. Good yields were obtained when the reaction was carried out using aliphatic aldehydes and bismuth nitrate as catalyst. However, when bignelli reaction was carried out using bismuth nitrate in acetonitrile under the effect of microwave radiation in the absence of solvent, product was obtained in quanitative yield [41], [42]. In 2011, Mansoor et al. described the synthesis of DHPM using triphenylphoshphine (10 mol%) as catalyst under solvent free conditions at 100 °C for 8 h from the use of similar three component substrates aromatic aldehydes 1, urea/thiourea 2, and β-ketoesters 3 [43]. Triphenyphosphine act as a lewis base by interacting with the electrophilic carbon of the aldehyde. The enolate of β-ketoester is formed by coordinating the aldehyde with PPh3, which helps in the deprotonation of β-ketoester. In 2014, Attri et al. utilized triethylammoniumacetae (TEAA) as a catalyst and reaction medium for the synthesis of 3,4-dihydropyrimidinone 20 and its derivatives under solvent free conditions using aldehydes 1, urea/thiourea 2, and β-ketoesters 3 as subtrates [44]. The reaction yield was obtained upto 92%. TEAA is relatively inexpensive, thermostable, non-toxic, recyclable and has vast applications as catalyst and reaction medium.
Scheme 4.

Synthesis of 3,4-dihydropyrimidin-2(1H)-ones using different catalyst under solvent free conditions.
Apart from the classical methods of bignelli reaction, focus has also been shifted to the catalytic asymmetric bignelli reaction in order to obtain enantiomerically pure DHPM [45]. It has been subdivided in two categories: metal-catalyzed enantioselective bignelli reaction and organocatalytic enantioselective bignelli reaction. In case of metal-catalyzed reaction, Muñoz-Muñiz and Juaristi reported enantioselective synthesis of DHPM 20, by utilsing CeCl3 or InCl3-catalysed bignelli reaction in the presence of chiral ligand. Different chiral ligands were employed with CeCl3 or InCl3 to achieve the best enantioselective synthesis. It was found that ligand (R,R)-24 showed best results in inducing enantioselectivity whereas other ligands as benzylamine (S)-25 did not revealed any stereoselective synthesis because it directly incorporates to the aldehyde component. When the reaction occurred between the intermediate 26 and ethylacetoacetate 3 at −78 °C to 0 °C, highest enantioselectivity was obtained upto 40% ee as described in Scheme 5 [45], [46]. In 2006, Chen et al. and co workers were able to synthesize a series of chiral-phosphoric acids which act as catalyst for the enantioselective synthesis of bignelli reaction. Chiral phosphoric acid 27, termed as H8-bino-based phosphoric acid was found to be effective in providing product 20 in yield upto 40–86% and 88–97% enantioselectivity was observed using three-component substrate as aldehydes 1, urea/thiourea 2, and β-ketoesters 3 [45], [47].
Scheme 5.

Enantioselective synthesis of 3,4-dihyropyrimidinones using chiral ligands.
Thorat and co-workers in 2014, have reported the metal free O-arylation of 4-aryl-6-methyl-pyrimidin-2(1H)-one sacffold of bignelli type to produce 2-aryloxy pyrimidine derivatives in appropriate yields using diaryliodonium salts as catalyst under metal free conditions [48]. The O-arylation of 4-aryl-6-methyl-pyrimidin-2(1H)-one 20 was carried out in presence of diphenyliodonium triflate 28 with K2CO3 as base in the presence of toluene and afforded 29 in 65% yield as described in Scheme 6 .
Scheme 6.

O-arylation of 4-aryl-6-methyl-pyrimidin-2(1H)-one.
Karami et al. described the one-pot reaction of aryl gloxals 30, urea 2 and ethylacetoacetate 3 using molybdate sulfuric acid as catalyst to furnish novel 5-acetyl-4-(aryloyl)-3,4-dihydropyrimidinones 31 in 70–85% yield under solvent free conditions [49]. The reaction is in accordance with green chemistry principles and 31 undergoes knorr-condensation with hydrazines to produce pyrimido[4,5-d]pyrazines. In 2014, Ábrányi-Balogh et al. have published 1-aryl-β-carboline-3-carbaldehydes 32 as versatile building blocks and is found to be useful starting material for bignelli type reaction [50]. Compound 32 was successfully reacted with urea 2 and four different β-dioxocompounds [pentane-2,4-dione 3a, ethyl acetoacetate 3b, ethyl isobutyrylacetate 3c, and ethyl benzoylacetate 3d]. The reaction was catalyzed with NiCl2.6H2O with ethanol containing catalytic amount of conc. HCl as solvent afforded the product as 2-oxo-1,2,3,4-tetrahydropyrimidine derivatives 33 in 36–90% yield as described in Scheme 7 .
Scheme 7.

Synthesis of 3,4-dihyropyrimidinone derivatives.
3. Biological activities
The biological investigation of dihydropyrimidines involve various mechanism like enzymatic action, receptor mediated mechanism and activity against the ion channel etc. The biological investigations have revealed that substitution of various groups on the ring imparts different activity. An outline of different activities of DHPMs is shown in Fig. 3 .
Fig. 3.

Biological activities of dihydropyrimidines.
3.1. Anti-inflammatory activity
In 2004, Kumar et al. synthesize and evaluated novel [4,6-(4-subsituted aryl)-2-thioxo-1,2,3,4-tetrahydroyrimidine-5-yl]-acetic acid derivatives as potential anti-inflammatory agents. The anti-inflammatory activity of all the compounds had been recorded on the basis of reference standard drug diclofenac sodium. All the compounds showed tendency to prevent edema and showed anti-inflammatory activity. The compounds are tested for the anti-inflammatory activity according to the method of Winter et al. [51] in albino rats employing the carageenan induced rat paw edema test. Percentage reduction in the inflammation after 3 h of administration of carageenan was recorded and test compounds were compared with that of the animals administrated with carangeenan using the reference standard diclofenac sodium. They synthesized three series of molecules with different functional groups which are mentioned by R & R1 in Fig. 4 . We observed different molecules which have shown anti-inflammatory activity. Compounds 36, 40 & 44 were found the most potent among the all molecules. The presence of 4-methoxy group at C-4 plays an important role in the activity of compounds. It was observed that 4-methoxy phenyl increases the activity of the compounds. On the other hand, C-6 phenyl group reduces the activity of the compounds but when the phenyl group was replaced by the p-chlorophenyl at C-6 & 4-methoxy phenyl at C-4 then the activity of the compound was found to be increased. When the methoxy group at C-4 was replaced by any other group then the activity decreased [52]. The structure and inhibitory percentage 50 value (IP50) of the compound are given in Fig. 4.
Fig. 4.

The structure of potent DHPMs having anti-inflammatory activity.
3.2. Anti-HIV activity
In 2015, Sari et al. synthesized dihydropyrimidine α,γ-diketobutanoic acid derivatives to tartgeting HIV integrase (3′-P and ST). They synthesized two series of new compounds. The compounds are shown in Fig. 5 . They synthesized twenty new molecules and evaluated for their enzymatic activity against HIV integrase [11]. Compound 46, 48, 53 & 55 inhibited strand transfer and sub-micromolar activities. They observed that C-6 substitution play an important role in the potency of the compounds. When the R2 was substituted with methyl or ethyl groups, an enhancement of activity was observed. On the other hand, when R2 was substituted with phenyl or isopropyl groups, a decremental effect in activity was observed. This proves that steric hindrance at C-6 was not tolerated. The structures of the potent compounds are given in Fig. 5, Fig. 6 and their IC50 values are shown in Table 1 , respectively. The different substitution R1, R2 and R3 are differentiated by the help of different color.
Fig. 5.

The structure of first generation anti-HIV compounds.
Fig. 6.

The structure of second generation Anti-HIV compounds.
Table 1.
Inhibition of integrase activities (3′-processing and strand transfer) of dihydropyrimidine α,γ-diketobutanoic acid derivatives.
| Compound No. | IC50 (μM) ± S.D. |
|
|---|---|---|
| 3′- processing Strand transfer (ST) | ||
| 46 | 16.9 ± 3.4 | 0.19 ± 0.12 |
| 47 | 17.4 ± 2.1 | 1.3 ± 0.1 |
| 48 | 19.1 ± 4.0 | 0.64 ± 0.16 |
| 49 | 35 ± 8 | 1.1 ± 0.4 |
| 50 | 72 ± 4.5 | 5.6 ± 4.6 |
| 51 | 16.3 ± 3.5 | 1.3 ± 0.5 |
| 52 | 16.6 ± 2.4 | 1.3 ± 0.5 |
| 53 | 22.5 ± 2.0 | 0.85 ± 0.12 |
| 54 | 21.1 ± 1.2 | 2.23 ± 0.27 |
| 55 | 29.3 ± 3.5 | 0.92 ± 0.14 |
| 56 | 30.7 ± 2.7 | 2.27 ± 0.28 |
| 57 | 22 ± 4 | 2.8 ± 0.9 |
3.3. Antitubercular activity
In 2010, Trivedi et al. synthesized novel dihydropyrimidines as a potential new class of antitubercular agents. They synthesized thirty new molecules by the multi-component Biginelli reaction. Initially nine compounds were screened with the help of Mycobacterium tuberculosis strain. They all were inhibited MTB upto 90–100%. In the secondary level, two compounds (58 and 59) inhibited MTB with MIC of 1 μg/mL and three compounds (61, 65 & 66) with MIC of 2 μg/mL. Among all the compounds, compounds 58 & 59 were found to be most potent compounds (MIC of 0.02 μg/mL and SI > 500) of the series. The compounds were found to be much better than existing drug INH (MIC: 0.03 μg/mL) in the in vitro studies. The compounds [53] gave a direction to develop an excellent lead as novel antitubercular active molecules. At fourth position of C-3 phenyl ring of pyrazolyl play an important role in the activity of the newly developed molecules. At C-3 position of phenyl ring of pyrazolyl substituted with the different electronegative element instead of methyl group, the compounds found to be more active [12]. The structures of potent compounds for tuberculosis are shown in Fig. 7 and their summarized data is given in Table 2 , respectively.
Fig. 7.

The structure of potent antitubercular compounds.
Table 2.
In vitro antitubercular screening data of dihydropyrimidines.
| Compound No. | % Inhibition | MIC μg/mL | S.I. |
|---|---|---|---|
| 58 | 100 | 0.02 | >500 |
| 59 | 100 | 0.02 | >500 |
| 60 | 92 | 3.13 | >3.2 |
| 61 | 96 | 1.56 | 5.7 |
| 62 | 94 | 3.13 | >3.2 |
| 63 | 91 | 3.13 | 3.0 |
| 64 | 90 | 6.25 | >1.6 |
| 65 | 98 | 1.56 | >6.4 |
| 66 | 97 | 1.56 | 4.7 |
| Isoniazid | – | 0.03 | – |
MIC: Microbial inhibitory concentration.
3.4. Antifungal activity
In 2012, Aly and Kamal developed a novel fused chromeno [2,3-d] pyrimidine and pyrano [2,3-d] pyrimidine derivatives. Screening of newly prepared compounds was done separately in vitro for their antifungal activity. The compounds were tested against the two fungal species, namely, Fungus, Aspergillus flavus and one yeast fungus Candida albicans on sabouraund dextrose agar plates. The antifungal activity measured by agar well diffusion method. Nine compounds were tested for their antifungal activity. The test was performed three times for each fungus. The tested compounds were compared with standard Amphotericin B to evaluate their potency. Zone of inhibition were determined for synthesized compounds and the result were summarized in Table 3 [53]. From the results obtained it have been concluded that the compound 72 was more potent than the standard drug Amphotericin B against the Aspergillus flavus fungus. On the other hand, it was found to be inactive against the Candida albicans fungus. The structures of potent compounds are shown in Fig. 8 and their summarized data is given in Table 3 respectively.
Table 3.
Antifungal activity of chemical substances tested.
| Compond No. | Inhibition zone diameter(mm) |
|
|---|---|---|
| Aspergillusflavus | Candida albicans | |
| 67 | 0 | 0 |
| 68 | 0 | 10 |
| 69 | 0 | 13 |
| 70 | 14 | 14 |
| 71 | 0 | 12 |
| 72 | 36 | 0 |
| 73 | 0 | 10 |
| 74 | 0 | 12 |
| 75 | 0 | 11 |
| Amphotericin B | 16 | 19 |
Fig. 8.

The structure of potent antifungal compounds.
3.5. Antibacterial activity
Rajanarendar et al. synthesized the antibacterial novel piperazine and morpholine linked substituted pyrimidine derivatives as antimicrobial agents. The newly synthesized compounds were estimated for in vitro antibacterial against the different types of Gram-positive, Gram-negative bacterial using broth dilution method. The results are shown in Table 4 . Ciprofloxacin was used as standard drug for comparison. The bacterial strains used in the present study are Bacillus subtilis (MTCC 441), Bacillus sphaericus (MTCC 511), Staphylococcus aureus (MTCC 96), Pseudomonas aeruginosa (MTCC 741), Klebsiella aerogenes (MTCC 39) and Chromobacterium violaceum (MTCC 2656). The results showed that compounds have average to good antibacterial activity and are more active than standard drug Ciprofloxacin. The activity was shown in terms of minimum inhibitory concentration (MIC). The SAR study disclosed that when the benzene ring is substituted by the electron withdrawing group like chloro and bromo it enhances the activity of compounds. Compound 76, 77, and 78 were found to be most potent compounds. They exhibit the same antibacterial activity compared to standard drug ciprofloxacin [54]. The structures of potent compounds are shown in Fig. 9 and their summarized data is given in Table 4 respectively (see Fig. 10 ).
Table 4.
Antibacterial activity data (MIC (in μg/mL) values) of 1-aryl-4-methyl-3,6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2,3,6,10b-tetrahydro-1H-pyrimido [5,4-c]quinolin-5-ones.
| Compound No. | Gram positive |
Gram negative |
||||
|---|---|---|---|---|---|---|
| B. subtilis | B. sphaericus | S. aureus | P. aeruginosa | K. aerogenes | C.violaceum | |
| 76 | 19 | 15 | 15 | 20 | 13 | 18 |
| 77 | 16 | 11 | 10 | 10 | 08 | 06 |
| 78 | 20 | 15 | 10 | 15 | 14 | 18 |
| 79 | 06 | 08 | 09 | 09 | 07 | 05 |
| 80 | 22 | 18 | 25 | 20 | 15 | 18 |
| 81 | 16 | 15 | 15 | 20 | 15 | 16 |
| 82 | 18 | 18 | 12 | 08 | 13 | 11 |
| 83 | 08 | 10 | 09 | 05 | 08 | 06 |
| Ciprofloxacin | 20 | 20 | 25 | 30 | 25 | 25 |
Negative control (acetone)-no activity. Values are indicated in lg/mL.
Fig. 9.

The structure of potent anti-bacterial compounds.
Fig. 10.

The structure of compounds possessing antibacterial activity.
Attri et al. also reported the antibacterial activity of the synthesized compounds (84–96) as shown in Fig. 10. These compounds were evaluated using disk diffusion assay and zone of inhibition was measured [44]. The assay method involves the pouring of nutrient agar (25 mL) into the petri dishes under aseptic conditions in a laminar flow hood. The plates were then kept for solidification, after solidification 100 μL of the fresh culture was spread onto the surface of the solidified medium. The plates were then left for drying in a laminar fashion. After drying, five plain sterile disks were placed in the plate and 5 μL of test solution of different concentrations (*250.000–15.625 ppm) was loaded onto the individual disks. Commercially available ampicillin (10μg/disk) was used in the control plate. The plates were then kept for incubation at 37 °C for 24 h. The plates were then taken out from incubator and zone of inhibition was recorded for all the test compounds and control. The experiments were conducted in triplicate for each treatment against each bacterium.
The MIC (Minimum inhibitory concentration) is defined as the lowest possible concentration of the test compound that is able to inhibit the organism and was measured using micro-dilution assay. It was determined on the basis of the serial dilution method by varying the concentration of test compounds ranging from 200 to 15.625 μg/mL. Requisite concentrations of test compounds were added to all the sterile Erlenmeyer flask, each containing 10 mL nutrient broth and were sonicated for 10 min. The flasks were inoculated with 1 mL of the freshly prepared bacterial suspension (E. coli, S. aureus, P. aeruginosa, K. pneumonia) in order to maintain the initial bacterial concentration of 103–104 CFU/mL. The flasks were then incubated in an orbital shaker at 200 rpm at 37 °C. Bacterial growth was then monitored using spectrophotometer by using an increase in absorbance at 600 nm. The experiments included positive control (flask containing test compound, nutrient media and absence of inoculum) as well as negative control (flask with inoculums, test compound and no test compounds). All the experiments were performed in triplicate.
All the compounds possessed moderate to good inhibitory activity. Compounds 86, 87, 88 and 89 were found to have MIC in the range of 31,250–15,625 ppm against E. coli which is comparable to the standard. Compounds 87, 88 and 92 were active against S. aureus with MIC values comparable to the standard while the compounds 94, 95 and 96 were completely inactive with MIC values of 250 ppm, whereas the remaining compounds were moderately active with MIC value ranging from 62.5 to 125.0 ppm. Compounds 84, 85, 86 and 88 showed good antibacterial activity with MIC value in the range of 15,625 to 31,250 ppm, while the compounds 87 and 93 showed moderate activities with MIC value of 62.5 ppm and rest of the compounds were inactive with MIC value of 250 ppm against P. aeruginosa. Similarly for K. pneumonia, compounds 86, 87, 88, 90, 91, 92 and 95 showed moderate antibacterial activities with MIC 31.25–62.50 ppm and the rest of the compounds were found to be inactive. Overall compounds 86, 87 and 88 showed the good antibacterial activity against all bacteria which is possibly attributed due to the presence of halogen atom in them. While compound 88 was found to be effective against bacteria with lower MIC in comparison to compound 86 and 87. This is due to the presence of two halogen atoms in compound 88 which tend to enhance the antibacterial activity.
3.6. Antifilarial activity
In 2008, Singh et al. Synthesized 2-sulfanyl-6-methyl-1,4-dihydropyrimidines as a antifilarial agents. All the synthesized compounds were evaluated in vitro for their macrofilaricidal activity against Brugia malayi according to the method of Murthy and Chatterjee [55]. Micro- and macro-filaricidal activities were evaluated by the method described by Lammler, Wolf, Chatterjee and Gaur [56]. It was concluded that all compounds showed in complete loss of motility of adult worms of B. malayi at 100 μM concentrations and they had shown 15.4–68.61% inhibition in MTT reduction assays while compound 103 do not show any inhibition in MTT reduction assay. Compound 98, 101 and 102 were affected both motility (irreversible loss) and MTT reduction (50% inhibition or more) and compounds 97, 99, 100, 101 and 103 either affected motility with <50% MTT reduction or only motility. When the compounds were screened at their 50% concentration also show the positive result. Compound 98 at 50 μM concentration resulted in complete loss of motility of filarial worms with no inhibition in MTT reduction assay, while at 25 μM concentration it showed only slack motility of filarial worm and displayed approximately 30% inhibition in MTT reduction assay. Compound 101 on the other hand displayed complete loss of motility in filarial worms at 50 μM concentration with 19% inhibition in MTT reduction assay. The most potent compound of the series was found to be compound 101 which resulted in complete loss of motility at two lower concentrations (50 and 25 μM) screened and good inhibition (around 70%) in MTT reduction assay. SAR study showed that when the 4-aryl-1,4-dihydropyridines substituted by less bulky electron-withdrawing group at the fourth position of phenyl ring (4-F-phenyl) compound 102 was the best compound of the series. Only three compounds 98, 101 and 102 were found to be most potent in vitro antiflarial activity [57]. The structures of synthesized compound are shown in Fig. 11 . All the data of potent compounds are summarized in Table 5 .
Fig. 11.

The structure of potent anti-filarial compounds.
Table 5.
In vitro macrofilaricidal activity of 2-sulfanyl-6-methyl-1,4-dihydropyrimidines against Brugia malayi.
| Compound No. | Concentration (μM) |
Motilitya | Percent inhibition in MTT reduction over control (means ± SD) |
|---|---|---|---|
| 97 | 100 | 0 | 18.97 ± 1.77 |
| 98 | 100 | 0 | 49.0 ± 11.50 |
| 50 | 0 | NI | |
| 99 | 100 | 0 | 43.14 ± 5.11 |
| 100 | 100 | 0 | 25.74 ± 3.44 |
| 101 | 100 | 0 | 52.11 ± 26.92 |
| 50 | 0 | 19.29 ± 9.49 | |
| 102 | 100 | 0 | 68.61 ± 5.13 |
| 50 | 0 | 68.4692 | |
| 103 | 100 | 0 | NI |
| DEC (citrate) | 100 | 3 | NI |
| 50 | 3 | NI |
Motility is expressed as highly motile (3), low (2), sluggish (1, irreversible) and dead (0); NI = no inhibition.
3.7. Antihyperglycaemic activity
In 2015, Bhosle et al. synthesized thiazolyl methoxyphenyl pyrimidine and evaluated antihyperglycaemic activity. Antihyperglycaemic activity was evaluated in male albino rats of Sprague Dawley strain. Present data showed that in normoglycemic rats, compound exhibit glycaemic control by decreasing the peak blood glucose and the area under oral sucrose tolerance (OSTT) curve. After 2 h of sucrose load which was indicative of an enhanced glucose utilization which may be provoke either by insulin production from the pancreatic β cells or by inhibiting the intestinal α-glucosidase enzyme responsible for breakdown polysaccharides into monomeric form and thus inhibit the postprandial hyperglycaemia. Amongst screened compounds 76, 77, 78 and 79 had reduced the blood glucose level in normal rats. The activity of the tested compounds was compared with standard drug metformin. Compounds number 78 and 79 have notably reluctance in the postprandial rise in blood glucose level of sucrose loaded rats. The main role for the activity of these compounds having functional group like nitro, fluro, methoxy on the benzenoid ring may be governing acidic behavior through mesomeric effect of the respective heterocyclic ring, which probably assisting for the activity [7]. The structures of potent compounds are shown in Fig. 12 and their summarized data is given in Table 6 respectively.
Fig. 12.

The structure of potent anti-hyperglycaemic compounds.
Table 6.
Effect of compounds and standard antidiabetic drug metformin on oral sucrose tolerance (OSTT) in post sucrose loaded normal rats.
| Compound No. | Dose (mg/kg) | %Improvement on OSSTT |
Significance |
|---|---|---|---|
| 104 | 100 | 0.07 | – |
| 105 | 100 | 1.05 | – |
| 106 | 100 | 8.22 | p<0.05 |
| 107 | 100 | 7.04 | p<0.05 |
| Metformin | 100 | 14.1 | p<0.01 |
3.8. Antihypertensive activity
In 2010, Alam et al. synthesized 1,4-dihydro-5-pyrimidine carboxamides as antihypertensive agents. They synthesized thirty molecules and all the molecules were screened for their antihypertensive activity by using tail-cuff method that measures the systolic blood pressure. The activity of the synthesized compounds was compared with standard drug nifedipine. Out of thirty compounds five compounds found to be most potent molecules. Structure activity relationship (SAR) study reveal that the different substitution at phenyl ring attached to the pyrimidine moiety played an important role in governing the antihypertensive activity. When the electron releasing groups like -OCH3 were substituted at position 3 and 4. Then there is increase in the antihypertensive activity. Substitutions at other phenyl rings were also important as the substituted benzyl derivatives were more active in the series. The para chloro-substituted derivatives were found to be more active than the other halogen. The phenyl ring attached to the amide linkage does not seem to have any significant effect on antihypertensive activity [58]. The structures of potent molecules are shown in Fig. 13 and their summarized data is given in Table 7 .
Fig. 13.

The structure of potent antihypertensive compounds.
Table 7.
Antihypertensive activity data shown by compounds at 10 mg/kg dose.
| Compound No. | Average systolic blood pressure (mmHg) at time (min) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 15 | 30 | 60 | 120 | 180 | 240 | 300 | 360 | 600 | 720 | 900 | |
| 108 | 193 ± 3 | 189 ± 2 | 183 ± 1 | 174 ± 2 | 161 ± 2 | 155 ± 1 | 145 ± 2 | 136 ± 1 | 130 ± 2 | 145 ± 1 | 156 ± 3 | 168 ± 4 |
| 109 | 194 ± 1 | 189 ± 1 | 183 ± 2 | 172 ± 1 | 163 ± 3 | 151 ± 2 | 139 ± 1 | 124 ± 2 | 120 ± 3 | 147 ± 2 | 154 ± 4 | 163 ± 2 |
| 110 | 193 ± 3 | 188 ± 1 | 182 ± 2 | 178 ± 1 | 169 ± 2 | 161 ± 3 | 152 ± 3 | 144 ± 2 | 129 ± 3 | 148 ± 2 | 157 ± 1 | 165 ± 2 |
| 111 | 194 ± 2 | 190 ± 1 | 186 ± 2 | 180 ± 1 | 174 ± 2 | 162 ± 1 | 154 ± 2 | 145 ± 1 | 131 ± 2 | 142 ± 1 | 160 ± 2 | 166 ± 3 |
| 112 | 195 ± 1 | 191 ± 2 | 188 ± 3 | 182 ± 3 | 170 ± 1 | 165 ± 2 | 156 ± 2 | 148 ± 1 | 130 ± 3 | 144 ± 1 | 164 ± 2 | 171 ± 2 |
| Nifedipine | 195 ± 3 | 192 ± 1 | 188 ± 2 | 180 ± 1 | 172 ± 1 | 160 ± 2 | 145 ± 1 | 132 ± 3 | 120 ± 2 | 148 ± 1 | 158 ± 2 | 162 ± 1 |
3.9. Analgesic activity
In 2005, Sondhi et al. synthesized some mono, bi- and tricyclic pyrimidine derivatives and evaluated for the analgesic activity. They synthesized ten molecules and screened by using phenyl quinone writhing assay [59]. Ibuprofen was taken as standard drug for comparison of tested compounds for analgesic activity. The synthesized compounds 113, 114 and 115 exhibited 100, 70 and 75% activity at 100 mg/kg dose respectively [60]. The structure of potent compounds are shown in Fig. 14 .
Fig. 14.

The structure of potent analgesic compounds.
3.10. Anticonvulsant activity
In 2010, Khanage et al. synthesized and evaluated some new pyrimidine derivatives containing 1,2,4-triazole. The synthesized compounds were screened for their anticonvulsant activity by maximal electroshock seizure method (MES). The albino mice were used in the study. The animals were stimulated by corneal electrodes to 50 mA current at a pulse of 60 Hz applied for 0.2 s. The test drug was given by the intraperitoneally. Destruction of the hind limb tonic extension spasm was recorded as the anticonvulsant activity. The test compounds were suspended in a 0.5% methyl cellulose-water mixture. In preparatory screening, each compound was administered through an intraperitoneally injection at three dose levels (30, 100 and 300 mg/kg) and the anticonvulsant activity was assessed after 0.5 h and 4 h intervals of administration. The anticonvulsant efficacy was evaluated by the maximal electroshock-induced seizure (MES) and screening data of compounds 116–125 are presented in Table 8 and their structures are given in Fig. 15 . All the screened compounds were compared with standard drugs phenytoin and carbamazepine. Compound 116, 117, 118, 123 and 125 were found to be most potent molecules among the series. The electron withdrawing groups substituted phenyl ring at sixth position of dihydropyrimidine shows marked increase in their anticonvulsant activity [61].
Table 8.
Anticonvulsant screening of compounds 116–125.
| Compound No. | MES screena |
|
|---|---|---|
| 0.5 h | 4 h | |
| 116 | 30 | 300 |
| 117 | 30 | 300 |
| 118 | 30 | 300 |
| 119 | 100 | 300 |
| 120 | 100 | 300 |
| 121 | 100 | 300 |
| 122 | 30 | 300 |
| 123 | 100 | 300 |
| 124 | 30 | 300 |
| 125 | 30 | 300 |
| Phenytoin | 30 | 30 |
| Carbamazepine | 100 | |
Doses of 30, 100 and 300 mg/kg were administered. The figures in the table indicate the minimum dose whereby bioactivity was demonstrated in half or more of mice. The animals were examined 0.5 and 4 h after injections were given.
Fig. 15.

The structure of potent anticonvulsant compounds.
3.11. Antioxidant activity
In 2009, Kumar et al. synthesized thirty two compounds and all the synthesized molecules were screened for their antioxidant activity by DPPH (2,2-diphenyl-1-picrylhydrazyl) method. Among the thirty two title compounds, compounds 126 & 127 found to be most potent molecules. The 3-nitro phenyl moiety at the fourth position of the dihydropyrimidine showed good antioxidant activity with IC50 value 58 and 63 μg concentrations, respectively. The DPPH free radicals formed in this assay will be reduced to a corresponding hydrazine when it reacts with hydrogen donors. The DPPH radical is purple in colour and upon reaction with hydrogen donors of the antioxidant changes to yellow colour. It is a discolouration assay, which is evaluated by the addition of the antioxidant or test compound to a DPPH solution in ethanol and the decrease in absorbance was measured. The assay was carried out in a 96-well microtitre plate. To 200 μl of each of DPPH ethanolic solution, 10 ml of each of the test compound (100 μg) or standard (ascorbic acid, 10 μg) solution was added separately to wells of the microtitreplate. The plates were incubated at 37 °C for 30 min and the absorbance of each solution was measured at 490 nm, using ELISA reader. The IC50 values (concentration which inhibits 50% of free radicals) for the compounds 126 and 127 were determined by serial dilution method at the concentrations below 100 μg. The structures of potent compounds (126 and 127) are shown in Fig. 16 with their IC50 value [62].
Fig. 16.

The structure of potent antioxidant compounds.
Attri et al. and coworkers synthesized 3,4-dihyropyrimidinone derivatives (84–96) and were evaluated for their antioxidant and antibacterial activity. The antioxidant activity was determined using DPPH (1,1-dipheny-2-picrylhydrazyl) free radical scavenging and (cupric reducing antioxidant capacity) CUPRAC assays. DPPH assay is basically used to determine the free radical scavenging property of antioxidant compounds [44]. Gallic acid and quercitin were used as standard compounds and the working solutions of test extracts and standards were prepared in methanol. When the dark colored DPPH radical solution in the presence of antioxidant compounds change to yellow colored diphenylpicrylhydrazine, the absorbance of the solution generally decreases. Different concentration of test solutions were prepared such as 100 μg/mL, 50 μg/mL, 25 μg/mL and 12.5 μg/mL and were pipetted to test tube and volume was adjusted to 3 mL using methanol. 1 mL of DPHH solution (0.1 mM) was mixed with 1 mL of the sample and standard separately. The samples were vortexed and incubated in dark for 30 min. Change in absorbance was measured at 517 nm using spectrophotometer. The absorbance was measured and radical scavenging activity was expressed as percent inhibition of DPPH racdical. It was calculated by the use of following equation:
A noticeable decline in the concentration of DPPH radical in terms of % inhibition was revealed due to the scavenging ability of test compounds. The percent inhibition of the test compound range from 64.4% to 17.3%. At 100 ppm, the standard gallic acid and quercitin exhibited inhibition percentage of 92.4% and 82.3% respectively, whereas compound 89 was found to show highest percent inhibition of 64.4% in comparison to all the remaining test compounds. Whereas at 12.5 ppm, gallic acid and quercitin possessed inhibition percent of 62.4% and 55.3% respectively, but all the test compounds showed a decreased percent inhibition. Compound 88 was found to posses the lowest inhibition percentage of 17.3%. Thus, finally all the compounds revealed moderate antioxidant activity in comparison to the standard compound. Compound 91 found to be the most promising antioxidant as described in Fig. 17 .
Fig. 17.

The structure of potent antioxidant compounds using DPPH assay.
CUPRAC assay is most widely used to determine the total antioxidant capacity of the antioxidant compound. The basic priniciple employed in this assay is the redox reduction of Cu (II) to Cu (I) which is monitored by change in absorbance at 450 nm. This redox reaction is a result of the CUPRAC reagent, Cu(II)-neocuproine (Cu(Nc)2 2+), with an antioxidant (A-OH), to form the CUPRAC chromophore, Cu(I)-neocuproine (Cu(Nc)2+ chelate [44], [63]. CUPRAC reagent solution was prepared by mixing 1 mL of 1.0 × 10−2 M copper (II) chloride, 1 mL of 7.5 × 10−3 M neocuprine solution and 1 mL of ammonium acetate buffer at pH 7.0. Sample solution and distilled water were added and mixed to obtain a total volume of 4 mL. The reduction capability is assessed by increased absorbance of the reaction mixture.
The results obtained from CUPRAC assay can easily be extended possibly to in vivo reactions of antioxidants. The assay is basically performed at physiological pH closer to 7. The absorbance of Cu (I)-chelate formed as a result of redox reaction with reducing antioxidant was measured at 450 nm. The color is possibly due to the formation of Cu (I)-Nc chelate. The increase in absorption indicates the higher antioxidant activity. All the compounds show moderate activity in comparison to the standard compounds. All the compounds were found to reduce copper ions from Cu (II) to Cu (I) in a concentration-dependent manner. At 100 ppm, compound 89 was found to have highest absorbance value of 0.87 in comparison to the standard gallic acid and quercitin having absorbance value of 1.4 and 1.2 respectively as described in Fig. 18 . Whereas at the lowest concentration 12.5 ppm, absorbance of test compounds range from 0.21 to 0.05 with compound 95 possessing the lowest absorbance value of 0.05. Thus in conclusion, it is stated that at lower concentration compounds do not show antioxidant activity.
Fig. 18.
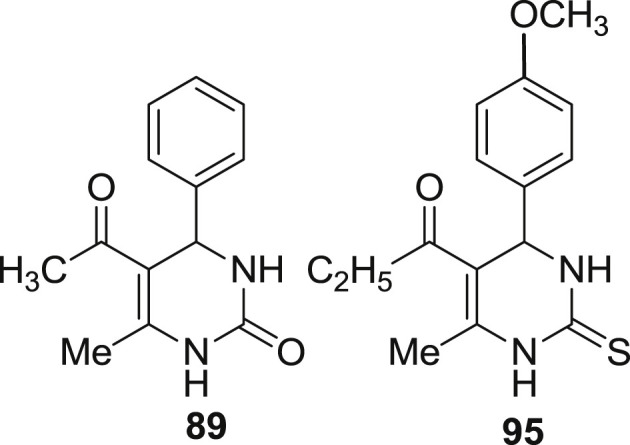
The structure of potent antioxidant compounds using CUPRAC assay.
3.12. Anticancer activity
The pyrimidines and their derivatives exhibit anticancer activity via interaction with different enzymes like tyrosinase, cytochrome P450, glutathione assisted and receptors like somaostatin, estrogen and progesterone receptors.
3.12.1. Colon cancer
In 2011, Agbaje et al. synthesized hexahydropyrimidine derivatives. They synthesized a series of seventeen molecules and the synthesized compounds were screened for the cytotoxicity against the COLO 320 cell line. Among the series of seventeen molecules only four (128, 129,130 and 131) compounds found to be potent analogs. The compound 128, 129 and 130 showed highest activity with IC50 values 11.5, 9.3 and 9.9 μM respectively. Compound 130 was found to be most potent molecule with IC50 = 8.1 μM. The anticancer studies showed that compounds 128, 129, 130 and 131 represent novel leads for further development [64]. The structures of potent molecules are shown in Fig. 19 with their IC50 values. The different substitutions on the basic ring are differentiated by the different colour.
Fig. 19.

The structure of potent compounds against colon cancer.
3.12.2. Breast cancer
In 2012, Yadlapalli et al. synthesized diarylpyrazole ligated dihydropyrimidines and synthesized compounds were screened for in vitro anticancer activity against the MCF-7 human breast cancer cell line. They synthesized twenty three compounds, in which six compounds found to be active. The compounds were screened by using SRB assay protocol [65]. All compounds were tested at four dose levels (1 × 10−7 M, 1 × 10−6 M, 1 × 10−5 M and 1 × 10−4 M) and each experiment was repeated thrice. Six compounds 132, 133, 134, 135, 136 and 137 were found to inhibit growth of MCF-7 cell line at GI50 (concentration of compound causing 50% inhibition of cell growth) exhibited excellent growth inhibition of MCF-7 cell lines. Compound 135 and 136 were found to be the most potent molecules among the six compounds. Compound 136 show the more affinity to inhibition of cell growth with GI50 of 33.2 μM and compound 136 also show a good dose response. Structure activity relationship (SAR) study suggests that the compounds containing thio-group are more potent than the compounds containing oxo-group. The SAR study showed that presence of thio-urea functional group in DHPMs enhances the anticancer activity of these types of scaffolds [66]. The structures of potent compound are given in Fig. 20 with their IC50 values.
Fig. 20.

The structure of potent molecules against breast cancer.
3.12.3. Blood cancer
In 2004, Holla et al. synthesized thiazolodihydropyrimidinones as an anticancer agent. They screened the synthesized compounds against the sixty human cancer cell lines. But they found that the synthesized compound show maximum activity against the leukemia cancer cell line. They were used different leukemia cancer cell lines like CCRF-CEM, HL-60[TB], K-562, MOLTA-4, RPMI-8226 and SR. The compound 138 displayed maximum inhibitory action against the SR and CCRF-CEM cancer cell line. Among the synthesized compound only one molecule showed the excellent potency against both cell lines. The Growth inhibition of 50% (GI50) value was found to be 3.34 μM and 3.24 μM respectively. The structure of the potent compound is shown in Fig. 21 . The SAR study suggests that the presence of 5-nitro-2-furfuryl moiety and 4-chlorophenyl groups as substituent tend to increase the anticancer activity [28].
Fig. 21.
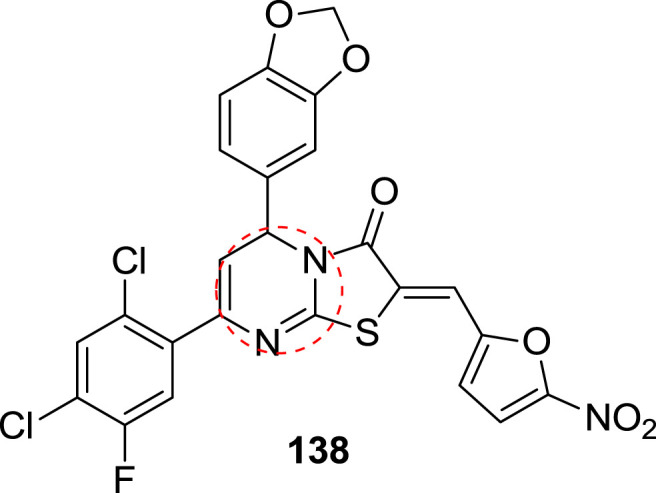
The structure of potent compound for blood cancer.
3.12.4. Anticancer activity against glioma cells
Treptow et al. have synthesized a new hybrid series of fatty acid dihyropyrimidinones which were further investigated for antitumor activity against two glioma cell lines (C6-rat and U-138-MG human) [37]. The relationship between antitumor activity and lipophilicity, unsaturation, number of carbon atoms, and functional group was also determined. A set of compounds derived from palmitic acid (set a), stearic (set b) and oleic acid (set c) was synthesized with the DHPM nucleus. Oxo-monastrol 141a-c and monastrol analogs 142a-c, as well as compounds 139a-c, 140a-c which lacked aromatic ring substitution were screened for cell viability in the C6 rat glioma cell line. The screening was conducted after 24 h of treatment with test compound at concentration ranging from 0 to 200 μM. Compound 139c was the only compound to reduce the C6 cell viability in a concentration dependent manner. Out of the compounds 141a-c, 142a-c only few were able to reduce the cell viability at 50 μM except compound 142b. However, compounds 139a-c, 141a-c, 142a-c were also screened in human glioma cell lines (UG-138-MG human) as described in Fig. 22 . The hybrid DHPM-fatty acid 141a-c and 142a-c were able to reduce the cell viability at 50 μM with 141a (hybrid oxo-monastrol-palmitic acid) being the most potent at 10 μM. Compound 139c again was found to decrease the cell viability in a concentration dependent manner. The result described that the hydroxyl group present in the aromatic ring are essential for the antitumor activity in hybrid DHPM-fatty acid against gliomas. It was also observed that all novel oxo-monastrol and monastrol fatty acid analogs were found to be more potent than monastrol itself. These fatty acid analogs tend to impart lipophilic character which is able to cross the blood brain barrier. This may be considered as a promising approach for overcoming resistance to chemotherapy and help in the development of new anti-tumor drugs.
Fig. 22.

DHPM fatty acid compounds active against glioma cells.
The cytotoxic activity of the compounds was evaluated using organotypic hippocampal slice cultures as models. This culture is composed of different neural cells and were prepared from 6 to 8 day old male Wistar rat using a McWallin tissue chopper and separated in ice-cold HBSS with pH maintained at 7.2. The slices were then placed into Millicell Culture insertsand further transferred to 6-well culture plate. The medium usually consists of 50% MEM, 25% HBSS, 25% horse serum which was changed after every three days and experiment was conducted after every 14 days. Cells were then treated with 200 μM concentrations of 139c, 141a, 141c, 142a and 142c for 24 h. Cellular death in organotypic hippocampal cultures was determined by flouroscent imaging analysis of PI uptake. 5 μM of PI was added to the cultures after 24 h of treatment and incubated for another 1 h. The most active compounds 139c, 141a, 141c, 142a and 142c did not showed any significant signs of neural cell death against glioma cell at 200 μM concentrations after 24 h. It was found that DHPM fatty acids 139c, 141c, 142a did not cause neural cell damage. Whereas, the compound 141a and 142c did not increase the neural cell death by at least 20-times and 4-times the active glioma concentration at10 μM and 50 μM respectively.
3.13. Transient receptor potential A1 (TRPA1) antagonist activity
In 2012, Gijsen et al. synthesized tricyclic 3,4-dihydropyrimidine-2-thione derivatives as potent TRPA1 antagonists. TRPA1 is believed to function as a mechanical and chemical stress sensor. The specific function of this protein may involve a role in signal transduction and growth control. All the synthesized compounds were tested on both the human and rat TRPA1 channel. In general, a similar activity was monitored for the human and rat channel, with most of the compounds, a slightly higher potency for rat TRPA1 was observed. At all concentrations the tested compounds behaved as true antagonists, in competition with the applied agonists. This is unlike many electrophilic ligands, which can display functional antagonism via desensitization of the channel, depending on their concentration. Introduction of various substituents on the aryl substituent of 143 had a strong effect on the potency of the tricyclic DHPMs. Ortho- and para-substituted compounds 144, 146 and 148 led to a drop in activity compared to unsubsituted compound 128. In variance, meta-substitution such as in 145, 147, 149, 150 and 151 increased the potency relative to compound 143. Especially meta-methoxy substituted compound 147 displayed a more than 10-fold enhanced potency and additional analogs around compound 147 were investigated. Combination of a meta-methoxy substituent with additional methoxy substituents as in 151–152 led to a loss in activity. Increase in the length of methoxy group proved to be better tolerated and compound 153 and 154 being the most potent compounds identified [67]. The structure and IC50 value of the compound are given in Fig. 23 and Table 9 respectively.
Fig. 23.

The structure of potent TRPA1 antagonists.
Table 9.
Anti-TRPA1screening of compound 128–139 in human and rat.
| Compound No. | hTRPA1 IC50 (μM) | rTRPA1 IC50 (μM) |
|---|---|---|
| 143 | 1.8 | 1.9 |
| 144 | 2.07 | 3.39 |
| 145 | 1.1 | 0.39 |
| 146 | >10 | 9.4 |
| 147 | 0.13 | 0.02 |
| 148 | 3.1 | 3.5 |
| 149 | 0.5 | 0.071 |
| 150 | 0.71 | 0.044 |
| 151 | >10 | 2.7 |
| 152 | >10 | 0.8 |
| 153 | 0.05 | 0.011 |
| 154 | 0.07 | 0.010 |
3.14. Anti-SARS activity
In 2010, Ramajayam et al. synthesized a series of pyrimidines and evaluated for the Severe acute respiratory syndrome (SARS) activity. The target compounds were tested for anti-SARS activity against SARS-CoV 3CLpro, by using previously developed assay method containing 0.05 μM SARS 3CLpro, 6 μM fluorogenic substrate Dabcyl-KTSAVLQSGFRKME-Edans and 50 μM of test compounds. Enhanced fluorescence of the reactions in the buffer of 20 μM Bis-Tris at pH 7.0 was monitored at 538 nm with excitation at 355 nm using a fluorescence plate reader. The compounds which inhibited more than 50% of the protease activity at 50 μM were selected for the next assay run at 10 μM for IC50 calculation. Only two compounds were found to be active against SARS-CoV 3CLpro among the fourteen molecules. The IC50 values of the active (155 &156) compounds are given 10.6 and 6.1 μM respectively. Compound 155 was found to be the most potent molecule. The SAR study showed that when the R′ position of the C-4 atom substituted by the nitro group and also R was replaced by Cl group. The potency of the molecule was increased markedly. When the both R′ position was replaced with the nitro group. The activity of the compound was decreased markedly [68]. Structure of molecules is shown in Fig. 24 with their IC50 values (see Fig. 25).
Fig. 24.

The structure of potent anti SARS compounds.
Fig. 25.

Structure of MCH1 receptor antagonist.
3.15. DHPM as melanin-concentrating hormone (MCH 1) receptor antagonists
Melanin-concentrating hormone is a cyclic 19-amino acid neuropeptide found mainly in fish and mammalian brain [69]. The biological functions of MCH are mediated by two receptors, MCH1 and MCH2. It plays vital role in regulating food intake, water balance, energy metabolism, attention, memory and psychiatric disorders. It mostly controls the food intake and stress in rodents. Mice generally lacks MCH gene encoding which is found to be lean, hypophagic with elevated metabolic rate. The overexpression of MCH gene in mice is suspectible to obesity and insulin resistance. Most of the in vitro and in vivo pharmacology of MCH1 receptor has been published which make it as an attractive target for the development of antagonist. Various MCH1 receptor antagonists have been developed out of which (+) SNAP-7941(157) has been found to have high affinity and selectivity towards MCH1 receptor (Fig. 25) [70].
The data for affinity of mammalian cell lines which express MCHR1 was found as Kb = 0.57 nM and more than 1000 times selectivity was observed in MCH (Ki = 15 ± 0.11 nM). When in vivo assays were performed on rats, the anorectic acid of 157 was observed by decreasing the palatable food intake of rats without working as malaise agent. Moreover, Diet-induced obese study (DIO) was done in rats, which revealed that 157 exhibited continous weight reduction in rats and also lead to 26% less weight than the vehicle-treated counterpart in 4 weeks period which is almost 2 times than the data obtained from the standard clinical drug fenfluramine.
Furthermore interestingly, 157 have also been found to have moderate antidepressant and antianxiety activity in different paradigms However, different statement and results have been made by different researchers on their antidepressant and anxiolytic activity. But recently Millan et al. have published that MCH1 antagonist tends to increase social recognition paradigm of rats and also enhances the level of acetylcholine in frontal cortex of rats by carrying out dialysis study at reasonable doses.
3.16. DHPM inhibitors of ROCK1 as potential therapeutic agent for cardiovascular disease
Rho-associated kinase isoform 1 (ROCK1) is a protein serine/threonine kinase enzyme associated with diverse cellular signaling functions such as smooth muscle contraction, cytoskeleton rearrangement, cell migration and cell proliferation. ROCK 1 is considered as a potential therapeutic target for the treatment of cardiovascular diseases and hypertension. Sehon et al. reported a new class of dihyropyrimidinone derieved amides (158–180) which were then evaluated for their activity against ROCK 1 [71]. The 4-OMe group present in the compound 158 was found to have better oral bioavailablity (35%) compared to compound 163 (0%) and therefore it was further studied. Compound 172 substituted with 2-F, 4-Cl group which are found to be optimal for the activity against ROCK1. It was found to possess good oral bioavailability at 58% and is 100 times more selective than other selected kinases. It revealed a remarkable IC50 value of 200 nM when the rat aortic contraction assay was performed. However, IC50 value against RSK1 and p70S6K was 510 and 2100 respectively. But it possessed significant P450 inhibition against CYP2D6 which appears to be general for the 4-OMe series and is insurmountable. Because of these issues, the focus was shifted to compound 163 which was substituted with pyridyl ring in which ROCK1 enzyme activity was maintained and CYP2D6 was substantially diminished but the oral bioavailability was decreased. Compound 176 and 178 were found to have improved oral bioavailability of 49% and 53% respectively as shown in Fig. 26 . Compound 176 consist of chloro group which showed improved half-life in rats (1.8 h), good selectivity upto 100 folds against a panel of 31 kinases, IC50 of 256 nM and has improved P450 profile. It was therefore selected for in vivo efficacy study and was used in a spontaneously hypertensive rat (SHR) model for hypertension. This revealed a drop in blood pressure of 25 mm of Hg at a single dose of 30 mg/kg.
Fig. 26.

Structure of active inhibitors against ROCK1.
3.17. α1a receptor antagonist activity
Benign prostatic hyperplasia (BPH) is the progressive enlargement of the prostate gland. It has been estimated that it may affect atleast 50% of the male population in the United States by 50 years of age. However, the incidence rate may increase to 85% by the age of 80 [73], [72]. BPH is a result of two mechanistic components: mechanical and dynamic component. Mechanical component is a result of increased prostatic mass which is mainly attributed due to the 5α-dihydrotestosterone. 5α-reductase is the enzyme responsible for the conversion of testosterone to 5α-dihydrotestosterone. Various inhibitors of this enzyme such as finasteride have been clinically proven to reduce the size of the prostate gland and decrease the symptoms of BPH. The dynamic component of BPH is attributed to the endogeneous adrenergic tone which restricts the flow through urethra. Adrenergic receptor α 1 antagonists such as terazosin, relieves symptoms of BPH by relaxing lower urinary tract tissue and finally reducing prostatic and uretheral tone. Tamsulosin is considered as the ‘prostate selective’ α 1 antagonists and is currently marketed due to better therapeutic index when compared to terazosin as shown in Fig. 27 . Recently it has been found that niguldipine, a calcium channel blocker was found to be potent α 1a receptor subtype antagonists with 100 times greater selectivity than other α 1receptor subtypes (see Fig. 28).
Fig. 27.

Currently available α1 antagonists for prostate cancer.
Fig. 28.

Compounds tested for α1a binding affinity.
Several modifications have been done over niguldipine skeleton and thereby led to the formation of dihydropyrimidinone moiety with two logical sites of attachment of the piperidine containing side chain with most important attachment being at the N-3 position of DHP. It was found that the exact structure of central moiety is not important but the mode of attachment of the piperidine containing side-chain via amide bond formation of the DHP C-5 carboxylate is crucial to produce potent and selective compounds. Thus, 4-aryldihydropyrimidinone heterocycle derivatives attached to aminopropyl-4-arylpiperidine via C-5 amide were synthesized and were then investigated for their in vitro and in vivo binding affinity of α 1 receptor antagonist. The binding affinity is expressed as Ki±SEM (nM).
It was found that among 4-arylpiperidines derivative (187–214), 2-cyanophenyl 187 (0.13 ± 0.015 nM) was found to be as potent as 4-fluorophenyl 191 (0.17 ± 0.030 nM) but has greater selectivity. However, combination of two, slightly increases the potency to similar level as that observed for 195 (0.07 ± 0.020 nM) which possess extra ordinary binding affinity to the α 1 receptor subtype. 2-pyridyl substituent was found to be highly selective but 5–100 times less potent than others except when combined with the 6-methoxymethyl DHP 202 (0.45 ± 0.16 nM). The 4-position of the piperidine was also examined for the receptor binding affinity. It revealed that hydroxy substituent leads to slight decrease in the binding affinity with modest increase in the selectivity when relative to the unsubstituted part (203 vs 191). However, cyano substitution possessed additional selectivity against the α 1b and α 1d receptor subtypes along with the α 1a binding affinity 204 (0.17 ± 0.040 nM) and 205 (0.16 ± 0.070 nM). The triflouromethyl group at the 6-position of DHP was not essential for binding (189, 193, 197 and 201) but hydrogen, methyl, methoxymethyl described excellent potency and 100 times greater selectivity relative to other α 1a subtype.
The polarity of substituents attached may limit absorption therefore, alkylation and acylation of the DHP nitrogens was examined for the effect on the binding affinity. Methylation of N-1 improves selectivity while methylation at N-3 decreased but both of them possessed similar potencies [181(0.18 ± 0.025 nM), and 206(0.14 ± 0.040 nM) vs 192(0.39 ± 0.05 nM)]. On combining N-1 methylated DHP with 4-cyanopiperdines, extraordinary selective compounds were obtained (201–213). However on acylation of N-3 with acetyl 207(2.2 ± 0.10 nM) and carbomethoxy 208(0.75 ± 0.010 nM), a decrease in both selectivity and potency was observed. Compound 194 was the only compound to exhibit 1000 time greater selectivity and α 1a receptor binding affinity (0.24 ± 0.050 nM) than any other compound synthesized.(see Fig. 29).
Fig. 29.

Compounds tested for α1a binding affinity.
Many of the compounds were also investigated for in vivo pharmacodynamic and pharmacokinetic parameters. The compounds were tested on rat models where the prostate of anesthetized rat was exposed. Compounds were tested for their inhibiton (AD50 (μg/kg) value) of contractile response induced by the α 1a selective agoinst A61063 [29], [26]. Compounds containing flouro and cyano substituent on the 4-arylpiperidine were found to be quiet potent with AD50 value below 2 μg/kg whereas the piperidine X = substituent followed a trend as OH < H < CN (203 vs 191 vs 204). The pharmacodynamic parameter i.e. duration of action for majority of compounds which was measured at agonist concentration and is 4 times greater AD50 and was found to be short and is unexplainable. Compounds with marked plasma-half lives were also found to have short duration of action as others. Methylation at N-1 nitrogen of DHP was found to have increased duration of action (181, 185, 209 and 211–214). The in vitro metabolism of most of the compounds (187, 188, 189, 194–196, 198, 209) was examined using human liver microsomal preparations (10–50 μM substrate, microsomal protein 2 mg/mL, for 1 h, LC-MS/MS detection). These experiments revealed rapid N-dealkylation of the piperidine and N-3 methyl of the DHP as well as the desaturation of DHP nucleus to the pyrimidine nucleus as primary metabolic pathways.(see Fig. 30).
Fig. 30.

The structure of compounds investigated for the in vitro and in vivo study.
Compound 194 and 209 were able to pass first-pass metabolism in rats and therefore their pharmacodynamic and pharmacokinetic parameters were evaluated in dog. Compound 194 was found to have long half-life in dogs (7–8 h) thus once a daily dosing is appropriate in humans. Pharmacodynamic efficacy and selectivity was done by canine intraurethral pressure (IUP) assay for compound 194. This leads to the simultaneous monitoring of the heart rate and blood pressure. When determined for inhibition of phenylepherine-induced changes in IUP, Compound 194 was found to be more potent than terazosin. It was also found to have longer duration of action of more than 6 h.
4. Conclusion
Dihydropyrimidinones are prototype acting via various mechanism that emerge out to be potent in several diseases. Dihydropyrimidinones revolutionized the chemistry of purines as well as pyrimidines by their different biological activities which make them a advantageous scaffold. Their antagonistic nature towards the natural pyrimidines makes them powerful candidates for the synthesis of various effective and successful molecules. A number of drugs like 5-fluorouracil, idoxuridine, methythiouracil, emivirin, aminophylline containing dihydropyrimidinones are already discovered and effectively used in the prevention or treatment of multiple diseases. The DPHMS and their derivatives are the versatile molecules, which have been explored for the various diseases as antimicrobial, antihypertensive, anti-HIV, anticancer etc. The researchers explored their SAR, as well as binding mode through molecular modeling studies. In addition, recognition of a rational picture towards the appropriate substitutions accountable for its effectiveness and toxicity may be a future skeleton so that the toxicity problems related with the dihydropyrimidinones can be recognized and overcome.
Conflict of interest
There is no conflict of interest.
Acknowledgements
We sincerely thank faculty of ISF College of pharmacy for his support and advice at all times.
References
- 1.Karelson M.M., Katritzky A.R., Szafran M., Zerner M.C. Quantitative predictions of tautomeric equilibria for 2-, 3-, and 4-substituted pyridines in both the gas phase and aqueous solution: combination of AM1 with reaction field theory. J. Org. Chem. 1989;54:6030–6034. [Google Scholar]
- 2.Soni R., Singh G., Kaur R., Kaur G., Gill R.K., Bariwal J. Chemistry & biology interface. Chem. Biol. 2014;4:163–175. [Google Scholar]
- 3.Sondhi S.M., Goyal R.N., Lahoti A.M., Singh N., Shukla R., Raghubir R. Synthesis and biological evaluation of 2-thiopyrimidine derivatives. Bioorg. Med. Chem. 2005;13:3185–3195. doi: 10.1016/j.bmc.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 4.Rovnyak G.C., Atwal K.S., Hedberg A., Kimball S.D., Moreland S., Gougoutas J.Z., O'Reilly B.C., Schwartz J., Malley M.F. Dihydropyrimidine calcium channel blockers. 4. Basic 3-substituted-4-aryl-1, 4-dihydropyrimidine-5-carboxylic acid esters. Potent antihypertensive agents. J. Med. Chem. 1992;35:3254–3263. doi: 10.1021/jm00095a023. [DOI] [PubMed] [Google Scholar]
- 5.Dragovich P.S., Fauber B.P., Corson L.B., Ding C.Z., Eigenbrot C., Ge H., Giannetti A.M., Hunsaker T., Labadie S., Liu Y. Identification of substituted 2-thio-6-oxo-1, 6-dihydropyrimidines as inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2013;23:3186–3194. doi: 10.1016/j.bmcl.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Agarwal N., Srivastava P., Raghuwanshi S.K., Upadhyay D., Sinha S., Shukla P., Ram V.J. Chloropyrimidines as a new class of antimicrobial agents. Bioorg. Med. Chem. 2002;10:869–874. doi: 10.1016/s0968-0896(01)00374-1. [DOI] [PubMed] [Google Scholar]
- 7.Bhosle M.R., Deshmukh A.R., Pal S., Srivastava A.K., Mane R.A. Synthesis of new thiazolylmethoxyphenyl pyrimidines and antihyperglycemic evaluation of the pyrimidines, analogues isoxazolines and pyrazolines. Bioorg. Med. Chem. Lett. 2015;25:2442–2446. doi: 10.1016/j.bmcl.2015.03.068. [DOI] [PubMed] [Google Scholar]
- 8.Lauro G., Strocchia M., Terracciano S., Bruno I., Fischer K., Pergola C., Werz O., Riccio R., Bifulco G. Exploration of the dihydropyrimidine scaffold for the development of new potential anti-inflammatory agents blocking prostaglandin E2 synthase-1 enzyme (mPGES-1) Eur. J. Med. Chem. 2014;80:407–415. doi: 10.1016/j.ejmech.2014.04.061. [DOI] [PubMed] [Google Scholar]
- 9.Said S.A., Amr A.E.-G.E., Sabry N.M., Abdalla M.M. Analgesic, anticonvulsant and anti-inflammatory activities of some synthesized benzodiazipine, triazolopyrimidine and bis-imide derivatives. Eur. J. Med. Chem. 2009;44:4787–4792. doi: 10.1016/j.ejmech.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Tale R.H., Rodge A.H., Hatnapure G.D., Keche A.P. The novel 3, 4-dihydropyrimidin-2 (1H)-one urea derivatives of N-aryl urea: synthesis, anti-inflammatory, antibacterial and antifungal activity evaluation. Bioorg. Med. Chem. Lett. 2011;21:4648–4651. doi: 10.1016/j.bmcl.2011.03.062. [DOI] [PubMed] [Google Scholar]
- 11.Naidu B.N., Sorenson M.E., Patel M., Ueda Y., Banville J., Beaulieu F., Bollini S., Dicker I.B., Higley H., Lin Z. Synthesis and evaluation of C2-carbon-linked heterocyclic-5-hydroxy-6-oxo-dihydropyrimidine-4-carboxamides as HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2015;25:717–720. doi: 10.1016/j.bmcl.2014.11.060. [DOI] [PubMed] [Google Scholar]
- 12.Trivedi A.R., Bhuva V.R., Dholariya B.H., Dodiya D.K., Kataria V.B., Shah V.H. Novel dihydropyrimidines as a potential new class of antitubercular agents. Bioorg. Med. Chem. Lett. 2010;20:6100–6102. doi: 10.1016/j.bmcl.2010.08.046. [DOI] [PubMed] [Google Scholar]
- 13.Dömling A. The discovery of new isocyanide-based multi-component reactions. Curr. Opin. Chem. Biol. 2000;4:318–323. doi: 10.1016/s1367-5931(00)00095-8. [DOI] [PubMed] [Google Scholar]
- 14.Beck B., Magnin-Lachaux M., Herdtweck E., Dömling A. A novel three-component butenolide synthesis. Org. Lett. 2001;3:2875–2878. doi: 10.1021/ol016328u. [DOI] [PubMed] [Google Scholar]
- 15.Armstrong R.W., Combs A.P., Tempest P.A., Brown S.D., Keating T.A. Multiple-component condensation strategies for combinatorial library synthesis. Acc. Chem. Res. 1996;29:123–131. [Google Scholar]
- 16.Patil A.D., Kumar N.V., Kokke W.C., Bean M.F., J.Freyer A., Brosse C.D., Mai S., Truneh A., Carte B. Novel alkaloids from the sponge Batzella sp.: inhibitors of HIV gp120-human CD4 binding. J. Org. Chem. 1995;60:1182–1188. [Google Scholar]
- 17.Kapoor T.M., Mitchison T.J. Eg5 is static in bipolar spindles relative to tubulin evidence for a static spindle matrix. J. Cell Biol. 2001;154:1125–1134. doi: 10.1083/jcb.200106011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russowsky D., Canto R.F., Sanches S.A., D’oca M.G., De Fatima A., Pilli R.A., K.Kohn L., Antônio M.A., De Carvalho J.E. Synthesis and differential antiproliferative activity of Biginelli compounds against cancer cell lines: monastrol, oxo-monastrol and oxygenated analogues. Bioorg. Chem. 2006;34:173–182. doi: 10.1016/j.bioorg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Chang R.S., Chen T.-B., O'Malley S.S., Pettibone D.J., Disalvo J., Francis B., Bock M.G., Freidinger R., Nagarathnam D., Miao S.W. In vitro studies on L-771,688 (SNAP 6383), a new potent and selective α 1A-adrenoceptor antagonist. Eur. J. Pharmacol. 2000;409:301–312. doi: 10.1016/s0014-2999(00)00854-2. [DOI] [PubMed] [Google Scholar]
- 20.Atwal K.S., Swanson B.N., Unger S.E., Floyd D.M., Moreland S., Hedberg A., O'Reilly B.C. Dihydropyrimidine calcium channel blockers. 3. 3-Carbamoyl-4-aryl-1, 2, 3, 4-tetrahydro-6-methyl-5-pyrimidinecarboxylic acid esters as orally effective antihypertensive agents. J. Med. Chem. 1991;34:806–811. doi: 10.1021/jm00106a048. [DOI] [PubMed] [Google Scholar]
- 21.Qian X., Wolff A.A., Bergnes G. Progress on mitotic kinesin inhibitors as anti-cancer therapeutics. Annu. Rep. Med. Chem. 2006;41:263–274. [Google Scholar]
- 22.Luszcki J.J., Jankiewicz K., Jankiewicz M., Czuczwar S.J. Pharmacokinetic and pharmacodynamic interactions of aminophylline and topiramate in the mouse maximal electroshock-induced seizure model. Eur. J. Pharmacol. 2007;562:53–59. doi: 10.1016/j.ejphar.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 23.Dragunow M. Adenosine receptor antagonism accounts for the seizure-prolonging effects of aminophylline. Pharmacol. Biochem. Behav. 1990;36:751–755. doi: 10.1016/0091-3057(90)90072-p. [DOI] [PubMed] [Google Scholar]
- 24.Robertson H., Riives M., Black D., Peterson M. A partial agonist at the anticonvulsant benzodiazepine receptor: reversal of the anticonvulsant effects of Ro 15–1788 with CGS-8216. Brain Res. 1984;291:388–390. doi: 10.1016/0006-8993(84)91275-7. [DOI] [PubMed] [Google Scholar]
- 25.Diasio R.B., Harris B.E. Clinical pharmacology of 5-fluorouracil. Clin. Pharmacokinet. 1989;16:215–237. doi: 10.2165/00003088-198916040-00002. [DOI] [PubMed] [Google Scholar]
- 26.Bose D.S., Sudharshan M., Chavhan S.W. New protocol for Biginelli reaction-a practical synthesis of Monastrol. Arkivoc. 2005;3:228–236. [Google Scholar]
- 27.Matthews J.M., Liotta F., Hageman W., Rivero R.A., Westover L., Yang M., Xu J., Demarest K. Discovery of a dihydropyrimidine series of molecules that selectively mimic the biological actions of calcitonin. Bioorg. Med. Chem. Lett. 2004;14:1155–1159. doi: 10.1016/j.bmcl.2003.12.071. [DOI] [PubMed] [Google Scholar]
- 28.Holla B.S., Rao B.S., Sarojini B., Akberali P. One pot synthesis of thiazolodihydropyrimidinones and evaluation of their anticancer activity. Eur. J. Med. Chem. 2004;39:777–783. doi: 10.1016/j.ejmech.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Yadlapalli R.K., Chourasia O., Perali R.S. A facile one-pot synthesis of 2-thioxo-dihydropyrimidines and polyfunctionalized pyran derivatives as mimics of novel calcium channel modulators. Tetrahedron Lett. 2012;53:6725–6728. [Google Scholar]
- 30.Shaabani A., Seyyedhamzeh M., Maleki A., Hajishaabanha F. Diketene as an alternative substrate for a new biginelli-like multicomponent reaction: one-pot synthesis of 5-carboxamide substituted 3, 4-dihydropyrimidine-2 (1H) ones. Tetrahedron. 2010;66:4040–4042. [Google Scholar]
- 31.Kappe C.O. Recent advances in the Biginelli dihydropyrimidine synthesis. New tricks from an old dog. Acc. Chem. Res. 2000;33:879–888. doi: 10.1021/ar000048h. [DOI] [PubMed] [Google Scholar]
- 32.[a] M. Reactions, Zhu, J., Bienaymé, H., Eds, in, Wiley-VCH: Weinheim, 2005.; [b] De Graaff C., Ruijter E., Orru R.V. Recent developments in asymmetric multicomponent reactions. Chem. Soc. Rev. 2012;41:3969–4009. doi: 10.1039/c2cs15361k. [DOI] [PubMed] [Google Scholar]; [c] Hao W.-J., Xu X.-P., Bai H.-W., Wang S.-Y., Ji S.-J. Efficient multicomponent strategy to pentacyclic pyrazole-fused naphtho [1, 8-fg] isoquinolines through cleavage of two carbon-carbon bonds. Org. Lett. 2012;14:4894–4897. doi: 10.1021/ol302452j. [DOI] [PubMed] [Google Scholar]
- 33.Gong K., Wang H., Wang S., Ren X. β-Cyclodextrin-propyl sulfonic acid: a new and eco-friendly catalyst for one-pot multi-component synthesis of 3,4-dihydropyrimidones via Biginelli reaction. Tetrahedron. 2015;71:4830–4834. [Google Scholar]
- 34.[a] Crini G. Review: a history of cyclodextrins. Chem. Rev. 2014;114:10940–10975. doi: 10.1021/cr500081p. [DOI] [PubMed] [Google Scholar]; [b] Challa R., Ahuja A., Ali J., Khar R. Cyclodextrins in drug delivery: an updated review. Aaps Pharmscitech. 2005;6:E329–E357. doi: 10.1208/pt060243. [DOI] [PMC free article] [PubMed] [Google Scholar]; [c] Aher L., Makhloufi-Chebli M., Dermeche L., Boutemeur-khedis B., Rabia C., Silva A.M., Hamdi M. Keggin and Dawson-type polyoxometalates as efficient catalysts for the synthesis of 3,4-dihydropyrimidinones: experimental and theoretical studies. Tetrahedron Lett. 2016;57:1492–1496. [Google Scholar]
- 35.Benazzouz A., Makhloufi-Chebli M., Khatir-Hamdi N., Boutemeur-Khedis B., M. Silva A., Hamdi M. A facile synthesis of new coumarin-3, 4-dihydropyrimidin-2 (1H) ones/thiones dyads. Tetrahedron. 2015;71:3890–3894. [Google Scholar]
- 36.Saher L., Makhloufi-Chebli M., Dermeche L., Boutemeur-Khedis B., Rabia C., Silva A.M., Hamdi M. Keggin and Dawson-type polyoxometalates as efficient catalysts for the synthesis of 3,4-dihydropyrimidinones: experimental and theoretical studies. Tetrahedron Lett. 2016;57:1492–1496. [Google Scholar]
- 37.Treptow T.G., Figueiró F., Jandrey E.H., Battastini A.M., Salbego C.G., Hoppe J.B., Taborda P.S., Rosa S.B., Piovesan L.A., D'oca C.D.R.M. Novel hybrid DHPM-fatty acids: synthesis and activity against glioma cell growth in vitro. Eur. J. Med. Chem. 2015;95:552–562. doi: 10.1016/j.ejmech.2015.03.062. [DOI] [PubMed] [Google Scholar]
- 38.Qiu Y., Sun H., Ma Z., Xia W. Efficient, stable, and reusable Lewis acid–surfactant-combined catalyst: one-pot Biginelli and solvent-free esterification reactions. J. Mol. Catal. A Chem. 2014;392:76–82. [Google Scholar]
- 39.Kaur G., Gupta M., Paul S., Rajnikanth K.V., Gupta SiO2-CuCl2: an efficient and recyclable heterogeneous catalyst for one-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones. J. Mol. Catal. A Chem. 2014;392:260–269. [Google Scholar]
- 40.Kuraitheerthakumaran A., Pazhamalai S., Gopalakrishnan M. Microwave-assisted multicomponent reaction for the synthesis of 3,4-dihydropyrimidin-2(1H)-ones and their corresponding 2(1H)-thiones using lanthanum oxide as a catalyst under solvent-free conditions. Arab. J. Chem. 2011;9:5461–5465. [Google Scholar]
- 41.Slimi H., Moussaoui Y., Ben salem R. Synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones/thiones via Biginelli reaction promoted by bismuth (III) nitrate or PPh3 without solvent. Arab. J. Chem. 2011;9:5510–5514. [Google Scholar]
- 42.Banik B.K., Reddy A.T., Datta A., Mukhopadhyay C. Microwave-induced bismuth nitrate-catalyzed synthesis of dihydropyrimidones via Biginelli condensation under solventless conditions. Tetrahedron Lett. 2007;48:7392–7394. [Google Scholar]
- 43.Mansoor S.S., Shafi S.S., Ahmed S.Z. An efficient one-pot multicomponent synthesis of 3, 4-dihydropyrimidine-2-(1H)-ones/thiones/imines via a Lewis base catalyzed Biginelli-type reaction under solvent-free conditions. Arab. J. Chem. 2011;9:5846–5851. [Google Scholar]
- 44.Attri P., Bhatia R., Gaur J., Arora B., Gupta A., Kumar N., Choi E.H. Triethylammonium acetate ionic liquid assisted one-pot synthesis of dihydropyrimidinones and evaluation of their antioxidant and antibacterial activities. Arab. J. Chem. 2017;10:206–214. [Google Scholar]
- 45.Wan J.-P., Lin Y., Liu Y. Catalytic asymmetric Biginelli reaction for the enantioselective synthesis of 3, 4-dihydropyrimidinones (DHPMs) Curr. Org. Chem. 2014;18:687–699. [Google Scholar]
- 46.Muñoz-Muñiz O., Juaristi E. An enantioselective approach to the Biginelli dihydropyrimidinone condensation reaction using CeCl3 and InCl3 in the presence of chiral ligands. Arkivoc. 2003;11:16–26. [Google Scholar]
- 47.Chen X.-H., Xu X.-Y., Liu H., Cun L.-F., Gong L.-Z. Highly enantioselective organocatalytic Biginelli reaction. J. Am. Chem. Soc. 2006;128:14802–14803. doi: 10.1021/ja065267y. [DOI] [PubMed] [Google Scholar]
- 48.Thorat P.B., Waghmode N.A., Karade N.N. Direct metal-free O-arylation of Biginelli 4-aryl-6-methyl-pyrimidine-2 (1H)-one derivatives using diaryliodonium salts. Tetrahedron Lett. 2014;55:5718–5721. [Google Scholar]
- 49.Karami B., Khodabakhshi S., Akrami S., Farahi M. Regiospecific strategies for the synthesis of novel dihydropyrimidinones and pyrimidopyridazines catalyzed by molybdate sulfuric acid. Tetrahedron Lett. 2014;55:3581–3584. [Google Scholar]
- 50.Ábrányi-Balogh P., Dancsó A., Frigyes D., Volk B., Keglevich G., Milen M. Convenient synthesis of 1-aryl-9H-β-carboline-3-carbaldehydes and their transformation into dihydropyrimidinone derivatives by Biginelli reaction. Tetrahedron. 2014;70:5711–5719. [Google Scholar]
- 51.Winter C.A., Risley E.A., Nuss G.W. Carrageenin-induced edema in hind paw of the rat as an assay for antiinflammatory drugs. Exp. Biol. Med. 1962;111:544–547. doi: 10.3181/00379727-111-27849. [DOI] [PubMed] [Google Scholar]
- 52.Bahekar S.S., Shinde D.B. Synthesis and anti-inflammatory activity of some [4, 6-(4-substituted aryl)-2-thioxo-1, 2, 3, 4-tetrahydro-pyrimidin-5-yl]-acetic acid derivatives. Bioorg. Med. Chem. Lett. 2004;14:1733–1736. doi: 10.1016/j.bmcl.2004.01.039. [DOI] [PubMed] [Google Scholar]
- 53.Aly H.M., Kamal M.M. Efficient one-pot preparation of novel fused chromeno [2, 3-d] pyrimidine and pyrano [2, 3-d] pyrimidine derivatives. Eur. J. Med. Chem. 2012;47:18–23. doi: 10.1016/j.ejmech.2011.09.040. [DOI] [PubMed] [Google Scholar]
- 54.Rajanarendar E., Reddy M.N., Murthy K.R., Reddy K.G., Raju S., Srinivas M., Praveen B., Rao M.S. Synthesis, antimicrobial, and mosquito larvicidal activity of 1-aryl-4-methyl-3, 6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2, 3, 6, 10b-tetrahydro-1H-pyrimido [5, 4-c] quinolin-5-ones. Bioorg. Med. Chem. Lett. 2010;20:6052–6055. doi: 10.1016/j.bmcl.2010.08.060. [DOI] [PubMed] [Google Scholar]
- 55.Murthy P., Chatterjee R. Evaluation of two in vitro test systems employing Brugia malayi parasite for prescreening of potential antifilarials. Curr. Sci. 1999;77:1084–1089. [Google Scholar]
- 56.Gaur R., Dixit S., Sahoo M., Khanna M., Singh S., Murthy P. Anti-filarial activity of novel formulations of albendazole against experimental brugian filariasis. J. Parasitol. 2007;134:537. doi: 10.1017/S0031182006001612. [DOI] [PubMed] [Google Scholar]
- 57.Singh B., Mishra M., Saxena N., Yadav G., Maulik P., Sahoo M., Gaur R., Murthy P., Tripathi R. Synthesis of 2-sulfanyl-6-methyl-1, 4-dihydropyrimidines as a new class of antifilarial agents. Eur. J. Med. Chem. 2008;43:2717–2723. doi: 10.1016/j.ejmech.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 58.Alam O., Khan S.A., Siddiqui N., Ahsan W., Verma S.P., Gilani S.J. Antihypertensive activity of newer 1, 4-dihydro-5-pyrimidine carboxamides: synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2010;45:5113–5119. doi: 10.1016/j.ejmech.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 59.Singh P., Junnarkar A., Rao C.S., Varma R., Shridhar D. Acetic acid and phenylquinone writhing test: a critical study in mice. Meth. Find. Exp. Clin. Pharmacol. 1983;5:601–606. [PubMed] [Google Scholar]
- 60.Sondhi S.M., Singh N., Johar M., Kumar A. Synthesis, anti-inflammatory and analgesic activities evaluation of some mono, bi and tricyclic pyrimidine derivatives. Bioorg. Med. Chem. 2005;13:6158–6166. doi: 10.1016/j.bmc.2005.06.063. [DOI] [PubMed] [Google Scholar]
- 61.Khanage S.G., Raju S.A., Mohite P.B., Pandhare R.B. Synthesis and pharmacological evaluation of some new pyrimidine derivatives containing 1,2 4-triazole. Adv. Pharm. Bull. 2012;2:213–222. doi: 10.5681/apb.2012.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar B.P., Sankar G., Baig R.N., Chandrashekaran S. Novel Biginelli dihydropyrimidines with potential anticancer activity: a parallel synthesis and CoMSIA study. Eur. J. Med. Chem. 2009;44:4192–4198. doi: 10.1016/j.ejmech.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 63.Apak R., Güclü K., Özyürek M., Celik S.E. Mechanism of antioxidant capacity assays and the CUPRAC (cupric ion reducing antioxidant capacity) assay. Microchim. Acta. 2008;160:413–419. [Google Scholar]
- 64.Agbaje O.C., Fadeyi O.O., Fadeyi S.A., Myles L.E., Okoro C.O. Synthesis and in vitro cytotoxicity evaluation of some fluorinated hexahydropyrimidine derivatives. Bioorg. Med. Chem. Lett. 2011;21:989–992. doi: 10.1016/j.bmcl.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 65.Skehan P., Storeng R., Scudiero D., Monks A., McMahon J., Vistica D., Warren J.T., Bokesch H., Kenney S., Boyd M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 66.Yadlapalli R.K., Chourasia O., Vemuri K., Sritharan M., Perali R.S. Synthesis and in vitro anticancer and antitubercular activity of diarylpyrazole ligated dihydropyrimidines possessing lipophilic carbamoyl group. Bioorg. Med. Chem. Lett. 2012;22:2708–2711. doi: 10.1016/j.bmcl.2012.02.101. [DOI] [PubMed] [Google Scholar]
- 67.Gijsen H.J., Berthelot D., De Cleyn M.A., Geuens I., Brône B., Mercken M. Tricyclic 3,4-dihydropyrimidine-2-thione derivatives as potent TRPA1 antagonists. Bioorg. Med. Chem. Lett. 2012;22:797–800. doi: 10.1016/j.bmcl.2011.12.068. [DOI] [PubMed] [Google Scholar]
- 68.Ramajayam R., Tan K.-P., Liu H.-G., Liang P.-H. Synthesis, docking studies, and evaluation of pyrimidines as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem. Lett. 2010;20:3569–3572. doi: 10.1016/j.bmcl.2010.04.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wan J.-P., Pan Y. Recent advance in the pharmacology of dihydropyrimidinone. Mini Rev. Med. Chem. 2012;12:337–349. doi: 10.2174/138955712799829267. [DOI] [PubMed] [Google Scholar]
- 70.Borowsky B., Durkin M.M., Ogozalek K., Marzabadi M.R., Deleon J., Heurich R., Lichtblau H., Shaposhnik Z., Daniewska I., Blackburn T.P. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat. Med. 2002;8:825–830. doi: 10.1038/nm741. [DOI] [PubMed] [Google Scholar]
- 71.Sehon C.A., Wang G.Z., Viet A.Q., Goodman K.B., Dowdell S.E., Elkins P.A., Semsus S.F., Evans C., Jolivette L.J., Kirkpatick R.B. Potent, selective and orally bioavailable dihydropyrimidine inhibitors of Rho kinase (ROCK1) as potential therapeutic agents for cardiovascular diseases. J. Med. Chem. 2008;51:6631–6634. doi: 10.1021/jm8005096. [DOI] [PubMed] [Google Scholar]
- 72.Barrow J.C., Nantermet P.G., Seknick H.G., Glass K.L., Rittle K.E., Gilbert K.F., Steele T.G., Homnick C.F., Freidinger R.M., Ransom R.W. In vitro and in vivo evaluation of dihydropyrimidinone C-5 amides as potent and selective α1A receptor antagonists for the treatment of benign prostatic hyperplasia. J. Med. Chem. 2000;43:2703–2718. doi: 10.1021/jm990612y. [DOI] [PubMed] [Google Scholar]
- 73.Berry S.J., Coffey D.S., Walsh P.C., Ewing L.L. The development of human benign prostatic hyperplasia with age. J. Urol. 1984;132:474–479. doi: 10.1016/s0022-5347(17)49698-4. [DOI] [PubMed] [Google Scholar]