

Abstract
The biological activity of Mannich bases, a structurally heterogeneous class of chemical compounds that are generated from various substrates through the introduction of an aminomethyl function by means of the Mannich reaction, is surveyed, with emphasis on the relationship between structure and biological activity. The review covers extensively the literature reports that have disclosed Mannich bases as anticancer and cytotoxic agents, or compounds with potential antibacterial and antifungal activity in the last decade. The most relevant studies on the activity of Mannich bases as antimycobacterial agents, antimalarials, or antiviral candidates have been included as well. The review contains also a thorough coverage of anticonvulsant, anti-inflammatory, analgesic and antioxidant activities of Mannich bases. In addition, several minor biological activities of Mannich bases, such as their ability to regulate blood pressure or inhibit platelet aggregation, their antiparasitic and anti-ulcer effects, as well as their use as agents for the treatment of mental disorders have been presented. The review gives in the end a brief overview of the potential of Mannich bases as inhibitors of various enzymes or ligands for several receptors.
Keywords: Aminomethylation, Biological activity, Drug design, Mannich bases, Structure–activity relationships
Graphical abstract
Highlights
-
•
Biological activities of structurally diverse Mannich bases are being reviewed.
-
•
Anticancer, antibacterial and antifungal activities are presented extensively.
-
•
Antimycobacterial, antimalarial and antiviral activities of Mannich bases are also covered.
-
•
Minor biological activities of Mannich bases are also summarized.
-
•
An overview of Mannich bases as inhibitors of enzymes and ligands for receptors is provided.
1. Introduction
The classical Mannich reaction, a three-component condensation between structurally diverse substrates (X–H) containing at least one active hydrogen atom, an aldehyde component (generally R1-CHO) and an amine reagent leads to a class of compounds generally known as Mannich bases 1 (Scheme 1 ). Because Mannich bases may be regarded as derivatives of the substrate obtained through substitution by an aminoalkyl moiety, Mannich reactions are also known as aminoalkylation reactions. In the particular instance when formaldehyde is employed as aldehyde component, the substrate is converted into the corresponding Mannich base through an aminomethylation process. Although primary amines and even ammonia (in the form of an ammonium salt) may be employed as amine reagents in aminomethylations or aminoalkylations, secondary aliphatic amines (R2NH) are the most commonly encountered as amine reagents in the Mannich reaction. As formaldehyde is used to a great extent as aldehyde component in the Mannich reaction, the structural diversity of Mannich bases stems primarily from the miscellaneous types of the substrates that can be subjected to aminomethylation, and secondarily from the variety of amine reagents that can be potentially employed in the Mannich reaction. Regardless of their structural diversity, the substrates should all have an activating functional group as a crucial structural feature that is required to render the substrate active in the Mannich reaction. The carbonyl function in ketones, the phenolic hydroxyl in phenols, the terminal carbon–carbon triple bond in alkynes, the heteroatom in heterocycles, or electron-withdrawing groups that substitute the carbon atom α to the carboxylate group in esters of aliphatic carboxylic acids are common examples of pairs of activating groups and corresponding substrates, but the list is far from being exhaustive. A general classification of the most common types of Mannich bases with respect of the substrates from which they derive and the nature of the atom substituted by the aminomethyl function is given in Fig. 1 . Under normal reaction conditions, substitution of a substrate with a single aminomethyl function results in mono-Mannich bases, but two aminomethyl groups may also be grafted onto a substrate containing more than one active hydrogen atom, leading to double Mannich bases such as 2–5 derived from dialkyl ketones, alkyl aryl ketones, 4-substituted phenols and pyrrole, respectively (Fig. 2 ). Also, the aminomethylation of substrate X–H with amine reagents other than secondary amines (such as ammonia, having three reactive hydrogen atoms at nitrogen, or primary amines R–NH2, having two reactive hydrogen atoms at nitrogen) may lead to tris-Mannich bases 6 and bis-Mannich bases 7, respectively (Fig. 2). In addition, the capability of some polyfunctional substrates to aminomethylate chemoselectively at a single potential reaction site under the appropriate reaction condition, or aminomethylate indiscriminately at multiple reaction sites, or even undergo aminomethylation simultaneously with ring closure, contributes considerably to the structural variety of the resulting Mannich bases. Two excellent, albeit rather old reviews provide more details on the synthesis and reactions of Mannich bases to the interested reader [1], [2].
Scheme 1.

General representation of the Mannich reaction.
Fig. 1.
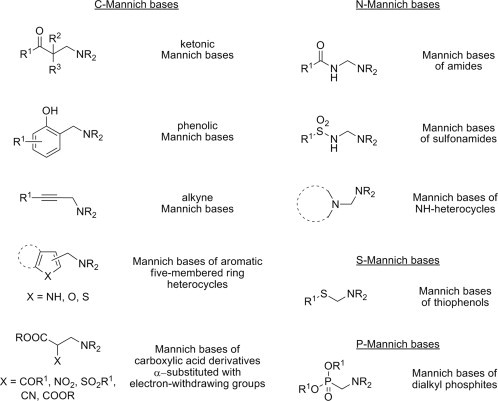
Examples of various types of Mannich bases.
Fig. 2.

Typical examples of double Mannich bases, bis-Mannich bases, and tris-Mannich bases.
Mannich bases have found numerous practical applications in the treatment of natural macromolecular materials such as leather, paper and textiles, the production of synthetic polymers, as additives used by the petroleum industry, as products used in water treatment, analytical reagents, cosmetics, dyes, etc. [3]. Nonetheless, the most important application of the Mannich reaction lies in the field of medicinal chemistry, and this claim is supported by the substantial number of papers published on this topic every year. First of all, Mannich bases could present interesting biological activities, many of these having yet to be discovered through a diligent screening process. Second, aminomethylation of drugs could be used to improve their delivery into the human body. Aminomethylation may increase the hydrophilic properties of drugs through the introduction of a polar function in their structure, the long-known rolicycline being one of the most common examples [4]. The solubility in water of a drug could be further enhanced through the quaternization of the nitrogen atom in its aminomethyl derivative and conversion into an ammonium salt. Alternatively, the lipophilic properties of a drug could be tailored through a Mannich reaction if the appropriate amine reagent is employed [5]. In addition, the aminomethylated drugs could act as prodrugs, releasing the active substance under controlled hydrolytic conditions via deaminomethylation [6] or deamination [7]. In spite of the tremendous potential of Mannich bases in medicinal chemistry, the wealth of information from studies concerning the structure–activity relationship (SAR) involving Mannich bases or the use of aminomethylated drugs as prodrugs does not appear to have inspired many recent literature reviews of consequence, to the best of our knowledge. The present review fills this void by providing a comprehensive coverage of the most relevant developments in the medicinal chemistry of Mannich bases generated exclusively through aminomethylation, the information being ordered according to the reported biological activity. Due the large number of articles published on this topic, the coverage of this review is limited to the last decade.
2. Anticancer and cytotoxic activity
Anticancer properties and cytotoxicity of ketonic Mannich bases (with an emphasis on Mannich bases of type 8 derived from acetophenones [8]) and of structurally related α,β-unsaturated ketones [9] were reviewed 15 years ago. These two groups of compounds were shown to exert their cytotoxic action through the alkylation of cellular thiols such as glutathione or cysteine, and may be useful in sensitizing tumor cells to antineoplastic agents, and even reverse drug resistance [10]. It is therefore no surprise that compounds having both a ketonic Mannich base moiety and an activated unsaturated carbon–carbon double bond in their structure (for example, Mannich bases of chalcones such as 9) have been considered as candidates for the evaluation of the sequential cytotoxicity theory [11]. This theory hypothesizes that the successive release of two or more cytotoxic agents will result in increased toxicity to malignant tissue rather than to normal cells [12]. In addition, Mannich bases 10 of enones, which are easily accessible from alkyl aryl ketones in one synthetic step, also demonstrated marked toxicity towards numerous cancer cell lines [13]. Furthermore, as ortho-phenolic Mannich bases undergo deamination easily to yield ortho-quinone methides, Mannich bases of chalcones derived from either phenolic aldehydes or ketones, a class of compounds for which structure 11 is prototypical, have been examined also as cytotoxic agents [14]. In the last decade, the quest for more potent anticancer agents amongst these four general types of cytotoxic Mannich bases 8–11 (Fig. 3 ) has steadily continued.
Fig. 3.
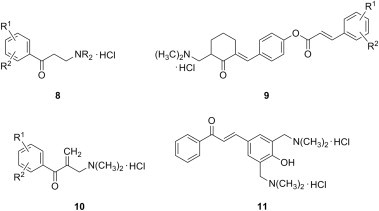
General types of ketonic Mannich bases with anticancer and cytotoxic properties.
Cytotoxicity of ketonic Mannich bases of type 8 with various substitution patterns in the aromatic ring and of a few types of their derivatives has been studied in detail. Gul et al. have shown that the structural modification of single Mannich bases 8 (R1 = R2 = H) derived from acetophenone and secondary aliphatic amines into double Mannich bases 12 (Fig. 4 ) generally results in increased cytotoxicity against mouse renal carcinoma (Renca) and transformed human T-lymphocyte (Jurkat) cell lines [15] to the extent that double Mannich bases were more cytotoxic than reference drugs 5-fluorouracil or melphalan. The cytotoxicity of these single and double Mannich bases 8 and 12, respectively, was reversed when the compounds were used in a brine shrimp bioassay, presumably due to the fast deamination of the double Mannich bases before they could reach their target [16]. Also, ketonic Mannich bases 8 with dimethylamino, 1-piperidinyl, 4-morpholinyl as amine moiety and featuring either an unsubstituted, variously monosubstituted phenyl rings, or a thiophene ring were evaluated with respect of their cytotoxicity towards Jurkat cells [17], [18] or androgen-independent prostate cancer (PC-3) cells [19], and the cytotoxicity for some of these compounds was 2.5- to 5.2-fold higher than that of the standard 5-fluorouracil. In addition, Mannich bases of type 8 derived from 4-aryloxyacetophenones were shown to display moderate cytotoxic properties towards murine L1210 cells as well as human Molt 4/C8 and CEM T-lymphocytes, and a number of these compounds possessed remarkable potencies towards seven human colon cancer cell lines [20].
Fig. 4.
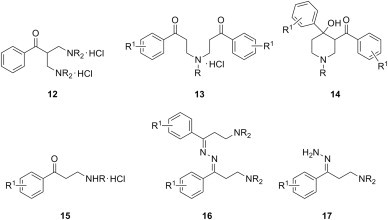
Ketonic double Mannich bases 5, ketonic bis-Mannich bases 6, piperidinols 7, secondary ketonic Mannich bases 8, and derivatives of ketonic Mannich bases with anticancer and cytotoxic properties.
The use of primary aliphatic amines in the Mannich reaction leads to reaction products with diverse structures (Fig. 4). Aminomethylation of acetophenones using methylamine as amine reagent afforded both bis-Mannich bases 13 (R = CH3) and piperidinols 14 (R = CH3), and the evaluation of their cytotoxicity towards Jurkat cells showed that bis-Mannich bases 13 were generally more potent than the corresponding piperidinols 14 or mono-Mannich bases 8 [21]. Besides their ability to alkylate cellular glutathione, compounds 13 (R = CH3) may exert their cytotoxic action through the inhibition of DNA topoisomerase I; as the corresponding piperidinols 14 were generally devoid of DNA topoisomerase I inhibitory action, the authors tentatively attribute the activity of bis-Mannich bases 13 to their linear structure and the possibility of formation of hydrogen bonds with DNA nucleotides [22]. On the other hand, aminomethylation of acetophenones using isopropylamine [23] and n-butylamine [24] as amine reagent yielded only secondary mono-Mannich bases 15 (R = CH(CH3)2, (CH2)3CH3), whose cytotoxicity was evaluated against Huh-7 hepatoma cells, human Jurkat and rat skeletal muscle derived myoblasts (L6) cells. Compared to reference drug 5-fluorouracil, these compounds were 2.1- to 2.8-fold more cytotoxic towards Huh-7 hepatoma cells, 2.6- to 4.2-fold more cytotoxic towards human Jurkat cells, and 1.2- to 2.2-fold more cytotoxic towards L6 cells. Aminomethylation of acetophenones using phenethylamine as amine reagent could led under carefully controlled reaction conditions either to mono-Mannich bases 15 (R = CH2CH2C6H5) [25] or to the corresponding piperidinols 14 (R = CH2CH2C6H5) [26]; the cytotoxicity of these compounds towards androgen-independent prostate cancer (PC-3) cells ranged from 8.2 to 32.1 μM, whereas the best compounds from each series had an average value for DNA topoisomerase I interference of approximately 40%.
Anticancer activity of ketonic Mannich bases has been compared with that of derivatives of the carbonyl function (Fig. 4). A series of azines 16 of ketonic Mannich bases 8 were designed as bifunctional cytotoxic agents, but their activity towards Jurkat cells [19] or PC-3 cells [17] was less potent or, in the best of cases, equipotent to the parent ketonic Mannich bases. On the other hand, hydrazones 17 were consistently more cytotoxic than the corresponding ketonic Mannich bases [18].
Noteworthy is the contribution of Gul et al. to the understanding of the mechanism of the cytotoxic action of these compounds. His group has provided evidence that connects the anticancer activity of ketonic Mannich bases of various structures or piperidinols 14 with their ability to alkylate glutathione [21], [27], [28], [29], whereas a few of the same compounds had mixed effects on thioredoxin, glutaredoxin, or heat shock proteins HSC70 and GRP75 [30].
Only a limited number of examples of Mannich bases of α,β-unsaturated ketones of type 9 with cytotoxic action are available in recent publications. A small series of Mannich bases 18 of 1-arylidene-2-tetralones (Fig. 5 ) were evaluated as cytotoxic agents using human Molt 4/C8 and CEM T-lymphocytes, as well as murine P388 and L1210 leukemic cells [31]. Compared to the parent α,β-unsaturated ketones, Mannich bases 18 were consistently more potent, with half maximal inhibitory concentration (IC50) values in the 0.2–10 μM range. Furthermore, Mannich bases 18 derived from aromatic aldehydes substituted with chlorine, carboxyl, methoxy or cinnamoyloxy groups exhibited significant potencies towards human tumor cell lines, with an emphasis on their antileukemic effect. In most instances, the compounds prepared in this study demonstrated selective toxicity to different cells, which further enhances their potential utility. In addition to compounds 18, simpler Mannich bases 19 derived from 2-benzylidenecyclohexanones were synthesized, and their evaluation against the same cell lines proved once more that Mannich bases were more cytotoxic than the corresponding 2-arylidenecyclohexanones, some of them showing growth-inhibiting properties (IC50 of approximately 2 μM) more potent than reference drug melphalan [32]. Because N-myristoyltransferase is expressed in larger quantities in tumors than it is in normal cells, this enzyme has been under consideration as a molecular target for cancer [33], [34]. However, the substantially high IC50 value of 500 μM towards N-myristoyltransferase for a representative compound of the series of candidates 19 suggests that the inhibition of this enzyme does not play an important role in the mechanism of the cytotoxic activity of these compounds. Several compounds 19 were also tested against murine cancer cells MAC13 (sensitive to most cytotoxic agents) and MAC16 (resistant to most cytotoxic agents), and they demonstrated high cytotoxicity against the latter, but also against normal murine cells C2C12 and 3T3 [35]. On the other hand, Mannich bases 20 showed no activity against MAC16, which points to the importance of the double bond conjugated to the carbonyl function. The exploration of possible mechanisms of cytotoxic action of these compounds revealed that compounds 19 may interfere with a number of essential cellular mechanisms by alkylation of thiols on enzyme or proteins, by disrupting mitochondrial electron transport, or by creating holes in the cell membrane, and thus promoting ATP leakage [35]. Finally, Mannich bases 21 had high cytotoxic activity against two human breast cancer cell lines (MCF-7 and MCF-7/Adr) cells and human leukemia HL-60 cells, showed glutathione binding ability, and exhibited inhibitory action on glutathione-s-transferase π, whereas their analogues obtained through the hydrogenation of the double carbon–carbon bond were slightly less active [36]. The nature of the dialkylamino group did not seem to affect the cytotoxic activity of these compounds, while the substitution of the aromatic rings with a methyl selectively increased the cytotoxic effect on breast cancer cells, but not on immortalized mammary epithelial (184B5) cells.
Fig. 5.
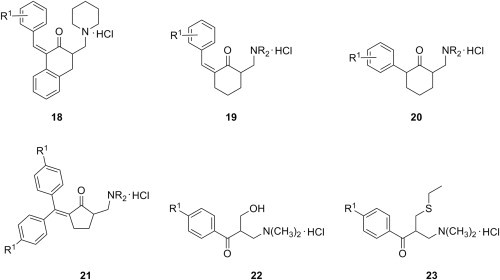
Cytotoxic Mannich bases of type 2 derived from α,β-unsaturated ketones, and potential prodrugs of Mannich bases of type 3 derived from enones.
The literature reporting the anticancer activity of Mannich bases of type 10 is even scarcer. Given the significant antineoplastic properties of compounds 10 [13], a novel series having substituents R1 in the phenyl ring that were carefully selected with a view to impart a variety of physicochemical properties has been designed and synthesized [37]. Since a gradual release of Mannich base 10 from niosomes had improved its bioactivity in vivo, amino alcohols 22 (Fig. 5), which may slowly undergo dehydration to yield the desired Mannich base 10, were also examined as cytotoxic agents. Finally, in order to explore the hypothesis that cytotoxicity would be retained even when a thiol is liberated, the synthesis of adducts 23 was carried out. Compounds 10, 22 and 23 (Fig. 5) were evaluated against both human WiDr colon cancer cells and human CRL-2522 foreskin fibroblasts. IC50 values lesser than 10 μM were obtained when compounds 10 were evaluated towards human WiDr colon cancer cells, and the corresponding candidates 22 also had IC50 values in the low micromolar range. On the other hand, conversion of Mannich bases 10 into the corresponding adducts 23 led to a 37-fold reduction in potency. In addition, compounds 10 and 22 demonstrated a preferential cytotoxicity to cancer cells compared to normal fibroblasts [37]. Further studies showed that compounds 10 and 22 are cytotoxic towards a large number of human tumor cell lines, two important features of many of these compounds being their lethal effects toward promyelocytic leukemic HL-60 cells and their selective toxicity for the aforementioned cancer cell line (selectivity index of 10 or more) [38]. Because divergence in the mechanism of action is required for drug candidates that are developed to be tumor-specific and spare normal tissues, it is noteworthy that a representative Mannich base 10 caused apoptosis and activated caspase-3, caspase-8, and caspase-9 in HL-60 cells, but not in HSC-2 cells.
Cytotoxic phenolic Mannich bases of chalcone analogues (type 11 in Fig. 3) are well represented in the recent literature. One of the strategies that are available for the synthesis of this type of Mannich bases consists in the aminomethylation of chalcone analogues derived from at least either a phenolic aldehyde or a phenolic ketone. In line with this strategy, a series of five mono-Mannich bases 24 (Fig. 6 ) were obtained through a Mannich reaction of chalcone analogues derived from 4-hydroxyacetophenone, employing piperidine as amine reagent [39]. Despite the use of an excess of both paraformaldehyde and piperidine, no double Mannich bases analogous to 11 were isolated. The evaluation of compounds 24 against androgene-independent prostate cancer (PC-3) cell line showed that although three of them were more potent than the parent chalcone analogues, the most cytotoxic Mannich base 24 was 2.5-fold less potent than the reference drug 5-fluorouracil. Based on correlations between cytotoxicity and Hammet constant on one hand and cytotoxicity and partition coefficient on the other hand, the authors hypothesized that an increase in the general hydrophobicity of the molecule would result in enhanced cytotoxicity. Therefore, a novel series of Mannich bases 24 was designed to incorporate a dibenzylaminomethyl residue as a replacement for the piperidinomethyl group [40]. Again, every attempt to obtain double Mannich bases by varying the reaction conditions failed, and only mono-Mannich bases could be isolated. When evaluated against androgene-independent prostate cancer (PC-3) cell line, these compounds consistently displayed lower cytotoxicity than that of the parent chalcone analogues. Unexpectedly, cytotoxicity in the series of Mannich bases with a dibenzylaminomethyl residue was also much lower than that of the corresponding Mannich bases in the series containing a piperidinomethyl motif, thus invalidating the hypothesis put forth by the authors in the previous study. Furthermore, no adduct between ethanethiol and a representative Mannich base 24 could be detected after 48 h, whereas the incubation of ethanethiol with the corresponding parent chalcone analogue under the same conditions resulted in formation of small amounts of adduct. The authors concluded that no active cyclohexadienone species are formed following a potential deamination of Mannich base 24, and that the introduction of the dibenzylaminomethyl group further reduces the ability of the chalcone moiety to undergo thiol addition.
Fig. 6.
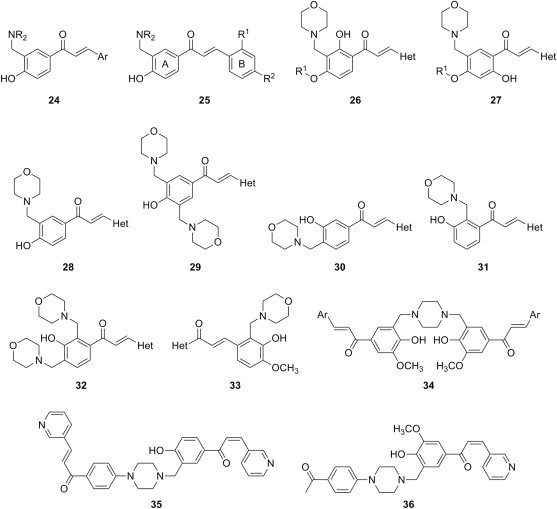
Examples of cytotoxic Mannich bases of type 4 derived from phenolic chalcone analogues.
With a view to explore the effect of variation of dialkylamino moiety on the cytotoxicity of phenolic Mannich bases of chalcone analogues, 27 candidates were synthesized through aminomethylation of three chalcone analogues derived from 4-hydroxyacetophenone and diverse secondary aliphatic amines [41]. Equimolar ratio of reactants afforded mono-Mannich bases of type 24, which were evaluated against hepatocellular carcinoma (HepG2), human lung carcinoma (SK-LU-1), and human breast cancer (MCF-7) cell lines. Mannich bases with 4-phenylpiperazine residue exhibited reduced cytotoxicity towards all three lines of cancer cells, whereas the candidates with 4-methylpiperazine or 4-ethylpiperazine residues were the most active in each series, but less cytotoxic than reference drug ellipticine. With respect to the substitution pattern in the B phenyl ring, Mannich bases 25 (Fig. 6) derived from 4-chlorobenzaldehyde (R1 = H, R2 = Cl) or 2-methoxybenzaldehyde (R1 = OCH3, R2 = H) were consistently more active than those derived from 4-methoxybenzaldehyde (R1 = H, R2 = OCH3). The screening identified five compounds whose IC50 values against MCF-7 cell line were lower than 2 μg/mL, whereas the most cytotoxic compound 25 (R1 = H, R2 = Cl, NR2 = 4-ethylpiperazinyl) had IC50 values lower than 2 μg/mL against all three cancer cell lines used in this study.
A larger library of phenolic Mannich bases of chalcone analogues featuring the dialkylaminomethyl moiety either in ring A or ring B of the chalcone system was synthesized through the Claisen–Schmidt condensation of the appropriately substituted Mannich bases of phenolic aldehydes or ketones with heterocyclic ketones or aldehydes, respectively [42]. The use of 4-alkoxy-2-hydroxyacetophenones as substrates in the Mannich reaction yielded a mixture of 5-aminomethylated derivative with the isomeric 3-aminomethylated derivative, the former being the major reaction product in all cases. The adept tailoring of the ratio between the substrate, formaldehyde and morpholine in the Mannich reaction of 4-hydroxyacetophenone resulted in the selective preparation of either single or double Mannich base from this substrate. On the other hand, 3-hydroxyacetophenone afforded a mixture of 4-morpholinylmethyl derivative with 2-morpholinylmethyl derivative and 2,4-bis(morpholinylmethyl) derivative, even when an equimolar ratio between the reagents was used. Isovanillin, which underwent aminomethylation only at position 2 with morpholine and piperidine as amine reagents, was employed as an example of a phenolic aldehyde substrate in the Mannich reaction. With the help of these intermediates, several small series of Mannich bases of heterocyclic chalcone analogues 26–33 (Fig. 6) were synthesized and evaluated for cytotoxic activity against four human cancer cell lines, namely PC-3, MCF-7, nasopharyngeal carcinoma (KB), and resistant nasopharyngeal carcinoma (KB-VIN). The rich diversity within this library comprised of structurally related entities allowed interesting insight on the cytotoxicity–structure relationship. First, the presence of two phenolic groups in ring A seems to enhance the cytotoxic activity, as proven by a candidate of type 26 (R1 = H, Het = 2-pyridinyl), which was the most potent in the entire library against all four types of cancer cell lines. Then, the presence of a methoxy group in series 27 (R1 = CH3) appears to be generally preferable to ethoxy and isopropoxy, whereas a comparison of the cytotoxicity of similar compounds of type 26 and type 27 usually favors the candidates in the latter series, which have the aminomethyl group para to the phenolic hydroxyl. An analysis of the cytotoxicity within series of candidates of type 28 demonstrated that six-membered heterocycles (particularly a 2-pyridinyl residue) are preferred to five membered heterocycles as ring B moieties, and this observation was validated by the inspection of anticancer activity of compounds in series 29. However, the presence of a second morpholinylmethyl group appears to be detrimental to the cytotoxic activity of candidates 29. Shuffling of hydroxy and morpholinylmethyl groups in Mannich bases 28 and 29 led to compounds in series 30–32, which are generally less cytotoxic than their counterparts derived from chalcone analogues having a 4-hydroxy substituent in ring A. Selective high cytotoxicity against MCF-7 cell line was displayed by the compounds in series 33 featuring the morpholinylmethyl moiety in ring B of the chalcone system. Overall, more than 80% of the Mannich bases in this library of Mannich bases of heterocyclic chalcone analogues 26–33 are cytotoxic (IC50 < 4 μg/mL), while four members of the library are highly cytotoxic (IC50 < 1 μg/mL) against all four cell lines, and other four against at least three cell lines, with MCF-7 and PC-3 being usually more sensitive than KB and KB-VIN cell lines. Later, novel Mannich bases of type 24 (Ar = 3-pyridinyl) were evaluated against several cancer cell lines, and the candidates showed cytotoxicity in the low micromolar range only towards promyelocytic leukemic cells (HL-60) and oral squamosa cell carcinomas (HSC-2, HSC-3 and HSC-4), whereas the IC50 values against non-malignant gingival fibroblasts, pulp cells and periodontal ligament fibroblasts were higher [43]. The tumor selectivity of these Mannich bases may be the result of their proven ability to cleave poly[ADP-ribose]polymerase-1 in HSC-2 cells, but not in gingival fibroblasts cells. In addition, the cytotoxic activity of Mannich bases of chalcone analogues with an aminomethyl moiety in B ring structurally similar to 33 against a panel of breast cancer (MCF7), melanoma (UACC62) and renal cancer (TK10) cell lines was described in a patent [44]. Most compounds were active, and some were potent mostly towards the first two cancer cell lines.
A series of Mannich bases 34 of bichalcone analogues (Fig. 6), in which the two chalcone units are linked through a bis(aminomethyl) function generated by the use of a bifunctional amine reagent such as piperazine, has also been synthesized and evaluated against 25 cancer cell lines [45]. Aminomethylation of acetovanillone with piperazine afforded a bis-Mannich base, which subsequently led to compounds 34 through a Claisen–Schmidt condensation with various aldehydes. Surprisingly, compound 34 (Ar = 2-pyridinyl) was selectively cytotoxic to human tongue squamous carcinoma (CAL-27) and human pharyngeal squamous carcinoma (FaDu) cell lines, whereas 3-pyridinyl and phenyl analogues were the most cytotoxic compounds towards all cell lines. Substitution of the phenyl ring (Ar = C6H5) with methoxy groups stripped Mannich bases 34 of their cytotoxicity towards most cell lines, whereas the decrease in cytotoxic activity induced by the presence of chlorine as substituent was not so drastic. Replacement of phenyl with 2-furanyl or 2-thiophenyl led to compounds that are selectively cytotoxic to one or more cell lines, but further substitution with methyl of these five-membered heterocycle renders them devoid of cytotoxicity against all lines. Despite a few notable examples of selectivity, the results obtained for this collection of compounds are not very encouraging, and they suggest that the incorporation of a second chalcone unit does not enhance the cytotoxicity of Mannich bases derived from chalcone analogues. Cytotoxic activity of Mannich bases of bichalcone analogues was further explored using candidates with a modified design. The synthetic strategy comprised the synthesis of mono-phenolic Mannich bases starting from 4-hydroxyacetophenone or acetovanillone as substrates and employing 1-(4-(piperazin-1-yl)phenyl)ethanone as amine reagent, then the bichalcone unit was generated through a Claisen–Schmidt condensation of both acetyl functions with aromatic aldehydes [46]. Only Mannich bases from bichalcone analogues featuring a pyridinyl moiety (such as candidate 35, Fig. 6) were active towards prostate cancer (DU145), non-small cell lung cancer (A549), ileocecal (HCT) and nasopharyngeal carcinoma (KB) cell lines with IC50 values between 0.7 and 4 μM; all other compounds had IC50 values greater than 20 μM. Compared to compound 35, Mannich base 36 having a single chalcone unit was less active (IC50 between 10 and 13 μM). An exploration of the mechanism of action for these compounds suggested that compound 36 most likely acted via the Fas/CD95 apoptosis signaling pathway.
Ferulic acid and its derivatives provide another example of a type of substrate containing an α,β-unsaturated system activated by an electron-withdrawing group, similar to that in phenolic chalcone analogues, from which phenolic Mannich bases can be synthesized. The growing body of evidence suggesting that 3′-azido-2′-deoxythymidine (AZT), a known antiviral, also possesses anticancer activity [47], [48], [49] has sparked a study aiming at cytotoxic evaluation of a series of conjugates of AZT and ferulic acid derivatives [50]. Thus, propargyl ester and N-propargylamide of ferulic acid chemoselectively underwent aminomethylation ortho to the phenolic hydroxyl with various secondary aliphatic amines, and the resulting phenolic Mannich bases 37 (X = O, NH) reacted through the terminal alkyne moiety with AZT via a Cu(I)-catalyzed click chemistry process to afford the corresponding 1,2,3-triazoles. Evaluation of cytotoxicity for both Mannich bases 37 (Fig. 7 ) and the related 1,2,3-triazoles against human breast adenocarcinoma (MDA-MB-231), lung adenocarcinoma (SK-LU-1) and colon adenocarcinoma (SW480) cell lines showed that only some of compounds 37 were cytotoxic. Despite the presence in their structure of an identical scaffold comprising a phenolic Mannich bases moiety and an activated carbon–carbon double bond motif that has been deemed responsible for the cytotoxic effect of Mannich bases 37, all of the corresponding 1,2,3-triazoles were inactive. Out of eight Mannich bases of propargyl ester of ferulic acid reported in this study, three were inactive against all three lines, while the rest had weak to moderate cytotoxicity, and the most active candidate (IC50 = 20–44 μg/mL) was Mannich base 37 (X = O) having a pyrrolidinylmethyl moiety. With the exception of Mannich base 37 (X = NH) with a piperidinylmethyl moiety, all others candidates derived from N-propargylamide of ferulic acid were inactive.
Fig. 7.
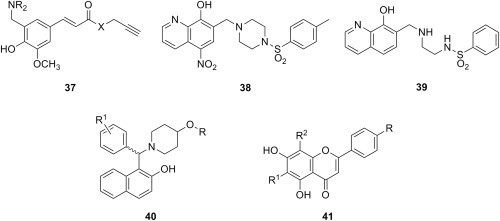
Phenolic Mannich bases with cytotoxic activity.
Other aminomethylated phenols with cytotoxic activity have been reported besides phenolic Mannich bases of chalcone analogues. Unfortunately, the wide structural diversity of phenolic substrates from which these phenolic Mannich bases originate and the lack of a systematic and comprehensive search for structure–cytotoxic activity relationships within a particular type of phenolic substrate undermine any efforts to discover good lead compounds for further development as drugs. Phenolic substrates subjected to the Mannich reaction with a view to obtain novel cytotoxic agents include both simple and very complex structures. Examples that illustrate structurally simple phenolic substrates are 1-naphthol and 8-hydroquinoline, whose Mannich bases with piperidine and 4-arylsulfonylpiperazines exhibited growth-inhibitory effects towards a panel of carcinoma cell lines, including HeLa (cervical epithelioid carcinoma cell), BT483 (mammary gland adenocarcinoma cell), SKHep (hepatocellular carcinoma cell), and CE81T (esophageal carcinoma cell) [51]. Although the cytotoxic effect of aminomethylated naphthols and 8-hydroxyquinoline derivatives has been known for some time [52], [53], a mechanistic study presented in the aforementioned report showed that these phenolic Mannich bases induce apoptosis by activation of caspase-dependent pathways. Furthermore, upon addition of copper ions, these compounds dramatically stimulate production of reactive oxygen species and activate various kinases, a group of enzymes that are known to be important in the oxidative stress-mediated cell death. Candidate 38 (Fig. 7) was the most potent in this small series, with a concentration for 50% cell growth inhibition of 0.71 μM; this value decreased to 0.06 μM in the presence of 50 μM copper. A more detailed study [54] of the structure–cytotoxic activity relationship within this class of compounds showed that either replacement of sulfonyl function in 38 with a methylene group, or replacement of piperazine ring with an ethylenediamino moiety (as in compound 39, Fig. 7) led to a significant increase in cytotoxic activity. The cell lines used in this study exhibited selective sensitivity towards different Mannich bases, which appeared to be modulated by the nature of the arylsulfonyl group. As for the 8-hydroxyquinoline part of the molecule, substitution at position 5 of the quinoline motif (especially with a nitro group) had a beneficial effect, whereas its replacement with phenol, 3-hydroxypyridine or 1-naphthol led to a decrease of cytotoxic activity [54]. Cytotoxicity of enantiomerically-enriched Mannich bases 40 (Fig. 7) derived from 2-naphthol, aromatic aldehydes and either 4-piperidinol (R = H) or its acetylated counterpart (R = COCH3) against murine leukemic L1210 and human lymphoblast Molt 4/C8 and CEM cell lines has been examined by another study [55]. All of the compounds were only moderately cytotoxic, with IC50 values in the middle micromolar range, and were also 10–70-fold less potent than that reference drug melphalan. Substitution of the aryl group in 40 with R1 = 4-dialkylaminoethoxy resulted in a series of compounds whose cytotoxicity potential against estrogen-responsive human MCF-7 breast cancer cells was found to be comparable to that of tamoxifen. However, removal of the 4-piperidinol moiety from Mannich bases 40 led to benzylnaphthols with enhanced cytotoxicity against MCF-7 cells, which is most likely due to their binding and antagonistic effects against human estrogen receptor alpha [55].
Flavones represent another type of phenolic substrate from which cytotoxic Mannich bases have been synthesized. Because flavones such as chrysin bear structural resemblances to androgens, Mannich bases 41 (R = R1 = H, R2 = aminomethyl; R = R2 = H, R1 = aminomethyl; R = H, R1 = R2 = aminomethyl) of chrysin (Fig. 7) have been designed as inhibitors of human aromatase, an enzyme which converts androgens to estrogens, and therefore represents a key target in the treatment of hormone-dependent tumors, including breast cancer [56]. Several single and double Mannich bases of chrysin were found to inhibit human aromatase more effectively than reference drug aminoglutethimide [57]. In addition, Mannich bases 41 (R = OH, R1 = H, R2 = aminomethyl) of apigenin (Fig. 7) have been prepared from aliphatic primary and secondary amines via chemoselective aminomethylation at C-8 in the benzopyran ring system [58]. Antiproliferative activity of these Mannich bases against four human cancer cell lines, namely human cervical (HeLa), human liver (HepG2), human lung (A549), and human breast (MCF-7) cancer cells, was determined using the standard 3-(4,5-dimethylthiazol-2-diphenyl-tetrazolium) bromide (MTT) assay. Pyrrolidine Mannich base of apigenin was the most promising compound in this series, its inhibition of cell proliferation being greater than 90% against all four cell lines at a concentration of 1 mg/mL.
Many natural or synthetic carbazoles, either simple or condensed with other heterocycles (such as pyridocarbazoles, indolocarbazoles, pyranocarbazoles, pyrrolocarbazoles, etc), have been reported as anticancer agents. A recent study examines the cytotoxicity of a series of oxazinocarbazoles, which were the major products arising from the Mannich reaction of N-substituted 2- or 4-hydroxycarbazoles with primary amines (Fig. 8 ) [59]. Under the appropriate reaction condition, 4-hydroxycarbazoles yielded 2,3,4,7-tetrahydro[1,3]oxazino[5,6-c]carbazoles 42, whereas 2-hydroxy-9-methylcarbazole led to a mixture of regioisomeric 2,3,4,7-tetrahydro[1,3]oxazino[6,5-b]carbazoles 43 and 2,3,4,7-tetrahydro[1,3]oxazino[5,6-a]carbazoles 44. Use of allylamine, 3,3-dimethylallylamine or benzylamine as amine reagents afforded 44 as the major product, while isomer 43 was the major component of the mixture when 2-pyridinylmethylamine was employed. In addition, small to moderate yields of bis-Mannich bases 45 or 46 were isolated from N-substituted 4-hydroxycarbazoles and 2-hydroxy-9-methylcarbazole, respectively, but only when 2-pyridinylmethylamine was employed as amine reagent. Evaluation of the antiproliferative action of these compounds against CEM (T cell leukemia), Jurkat (acute T cell leukemia), Raji (Burkitt's lymphoma), MCF-7 (breast cancer cells) and Caco-2 (colorectal cancer) using the WST-1 colorimetric assay showed, after the primary screening at 100 μM, that bis-Mannich bases 45 and 46 were less active than the oxazinocarbazoles. In the series of compounds 42, the best antiproliferative effect was observed for candidates having either an allyl or a prenyl group at the carbazole nitrogen atom and an allyl group at the oxazine nitrogen atom. Thus, candidate 42 (R1 = prenyl, R2 = allyl) had IC50 = 12 μM against CEM, Jurkat and Raji cell lines. The majority of candidates 43 and 44 exhibited significant antiproliferative action at 100 μM, and compound 43 (R = 2-pyridinylmethyl) was the most active towards Jurkat and Raji cell lines (IC50 = 12 μM) [59]. Aminomethylated derivatives of hydroxycarbazoles have been also mentioned in a different study [60], which described the synthesis of a small series of phenolic Mannich bases 47 (Fig. 8) obtained from 5-substituted 2-hydroxy-5H-benzo[b]carbazole-6,11-diones along with their in vitro anticancer evaluation at National Cancer Institute (NCI) using an in-house developed screening panel of approximately 60 cell lines derived from nine different types of cancer. Only one Mannich base 47 (R = 4-H3COC6H4) was more active than the parent benzocarbazoledione, and a COMPARE analysis [61] revealed that its mechanism of action is novel and does not resemble the known mechanisms of action for standard anticancer drugs, but this candidate was not selected for further studies concerning its interaction with DNA. In a related report, phenolic Mannich bases 48 of 5-hydroxy-1H-naphtho[2,3-g]indoles were found to be inactive in a similar screening [62].
Fig. 8.

Cytotoxic oxazines, double phenolic Mannich bases, and tertiary phenolic Mannich bases derived from hydroxycarbazoles.
Quinones are a class of compounds that have been widely investigated as anticancer agents [63]. Besides anthracycline antibiotics, other natural hydroxyquinones such as plumbagin [64], [65], juglone [66], [67] or lapachol [68] and their derivatives have been reported to exhibit significant cytotoxicity. The Mannich bases of another natural phenolic quinone, namely lawsone, and their Pt(II) complexes 49 (Fig. 9 ) have been synthesized and shown to be highly cytotoxic towards six cancer cell lines: MDA-MB-435 (melanoma), HL-60 (promyelocytic leukemia), HCT-8 (colon), SF-295 (brain), OVCAR-8 (ovary) and PC-3 (prostate) [69]. The ligands and the complexes that have long alkyl chains (R = n-heptyl or n-decyl) were the most active (the complexes were actually more cytotoxic than cisplatin), whereas the neutral complexes (X = Cl) were generally more cytotoxic than the corresponding charged complexes (X = H2O or NH3). Examination of the mechanism of action for some of these complexes has shown that aqua complexes 49 were more efficient inhibitors of ethidium bromide intercalation into DNA than amino complexes, and candidate 49 (R = (CH2)3CH3, X = H2O) was more efficient than cisplatin [70]. The same aqua complex also induced DNA strand breaks, while the corresponding amino complex was ineffective. The ability of these complexes to inhibit topoisomerase I was also examined. Most chlorido and amino complexes were as active as reference drug camptothecin in the DNA relaxation assay, and did not cause major unwinding of DNA, with the exception of complex 49 (R = n-decyl, X = Cl). In addition, cellular platinum accumulation was shown to increase with the increase of the length of the alkyl chain of the amino moiety in the Mannich base ligand. The chlorido Pt(II) complexes were oxidized to the corresponding chlorido Pt(IV) complexes, but the cytotoxicity of these new complexes was comparable to the cytotoxicity of the parent Pt(II) complexes, presumably owing to the rapid reduction of Pt(IV) complexes before entering the cancer cells [71].
Fig. 9.
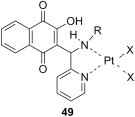
Cytotoxic Pt(II) complexes of Mannich bases generated from lawsone as substrate.
Furthermore, miscellaneous aminomethylated phenols from structurally diverse substrates have been reported as anticancer agents (Fig. 10 ). Thus, a small series of seven phenolic Mannich bases 50 were synthesized through aminomethylation of naturally occurring antibiotic lasalocid, with simultaneous elimination of the carboxyl group neighboring the phenolic hydroxyl [72]. Antiproliferative effect of Mannich bases 50 was evaluated against MCF-7 (human breast adenocarcinoma), A549 (human lung adenocarcinoma), HT-29 (human colon carcinoma) and P388 (murine leukemia) using either MTT or sulforhodamine B (SRB) assay, and four candidates were more cytotoxic towards A549, HT-29 and MCF-7 cell lines that anticancer drug cisplatin. These candidates also presented higher selectivity towards cancer cells than towards BALB/3T3 (normal murine embryonic fibroblast) or HLMEC (human lung microvascular endothelial) cell lines. The lack of activity of the other three Mannich bases was attributed to the aralkyl or long alkyl chains in the amine moiety of these compounds [72].
Fig. 10.
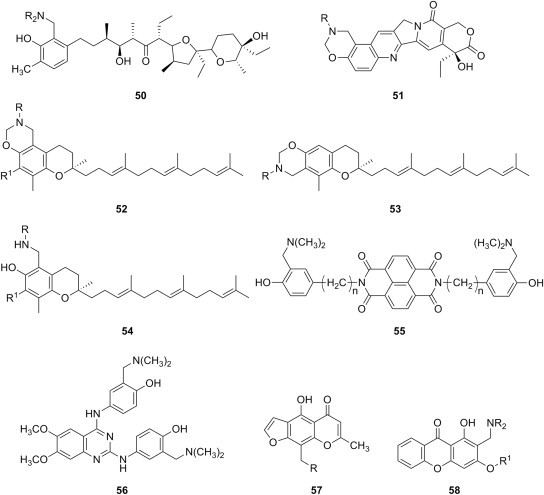
Cytotoxic phenolic Mannich bases obtained from miscellaneous phenolic substrates.
Camptothecin, another naturally occurring phenolic substrate and lead compound for a plethora of cytotoxic substances, was converted into the oxazino derivatives 51 (Fig. 10) by means of the Mannich reaction using primary aliphatic and aromatic amines [73]. Evaluation of these novel hexacyclic camptothecin derivatives towards nine human cancer cell lines (BXPC-3, NCI-446, MCF-7, HEPG-2, A549, A2780, Bel7402, HT-29, and KB), using MTT assay and camptothecin and topotecan as reference compounds, showed that most of them exhibit cytotoxicity towards several cell lines that is superior or comparable to topotecan, while only a few of the candidates presented cytotoxicity comparable to camptothecin. Because candidates 51 (R = C2H5 or n-C3H7) were the most potent antiproliferative agents in this series, the presence of a small alkyl group at the nitrogen in the oxazine moiety seems to be preferable for a high cytotoxicity [73].
A series of oxazino derivatives 52 (R1 = CH3) (Fig. 10) were also prepared from γ-tocotrienol and primary amines via the Mannich reaction, whereas δ-tocotrienol afforded under the same conditions a mixture of oxazino derivatives 52 (R1 = H) and 53, in which the former is the major component [74]. No reaction occurred when secondary amines were used instead, but two phenolic Mannich bases 54 were obtained indirectly from the corresponding oxazino derivatives 52. Out of 42 candidates in this library, thirty compounds had greater antiproliferative activity against the highly metastatic + SA mouse mammary epithelial cancer cells than that of the parent tocotrienols (IC50 = 3 μM), and seven candidates had IC50 values in the nanomolar range. Mannich bases 54 were less active than the corresponding oxazino derivatives 52 against NCI's standard panel of 60 cell lines, which suggests that the oxazine ring is an essential pharmacophore for the cytotoxic activity of these tocotrienol derivatives. Generally, the oxazino derivatives 52 of δ-tocotrienol were more active than the corresponding isomers 53 derived from γ-tocotrienol, and a long alkyl chain at the nitrogen atom (preferably with a terminal hydroxyl group) proved beneficial for the antiproliferative activity. The same conclusions were drawn after the evaluation of structure–antimigratory activity relationship using the highly metastatic MDAMB-231 breast cancer cell line [74].
Novel G-quadruplex ligand/alkylating hybrid structures 55 (Fig. 10) were obtained by tethering a naphthalene diimide core having G-quadruplex recognizing properties to phenolic Mannich bases using flexible spacer [75]. The assessment of cytotoxic effects of these compounds 55 (n = 1, 2, or 3) and their corresponding methiodides against human embryonic kidney 293T cell line by MTT assay suggests that the length of the spacer between the core and the phenolic Mannich bases moiety modulates the cytotoxicity: candidates 55 having two-carbon atoms and one-carbon atom spacers were the most active (IC50 4.5 and 10.5 μM, respectively), whereas the Mannich base 55 with a three-carbon atoms spacer was less active. Cytotoxicity of these compounds parallels their ability to alkylate DNA, and the grafting of the alkylating Mannich base moiety to the central core contributes to the enhancement of the G-quadruplex folding induction and stabilization. A similar G-quadruplex ligand/alkylating hybrid structure was shown to significantly slow the growth of melanoma cells by causing telomere dysfunction and down-regulation of telomerase expression [76], which suggests that these hybrids could be possible candidates for the development of novel targeted anticancer therapies. In connection to this, the methiodide of double Mannich base 56 (Fig. 10) with a quinazoline core was also shown to cross-link linear DNA at concentrations as low as 1 μM, and to inhibit DNA transcription almost completely at 10 μM [77].
Phenolic Mannich bases 57 of norvisnagin (Fig. 10) were prepared through direct aminomethylation, and their ability to interact with DNA was evaluated using both a qualitative binding assay and a colorimetric microassay based on the displacement of methyl green from DNA [78]. Three of the candidates 57 (R = pyridinyl-2-amino, diethylamino, and methylamino) showed moderate DNA binding affinity, and could be potentially cytotoxic. Also, Mannich bases 58 (R1 = H) of 1,3-dihydroxyxanthone (Fig. 10) displayed moderate to good cytotoxicity against lung cancer (NCI-H460), tongue squamosa cell carcinoma (TCA-8113), liver cancer (BEL-7402), hepatocarcinoma (HepG2), gastric carcinoma (SGC-7901) and urinary bladder carcinoma (T24) in an MTT assay [79].
Several reports of Mannich bases of indoles as cytotoxic agents are also available in recent literature. Cytotoxicity of a few indole Mannich bases 59 derived from 4-substituted piperazines (Fig. 11 ) was evaluated against liver (HUH7), breast (MCF7) and colon (HCT116) cancer cell lines using SRB assay, and some of these compounds had IC50 values in the lower micromolar range. Compound 59 (R = 3,4-dichlorobenzyl) was cytotoxic towards all three cancer cell lines, and fared better than reference drug 5-fluorouracil (5-FU) [80]. On the other hand, N-Mannich bases 60 of 3-methylindole did not inhibit the growth of cancer cells or had high IC50 values. In spite of this fact, a subsequent study [81] was dedicated exclusively to the investigation of cytotoxicity of a novel series of N-Mannich bases of type 60 against the same cancer cell lines. The broadening of the nature of substituent at position 4 of piperazine proved favorable, as several compounds reported in this later study presented cytotoxic activity comparable to reference drug 5-FU. A comparison between the morphological features of cancer cells for which apoptosis was induced either by a selected 3-metylindole N-Mannich base 60 or by paclitaxel suggests that compounds 60 (R1 = CH3) and paclitaxel share the same mechanism of action. Design of novel cytotoxic Mannich bases in which indole itself was the substrate for aminomethylation was also revisited using an extended panel of 4-substituted piperazines as amine reagents in aminomethylation [82]. The novel candidates 59 proved to be cytotoxic towards the same cancer cell lines (HUH7, MCF7, and HCT116); however, no cytotoxicity was observed for the candidates with an electron-withdrawing group (such as 4-nitrophenyl, benzoyl or acetyl) as substituent at position 4 of piperazine.
Fig. 11.

Cytotoxic indole and azaindole Mannich bases.
Other C-Mannich bases of indole derivatives have been claimed as potent inhibitors of isoprenylcysteine carboxyl methyltransferase (Imct), an enzyme that plays an important role in the post-translational modification of proteins that are involved in the regulation of cell growth, and therefore represents a potential therapeutic target in oncogenesis. Among the few small molecules inhibitors of Imct that were discovered so far, the most promising appears to be cysmethynil, an indol-3-ylacetamide derivative which impairs growth factor signaling and induces cell cycle arrest and autophagy. Because cysmethynil suffers from poor water solubility and strong binding to plasma proteins, rational modification of this hit compound has been expected to yield more potent Imct inhibitors with improved bioavailability. Replacement of the acetamide function in cysmethynil with various aminomethyl moieties led to compounds 61 (R = dialkylamino) with an inhibitory effect on Icmt that was generally 2–3-fold more potent than that of the parent inhibitor [83]. Evaluation of the effects of these candidates on viability of MDA-MB-231 human breast cancer cells using the colorimetric tetrazolium assay confirmed the results for Icmt inhibition. Thus, compounds 61 (Fig. 11) were found to be more cytotoxic (IC50 3–13 μM) than cysmethynil (IC50 22 μM). Other modifications of the lead compound 61 (R = diethylamino) either preserved the potency of the candidates (e.g., shuffling of the methyl group on the phenyl ring), or resulted in a potency decrease (e.g., replacement of n-octyl with prenyl). However, the replacement of m-tolyl moiety in 61 with more polar heteroaromatic rings led to submicromolar IC50 values in the Icmt inhibition assay, and to IC50 values in the antiproliferative assay on breast MDA-MB-231 and prostate PC3 cell lines that are 2–3-fold lower than that of the lead 61 (R = diethylamino) [84]. Compound 62 (Fig. 11) was the most potent compound in this series, and presented a series of improvements of the drug-like profile over cysmethynil, such as good solubility in water, acceptable permeability through an artificial membrane, and limited tendency to form light scattering aggregates.
Using naphtho[2,3-f]indole-5,10-dione as scaffold, a series of 3-aminomethylated derivatives was synthesized, and four candidates were evaluated as antiproliferative agents against the standard panel of 60 human cancer cell lines at NCI [85]. Compounds 63 (R = primary or secondary aliphatic amine residue, R1 = OH) (Fig. 11) were less potent than doxorubicin against any of the cell lines, but multidrug resistant breast cancer cells were found to be more sensitive to 63 than to doxorubicin. In addition, Mannich bases 63 showed potency for cancer cell lines that are otherwise resistant to anticancer drugs, such as the P-glycoprotein-positive subline of K562 leukemia cells or the p53-null subline of HCT116 colon carcinoma cell line. Replacement of phenolic hydroxyl groups R1 in 63 (R = dimethylamino) by 2-aminoethyleneamino moieties led to mixed results, as the potency improved for some of the cell lines and declined for others, but the sensitivity of multidrug resistant breast cancer cells to these modified candidates was completely lost [86]. Furthermore, candidate 63 (R = quinuclidin-3-ylamino, R1 = OH) was shown to inhibit topoisomerase I-mediated relaxation of DNA, but the suppression of the topoisomerase I activity is presumably the leading although probably not the only factor contributing to cytotoxicity of Mannich bases of naphtho[2,3-f]indole-5,10-diones [87]. Preobrazhenskaya et al. have also shown that a series of single and double Mannich bases 64 (R1 = H or dialkylaminomethyl) of 3,4-bis(indol-1-yl)maleimides (Fig. 11), structurally related to rebeccamycin or staurosporine, were highly cytotoxic towards К562 and HCТ116 cell lines, but their cytotoxicity does not correlate well with their ability to either inhibit protein kinase C-α or constrain activation of multiple drug resistance [88].
Mannich bases of an indole isostere, namely 5H-pyrrolo[3,2-d]pyrimidine, have been designed as inhibitors of phosphatidylinositol-3-kinase α (PI3Kα), a lipid kinase that modulates activity of the PI3K downstream effectors Akt and mTOR. Since the consequences of biological activation of Akt include tumor progression, proliferation, survival, growth, invasion, angiogenesis, and metastasis, PI3Kα represents an attractive target for development of anticancer drugs. Although aminomethylated pyrrolopyrimidine 65 (Fig. 11) was an efficient inhibitors of PI3Kα (IC50 = 20 nM) and showed good selectivity for PI3Kα over mTOR (170-fold), this compound exhibited low cytotoxicity towards PC3 cancer cells [89]. In addition, all the other analogues of Mannich base 65 were found to be even weaker inhibitors of PI3Kα than the lead compound.
Isatin is nowadays a well recognized and privileged scaffold in the design of cytotoxic and anticancer compounds [90]. Several isatin-containing substrates, namely isatin and its 5-halogenated analogues, the corresponding imine derivatives obtained from sulfadiazine, sulfadoxine and trimethoprim, and a hydrazone derived from isoniazid, were aminomethylated using gatifloxacin as amine reagent [91]. The resulting Mannich bases were tested against NCI's standard panel of 60 cell lines using SRB assay, and compound 66 (Fig. 12 ) emerged as an efficient anticancer agent that was generally more potent than reference drug etoposide against most cell lines in the panel. Another library of Mannich bases derived from isatin, 4-halogenated isatins and their Schiff bases with 2-amino-6-methylbenzothiazole was evaluated for cytotoxic effects on three breast cancer cell lines (MDA-MB468, MDAMB231 and MCF7) using SRB assay [92]. The introduction of a halogen as substituent at position 4 of isatin Mannich bases led to an increase in cytotoxicity, and modification of isatins into Schiff bases followed by aminomethylation resulted in Mannich bases further enhanced the cytotoxicity of these candidates. Compounds 67 and 68 (Fig. 12) were the most potent in each series (IC50 < 20 μM), and more potent than reference drug cisplatin, while their cytotoxicity against normal cells was low. As far as their mechanism of action is concerned, compound 67 induced cell cycle arrest in G2/M phase at concentrations similar to those observed for cell growth inhibition, whereas compound 68 did not [92]. Furthermore, another collection of Mannich bases of isatin imines generated either from 2-aminobenzimidazole or 2-amino-4,5-dihydrothiazole was evaluated against MCF-7 human breast adenocarcinoma cell line using SRB assay, but the cytotoxicity of these compounds was moderate (IC50 > 20 μM) and inferior to that of doxorubicin [93]. Generally, aminomethylated Schiff bases of isatin derived from 2-aminobenzimidazole were more cytotoxic than their counterparts derived from 2-amino-4,5-dihydrothiazole, compound 69 (Fig. 11) being the most potent in this collection (IC50 = 22.6 μM). Cytotoxic Mannich bases of isatins were also designed employing hybridization of isatin with a 4-aminoquinoline scaffold to generate the substrate subjected to aminomethylation [94]. The cytotoxicity of these compounds towards breast cancer cell lines MDA-MB468 and MCF7 was moderate (IC50 values between 15 and 65 μM), but the activity improved slightly in the series of the corresponding thiosemicarbazones (IC50 in the range of 10–55 μM). Mannich bases 70 and 71 (Fig. 12) were the most potent candidates in every series (2–3-fold more cytotoxic than cisplatin), and they preferentially inhibited the growth of cancer cell over normal cells. Studies using flow cytometry also suggest that these compounds induce cancer cell death by apoptosis.
Fig. 12.
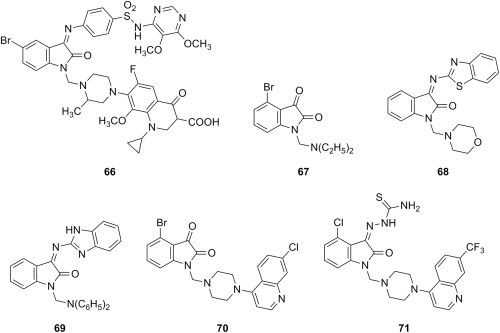
Cytotoxic Mannich bases of isatin derivatives.
Beside isatin derivatives, several other classes of NH-azoles have been aminomethylated with a view to synthesize cytotoxic Mannich bases. 2,3-Dihydro-1,3,4-oxadiazole-2-thiones appear to be the preferred substrate within this category, most likely owing to their straightforward preparation. Starting from methyl salicylate, good yields of the corresponding oxadiazolethione Mannich bases 72 (Fig. 13 ) were obtained in three steps [95]. Most compounds in this collection arise from primary aromatic amines diversely substituted in the aromatic ring, whereas Mannich bases 72 derived from secondary aliphatic amines are poorly represented. Selected candidates from this series have been initially evaluated by NCI against a panel consisting of NCI-H460 (lung), MCF7 (breast), and SF-268 (glioblastoma) cancer cell lines using SRB assay. Seven of these thirteen Mannich bases 72, most of them having either chlorine or carboxy group as substituent in the aromatic ring of the amine moiety, reduced the growth of NCI-H460 cell line to 30% or less, and they were further selected for the standard 60-cell lines panel assay. Compounds 72 (R = H, R1 = 3-ClC6H4 or 4-ClC6H4) presented higher cytotoxicity than reference drugs 5-FU or cyclophosphamide against most cancer cell lines in this panel [95]. The ability of several oxadiazolethione Mannich bases 73 featuring variously substituted aromatic ring at position 5 of the oxadiazolethione ring (R1 = H and R = NO2, OH, CH3, or R = R1 = Cl) to inhibit the growth of tumors in vivo has been also investigated [96]. Tumor volume and tumor weight in mice injected with Ehrlich ascites carcinoma cells were reduced by 52–74% at a dose of 50 mg candidates 73 (Fig. 13) per kg body weight, whereas similar dose of reference drug 5-FU inhibited tumor formation by 93%. Mannich bases 73, especially those having hydroxyl, methyl or chloro substituents on the phenyl ring at position 5, were the most potent. Also, the counts of red blood cells and leukocytes, as well as hemoglobin levels, have been restored almost to the normal values in mice treated with Mannich bases 73 [96]. Aminomethylated oxadiazolethiones 74 (Fig. 13) were generally more cytotoxic against colon carcinoma (HT29) and less cytotoxic against breast cancer (MCF7) cells using SRB assay, but the most potent candidate 74 (NR2 = NHC6H4CH3-4) was 5-fold less cytotoxic than reference drug doxycycline [97]. Cytotoxicity of a series of Mannich bases 75 (Fig. 13) of a norharman–oxadiazolethione structural hybrids was evaluated against a panel comprising melanoma (UACC-62), breast (MCF7), ovarian resistant (NCI/ADR), renal (786-0), lung (NCI-460), prostate (PCO-3), ovarian (OVCAR) and colon (HT-29) cell lines using SRB assay [98]. Several of candidates 75 (R = H or N(CH3)2, R1 = isopropylamino or benzylamino) exhibited a broad spectrum cytotoxic activity, and aminomethylation of the parent oxadiazolethiones significantly enhanced the cytotoxicity of each resulting Mannich bases 75 compared to that of the corresponding substrate. Furthermore, Mannich bases 76 of a fluoroquinolone–oxadiazolethione hybrid were prepared using either secondary aliphatic amines or substituted arylamines as amine reagents [99]. The in vitro evaluation of cytotoxicity against Hep3B cancer cells using MTT assay showed that compounds 76 (Fig. 13) were more potent than the parent fluoroquinolone pefloxacin. Also, Mannich bases in this series derived from aliphatic amines were generally more cytotoxic than those derived from arylamines, and candidate 76 (R = N(CH3)2) was even more potent than reference drug bisantrene.
Fig. 13.
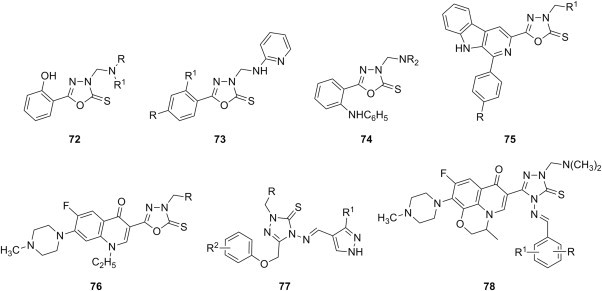
Mannich bases of 2,3-dihydro-1,3,4-oxadiazole-2-thiones and 2,3-dihydro-1,2,4-triazole-3-thiones as cytotoxic agents.
2,3-Dihydro-1,2,4-triazole-3-thiones could also act as substrates for the preparation of cytotoxic N-Mannich bases. Schiff bases obtained from 5-aryloxymethyl-4-amino-3-mercapto-1,2,4-triazoles and 3(5)-substituted pyrazole-4-carboxaldehydes were aminomethylated using either morpholine or diphenylamine to afford Mannich bases 77 (Fig. 13), whose cytotoxicity against HepG2 cell line was evaluated using MTT assay [100]. Out of five tested candidates, Mannich bases 77 having a morpholine moiety were more potent than those with a diphenylamine moiety, but they were still 2–3-fold less cytotoxic than reference drug doxorubicin. Eleven dimethylamine Mannich bases 78, that were obtained through aminomethylation of Schiff bases derived from a 4-amino-1,2,4-triazole-3-thione having at position 5 a moiety originating from fluoroquinolone ofloxacin, were screened for cytotoxicity against murine leukemia cell line (L1210) and human leukocytoma cell line (HL60) [101]. Mannich bases 78 (Fig. 13) were generally more cytotoxic than the corresponding parent Schiff bases, and candidates 78 with a hydroxyl group in the aromatic ring of the azomethine function (R = OH) were the most cytotoxic compounds in the series (IC50 values in the range of 0.14–0.83 μM).
Mannich bases of thiazolidinone derivatives have also been investigated as cytotoxic agents. C-Aminomethylation of two 4-thiazolidinones using secondary aliphatic amines afforded Mannich bases 79 (X = O, CH2) (Fig. 14 ), which were evaluated against colon (HCT116) and breast (T47D) cancer cell lines by SRB assay, but their cytotoxicity was generally moderate to low (IC50 values between 13 and 50 μM) [102]. In addition, N-aminomethylation of two thiazolidine-2,4-diones (R = Cl, OCH3) with morpholine, piperidine and variously 1-substituted piperazines yielded Mannich bases 80 (Fig. 14), which were investigated at NCI against the standard 60-cell lines panel using SRB assay, and proved to be virtually inactive (growth inhibition for the most sensitive cell line between 18 and 33%) [103].
Fig. 14.
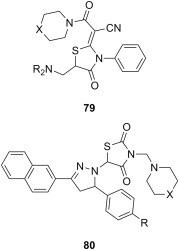
Mannich bases of thiazolidinone derivatives as cytotoxic agents.
Several examples of P-Mannich bases derived from organic esters of phosphorous acid that were disclosed as cytotoxic agents are available in the literature. Thus, α-aminophosphonates 81 were synthesized from alkyl phosphites (R3 = CH3, C2H5, n-C3H7, i-C3H7, n-C4H9), fluorine-substituted benzaldehydes and 2-aminobenzothiazoles, and their cytotoxicity was evaluated against PC3 (prostate), A375 (melanoma), A431 (epidermoid carcinoma), and Bcap-37 (breast) cancer cells using MTT assay [104]. Most Mannich bases 81 (Fig. 15 ) exhibited low to moderate growth inhibition of A375 and Bcap-37 cells, but their cytotoxicity towards PC3 and A431 cells was generally greater. The nature of the fluorine substituent appears to influence cytotoxicity, as candidates 81 having fluorine directly attached to the aromatic ring are more potent than those with a trifluoromethyl substituent. An improvement of cytotoxicity with the increase of the length of the alkyl residue R3 from the initial phosphite was also noted. P-Mannich bases 82 (R1 = CH3, C2H5, i-C3H7) (Fig. 15) were prepared using thieno[3,2-c]pyridine-2-carboxaldehyde as aldehyde component in the Mannich reaction of dialkyl phosphites with variously substituted arylamines [105]. The majority of candidates 82 showed good cytotoxicity against esophageal cancer cells (EC109), but they were generally more potent against hepatocellular liver carcinoma cells (HepG2) at a concentration of 50 μg/mL. A few α-aminophosphonates 83 (Fig. 15) were obtained through a Mannich-type process from diphenyl phosphite, 3-acetylpyridine and variously substituted anilines, and proved to be cytotoxic to HepG liver carcinoma cell line (IC50 ∼ 15 μM) and MCF7 breast adenocarcinoma cell line (IC50 ∼ 20 μM) using MTT assay [106]. Although these candidates were approximately 7-fold less cytotoxic than reference drug doxorubicin, their LD50 values were greater than the corresponding IC50 values, making them safe to use. Aminomethylation of diethyl phosphite with either aromatic aldehydes and aromatic diamines or terephthaldehyde and various amines led to bis(α-aminophosphonates) 84 or 85 (Fig. 15), respectively, whose cytotoxicity against Jurkat (T-cell lymphoma), Raji (Burkit's lymphoma) and MCF-7 (breast cancer) cell lines was determined using MTT assay [107]. In this structurally diverse series of compounds, a few were devoid of cytotoxicity while most of them were moderately cytotoxic. Compound 85, derived from tryptamine as amine reagent in the Mannich reaction, emerged as the most potent in this series, its cytotoxicity being comparable to that of doxorubicin.
Fig. 15.
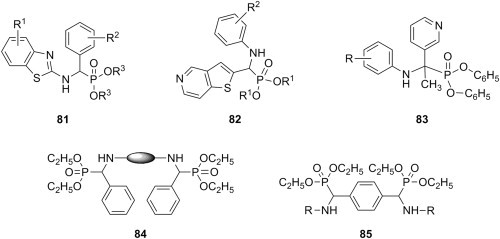
Cytotoxic P-Mannich bases.
Furan belongs to the category of electron-rich heterocycles known to undergo electrophilic substitutions such as the Mannich reaction with great ease. For example, aminomethylation of synthetic lactone of natural furanditerpene 6α,7β-dihydroxyvouacapan-17β-oic acid as substrate and using various secondary aliphatic amines led to the furan Mannich bases 86 (Fig. 16 ) [108]. Antiproliferative effect of these compounds was more potent than that of the parent lactone against a panel of nine cancer cell lines (melanoma (UACC-62), breast (MCF7), ovarian expressing the resistance phenotype for adryamycin (NCI-ADR/RES), kidney (786-0), lung, non-small cells (NCI-H460), prostate (PC3), ovarian (OVCAR-03), colon (HT-29), and K562 erythromyeloblastoid leukemia), as determined by SRB assay. Mannich bases 86 were also equipotent to reference drug doxorubicin against at least one, if not many, of these cancer cell lines, often with IC50 values as low as 1 μg/mL. In addition, 1,2,4-triazolo[1,5-a]pyrimidine-7-amines having at position 2 a side chain capped with a furan ring were aminomethylated with various secondary aliphatic amines to yield a large series of furan Mannich bases 87 (Fig. 16) [109], [110]. Cytotoxicity of compounds 87 against liver cancer (Bel-7402) and fibro sarcoma (HT-1080) cells was established using MTT assay, and the results suggest that both the substitution of the arylamine moiety and the nature of the aliphatic amino residue in the aminomethyl function have a significant influence on the potency of these candidates. In particular, the presence of a 4-trifluoromethyl group or a 4-fluoro-3-trifluoromethyl substitution pattern in the arylamine moiety led to high cytotoxicity against both cell lines at levels comparable to that of reference drug cisplatin. Also, the presence of dimethylamino, 1-piperidinyl or 1-pyrrolidinyl moieties as aliphatic amino residues in the aminomethyl group of Mannich bases 87 appears to result in significant antiproliferative effects, while 4-morpholinyl or 4-methylpiperazinyl moieties drastically decrease or even abolish cytotoxicity.
Fig. 16.
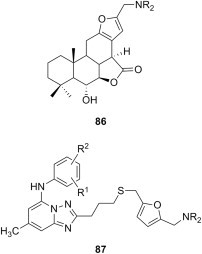
Cytotoxic furan Mannich bases.
Titanium-based chemical entities enjoy the reputation of having a tremendous potential against solid tumors. Recently, a number of studies presented the synthesis and the cytotoxicity for a series of titanocenes, some of them featuring various aminomethylated five-membered heterocycles as substituent of either one or both cyclopentadiene moieties [111], [112], [113], [114]. Thus, titanocenes 88 and 89 (Fig. 17 ) containing mono- and bis-aminomethylated pyrroles, respectively, or titanocenes 90 and 91 (Fig. 17) having a dimethylaminomethyl group at position 1 and 3 of indole, respectively, as well as titanocene 92 (Fig. 17) presenting an aminomethylated imidazole ring, have been screened against pig kidney epithelial cells (LLC-PK1) or human renal cancer cells (Caki-1) and found to have IC50 values quite similar to that of cisplatin (in the range of 5–10 μm). The presence of at least one aminomethyl function in their structure is claimed to be crucial for the high cytotoxicity of these titanocenes. The aminomethyl groups are able to coordinate the titanium center, and could therefore stabilize the mono- or dication formed through hydrolysis of either one or both chlorine atoms inside the cell. This results in enhanced interactions between titanocene and DNA, leading to cell death at low concentrations.
Fig. 17.
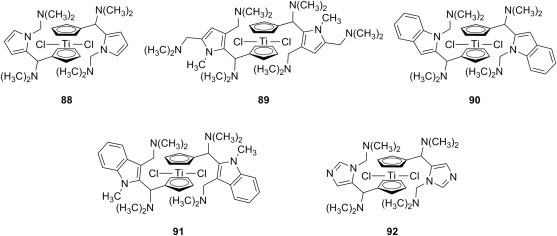
Cytotoxic Mannich bases of pyrrole-, indole- and pyrazole-containing titanocenes.
Aminomethylation of terminal alkynes has also been employed for the generation of Mannich bases with potential cytotoxic effect. 10-(Prop-2-ynil)phenothiazines underwent aminomethylation with secondary aliphatic amines to give propargylamines 93 (R1 = H, Cl, CF3) (Fig. 18 ), which were first evaluated for cytotoxic activity using two hematological tumor cell lines, namely HL60 (promyelocytic leukemia) and the CCRF/CEM (lymphocytic leukemia), and were afterwards tested, in combination with doxorubicin, for ability to revert activity in the corresponding multidrug resistant variants, HL60R and CEM/VBL300 [115]. Although most compounds 93 were devoid of significant antiproliferative effect on the sensitive cell lines, a few of them were highly cytotoxic for the resistant cell lines, and appear to arrest cells in G1 phase of the cell cycle, unlike classic anticancer agents. Furthermore, several Mannich bases 93 were able to restore sensitivity to doxorubicin of the resistant cell lines, an effect that was concentration-dependent and reached maximum at 10 μM. Propargylamines 93 seem to induce apoptosis by activating the caspase cascade, although neither the extrinsic nor the intrinsic pathways appear to be involved in apoptosis [115]. Betulin derivatives bearing an ethynyl function have also served as starting materials for acetylenic Mannich bases with cytotoxic potential. For example, propargylamines 94 (Fig. 18) have been obtained from alkynes prepared through addition of an organometallic derivative of acetylene to the carbonyl function in betulonic acid esters [116], and alkynes synthesized from betulin through a sequence comprising the oxidation of the primary alcohol function to aldehyde, followed by addition of an organometallic derivative of acetylene to aldehyde carbonyl, afforded propargylamines 95 (Fig. 18) [117]. Cytotoxicity of these Mannich bases was evaluated on a panel of nine human cancer cell lines using SRB assay, and the results prove that some of these compounds show considerable toxicity (IC50 values as low as 4 μM). Introduction of the aminomethyl group significantly improved the cytotoxicity of propargylamines 94 and 95 compared to that of the parent alkynes, presumably by enhancing their solubility and bioavailability. Highly hydrophobic and sterically hindered amino moieties, such as dicyclohexylamino or dibenzylamino, led to a decrease in cytotoxicity of the corresponding aminomethylated alkynes. Mannich bases of this type were shown to act by triggering apoptosis, although a complementary process of autophagy could also be involved.
Fig. 18.
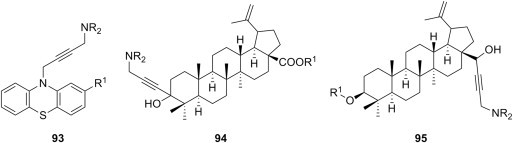
Cytotoxic alkyne Mannich bases.
In an attempt to circumvent the resistance mechanism developed by cancer cells after prolonged administration of doxorubicin and address the issues of poor solubility, short lifetime and high toxicity of prodrug doxoform [118], a second-generation, water-soluble prodrug of doxorubicin was developed by conjugation of the active drug with salicylamide by means of a Mannich reaction [119]. Doxorubicin–salicylamide conjugate doxaliform 96 (Fig. 19 ) has a half-life of approximately one hour, and was more cytotoxic than doxorubicin against MCF-7 sensitive (4-fold) and MCF-7/Adr resistant (10-fold) breast cancer cells. Furthermore, doxaliform is amenable to functionalization with a view to provide a site for attachment of a releasable targeting group that might direct the conjugate to a specific receptor that is overexpressed by cancer cells. Because many breast cancer cells overexpress estrogen receptor α, this receptor was chosen for targeting by doxasaliform having tethered a hydroxytamoxifen moiety, as in prototype 97 (Fig. 19) [120]. Cytotoxicity of these candidates as a function of the length of the tether showed that a triethylene glycol unit provides a lead compound whose growth inhibition of four selected breast cancer lines (MCF-7, MCF-7/Adr, MDA-MB-231 and MDA-MB-435) was enhanced up to 140-fold relative to doxorubicin. Later work confirmed that uptake of hydroxytamoxifen-targeted doxorubicin–salicylamide conjugate is mediated by both the antiestrogen binding site and estrogen receptor [121]. Also, several doxorubicin–formaldehyde conjugates tethered to the nonsteroidal antiandrogen cyanonilutamide were designed, synthesized and evaluated as androgen receptor-targeted ligands for specific delivery of the conjugate to prostate cancer cells [122]. Such a construct was later used in studies intended to evidence binding to androgen receptor in live PC3 prostate cancer cells and the subsequent translocation of the construct bound to the receptor to the nucleus, but the results were not very promising [123]. Other efforts [124] were directed towards the conjugates of doxasaliform with the cyclic peptide N–Me-VRGDf (known as Cilengitide), which is a potent antagonist of αVβ3 integrin involved in many cell-matrix recognition and cell adhesion phenomena, and plays an important role in angiogenesis and tumor metastasis. Although the complete construct maintained a high affinity for αVβ3 integrin, the IC50 for growth inhibition of MDA-MB-435 cells was 2-fold greater than that of doxasaliform; the poor results have been tentatively blamed on limitation of drug delivery caused by the specific reduced abundance of receptors in this type of cell. Eventually, because doxasaliform is not as active as prodrugs doxoform or doxazolidine, this line of research was terminated. However, the topic was later revisited by other authors, who reported the synthesis of constructs derived from either doxorubicin–salicylamide, daunorubicin–salicylamide or their 2-acyloxymethyl derivative, and amino-terminated poly(ethylene glycol), and their use for in vitro and in vivo studies [125]. These constructs presented cytotoxicities comparable to those of the parent drugs, and the lifetime of one of these constructs was determined to be longer than that of doxorubicin. Also, the construct was more efficient than doxorubicin at reducing the weight of S-180 xenografted tumors [125].
Fig. 19.
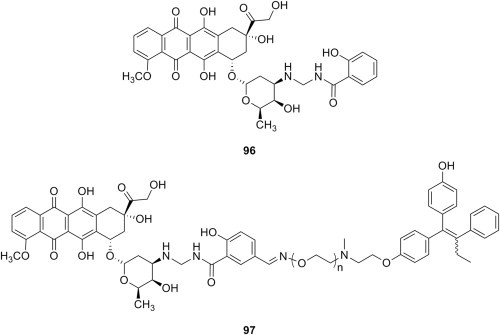
Prodrugs of doxorubicin obtained through the Mannich reaction.
Cytotoxic Mannich bases derived from miscellaneous, structurally unrelated substrates are grouped in the last paragraph of this section. 2-Aminomethylated 9-alkyl-1,2,3,4-tetrahydrocarbazole-1-ones 98 (Fig. 20 ) show moderate to potent cytotoxicity towards A549 (human lung adenocarcinoma), SGC (human gastric cancer), K562 (human myelogenous leukemia), HCT116 (human colorectal carcinoma), and KB-VCR (human oral cancer) cells using MTT assay [126]. One of candidates 98 (R = C2H5, R1 = CH3) was more cytotoxic than reference drug taxol against A549 cell line (IC50 = 70 nM), and at least one of its mechanism of action appears to be the inhibition of tubulin polymerization. A series of 3-aminomethyl imidazo[2,1-b]benzothiazoles 99 (Fig. 20) were evaluated for antiproliferative activity against hepatocellular carcinoma (HepG2), human breast (MCF-7) and human cervical (HeLa) cancer cell lines using MTT assay [127]. Mannich bases 99 inhibited the proliferation of cancer cells at concentrations lower than 10 μM, arrested the cell cycle at G2/M phase while downregulating cyclin B and upregulating Chk2 protein, and appeared to induce apoptosis based on the elevated levels of caspase-3. 6-Cinnamoyl-benzoxazol-2-ones (R1 = H, CH3O) were the substrates used for the synthesis of Mannich bases 100 (Fig. 20), which had high to moderate cytotoxicity (IC50 between 5 and 40 μM) towards human pre-B-cell leukemia cell line BV-173 and chronic myeloid leukemia K-562, and appear to exert their cytotoxic action at least in part through induction of apoptosis [128]. Replacement of the methoxy substituent in compounds 100 with chlorine led to a marginal improvement of IC50 values [129]. A series of Mannich bases 101 (R = H, CH3; X = CH2, O, N-R1) (Fig. 20) of fused pyridazinone derivatives were synthesized and tested in vitro using SRB assay to determine their growth inhibitory properties at a single 10 μM dose against sixty different human tumor cell lines. Most of them showed no activity of all, but two candidates moderately inhibited the growth of non-small cell lung cancer (EKVX and Hop-92) and glioblastoma (SNB-75) cell lines [130].
Fig. 20.
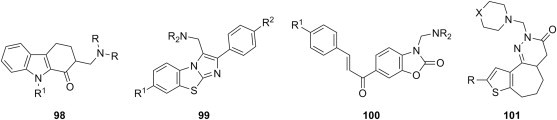
Cytotoxic Mannich bases obtained from miscellaneous substrates.
3. Antibacterial activity
An impressive number of articles dealing with the antibacterial activity of Mannich bases have been published in the last decade. From a structural point of view, Mannich bases reported in these studies are derived from nearly all of the major types of substrates capable of undergoing aminomethylation. These Mannich bases have been evaluated against both Gram-positive and Gram-negative bacteria belonging to various families. The evaluation of the antibacterial activity was performed using different screening methods, and not always the standardized versions, and this complicates the interpretation and the comparison of results obtained in different studies. Owing to the particular relevance of tuberculosis, the reports concerning the screening of Mannich bases against Mycobacterium species are presented in a separate section.
Ketonic Mannich bases with antibacterial potential are at the heart of several studies within the period of time covered by this review. Aminomethylation of α,β-unsaturated cyclic ketones with variable ring sizes afforded a library of compounds 102 (n = 1–4) (Fig. 21 ) derived from secondary cyclic aliphatic amines and having on the aromatic ring either no substituent at all, or a methyl group, or a variable number of methoxy groups [131]. Evaluation of the antibacterial activity of these Mannich bases against a panel of both Gram-positive and Gram-negative bacteria using the serial dilution method showed that the candidates derived from seven- or eight-membered α,β-unsaturated cyclic ketones were the least active of all, and that the nature of the amine moiety, or the number and the position of the methoxy groups on the aromatic ring, did not influence the antibacterial activity. None of the compounds were active against Pseudomonas aeruginosa, and only a few were moderately active against Escherichia coli. Mannich bases 102 affected Gram-positive bacteria (minimum inhibitory concentration (MIC) < 12.5 μg/mL in most cases), but the candidates were less potent than reference antibiotics. Another set of Mannich bases 103 (Fig. 21) obtained from benzo-fused cyclic ketones (indanone, 1-tetralone, benzosuberone) were tested under the same conditions against an extended panel of Gram-positive and Gram-negative bacteria [132]. In this collection, both the nature of the amine moiety and the alkoxy substituent R1 influence the antibacterial activity, to a greater extent for Gram-positive bacteria and to a lesser extent for Gram-negative bacteria. Indanone derivatives were active against both classes of bacteria (MIC between 1.56 and 25 μg/mL), whereas tetralone and benzosuberone derivatives were mostly active against Gram-positive bacteria (presumably due to the higher outer membrane permeability of these bacteria). Also, a statistical comparison between the antibacterial activity of Mannich bases 102 and Mannich bases 103 showed that the latter were more active than the former [131], [132]. Based on oxazolidinone antibacterials linezolid and eperezolid, a small series of Mannich bases 104 (R = NHCOCH3 or NHCSCH3) (Fig. 21) derived from cyclohexanone or benzo-fused cyclic ketones was synthesized and evaluated against three selected Gram-positive microorganisms using the standard serial dilution method [133]. All of the compounds having an acetamido group had low antibacterial activity, but the replacement of this group with thioacetamido restored the activity up to the level exhibited by the parent oxazolidinone antibacterial. Further modification (R = NHCSNH2) produced a candidate whose antibacterial activity was superior to that of linezolid. 4,6-Dimethoxybenzofuran-3(2H)-one double Mannich bases 105 (Fig. 21) had moderate to high antibacterial activity, as determined using disc diffusion method at a concentration of 0.5% in DMSO [134]. The majority of compounds 105 were active against E. coli, and those derived from pyrrolidine, diethylamine and di-n-butylamine as amine reagents showed significant antibacterial activity (zone of inhibition 17–22 mm) towards most bacteria, comparable to that of reference drug gentamycin (zone of inhibition 18–24 mm). On the other hand, compound 105 derived from N-ethylmethylamine was inactive (no inhibition at concentrations higher than 100 μg/mL). Also, structurally diverse types of Mannich bases derived from acetophenones or 2-acetylthiophene, such as mono-Mannich bases of type 8 (R = CH3) (Fig. 3), bis-Mannich bases of type 13 (R = CH3) (Fig. 4), and piperidinols of type 14 (R = CH3) (Fig. 4), as well as their azine derivatives 16 (Fig. 4), are devoid of antibacterial activity against a wide range of human- and plant-pathogenic bacteria [135]. Methiodide 106 (Fig. 21) of dimethylamine Mannich base of acetophenone was the sole active compound in this series, and only against Staphylococcus aureus, but the diameter of the inhibition zone measured 9 mm (compared to 25 cm in the case of reference drug ofloxacin), and its MIC was 500 μg/mL.
Fig. 21.
Antibacterial ketonic Mannich bases.
The antibacterial activity of ketonic Mannich bases generated from arylamines and aromatic aldehydes was also investigated by several groups. Starting from cyclohexanone, a set of eleven compounds 107 was obtained, and screening against four common bacteria using serial dilution method showed that many of these candidates have excellent antibacterial activity, comparable to that of ceftriaxone [136]. However, the lack of rational design and systematic use of the aldehyde and amine components used to generate Mannich bases 107 (Fig. 21) preclude any logical conclusions regarding SAR in this collection. Use of four differently substituted acetophenones, 4-fluorobenzaldehyde and several para-substituted anilines led to Mannich bases 108 (Fig. 21), which were screened against four common bacteria [137]. Most of these compounds were inactive, but candidate 108 (R1 = 2-Br, R2 = F) was more potent than reference drug ampicillin against all four microorganisms, whereas another candidate 108 (R1 = 2-Br, R2 = H) was more potent than ampicillin against E. coli and S. aureus. Arylamine Mannich bases 109 and 110 (Fig. 21) were obtained from 3-acetylcoumarine and 2-acetylbenzofuran, respectively, and evaluated against E. coli, S. aureus and P. aeruginosa using disc diffusion method [138]. Since most compounds in this series are derivatives of 4-aminobenzoic acid, it has been expected that at least a few of these compounds have good antibacterial activity. Three candidates 109 (R = H, Cl or F) had antibacterial activity comparable to that of reference drug streptomycin, but the rest were almost inactive. The series of candidates 110 provided only one remarkably active Mannich base (R = N(CH3)2), but the rest of the compounds with a benzofuran moiety are generally more active than coumarine-derived candidates 109. 3-(Arylamino)-1-ferrocenyl-1-propanones 111 (Fig. 21) showed broad spectrum activity against both Gram-positive and Gram-negative bacteria, the highest degree of growth inhibition being obtained for S. aureus [139]. MIC values varied between 0.2 and 12.5 mg/mL, making these compounds approximately 100-fold less active than reference drug tetracycline. Because compounds 112 (Fig. 21) with a similar structure but with a phenyl ring instead of ferrocene have been reported to possess moderate to high antibacterial activity [140], it appears that the replacement of phenyl in 112 with ferrocene could be responsible for the substantial decrease in activity recorded in the case of compounds 111.
Aminomethylated phenols have also been reported to have antibacterial activity. Mannich bases 113 (Fig. 22 ) obtained from 4-t-butylcatechol and secondary aliphatic amines have been screened for antibacterial activity against seven types of bacteria, but their potency was rather low to moderate [141]; their corresponding copper(II) complexes fared better, as two of them had MICs comparable to that of chloramphenicol (12.5 μg/mL). The Mannich reaction of a derivative of another dihydroxylic phenol with either secondary aliphatic amines or primary arylamines afforded compounds 114 (Fig. 22), whose zones of inhibition for three common bacteria, determined at two concentrations (100 and 200 μg/mL) using disc diffusion method, were smaller than those observed for reference drug ofloxacin at 20 μg/mL [142]. Because chemical modification of natural antibiotic novobiocin by means of aminomethylation was hypothesized to increase permeability through the outer membrane of Gram-negative bacteria, phenolic Mannich bases 115 (Fig. 22) were designed, synthesized and evaluated against S. aureus (one of novobiocin's usual targets), Francisella tularensis and E. coli [143]. Candidates 115 were significantly less potent than novobiocin against these three microorganisms, the antibacterial activity of the most potent of these compounds being almost 100 times weaker than that of novobiocin. Thirteen novel phenolic Mannich bases 116 (Fig. 22) derived from lawsone, benzaldehydes and primary aliphatic amines, together with their corresponding copper(II) complexes, were tested for antibacterial activity against seven types of bacteria [144]. With a few exceptions, Mannich bases 116 were generally more potent than the corresponding complexes, presumably due to the greater solubility of the pro-ligands. Two Mannich bases 116 had antibacterial activity comparable or higher to that of chloramphenicol against Bacillus subtilis, E. coli and S. aureus, whereas the other compounds inhibited bacterial growth at concentrations greater than 200 μmol/L.
Fig. 22.
Antibacterial phenolic Mannich bases and fused 1,3-oxazines.
Antibacterial activity of fused oxazines obtained through the Mannich reaction of phenols and naphthols with primary amines has also been examined. 6-Acetyl-1,3-benzoxazines 117 (Fig. 22) were screened at 50 μg/mL against three common microorganisms using disc diffusion method, and they showed good growth inhibitory activity towards S. aureus, but only moderate potency against E. coli and B. subtilis [145]. The antibacterial activity was evaluated for 1,3-benzoxazines 118 (Fig. 22) substituted with chloro and methyl groups in the aromatic ring, bis(1,3-benzoxazines) 119 (Fig. 22), naphtho[1,2-e]-1,3-oxazines 120 (Fig. 22), as well as naphtho[2,1-e]-1,3-oxazines 121 (Fig. 22) using serial dilution method [146]. Regardless of the aliphatic or aromatic nature of the moiety at the nitrogen atom, most compounds, and especially naphthoxazines 120 and 121, had MIC values greater than 50 μg/mL, but two benzoxazines 118 derived from 4-chlorophenol and aromatic amines were more potent than ampicillin against E. coli, whereas compound 119 derived from 2,4-dichlorophenol was equipotent to gentamycin against S. aureus. In addition, benzoxazines 118 and naphthoxazines 120 and 121 having benzazole moieties at the nitrogen atom were tested against four common types of bacteria [147]. Although the majority of the compounds were inactive, the antibacterial activity of few candidates was 2-fold–15-fold greater than that of reference drug tetracycline; the enhancement of activity for these candidates has been associated with the presence of a benzimidazole system at the nitrogen atom and at least one halogen in the phenolic starting material. In contradiction to the previously mentioned results, a number of naphtho[2,1-e]-1,3-oxazines 121 obtained from variously substituted arylamines were found to be quite active as antibacterial agents towards E. coli and B. subtilis [148]. These compounds were generally more active against the latter pathogen, and the candidates derived from 4-fluoroaniline, 4-ethoxyaniline and 2,4,6-tribromoaniline were even more potent than reference drug streptomycin.
The Mannich reaction of phenols fused with heterocycles has also been investigated for the preparation of antibacterials. Mannich bases of 8-hydroxyquinoline have been quaternized at the heterocyclic nitrogen atom with bromoacetic acid esters of superior alcohols (R = n-C12H25, n-C16H33, n-C18H37) to give compounds 122 (X = O, CH2) (Fig. 22) [149]. The antibacterial activity of candidates 122, determined for three concentrations (1, 2.5 and 5 mg/mL) using disc diffusion method, showed that the compounds were active towards both Gram-positive and Gram-negative bacteria, but the presence in their structure of long alkyl chains rather than the aminomethyl function is most likely responsible for their activity. Phenolic Mannich bases 123 (R1 = H, Br) (Fig. 22) were evaluated against E. coli and Bacillus cirroflagellosus at 1 mg/mL using disc diffusion method, but their antibacterial activity was generally weak, with the exception of candidates derived from morpholine as amine reagent [150]. Aminomethylated derivatives 41 (R = OH, R1 = H, R2 = aminomethyl) of apigenin (Fig. 7) also manifested antibacterial activity in various degrees; the most active candidates were the Mannich base derived from cyclohexylamine, which was as potent as tetracycline against B. subtilis (MIC 3.9 μg/mL), and the Mannich base derived from morpholine, which had the same antibacterial activity as ampicillin against S. aureus (MIC 2 μg/mL) [58].
Phenolic Mannich bases derived from 3-hydroxy-4H-pyran-4-ones received special attention as potential antibacterials. Allomaltol (5-hydroxy-2-methyl-4H-pyran-4-one) was converted into Mannich bases 124 (Fig. 23 ), whose antibacterial activity against four different Gram-positive bacteria was only moderate (MIC > 32 μg/mL) [151]. On the other hand, the antibacterial activity of Mannich bases 125 (Fig. 21) derived from chlorokojic acid was superior to that of analogous 124, and the candidates were shown to possess a broad spectrum, while being more active against standard strains (MICs between 1 and 32 μg/mL) than towards clinical, drug-resistant isolates [152]. There is no striking difference between the antibacterial activity of aminomethylated derivatives 125 having 4-benzylpiperazines and those having 4-substituted piperidines as amine moiety. The growth inhibition of Gram-positive bacteria (with the exception of Enterococcus faecalis) occurred at MIC values that were lower than those required for the growth inhibition of Gram-negative bacteria. Mannich base 125 with 1-(4-chlorobenzhydryl)piperazine as amine moiety seems to be best compound in this series, but its activity was consistently lower than that of any reference antibacterial drug. Subsequently, a novel series of Mannich bases 125 was synthesized, this time using various 4-(substituted aryl)piperazines as amine reagent, but the compounds in this collection were weaker antibacterials than analogous 125 derived from 4-benzylpiperazines [153]. Screening of another novel set of compounds 125 with either 4-benzylpiperazines of 4-arylpiperazines did not identify any compounds with improved or remarkable antibacterial activity, as the best candidates in this study had MIC values between 4 and 16 μg/mL [154]. However, when piperazinyl-substituted fluoroquinolones were employed as amine reagents in the Mannich reaction of 3-hydroxy-4H-pyran-4-ones as substrates, the resulting hybrids 126 (R = Cl) (Fig. 23) had a broad spectrum and were highly active against a panel comprising both Gram-negative and Gram-positive bacteria [155]. Generally, aminomethylated derivatives of chlorokojic acid (126, R = Cl) were more potent antibacterials than aminomethylated derivatives of kojic acid (126, R = OH). Mannich base 126 (R = Cl) derived from fluoroquinolone ciprofloxacin was the most potent candidate in the series. Replacement of the quinolone scaffold in candidates 126 derived from norfloxacin with a naphthyridone moiety (as in Mannich bases 126 derived from enoxacin) resulted in decreased antibacterial activity. Among the bacteria used in this study, E. coli and Klebsiella pneumoniae were the most sensitive microorganisms to hybrids 126. Docking of the most active compound into topoisomerase II DNA-gyrase active site (which is the traditional target of quinolones) showed that Mannich base 126 derived from ciprofloxacin binds in a manner similar to ciprofloxacin to the enzyme's active site, and that the 3-hydroxypyran-4-one moiety is anchored with additional hydrogen bonding within the binding pocket, thus increasing the stability of the inhibitor–enzyme complex.
Fig. 23.
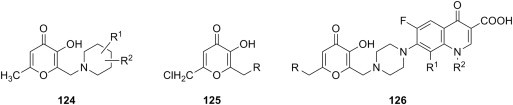
Antibacterial phenolic Mannich bases derived from 3-hydroxy-4H-pyran-4-ones.
Mannich bases of isatin derivatives have also been investigated as antibacterial agents. Aminomethylation of isatin semicarbazone with (hetero)arylamines afforded Mannich bases 127 (Fig. 24 ), whose antibacterial activity was moderate towards E. coli and good towards S. aureus [156]. In addition, a series of Mannich bases 128 (R = secondary aliphatic amines) (Fig. 24) derived from a hydrazone of isatin incorporating the biologically relevant moieties of 1,3,5-triazine, sulfa drugs and azacarbazole had moderate to good antibacterial activity against the previously mentioned microorganisms [157]. Hydrazidones obtained from isatin and hydrazides of variously substituted acetic acids have also been aminomethylated with a view to synthesize antibacterial Mannich bases. For example, Mannich bases 129 (R1 = 2-oxo-3-phenyl-2H-1,8-naphthyridin-1-yl) (Fig. 24) derived from substituted isatins and morpholine, piperidine or 1-methylpiperazine generally showed poor antibacterial activity against the two aforementioned types of bacteria, with the exception of compound 129 (R = 4-methylpiperazinyl, R2 = 5-Br), whose inhibition determined by disc diffusion method was approximately half of that for reference drug gentamycin [158]. Another collection of Mannich bases 129 (R1 = 4- or 4,5-substituted 2-ethylphenyloxy) generated from substituted isatins as substrates and morpholine or piperidine as amine reagents exhibited weak to moderate antibacterial activity towards E. coli, S. aureus and Shigella flemeri (zone of inhibition 12–20 mm) compared to norfloxacin (zone of inhibition 26–28 mm) [159]. Schiff bases generated from isatin and various (hetero)aromatic amines represent another type of isatin-derived substrate that was subjected to aminomethylation to obtain potential antibacterial agents. The use of isatin Schiff bases in the Mannich reaction with 2-((2,6-dichlorophenyl)amino])phenylacetic acid as the amine reagent yielded compounds 130 (R1 = substituted phenyl) (Fig. 24), which proved to be 3–12-fold less potent than reference drug ciprofloxacin against an array of bacteria [160]. When 3-chloro-4-fluoroaniline was used to generate Schiff bases from isatin and 5-chloroisatin, aminomethylation with secondary aliphatic amines gave Mannich bases 131 (R1 = 3-Cl-4-FC6H3, R2 = H or 5-Cl) (Fig. 24); these compounds had moderate to good activity against E. coli, S. aureus, P. aeruginosa and Salmonella typhi in disc diffusion assay, and showed also moderate to good β-lactamase inhibitory activity [161]. Schiff base of 5-chloroisatin and trimethoprim was aminomethylated to give compounds 131 (R1 = 4-amino-5-(3,4,5-trimethoxybenzyl)pyrimidin-2-yl), but only Mannich bases 131 derived from ciprofloxacin, lomefloxacin or gatifloxacin as amine reagents in the Mannich reaction were at least as potent, if not more potent, than the corresponding parent fluoroquinolones against a large panel of bacteria [162]. Antibacterial activity of Mannich bases 131 (R1 = 5-benzyl-3-thioxo-2H-1,2,4-triazol-4-yl) derived from Schiff bases of isatin or 5-halogen-substituted isatins and an aminotriazolethione was generally good against four common microorganisms, as the size of the inhibition zones for some of the candidates (especially those derived from halogenated isatins and having piperidine or morpholine residues in the aminomethyl group) was comparable to the size of the inhibition zone for reference drug ciprofloxacin [163]. Recently, Mannich bases 132 (Fig. 24) obtained using Schiff bases derived from 5-fluoroisatin and 4-arylideneaminoanilines as substrates and ciprofloxacin as amine reagent were shown to be generally less potent antibacterials than reference drug ciprofloxacin, although some candidates had MIC values comparable to those of ciprofloxacin against the investigated bacteria [164].
Fig. 24.
Antibacterial phenolic Mannich bases generated from isatin derivatives.
A large number of papers published in the last decade report the antibacterial activity of Mannich bases of 2,3-dihydro-1,2,4-triazole-3-thiones. From a structural point of view, Mannich bases from triazolethiones that were synthesized through aminomethylation in these reports fall into two major categories: 5-substituted 2-aminomethyl-2H-1,2,4-triazole-3-thiones 133 and 5-substituted 2-aminomethyl-4-arylideneamino-2H-1,2,4-triazole-3-thiones 134. The relevant information from these studies (i.e., substituents at position 4 and 5 of the triazole ring, amine reagents used in aminomethylation, microorganisms employed in the antibacterial evaluation) is presented in a condensed manner in Table 1 . In the series of compounds 133a, the nature of the amine moiety in the aminomethyl group appears to be critical for the antibacterial activity, as Mannich bases from piperazines were consistently more potent than Mannich bases from arylamines, and some of them were even more potent than reference drug ampicillin [165]. A comparison between antibacterial activities of 133a and structurally related 134b suggests that the replacement of substituent at position 4 of the triazole ring in 133a with hydroxybenzylideneamino in 134b further enhances the antibacterial activity. Although it was shown that substitution with chlorine of the phenyl at position 5 of the triazole ring improves the antibacterial activity in the series of compounds 133b and 133c, and that aminomethylation also increases the bacterial growth of Mannich bases 133c compared to the parent triazolethione, no candidates with substantial antibacterial activity could be singled out from these two collections [166], [167], [168]. However, use of ciprofloxacin as amine reagent in the synthesis of Mannich bases 133c and 133d led to a substantial increased antibacterial activity of these compounds, and some of them were as potent as reference drugs ciprofloxacin and vancomycin against several types of bacteria and methicillin-resistant S. aureus (MRSA), respectively [169]. Mannich bases 133e or 134f having a diphenylsulfone moiety at position 5 of the triazole ring had only weak antibacterial activity [170]. A small number of Mannich bases 133f, especially those having a halogen as substituent of the phenyl at N-4 of the triazole ring, inhibited the growth of Gram-positive bacteria [171]. Mannich bases 133g, especially candidates with morpholine, 4-benzylpiperazine, N-methylpiperidine and trifluoromethylphenylpiperazine in the aminomethyl group, had very good antibacterial activity (MIC 1.56–12 μg/mL) [172], whereas analogous 133h having 2-furyl instead of 2-thiophenyl were less potent (inhibition zones of 6–13 mm, compared to 10–35 mm for ampicillin) [173]. The three Mannich 133i bases reported in a study gave interesting results, as the candidates derived from 1-methylpiperazine (inhibition zones of 23–30 mm for all bacteria) and 2-(4-morpholinyl)ethylamine (inhibition zones of 14–22 mm for some of the microorganisms under evaluation) were more potent than reference drug ampicillin, whereas the bis-Mannich base 133i derived from piperazine was inactive [174]. However, further elaboration of 133i into compound 133p abolished the antibacterial activity [174]. Mannich bases 133j, which feature a 3-pyridinyl moiety at position 5 of the triazole ring, were generally poorer antibacterials than reference drug ampicillin, although there were isolated examples amongst these candidates that show more potency than ampicillin against certain bacteria [175]. Furthermore, the replacement of 4-pyridinyl with 2-quinolinyl as substituent at position 5 of the triazole ring, and modification of phenyl into benzyl as substituent at position 4 of the triazole ring led to inactive Mannich bases 133k [176]. Both types of Mannich bases 133l and 134l, featuring a 1,2,4-triazole moiety at position 5 of the triazolethione scaffold, showed good antibacterial activity, but compounds 134l were generally more potent than 133l, and two of candidates 134l actually had MIC values comparable to those of reference drug ciprofloxacin [177]. Mannich bases 133m and 133n were devoid of effective antibacterial activity [178], [179], as was structurally related candidate 133o (a singular and notable exception is this compound's activity against Bacillus cereus) [179]. Mannich bases 133q were also inactive as antibacterials, except for a few candidates that were more potent against P. aeruginosa than reference drug ampicillin [180]. Every member of the series of Mannich bases 134a was active against the tested bacteria, but not as potent as reference drug nitrofurazone [181]. In addition, the nature of substituent at position 5 of the triazole ring in the series of compounds 134a had no noticeable influence on their antibacterial activity, but the data (based on a single example) suggest that presence of chlorine in the aromatic ring of substituent X of the pyrazole moiety could improve the activity. Mannich bases 134c were mostly active against S. aureus and B. subtilis, and candidates derived from ethyl piperidine-4-carboxylate were generally more potent than those derived from piperazines [182]. It should be noted that the presence of halogen in the structure of 134c (R2 = 2,6-difluorophenyl or 2,6-dichlorophenyl) did not enhance the antibacterial activity of these Mannich bases. In contrast, substitution with halogen in compounds 134d (e.g., R2 = 2,4-dichlorophenyl) led to the most potent candidates in the series, and their antibacterial activity was comparable to that of reference drug ciprofloxacin [183]. Use of 2,4-dichloro-5-fluorophenyl moiety at position 5 of the triazole ring in Mannich bases 134e also produced candidates with good to excellent antibacterial activity [184]. Compounds 134e having secondary aliphatic amines in the aminomethyl group were generally more potent than those having arylamines, even if the anilines used in aminomethylation were substituted with additional halogen atoms [184]. Although Mannich base 134g had no antibacterial activity, a close analogue of 134g such as compound 135 (Fig. 25 ) was 1.5–3-fold more potent than ampicillin against most bacteria in the panel, with the exception of S. aureus [185]. Replacement of 4-methylpiperazin-1-yl with 4-morpholinyl in the aminomethyl group of Mannich base 134g, and use of substituted phenyl other than 4-fluorophenyl as R2 afforded structurally related candidates 134h with moderate antibacterial activity [186]. Antibacterial activity of Mannich bases 134i was moderate to good, but even the most potent compounds in this series (all of them having a 4-methylpiperazin-1-yl moiety in the aminomethyl group) were at least 4-fold less active than reference drug ciprofloxacin [187]. Mannich bases 134j also had moderate to good antibacterial activity (inhibition zones of 12–22 mm), but were less potent than tetracycline (inhibition zones of 25 mm) [188]. Overall antibacterial activity was reasonable in the series of Mannich bases 134k, while a few compounds had MIC values against various bacteria close to those determined for reference drugs [189]. A few Mannich bases 134m were more potent against P. aeruginosa than reference drugs levofloxacin and amikacin, while most candidates exhibited broad, moderate to good antibacterial activity [190].
Table 1.
Antibacterial Mannich bases derived from 2,3-dihydro-1,2,4-triazole-3-thiones: substitution patterns and tested microorganisms.
 | |||||
|---|---|---|---|---|---|
| R1 | R2 | NR2 | Microorganisms | Reference | |
| a |  |
CH3 4-ClC6H4 |
4-substituted piperazines substituted arylamines |
S. aureus B. subtilis Micrococcus luteus E. coli P. aeruginosa |
[165] |
| b |  |
4-BrC6H4 | secondary aliphatic amines |
S. aureus Staphylococcus epidermidis B. subtilis Bacillus cereus M. luteus |
[166] |
| c |  |
substituted phenyl | secondary aliphatic amines, ciprofloxacin |
S. aureus S. epidermidis B. subtilis B. cereus M. luteus |
[169], [167], [168] |
| d | 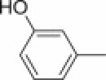 |
substituted phenyl | ciprofloxacin |
S. aureus S. epidermidis B. subtilis B. cereus M. luteus E. coli P. aeruginosa Proteus mirabilis |
[169] |
| e | 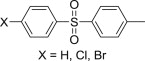 |
n-C3H7 | morpholine |
Acinetobacter baumanii Citrobacter freundii P. aeruginosa E. faecalis S. aureus S. epidermidis B. subtilis |
[170] |
| f | substituted phenyl | secondary aliphatic amines |
E. coli K. pneumoniae P. aeruginosa S. aureus |
[171] | |
| g | C2H5 CH2CH CH2 C6H5 |
secondary aliphatic amines |
E. coli K. pneumoniae P. aeruginosa S. aureus |
[172] | |
| h | CH2C6H5 | secondary aliphatic amines |
E. coli Enterobacter aerogenes Yersinia pseudotuberculosis P. aeruginosa S. aureus E. faecalis B. cereus |
[173] | |
| i | C6H5 | piperazine 1-methylpiperazine 2-(4-morpholinyl)ethylamine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[174] | |
| j | C6H5 CH2C6H5 |
morpholine thiomorpholine 4-(4-fluorophenyl)piperazine 1-(3-aminopropyl)imidazole furfurylamine |
E. coli Y. pseudotuberculosis P. aeruginosa S. aureus E. faecalis B. cereus Serratia marcescens Klebsiella oxitoca Arthrobacter oxydans Proteus vulgaris |
[175] | |
| k |  |
CH2C6H5 | secondary and primary aliphatic amines |
E. coli E. aerogenes Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[176] |
| l |  |
C2H5 CH2C6H5 |
morpholine 1-methylpiperazine |
B. subtilis Streptococci K. pneumoniae E. coli |
[177] |
| m | 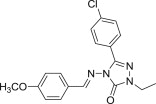 |
C6H5 | morpholine, piperazine |
E. coli K. pneumoniae Y. pseudotuberculosis E. aerogenes P. aeruginosa S. aureus E. faecalis B. cereus |
[178] |
| n | 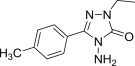 |
C6H5 | 1-methylpiperazine 2-(4-morpholinyl)ethylamine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[179] |
| o | 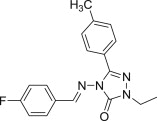 |
4-FC6H4 | 1-methylpiperazine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[179] |
| p | 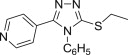 |
4-CH3C6H4 | 1-methylpiperazine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[174] |
| q | 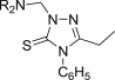 |
C6H5 | morpholine 2-(4-morpholinyl)ethylamine furfurylamine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[180] |
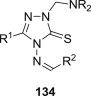 | |||||
| a | CH3 C3H7 C6H5 |
 |
1-methylpiperazine |
S. aureus B. subtilis E. coli P. aeruginosa |
[181] |
| b |  |
4-HOC6H4 2-HOC6H4 |
1-substituted piperazines |
S. aureus B. subtilis M. luteus E. coli P. aeruginosa |
[165] |
| c |  |
substituted phenyl | 1-substituted piperazines ethyl piperidine-4-carboxylate |
S. aureus B. subtilis M. luteus E. coli P. aeruginosa |
[182] |
| d | substituted phenyl | morpholine 1-methylpiperazine |
S. aureus P. aeruginosa K. pneumoniae E. coli |
[183] | |
| e |  |
4-ClC6H4 4-(CH3)2NC6H4 3,4-(OCH2O)C6H3 |
morpholine 1-ethylpiperazine substituted primary arylamines |
S. aureus E. coli P. aeruginosa K. pneumoniae |
[184] |
| f |  |
substituted phenyl, 2-furyl | morpholine |
A. baumanii C. freundii P. aeruginosa E. faecalis S. aureus S. epidermidis B. subtilis |
[170] |
| g | 4-FC6H4 | 1-methylpiperazine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[185] | |
| h | substituted phenyl, 2-thienyl, 2-furyl | morpholine |
S. aureus B. cereus E. coli K. pneumoniae |
[186] | |
| i | 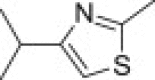 |
substituted phenyl | morpholine 1-methylpiperazine |
S. aureus S. faecalis B. subtilis K. pneumoniae E. coli P. aeruginosa |
[187] |
| j | 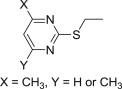 |
substituted phenyl, 6-methoxy-2-naphthyl, 8-quinolinyl | 1-methylpiperazine |
P. aeruginosa S. marcescens S. aureus E. coli |
[188] |
| k |  |
substituted phenyl | secondary aliphatic amines |
E. coli Salmonella typhimurium Listeria monocytogenes S. aureus P. aeruginosa Streptococcus pyogenes |
[189] |
| l |  |
C6H5 4-O2NC6H4 4-CH3OC6H4 |
morpholine 1-methylpiperazine |
B. subtilis Streptococci K. pneumoniae E. coli |
[177] |
| m | 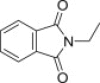 |
substituted phenyl | morpholine |
E. coli P. aeruginosa K. pneumoniae S. aureus Enterobacter cloacae |
[190] |
Fig. 25.
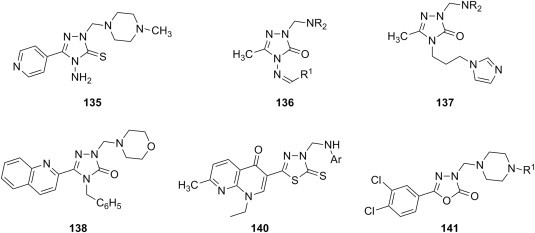
Antibacterial Mannich bases derived from 2,3-dihydro-1,2,4-triazole-3-ones, 2,3-dihydro-1,3,4-oxadiazole-3-ones, and 2,3-dihydro-1,3,4-thiadiazole-3-thiones.
Evaluation of Mannich bases of 2,3-dihydro-1,2,4-triazole-3-ones as antibacterial agents received little attention in recent literature. A small series of six Mannich bases 136 (Fig. 25) derived from 4-(substituted (hetero)arylidendeamino)-5-methyl-1,2,4-triazole-2-ones was synthesized and tested against seven types of bacteria, and the antibacterial activity of the candidates ranged from moderate to excellent [191]. All compounds were more potent than reference drug ampicillin against E. coli and P. aeruginosa, and candidate 136 (R1 = 2-hydroxyphenyl, NR2 = 2-(4-morpholinyl)ethylamino) was at least as active as ampicillin against all types of bacteria used in the study, with the exception of S. aureus. Out of Mannich bases of type 137 (Fig. 25), only those derived from morpholine or piperidine were excellent, broad spectrum antibacterials, whereas other analogues were either inactive, or weak antibacterials, or moderately active against the tested bacteria [192]. A single Mannich base 138 (Fig. 25) of 1,2,4-triazole-3-ones having a 2-quinolinyl moiety as substituent at position 5 of the triazole ring was synthesized and evaluated for antibacterial activity, and its effect on E. coli and P. aeruginosa was comparable to that of ampicillin [176]. Three Mannich bases having a 3-pyridinyl moiety at position 5 of 1,2,4-triazole-3-one scaffold, analogous to compounds 133j derived from 1,2,4-triazole-3-thione, had moderate to good activity against most bacteria in the panel, but were not superior to reference drug ampicillin [175].
Antibacterial activity of Mannich bases 139 of 2,3-dihydro-1,3,4-oxadiazole-2-thiones has been investigated extensively. The structural features of the compounds reported in these studies have been summarized in Table 2 , along with the microorganisms used for screening. Mannich bases 139a had moderate to good antibacterial activity (MIC values between 6.25 and 25 μg/mL), but none was as potent as reference drug ciprofloxacin, and no definite SAR could be inferred from the biological results [193]. The growth inhibition of Mannich bases 139b appears to be modulated by the nature of the amine in the aminomethyl residue; thus, candidates derived from 3-trifluoromethylaniline were the most potent in this series and had antibacterial activity comparable by that of reference drug ciprofloxacin, followed in order by Mannich bases 139b derived from morpholine, 2-chloro-5-methylaniline and 4-chloro-2-trifluoroacetylanline [194]. In the case of Mannich bases 139c, candidates derived from secondary aromatic amines were inactive, whereas the remaining compounds were less active against Gram negative bacteria, and more active against Gram positive bacteria (MIC values 2–8-fold lesser than those of reference drug ampicillin) [195]. Both Mannich bases 139d reported in this study [185] had moderate to good antibacterial activity, and both were more potent than ampicillin against E. coli and E. faecalis, but the candidate derived from 1-methylpiperazine was generally more potent than Mannich base derived from 2-(4-morpholinyl)ethylamine. In the case of thiazole-containing Mannich bases, antibacterial activity of compounds 139e was moderate (MIC values between 16 and 62.5 μg/mL) [187], whereas compounds two of compounds 139f emerged as effective antibacterials, especially against Gram-negative bacteria, with MIC values 2–4-fold lesser than those for reference drug chloramphenicol [196]. Several Mannich bases 139g (NR2 = NHC6H4F-4, NHC6H4Cl-4, NHC6H4CF3-2, NHC6H3F2-2,5) had antibacterial activity comparable to that of reference drug penicillin, and, for some bacteria, even comparable to reference drug streptomycin [197]. Mannich base 139h (NR2 = 1-piperazinyl) was equipotent to reference drug ciprofloxacin against S. aureus, but the antibacterial activity of the rest of candidates in this series was weaker than that of ciprofloxacin against the tested bacteria [198]. The compounds reported in a study, including Mannich bases 139i, are claimed to exhibit moderate antibacterial activity, but lack of access to the original information (which is only provided in the online edition as Supplemental Material, and is missing) makes data assessment impossible [199]. Phenylpiperazine-containing Mannich base 139j was generally more potent that candidate 139j derived from ethyl isonipecotate, but both had moderate antibacterial activity compared to reference drug ampicillin [176]. Three out of eight synthesized Mannich bases 139k were screened for antibacterial activity, but their growth inhibition was only weak to moderate (inhibition zones of 12–26 mm) compared to that of reference drug chloramphenicol (inhibition zones of 37–44 mm) [200]. None of the 21 synthesized Mannich bases 139l presented any antibacterial activity at the maximum concentration tested (250 μg/mL) [98]. Mannich base 139m was the only compound with antibacterial activity in the structural diverse library evaluated in the study, but its effects were moderate at best [189]. Screening results for Mannich bases 139n were also disappointing, as they only barely inhibited the growth of E. coli at a concentration of 10 mg/mL [174]. Out of two synthesized Mannich bases 139o, the candidate having a morpholine residue in the aminomethyl group displayed excellent antimicrobial activity against all the tested microorganisms, while the candidate having a methylpiperazine residue had good or moderate activities against most bacteria in the panel, with the exception of E. coli and K. pneumoniae [190]. The growth inhibition exerted by Mannich bases 139p was moderate compared to reference drugs, and it was uninfluenced by the nature of the amine moiety in the aminomethyl group [177]. Nalidixic acid-based oxadiazolethione Mannich bases 139q exhibited very weak antibacterial activity (MIC values > 750 μg/mL) compared to reference drug streptomycin (MIC = 5 μg/mL), and only towards B. subtilis, but some of the structurally related Mannich bases 140 of 2,3-dihydro-1,3,4-thiadiazole-2-thione (Fig. 25) were quite active (MIC values as high as 6.25 μg/mL) against the microorganisms used in the study [203]. Finally, Mannich bases 141 having a 3,4-dichlorophenyl moiety at position 5 of 2,3-dihydro-1,3,4-oxadiazole-2-one ring (Fig. 25) were inactive against S. aureus, E. coli, P. aeruginosa and Bacillus megaterium [204].
Table 2.
Antibacterial Mannich bases derived from 2,3-dihydro-1,3,4-oxadiazole-3-thiones: substitution patterns and tested microorganisms.
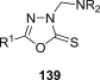 | ||||
|---|---|---|---|---|
| R1 | NR2 | Microorganisms | Ref. | |
| a |  |
secondary aliphatic amines |
S. aureus E. coli P. aeruginosa K. pneumoniae |
[193] |
| b |  |
morpholine primary arylamines |
S. aureus E. coli P. aeruginosa K. pneumoniae |
[194] |
| c | piperazines primary arylamines secondary arylamines |
S. aureus B. subtilis E. coli P. aeruginosa |
[195] | |
| d | 1-methylpiperazine 2-(4-morpholinyl)ethylamine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[185] | |
| e |  |
morpholine 1-methylpiperazine |
S. aureus S. faecalis B. subtilis K. pneumoniae E. coli P. aeruginosa |
[187] |
| f | substituted arylamines |
S. aureus S. pyogenes E. coli P. aeuroginosa |
[196] | |
| g |  |
primary arylamines |
B. subtilis S. aureus P. aeruginosa Klebsiella aerogenes Bacillus sphaericus Chromobacterium violaceum |
[197] |
| h |  |
primary arylamines diphenylamine piperazines |
S. aureus S. pyogenes |
[198] |
| i |  |
primary arylamines |
S. aureus E. coli B. subtilis P. aeruginosa |
[199] |
| j |  |
1-phenylpiperazine ethyl piperidine-4-carboxylate |
E. coli E. aerogenes Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[176] |
| k |  |
primary arylamines |
E. coli M. luteus S. aureus |
[200] |
| l | 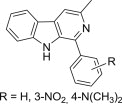 |
primary aliphatic amines secondary aliphatic amines |
S. aureus B. subtilis E. coli P. aeruginosa |
[98] |
| m | 1-phenylpiperazine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[201] | |
| n |  |
2-(4-morpholinyl)ethylamine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus B. cereus |
[174] |
| o |  |
morpholine 1-methylpiperazine |
E. coli Y. pseudotuberculosis P. aeruginosa E. faecalis S. aureus K. pneumoniae E. aerogenes B. cereus |
[202] |
| p |  |
morpholine 1-methylpiperazine |
B. subtilis Streptococci K. pneumoniae E. coli |
[177] |
| q |  |
primary arylamines |
S. aureus B. subtilis E. coli K. pneumoniae P. aeruginosa |
[203] |
Two studies presented the synthesis and antibacterial evaluation of C-Mannich bases 142 of thiazolidinones (Fig. 26 ) having a thiazolyl-2-imino substituent at position 2 [205], [206]. While candidates 142 having alkyl or aryl groups as R1 substituent at C-5 of thiazolidinone ring showed no antibacterial activity against a panel of eight bacteria, some of their analogues unsubstituted at C-5 (R1 = H) had weak antibacterial activity against S. aureus (MIC values of 20–40 μg/mL). Two of Mannich bases 143 (R1 = 2-HOC6H4) of thiazolidinones featuring a 6,8-dibromo-4-oxoquinazolin-3-yl moiety (Fig. 26) were 2-fold less potent against S. typhimurium (when NR2 = diethylamino) and 4-fold less potent against B. cereus (when NR2 = 1-piperidinyl) than reference drug ciprofloxacin [207]. Antibacterial activity of Mannich bases 144 (Fig. 26) was found to be more potent (MIC values between 6.25 and 100 μg/mL) than that of parent thiazolidinones, and the SAR suggests that the nature of substituent R1 influences significantly the ability of these compounds to inhibit the growth of various bacteria [208].
Fig. 26.
Antibacterial Mannich bases derived from thiazolidinones and pyrimidine-2-(thi)ones.
Aminomethylation of pyrimidine-2-thiones and pyrimidine-2-ones, substrates that can be easily obtained in a considerable structural variety through a simple Biginelli reaction, has also been investigated in relation with the antibacterial activity of the resulting Mannich bases. Thus, pyrimidine-2-thione Mannich base 145 (Fig. 26) was obtained using natural alkaloid cytosine as amine reagent in aminomethylation, and its evaluation as antibacterial agent showed that it is a potent growth inhibitor of S. aureus and B. subtilis, and it has weak activity against P. aeruginosa and E. coli [209]. Double Mannich bases 146 (Fig. 26) of pyrimidine-2-thiones (X = S) were good antibacterial agents (zones of inhibition of 96 to 88% of the zone of inhibition for reference drug streptomycin) against E. coli and B. subtilis [210], and analogous double Mannich bases 146 of pyrimidine-2-ones (X = O) were even more potent, with the exception of the candidate having a 4-methoxyphenyl moiety at position 4 of the pyrimidine ring (R = 4-OCH3, R1 = H) [211].
Taking advantage of the lability of the proton located at the nitrogen atom in the amide function, several N-Mannich bases 147 of 5-chloro-2-methoxybenzamide (Fig. 27 ) obtained from either sulfonamides or common secondary amines as amine reagents have been synthesized and evaluated for antibacterial activity against a panel of bacteria [212]. Candidates 147 derived from sulfonamides were more potent than the parent compounds, whereas Mannich base 147 derived from morpholine was an antibacterial agent with broad spectrum and good activity. N-Mannich bases 148 of glutarimides (R1 = H, c-C5H9, c-C6H11, 4-ClC6H4) (Fig. 27) have been screened for antibacterial activity, and claimed to be potent very potent, although no comparison to currently-in-use antibacterials was available [213], [214]. Compounds 148 derived from sulfonamides showed improved growth inhibition activity of the tested microorganisms over the corresponding parent amines, whereas analogues generated from secondary aliphatic amines were generally equipotent to those obtained from sulfonamides [213]. Also, N-Mannich bases 149 of succinimide (R1 = H or C6H5, NR2 = NHC6H5, 2-pyridinyl) (Fig. 27) and an N-Mannich base from phthalimide were weak to moderate antibacterials against E. coli, S. typhi and B. subtilis [215].
Fig. 27.
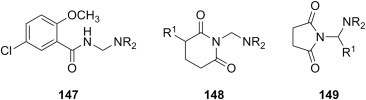
Antibacterial Mannich bases derived from amides and imides.
Well-known antibiotics have also been subjected to aminomethylation with a hope of increasing their antibacterial activity or improving their bioavailability. Hybrids 150 of tetracycline and sulfonamides (Fig. 28 ) have been generated using the Mannich reaction, and their screening against Salmonella enteritidis and Pasturella multocida showed that their antibacterial activity is higher than that of the parent sulfonamides; unfortunately, no comparison with the activity of tetracycline or a reference drug is provided in the study [216]. Aminomethylation is one of the modifications that have also been explored in the case of vancomycin. After an N-decylaminoethyl moiety was introduced into the structure of vancomycin with a view to restore antibacterial activity against vancomycin-resistant enterococci while retaining potency against MRSA, grafting a hydrophilic group has been considered as a way to reduce the overall lipophilicity of the N-decylaminoethyl-modified vancomycin and to restore the initial favorable distribution properties of vancomycin. Several types of amine reagents (amino acids, amino sugars, phosphonic acids) have been considered for the synthesis of Mannich bases 151 (Fig. 28), and some of these modifications led to candidates active against vancomycin-resistant bacteria, and ultimately to the discovery of telavancin (TD-6424) as a lead compound with improved absorption, distribution, metabolism and excretion (ADME) properties over N-decylaminoethylvancomycin [217]. Telavancin 151 (NR2 = NHCH2PO3H2) (Fig. 28) was subsequently screened against a large number of predominantly Gram-positive isolates with developed antibacterial resistance, and showed to be highly active against methicillin-resistant staphylococci (MIC90 0.5–1 μg/mL), streptococci (MIC values ≤ 0.12 μg/mL), and VanB-type enterococci (MIC values ≤ 2 μg/mL) [218]. With respect to its mechanism of action, telavancin inhibited late-stage peptidoglycan biosynthesis in a substrate-dependent fashion, bound to cell wall with high affinity, and perturbed bacterial cell membrane potential and permeability [219]. Aminomethylation of vancomycin has also been used as the first step in the construction of glycopeptide/β-lactam heterodimers via linkage of aminomethylated vancomycin and various cephalosporin cores through an adipic acid moiety [220]. These heterodimers were all found to exhibit excellent potency against a range of important Gram-positive bacteria, and a subset of these candidates also demonstrated rapid bactericidal activity against MRSA, whereas two of the most attractive compounds have been shown to exhibit in vivo efficacy 40 fold greater than that of vancomycin. Finally, a patent claims a series of Mannich bases of vancomycin as compounds suitable for oral delivery and having enhanced antibacterial potency [221].
Fig. 28.

Antibacterial Mannich bases derived from well-known antibiotics.
Antibacterial activity of Mannich bases derived from miscellaneous substrates is covered in the last paragraph of this section. Mannich bases 152 (R = CH3, OC2H5) (Fig. 29 ) derived from either ethyl acetoacetate or diethyl malonate as substrates, variously substituted benzaldehydes as aldehyde components and benzyl carbamate as amine reagent had poor to moderate activity against five types of common bacteria [222]. Hydrazidones derived from isoniazid and 2-alkoxybenzaldehydes have been aminomethylated at position ortho to the alkoxy group to give Mannich bases 153 (Fig. 29). A few members of this collection having an ethoxy group demonstrated at concentration of 100 μg/mL antibacterial activity superior to that of reference drug amoxicillin at 50 μg/mL [223], whereas candidates having a propoxy group were less potent [224]. Mannich bases 154 derived from 7-methyl-2-(substituted aryl)imidazo[1,2-a]pyridines (Fig. 29) and having two halogens as substituents in the phenyl ring at position 2 of the fused heterocyclic system were the most potent in this series against E. coli, P. aeruginosa, S. aureus and Streptococcus pyogenes [225]. Mannich bases 155 derived from an imidazo[2,1-b]-1,3,4-thiadiazole ring system (Fig. 29) showed weak activity against Gram-positive bacteria and moderate activity against Vibrio cholera, but their growth inhibition activity of E. coli was comparable to reference drug norfloxacin [226]. Evaluation of the antibacterial activity of Mannich bases derived from 2-alkyl-3-hydroxy-pyridine-4(1H)-ones showed that only one candidate 156 (NR2 = 1-piperidinyl, R1 = CH3) (Fig. 29) had moderate activity against S. enteritidis (16 μg/mL) and S. aureus (16 μg/mL) compared to reference drug ciprofloxacin (1 μg/mL and 0.5 μg/mL, respectively, for the mentioned microorganisms) [227]. Mono-Mannich bases 157 (R1 = CH3) and double Mannich bases 158 generated from sydnones (Fig. 29) were both found to inhibit moderately the growth of four standard microorganisms, regardless of the nature of the amine reagent used in aminomethylation, but no useful conclusions regarding SAR in this series could be drawn for further development [228]. Also, most Mannich bases 157 (R1 = OCH3) had weak to moderate activity against both Gram-positive and Gram-negative bacteria, with the exception of candidate 157 (R1 = OCH3, NR2 = 4-nitrobenzothiazole-2-ylamino), which was as potent as reference drugs ciprofloxacin and ampicillin against Gram-positive bacteria [229]. In contrast, aminomethylation of another sydnone derivative using various primary and secondary aliphatic amines was reported to occur in the at N-1 in the pyrazoline ring to give Mannich bases 159 instead (Fig. 29), which had good antibacterial activity compared to reference drug ampicillin against four common bacteria, and particularly against B. subtilis [230]. The copper(II) complex of pyrazole C-Mannich base 160 (Fig. 29) inhibited the growth of B. subtilis at concentrations as low as 1.25 mM, most likely by promoting mutagenesis and inducing cell death [231]. Screening of a few of pyrazolone N-Mannich bases 161 (Fig. 29) against various bacteria resulted in inhibition zones that were comparable to those of reference drug ciprofloxacin, but the MIC values for these candidates were ultimately far greater from those of ciprofloxacin [232]. Some of the acetylenic Mannich bases 162 (R1 = C6H5, 4-ClC6H4; R2 = C6H5, 1-C10H7) (Fig. 29) had antibacterial activity comparable to reference drug ofloxacin against S. aureus, fewer showed good activity against E. coli, and only one candidate was equipotent to ofloxacin against P. aeruginosa [233]. Various substrates (e.g., benzimidazole, 3-methylpyrazole-5(4H)-one, succinimide, phthalimide or naphthalimide) were aminomethylated using norfloxacin as amine reagent, and some of the corresponding Mannich bases were more potent than the parent fluoroquinolone, especially against E. coli and P. aeruginosa [234]. Sulfonamides and primary or secondary aliphatic amines were employed as amine reagents in aminomethylation of isoniazid to give Mannich bases 163 (Fig. 29), and some of the candidates had antibacterial activity that was superior to that of the parent sulfonamides, whereas Mannich base 163 (NR2 = NHCH3) was the most potent compound in the series against S. aureus and B. anthracis [235]. Use of hydantoins as substrates in the Mannich reaction led to 3-aminomethylated derivatives 164 or to 1-aminomethylated derivatives 165 (Fig. 29), depending on the substituent on N-3 of imidazole ring, and some of these Mannich bases had moderate to good antibacterial activity against four common bacteria [236]. Aminomethylation and aminoalkylation of saccharin using various types of amine reagents led to Mannich bases 166 (Fig. 29) with moderate to excellent antibacterial activity against S. aureus (zones of inhibition between 12 and 50 mm, compared to 31 mm for reference drug chloramphenicol) [237]. A note reports in a concise manner the synthesis and antibacterial activity of Mannich bases 167 (Fig. 29) derived from a phenothiazine and primary aromatic amines, and some of the compounds in this study were potent and had a broad spectrum against the tested bacteria [238]. Mono-Mannich base 168 of quinoxaline-2,3(1H,4H)-dione (Fig. 29) was screened at concentration of 100 μg/disc, and was found to be more potent than reference drug nalidixic acid at concentration of 50 μg/disc against four common bacteria, with the exception of Proteus vulgaris [239]. Moderate to good antibacterial activity was determined for Mannich bases 169 of 3-aryl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (Fig. 29), but none of the candidates was as potent as reference drug amikacin against the four common bacteria used in this study [240]. In addition to their anticancer activity evaluation, P-Mannich bases 83 (Fig. 15) were also screened for antibacterial activity, and the MIC values were generally approximately 10 μg/mL, regardless of candidate's structure and type of bacteria, but no comparison with reference drugs was provided in the study [106]. A paper claiming the synthesis of S-Mannich bases 170 of 4,6-diaryl-2-mercaptopyrimidine and secondary aliphatic amines (Fig. 29) also reports their antibacterial activity weak to moderate, as the most potent candidates are 3- to 7-fold less potent than reference drug ciprofloxacin against four types of bacteria [241].
Fig. 29.
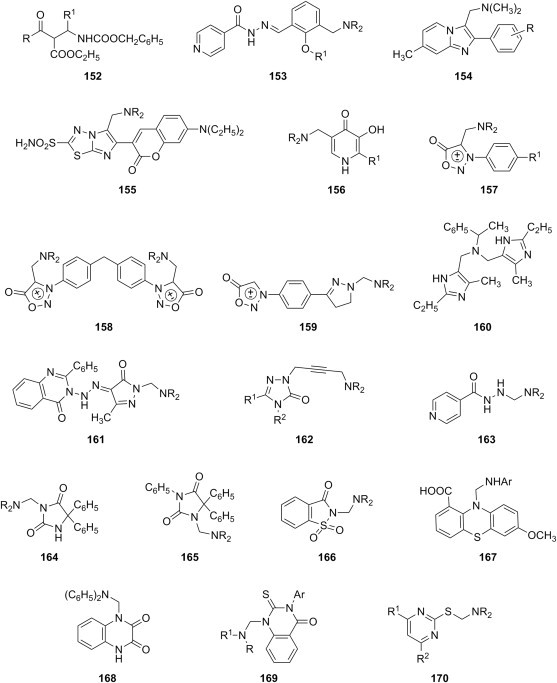
Antibacterial Mannich bases derived from miscellaneous substrates.
4. Antimycobacterial activity
Tuberculosis is a chronic disease whose magnitude and gravity can be gleaned from a 2010 study by World Health Organization stating that in 2009 alone, 9.4 million new cases of tuberculosis were reported, and 1.7 million deaths were caused by tuberculosis, out of which 0.38 million people were infected with both human immunodeficiency virus and Mycobacterium [242]. Continuous rise of the number of reported cases of drug-resistant and latent tuberculosis is also alarming and calls for urgent discovery of novel classes of compounds with antimycobacterial activity capable of overcoming the resistance of Mycobacterium strains to current drugs.
Mannich bases are among these novel classes of antimycobacterial compounds that have recently caught the attention of researchers in the field. Several examples of papers dealing with antitubercular ketonic Mannich bases are available. Thus, dimethylamine Mannich bases 171 of 2-benzylidenecyclohexanones (Fig. 30 ) were reported to have MIC values between 0.39 μg/mL and more than 12.5 μg/mL against Mycobacterium tuberculosis H37Rv (for candidate 171 (n = 0, R1 = R2 = H) and candidate 171 (n = 1, R1 = F, R2 = H), respectively), which is approximately 20–25% of the efficiency of reference drug rifampin against this microorganism [243]. In addition, the ability of the same candidates to inhibit the growth of Mycobacterium avium at concentration of 12.5 μg/mL ranged from 0 to 87%. However, no significant correlations between the structural patterns of Mannich bases 171 (e.g., presence or absence of the cinnamoyl double bond, substitution pattern in the phenyl ring, electronic, hydrophobic or steric properties of the substituents in the aromatic ring) and their antimycobacterial activity could be established. Mannich base 171 (n = 0, R1 = OCH3, R2 = H) emerged from this series as a viable hit compound, having a MIC value of 0.78 μg/mL and an IC50 value for Vero cells of 16.4 μg/mL. This compound presents therefore a greater toxicity towards Mycobacterium over normal mammalian cells, which has been tentatively explained by its ability to affect respiration in isolated rat liver mitochondria, and had MIC values against several drug-resistant strains of Mycobacterium that were similar or identical to the values determined for the H37Rv strain [244].
Fig. 30.
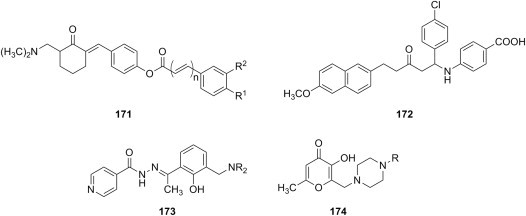
Antimycobacterial Mannich bases derived from ketones, phenols and allomaltol.
Isocitrate lyase is an enzyme in the glyoxylate cycle that catalyzes the cleavage of isocitrate to succinate and glyoxylate, and, together with malate synthase, bypasses the two decarboxylation steps of the tricarboxylic acid cycle [245]. Glyoxylate cycle is essential for the survival of pathogens, including M. tuberculosis, inside the host [246], and isocitrate lyase has been identified only in bacteria, fungi, and plants, but no analogous enzyme has been documented in humans. Therefore, isocitrate lyase has been pursued as a promising target for the development of novel antimycobacterial agents [247]. A high-throughput screening of a large library of ketonic Mannich bases against this enzyme led to the identification of candidate 172 (Fig. 30) with a potent inhibitory activity (IC50 = 53.5 μg/mL), but this compound's activity against M. tuberculosis has not been actually evaluated [248].
Phenolic Mannich bases 173 (Fig. 30) derived from the hydrazone generated from isoniazid and 2′-hydroxyacetophenone were screened against M. tuberculosis H37Rv, and found to inhibit its growth with MIC values ranging from 0.56 to 4.61 μM [249]. Six compounds in this series were more potent than isoniazid (MIC = 2.04 μM), and the most potent candidate 173 (NR2 = 1-piperazinyl) was shown to be equipotent to isoniazid in decreasing bacterial load in lungs of infected mice at a dose of 25 mg/kg. Anti-Mycobacterium activity of novobiocin Mannich bases 115 (Fig. 22) was evaluated, and two of these candidates (NR2 = N-acetoxyacetyl-N-methyl and N-acetaminoacetyl-N-methyl) showed an increased potency over novobiocin, although no comparison to established antimycobacterial drugs was provided [143]. Phenolic Mannich bases 174 derived from allomaltol and piperazines (Fig. 30) had good activity against Mycobacterium smegmatis, with MIC values between 4 and 16 μg/mL [250]. Although the growth inhibition of Mycobacterium by the candidates in the library was moderate to good (MIC values between 4 and 128 μg/mL), Mannich bases derived from 1-phenylpiperazine and its analogues substituted with halogens in the aromatic ring were among the most potent compounds in the series. Departure from substitution of piperazine in candidates 174 with bulky, aromatic rings directly linked at N-4 generally led to decreased potency against Mycobacterium, although some substituents (such as t-butoxycarbonyl, ethoxycarbonyl, 2-hydroxyethyl, cyclohexyl, benzoyl, furfuryl or 2-(dimethylamino)ethyl) appear to be tolerated better than others (e.g., isopropyl, allyl, 2-methoxyethyl, 2-ethoxyethyl, phenethyl or benzyl). An extension of this study, aimed at evaluating against M. tuberculosis H37Rv the library of Mannich bases of type 124 (Fig. 23) obtained by replacement of piperazines in compounds 174 with piperidines, led to valuable insight on the SAR for this particular type of compounds [251]. Inspection of the SAR in the subset of Mannich bases 124 suggested that the position of the substituents and, to a lesser extent, the nature of these substituents plays an important role in their antimycobacterial activity, as the most potent candidates in this subset are generally those substituted at position 4 of piperidine ring [251]. Replacement of allomaltol with chlorokojic acid as substrate afforded piperazine Mannich bases 125 (R = 4-substituted piperazin-1-yl) (Fig. 23), analogous to both 124 and 174 and having comparable antimycobacterial activity (MIC values between 4 and 32 μg/mL), although M. avium rather than M. tuberculosis exhibited the greatest sensitivity towards the candidates 125 [154].
Many papers reporting the antimycobacterial activity of Mannich bases of pyrrole derivatives have been published in the last decade. The intensive research in this field stems from the discovery by Italian researchers at Università “La Sapienza” in Rome of BM212 175 (Fig. 31 ) as a potent agent, not only against several collection strains and clinical isolates of M. tuberculosis, but also against non-tuberculosis mycobacterial strains and drug-resistant mycobacteria, with MIC values that are comparable to those of reference drugs isoniazid and streptomycin [252]. Although the quest for a more potent antimycobacterial agent through systematic modification of lead compound BM212 was not successful at first, it provided experimental proofs for the importance of the presence and nature of substituents at positions 1 and 5 of pyrrole ring [253]. Subsequently, novel pyrrole Mannich bases 176 analogous to BM212 (Fig. 31), derived from 1-methylpiperazine but also from thiomorpholine as amine reagents, and having either chlorine [254] or chlorine and fluorine [255] as substituents of the phenyl rings in positions 1 and 5 of pyrrole, have been synthesized. Two candidates 176 (R1 = F, R2 = H or R1 = H, R2 = F) showed an improved activity (MIC values of 0.4 and 0.5 μg/mL) against M. tuberculosis over Mannich base 175 (MIC = 0.7 μg/mL) [255]. Investigation of structure–antimycobacterial activity relationship in analogues of 175 and 176 was further pursued with a view to optimize the structure of these candidates. Thus, synthesis of novel Mannich bases of pyrrole using iterative introduction of more lipophilic 1-naphthyl or 2-fluoro- or 2-chlorophenyl moieties at either one of positions 1 and 5 in pyrrole ring of these lead compounds [256], or a recombination through shuffling of any of the substituents employed so far on the phenyl rings at these two positions of the pyrrole ring [257] was undertaken. These studies confirmed the superior antimycobacterial activity of Mannich bases containing a thiomorpholinyl moiety compared to the activity of the corresponding analogues with a 4-methylpiperazinyl moiety, evidenced the importance of fluorine substitution (especially at position 2 of the phenyl ring) for antimycobacterial activity, and proved the decrease of activity with the introduction of 1-naphthyl moieties in the structure of lead compounds 175 and 176. Unfortunately, no novel candidates, more potent than Mannich bases 175 and 176, have been identified during these investigations. However, replacement of one halogen in the structure of lead compound 175 with more lipophilic substituents (methyl, ethyl n-propyl, i-propyl) afforded candidates with improved antimycobacterial activity (MIC values between 0.125 and 0.5 μg/mL) over 175 and even over reference drug streptomycin [258], [259]. In order to further increase the lipophilicity, replacement of the methyl group at position 2 of the pyrrole ring with ethyl was also examined, but these novel Mannich bases 177 (X = S or NCH3) (Fig. 31) were generally less active against several M. tuberculosis strains than their methyl-substituted analogues, although their cytotoxicity towards normal cells was lower than that of the methyl-substituted counterparts [260]. Further exploration of the relationship between the nature of substituents on phenyl rings at positions 1 and 5 of pyrrole ring and the antimycobacterial activity of pyrrole Mannich bases led to the identification of a novel lead compound 178 (Fig. 31) with a very high activity toward both M. tuberculosis 103471 and H37Rv strains (MIC = 0.125 μg/mL) [261]. Antimycobacterial activity of Mannich base 178 was comparable to that of reference drugs streptomycin or rifampin, while the candidate demonstrated low cytotoxicity towards normal cells (selectivity index IC50/MIC > 1000) [261]. Subsequently a series of morpholine derivatives were designed and synthesized with a view to lower the clearance rate in mouse microsomal fractions associated with the presence of thiomorpholine [262]. Although the replacement of thiomorpholine with morpholine generally resulted in a decrease in potency, candidate 179 (Fig. 31) was found to be equipotent to any of the lead compounds, showed improved microsomal clearance and lower cytotoxicity to normal cells, and was ultimately selected for in vivo pharmacokinetic and efficacy studies in an acute murine tuberculosis infection model. Mannich base 179 had an efficacious dose that results in a 99% colony-forming unit reduction in the lung of 49 mg/kg, which is within the range of commonly employed tuberculosis drugs. A recent study showed that mutations in the mmpl3 gene in Mycobacterium strains are responsible for resistance to BM212, and suggested that products of this gene are cellular targets for BM212, which makes Mmpl3, a member of the MmpL (mycobacterial membrane protein, large) family, a new potential druggable target for the treatment of tuberculosis [263]. Point mutations in mmpl3 gene that confer resistance to BM212 and other analogous pyrrole Mannich bases have been later identified in several Mycobacterium mutants [262]. A large number of antimycobacterial pyrrole Mannich bases were also employed to obtain a final multiprobe 3-D QSAR model, which was shown to offer good predictions for antimycobacterial activity of an external, unrelated test set of compounds [264]. Accounts at various stages of the endeavors concerning the synthesis of pyrrole Mannich bases as antimycobacterial agents, along with the related biological results obtained by the Italian researchers, have also made the object of several author's reviews along the years [265], [266], [267], [268]. A patent claiming the antimycobacterial activity of pyrrole Mannich bases derived from 1-arylpiperazines is also worth mentioning [269]. On the other hand, attempts to make a more drastic departure from these already established structural features of antimycobacterial pyrrole Mannich bases (such as the introduction of an oxazolidinone moiety reminding of antibacterial linezolid) led to a series of compounds 180 (X = acetamido of 4-(hetero)aryl-1,2,3-triazol-1-yl) (Fig. 31) that were at best 15–65-fold less potent than Mannich base 178 [270]. In addition, pyrrole Mannich bases 181 (Fig. 31) were shown to act as inhibitors (IC50 values ranging from 1.5 to 15 μM) of Mycobacterium protein tyrosine phosphatase B, an enzyme that is essential for the survival of bacteria in the host, but no in vitro evaluation using Mycobacterium strains was performed [271].
Fig. 31.
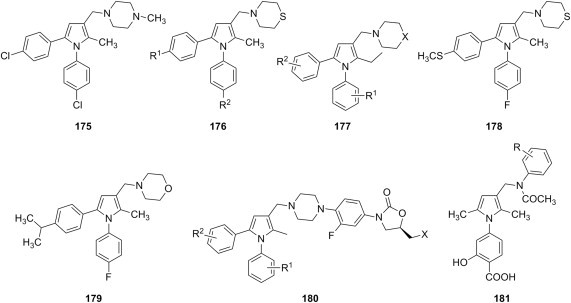
Antimycobacterial pyrrole Mannich bases.
Mannich bases of isatin and its derivatives have been evaluated as antimycobacterial agents as well. Several isatin Mannich bases 182 (Fig. 32 ) containing a semicarbazide moiety were equipotent (MIC = 0.625 μg/mL) to isoniazid against M. tuberculosis, and two of the candidates were more potent in vitro than either isoniazid or rifampicin against a multidrug-resistant Mycobacterium strain (MIC = 6.25 μg/mL) [272]. Schiff bases obtained from 5-substituted isatins (R1 = F, Cl, F) and lamivudine were aminomethylated using fluoroquinolones (R2 = ethyl, cyclopropyl; R3 = H, CH3) as amine reagents, and the resulting Mannich bases 183 (Fig. 32) showed 92–100% growth inhibition of M. tuberculosis H37Rv at a concentration of 6.25 μg/mL; inhibition for one selected Schiff base was 82%, and lamivudine alone did not inhibit the growth of this Mycobacterium strain at the given concentration [273]. Several morpholine Mannich bases 184 of 4-substituted thiosemicarbazones of 5-trifluoromethoxyisatin (R = c-C6H11, 4-FC6H4, 4-ClC6H4, 4-BrC6H4) (Fig. 32) were potent antimycobacterial agents (MIC values between 3 and 5 μg/mL) [274]. Replacement of the trifluoromethoxy group in 184 with nitro led to a slight decrease in potency, which became more drastic when fluorine replaced the trifluoromethoxy group, whereas the substitution of morpholine with piperidine generally decreased the antimycobacterial activity of the candidates [275]. Using fluoroquinolone gatifloxacin as amine reagent, sixteen Mannich bases derived from isatins, isatin semicarbazones, isatin thiosemicarbazones, isatin hydrazones with isoniazid, and isatin Schiff bases with sulfadiazine were prepared and tested for antimycobacterial activity [276]. MIC values recorded for 90% growth inhibition of Mycobacterium by these compounds ranged from 0.78 μg/mL to 12.5 ng/mL, whereas MIC values between 0.78 and 0.1 μg/mL were determined for multidrug-resistant strains. Promising results were obtained when the most potent compound in this series was tested in a murine model at a dose of 50 mg/kg, and the analysis of data for inhibition of M. tuberculosis DNA gyrase suggested that this enzyme could be the target of these isatin–fluoroquinolones hybrids. Good results were obtained from the evaluation as antimycobacterial agents of Mannich bases obtained from 5-substituted isatins and their semicarbazones and thiosemicarbazones as substrates and ciprofloxacin as amine reagent (MIC values between 1.2 and 10.3 nM) [277], and Mannich bases generated from 5-substituted isatins, the corresponding semicarbazones, thiosemicarbazones and oxime ethers using 8-methoxyciprofloxacin as amine reagent had comparable potencies [278]. Analogous Mannich bases derived from moxifloxacin as amine reagent were also potent (MIC value of 1 μg/mL for M. tuberculosis, and between 4 and 16 μg/mL for multidrug-resistant Mycobacterium strains), but all of these isatin–moxifloxacin conjugates were generally less potent antimycobacterials than parent fluoroquinolone [279].
Fig. 32.

Antimycobacterial Mannich bases of isatin derivatives.
Evaluation as antimycobacterial agents of aminomethylated 2,3-dihydro-1,3,4-oxadiazole-2-ones, 2,3-dihydro-1,3,4-oxadiazole-2-thiones and 2,3-dihydro-1,2,4-triazole-3-thiones has led to interesting results. A direct comparison between Mannich bases 185 (X = O) of 2,3-dihydro-1,3,4-oxadiazole-2-one and Mannich bases 185 (X = S) of 2,3-dihydro-1,3,4-oxadiazole-2-thione (Fig. 33 ), having both a 4-pyridinyl substituent at position 5 of the azole ring and derived from cyclic secondary aliphatic amines, showed that the former were significantly more potent against an isoniazid-sensitive M. tuberculosis H37Rv strain than the latter, thus suggesting that the presence of oxygen is crucial for the antimycobacterial activity of this type of compounds [280]. In a continuation of the initial study, the search for better antimycobacterial agents was restricted to Mannich bases of 2,3-dihydro-1,3,4-oxadiazole-2-ones, while the variety of amine reagents employed in aminomethylation was expanded to include N-(substituted benzyl)methylamines, a structural modification that preserved the good activity of the candidates (MIC values of 4–8 μg/mL) [281]. However, replacement of the 4-pyridinyl substituent at position 5 of Mannich bases 185 (X = O) with phenyl, 3-pyridinyl of pyrazinyl resulted in a significantly decreased antimycobacterial activity, whereas replacement with 2-pyridinyl did not generally affect the activity of the candidates. Bis-Mannich bases 186 (Fig. 33) derived from dapsone as amine reagent, benzaldehyde and furan-2-carboxaldehyde as aldehyde reagents and variously 5-substituted 1,3,4-oxadiazole-2-thiones as substrates had excellent antimycobacterial activities against both isoniazid-sensitive and isoniazid-resistant strains of M. tuberculosis, as they were 5-fold more potent than isoniazid against the sensitive strain and 10-fold more potent than isoniazid against the resistant strain [282]. A study allowed another direct comparison, this time between Mannich bases 139e of 2,3-dihydro-1,3,4-oxadiazole-2-thiones (Table 2) and Mannich bases 134i (Table 1) of 2,3-dihydro-1,2,4-triazole-3-thiones having the same 4-isopropylthiazole-2-yl substituent at position 5 of the aforementioned azoles. Some of candidates 134i were 2- to 4-fold more potent than the most potent Mannich base 139e against M. tuberculosis H37Rv strain, but they were still 16-fold less potent than isoniazid [187]. On the other hand, Mannich bases 187 (R1 = H, OH; R2 = C2H5, CH2CH CH2, C6H5, 4-BrC6H4; R3 = CH2C6H5, C6H5, substituted phenyl, 2-pyrimidinyl) derived from 1,2,4-triazole-3-thiones (Fig. 33) had low antimycobacterial activity (MIC values between 25 and 100 μg/mL) [283]. The majority of Mannich bases derived from either 2,3-dihydro-1,2,4-triazole-3-thione or 2,3-dihydro-1,2,4-triazole-3-one having a 3-pyridinyl moiety at position 5 of the triazole ring were more potent antimycobacterials (MIC between 16 and 32 μg/mL) than reference drug streptomycin against M. smegmatis [175]. In addition, 2,3-dihydro-1,2,4-triazole-3-one Mannich base 137 (NR2 = 1-piperidinyl) (Fig. 25) was equipotent to reference drug streptomycin (MIC = 4 μg/mL) against M. smegmatis [192].
Fig. 33.
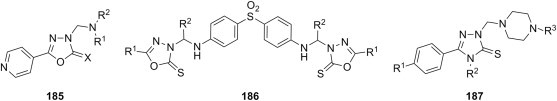
Antimycobacterial Mannich bases of 2,3-dihydro-1,3,4-oxadiazole-2-ones, 2,3-dihydro-1,3,4-oxadiazole-2-thiones and 2,3-dihydro-1,2,4-triazole-3-thiones.
Mannich bases 188 of imidazo[2,1-b]-1,3,4-thiadiazole derivatives (Fig. 34 ) have been screened for antimycobacterial activity. Evaluation of a library of Mannich bases 188 (R1 = c-C6H11, 2-furyl, 2-thienyl; R2 = H, Br; NR2 = 1-pyrrolidinyl, 1-piperidinyl, 4-morpholinyl) against M. tuberculosis H37Rv showed that all of the candidates had MIC >6.25 μg/mL [284], but a second collection of aminomethylated imidazo[2,1-b]-1,3,4-thiadiazole derivatives 188 (R1 = n-C3H7, c-C6H11, 2-thienyl; R2 = Cl, CH3, OCH3; NR2 = 1-pyrrolidinyl, 1-piperidinyl, 4-morpholinyl) consisted of Mannich bases with good antimycobacterial activity (52–99% growth inhibition) at a concentration of 6.25 μg/mL [285]. Amongst Mannich bases 188 (R1 = 4-CH3OC6H4CH2), pyrrolidine-containing derivatives were consistently the best agents against M. tuberculosis H37Rv [286]. Also, compounds 155 (NR2 = 1-pyrrolidinyl, 1-piperidinyl, 4-morpholinyl) (Fig. 29) had moderate antimycobacterial activity (MIC = 25 μg/mL) [226].
Fig. 34.
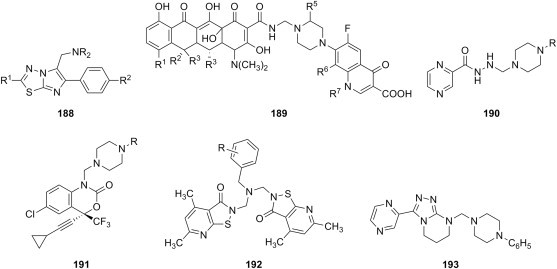
Antimycobacterial Mannich bases obtained from miscellaneous substrates.
In addition to the types of substrates presented so far, miscellaneous other substrates have been subjected to the Mannich reaction with a view to synthesizing compounds with antimycobacterial activity. Aminomethylation of the amide function in tetracyclines using fluoroquinolones as amine reagents afforded Mannich bases 189 (Fig. 34) with excellent activity against M. tuberculosis (MIC values between 0.2 and 1.56 μg/mL) [287]. Pyrazinamide was also aminomethylated at the amide function using piperazines (including four fluoroquinolones) as amine reagents; the resulting Mannich bases 190 (Fig. 34) were at least as potent as reference drug pyrazinamide against M. tuberculosis, and showed a significantly improved antimycobacterial activity over pyrazinamide against multidrug-resistant Mycobacterium strain [288]. Anti-HIV drug efavirenz was aminomethylated using either common piperazines or piperazine-containing fluoroquinolones as amine reagents to afford hybrids 191 (Fig. 34), but only the Mannich bases derived from fluoroquinolones had good activity against M. tuberculosis (no comparison with standard antimycobaterial drugs was available though) [289]. Investigation of antimycobacterial activity of a small number of indole Mannich bases 59 obtained from 1-arylpiperazines (Fig. 11) uncovered a few candidates that inhibited the growth of M. tuberculosis H37Rv almost completely at a concentration of 6.25 μg/mL [290]. The nature of substituents in the aryl moiety at N-4 of piperazines employed in aminomethylation appears to modulate the activity, as other candidates 59 in this collection were almost inactive at the same concentration. When screened at a concentration of 6.25 μg/mL, bis-Mannich bases 192 (Fig. 34) failed to inhibit significantly the growth of M. tuberculosis H37Rv [291]. Out of a series of five candidates, Mannich base 193 (Fig. 34) was the most potent against several Mycobacterium strains, with MIC = 12.5 μg/mL for M. tuberculosis H37Rv and MIC values of 25 μg/mL for several drug-resistant clinical isolates [292]. In addition, several Mannich bases 159 (Fig. 29) had good antimycobacterial activity (MIC < 5 μg/mL) [230], while Mannich base 168 (Fig. 29) was moderately active against M. tuberculosis H37Rv [239]. Finally, pyrazolone Mannich base 161 (NR2 = NHCOC6H5) (Fig. 29) was more potent than reference drugs ethambutol and ciprofloxacin, but inferior to isoniazid, against M. tuberculosis H37Rv [232].
5. Antifungal activity
The incidence of fungal infections of any variety has been steadily increasing over the last decades, and it has become one of the main causes of morbidity and mortality, especially in patient with compromised immune systems [293]. Development of resistance to currently-in-use antifungal drugs also represents a major concern, and the discovery of novel antifungal agents, preferably outside any of the six major classes that are presently available for treatment, is one of the highest priorities for pharmaceutical industry. The latest advances in the evaluation of Mannich bases with various structures as potential antifungals are presented in this section.
First reports on the antifungal activity of ketonic Mannich bases are relatively recent. A few mono-Mannich bases of type 8 (Fig. 3) and double Mannich bases 12 (Fig. 4) of acetophenone (R1 = R2 = H; NR2 = dimethylamino, 1-piperidinyl, 4-morpholinyl) have been shown to have weak antifungal activity, but the corresponding methiodides were more potent than reference drug amphotericin B against yeasts Saccharomyces cerevisiae and Candida krusei, and against dermatophytes Trichophyton mentagrophytes, Trichophyton rubrum and Microsporum canis [294]. Thus, quaternization of the nitrogen atom in Mannich bases of type 8 appears to be a chemical modification leading to efficient antifungals [135]. Various substitution patterns in the phenyl ring in ketonic Mannich bases 8 also did not improve significantly the antifungal activity of these compounds [295]. Bis-Mannich bases 13 (R = C2H5) derived from various acetophenones (Fig. 4) were only active against Aspergillus fumigatus, but the antifungal activity of the related piperidinols 14 (R = C2H5) (Fig. 4) was broader and slightly more potent compared to that of corresponding mono-Mannich bases 8 and bis-Mannich bases 13, especially against dermatophytes T. rubrum and M. canis [295]. However, a later study showed that a minor modification such as replacement of ethyl with methyl as substituent at nitrogen increased the antifungal activity of bis-Mannich bases 13 (R = CH3), while it decreased the potency of piperidinols 14 (R = CH3) against T. rubrum and M. canis [296]. Use of phenethylamine as amine reagent in the Mannich reaction with various acetophenones afforded secondary ketonic mono-Mannich bases 15 (R = CH2CH2C6H5) (Fig. 4) along with piperidinols 14 (R = CH2CH2C6H5), but these compounds had relevant activity (most MIC values were either 12.5 or 25 μg/mL) only against M. canis, a dermatophyte whose growth was not inhibited by reference drug nystatin in the concentration range used in this study [297]. Most members of a library of Mannich bases 10 of enones had antifungal effects 2–16-fold more potent than amphotericin B against at least one (if not many) selected plant fungi [298]. Ketonic Mannich bases derived from arylamines as amine reagents have also been reported to have antifungal effects. Thus, Mannich bases 108 (R1 = 2-Br, R2 = H or F) (Fig. 21) were almost twofold more potent than reference drug ampicillin against Candida albicans [137], whereas candidates 109 (R = F or Cl) derived from 3-acetylcoumarin and 110 (R = Cl or N(CH3)2) derived from 2-acetylbenzofuran (Fig. 21) had MIC values comparable to those of reference drug fluconazole against Aspergillus flavus, Chrysosporium keratinophilum, and C. albicans [138].
Evaluation of Mannich bases 194 (Fig. 35 ) derived from cyclic ketones (n = 1–4) showed that these compounds had good anti-Candida and anti-Saccharomyces activity, but they were less potent against Aspergillus strains, and none of the compounds were as potent as reference drug amphotericin B [299]. The anti-Candida potency of candidates 194 generally diminished with the increasing size of the ring, while the substitution pattern of the aromatic ring or the nature of the amine in the aminomethyl moiety did not influence the antifungal activity considerably. In addition, the α,β-unsaturated ketone system is a structural requirement for the antifungal activity of Mannich bases 194, as the amino alcohols obtained through the reduction of the carbonyl group in 194 were mostly inactive towards all the fungi used in this study. Inhibition of ergosterol synthesis does not appear to be the mechanism by which Mannich bases 194 exert their anti-Candida activity, although they are able to influence the development of pseudohyphae and induce noticeable changes in the protein composition in C. albicans [299]. Inspection of SAR disclosed in the previous study also proved valid for Mannich bases 195 derived from cyclic ketones fused with an aromatic ring (Fig. 35), except that the MIC values for compounds 195 were consistently better than those for Mannich bases 194 [300]. Transcript profiling of C. albicans cells treated with candidate 195 (n = 0, NR2 = 4-morpholinyl) showed that the transcriptional response is typical for oxidative stress and similar to that of a C. albicans Cap1 transcriptional activator, which suggests that the ability of Mannich bases 195 to directly trigger oxidative stress may be at least in part the reason for their antifungal activity [300]. Mannich bases 196 of thiocroman-4-ones (Fig. 35) obtained either from secondary aliphatic amines or from primary arylamines had good activity against several types of fungi, and two candidates 196 (NR1R2 = N(CH3)2, R3 = 6-Cl, R4 = 5-F or 8-Cl) were more potent than reference drug ketoconazole against the fungi investigated in the study [301]. Antifungal activity of double Mannich bases 105 (Fig. 21) against several fungi was also good, although the potency of compounds 105 against C. albicans was only moderate [134].
Fig. 35.
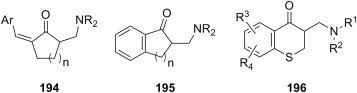
Antifungal ketonic Mannich bases.
Several studies reporting the antifungal activity of phenolic Mannich bases are available. The hydrazone obtained from 2,4-dinitrophenylhydrazine and salicylaldehyde was aminomethylated using various secondary amines to give Mannich bases 197 (Fig. 36 ) [302] The most potent anti-Candida candidates 197 (NR2 = diphenylamino, 4-morpholinyl, 1-piperazinyl) were 5–10-fold weaker than reference drug clotrimazole, whereas the growth of Aspergillus niger was inhibited by the most potent compounds at MIC values in the range of 1.56–3.12 μg/mL [302]. Most aminomethylated 4-t-butylcatechols 113 (Fig. 22), along with their copper(II) complexes, have significant antifungal activity (radial inhibition of mycelial growth ≥ 70%) against several plant pathogenic fungi, which is comparable, or even higher in a several cases, to that of reference drugs nystatin and terbinafine [141]. Even at a concentration 10-fold greater than that of reference drug voriconazole, only three of Mannich bases 114 (NR1R2 = 4-methoxyphenylamino, 1-piperidinyl, 4-methylpiperazinyl) (Fig. 22) barely showed anti-Candida activity that was comparable to that of voriconazole [142]. Aminomethylation of mulundocandin, an antifungal lipopeptide belonging to echinocandin antifungals, was undertaken with a view to improve its solubility in water; the resulting mixture of Mannich bases 198 (R1 = CH2NR2, R2 = H) and double Mannich bases 198 (R1 = R2 = CH2NR2) (Fig. 36) was separated before the antifungal activity of the compounds was evaluated in vitro, but only one candidate showed good inhibition against C. albicans and A. fumigatus [303]. Nonetheless, in vivo testing of the collection was promising, as many compounds showed anti-Candida activity comparable to parent mulundocandin, while a significant improvement in anti-Candida activity compared to mulundocandin was observed for mono-Mannich base 198 (R1 = 4-phenylpiperazinylmethyl, R2 = H), which was equipotent to fluconazole. Three members of a collection of 6-acetyl-1,3-benzoxazines 117 (R1 = CH3, R = 4-Br; R1 = H, R = 2-OCH3 or 4-F) (Fig. 22) had inhibition zones against A. niger comparable to that of reference drug fluconazole [145]. Benzoxazines 118 and 119, as well as naphthoxazines 120 and 121 (Fig. 22) were either inactive or had weak activity against Candida spp., A. fumigatus and Cryptococcus neoformans, but a few compounds in this library had moderate activity against Sporothrix schenckii and T. mentagrophytes [146]. In contrast, naphtho[2,1-e]-1,3-oxazine 121 (R = 4-C2H5OC6H4) was more potent than reference drug griseofulvin against C. albicans, whereas naphtho[2,1-e]-1,3-oxazine 121 (R = 3-CH3OC6H4) was equipotent to griseofulvin against A. niger [148]. Examples of Mannich bases from phenols fused with heterocyclic rings that were investigated for antifungal activity include compounds 123 (Fig. 22), which had only moderate to weak activity against A. niger and Penicillium spp. compared to reference drug griseofulvin [150], and compounds 122 (Fig. 22), which were efficient against Aspergillus spp. and C. albicans at concentrations as high as 5 mg/mL [149]. Mannich bases 125 of chlorokojic acid (Fig. 23) had good anti-Candida activity (MIC values between 4 and 16 μg/mL) [152], [153], [154], but the antifungal activity against dermatophytes was slightly poorer [304]. In contrast, Mannich bases 174 (Fig. 30) derived from allomaltol and 4-substituted piperazines had poor antifungal activity against C. albicans and Candida parapsilosis compared to that of reference drug fluconazole, but some members of the library were equipotent to fluconazole against C. krusei [305].
Fig. 36.
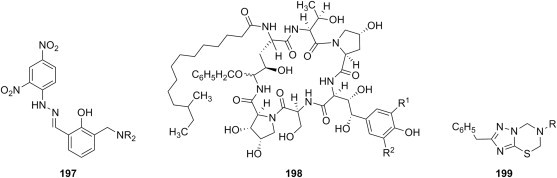
Antifungal phenolic Mannich bases 190 and 191 and cyclic bis-Mannich base 192 derived from 2,3-dihydro-1,2,4-triazole-3-one.
Antifungal activity of various Mannich bases of isatin derivatives has been examined jointly with their antibacterial activity. Mannich bases 127 (Fig. 24) obtained from isatin semicarbazone as substrate and (hetero)arylamines as amine reagents had good anti-Candida activity (75–87% of the activity of reference drug fluconazole) [156], whereas Mannich bases 128 (R = secondary aliphatic amines) (Fig. 24) had moderate antifungal activity against Aspergillus spp., and consistently lower than that of reference drug fluconazole [157]. For the most active Mannich bases 129 (R1 = 4- or 4,5-substituted 2-ethylphenyloxy, R = 1-piperidinyl or 4-morpholinyl), the antifungal potency against C. albicans and A. niger was 2/3 of the potency of reference drug griseofulvin [159]. A couple of Mannich bases 130 (R1 = substituted phenyl) (Fig. 24) were moderately active against A. niger, but the activity against C. albicans and Penicillium notatum was generally poor [160]. Mannich bases 131 (R1 = 5-benzyl-3-thioxo-2H-1,2,4-triazol-4-yl) (Fig. 24) were moderately potent against C. albicans and A. niger compared to reference drug fluconazole [163]. Finally, in their evaluation against A. niger, Mannich base 132 (Ar = C6H4OH-4) (Fig. 24) was equipotent to reference drug ketoconazole against A, fumigatus, whereas Mannich base 132 (Ar = C6H4N(CH3)2-4) was twofold more potent than ketoconazole, and three other analogues were equipotent to ketoconazole [164].
Evaluation of Mannich bases of 2,3-dihydro-1,2,4-triazole-3-thiones (Table 1) as antifungal agents has been reported in conjunction with their antibacterial activity. In the series of Mannich bases 133a, with the exception of candidate 133a (R = C6H4Cl-4, NR2 = NHC6H3F2-2,6) whose anti-Candida activity was 2/3 of that of reference drug clotrimazole, all other compounds were inactive, and so were Mannich bases 133b investigated in the same study [165]. Compared to reference drug fluconazole, most active Mannich bases 133h were 3-fold less potent against C. albicans and S. cerevisiae [173], Mannich bases 133j were either inactive or showed low antifungal activity [175], whereas Mannich bases 133k were inactive against C. albicans and S. cerevisiae [176]. Mannich bases 133i lacked any anti-Candida activity [174], as did Mannich bases 133n and 133o [179], and most Mannich bases 133q [180]. The majority of Mannich bases 134a were equipotent to reference drug fluconazole against C. albicans [181], and a small number of candidates 134c were moderately active against the same fungus, whereas the rest were inactive [182]. While most Mannich bases 134d were moderately active against Aspergillus spp., C. albicans and Penicillium marneffei, two candidates 134d (R2 = 4-ClC6H4 or 2,4-Cl2C6H3) derived from 1-methylpiperazine as amine reagent were equipotent to reference drug ciclopiroxolamine [183]. Several Mannich bases 134e were also equipotent to reference drug fluconazole against Aspergillus spp., T. mentagrophytes and P. marneffei [184]. Neither compound 134g, nor candidate 135 (Fig. 25) exhibited any anti-Candida activity [185], whereas Mannich bases 134i had weak to moderate activity against Candida tropicalis, S. cerevisiae and A. niger [187]. The antifungal activity of Mannich bases 134j against Aspergillus spp. and Penicillium spp. was moderate to good, and Mannich base 134j (X = Y = CH3, R2 = 4-NO2C6H4) was more potent than reference drug fluconazole against C. albicans [188]. A few Mannich bases 134k also showed good antifungal activity against C. albicans and Fusarium solani [189]. The best anti-Candida activity in the series of Mannich bases 134m was recorded for the candidate with R2 = C6H4OCH3-4 (85% of the inhibition zone of reference drug fluconazole) [190]. In addition, 1,2,4-triazolo[5,1-b]-1,3,5-thiadiazines 199 (Fig. 36) have been synthesized through a double Mannich reaction starting from 5-benzyl-2H-1,2,4-triazole-3(4H)-thione and primary aliphatic and aromatic amines, and these compounds had good antifungal activity against Chrysosponium tropicum and T. rubrum, but were completely inactive against A. fumigatus, A. flavus and Microsporum nanum [306]. Mixed results were obtained for other fungi, as a few candidates 199 were more potent than reference drug clotrimazole against A. niger, but were moderately potent against C. albicans or Fusarium oxysporum, whereas the rest of the compounds in the series were totally inactive against these three fungi. As far as the aminomethyl derivatives of 2,3-dihydro-1,2,4-triazole-3-ones are concerned, candidates 136 and 138 (Fig. 25) had no activity against C. albicans and S. cerevisiae [176], [191], whereas in the series of Mannich bases 137 featuring an imidazole moiety (Fig. 25), candidate 137 (NR2 = 1-piperidinyl) was twofold more potent against S. cerevisiae and twofold less potent against C. albicans than reference drug fluconazole [192].
Antifungal activity of aminomethylated 2,3-dihydro-1,3,4-oxadiazole-2-thiones (Table 2) has been reported in the same studies dealing with these compounds' antibacterial activity. Several Mannich bases 139a were equipotent to reference drug ciclopiroxolamine against T. mentagrophytes, A. flavus and A. fumigatus (MIC 6.25 μg/mL), but not against P. marneffei [193]. Mannich bases 139b derived from either morpholine or 3-trifluoromethylaniline showed high activity against P. marneffei, T. mentagrophytes, A. flavus and A. fumigatus compared to reference drug ciclopiroxolamine, whereas those derived from other primary aromatic amines had only weak antifungal activity [194]. Only a few of Mannich bases 139c derived from piperazines had moderate anti-Candida activity, and the rest of the collection was inactive [195]. Oxadiazolethione Mannich bases 139d [185] and 139j [176], having at position 5 of the oxadiazole ring either a pyridine or a quinoline moiety, had no noticeable anti-Candida activity, and the activity of Mannich bases 139e against S. cerevisiae, C. tropicalis and A. niger was moderate at best [187]. Most Mannich bases 139f had weak antifungal activity against C. albicans, A. niger and Aspergillus clavatus, but one candidate 139f (NR2 = NHC6H4OCH3-4) was 2-fold more potent than reference drug ketoconazole against the first two fungi, and almost equipotent to ketoconazole against the last fungus [196]. Several Mannich bases 139g had similar MIC values as reference drug amphotericin B against C. albicans, A. fumigatus, T. rubrum and T. mentagrophytes [197]. Most imidazole-containing Mannich bases 139h exhibited better antifungal activity against C. albicans, T. rubrum and T. mentagrophytes than reference drug fluconazole [198]. The compounds reported in a study, including Mannich bases 139i, are claimed to exhibit significant antifungal activity against C. albicans, but lack of access to the original information (which is only provided in the online edition as Supplemental Material, and is missing) makes data assessment impossible [199]. Three Mannich bases 139k were moderately active against Aspergillus spp. and Curvularia lunata [200], while Mannich bases 139l [98] and 139o [202] had no anti-Candida activity, and Mannich base 139m showed no antifungal activity against C. albicans and S. cerevisiae [201]. In addition, several aminomethylated 2,3-dihydro-1,3,4-oxadiazole-2-ones 141 were more potent than reference drug nystatin against fungal plant pathogens Aspergillus spp. (R1 = 2-FC6H4), F. oxysporum (R1 = 4-F3CC6H4), Botrytis cinerea (R1 = 2-FC6H4, 4-FC6H4, 2-ClC6H4, 4-CNC6H4) and Penicillium spp. (R1 = 4-FC6H4) [204].
Anti-Candida activity of C-Mannich bases 142 of thiazolidinone (Fig. 26) was found to be 16-fold weaker than that of reference drug terbinafine [205]. While three Mannich bases 143 were moderately active against C. albicans and A. flavus, one candidate 143 (R1 = C6H5, NR2 = N(C2H5)2) (Fig. 26) showed against the latter fungus a growth inhibition potency (MIC = 1.56 μg/mL) which was half of that determined for reference drug fluconazole [207]. Also, aminomethylated thiazolidinones 144 (Fig. 26) had moderate activity against Aspergillus spp. and C. albicans compared to reference drug ketoconazole [208].
Pyrimidine derivatives that have been aminomethylated with a view to evaluate the antifungal activity of the resulting Mannich bases include pyrimidin-2-ones, pyrimidin-2-thiones and (thio)barbituric acid. Double N-Mannich bases 146 (X = O or S) (Fig. 26) had 67–77% of the anti-Candida activity of the clotrimazole-containing commercial drug Imidil [210], [211]. C-Mannich bases 200 of (thio)barbituric acid (Fig. 37 ), obtained using furan-2-carboxaldehyde or indole-3-carboxaldehyde as aldehyde components and 2-aminopyridine or 4-aminoantipyrine as amine reagents in the Mannich reaction, showed good activity against Aspergillus spp. compared to reference drug salicylic acid [307].
Fig. 37.

Antifungal Mannich bases derived from (thio)barbituric acid.
Antifungal evaluation of Mannich bases from miscellaneous substrates that do not belong to any of the previously mentioned classes is the topic of several studies. P-Mannich bases 83 (Fig. 15) showed good activity against C. albicans and S. cerevisiae (MIC = 10 μg/mL) compared to reference drug amphotericin B (MIC = 15 μg/mL) [106]. Various Mannich bases of type 149 (Fig. 27) derived from imides presented anti-Aspergillus activity that ranged from weak to good, but no correlations between structure and activity for the small number of compounds in this series could be established [215]. Anti-Candida activity of the most potent Mannich bases 152 (Fig. 29) was only half of the activity of reference drug fluconazole [222]. Aminomethylated hydrazidones 153 (R1 = C2H5) derived from isonicotinic acid hydrazide (Fig. 29) had good anti-Candida activity [223], but their analogues 153 (R1 = n-C3H7) were less active, and their activity against A. niger was even poorer [224]. Several dimethylamine Mannich bases 154 derived from 7-methyl-2-(substituted aryl)imidazo[1,2-a]pyridines (Fig. 29) were 4-fold–2-fold more active against Aspergillus spp. and C. albicans than reference drug griseofulvin [225]. When evaluated against the same fungi, Mannich bases 157 (R1 = CH3) and 158 of sydnones (Fig. 29) were consistently less active than both reference drugs griseofulvin and nystatin [228], and the majority of Mannich bases 157 (R1 = OCH3) behaved similarly, with the exception of candidate 157 (R1 = OCH3; NR2 = 4-nitrobenzothiazole-2-ylamino), which was more potent than reference drugs fluconazole and nystatin against C. albicans [229]. All of the Mannich bases 156 derived from 2-alkyl-3-hydroxy-pyridine-4(1H)-ones (Fig. 29) were inactive against Aspergillus spp. and C. albicans [227]. A few aminomethylated pyrazolines 159 (Fig. 29) had anti-Candida activity (MICs between 1.5 and 4 μg/mL) comparable or even better than that of reference drug clotrimazole (MIC = 2 μg/mL) [230], while several acetylenic Mannich bases 162 (Fig. 29) were equipotent to reference drug ketoconazole against C. albicans [233]. Mannich bases 164 and 165 derived from hydantoins (Fig. 29) were generally moderate growth inhibitors for C. albicans and A. niger [236], and phenothiazine Mannich bases 167 (Fig. 29) were all active against A. niger (sizes of inhibition zone between 12 and 21 mm) [238]. Antifungal activity of Mannich base 168 (Fig. 29) of quinoxaline-2,3(1H,4H)-dione (100 μg/disc) against C. albicans and A. niger was superior to that of reference drug clotrimazole (50 μg/disc) [239], while antifungal activity of Mannich bases 169 of 3-aryl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (Fig. 29) against the same fungi was generally moderate, with the exception of candidates 169 (Ar = 4-C2H5OC6H4, R = 4-OHC6H4, R1 = COCH3; Ar = 4-C2H5OC6H4, R = 2-HOOCC6H4, R1 = 2-(NaOOCCH2)C6H4), which were equipotent to reference drug fluconazole against C. albicans (MIC 6.25 μg/mL) [240].
6. Antimalarial activity
As much as 20% of the world population live in areas with high risk of contracting malaria, and millions of people suffer from acute malaria each year, while approximately half million died from the disease in 2012 [308]. Malaria has been largely eradicated in many parts of the world, but the total number of recorded cases is still on the rise. One of the causes for this grave situation is the proliferation of malaria parasites that are rapidly becoming resistant to antimalarial drugs, especially those that are most frequently used, such as chloroquine. The development of novel antimalarials has stagnated since the 1970s owing to lack of interest in developed countries for this topic, and limited market potential for these drugs. Initiation of numerous programs supported by public funding led to a surge of interest in drugs for neglected diseases, and antimalarials are certainly amongst these drugs.
Use of the Mannich reaction for the synthesis of antimalarials has a long and rich history, the quest for aminomethylated substrates for treatment of malaria starting with amodiaquine 201 (Fig. 38 ), the first Mannich base that was used to treat malaria. Many of these early studies focused on derivatives of 4-aminoquinolines, and the SAR within this class of antimalarials suggested that the chlorine atom at position 7 of the quinoline scaffold and the phenolic Mannich bases moiety are structural features that are essential for the antimalarial activity of these compounds. Nowadays, the use of amodiaquine has declined owing to its considerable toxicity as a result of its in vivo conversion into a reactive quinone-imine metabolite by cytochrome P450. It was therefore hypothesized that swapping the phenolic hydroxyl and the aminomethyl side chain in amodiaquine should prevent the formation of toxic metabolites, and help circumvent the adverse side effects associated with the use of amodiaquine. This hypothesis led to the design and synthesis of several amodiaquine analogues that retain the antimalarial activity, while lacking the potential to form a quinone-imine derivative [309]. Out of these analogues, Mannich base 202 (the isomer of amodiaquine henceforth named isoquine, Fig. 38) expressed in vitro activity against both chloroquine sensitive HB3 strain and highly chloroquine-resistant K1 strain of Plasmodium falciparum below 10 nM, and was 20 times more potent that chloroquine. Isoquine (as its diphosphate salt) was subsequently tested in vitro against the murine Plasmodium yoelii NS strain, and found to be 3-fold more potent than amodiaquine when administered orally. Moreover, no toxic metabolites or any excessive accumulation of isoquine have been noticed in animal models, but unacceptably high first pass metabolism of isoquine to N-dealkylated metabolites in four animal species compromised isoquine's activity against parasites, and complicated the development process of isoquine into a marketable drug. A novel candidate, N-tert-butyl isoquine (GSK369796) 203 (Fig. 38), was identified using isoquine as lead, and shown to exhibit low nM activity against chloroquine-resistant and sensitive parasites, as well as in vivo oral activity equivalent to amodiaquine, but with a significantly improved antimalarial exposure profile [310]. In addition, N-tert-butyl isoquine had a better overall preclinical safety profile than chloroquine or amodiaquine, and can be synthesized in a scalable and cost-effective way. The study of disposition of candidates 202 and 203 showed that isoquine 202 undergoes in vivo oxidative N-dealkylation to desethyl-isoquine, a metabolite that had an improved blood clearance over isoquine; unfortunately, desethyl-isoquine also had a more potent inhibitory action of CYP1A2 than isoquine [311]. On the other hand, N-tert-butyl isoquine 203 had human plasma protein binding ability and inhibition of CYP2D6 similar to those of isoquine and desethyl-isoquine, but a reduced rate of N-dealkylation and a higher oral bioavailability compared to isoquine and desethyl-isoquine, and these properties made N-tert-butyl isoquine 203 a better candidate than isoquine 202 for further development. A study of the in vitro activity of isoquine 202 against P. falciparum clinical isolates collected in Kenya in relation to amino acid changes in pfcrt and pfmdr1, the two genes associated with 4-aminoquinoline resistance, showed that isoquine possesses high activity against field isolates (IC50 was 9 nM, compared with 56 nM for chloroquine, and 8 nM for amodiaquine), and that isoquine's activity could be correlated with polymorphisms in pfcrt, but not in pfmdr1 [312]. Starting from N-tert-butyl isoquine 203 as a template, three benzoxazines 204 (R = C2H5, n-C3H7, CH2C6H5) (Fig. 38) were designed and generated through a double Mannich reaction, but despite the good anti-Plasmodium activity against both chloroquine-sensitive (IC50 between 21 and 38 nM) and chloroquine-resistant (IC50 between 31 and 72 nM) strains, the rapid ring opening of benzoxazine at low pH makes them unsuitable for further development [313]. Novel analogues 205 (NR2 = aliphatic primary and secondary amines, aromatic primary and secondary amines) of amodiaquine have been tested in vitro against chloroquine-sensitive strains of P. falciparum, and while all of the compounds were active, only Mannich base 205 (NR2 = 4-morpholinyl) (Fig. 38) showed good activity (MIC = 63 ng/mL) compared to chloroquine (MIC = 250 ng/mL) [314]. Antimalarial activity of analogues 206 of isoquine (Fig. 38) has also been evaluated, but regardless of the nature of the amine in the aminomethyl moiety, their activity was considerably lower than that of reference drug chloroquine [315], [316]. Use of primary amines derived from bicyclic scaffolds for the synthesis of novel isoquine analogues led to the discovery of 207 (Fig. 38), which was approximately twofold more potent that either chloroquine or isoquine against chloroquine-sensitive and resistant P. falciparum strains [317]. A series of isotebuquine analogues 208 (R = Cl or CF3, R1 = H), the corresponding double Mannich bases 208 (R1 = CH2NHC(CH3)3), as well as N-oxides derived from a mono-Mannich base and a double Mannich base of type 208 (Fig. 38) were also evaluated against chloroquine-sensitive and resistant P. falciparum strains [318]. While only a few of candidates 208 were more potent than chloroquine against the chloroquine-sensitive strain, the majority of these Mannich bases were more potent than chloroquine against the chloroquine-resistant strain, and Mannich bases having one aminomethyl function were generally more potent than double Mannich bases.
Fig. 38.

Antimalarial phenolic Mannich bases based on 4-amino-7-chloroquinoline.
Pyronaridine 209 (Fig. 39 ) is another example of antimalarial drug that feature a phenolic Mannich bases moiety in its structure, but the aminoquinoline scaffold that was typical for amodiaquine and its congeners has been replaced in pyronaridine by a naphthyridine ring system. Pyronaridine was first synthesized in 1970 at the Chinese Institute of Parasitic Disease, and has been used solely in China as treatment for malaria with good results for over 30 years [319]. Beside the difficultly accessible literature in Chinese, there are several recent studies that provide information on its physicochemical properties [320] and ADME properties in rats [321], interaction with other antimalarials [322], potential use in combination therapy with artesunate [323], or its mechanism of action based on inhibition of β-hematin formation and subsequent glutathione-dependent hematin degradation process [324]. Based on the observation that substitution of the quinoline scaffold in amodiaquine with a napthyridine ring system in pyronaridine preserved the antimalarial action, Görlitzer initiated an ambitious synthetic program aiming at exploring the effect on the antimalarial properties of replacement of quinoline in quinoline-based antimalarials with other fused heterocyclic systems containing one pyridine ring. Thus, single (R1 = H) and double Mannich bases (R1 = CH2NR2) structurally related to amodiaquine and pyronaridine, respectively, and featuring pyrido[3,2-b]indol-4-yl (compounds 210, X = NH) [325], benzofuro[3,2-b]pyridin-4-yl (compounds 210, X = O) [326], benzothieno[3,2-b]pyridin-4-yl (compounds 210, X = S) [327], thieno[2,3-c]quinolin-4-yl (compounds 211) [328], thieno[3,2-c]quinolin-4-yl (compounds 212) [329], thieno[3,4-c]quinolin-4-yl (compounds 213) [330], benzo[c][2,7]naphthyridin-5-yl (compounds 214) [331] and benzo[h][1,6]naphthyridin-5-yl (compound 215) [332] moieties were synthesized and evaluated against chloroquine-sensitive and resistant P. falciparum strains (Fig. 39). As expected, all of the Mannich bases presented in these studies exhibited good antimalarial activity, and double Mannich bases were generally more potent than single Mannich bases. However, in spite of the variety of fused heterocyclic systems that were investigated, no compound with antimalarial activity at least comparable to that of chloroquine emerged, with the exception of one pyronaridine analogue (210, X = NH, R1 = CH2NR2, NR2 = 1-pyrrolidinyl). This candidate had IC50 = 50 nM against chloroquine-sensitive strain 3D7, IC50 = 38 nM against chloroquine-resistant strain Dd2, and its evaluation against Plasmodium winckei in a murine malaria model (intraperitoneal dosage of 100 mg/kg) showed that no intact parasite could be noticed after three days of treatment [325].
Fig. 39.
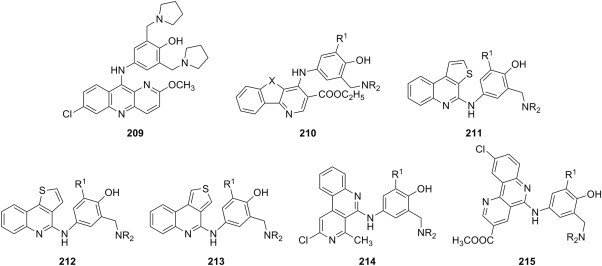
Antimalarial phenolic Mannich bases having tricyclic heterofused pyridines scaffolds.
The phenolic Mannich base moiety in the previously mentioned antimalarial agents can be also found in candidates that were designed through its incorporation into hybrids with dual mode of action. Thus, the combination of a tetraoxane and a phenolic Mannich base moiety afforded hybrids 216 called mannoxanes (Fig. 40 ), which are active at low nanomolar concentrations and surpass the ability of artesunate, tetraoxane RKA 182 and a peroxide/amodiaquine combination to cure malaria in mice at 10 mg/kg [333]. Hybridization of artemisin with a phenolic Mannich base moiety led to candidates 217 (Fig. 40), which are up to 3 times more potent than artemisin against P. yoelii nigeriensis, could be easily converted into water soluble forms, can be administered orally, are presumed to have good bioavailability, and lack the capability to form neurotoxic metabolite dihydroartemisinin [334]. Hybrids 218 (Fig. 40) are structurally very similar to 217, and have been shown to be more active than sodium artesunate against chloroquine-sensitive NF54 and chloroquine-resistant K1 P. falciparum strains [335]. Screening of a collection of compounds containing a 2-hydroxyethylamino motif in their structures for inhibition of parasite growth in red blood cells infected with chloroquine-sensitive NF54 P. falciparum strain identified a hit compound, whose further elaboration led to several hybrids featuring several types of phenolic Mannich moieties as the second pharmacophore [336]. Their evaluation against NF54 strain, K1 strain, and the rodent parasite Plasmodium berghei showed that the introduction of the phenolic Mannich bases moiety resulted in candidates that were very potent against the parasites in the 72 h assay. For example, compound 219 (Fig. 40) had IC50 NF54 = 42 nM and IC50 K1 = 9.5 nM, as determined by measuring incorporation of the nucleic acid precursor [3H]hypoxanthine after 72 h of incubation in the presence of 50% serum, and IC50 P. berghei = 140 nM after 24 h.
Fig. 40.
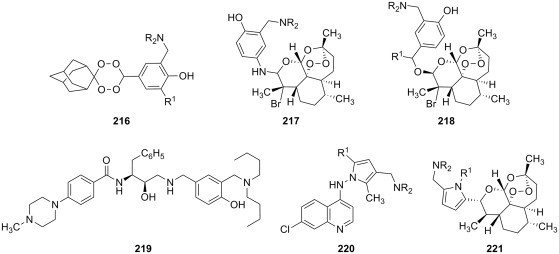
Hybrid antimalarials with dual mode of action featuring phenolic and pyrrole Mannich bases.
The design of hybrids with dual mode of action has also been broadened to include pyrrole Mannich bases. Thus, hybrids 220 from 4-amino-7-chloroquinoline and pyrrole Mannich bases (Fig. 40) were evaluated against chloroquine-sensitive D10 and chloroquine-resistant W2 P. falciparum strains, but they were all less potent than isoquine against both strains [317]. However, candidate 220 (R1 = 4-ClC6H4, NR2 = N(C2H5)2) was equipotent to chloroquine against D-10 strain, and four of the candidates were also more potent than chloroquine against W2 strain [317]. Several candidates from a small library of Mannich bases 221 (R1 = H or CH3) of C-10 pyrrole analogues of artemisin (Fig. 40) had antimalarial activity that was superior to both artemisin and chloroquine against chloroquine-sensitive 3D7 P. falciparum strain, and selected Mannich bases from this library were more potent than the aforementioned reference drugs against chloroquine-resistant K1 P. falciparum strain [337]. All of the compounds tested displayed good activity in Peter's 4 day suppressive test using 30 mg/kg over days 1–3 post-infection, and candidates 221 (R1 = H, NR2 = 4-morpholinyl; R1 = CH3, NR2 = 4-morpholinyl; R1 = CH3, NR2 = 4-methyl-1-piperazinyl) showed complete elimination of parasites. These three Mannich bases were further investigated in vivo, and had effective doses between 4.3 and 5.3 mg/kg that eliminate 90% of P. berghei in mice, whereas candidate 221 (R1 = CH3, NR2 = 4-methyl-1-piperazinyl) reached 100% clearance of parasitemia 24 h after the last treatment and increased mouse survival to 9 days, a biological profile that render it superior to clinically used sodium artesunate [337].
Phenolic Mannich bases of chalcone analogues (such as 222, Fig. 41 ) have been claimed to possess antimalarial activity against chloroquine-sensitive D10 P. falciparum strain at concentrations ranging from 0.3 to 3 μg/mL [44]. Taking into account the structural similarity between phenolic Mannich bases 222 and ketonic Mannich bases of α,β-unsaturated ketones, a possible mechanism action for compounds 222 could be the inhibition of thioredoxin reductase in P. falciparum [338]. Phenolic Mannich bases 223 of 2,4-dihydroxybenzaldehyde, its thiosemicarbazone and semicarbazone (Fig. 41), as well as related quinoline-containing derivatives 224 (Fig. 41) have been screened against chloroquine-resistant W2 P. falciparum strain [339]. Only candidates 223 derived from 1-(7-chloroquinolin-4-yl)piperazine had antimalarial activity, while the remaining Mannich bases 223 were inactive. All of the candidates 224 were potent antimalarial agents, with Mannich base 224 (NR2 = 4-(7-chloroquinolin-4-yl)piperazin-1-yl) featuring two quinoline motifs in its structure being the most potent (IC50 = 77 nM) [339]. Imines of 4(1H)-pyridones 225 (R1 and R2 = H or CH3) (Fig. 41) having a phenolic Mannich base moiety were active against chloroquine-resistant W2 P. falciparum strain (IC50 values between 9 and 33 μM) and atovaquone-resistant FCR3 P. falciparum strain (IC50 values as low as 8 μM), but their potency was lower than that of reference drugs chloroquine or atovaquone [340]. In addition, aminoalkylation of lawsone using various aldehydes and N-alkyl-ferrocenylmethylamines afforded Mannich bases 226 (R1 = alkyl) (Fig. 41) structurally resembling antimalarial drug atovaquone, but evaluation of three candidates 226 (R1 = n-C6H13, n- C7H15, n- C8H17) against P. falciparum showed that they were less potent than atovaquone itself [341].
Fig. 41.
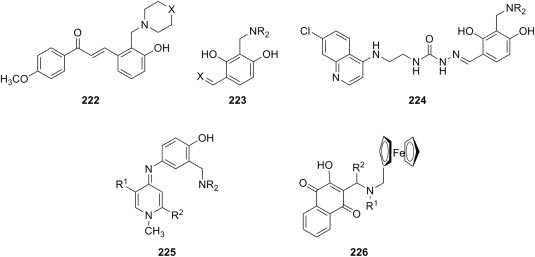
Antimalarial phenolic Mannich bases derived from miscellaneous substrates.
7. Antiviral activity
Because of their self-limiting nature, most viral diseases (with the exception of those caused by human immunodeficiency virus, HIV) do not require specific therapy, although treatments are used to bring the condition or its symptoms to an end more quickly. Currently available drugs are designed to target four main groups of viruses, namely herpes viruses, hepatitis viruses, influenza viruses, and HIV. Recent investigations in the antiviral activity of Mannich bases deal mostly with aminomethylated phenols and aminomethylated isatins, although examples of Mannich bases with antiviral properties derived from other types of substrates are available in literature.
Phenolic Mannich base arbidol 227 (Fig. 42 ) [342] is a broad-spectrum antiviral agent commonly used in Russia to treat acute respiratory viral infections, which acts by inhibiting virus-mediated fusion with target membrane and subsequent blockage of virus entry into target cells [343], [344]. Using arbidol as lead compound, a group at Shenyang Pharmaceutical University led by Gong initiated a program aiming at investigating structure–anti-hepatitis B relationship for a series of phenolic Mannich bases derived from various heterocyclic ring systems. In their first report on this topic, the Chinese researchers presented a series of analogues of arbidol 228 (X = SO, R1 = CH3, c-C3H5, R2 = Br) (Fig. 42) having various substituents R2 in the aromatic sulfoxide moiety, and presenting amino residues different than the original dimethylamino group in arbidol (e.g., secondary aliphatic amines, NH-azoles belonging to imidazoles and 1,2,4-triazoles) in the aminomethyl function in position 4 [345]. SAR investigation was based on the ability of these compounds to inhibit replication of hepatitis B virus (HBV) and the production of surface antigen of the hepatitis B virus (HBsAg) and extracellular form of hepatitis B core antigen (HBeAg) in HepG2.2.15 cells infected with HBV, and showed that half of these Mannich bases exhibited inhibitory effects on HBV that were superior to reference drug lamivudine. Replacement of the original methyl group at N-1 in 228 with cyclopropyl led to an increase in antiviral activity, but enhanced the cytotoxicity of these candidates as well, whereas the presence of fluorine or chlorine as substituents in the aromatic sulfoxide moiety resulted in an improvement of the anti-HBV activity. Departure from the original phenylmercapto function in arbidol reduced the cytotoxicity of Mannich bases 228 (X = SO), leaving in the same time the anti-HBV activity practically unchanged. Also, the nature of the amino group in the aminomethyl function appears to have little effect on the anti-HBV activity of the Mannich bases reported in this study [345]. The relationship between the presence of a linker (containing two or three atoms between the sulfur atom in the side chain and the phenyl moiety) and the anti-HBV activity of Mannich bases 228 (X = SOCH2CH2Y, Y = CH2, O or null, R1 = CH3, R2 = Br) was also explored, and candidates having a phenethyl residue proved to be inactive, whereas candidates with a 3-phenylpropyl residue presented remarkable anti-HBV activity [346]. A subsequent study [347] of the anti-HBV activity of arbidol analogues 228 (X = SO or SO2, R1 = CH3, c-C3H5, R2 = H) unsubstituted at position 5 of the indole scaffold claimed that the nature of the amino function plays an important role, and that pyrrolidine- and imidazole-containing candidates exhibited better anti-HBV activity than candidates having different amino residues. For another set of Mannich bases 228 (X = SO or SO2, R1 = CH3, R2 = H), the authors suggested that the presence of 4-methylpiperazin-1-yl residue as amino moiety, rather than 1-imidazolyl, 2-methyl-1-imidazolyl or 1-piperidinyl, afforded more potent anti-HVB agents [348]. Replacement of the sulfinyl group in the side chain with sulfonyl appeared to enhance the antiviral activity while reducing cellular toxicity of these Mannich bases [347], although the results in the later study [348] supported the opposite conclusion. Substitution of the indole scaffold with quinoline in Mannich bases 229 (X = S or SO) (Fig. 42) resulted in potent inhibition of HBV DNA replication (10- to 66-fold compared to reference drug lamivudine) [349]. Analysis of SAR for compounds 229 revealed that the presence of 4-fluorophenyl moiety linked to sulfur in the side chain at position 2 of the quinoline scaffolds, in conjunction with amine moieties such as 1-pyrrolidinyl, 1-piperidinyl, 1-imidazolyl (but not 4-methylpiperazinyl or morpholinyl) in the aminomethyl function are required for good anti-HBV activity. Evaluation of Mannich bases 230 (Fig. 42) derived from a tetracyclic ring system comprising the quinoline scaffold fused with a benzothiopyrane ring system that has various substitution patterns in the aromatic ring identified a significant number of candidates with good inhibition of HBV replication [350], [351]. The search for anti-HBV agents with similar structures continued with the evaluation of a series of phenolic Mannich bases 231 (R1 = CH3, C2H5, CH(CH3)2, R2 = H or F, X = S or SO) of 5-hydroxybenzimidazoles substituted at position 2 with 4-alkylmercaptophenyl residues (Fig. 42) [352]. These compounds exhibited inhibitory effect on the secretion of HBsAg that was superior to reference drug lamivudine, but lacked the ability to inhibit the replication of HBV DNA in HepG2.2.15 cells at concentrations that were inferior to those corresponding to 50% of their cytotoxicity. The presence of fluorine at position 6 of benzimidazole scaffold and the presence of sulfinyl function appears to be critical for the inhibition of HBsAg secretion of Mannich bases 231. Evaluation of another series of Mannich bases 232 (X = S, SO, SO2, R1 = H, 4-F, 4-OCH3, 4-CH3) of 5-hydroxybenzimidazoles (Fig. 42) showed that the thioethers in this series did not inhibit the replication of HBV DNA, but the candidates with 4-methylpiperazin-1-yl as amino moiety were good inhibitors of secretion of HBsAg, and the inhibitory effect significantly decreased upon oxidation of sulfur in thioethers to sulfinyl and sulfonyl [353]. Phenolic Mannich bases 233 (R1 = H, 4-F, 4-Cl, 4-Br, 4-CH3, 3-OCH3) derived from 6-bromo-8-hydroxyimidazo[1,2-a]pyridine-3-carboxylates (Fig. 42), especially those having 4-morpholinyl or 4-methylpiperazin-1-yl moieties in the aminomethyl function, inhibited the replication of HBV DNA, but had no inhibitory effect on secretion of HBsAg or HBeAg [354]. The nature of substituents in the arylmercapto residue modulates the anti-HBV activity, as the decrease of bulkiness of the substituents (from bromine to fluorine, for example) and the presence of lipophilic substituents (e.g., a methyl group) enhance the inhibition of replication of HBV DNA, whereas the presence of a 3-methoxy group further increases the activity.
Fig. 42.
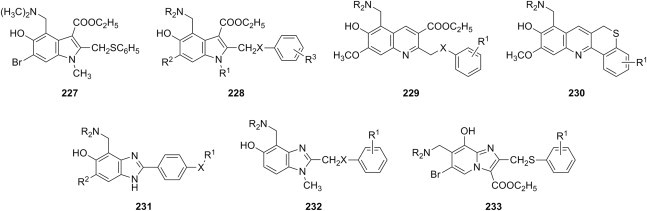
Phenolic Mannich bases inspired by arbidol and useful as anti-hepatitis B agents.
Phenolic Mannich bases 125 derived from chlorokojic acid (Fig. 23) have been evaluated against herpes simplex virus type-1 (HSV-1) and parainfluenza-3 virus (PI-3). Amongst the candidates reported in one study, only two Mannich bases had inhibitory concentration in the range of 0.1–0.8 μg/mL against HSV-1, whereas all of the compounds were active in various degrees against PI-3 [152]. All of the Mannich bases 125 derived from 1-arylpiperazines as amine reagents inhibited both HSV-1 and PI-3, and one candidate 125 (R = 4-(4-methoxyphenyl)piperazin-1-yl) was as potent as reference drug acyclovir against HSV-1 [153]. In addition, candidate 125 (R = 4-(3-chlorophenyl)piperazin-1-yl) presented remarkable activity (0.025–0.4 μg/mL) against PI-3.
Other aminomethyl derivatives of various phenolic substrates have been tested against different viruses. Only one phenolic Mannich base 57 (R = NHCH3) of norvisnagin (Fig. 10) was moderately active against HSV-1 [78]. Evaluation of aminomethylated 7-hydroxycoumarin derivatives 234 (Fig. 43 ) against Flaviviridae and other viruses led to mixed results [355]. Phenolic Mannich bases 234 (R1 = H, R2 = H or CH3) were generally inactive against bovine viral diarrhea virus (BVDV), yellow fever virus (YFV) or respiratory syncytial virus (RSV), and a few other candidates that were active against BVDV or YFW were also cytotoxic. O-Alkylated phenolic Mannich bases 234 (R1 = n-C3H7, R2 = H or CH3), and especially candidates having a methyl group at position 4 of the coumarin ring system, were active against BVDV, but they were inactive against YFV or RSV. O-Acylated phenolic Mannich bases 234 (R1 = COC6H5, R2 = H or CH3) were generally inactive against all three viruses, but one candidate 234 (R1 = COC6H5, R2 = H, NR2 = 1,2,3,4-tetrahydroisoquinolin-2-yl) had a remarkable activity against RSV, comparable to that of reference drug 6-azauridine, and had also very low toxicity. Compounds 234 were not active against a panel of viruses comprising HIV-1, coxsackievirus B2, SB-1 strain of Marek's disease virus, vesicular stomatitis virus and a reovirus. In addition, screening of an extensive library of compounds identified Mannich bases 235 of 5-chloro-8-hydroxyquinoline (Fig. 43) as reactivators of latent HIV-1, which could prove helpful in eradicating the latent reservoir of HIV-1 in resting memory CD4+ T cells, either alone or in combination with other treatments [356].
Fig. 43.
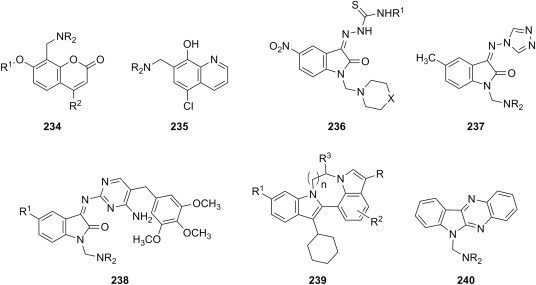
Miscellaneous antiviral Mannich bases.
Antiviral activity of Mannich bases of isatin and its derivatives is the topic of several investigations. Screening of twelve Mannich bases 236 derived from semithiocarbazones of 5-nitroisatin (Fig. 43) against a panel of other viruses afforded no compound with antiviral properties, with the exception of candidate 236 (X = O, R1 = allyl), which had weak activity against YFV (strain 17D) at subtoxic concentrations. This candidate was more potent than reference drug ribavirin, but was also more cytotoxic [357]. Based on results of molecular studies aiming at designing inhibitors of HIV reverse transcriptase, a series of Mannich bases 237 (Fig. 43) have been synthesized [358]. Candidates 237 having secondary aliphatic amino moieties in the aminomethyl function showed 92 to 69% inhibition of the enzyme, whereas Mannich bases 237 (NR2 = NHC6H5) showed no inhibition. Aminomethylation of Schiff bases obtained from 5-substituted isatins (R1 = F, Cl, F) and lamivudine using fluoroquinolones (R2 = ethyl, cyclopropyl; R3 = H, CH3) as amine reagents gave Mannich bases 183 (Fig. 32), which were less potent against HIV than the parent Schiff bases, and the most potent candidates 183 (R1 = F) were also 10-fold less potent than lamivudine itself against HIV [273]. Starting from Schiff bases of trimethoprim, Mannich bases 238 (Fig. 43) were obtained using fluoroquinolones as amine reagents. Their evaluation against HIV and hepatitis C virus (HCV) showed that Mannich bases 238 (R1 = Cl) inhibited replication of HIV in MT-4 cells at effective concentrations (EC50) ranging from 9.4 to 56 μg/mL, and most compounds were active against HCV RNA replication (80% inhibition at 50 μg/mL) [162]. Mannich base 238 (R1 = CH3, NR2 = 4-(4-chlorophenyl)piperazin-1-yl) and three Mannich bases 238 (R1 = CH3) derived from fluoroquinolones as amine reagents showed inhibition against replication of HIV in MT-4 cells at EC50 ranging from 11.6 to 28.4 μg/mL [359]. In addition, all of the compounds 238 (R1 = CH3) were active against HCV RNA replication (65% inhibition at 50 μg/mL) [359]. Furthermore, a study reports candidate 238 (R1 = H, NR2 = 4-(4-nitrophenyl)piperazin-1-yl) as inhibitor of Japanese encephalitis virus and West Nile virus in vitro, and a remarkable inhibitor of Japanese encephalitis virus in a murine model [360].
Investigations into the antiviral activity of Mannich bases derived from substrates other than phenols or isatins are available in literature as well. The majority of Mannich bases 80 (R = Cl, OCH3) (Fig. 14), obtained through N-aminomethylation of thiazolidine-2,4-diones with morpholine, piperidine and variously 1-substituted piperazines, showed no activity against severe acute respiratory syndrome (SARS) coronavirus [103]. Also, all of these Mannich bases had antiviral activity against types A and B of influenza virus, but virus inhibition occurred almost at cytotoxic concentration. Aminomethylation of the amide function in tetracyclines using fluoroquinolones as amine reagents afforded Mannich bases 189 (Fig. 34), and four candidates derived from tetracycline and one derived from minocycline were found to inhibit HIV replication with EC50 values below 20 μM, while their toxicity against mock infected CEM cell line was greater than 140 μM [287]. In addition, all Mannich bases 189 showed moderate inhibition of both 3′-processing and strand transfer steps of HIV-1 integrase. Evaluation of efavirenz Mannich bases 191 (Fig. 34) led to the discovery of three candidates that were at least as potent as the parent compound against HIV [289]. A large number of indole Mannich bases 239 (R = aminomethyl, 3-amino-1-propyn-1-yl) (Fig. 43) derived from a pentacyclic core, along several acetylenic Mannich bases with the same core, have been claimed to be active against HCV and members of the Flaviviridae family of viruses [361]. None of the Mannich bases 240 (Fig. 43) derived from indophenazine as substrate and sulfonamides, anthranilic acid or 2-aminopyridine as amine reagents either showed any activity against HIV above their cytotoxic concentrations, but some presented weak activity against HSV, VSV, or vaccinia virus [362].
8. Anticonvulsant activity
Anticonvulsants are drugs used in the treatment of seizures in epilepsy, a common chronic neurological disorder that affects around 50 million people of all ages worldwide. The discovery of novel antiepileptic drugs relies either on rational design based on the use of well-known pharmacophores (e.g., imides), or on random screening of libraries of compounds.
Ketonic Mannich bases 8 (R1 = 4-substituted aryloxy, 4-substituted arylthio, R2 = H, NR2 = N(C2H5)2; R1 = 4-fluorophenyloxy, R2 = H, NR2 = dimethylamino, 1-piperidinyl, 4-morpholinyl, 1-pyrrolidinyl) (Fig. 3) and the related piperidinols 14 (R1 = 4-substituted aryloxy, R = C2H5) (Fig. 4) were examined for anticonvulsant activity in the maximal electroshock (MES) and subcutaneous pentylenetetrazole (scPTZ) screens [20]. None of the compounds provided protection in the scPTZ screen, but several candidates 8 and 14 demonstrated anticonvulsant activity in the MES screen below their neurotoxic levels (30 mg/kg). Four ketonic Mannich bases of type 8 derived from acetophenone or 4-hydroxyacetophenone as substrates and common secondary aliphatic amines as amine reagents, as well as the corresponding azines 16 (Fig. 4), were assessed as anticonvulsants using the same two tests. The results showed that anticonvulsant activity of candidates 8 was superior to that of azines 16, and compounds derived from 4-hydroxyacetophenone were active at a dose of 300 mg/kg in the MES test, but no candidate showed anticonvulsant activity in the cPTZ test [363]. Bis-Mannich bases 13 (R1 = H, 4-Cl, 4-CH3, 2-thienyl, R = C2H5) and the corresponding piperidinols 14 were protective in the MES test at 30 mg/kg and/or above, while candidate 14 (R1 = 4-Cl, R = C2H5) was protective in the scMet test at 300 mg/kg after 4 h [364]. The presence of a 4-chlorophenyl moiety appears to be important for the anticonvulsant activity of these compounds, and analogues having the same moiety were identified as good anticonvulsant agents in a previous study [365].
Anticonvulsant activity of phenolic Mannich bases of 3-hydroxy-4-pyranones has been investigated extensively by Aytemir's group. Screening of derivatives of allomaltol 174 having a 4-substituted piperazin-1-ylmethyl group (Fig. 30) using MES and scPTZ tests showed that candidate 174 (R = 3-CF3C6H4) provided excellent protection against pentylenetetrazole-induced seizures, but was neurotoxic at high dose (300 mg/kg), while candidate 174 (R = 4-ClC6H4) had high anticonvulsant activity in MES test at all doses after 30 min, without being neurotoxic [305]. Evaluation of another series of Mannich bases of allomaltol revealed that candidate 174 (R = 2,3-(CH3)2C6H3) was active in scPTZ test at 300 mg/kg after 4 h, along with candidate 174 (R = 3-ClC6H4), which was active in MES test at 300 mg/kg after 5 h [366]. Although Mannich bases of allomaltol obtained using morpholine or 4-(1-piperidinyl)piperidine as amine reagents had no anticonvulsant activity [366], a subsequent study dealing with novel Mannich bases generated from piperidines as amine reagents reported that candidates 124 (R1 = 3-CH3, R2 = 5-CH3; R1 = 4-HOCH2CH2, R2 = H; R1 = 4-C6H5CH2, R2 = H) (Fig. 23) were protective in scPTZ test, while only candidate 124 (R1 = 4-HOCH2CH2, R2 = H) provided protection in MES test at 300 mg/kg [367]. Use of other piperidines as amine reagent in the Mannich reaction with allomaltol as substrate led to candidates 124 (R1 = 4-(un)substituted phenyl, R2 = OH, CN, COCH3), and two of these Mannich bases (R1 = 4-BrC6H4, R2 = OH; R1 = 4-ClC6H4, R2 = OH) were active in scPTZ test at 300 mg/kg after 4 h, while all of them were active in MES test either after 0.5 h or after 4 h [368]. In contrast, when allomaltol was replaced with kojic acid as substrate in the Mannich reaction, but the same piperidines were used as mine reagents, the resulting candidates 241 (NR2 = 4,4-disubstituted piperidines) (Fig. 44 ) were all active in scPTZ test either after 0.5 h or after 4 h, but only two of them (R1 = 4-BrC6H4, R2 = OH; R1 = 4-ClC6H4, R2 = OH) were active in the MES test at 300 mg/kg after 0.5 h, and one candidate 241 (R1 = C6H5, R2 = COCH3) was active at any dose after 4 h [368]. Mannich bases 241 (NR2 = 4-substituted piperazines) of kojic acid as substrate and piperazines as amine reagents were generally better anticonvulsants than analogous Mannich bases 174 of allomaltol, or than Mannich bases 241 derived from kojic acid and 4,4-disubstituted piperidines [369]. The authors tentatively explain the enhanced anticonvulsant activity of Mannich bases of kojic acid compared to the activity of Mannich bases of allomaltol through the possible formation of an extra hydrogen bond in the former compounds. However, Mannich bases derived from chlorokojic acid were also good anticonvulsants, and they present, like Mannich bases of allomaltol, only one hydrogen bond in their structure [154].
Fig. 44.
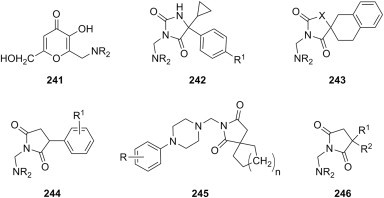
Anticonvulsant Mannich bases derived from kojic acid, hydantoins and succinimides.
Hydantoin represents the core structure of the old generation of antiepileptic drugs, such as phenytoin, and substitution with an aminomethyl moiety was shown to improve activity against MES seizures in mice [370]. Evaluation of a library of N-Mannich bases 242 (Fig. 44) derived from 5-cyclopropyl-5-arylhydantoins having 4-substituted piperazines in the aminomethyl function showed that the majority of candidates were effective in the MES or/and scPTZ screens, and quantitative studies in rats after oral administration showed that three Mannich bases 242 (R1 = H, R2 = C6H5, C6H5CH2, 3-CH3C6H4CH2) were more potent than phenytoin in MES test [371]. Results from another study [372] showed that candidates 242 were generally more active in MES test than in scPTZ screen, and chlorine-substituted Mannich bases (R1 = Cl) were generally less active than those unsubstituted in the aromatic ring (R1 = H), whereas candidates derived from 1,2,3,4-tetrahydroisoquinoline as amine reagent in the Mannich reaction were less potent than those derived from morpholine or piperazines. Compared to candidates 242 obtained from arylpiperazines as amine reagents, Mannich bases having alkylene, alkenylene, carbonyl or ester linkers between the piperazine moiety and phenyl ring presented enhanced anticonvulsant protection to pentylenetetrazole-induced seizures, which was noticeable not only after 0.5 h, but after 4 h as well [372]. Taking into account the established anticonvulsant properties of many spirohydantoins [373], [374], [375], Mannich bases 243 (X = NH) of spirohydantoins (Fig. 44) have been synthesized and evaluated as anticonvulsants, and while some of them were effective in MES or/and scPTZ screens, and were more potent than reference drug phenytoin, their high neurotoxicity precluded further testing [376].
Besides hydantoin, succinimide presents the structural requirements for the core structure of good anticonvulsant agents (namely, a nitrogen-containing heteroatomic system with a least one carbonyl group), and the well-established drug ethosuximide is an example for this class of antiepileptic drugs. The group of Obniska has been developing novel anticonvulsants for a long time, and Mannich bases derived from variously substituted succinimides has been one of the classes of compounds that provided some of the most interesting results reported by these researchers. Based on the significant number of candidates that have been synthesized, aminomethylated derivatives 244 of 3-phenylsuccinimides (Fig. 44) have been amongst the most investigated compounds within this class. Mannich bases of 3-arylsuccinimides derived from 4-(substituted aryl)piperazines or benzylpiperazines as amine reagents, such as 244 (R1 = 2-ClC6H4, 3-BrC6H4) [377], 244 (R1 = 2-CH3C6H4, 3-CF3C6H4) [378], 244 (R1 = 3-CH3C6H4) [379], 244 (R1 = 2-ClC6H4, 3-ClC6H4, 4-ClC6H4) [380], [381] or 244 (R1 = 2-FC6H4, 2-BrC6H4) [382], generally showed protection in MES screen, but some of them were also effective in scPTZ screen. Moreover, a few candidates presented activity not only 0.5 h after administration, but also after 4 or 5 h, which is indicative of quick onset and long duration of anticonvulsant activity. Mannich bases 244 of 3-arylsuccinimides derived from other secondary aliphatic amines, such as morpholine, 4-benzylpiperidine, 4-cyclohexylpiperazine, were generally effective in both screens [381], [382]. Despite the large number of compounds evaluated in these studies, no consistent SARs could be established. Compounds with good anticonvulsant properties emerged from almost all of these studies, but none of these anticonvulsant Mannich bases was deemed sufficiently promising to advance to clinical studies. Aminomethylated spirosuccinimides have also been investigated as anticonvulsants. Candidates 243 (X = CH2) derived from 4-(3-trifluoromethylphenyl)piperazine and 4-(3-chlorophenyl)piperazine as amine reagents in the Mannich reaction were the most potent anticonvulsants in this series in MES test, and replacement of substituted aryl with 2-hydroxyethyl rendered the candidates active in both tests [376]. The activity of Mannich bases 245 of simpler spirosuccinimides (Fig. 44) appears to depend on the nature of the substituent at N-4 of the piperazine moiety. Thus, candidates 245 (n = 1) derived from 4-(3-chlorophenyl)-, 4-(2-methoxyphenyl)-, 4-(2-fluorophenyl)- and 4-(4-fluorophenyl)-piperazine [377] and candidates 245 (n = 1 or 2) derived from 4-(2-methylphenyl)piperazine [383] were devoid of anticonvulsant activity. On the other hand, Mannich bases 245 (n = 1 or 2) obtained from 4-(3-trifluoromethylphenyl)piperazine as amine reagent were efficient in MES screen, but not in scPTZ screen [383]. Evaluation as anticonvulsant agents of Mannich bases 246 of succinimides 3,3-disubstituted with identical (R1 = R2 = CH3 or C6H5) or different (R1 = CH3, R2 = C6H5; R1 = CH3, R2 = C2H5; R1 = CH3, R2 = C2H5) substituents (Fig. 44) has also been the topic of several recent papers [384], [385], [386], [387]. Generally, Mannich bases obtained from 3,3-diphenylsuccinimide were more potent than those obtained from 3-alkyl-3-phenylsuccinimides, which, in turn, were more potent than Mannich bases derived from 3,3-dialkylsuccinimides, which were actually inactive in most cases. Candidates generated from 4-arylpiperazines as amine reagents in the Mannich reaction were efficient only in MES test, and the nature and position of the substituent in the aromatic ring attached to piperazine modulate the anticonvulsant activity of these compounds. However, Mannich bases 246 derived from 4-(2-hydroxyethyl)piperazine or 4-benzylpiperidine were efficient in both screens [384]. The effect of Mannich bases 246 with potent anticonvulsant activity on NaV1.2 sodium channel currents was also investigated as a potential mechanism of action for these compounds, and the results showed that the anticonvulsant activity of these candidates correlates nicely with their effectiveness as sodium channel blockers [385], [386]. In addition, there was no direct correlation between anticonvulsant properties and 5-HT1A, 5-HT2A, 5-HT1A and/or 5-HT7 serotonin receptor affinity [383], [387].
Besides hydantoins and succinimides, other substrates featuring the ureido motif in their structure have been aminomethylated with a view to obtain anticonvulsant agents. Barbituric acid and its thio analogue have been subjected to the Mannich reaction with two amine reagents having a quinazolinone moiety, and the resulting candidates 247 and 248 (X = O or S, R = 6-Br, 6-I, 6,8-Br2) (Fig. 45 ) provided good protection (40–80%, and 50–90%, respectively) in both MES and scPTZ screens at a dose of 50 mg/kg, while lacking neurotoxicity, or sedative and hypnotic effects [388]. Mannich bases 169 of 3-aryl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (Fig. 29) showed significant anticonvulsant activity in MES test, as some of these candidates afforded results comparable to those obtain for reference drug phenytoin [240].
Fig. 45.
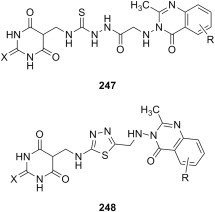
Mannich bases with anticonvulsant activity derived from (thio)barbituric acid.
Several studies concerning the anticonvulsant activity of fused seven-membered ring systems containing two heteroatoms are available in literature. Aminomethylated 2,3-dihydro-1,5-benzoxazepines 249 (Fig. 46 ) provided protection in the MES test in the range of 30–90% at a dose of 30 mg/kg; candidate 249 (R1 = 3-OCH3-4-OH, R2 = 2-OCH3) was equipotent to reference drugs phenytoin and lamotrigine in MES screen, and was also equipotent to reference drug valproate in scPTZ screen [389]. Another study allowed a direct comparison between the anticonvulsant activities of analogous aminomethylated 2,3-dihydro-1,5-benzoxazepines 250 (X = O) and 2,3-dihydro-1,5-benzothiazepines 250 (X = S) (Fig. 46) [390]. At a dose of 30 mg/kg, the latter provided more protection (20–90%) in MES test than the former, with Mannich base 250 (X = S, R1 = 2-Cl, R2 = OCH3) being the most active compound in this series. Ketonic Mannich bases 251 (R = H, Cl, NO2; R1–R2 = R3–R4 = (CH2)4 or R1 = R2 = R3 = CH3, R4 = H) having a 2,3-dihydro-1,5-benzodiazepine scaffold as the amino moiety (Fig. 46) were evaluated using a model in which the seizures were induced chemically, and some of these compounds provided protection after 0.5 h, whereas only candidates 251 derived from acetophenone (R = H) provided protection up to 2 h [391].
Fig. 46.
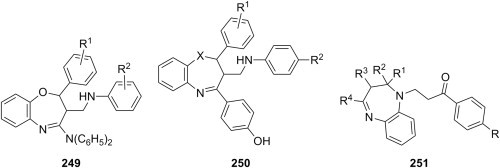
Anticonvulsant Mannich bases with an azepine scaffold.
9. Anti-inflammatory activity
Inflammation is part of the complex, nonspecific immune response of vascular tissue that occurs in reaction to any type of injury, such as pathogens, irritants, damaged cells, etc. The initial response of the body to harmful stimuli is initiated by cells already present in all tissues, and is achieved by the increased movement of plasma and leukocytes (especially granulocytes) from blood into the injured tissue, where they release a series of cell-derived mediators, triggering afterwards a cascade of biochemical events that propagate the inflammatory response. Nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly used for treatment of inflammation, along with its symptoms (fever, pain, swelling), but they present significant side effects after long-term usage, such as gastrointestinal lesions, kidney injury, and cardiovascular risk.
Along with many other classes of compounds, Mannich bases of various structures have been investigated as part of the ongoing search for novel anti-inflammatory drugs. Ketonic Mannich bases 252 (R1 = H, CH3, SO2CH3) derived from 2-arylidenecyclohexanones and aromatic amines (Fig. 47 ) have been evaluated using xylene-induced ear swelling test and carrageenan-induced paw edema, and candidate 252 (R = H) was superior to reference drug ibuprofen in both tests, whereas several candidates 252 (R = CH3) were less efficient, but still more effective than ibuprofen, in the same tests [392]. Administration of ketonic Mannich base 253 (Fig. 47) to rats with a chronic inflammatory process induced by insertion of cotton pellets led to increased phagocytic capacity of peripheric neutrophils, enhanced activity of the serum complement system, and higher catalase activity, while a slightly decrease in the activity of superoxide dismutase was observed [393]. A few double Mannich bases 105 of 4,6-dimethoxybenzofuran-3(2H)-one (Fig. 21) inhibited the production of pro-inflammatory cytokines TNF-α and IL-6 by 76–100% at a concentration of 10 μM, but they were more cytotoxic than reference drug dexamethasone, which had comparable effects with candidates 105 on the production of the same cytokines at only 1 μM [134]. Investigation of the anti-inflammatory activity of Mannich bases 254 (R = H, F, Cl, Br, OR2; R2 = OCH3 and OC4H9-n) having amino acids isoleucine and methionine as amine moieties (Fig. 47) was performed using both carrageenan-induced paw edema model and pellet-induced granuloma model, and resulted in the discovery of two anti-inflammatory agents 254 (R = OCH3 and OC4H9-n; R1 = CH2CH2SCH3) derived from methionine [394]. At a dose of 25 mg/kg, these two Mannich bases 254 had 81% and 71%, respectively, of the efficiency of the reference drug diclofenac (dose of 10 mg/kg) on the reduction of paw swelling. At the same dose, these two candidates 254 also showed 88% and 79%, respectively, of the activity of reference drug indomethacin (dose of 3 mg/kg) on chronic inflammation. A continuation of this study was undertaken with a view to broaden the nature of amino acids (cysteine, threonine, methionine, isoleucine, asparagine, and glutamine) as amine moieties in Mannich bases 254, and also the nature of the alkoxy substituents in the aromatic ring, but these novel candidates had no anti-inflammatory activity at 5 mg/kg, with the exception of two candidates 254 (R = OC2H5; R1 = CH2SH, CH2CONH2), which displayed mild activity in the acute inflammation model [395].
Fig. 47.

Ketonic Mannich bases with anti-inflammatory activity.
Only one example of Mannich bases obtained from simple phenols as potential anti-inflammatory agents is available in recent literature. Thus, evaluation of four phenolic Mannich bases 255 (Fig. 48 ) using formalin-induced acute inflammation model in mice revealed that one candidate had reliable activity upon parenteral administration (50 mg/kg), whereas reference drug diclofenac sodium exhibited only two-thirds of its activity, albeit at 8 mg/kg [396]. Phenolic Mannich bases 256 of resveratrol analogues containing a pyridinyl moiety and either one, two or three aminomethyl residues (Fig. 48) were tested using xylene-induced ear edema in mice at 200 mg/kg, and two of them had 80% of the efficiency of reference drug ibuprofen, whereas the remaining candidates were less potent [397]. The anti-inflammatory activity of Mannich bases of polyhydroxylic phenols was also investigated. At a dose of 10 mg/kg, two candidates 113 (NR1R2 = 1-piperidinyl and 4-morpholinyl) derived from 4,6-diacetylresorcinol (Fig. 22) had a slightly lower activity than that of reference drug indomethacin in carrageenan-induced paw edema [398]. Three candidates 257 (R1 = 3-Cl, NR2 = 4-methylpiperazin-1-yl; R1 = 2-Br, NR2 = 1-piperidinyl or 1-pyrrolidinyl) (Fig. 48) were more efficient at inhibiting the production of TNF-α at a concentration of 10 μM than reference drug dexamethasone at 1 μM [399]; under the same experimental conditions, most Mannich bases in the study were also more efficient at inhibiting the production of IL-6 than dexamethasone. The same researchers also investigated the action on enzymes that are involved in inflammation (such as cyclooxygenases (COX), trypsin and β-glucuronidase) of phenolic Mannich bases 258 (R1 = 2-F, 2-Cl, 2-Br, 3-F, 3-Cl, 3-Br) of chalcone analogues (Fig. 48) having the same substitution pattern in ring A as candidates 257 [400]. With one exception, none of the candidates inhibited the activity of trypsin, whereas most Mannich bases in this study inhibited the activity of β-glucuronidase. Candidates 258 derived from chalcone analogues having a halogen at position 3 of the B ring were generally more potent than those derived from chalcone analogues having a halogen at position 2 of the B ring. In addition, a few Mannich bases 258 were poor inhibitors of COX-1, but excellent inhibitors of COX-2, and the majority of the candidates were more efficient at inhibiting COX-2 than reference drug aspirin. In a different study [401], two out of four phenolic Mannich bases of other chalcone analogues were found to reduce rat paw edema induced by carrageenan, and although these compounds were found to be good inhibitors of trypsin this time, no satisfactory correlation with their antioxidant, free radical scavenging, or lipooxygenase inhibition could be established. Because inducible nitric oxide synthase (iNOS) generates high levels of nitric oxide that modulates inflammations through multiple pathways, the inhibition of this enzyme by phenolic Mannich bases of heterocyclic analogues of chalcones with various structures was also investigated. This type of phenolic Mannich bases was found to strongly inhibit NO production, with IC50 values ranging between 10.5 and 0.018 μM [402]. Benzoxazines 117 (Fig. 22), easily obtained from 4-hydroxyacetophenones through a double Mannich reaction, inhibited the swelling of rat paw in various degrees when administered orally in doses equimolar to 20 mg/kg of indomethacin [145]; compared to indomethacin, one candidate 117 (R = 4-F) was more efficient, while another candidate 117 (R = 4-OCH3) was equipotent. Chalcone analogues 259 (R = 4-OCH3 or 4-CH3) derived from these benzoxazines (Fig. 48) had a more pronounced anti-inflammatory activity compared to candidates 117, and all of them displayed 70–90% of the efficiency of reference drug indomethacin [403]. Phenolic Mannich bases of naturally-occurring benzopyranones have also been tested as anti-inflammatory agents. Most Mannich bases 260 derived from 7-hydroxycoumarin (Fig. 48) were superior to reference drug indomethacin at reducing rat paw edema induced by carrageenan, and two candidates 260 (NR2 = 4-morpholinyl, 1-piperazinyl) were 1.6 times more efficient than indomethacin at 10 μm without significant inhibition of COX-1 [404]. Irisolidone is an isoflavone which was isolated from Pueraria spp., and was found to exert its anti-inflammatory action through suppression of iNOS gene expression and pro-inflammatory cytokines in activated microglia [405]. Chemical modification of irisolidone by means of the Mannich reaction using primary aliphatic and aromatic amines as amine reagents led to candidates 261 (Fig. 48) with enhanced ability to inhibit nitric oxide production compared to parent irisolidone [406].
Fig. 48.
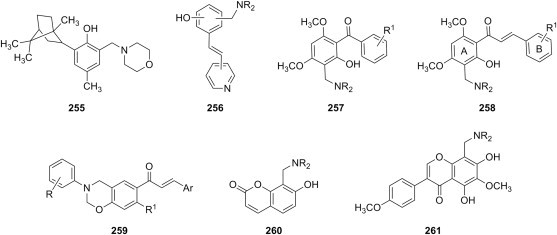
Phenolic Mannich bases and 1,3-oxazines as anti-inflammatory agents.
Anti-inflammatory activity of Mannich bases of 2,3-dihydro-1,2,4-triazole-3-thiones has also been investigated. Several candidates 133f (Table 1) have been evaluated using carrageenan-induced rat paw edema model, and the activity of one of them (133f, R2 = 2-CH3C6H4, NR2 = 4-morpholinyl) was comparable to that of reference drug ibuprofen [171]. Mannich bases 133i with secondary aliphatic amino moieties (dimethylamino, diethylamino, 1-pyrrolidinyl) in the aminomethyl function were as efficient as reference drug celecoxib in reducing rat paw edema both after 1 h and after 2 h, and they also had lower IC50 values (around 1 nM) for the inhibition of COX-2 compared to celecoxib (1.9 nM) [407]. As far as 5-substituted 2-aminomethyl-4-arylideneamino-2H-2,3-dihydro-1,2,4-triazole-3-thiones 134 (Table 1) are concerned, the majority of Mannich bases 134c had, upon administration of either 20 mg/kg or 40 mg/kg, a lower anti-inflammatory activity in carrageenan-induced rat paw edema model than reference drug indomethacin (5 mg/kg), although one candidate (134c, R2 = 2,6-Cl2C6H3, NR2 = 4-phenylpiperazin-1-yl) inhibited edema formation at the higher dose almost as efficiently as indomethacin [182]. Starting from ibuprofen, two separate studies reported the synthesis and anti-inflammatory activity of Mannich bases 262 having different arylideneamino moieties at position 4 of the 2,3-dihydro-1,2,4-triazole-3-thione scaffold (Fig. 49 ). The most potent compounds in this series were those having either a 4-morpholinyl or a 4-methylpiperazin-1-yl residue in the aminomethyl function. When common 4-substituted benzaldehydes (R2 = 4-ClC6H4, 4-CH3C6H4, 4-BrC6H4, 4-NO2C6H4) were used to generate the azomethine moiety, several candidates 262 had anti-inflammatory activity comparable with that of reference drug diclofenac, and were generally more efficient than ibuprofen at every time interval up to 3 h [408]. Also, the most potent compounds in this series were those having either a 4-morpholinyl or a 4-methylpiperazin-1-yl residue in the aminomethyl function. On the other hand, when 3-aryl-4-formylsydnones were used to generate the azomethine moiety, the resulting Mannich bases 262 were consistently more potent than the analogues in the previously mentioned series of compounds, but all of these candidates were less efficient than reference drug indomethacin in reducing rat paw edema [409]. Several Mannich bases 134h showed a peak in their anti-inflammatory activity at 1 h post-injection (from 50% to 130% edema inhibition compared to indomethacin), whereas the anti-inflammatory activity of other candidates 134h peaked at 4 h post-injection (from 84% to 100% edema inhibition compared to indomethacin) [186]. All of the Mannich bases 263 (Fig. 49) had only moderate anti-inflammatory activity, with the most potent compound in the series showing approximately 80% of the efficiency of reference drug indomethacin [410]. A few Mannich bases 264 and other structurally related aminomethylated 2,3-dihydro-1,2,4-triazole-3-thiones (Fig. 49) reduced significantly the inflammatory response (maximum inhibition between 30 and 59%) compared to reference drug diclofenac (inhibition between 42 and 63%), and some of them presented fast onset of their anti-inflammatory action, while others had a long-lasting anti-inflammatory effect [411].
Fig. 49.
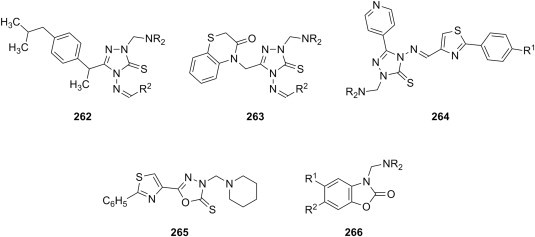
Anti-inflammatory Mannich bases of 2,3-dihydro-1,2,4-triazole-3-thiones, 2,3-dihydro-1,3,4-oxadiazole-2-thiones and 2-benzoxazolinones.
The anti-inflammatory activity of Mannich bases of 2,3-dihydro-1,3,4-oxadiazole-2-thiones has been scarcely examined. Several Mannich bases 139a (Table 2), especially those derived from ibuprofen as starting material for the 1,3,4-oxadiazole-2-thione substrate and generated from 4-arylpiperazines as amine reagents in the aminomethylation step, were as efficient as reference drug diclofenac at reducing rat paw edema [193]. Also, Mannich base 265 (Fig. 49) had anti-inflammatory activity comparable to that of reference drug indomethacin in carrageenan-induced rat paw edema test [412].
Several papers published by Gökhan's group have reported the anti-inflammatory activity of Mannich bases of 2-benzoxazolinones. One of their studies showed that candidates 266 (R1 = CH3, R2 = 2- or 4-ClC6H4CO) having an acyl moiety at position 6 of the benzoxazolidinone scaffold were more potent than their analogues 266 (R1 = 2- or 4-ClC6H4CO, R2 = H) with the same acyl group at position 5 (Fig. 49); in addition, the anti-inflammatory activity of two of these candidates was equipotent to that of reference drug indomethacin [413]. Substitution with fluorine in the phenyl ring of the acyl moiety appears to be less favorable for the anti-inflammatory activity of candidates 266 (R1 = H, R2 = difluorobenzoyl) [414]. Mannich bases 266 (R1 = CH3, R2 = H) having 4-arylpiperazine residues in the aminomethyl function were all less potent than reference drug indomethacin in carrageenan-induced rat paw edema test, but the results suggest that the nature of the substituent in the aryl group of piperazines plays an important role in the anti-inflammatory activity of these compounds [415]. A few Mannich bases 266 (R1 = NO2, R2 = H) had an anti-inflammatory effect comparable to that of reference drug indomethacin 3 h after administration, but their efficiency in reducing the swelling reached a plateau afterwards, whereas that of indomethacin continued to increase in time [416].
Two studies have reported the anti-inflammatory activity of Mannich bases of isatins. Aminomethylation of derivatives of 5-methylisatin thiosemicarbazone afforded Mannich bases 267 (R1 = R2 = H or CH3, NR2 = secondary aliphatic amines) (Fig. 50 ), which showed moderate anti-inflammatory activity (18–44% inhibition of edema at a dose of 100 mg/kg) compared to reference drug diclofenac (65% inhibition of edema at a dose of 45 mg/kg) [417]. Several Mannich bases 268 (Fig. 50), which were obtained from a derivative of isatin hydrazone, had anti-inflammatory activity comparable to that of reference drug diclofenac, and the highest level of activity was observed after 2 h [418]. A correlation in this series between anti-inflammatory activity and the nature of the amino moiety in the aminomethyl function was noticed, as the activity decreased with the increasing lipophilicity of the amino group.
Fig. 50.
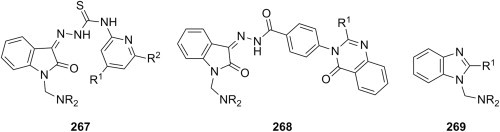
Mannich bases with anti-inflammatory activity derived from isatin derivatives and benzimidazoles.
A series of N-Mannich bases 269 of benzimidazole derivatives (Fig. 50) were evaluated as anti-inflammatory agents using formalin-induced paw edema method. Compared with reference drug diclofenac (50 mg/kg), they all caused significant reduction of paw edema, albeit at different doses (200 mg/kg for Mannich base 269 (R1 = H or CH3) and 40 mg/kg for Mannich bases 269 of 2-styrylbenzimidazole) [419]. Also, several Mannich bases 269 (R1 = C2H5), derived from both secondary aliphatic amines and primary arylamines, showed moderate anti-inflammatory activity 4 h after administration (33–57% of the activity of reference drug aspirin at the same dose of 100 mg/kg) [420].
Mannich bases obtained from substrates other than those mentioned so far have been examined as anti-inflammatory agents as well. Thus, N-Mannich bases 270 (NR2 = 1-pyrrolidinyl, 1-piperidinyl, 4-morpholinyl) of pyrimido[1,6-a]azepine derivatives (Fig. 51 ) had moderate activity (46–58% reduction of edema compared to that recorded for reference drug diclofenac) [421], whereas N-Mannich bases 271 of a tricyclic system derived from pyrimido[1,6-a]azepine (Fig. 51) were less potent (anti-inflammatory activity of approximately 40% of that of diclofenac) [422]. Two aminomethylated pyridazinones bearing a thiophene ring, namely compounds 272 and 273 (Fig. 51), showed 65% and 95% reduction of rat paw edema, respectively, compared to reference drug indomethacin [423]. Even when administered in a higher doses relative to that of reference drug, none of the Mannich bases 274 (R = H, CH3, OCH3, Cl, X = O or CH2) derived from isoxazolines (Fig. 51) was as efficient as indomethacin in reducing rat paw edema [424]. Out of the two acetylenic Mannich bases 275 (X = O or CH2) with a betulonic acid scaffold (Fig. 51) that were investigated as anti-inflammatory agents, only the piperidine derivative was almost as efficient as reference drug indomethacin in reducing rat paw edema; the morpholine analogue had only half of the activity of the piperidine Mannich base [425].
Fig. 51.
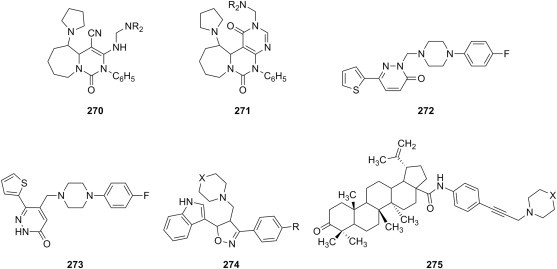
Anti-inflammatory Mannich bases derived from miscellaneous substrates.
10. Analgesic activity
As pain is a symptom of inflammation, the anti-inflammatory and analgesic activities of novel candidates are usually evaluated at the same time. Therefore, it is not surprising that many of the studies that report the anti-inflammatory activity of Mannich bases also offer information on their analgesic potential.
Several ketonic Mannich bases 252 (R1 = H, CH3, SO2CH3) derived from 2-arylidenecyclohexanones and aromatic amines (Fig. 47) were equipotent to reference drug ibuprofen in both acetic acid-induced writhing test and hot plate test [392]. One of the most potent candidate was compound 252 (R = R1 = H) derived from p-toluidine, but a few Mannich bases derived from 2-(4-methylsulfonylbenzylidene)cyclohexanone were also efficient as pain relievers.
Phenolic Mannich bases of 1- and 2-naphthols substituted in either rings of naphthalene system with various functions, which were synthesized using preformed aminomethylation reagents (e.g., imonium salts obtained from aromatic aldehydes and secondary aliphatic amines), were claimed to have analgesic activity [426]. The claim is difficult to assess, because only two candidates have been evaluated, and no comparison with established analgesics was provided. In addition, while candidate 276 (X = CH2) (Fig. 52 ) was efficient (92% inhibition of the writhing reaction), the second candidate 276 (X = O) offered only modest protection against pain (30% inhibition of the writhing reaction).
Fig. 52.

Analgesic Mannich bases derived from 2-naphthol and 2H-4,6-dimethyl-3-oxo-2,3-dihydroisothiazolo[5,4-b]pyridine.
Mannich bases of 2,3-dihydro-1,2,4-triazole-3-thiones with analgesic activity have been reported in several studies. Candidates 133f (R2 = 2-CH3C6H4, NR2 = 4-morpholinyl; R2 = 4-CH3OC6H4, NR2 = 4-methylpiperazinyl) (Table 1), which had good anti-inflammatory activity, were also tested for analgesic activity; their efficiency, determined using tail flick method in albino rats, was comparable to that of reference drug ibuprofen [171]. Several Mannich bases 262 (Fig. 49) showed analgesic activity (tail flick latency between 6.8 and 7.1 s) that was comparable with that of reference drug pentazocine (tail flick latency of 7.45 s), while the rest of the compounds were moderately active [409]. Mannich bases 263 (Fig. 49) were less efficient in the tail flick test, as the reaction time for the most potent compound in the series was approximately 80% of the latency provided by reference drug pentazocine [410]. Two candidates 139a (Table 2), both derived from ibuprofen as starting material for the 2,3-dihydro-1,3,4-oxadiazole-2-thione substrate and generated using either ethyl piperidine-4-carboxylate or 4-(4-fluorophenyl)piperazine as amine reagents in the aminomethylation step, were more efficient analgesics than reference drug diclofenac in hot plate test [193], while Mannich base 265 (Fig. 49) was equipotent to reference drug diclofenac in acetic acid-induced writhing test [412].
Analgesic activity of Mannich bases of 2-benzoxazolinones was also determined for the same candidates that were investigated for anti-inflammatory activity. In the library of Mannich bases 266 derived from 2-benzoxazolinones (Fig. 49) carrying a benzoyl moiety in the aromatic ring (R1 or R2 = substituted benzoyl), no significant difference in analgesic activity between the 5-benzoylated and the 6-benzoylated analogues could be observed [413]. Although most candidates provided moderate to low protection in either tests used to determine their analgesic activity (namely acetic acid-induced writhing test and p-benzoquinone-induced abdominal constriction test), two Mannich bases derived from 6-(substituted benzoyl)-2-benzoxazolinones were found to be as efficient as reference drug aspirin. Thus, analgesic activity of Mannich base 266 (R1 = H, R2 = 2,6-F2C6H3CO, NR2 = 4-(4-acetylphenyl)piperazin-1-yl) was equipotent to that of aspirin [414], while analgesic activity of Mannich base 266 (R1 = CH3, R2 = 4-ClC6H3CO, NR2 = 4-(4-fluorophenyl)piperazin-1-yl) was slightly poorer than that of aspirin [413]. In the series of Mannich bases 266 derived from 5-methyl-2-benzoxazolinone, several compounds showed better analgesic activity compared to reference drug aspirin, and the analgesic activity for all the compounds was consistently higher than their anti-inflammatory activity, suggesting that these Mannich bases might exert their analgesic activity centrally [415]. Two Mannich bases 266 derived from 5-nitro-2-benzoxazolinone were also found to possess analgesic activity comparable to that of aspirin [416].
Mannich bases 267 of derivatives of 5-methylisatin thiosemicarbazone (Fig. 50) were moderately efficient (16–53% protection) in preventing acetic acid-induced writhing in mice at a dose of 100 mg/kg, while reference drug diclofenac provided 74% protection in the same test at a dose of only 45 mg/kg [417]. Mannich bases 268 of isatin hydrazone (Fig. 50) carrying a quinazoline moiety and derived from acyclic secondary aliphatic amines (dimethylamine, diethylamine) were the most potent; their analgesic effect was superior or comparable to that of reference drug diclofenac 2 h after administration, but it decreased rapidly afterwards [418].
N-Mannich bases 269 (Fig. 50) obtained from benzimidazole using dimethylamine or diethylamine as amine reagents were found to be moderate analgesics at a dose of 200 mg/kg relative to paracetamol (100 mg/kg), while Mannich base 269 derived from 2-methylbenzimidazole as substrate and diphenylamine as amine reagent was a poor analgesic candidate [419]. Owing to their toxicity, Mannich bases derived from 2-styrylbenzimidazole were administered at a lower dose (20 and 40 mg/kg), and their analgesic activity ranged from moderate (NR2 = 4-morpholinyl) to very good (NR2 = diethylamino or 1-piperidinyl) when compared to paracetamol [419]. N-Mannich bases 269 of 2-ethylbenzimidazole had analgesic activity comparable to that of reference drug pentazocine only when administered in doses that were 25 times greater than that of pentazocine [420]. Other Mannich bases 269 derived from 2-substituted benzimidazoles (R1 = CH2NHNHC6H5 or 2-HOC6H4) also showed moderate to good analgesic activity in acetic-acid-induced writhing test (62–84% of the analgesic activity of diclofenac at the same dose) [427].
Evaluation of a first series of N-Mannich bases 277 derived from 2H-4,6-dimethyl-3-oxo-2,3-dihydroisothiazolo[5,4-b]pyridine (Fig. 52) led to identification of weak to moderate analgesic agents [428]. Amongst them, candidates derived from 4-aryl- or 4-benzylpiperidine and those having a 4-(2-substituted phenyl)piperazine as the amine moiety were the most efficient analgesics in writhing and hot plate tests. A second series of Mannich bases 277 was subsequently synthesized, and some of the candidates displayed significant activity in the writhing test, with analgesic activity 2 to 10 times more potent than that of aspirin and 1.5 to 10 times weaker than that of morphine, used as reference drugs in the study [429].
11. Antioxidant activity
Excess of reactive oxygen species produced in living organisms can cause oxidative stress and damage cells by initiating chain reactions that lead to lipid peroxidation, DNA damage or protein oxidation. Besides the complex system of antioxidant metabolites and enzymes that naturally prevent cell damage, exogenous antioxidants may sometimes be required to keep reactive oxygen species at an optimum level. Use of the Mannich reaction to generate novel chemical entities capable of acting as antioxidants is presented in this section.
Ketonic Mannich bases 109 and 110 (Fig. 21), which were obtained from arylamines as amine reagents and 3-acetylcoumarine and 2-acetylbenzofuran as substrates, respectively, were tested for antioxidant activity by evaluating their ability to scavenge 1,1-diphenyl-picrylhydrazyl (DPPH) radical [138]. The most efficient candidates in each series, namely 109 (R = H) and 110 (R = N(CH3)2), showed moderate potency in scavenging DPPH radical (approximately 65%) compared to standard butylated hydroxytoluene (90%). Substitution of the aromatic ring with electron-withdrawing groups appears to reduce the antioxidant ability of these Mannich bases.
A large number of phenolic Mannich bases have been reported in the literature as potential antioxidants. Antioxidant activity of two phenolic Mannich bases 278 derived from thymol (Fig. 53 ) has been assessed by means of xanthine oxidase inhibition test for the cell-free system, and by inhibition of lipid peroxidation using rat-liver homogenate [430]. Both candidates, and especially Mannich base 278 having morpholine as amine moiety, presented enhanced antioxidant activity in both tests compared to parent thymol. Three phenolic Mannich bases 114 (NR1R2 = 4-CH3OC6H4NH, 1-piperidinyl and 4-morpholinyl) derived from 4,6-diacetylresorcinol (Fig. 22), designed primarily as anti-inflammatory agents, were also evaluated for their ability to inhibit lipid peroxidation; results showed that these candidates were more efficient than indomethacin at preventing lipid peroxidation, and that the antioxidant activity may be correlated with their ulcerogenic activity [398]. Only three Mannich bases 257 (Fig. 48), all of them having piperidine as the amine moiety (NR2 = 1-piperidinyl), showed moderate to high ability in scavenging DPPH radical (49–74% of DPPH scavenging ability of standard gallic acid), while the rest of the candidates were either poor scavengers of DPPH radical, or failed to react with DPPH radical at all [399]. Although all of the phenolic Mannich bases 197 showed efficiency as antioxidants in various degrees, candidates 197 (NR2 = diphenylamino, 4-morpholinyl, 1-piperazinyl) (Fig. 36) were the most potent scavengers of hydrogen peroxide, their activity being comparable to that of standard ascorbic acid, but inferior to that of standard butylated hydroxyanisole [302]. Selected tacrine–8-hydroxyquinoline hybrids 279 (R1 = R2 = H, n = 7; R1 = CH3, R2 = H, n = 6; R1 = H, R2 = Cl, n = 7) (Fig. 53), that were developed primarily as agents for the treatment of Alzheimer's disease, exhibited also potent peroxyl radical absorbance capacities (2.6–4.7 Trolox equivalents/μmol of Mannich base), as determined by means of ORAC-FL method (oxygen radical absorbance capacity by fluorescence) [431].
Fig. 53.
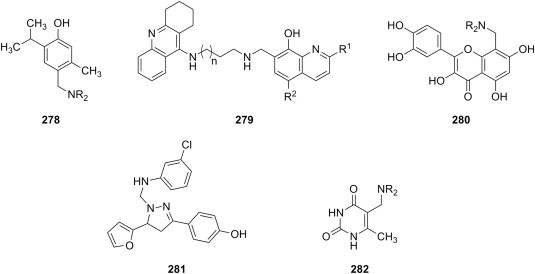
Antioxidant Mannich bases generated from various substrates.
Phenolic Mannich bases of natural flavanones have been designed and synthesized with the view to improve antioxidant efficiency, bioavailability and water solubility of parent flavanones. Only three Mannich bases 41 (R = OH, R1 = H, R2 = 1-pyrrolidinyl, diethylamino or diisopropylamino) of apigenin (Fig. 7) exhibited antioxidant activity greater than that of apigenin, while the rest of candidates 41 presented antioxidant activity comparable to that of apigenin [58]. Antioxidant activity of these apigenin derivatives was concentration-dependent, and DPPH radical scavenging ability of the most potent Mannich bases 41 was almost the same as that of the standard ascorbic acid at a concentration of 1.25 mg/mL. Scutellarein was also aminomethylated to give Mannich bases 41 (R = R1 = OH, R2 = aminomethyl), which had the ability to scavenge 50% of DPPH radical in the sample at concentrations ranging from 24 to 33 μM, but no comparison with a well-established antioxidant was provided in the study [432]. Mono- and bis-Mannich bases 280 (NR2 = 4-morpholinyl, 6,7-dihydroxyisoquinolin-2-yl, Z–CH2–quercetin, where Z = piperazine, N,N′-dimethylethylenediamine) of quercetin (Fig. 53) were synthesized and evaluated as inhibitors of photooxidation of A2E (a pigment of the lipofuscin of retinal pigment cells, thought to play a role in macular degeneration) [433]. These candidates showed sufficient antioxidant ability to inhibit noncellular and intracellular photooxidation of A2E, and were superior as antioxidants to quercetin itself. On the other hand, phenolic Mannich bases obtained from other known antioxidants such as sesamol or 1-(2-hydroxy-4-methoxyphenyl)-3-phenylprop-2-en-1-one were ineffective inhibitors of A2E photooxidation at 200 μM [433].
Phenolic Mannich bases of chalcone analogues such as candidates 258 (R1 = 2-F, 2-Cl, 2-Br, 3-F, 3-Cl, 3-Br) (Fig. 48) were modest to poor scavengers of DPPH radical (3–58% reduction of DPPH absorption) compared to standard quercetin (86% reduction of DPPH absorption) [400]. Results for DPPH scavenging ability of phenolic Mannich bases derived from other chalcone analogues obtained in a different study are also in line with the antioxidant data recorded for Mannich bases 258, as the greatest reduction of DPPH absorption was only 13% at concentrations as high as 1 mM of Mannich base [401]. However, one candidate in this series scavenged superoxide radical efficiently, while another candidate was a potent inhibitor of heme dependent lipid peroxidation [401].
Several Mannich bases 133g derived from 5-(2-thienyl)-2,3-dihydro-1,2,4-triazol-3-thiones (Table 1) were as efficient antioxidants as standard ascorbic acid (over 90% reduction of DPPH radical), and showed enhanced antioxidant activity over the parent 2,3-dihydro-1,2,4-triazole-3-thiones [172]. A series of Mannich bases 139 (R1 = 2,3-dihydrobenzo[b][1,4]dioxin-6-yl, NR2 = substituted primary arylamines) (Table 2) were designed as antioxidant agents, and were evaluated using scavenging DPPH radical assay, scavenging 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) radical cation (ABTS) assay, and ferric reducing antioxidant power assay [434]. More than half of these Mannich bases showed greater DPPH radical scavenging ability than butylated hydroxytoluene (IC50 = 44 μM), and seven of them exhibited greater antioxidant activity than Trolox (IC50 = 30 μM) in the same assay. A few candidates 139 with high DPPH scavenging activity, along with other Mannich bases 139, were also very good scavengers of ABTS radical cation (their activity was greater than that of Trolox). In ferric reducing antioxidant power assay, three Mannich bases 139 showed better activity than Trolox, and other seven were more potent than butylated hydroxytoluene. Mannich bases 139 (R1 = 2,3-dihydrobenzo[b][1,4]dioxin-6-yl, NR2 = 2,6-F2C6H4 or NR2 = 3,4,5-F3C6H3) were the only compounds to exhibit high potency in all three assays, and they were also more efficient than Trolox in inhibiting lipid peroxidation in mice liver microsomes homogenate [434].
N-Mannich bases 149 of succinimide (R1 = H or phenyl, NR2 = NHC6H5, 2-pyridinyl) (Fig. 27) and an N-Mannich base of phthalimide showed moderate DPPH scavenging activity compared to standards vitamin C and vitamin E, while their ability to inhibit peroxidation of linoleic acid was moderate to weak [215]. Also, several N-Mannich bases 166 of saccharin (Fig. 29) were antioxidants as potent as standard ascorbic acid in the ABTS assay, and more potent antioxidants than saccharin itself [237]. N-Mannich base 281 derived from a 3,5-disubstituted pyrazoline (Fig. 53) had DPPH scavenging and nitric oxide scavenging activities comparable to standard antioxidants ascorbic acid and rutin; the presence of the phenolic hydroxyl group could be responsible for the antioxidant activity of this compound [435]. Finally, antioxidant activity of two C-Mannich bases 282 of uracil (Fig. 53) was determined by means of measuring the rate of oxidation of 2-propanol [436]; addition of compounds 282 to the reaction mixture reduces the rate of oxidation to levels comparable to the rate of oxidation in the presence of butylated hydroxytoluene.
12. Agents for blood pressure regulation
A small number of studies deal with the investigation of Mannich bases as antihypertensive agents. Phenolic Mannich bases appear to be particularly remarkable as blood pressure-lowering substances. Several single and double Mannich bases of various phenols and having thiomorpholine as amine moiety showed a gradual effect on systolic, diastolic and mean arterial pressure, starting at a dose 0.1 mg/kg; in the case of reference drugs captopril, losartan and omapatrilat, the same effect was noticeable at 0.001 mg/kg [437]. In a manner similar to the aforementioned reference drugs, these phenolic Mannich bases also induced a gradual (but significant) reduction of heart rate in anesthetized mice. Double Mannich base 283 (Fig. 54 ) emerged as a valuable candidate, with one of the lowest effective dosage in this collection, while exhibiting the highest antihypertensive activity in conscious spontaneous hypertensive rat model [437]. The same researchers examined the antihypertensive effect of double Mannich base 284 with a 1,4-dihydropyridine moiety in its structure (Fig. 54), but the results showed that the activity of this candidate was inferior to that of double Mannich base 283, presumably due to the change in bulkiness of the substituent para to the phenolic hydroxyl [438]. The presence of two aminomethyl functions in the structure of 284 may have been essential in preserving the antihypertensive activity of this candidate to a reasonable level, while replacement of thiomorpholine in 283 with morpholine in 284 could be also the cause for the poorer antihypertensive properties of candidate 284 relative to those of Mannich base 283.
Fig. 54.
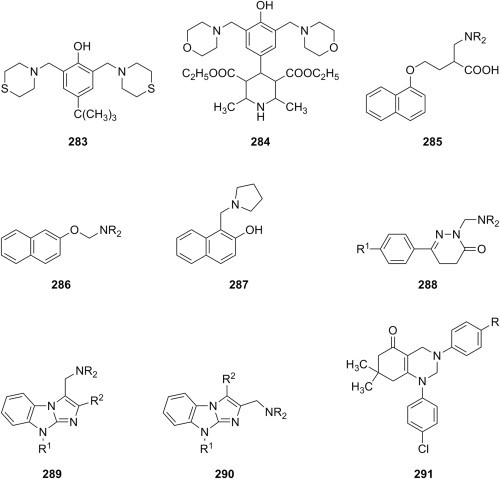
Mannich bases as agents for blood pressure regulation.
In an attempt to synthesize aminomethylated 4-(naphthyloxy)butanoic acids (e.g., compound 285, Fig. 54) as analogues of propranolol, Tandon et al. claim to have obtained O-Mannich bases 286 of 2-naphthol (Fig. 54) instead of the desired compound in the case of 4-(naphthalen-2-yloxy)butanoic acid; however, amino acids 285 were obtained when 4-(naphthalen-1-yloxy)butanoic acid was subjected to aminomethylation [439]. At a concentration of 5 mg/kg, candidate 285 (NR2 = 1-piperidinyl) lowered blood pressure in anesthetized cats similarly to propranolol, while compound 286 (NR2 = 4-phenylpiperazin-1-yl) was superior to propanolol at the same dose (fall of blood pressure of 60 mm Hg, duration of the effect of 120 min). It is also worth mentioning that the antihypertensive effect of a C-Mannich base of 2-naphthol, namely 1-pyrrolidinylmethyl-2-naphthol 287 (Fig. 54), was reported in an earlier publication [440].
A large collection of Mannich bases 288 (R1 = H, CH3, C2H5, CH2CH(CH3)2, C6H5, OCH3, Cl) (Fig. 54) was evaluated for antihypertensive activity by the non-invasive tail cuff method, and several candidates were found to significantly reduce mean arterial blood pressure, albeit at higher doses than reference drug hydralazine [441]. The presence of alkyl groups as substituent R1 seems to be favorable for the antihypertensive activity, whereas the nature of the amine moiety does not appear to influence the activity.
Several Mannich bases 289 (R1 = CH3, C2H5, n-C4H9, CH2C6H5; R2 = CH3, C6H5) of imidazo[1,2-a]benzimidazoles (Fig. 54) were found to reduce arterial blood pressure in anesthetized rats with 20% at doses that were lower than the dose at which reference drug dibazole produces the same effect (22 mg/kg) [442]. Analogous aminomethylated imidazo[1,2-a]benzimidazoles 290 (R1 = CH3, n-C4H9, CH2C6H5) (Fig. 54) were more efficient antihypertensive agents than Mannich bases 289, as they reduce arterial blood pressure with 20% at doses lower than those recorded for candidates 289.
Several octahydroquinazoline derivatives 291 (R = H, 2-F, 4-F, 4-Cl, 4-Br, 4-CH3, 4-OH) (Fig. 54) were obtained through a double Mannich reaction starting from 3-(4-chlorophenylamino)-5,5-dimethyl-2-cyclohexenone and arylamines, and were shown to produce insignificant changes in both arterial blood pressure and heart rate at a dose of 5 mg/kg [443]. However, derivatives 291 (R = OCH2CONHN CHC6H4R1, R1 = H, 4-CH3, 4-NO2) of a candidate in the initial series afforded significant decreases in both systolic and diastolic arterial blood pressure, with rapid onset of action (5 min) and minor decrease of heart rate in anesthetized male adult albino rats [443]. On the other hand, candidate 291 (R = 4-Cl) produced a time-dependent significant increase in both systolic and diastolic arterial blood pressure, without causing tachycardia for 30 min, which makes this compound useful for treatment of hypotension.
13. Antiparasitic activity
A small number of studies report the activity of Mannich bases against parasites other than Plasmodium spp. Among these parasites, which are the cause of parasitic infections especially in developing countries, different species from the Trypanosomatidae family are pathogenic to humans and cause African trypanosomiasis (sleeping sickness, Trypanosoma brucei), American trypanosomiasis (Chagas' disease, Trypanosoma cruzi) or leishmaniasis (Leishmania spp.). In addition, one study investigated the activity of Mannich bases against Entamoeba histolytica, and one study reported the anti-Schistosoma activity of Mannich bases derived from praziquantel.
All parasitic protozoa belonging to Trypanosoma spp. have a unique thiol metabolism based on the flavoenzyme trypanothione reductase. This enzyme could be therefore considered a promising target for rational drug design against African sleeping sickness, Chagas' disease, and different forms of leishmaniasis, owing to its absence of in the mammalian host, the structural differences to related host enzymes, and its essential role for parasite survival. Because unsaturated ketonic Mannich bases react readily with thiols, and because several such compounds were shown to be efficient mechanism-based inhibitors of P. falciparum thioredoxin reductase [338], a study concerning the ability of these compounds to interact with both trypanothione reductase and free trypanothione was undertaken [444]. Candidates 292 (NR2 = dimethylamino, 1-piperidinyl, 4-morpholinyl) (Fig. 55 ) inactivated trypanothione reductase, but only in the presence of NADPH, suggesting that reduction of this enzyme prior to its interaction with the Mannich base is essential. The divinyl ketone arising from the deamination of candidates 292 is the actual inhibitor, and its activity against trypanothione reductase was higher than that of parent Mannich bases; unfortunately, this divinyl ketone was also too reactive to be considered a drug candidate. Mannich bases 292 displayed only modest activity against all strains of intracellular parasites, which may be explained by reaction with glutathione present in millimolar concentrations in the cytosol of the mammalian host cells. Nonetheless, they showed a significant effect against extracellular T. b. rhodesiense, which might (at least partially) be due to their high reactivity toward trypanothione reductase and trypanothione. With trypanothione reductase validated as a drug target in trypanosomiasis, design and synthesis of novel unsaturated Mannich bases based on melaminophenyl arsenical drug melarsoprol was subsequently pursued [445]. Candidates 293 (R1 = Cl, H, CH3) (Fig. 55) were efficient in vitro trypanothione reductase inhibitors, and while they did not display any significant activity in cell-based assays against T. cruzi, they were active towards T. brucei. The presence of the melamine residue para to the enone motif did not result in any improvement in the trypanocidal potency of 293 (R1 = Cl) compared to hit compounds 292. However, the presence of the melamine residue meta to the enone motif significantly lowered the IC50 values of Mannich bases 293 against the human cell line used in the study, which is indicative of these compounds' high cytotoxicity. On the other hand, phenolic Mannich base 294 (Fig. 55) was basically inactive towards all parasites. Mannich bases 293 were taken up effectively into cells despite the absence of P2 and HAPT1 carriers, which suggests that the main route of entry for these compounds was not through aminopurine transporters.
Fig. 55.
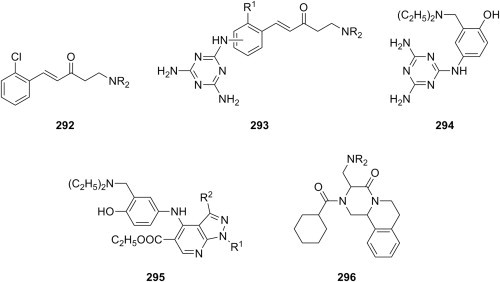
Antiparasitic Mannich bases.
Stemming from the observation that heterocyclic pyrazolopyridine ring system can be considered analogous to quinoline, and because aminoquinoline derivatives are efficient antimalarials, several derivatives featuring the pyrazolopyridine scaffold were designed and tested against Leishmania amazonensis, in the evolutive form of promastigotes [446]. Thus, both Mannich bases 295 (R1 = C6H5, R2 = CH3, C6H5) (Fig. 55) are active antileishmanial agents with IC50 values of 390 and 120 nM, whereas aminoquinoline derivative amodiaquine had IC50 = 890 nM. From the structure–activity point of view, the results suggest that pyrazolopyridine ring system is a bioisostere of quinoline, and that the presence of the carbethoxy group in the structure of candidates 295 does not influence significantly the biological activity.
Amebiasis is a contagious disease of the human gastrointestinal tract that is caused by parasitic protozoa E. histolytica. All the candidates in a small library of piperazine Mannich bases 139d of 5-(4-pyridinyl)-2,3-dihydro-1,3,4-oxadiazole-2-thione (Table 2) were active against HM1:IMSS strain of E. histolytica, but only three of them were more potent than reference drug metronidazole (IC50 = 1.81 μM) [447]. The amine residues in the aminomethyl function of these active Mannich bases were 4-ethylpiperazin-1-yl (IC50 = 327 nM), 4-(4-fluorophenyl)piperazin-1-yl (IC50 = 245 nM), and 4-(2-methoxyphenyl)piperazin-1-yl (IC50 = 1.06 μM). In addition, cytotoxicity of these antiamoebic candidates was low (in the concentration range of 2.5–250 μM).
Out of four newly synthesized Mannich bases 296 of praziquantel (Fig. 55), two candidates (NR2 = N(C3H7-n)2 and NHCH2CH2OH) exhibited significant in vitro anti-Schistosoma activity (100% worm killing at 40 μM and 30 μM, respectively), but they were not as efficient as praziquantel itself (100% worm killing at 10 μM) [448].
14. Inhibitors of platelet aggregation
Platelets are crucial for hemostasis, as they connect to one another through receptor bridges, form aggregates, and finally plug the tear in the interrupted endothelium. However, the aggregation of platelets leading to formation of clots can also be triggered by irregularities on the vessel wall, resulting in abnormal clot formation, which is the primary factor in the development of thrombotic disorders such as unstable angina, myocardial infarction, stroke and peripheral vascular diseases, especially when it occurs in the coronary artery. Platelet aggregation is also initiated by endogenous substances, such as collagen, thrombin, prostaglandin endoperoxides, thromboxanes, arachidonic acid, adenosine diphosphate (ADP), etc. Inhibition of platelet aggregation represents a promising strategy for the treatment of thrombotic diseases, and several studies have reported the activity of Mannich bases as inhibitors of platelet aggregation.
Several types of phenolic Mannich bases represented by structures 24–32 (Fig. 6) were also evaluated as inhibitors of platelet aggregation induced by ADP or collagen at concentrations of 20 μM or 10 μg/mL, respectively [449]. Numerous candidates having various structures showed good inhibitory effects and were more potent than reference drug clopidogrel, whereas nine compounds in this collection exhibited inhibitory activity in the range of 90–100% in both models. Good platelet aggregation inhibitory activity was associated with the presence of a pyridyl moiety as ring B in the structure of chalcone analogues, and also by the presence in ring A of a hydroxy group meta to the carbonyl function.
Phenolic Mannich bases 41 (R = R1 = OH, R2 = aminomethyl) of scutellarein (Fig. 7) were investigated as thrombin inhibitors, and their influence on several parameters such as prothrombin time, activated partial thromboplastin time, thrombin time and fibrinogen was determined [432]. All of these candidates showed greater thrombin inhibitory activity compared to parent scutellarein. Mannich base derived from scutellarein and containing a morpholinyl residue in the aminomethyl function was the most potent of all, and was subsequently selected for molecular docking experiments with thrombin. These experiments revealed that the morpholinylmethyl group occupies deep pocket S3 of the thrombin binding site, whereas the scutellarein part of the inhibitor's molecule is anchored by three hydrogen bonds within the active site.
The effect of Mannich bases 289 (R1 = CH3, C2H5, n-C4H9, CH2C6H5; R2 = CH3, C6H5) and 290 (R1 = CH3, n-C4H9, CH2C6H5; R2 = H, COOC2H5) of imidazo[1,2-a]benzimidazoles (Fig. 54) on ADP-induced aggregation of rabbit thrombocytes was studied in vitro [442]. Several candidates 289 and most candidates 290 were found to have lower effective concentrations (EC50) at which they decrease the degree of aggregation by half than reference drug acetylsalicylic acid.
Cellular effects of thrombin, including platelet aggregation, are mediated through the activation of thrombin receptor PAR-1 belonging to the family of four G-protein-coupled receptors called protease-activated receptors. PAR-1 has become an attractive drug discovery target, and peptide-mimetics or small organic molecules with PAR-1 antagonist properties have already been designed and synthesized. An indole Mannich base motif has been incorporated in the structure of a series of novel peptide-mimetics 297 (R = 4-CH3OC6H4CH2, 3,4-F2C6H3CH2, naphthalen-1-ylmethyl, naphthalen-2-ylmethyl) (Fig. 56 ) capable to bind to PAR-1 [450]. The ability of these candidates to inhibit PAR-1-induced platelet aggregation has been tested by measuring the degree of aggregation of human platelets, and also by establishing the degree of inhibition of contraction of aortic rings. Compounds 297 (R = 4-CH3OC6H4CH2, 3,4-F2C6H3CH2) belonging to the series of 6-aminoindole derivatives were the most potent candidates, with values of inhibitory concentration that were about 5 times lower than reference compound RWJ54003 for 75% inhibition of platelet aggregation. In addition, these results were confirmed by the enhanced inhibitory activity of these two candidates on the contraction of aorta rings when compared to the activity of RWJ54003, an effect that became more evident with the increase of PAR-1 dose. Candidates in the series of 5-aminoindole derivatives were found inactive or weak inhibitors in both assays.
Fig. 56.
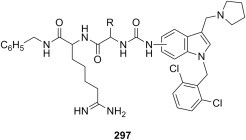
Inhibitor of PAR-1-induced platelet aggregation 289.
15. Anti-ulcer activity
Anti-ulcer agents are a class of drugs used to treat ulcers in the stomach and the upper part of the small intestine. Acid peptic disease is a chronic pathology that affects millions of people worldwide, and it has been estimated that 10% of the world population will develop this condition in their lifetime [451]. An imbalance between aggressive factors (such as gastric acid, Helicobacter pylori infection, excessive intake of anti-inflammatory drugs, immoderate consumption of alcohol, high concentrations of reactive oxygen species) on one hand, and protective factors (e.g., mucus, bicarbonate anion, prostaglandins, good blood flow, efficient cellular repair, endogenous and exogenous antioxidants) on the other hand is considered to be the cause of ulcers. Treatment of ulcers and their symptoms relies on pain relievers, antiacids and cytoprotective agents to allow the healing of ulcers, and agents to prevent the recurrence of ulcers, such as proton pump inhibitors, muscarinic antagonist pirenzepine or H2 receptor antagonist cimetidine.
The ability of Mannich bases to act as anti-ulcer agents has been reported in a small number of studies. Anti-ulcer activity of S-aminomethyl derivatives 170 generated from 4,6-diaryl-2-mercaptopyrimidines (Fig. 29) as substrates and secondary aliphatic amines as amine reagents in the Mannich reaction was evaluated in vivo using aspirin-induced ulcer model in albino rats [241]. Based on the ulcer score, the mean ulcer index and the degree of protection were calculated and compared to those of reference drug omeprazole. Five candidates 170 (R1 = 4-ClC6H4, R1 = 3-NO2C6H4 or 4-CH3OC6H4) offered more than 50% protection, while other five Mannich bases 170 (R1 = 4-ClC6H4, R1 = 4-ClC6H4, 3-NO2C6H4 or 4-(H3C)2NC6H4) had only moderate anti-ulcer activity (approximately 30% protection) compared to omeprazole (99% protection).
Aminomethylation of 1,4-dihydropyrimidines with sulfanilamide as amine reagent afforded Mannich bases 298 (Fig. 57 ), whose ability to reduce the volume of acid secretion was determined by pyloric ligation method [452]. Three candidates 298 (R = 4-CH3OC6H4, 3-CH3O-4-HOC6H3, 2-furyl) presented good anti-ulcer activity (ulcer indices in the range of 0.18–0.35 at a dose of 10 mg/kg), while reference drug omeprazole had an ulcer index of 0.08 at a dose of 1 mg/kg.
Fig. 57.
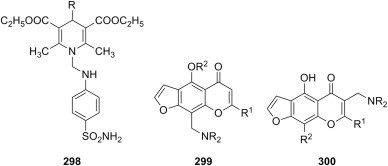
Various Mannich bases as antiulcer agents.
A small number of furo[3,2-g]flavones of type 299 (R1 = C6H5, 4-ClC6H4, 3-pyridinyl; R2 = H or CH3) and 300 (R1 = C6H5, 3-pyridinyl) (Fig. 57), aminomethylated either at position 6 or 9, have been evaluated as gastroprotective agents using the ethanol-induced gastric ulcer model in rats [453]. The mean values of the protection index for these Mannich bases were in the range of 20–44%, and no comparison with an anti-ulcer reference drug was provided in the study. The best three candidates 299 (R1 = 3-pyridinyl, R2 = CH3, NR2 = 4-methylpiperazin-1-yl), 300 (R1 = 3-pyridinyl, R2 = OCH3, NR2 = 4-methylpiperazin-1-yl) and 300 (R1 = C6H5, R2 = H, NR2 = 4-morpholinyl) have a methoxy group at position 4 as a common structural feature; the corresponding Mannich bases having a hydroxyl group instead of methoxy were significantly less active.
16. Agents for treatment of mental disorders
Mental disorders comprise a broad range of problems, with different symptoms, generally characterized by some combination of abnormal thoughts, emotions, behavior and relationships with others. Common neurological conditions labeled as mental disorders include depression and anxiety, while schizophrenia and bipolar disorder stand out as mental disorders that are severe and disabling. Untreated mental, neurological and substance use disorders exact a high toll, accounting for 13% of the total global burden of disease, while unipolar depressive disorder is the third leading cause of disease burden, accounting for 4.3% of the global burden of disease. Current predictions indicate that depression will be the leading cause of disease burden globally by 2030 [454]. A range of different types of treatment for mental disorders are available, and the most suitable treatment depends both on the disorder and on the individual. A major option for many mental disorders is psychotherapy, while another option is psychiatric medication. Amongst several groups of drugs that are currently employed in psychiatric medication, antidepressants treat clinical depression as well as anxiety and a range of other disorders, anxiolytics (including sedatives) are used for anxiety disorders and related problems such as insomnia, mood stabilizers are used primarily in bipolar disorder, and antipsychotics are used for psychotic disorders, notably for positive symptoms in schizophrenia.
Selective serotonin reuptake inhibitors (SSRIs) play an important role in pharmacotherapeutic treatment of depression. In an attempt to modify the general SSRI structural motif of γ-phenoxypropylamine, two ketonic Mannich bases 301 (R = Cl, Br) (Fig. 58 ) derived from 4-chloro- and 4-bromoacetophenone as substrates and 4-benzylpiperidine as amine reagent were synthesized, the carbonyl function in these amino ketones was subsequently reduced, and the resulting secondary hydroxyl was then converted into an ether group through reaction with 1-chloro-4-trifluoromethylbenzene [455]. Although the designed SSRI analogues targeted in this study showed no antidepressant activity, the intermediate ketone Mannich bases 301 were as effective as reference drugs fluoxetine, sertraline or imipramine at similar dosage in a validated experimental model of depression in mice such as the forced swimming test.
Fig. 58.
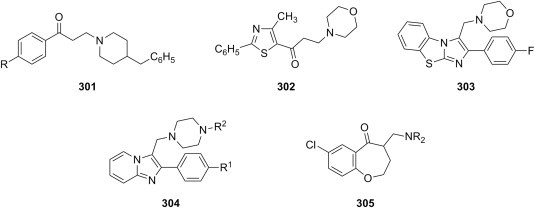
Mannich bases as agents for treatment of mental disorders.
An innovative computer-assisted approach based on the Prediction of Activity Spectra for Substances (PASS) has been applied for the discovery of new anxiolytics [456]. An initial database comprising 5494 structures was generated by virtual combinatorial design of highly diverse chemical compounds, including different types of heterocycles such as thiazoles, pyrazoles, isatins, fused imidazoles, with the view to increase the probability of finding new chemical entities as anxiolytics. Out of the eight hits obtained from this database, four candidates were Mannich bases. Ketonic Mannich base 302, Mannich base 303 derived from an imidazo[2,1-b]benzo[d]thiazole and two Mannich bases 304 (R1 = F, R2 = CH3; R1 = NO2, R2 = C6H5) derived from imidazo[1,2-a]pyridines (Fig. 58) were synthesized and tested as potential anxiolytics using the conflict situation test. All of the candidates showed an anxiolytic effect that was comparable or greater than that of reference drug medazepam. Mannich base 304 (R1 = NO2, R2 = C6H5) was the most potent anxiolytic in this study, being two times more potent than medazepam.
A collection of compounds sharing a benzoxepin scaffold as common structural feature was evaluated for their sedative–hypnotic effect using phenobarbital-induced sleep test [457]. Among these compounds, three ketonic Mannich bases 305 (NR2 = 1-piperidinyl, 4-methylpiperazin-1-yl, 4-morpholinyl) (Fig. 58) decreased the onset of phenobarbital-induced sleep and prolonged the duration of hypnosis when administered in a dose equimolar to that of reference drug phenobarbital. The hypnotic activity of candidates 305 (NR2 = 1-piperidinyl, 4-methylpiperazin-1-yl) was comparable to that of phenobarbital, while Mannich base 305 (NR2 = dimethylamino) exhibited only moderate activity, and Mannich base 305 (NR2 = 4-(2-chlorophenyl)piperazin-1-yl) was virtually inactive. Therefore, the nature of the amino residue in these Mannich bases seems to have a significant impact on their hypnotic activity.
17. Miscellaneous biological activities
In addition to biological activities of Mannich bases presented so far, isolated studies were found to report various other biological activities for Mannich bases. These singular results are covered in this section, without any attempt to arrange them in a systematic order.
Two phenolic Mannich bases derived from 2,4- and 2,6-di-t-butylphenol as substrates and dimethylamine as amine reagent were evaluated as hepatoprotective agents against experimental toxic hepatitis induced by tetrachloromethane. The degree of liver damage was evaluated in terms of alanine aminotransferase activity in blood serum and malonic dialdehyde content in liver homogenates. Even at a dose of 10% of the corresponding LD50, the ability of these two candidates to diminish the hepatotoxic action of tetrachloromethane was superior to that of reference compound emoxypine [458]. In addition, in an assay using the same tetrachloromethane-induced hepatitis model, two acetylenic Mannich bases 275 (X = O or CH2) with a betulonic acid scaffold (Fig. 51) were shown to decrease alkaline phosphatase activity and lower alanine aminotransferase and aspartate aminotransferase activities in blood serum compared to control [425].
Acetylenic Mannich base 306 (Fig. 59 ) was tested on guinea pig spontaneously beating atria with a view to evaluate its negative chronotropic activity, but the potency of this candidate was approximately four times lower than that of bradycardic agent zatebradine, and was therefore excluded from subsequent testing as blocker of hyperpolarization-activated current [459].
Fig. 59.
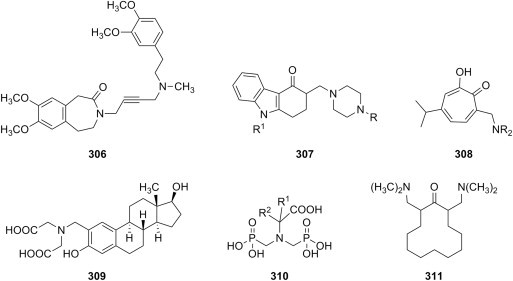
Mannich bases with various biological activities.
Ondansetron analogues 307 (R1 = CH3) having a piperazine moiety instead of imidazole (Fig. 59) were synthesized and evaluated as anti-emetic agents using a retching model [460]. All the candidates were effective to some extent when administered at a dose of 8 mg/kg, but only Mannich base 307 (R = 2-pyrimidinyl) had anti-emetic activity comparable to that of reference drug ondansetron at low dosage (2 mg/kg).
Several 7-aminomethylated β-thujaplicin analogues 308 (Fig. 59) were tested against oxidative stress-induced death of HT22 cells following exposure to glutamate [461]. Piperazine-containing Mannich bases 308 were more potent than parent β-thujaplicin in protecting glutamate-challenged HT22 cells, with EC50 values ranging from 0.08 to 1.7 μM. Because the most potent candidates were those having a chroman moiety as substituent at N4 in the piperazine residue, the high in vitro neuroprotective activity of these Mannich bases may be due to the potent antioxidant effect imparted by the chroman moiety. Analogous morpholine Mannich base 308 was also active in protecting HT22 cells from oxytosis, but it was less active than the piperazine-containing β-thujaplicin derivatives. The presence of the isopropyl group also seems to be important for the neuroprotective activity of these compounds, since an analogous Mannich base derived from tropolone was not active.
Estrogen deficiency after menopause is one of the most common causes of osteoporosis. Hormone replacement therapy is widely used to prevent bone loss, although it presents potential drawbacks such as increased risk of uterine bleeding and/or hyperplasia, increased risk of endometrial, breast or ovarian cancer, higher occurrence of myocardial infarction, cardiovascular disease, etc. Conjugate 309 of 17β-estradiol and iminodiacetic acid (Fig. 59) was designed as an estrogen-containing, bone-seeking agent that could prevent bone loss with lesser side effects, and was subsequently synthesized by means of the Mannich reaction [462]. Mannich base 309 showed significant affinity for bone, but lower affinity for ovary and uterus than 17β-estradiol, while it maintained 92% of the affinity of 17β-estradiol for osteoblast estrogen receptors. Candidate 309 did not induce uterine hypertrophy, and lower levels of biochemical markers of bone turnover (e.g., osteocalcin, alkaline phosphatase, C-terminal telopeptide fragment of type I collagen C-terminus) were found in the group of rats treated with 309 than in ovariectomized rats, which suggests decreased bone turnover. Administration of candidate 309 improved bone mineral density and trabecular architecture after ovariectomy, but did not suppress body weight increase. These results suggest that compound 309 is effective in preventing ovariectomy-induced bone loss while exhibiting exhibited fewer adverse side effects than 17β-estradiol, making Mannich base 309 a better choice for the prevention of postmenopausal osteoporosis.
Three bis-P-Mannich bases 310 (R1 = R2 = CH3; R1–R2 = (CH2)3; R1–R2 = (CH2)4) (Fig. 59), derived from diethyl phosphite as substrate and amino acids as amine reagents in the Mannich reaction, have been used to investigate the induction of adipogenic or osteogenic differentiation of mesenchymal stem cells isolated from human adipose tissue (hMADS cells) [463]. Candidates 310 did not affect the adipocyte differentiation, but induced the inhibition of osteoblast formation without any detectable cytotoxic effect, whereas reference drug sodium alendronate elicited a cytotoxic effect even at the lowest concentration used in the study (10−7 M).
Therapeutic efficacy and toxicity profiles of ketonic double Mannich base 311 (Fig. 59), a putative immunosuppressant which preferentially inhibited JAK3 as opposed to several other kinases, were examined [464]. Candidate 311 blocked IL-2-induced activation of JAK3 and its downstream substrates STAT5a/b, while it failed to inhibit several other enzymes, including growth factor receptor tyrosine kinases, Src family members, and serine/threonine protein kinases. Mannich base 311 alone prolonged kidney allograft survival and induced transplantation tolerance, and its combination with cyclosporin A presented therapeutic synergism. Candidate 311 showed no nephrotoxicity, did not affect hematopoiesis or lipid metabolism, and was not metabolized by the cytochrome P450 3A4 isoform. Therefore, because Mannich base 311 prolongs allograft survival without several toxic effects associated with current immunosuppressive drugs, it may provide significant clinical benefits for transplant patients.
18. Mannich bases as enzyme inhibitors
An important number of studies report the activity of Mannich bases as enzyme inhibitors. Although the studies covered in this section usually validate the importance of the topic through a rational and evidence-supported connection between the enzyme under scrutiny and a certain medical condition, they only examine the action of Mannich bases as enzyme inhibitors, and do not provide any experimental evidence that these Mannich bases could be actually useful agents for the treatment of specific medical conditions.
18.1. Inhibitors of cholinesterases
Starting from tacrine, the first drug approved for the treatment of Alzheimer's disease and a potent inhibitor of both acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE), multifunctional compounds 279 (Fig. 53) that combine neuroprotective, antioxidant, metal-binding properties, and dual inhibition of AChE and BuChE in a single small molecule have been designed and synthesized through the Mannich reaction [431]. Tacrine–8-hydroxyquinoline hybrids 279 were potent inhibitors of both AchE and BuChE of bovine origin with IC50 ranging from submicromolar to nanomolar concentrations. Candidates containing an unsubstituted 8-hydroxyquinoline moiety and a methylene tether of 7–10 carbon atoms had the most potent inhibitory activities. Selected Mannich bases 279 were evaluated as inhibitors of human cholinesterases, and they exhibited IC50 values in the range of 0.5–5.5 nM against AChE, and in the range of 6.5–55 nM against BuChE. Mannich base 279 (R1 = R2 = H, n = 7) was the most potent dual inhibitor of human AChE and BuChE in this library [431].
Other Mannich base hybrids 312 (Fig. 60 ) were designed as dual binding site inhibitors of AChE by combining tacrine (a recognized inhibitor of catalytic binding site of AChE) with the indanone moiety of donepezil (known to be responsible for the binding of this drug to the peripheral site of AChE) [465]. Their evaluation as inhibitors of AChE (bovine erythrocyte) and BuChE (human serum) using Ellman's method showed that the activity of two of these candidates was modest, while the third candidate 312 (Z = H, R = OCH3, X = (CH2)3) exhibited moderate activity against AChE (25 nM) and good activity against BuChE (0.6 nM). Apparently, the difference in activity between these candidates is due to the lengthening by one methylene group of the alkyl chain linking the tacrine and indanone moieties.
Fig. 60.
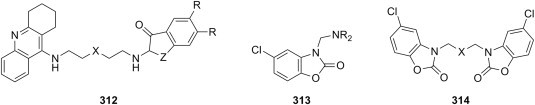
Mannich bases as inhibitors of cholinesterases.
2-Benzoxazolone mono-Mannich bases 313 (Fig. 60) derived from secondary aliphatic amines or primary arylamines, and bis-Mannich bases 314 derived either from primary aliphatic amines (X = NR, R = C2H5, R1C6H4CH2CH2, R1 = H, 4-Cl, 3,4-(CH3O)2) or piperazine have been evaluated for AChE inhibitory activity using Ellman's method. The degree of inhibition was in the range of 70–82% at a concentration of 1 mM, but decreased to 8–34% at 0.1 mM, whereas the degree of AChE inhibition by tacrine was virtually the same at both concentrations (greater than 99%) [466]. The most potent inhibitor in this series at both concentration was bis-Mannich base 314 derived from piperazine.
Inhibitory activity against AChE and BuChE of phenolic Mannich bases 58 (R1 = H, CH3, allyl, prenyl; NR2 = N(CH3)2, N(C2H5)2, 1-pyrrolidinyl, 1-piperidinyl, 4-morpholinyl) of xanthone derivatives (Fig. 10) was found to be moderate to good compared to that of reference drug galantamine [467]. Candidates 58 derived from diethylamine as amine reagent in the Mannich reaction were generally the most potent inhibitors of both enzymes. The nature of alkyl moiety R1 modulates the selectivity of inhibitors 58 towards one of these enzymes; thus, the most potent inhibitors of AChE feature a methyl group as R1, whereas the increasing bulkiness of this substituent appears to favor the inhibition of BuChE. Mannich base 58 (R1 = prenyl, NR2 = N(C2H5)2) was the most potent dual inhibitor in this series, and molecular docking studies were performed with the view to garner information on the binding mode of this inhibitor with both enzymes.
18.2. Inhibitors of α-glucosidase
Several phenolic Mannich bases 315 of naturally occurring flavanone oroxylin A (Fig. 61 ) were more potent inhibitors of intestinal α-glucosidase than parent oroxylin A, but the same candidates were less potent than oroxylin A against yeast α-glucosidase [468]. Nonetheless, the most potent inhibitor of intestinal α-glucosidase in this series was still 5.5 less potent than reference drug acarbose. Because all of the candidates 315 which failed to inhibit both enzymes were derived from acyclic secondary amines as amine reagents in the Mannich reaction, the alicyclic nature of secondary amines may responsible for the good inhibitory activity against α-glucosidases in this series.
Fig. 61.

Mannich bases as inhibitors of α-glucosidase.
Ketonic Mannich bases 316 derived from nabumetone (Fig. 61) possess modest inhibitory activity against intestinal α-glucosidase (approximately 20 mM), in addition to modest inhibitory activity against another therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance, namely protein-tyrosine phosphatase 1B [469]. However, two Mannich bases 316 (R = H or 4-OH) were good agonists of peroxisome proliferator-activated receptors, which are major regulators of lipid and glucose metabolism. A novel series of inhibitors was designed by retaining the nabumetone moiety, while sulfanilamide and 3-amino-5-methylisoxazole served as amine reagents in the synthesis of ketonic Mannich bases of type 317 and 318, respectively (Fig. 61) [470]. Candidates 317 were generally more potent inhibitors of α-glucosidase than analogous 318, suggesting that sulfonamide moiety is a more potent pharmacophore than aminoisoxazole. Mannich base 317 (R = 4-Br) inhibited 80% of α-glucosidase activity at a dose of 10 μg/mL. Replacement of the sulfonamide moiety with 4-aminobenzoic acid, with a view to mimic the acidic, hydrophilic part of molecules with known antidiabetic activity, led to a novel series of Mannich bases 319 (R1 = H). A second series of candidates 319 (R1 = C2H5) (Fig. 61) was also evaluated as α-glucosidase inhibitors, but their activity was generally poorer than that of Mannich bases 319 (R1 = H) derived from 4-aminobenzoic acid [471]. For example, the most potent α-glucosidase inhibitor (67% inhibition) in the series derived from 4-aminobenzoic acid was candidate 319 (R = 4-Cl, R1 = H), while the degree of α-glucosidase inhibition of analogous 319 (R = 4-Cl, R1 = C2H5) derived from ethyl 4-aminobenzoate reached only 24%. As part of their search for antidiabetic agents within this type of ketonic Mannich bases, Yang et al. also published a series of less accessible studies in Chinese, which are referenced in the aforementioned articles.
18.3. Inhibitors of purine nucleoside phosphorylase
Purine nucleoside phosphorylase (PNP) catalyzes the phosphorolytic cleavage of inosine, guanosine and their 2′-deoxy analogues to (deoxy)ribose 1-phosphate and hypoxanthine or guanine. Genetical deficiency in PNP leads to complete T-cell deficiency early in life and eventual death of infants from virus infections, and PNP has been identified as a target for the treatment of T-cell lymphoma, rheumatoid arthritis, psoriasis, multiple sclerosis, and other T-cell mediated disorders. A transition state analogue is a compound that occupies the catalytic site of an enzyme and induces the slow conformational changes that would normally occur to place the catalytic site in the appropriate geometry for the search that would locate the transition state with the normal substrate of the enzyme. Schramm et al. have developed a series of Mannich bases of 9-deazahypoxanthine as transition state analogues for purine nucleoside phosphorylase inhibition, generally called immucilins. Although aminomethylated purines and analogues have been present within the first- and second-generation of PNP inhibitors [472], [473], they have been obtained through reductive amination, and not through direct Mannich reaction. Because a more expeditious synthesis of second-generation PNP inhibitors by means of the Mannich reaction has been later developed [474], the third generation of these inhibitors included several examples of Mannich bases 320 (Fig. 62 ) obtained through direct aminomethylation of substrate 9-deazahypoxanthine with amino alcohols such as ethanolamine, 3-amino-propan-1-ol, 4-amino-butan-1-ol, diethanolamine and 3-(2-hydroxyethylamino)propan-1-ol as amine reagents [475]. Unfortunately, all these candidates obtained through direct aminomethylation proved to be modest or poor inhibitors of PNP (dissociation constants K i in the range of 780 to 120,000 pM) compared to other Mannich bases previously investigated, such as 321 (DADMe-immucilinH, K i = 8.5 pM) or 322 (DADMe-immucilinG, K i = 7.0 pM) (Fig. 62). Nonetheless, this study identified two acyclic, simplified alternatives of immucilin analogues, namely 323 (K i = 9.0 pM) having an open-chain amino alcohol with two asymmetric carbon atoms, and 324 (K i = 5.0 pM) derived from achiral dihydroxyaminoalcohol seramide (Fig. 62), as potent inhibitors of PNP. Because the enantiomers of both immucilinH and DADMe-immucilinH 321 have been shown to have PNP binding properties that differ considerably [476], and since access to enantiomerically pure starting azasugar employed for the generation of 322 requires a suitable enzyme-based route, the discovery of the two simpler Mannich bases 323 and 324 represents a significant step forward in the development of clinically useful PNP inhibitors. Sulfur-containing transition state analogues have been developed as well, originally as specific inhibitor of PNP from P. falciparum, but it was later shown that sulfur-containing candidates analogous to DADMe-immucilinH 320 or Mannich base 323 also bind to human native PNP [477]. Although human PNP tolerates substitution of 5′-hydroxyl in transition state analogues 321 and 324 with alkylthio and arylthio groups, the slow-onset nature of inhibition is lost, and inhibitor dissociation constants increase by two to three orders of magnitude. Fluorine-containing analogue 325 (Fig. 62) of DADMe-immucilinH 321 has also been evaluated as human PNP inhibitor, and its enantiomers exhibit slow-onset binding constants of 32 pM for [(3S,4S)-325] and 1.82 nM for [(3R,4R)-325] [478]. In addition, direct Mannich reaction has been used to generate in a similar fashion potent transition state inhibitors for other enzymes, such as methylthioadenosine phosphorylase methylthioadenosine nucleosidase [479], [480] or purine specific nucleoside hydrolase [481].
Fig. 62.
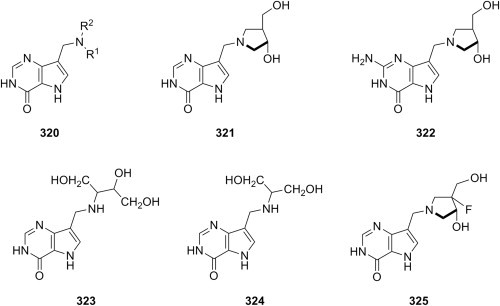
Mannich bases as inhibitors of purine nucleoside phosphorylase.
18.4. Inhibitors of various other enzymes
A large library of α-, β- and γ-amino ketones was screened with a view to discover inhibitors of transglutaminase, but only β-amino ketones (ketonic Mannich bases) strongly inhibited this enzyme [482]. Mannich bases derived from aliphatic ketones as substrates, or those derived from alkyl aryl ketones as substrates and primary amines as amine reagents were weak inhibitors (IC50 > 30 μM) of transglutaminase. Potent transglutaminase inhibitory activity resided with ketonic Mannich bases derived from alkyl aryl ketones as substrates and secondary aliphatic amines as amine reagents. Both the nature of the aryl moiety in the substrate and the nature of the alkyl groups attached to the nitrogen atom influenced transglutaminase inhibitory activity of these candidates. Generally, heteroaryl-substituted substrates (2-thienyl, 2-benzothienyl, 2-furyl, 3-furyl, pyridinyls, pyrazinyl) fared better than substrates having aryl moieties (phenyl, naphthyls), and t-butyl, isopropyl, benzyl or 2-hydroxyethyl groups (but not phenyl) as alkyl substituent at nitrogen afforded potent transglutaminase inhibitors, such as candidate 326 (IC50 = 81 nM) (Fig. 63 ). Because disulfides are well-known transglutaminase inhibitors, a novel series of β-amino ketones 327 (R = H, n = 0; R = COOCH3, n = 0; R = COOCH3, n = 1) (Fig. 63) derived from cystamine, dimethyl cystine ester, and dimethyl homocystine ester as amine reagents in the Mannich reaction were synthesized and found to be approximately 300 times more active than the starting disulfides [483]. Mannich bases derived from 2-acetylthiophenes were once more the most potent inhibitors in this series, while the nature of the disulfide-containing amine moiety did not significantly influence the activity of candidates 327 having the same heterocyclic or carbocyclic moiety R1.
Fig. 63.
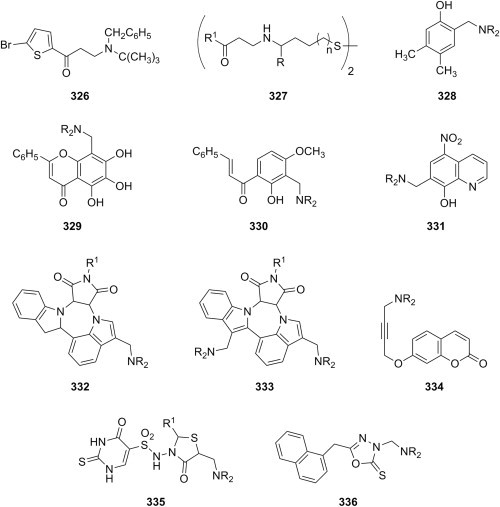
Mannich bases as inhibitors of various enzymes.
Mannich bases 328 derived from 3,4-dimethylphenol (Fig. 63) have been evaluated in vitro as inhibitors of two human carbonic anhydrase (hCA, EC 4.2.1.1) isozymes I and II using the hydratase and esterase assays, respectively [484]. Only two candidates exhibit weak hCA II inhibitory effects on esterase activity, whereas other two Mannich bases 328 could be used as carbonic anhydrase activators.
Phenolic Mannich bases 280 derived from quercetin (Fig. 53), 329 derived from baicalein and 330 derived from 1-(2-hydroxy-4-methoxyphenyl)-3-phenylprop-2-en-1-one (Fig. 63) were screened for inhibition of cyclin-dependent kinases using a biochemical assay method based on fluorescence resonance energy transfer [485]. Candidates 280 and 330 are essentially devoid of inhibitory activity of cyclin-dependent kinases; on the other hand, baicalein Mannich bases 329 were 6–20 times more potent than the parent flavone, suggesting that the presence of a nitrogen atom at C-8 is crucial for the inhibition of cyclin-dependent kinases, while the presence of a second heteroatom in the amine moiety (e.g., oxygen in morpholine, sulfur in thiomorpholine, nitrogen in N-methylpiperazine) further increases the potency of these candidates.
Virtual screening of a commercial library containing drugs and drug-like molecules against a 3D model of heparanase led to the identification of several candidates with high scores for binding affinity; they were subsequently tested in NMR competitive inhibition experiments using suramine as a known heparanase inhibitor [486]. One of these candidates is amodiaquine 201 (Fig. 38), and a subset of fourteen representative compounds that retained the characteristic 4-phenylamino-chloroquinoline scaffold of amodiaquine, but presented different functional groups in the phenylamino moiety, was further selected for NMR competitive inhibition experiments. Although no candidate with improved heparanase inhibitory activity over that of amodiaquine emerged from this subset, several structural requirements for the binding of an inhibitor to the active site of the enzyme were obtained.
Four Mannich bases 331 of nitroxoline (Fig. 63) were found to inhibit efficiently the activity of methionine aminopeptidase-1 from Burkholderia pseudomallei with IC50 values ranging from low micromolar to 30 nM, and a few candidates in this series also showed in vitro growth inhibition of Burkholderia thailandensis [487].
Hexacyclic indole Mannich bases 332 (R1 = H or CH3, NR2 = N(C2H5)2, 1-pyrrolidinyl) and 333 (R1 = CH3, NR2 = 4-morpholinyl) derived from annelated maleimidoindolodiazepines (Fig. 63) were tested against a panel of 25 human protein kinases, but they were inactive against all of the tested enzymes at micromolar concentrations [488].
Out of four synthesized acetylenic Mannich bases 334 (NR2 = N(C2H5)2, 1-pyrrolidinyl, N-allyl-N-methyl, N-(2-dimethylaminoethyl)-N-methyl) derived from 7-(prop-2-ynyloxy)chromen-2-one (Fig. 63), three were inhibitors of squalene-hopene cyclase [489]. Candidate 334 (NR2 = N-allyl-N-methyl) was half as potent as the Hoffman–La Roche's anticholesteremic drug Ro 48-8071, which is an effective inhibitor of both squalene- and oxidosqualene cyclases that features the same secondary amino group in its structure.
Thiazolidinone C-Mannich bases 335 (R1 = 4-CH3C6H4, 4-NO2C6H4, 2-furyl, 3,4,5-(CH3)3C6H2; NR2 = N(C2H5)2, 4-morpholinyl, 4-methylpiperazinyl) (Fig. 63) were evaluated as inhibitors of Schistosoma mansoni cercarial elastase, but they were all inactive [490].
Oxadiazolethione N-Mannich bases 336 having a naphthalen-1-ylmethyl residue at C-5 (Fig. 63) were all active against mushroom tyrosinase, and 6 to 10 times more potent than reference compound kojic acid [491]. The presence of a thione group able to chelate with copper, and the ability of the cyclic secondary amine to form extensive hydrophobic contacts within the binding site of tyrosinase appear to be responsible for the activity. Another oxadiazolethione N-Mannich base, namely 139m (Table 2), presented moderate inhibitory activity against urease within the series of compounds investigated, but no comparison with the activity of a well-established urease inhibitor was provided [201].
19. Mannich bases as receptor ligands
19.1. Ligands of 5-hydroxytryptamine receptors
5-Hydroxytryptamine receptors (5-HT receptors), also known as serotonin receptors, belong to the group of G protein-coupled receptors (with the exception of 5-HT3, which is a ligand-gated ion channel). They are found in the central and peripheral nervous systems, and their function is to mediate both excitatory and inhibitory neurotransmission. 5-HT receptors are activated by their natural ligand, the neurotransmitter serotonin.
Mannich bases 245 (n = 1 or 2) (Fig. 44) were evaluated as ligands for the types of 5-HT receptors that could be involved in the mediation of the anticonvulsant effect of these compounds, but the candidates exhibited only moderate to low affinity for both 5-HT1A (K i = 81–370 nM) and 5-HT2A receptors (K i = 126–1370 nM) [383]. The nature of the spiranic cycloalkyl group did not influence serotonin receptor affinity significantly, although cyclohexyl-substituted candidates 245 were slightly more active than cyclopentyl-substituted analogues. The nature of the substituent in the aryl moiety at N-4 of piperazine ring modulates the selectivity towards one of these 5-HT receptors; thus, Mannich bases 245 (R = 2-OCH3) showed the highest affinity for 5HT1A receptors and the lowest affinity for 5-HT2A receptors, while the highest 5-HT2A receptor affinity was shown by Mannich bases 245 (R = 3-CF3). In addition, computer-aided design of novel Mannich bases of imidazoline-2,4-diones led to the identification of several compounds with potential dual affinity for 5HT1A receptors and serotonin transporter, whose synthesis and evaluation finally led to candidates 337 (R = H or F) (Fig. 64 ) having the desired pharmacological profile [492].
Fig. 64.
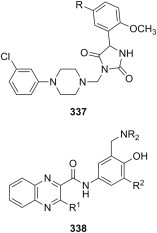
Mannich bases as ligands of 5-hydroxytryptamine receptors.
A series of quinoxaline-2-carboxamides N-substituted with phenolic Mannich bases moieties having either one or two aminomethyl functions have been synthesized and tested for 5-HT3 receptor antagonistic activity in longitudinal muscle-myenteric plexus preparation from guinea pig ileum against 5-HT3 agonist, 2-methyl-5-HT [493], [494]. Candidates 338 (R1 = Cl) (Fig. 64) exhibited mild to moderate antagonist activity, and double Mannich bases were generally less potent than their mono-Mannich bases counterparts, but Mannich base 338 (R1 = Cl, R2 = H, NR2 = 4-methylpiperazin-1-yl) had 5-HT3 receptor antagonistic activity comparable to that of reference compound ondansetron [493]. Replacement of chlorine with methoxy led to less potent candidates 338 (R1 = OCH3), whereas candidates with an ethoxy group at position 3 of quinoxaline ring were even less potent [494]. The presence of piperazines in the aminomethyl moiety of candidates 338 seemed to be more favorable for 5-HT3 receptor antagonistic activity than the presence of other cyclic secondary amines.
19.2. Ligands of dopamine receptors
Dopamine receptors are G protein-coupled receptors that are widely distributed in the brain, and whose primary endogenous ligand is the neurotransmitter dopamine. There are at least five subtypes of dopamine receptors, but D1-2 receptor subtypes usually predominate, as they are 10–100 times more numerous than D3-5 subtypes.
Discovery of L-745,870 339 [495] and FAUC 113 340 [496] (Fig. 65 ) as potent and selective D4 receptor ligands has fueled the search for novel chemical entities having the ability to bind to dopamine receptors. Based on the observation that these two ligands and many other dopamine receptor ligands feature a methylene bridge between the piperazine ring and the (hetero)aromatic part of the molecule, various analogues have been designed and synthesized. However, the majority of these analogues were obtained either through nucleophilic displacement by an appropriately substituted piperazine of a halogen atom from a halomethyl derivative of a selected (hetero)aromatic substrate, or through reductive amination of a formyl-substituted (hetero)aromatic substrate in the presence of a piperazine. Literature search revealed a few examples when the Mannich reaction was employed to synthesize the ligands to be evaluated for dopamine receptor binding ability. In addition to pyrrolo[2,3-b]pyridine and pyrazolo[1,5-a]pyridine ring systems used to in the generation of ligands 339 and 340, imidazo[1,2-a]pyridine is another example of an azaindole ring system that was aminomethylated to yield Mannich bases 341 (Fig. 65) [497]. Receptor binding profiles of candidates 341 and several mono-Mannich bases 342 derived from pyrrole (Fig. 65) were determined in vitro by measuring their ability to compete with [3H]spiperone for the cloned human dopamine receptor subtypes D2 long, D2 short, D3 and D4.4, while D1 receptor affinities were measured employing an assay that used D1 selective ligand [3H]SCH 23390 and porcine striatal membranes. All of the Mannich bases 341 and 342 were selective ligands for the D4 receptor subtype, and the candidates with an imidazo[1,2-a]pyridine scaffold were more potent than their counterparts having a pyrrole ring. The most potent Mannich bases 341 (R = 2-C2H5O, 2,3-Cl2, 3-CF3) showed an affinity for the dopamine D4 receptor in the low nanomolar range, and their selectivity for this receptor subtype exceeded that of reference drug clozapine [497].
Fig. 65.
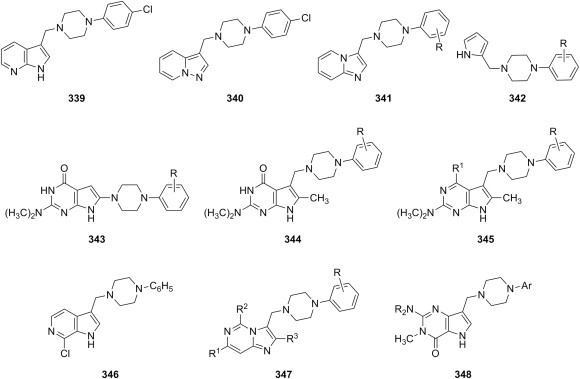
Piperazine Mannich bases as ligands of dopamine receptors.
Several small series of candidates comprising Mannich bases 343, 344 and 345 (R1 = H or Cl) having a pyrrolo[2,3-d]pyrimidine scaffold (Fig. 65) were designed and evaluated as ligands for dopamine receptor subtypes D1, D2 long, D2 short, D3 and D4.4, but they had no appreciable affinity for any of the dopamine receptors tested [498]. The strongest binding in these series was recorded for candidates 343 (R = 2-CH3O) and 345 (R1 = H, R = 2-CH3O) at dopamine D4.4 receptor, with K i values of 2.4 and 1.9 nM, respectively.
Novel heteroaromatic scaffolds have been employed in the synthesis of Mannich bases 346, 347 and 348 (Fig. 65) as potential D4 receptor ligands [499]. Candidate 346 derived from pyrrolo[2,3-c]pyridine ring system bound selectively at D4.4 receptor (K i = 6.4 nM). Evaluation of Mannich bases 347 generated from imidazo[1,2-c]pyrimidines and having various substituents at positions 2, 5 and 7 led to different results, depending on the nature and number of these substituents. Thus, Mannich base 347 (R1 = R2 = R3 = H) derived from the unsubstituted scaffold and all of the Mannich bases 347 (R1 = CH3, R2 = R3 = H) obtained from 7-methylimidazo[1,2-c]pyrimidine had no effect on dopamine D4 receptor. The results for the binding assay for Mannich bases derived from dimethyl-substituted imidazo[1,2-c]pyrimidines showed that candidates 347 (R1 = R3 = CH3, R2 = H) have binding affinities to dopamine D4 receptor in the range of reference drug clozapine, while candidate 347 (R1 = R2 = CH3, R3 = H) is a weak ligand. Further modifications, such as introduction of a carbethoxy group in the structure of candidates 347 (R1 = R2 = CH3, R3 = COOC2H5), or two methoxy groups in Mannich bases 347 (R1 = R2 = OCH3, R3 = H), led to compounds without affinity for dopamine D4 receptor. In the series of Mannich bases derived from 2,5,7-trimethylimidazo[1,2-c]pyrimidine, only candidate 347 (R1 = R2 = R3 = CH3, R = 2-OCH3) had good binding properties for the dopamine D4 receptor, whereas another candidate 347 (R1 = R2 = R3 = CH3, R = 4-OCH3) exhibited a slight selectivity for dopamine D3 receptor. Use of pyrrolo[3,2-d]pyrimidine as scaffold led to some of the most potent ligands of dopamine D4 receptor in this study (K i values between 2.4 and 3.8 nM). The nature of the amino group at position 2 of pyrrolo[3,2-d]pyrimidine scaffold modulates the activity; thus, Mannich bases 348 (NR2 = 1-pyrrolidinyl) had good affinities for dopamine D4 receptor, but replacement of pyrrolidine with morpholine resulted in significant loss of dopamine D4 receptor binding affinity.
Two novel indole Mannich bases 59 (R = 2-C2H5OC6H4, 2,3-Cl2C6H3) (Fig. 11) were synthesized through direct aminomethylation and evaluated as ligands for several dopamine receptor subtypes, along with other heteroarylmethylpiperazines [500]. Both candidates were potent and selective dopamine D4 receptor ligands, and Mannich base 59 (R = 2-C2H5OC6H4) was the most potent compound in this library (K i = 0.03 nM). In addition, Mannich base 59 (R = 2,3-Cl2C6H3) was the most potent candidate having a 4-(2,3-dichlorophenyl)piperazine moiety, suggesting the importance of indole as (hetero)aromatic moiety in this type of dopamine D4 receptor ligands.
The effect on the affinity to D2, D3 and D4 dopamine receptor subtypes of replacement of 4-arylpiperazinyl in Mannich bases with other monocyclic or bicyclic amine residues was also investigated [501]. Mannich bases 349 (X = CH or N) (Fig. 66 ) derived from 3-(4-chlorophenyl)-8-azabicyclo[3.2.1]octan-3-ol as amine reagent in the Mannich reaction had modest affinity and moderate selectivity for dopamine D3 receptor subtype compared to D2 and D4 receptor subtypes. Pyrrolidinol analogues 350 (X = CH or N) (Fig. 66) had poor binding affinities to all subtypes of dopamine receptors, while homopiperazine analogue 351 (Fig. 66) had moderate binding affinity to dopamine D4 receptor subtype (K i = 18.6 nM). Candidate 352 derived from 3,9-diazabicyclo[4.2.1]nonane ring system as amine reagent in the Mannich reaction (Fig. 66) bound poorly to all types of dopamine receptor investigated in this study. Although candidate 353 (Fig. 66) derived from 2,5-diazabicyclo[2.2.1]heptane ring system as amine reagent in the Mannich reaction had the highest D3 receptor affinity of all the compounds tested in this paper (K i = 11 nM), it also had poor selectivity towards D3 (K i D2 = 62 nM, K i D4 = 69 nM). Mannich base 354 derived from 2-(2-pyrimidinyl)piperazine (Fig. 66) had modest binding affinity to dopamine D4 receptor subtype (K i = 56 nM), but good selectivity over D2 and D3 subtypes.
Fig. 66.

(Aza)indole Mannich bases derived from various amines as ligands of dopamine receptors.
19.3. Inhibitors of the interaction between thyroid receptors and their coregulators
Thyroid receptors are nuclear receptors belonging to a superfamily whose members function as hormone activated transcription factors. The regulation of transcription for thyroid receptors is controlled by their natural ligand, the thyroid hormone. Both unliganded and liganded thyroid receptors can bind to and regulate genes under the control of thyroid response elements, but the liganded thyroid receptor complex can also recruit a particular coactivator protein out of many available, with a view to steer the course of transcriptional regulation. High-throughput screening of a library of compounds identified a number of hits for inhibitors of the interaction between thyroid receptor and its coactivator SRC2 (steroid receptor coactivator 2), and two of these hit compounds were ketonic Mannich bases 355 and 356 (Fig. 67 ) [502]. These candidates represent a new class of thyroid receptor antagonists that have exceptional selectivity towards β isoform and are active in the presence of thyroid hormone, while being able to silence its signaling. Mannich bases 355 and 356 are most likely covalently bound to thyroid receptor as a result of an alkylation process, thus inhibiting its hormone-induced gene transcription. Subsequent preliminary SAR showed that a hydrophobic moiety para to the ketone function in other ketonic Mannich bases analogous to 355 and 356 is an important structural feature of potent inhibitors of the thyroid receptor–coactivator interaction [503]. A second series of candidates explored the effect that the nature of the amino group in ketonic Mannich bases analogous to 355 and derived from 4-n-hexylacetophenone has on the interaction between coregulatory protein SRC2 and both isoforms of thyroid receptors. All the synthesized candidates inhibited the interaction in the low micromolar range, with a roughly 2-fold selectivity towards isoform β. These ketonic Mannich bases most likely function as prodrugs for the true active species, namely the α,β-unsaturated ketones resulted through deamination. Therefore, a collection of enones having various substituents at the carbon–carbon double bond was also tested with a view to provide an insight on the mechanism of action of ketonic Mannich bases as inhibitors of this interaction. The results showed that the inhibition of the interaction between thyroid receptor and coactivator protein SRC2 depends strongly on the substitution pattern at the carbon–carbon double bond in these α,β-unsaturated ketones. The most potent was the unsubstituted enone 357 (Fig. 67), which formally arises from 355 through deamination, whereas all other substituted enones were weaker inhibitors than 357. Subsequent investigations confirmed that enone 357 is the actual inhibitor, and that compound 357 is generated within the binding site, where it remains bound until it reacts with one of the local cysteine residues [504]. In addition, an attempt to examine a complex between thyroid receptor β and Mannich base 355 by X-ray crystallography led to the detection in the activation function-2 cleft of thyroid receptor β of enone 357 instead of Mannich base 355. Although enone 357 did not react rapidly with thyroid hormone, it positioned close to several cysteine residues. Several other experiments showed that, once formed, enone 357 slowly alkylates nearby Cys298 residue to occlude activation function-2 pocket of thyroid receptor β. Significant improvement of pharmacological properties (especially potency and therapeutic index) of inhibitors of thyroid hormone receptor coactivator binding based on ketonic Mannich bases was achieved with a series of novel candidates having in the aromatic ring electron withdrawing substituents associated with high efficacy, and a sulfonyl group to reduce cytotoxicity. Also, piperazinone and 4-acetylpiperazine were used as preferred amine moieties with a view to minimize ion channel activity of these inhibitors [505]. The most potent compounds 358 and 359 (R = 2,3-Cl2 or 2,5-Cl2) (Fig. 67) showed outstanding thyroid hormone receptor coactivator interaction inhibitory potency, good therapeutic index, while no inhibition of KCl-stimulated depolarization of HEK293-hERG cells was observed for these Mannich bases.
Fig. 67.
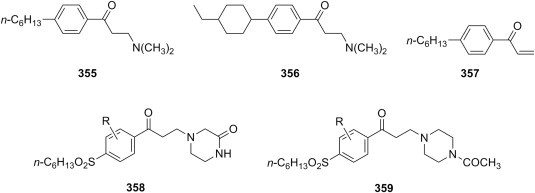
Mannich bases as inhibitors of the interaction between thyroid receptors and their coregulators.
19.4. Ligands of various receptors
Androgen receptor is a ligand-regulated transcription factor in the nuclear receptor superfamily. The receptor, which represents an important molecular target for the treatment of prostate cancer, is activated through the binding of its two natural endogenous ligands, testosterone or 5α-dihydrotestosterone. Subsequent activation of androgen-responsive genes can be blocked by androgen receptor antagonists through competitive inhibition. High-throughput screening of a structurally diverse chemical library comprising 16,000 synthetic and natural compounds led to the identification of 130 hit compounds that exhibited more than 85% competitive inhibition of 5α-dihydrotestosterone binding to androgen receptor [506]. Mannich base 360 (R1 = R2 = CH3, R3 = C6H5) (Fig. 68 ) was selected for further development owing to its consistent androgen receptor antagonist activities in a cell-based assay using monkey kidney fibroblast CV-1 cells, and forty-nine analogues were designed based on its β-amino ketone core structure. Nine of these analogues showed higher binding affinity to androgen receptor than 5α-dihydrotestosterone, and evaluation of the enantiomers of candidate 360 (R1 = NO2, R2 = Cl, R3 = 2-furyl) showed that the androgen receptor-modulating activity and binding resided with the dextrorotatory isomer. SAR within this library of ketonic Mannich bases revealed that the presence of chlorine as substituent R2, preferably para to the amino group, is important for androgen receptor antagonistic effects, while the presence of an electron-withdrawing group as substituent R1 is crucial for high-affinity binding to the receptor. Good androgen receptor binding activity was found among analogues having phenyl, 2-furyl and 2-thienyl as substituent R3, and Mannich bases with heteroaryl moiety exhibited less cytotoxicity towards HeLa cells than Mannich bases with R3 = (substituted)phenyl. Candidate 360 (R1 = NO2, R2 = Cl, R3 = 2-furyl) inhibited the growth of SC3 cells (a mouse mammary cell line that responds well to androgens), while its effects on the proliferation of PC3 cells (a human prostate cancer cell line that lacks measurable androgen receptors) were very weak. Mannich bases of type 360 were subsequently disclosed as selective non-steroidal antagonists of another member of the nuclear receptor superfamily, namely progesterone receptor, and SAR within a collection of Mannich bases 360 showed that coordinating effects of substituents R1 and R3 can modulate the potency and selectivity for progesterone receptor over androgen receptor [507].
Fig. 68.
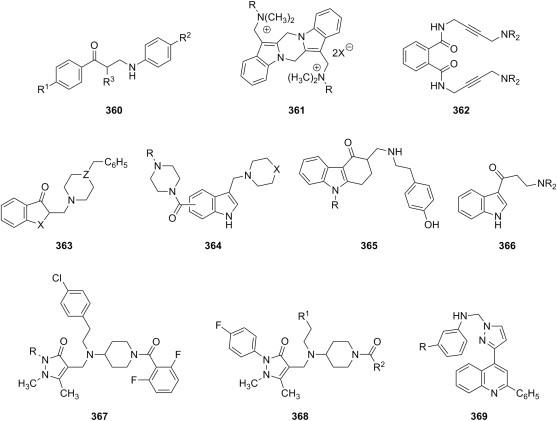
Mannich bases as ligands of various receptors.
Mannich bases that modulate the activity of acetylcholine receptors have been the topic of two recent studies. Two quaternary salts 361 (R = CH3, allyl) derived from bis-aminomethylated pyrazinodiindole (Fig. 68) were synthesized in order to probe their binding to the allosteric binding domain of muscarinic receptor M2 [508]. These quaternary salts showed only slightly higher binding affinity to muscarinic M2 receptor subtype than the corresponding parent double Mannich bases, which in turn exhibited relatively poor allosteric potency. Due to the structural similarity of candidates 361 and neuromuscular-blocking agents, their binding affinity to the muscle-type of the nicotinic acetylcholine receptors was also determined. Thus, compound 361 (R = CH3) had a 34-fold higher affinity for the muscle type nicotinic acetylcholine receptor than for the allosteric site of M2 receptors. Double Mannich bases 362 (NR2 = 1-piperidinyl, 2-methyl-1-piperidinyl, 1-azepanyl) derived from phthalamide (Fig. 68) had a competitive antagonistic effect to acetylcholine on isolated guinea pig ileum that was poorer than that of reference compound atropine, and behaved as non-competitive antagonists at higher concentration [509]. In addition, candidates 362 (NR2 = 1-piperidinyl, 2-methyl-1-piperidinyl, 1-azepanyl) antagonize the motor effect of oxotremorine in mice, while Mannich base 362 (NR2 = 2,6-dimethyl-1-piperidinyl) was inactive in both tests.
Indole Mannich base 59 (R = phenethyl) (Fig. 11) had moderate binding affinity to both sigma-1 (K i = 14.2 nM) and sigma-2 (K i = 55.8 nM) receptors, and can be considered as a potent, but non-selective ligand for sigma receptors [80]. Ketonic Mannich bases 363 (Fig. 68) derived from cyclic ketones such as α-tetralone (X = CH2CH2), chromanone (X = OCH2), indanone (X = CH2) or benzosuberone (X = (CH2)3) as substrates and either 4-benzylpiperidine (Z = CH2) or 4-benzylpiperazine (Z = N) as amine reagents in the Mannich reaction were also evaluated for their affinity towards sigma-1 and sigma-2 receptors [510]. Generally, 4-benzylpiperazine-containing Mannich bases were higher-affinity sigma receptor ligands than the corresponding 4-benzylpiperidine-containing analogues. Compared to other alkylpiperidines or alkylpiperazines that were evaluated in this study, ketonic Mannich bases 363 had only moderate affinity for sigma receptors. The presence of the carbonyl group in their structure appears to be detrimental, especially for the affinity to sigma-1 receptors, and the binding affinity seems to be influenced by the size of the saturated ring fused to the aromatic ring. Thus, six-membered candidates 363 are more potent than Mannich bases 363 derived from indanone or benzosuberone. However, candidate 363 (X = CH2CH2, Z = CH2) was one of the most selective compounds in this study, with a selectivity towards sigma-2 receptors of approximately 0.1 (K i-σ1 = 24 nM, K i-σ2 = 3.4 nM). In contrast, Mannich bases 363 derived from 4-benzylpiperazine were consistently better ligands for sigma-1 than for sigma-2 receptors.
Mannich bases of indoles substituted in the carbocyclic ring with a carboxamide group were evaluated as human histamine H3 receptor antagonists [511]. Preliminary analysis of the binding affinity of candidates 364 (R = i-C3H7, c-C4H7, X = CH2, O, NC3H7-i, NC4H7-c) (Fig. 68) to human H3 receptor with respect to different positional isomers within this series established a correlation between the 3,6-substitution pattern in 364 and good human H3 receptor affinity, while the 3,4-substitution pattern was the least favorable for H3 receptor binding affinity. The presence of piperidine and morpholine in the aminomethyl group led to compounds with excellent histamine H3 receptor affinity, whereas the presence of 4-substituted piperazines in the aminomethyl group was not well tolerated. Several Mannich bases 364 of indoles with a 3,7-substitution pattern also presented reasonable potency as human H3 antagonists; in this case, the presence of a cyclobutyl residue in the piperazine ring in the carboxamide group seems to be more favorable for human histamine H3 binding affinity than substitution with an isopropyl residue.
In the search for new ligands able to discriminate among the three α1-adrenergic receptor subtypes, a series of Mannich bases 365 (R = CH3, n-C3H7, CH2C6H5, CH2C6H11-c, C6H5) derived from 1,2,3,4-tetrahydro-4H-carbazol-4-ones (Fig. 68), that are structurally related to the potent α1-adrenergic receptor antagonist HEAT, were synthesized and evaluated [512]. As a general trend, all candidates showed lower affinities than HEAT for all three α1-adrenergic receptor subtypes, and replacement of tyramine with 4-(2-substituted phenyl)piperazines did not enhance the affinity towards any subtype in particular. In addition, Mannich bases 366 derived from 3-acetylindole and the same amines (Fig. 68) were designed as “open chain” analogues of candidates 365, and their evaluation showed a slight improvement in the affinity for all three α1-adrenergic receptor subtypes.
Starting from a hit obtained by screening a large library of compounds, and employing subsequent stepwise refinement of structural features, a series of Mannich bases 367 (R = (substituted)phenyl) of pyrazolones (Fig. 68) was designed as ligands of CC chemokine receptor 3 (CCR3), a potential molecular target for treatment of allergic asthma [513]. The evaluation of binding to human CCR3 receptor of candidates 367 showed that most compounds display IC50 values around 100 nM, and that only two of these Mannich bases (R = 4-FC6H4 and 2-pyridinyl) bound more tightly to CCR3 than the initial hit compound (IC50 = 32 nM). Because the lead compound 367 (R = 4-FC6H4, IC50 = 20 nM) suffered from poor water solubility and metabolic stability, less lipophilic analogues were prepared by the introduction of ionizable groups in different parts of the molecule. Replacement in the structure of 367 (R = 4-FC6H4) of the 4-chlorophenyl moiety with 4-fluorophenyl, 4-nitrophenyl, 4-sulfonamidophenyl, 4-aminophenyl or pyridinyl moieties, as well as substitution of the 2,6-diflurophenyl moiety with pyridinyl or pyridinyl-N-oxide led to a second series of pyrazolone Mannich bases 368 (Fig. 68) with improved solubility in water and enhanced metabolic stability. Although structural modulations in the R1 moiety generally decreased the binding affinity of candidates 368 to CCR3, modifications in the R2 part afforded potent compounds (IC50 = 12 nM).
Two N-Mannich bases 369 (R = H or F) of a pyrazole derivative (Fig. 68) were reported in a study that examined the interaction of a series of 4-heteroaryl-2-phenylquinolines with neurokinin NK-2 and NK-3 receptors, but they were inactive (K i > 10 μM) [514].
20. Conclusions
Throughout the last decade, a large number of novel Mannich bases have been synthesized and evaluated as potential treatments for a multitude of diseases and medical conditions, as prodrugs, or as molecules eliciting a response from biological targets. The vast amount of data concerning the structure–activity relationship for Mannich bases derived from structurally diverse substrates has added to previous knowledge, thus allowing better insight into the design of more effective drug candidates based on the aminomethylation reaction in the future. Tremendous progress has been made in the field of anticancer agents and antimicrobials, and the last decade has witnessed the chemical modification of many biologically-active, naturally-occurring substrates or well established drugs by means of the Mannich reaction with a view to improve their biological activity. As such, the Mannich reaction has earned its rightful place as a powerful tool in medicinal chemistry, both for the synthesis of novel chemical entities endowed with various and interesting biological properties, and for the modification of physico-chemical properties of a candidate, that ultimately influence the candidate's biodisponibility, performance and pharmacological activity as a drug.
Conflict of interest
The author declares that there is no conflict of interest.
References
- 1.Tramontini M. Advances in the chemistry of Mannich bases. Synthesis. 1973:703–775. [Google Scholar]
- 2.Tramontini M., Angiolini L. Further advances in the chemistry of Mannich bases. Tetrahedron. 1990;46:1791–1837. [Google Scholar]
- 3.Tramontini M., Angiolini L. CRC Press; Boca Raton: 1994. Mannich Bases: Chemistry and Uses. [Google Scholar]
- 4.Siedel W., Soder A., Lindner F. Die Aminomethylierung der Tetracycline. Zur Chemie des Reverin, Munch. Med. Wochenschr. 1958;100:661–663. [PubMed] [Google Scholar]
- 5.Saab A.N., Sloan K.B., Beall H.D., Villaneuva R. Effect of aminomethyl (N-Mannich base) derivatization on the ability of S6-acetyloxymethyl-6-mercaptopurine prodrug to deliver 6-mercaptopurine through hairless mouse skin. J. Pharm. Sci. 1990;79:1099–1104. doi: 10.1002/jps.2600791212. [DOI] [PubMed] [Google Scholar]
- 6.Huttunen K.M., Rautio J. Prodrugs – an efficient way to breach delivery and targeting barriers. Curr. Top. Med. Chem. 2011;11:2265–2287. doi: 10.2174/156802611797183230. [DOI] [PubMed] [Google Scholar]
- 7.Simplicio A.L., Clancy J.M., Gilmer J.F. β-Aminoketones as prodrugs with pH-controlled activation. Int. J. Pharm. 2007;336:208–214. doi: 10.1016/j.ijpharm.2006.11.055. [DOI] [PubMed] [Google Scholar]
- 8.Dimmock J.R., Kumar P. Anticancer and cytotoxic properties of Mannich bases. Curr. Med. Chem. 1997;4:1–22. [Google Scholar]
- 9.Dimmock J.R., Elias D.W., Beazely M.A., Kandepu N.M. Bioactivities of chalcones. Curr. Med. Chem. 1999;6:1125–1149. [PubMed] [Google Scholar]
- 10.Pati H.N., Das U., Sharma R.K., Dimmock J.R. Cytotoxic thiol alkylators. Mini Rev. Med. Chem. 2007;7:131–139. doi: 10.2174/138955707779802642. [DOI] [PubMed] [Google Scholar]
- 11.Dimmock J.R., Kandepu N.M., Nazarali A.J., Motaganahalli N.L., Kowalchuk T.P., Pugazhenthi U., Prisciak J.S., Quail J.W., Allen T.M., LeClerc R., Santos C.L., De Clercq E., Balzarini J. Sequential cytotoxicity: a theory evaluated using novel 2-[4-(3-aryl-2-propenoyloxy)phenylmethylene]cyclohexanones and related compounds. J. Med. Chem. 2000;43:3933–3940. doi: 10.1021/jm000058o. [DOI] [PubMed] [Google Scholar]
- 12.Dimmock J.R., Sidhu K.K., Chen M., Reid R.S., Allen T.M., Kao G.Y., Truitt G.A. Evaluation of some Mannich bases of cycloalkanones and related compounds for cytotoxic activity. Eur. J. Med. Chem. 1993;28:313–322. [Google Scholar]
- 13.Dimmock J.R., Vashishtha S.C., Patil S.A., Udupa N., Dinesh S.B., Devi P.U., Kamath R. Cytotoxic and anticancer activities of some 1-aryl-2-dimethylaminomethyl-2-propen-1-one hydrochlorides. Pharmazie. 1998;53:702–706. [PubMed] [Google Scholar]
- 14.Dimmock J.R., Kandepu N.M., Hetherington M., Quail J.W., Pugazhenthi U., Sudom A.M., Chamankhah M., Rose P., Pass E., Allen T.M., Halleran S., Szydlowski J., Mutus B., Tannous M., Manavathu E.K., Myers T.G., de Clercq E., Balzarini J. Cytotoxic activities of Mannich bases of chalcones and related compounds. J. Med. Chem. 1998;41:1014–1026. doi: 10.1021/jm970432t. [DOI] [PubMed] [Google Scholar]
- 15.Gul H.I., Vepsalainen J., Gul M., Erciyas E., Hanninen O. Cytotoxic activities of mono and bis Mannich bases derived from acetophenone against Renca and Jurkat cells. Pharm. Helv. Acta. 2000;74:393–398. doi: 10.1016/s0031-6865(00)00022-4. [DOI] [PubMed] [Google Scholar]
- 16.Gul H.I., Gul M., Hanninen O. Cytotoxic activities of some mono and bis Mannich bases derived from acetophenone in brine shrimp bioassay. Arzneimittelforschung. 2002;52:840–843. doi: 10.1055/s-0031-1299977. [DOI] [PubMed] [Google Scholar]
- 17.Gul H.I., Gul M., Vepsalainen J., Erciyas E., Hanninen O. Cytotoxicity of some azines of acetophenone-derived Mannich bases against Jurkat cells. Biol. Pharm. Bull. 2003;26:631–637. doi: 10.1248/bpb.26.631. [DOI] [PubMed] [Google Scholar]
- 18.Kucukoglu K., Gul M., Atalay M., Mete E., Kazaz C., Hanninen O., Gul H.I. Synthesis of some Mannich bases with dimethylamine and their hydrazones and evaluation of their cytotoxicity against Jurkat cells. Arzneimittelforschung. 2011;61:366–371. doi: 10.1055/s-0031-1296212. [DOI] [PubMed] [Google Scholar]
- 19.Gul H.I., Das U., Pandit B., Li P.K. Evaluation of the cytotoxicity of some mono-mannich bases and their corresponding azine derivatives against androgen-independent prostate cancer cells. Arzneimittelforschung. 2006;56:850–854. doi: 10.1055/s-0031-1296797. [DOI] [PubMed] [Google Scholar]
- 20.Vashishtha S.C., Zello G.A., Nienaber K.H., Balzarini J., De Clercq E., Stables J.P., Dimmock J.R. Cytotoxic and anticonvulsant aryloxyaryl Mannich bases and related compounds. Eur. J. Med. Chem. 2004;39:27–35. doi: 10.1016/j.ejmech.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 21.Gul M., Gul H.I., Das U., Hanninen O. Biological evaluation and structure-activity relationships of bis-(3-aryl-3-oxo-propyl)-methylamine hydrochlorides and 4-aryl-3-arylcarbonyl-1-methyl-4-piperidinol hydrochlorides as potential cytotoxic agents and their alkylating ability towards cellular glutathione in human leukemic T cells. Arzneimittelforschung. 2005;55:332–337. doi: 10.1055/s-0031-1296868. [DOI] [PubMed] [Google Scholar]
- 22.Canturk P., Kucukoglu K., Topcu Z., Gul M., Gul H.I. Effect of some bis Mannich bases and corresponding piperidinols on DNA topoisomerase I. Arzneimittelforschung. 2008;58:686–691. [PubMed] [Google Scholar]
- 23.Mete E., Gul H.I., Cetin-Atalay R., Das U., Sahin E., Gul M., Kazaz C., Dimmock J.R. The design and cytotoxic evaluation of some 1-aryl-3-isopropylamino-1-propanone hydrochlorides towards human Huh-7 hepatoma cells. Arch. Pharm. (Weinheim) 2011;344:333–339. doi: 10.1002/ardp.201000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gul M., Mete E., Atalay M., Arik M., Gul H.I. Cytotoxicity of 1-aryl-3-butylamino-1-propanone hydrochlorides against Jurkat and L6 cells. Arzneimittelforschung. 2009;59:364–369. doi: 10.1055/s-0031-1296409. [DOI] [PubMed] [Google Scholar]
- 25.Mete E., Gul H.I., Kazaz C. Synthesis of 1-aryl-3-phenethylamino-1-propanone hydrochlorides as possible potent cytotoxic agents. Molecules. 2007;12:2579–2588. doi: 10.3390/12122579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mete E., Gul H.I., Canturk P., Topcu Z., Pandit B., Gul M., Li P.-K. Biological activity of 1-aryl-3-phenethylamino-1-propanone hydrochlorides and 3-aroyl-4-aryl-1-phenethyl-4-piperidinols on PC-3 cells and DNA topoisomerase I enzyme. Z. Naturforsch. C: J. Biosci. 2010;65C:647–652. doi: 10.1515/znc-2010-11-1203. [DOI] [PubMed] [Google Scholar]
- 27.Gul M., Gul H.I., Vepsalainen J., Erciyas E., Hanninen O. Effect of acetophenone derived Mannich bases on cellular glutathione level in Jurkat cells. A possible mechanism of action. Arzneimittelforschung. 2001;51:679–682. doi: 10.1055/s-0031-1300100. [DOI] [PubMed] [Google Scholar]
- 28.Gul H.I., Gul M., Erciyas E. Syntheses and stability studies of some Mannich bases of acetophenones and evaluation of their cytotoxicity against Jurkat cells. Arzneimittelforschung. 2002;52:628–635. doi: 10.1055/s-0031-1299942. [DOI] [PubMed] [Google Scholar]
- 29.Gul M., Gul H.I., Hänninen O. Effects of Mannich bases on cellular glutathione and related enzymes of Jurkat cells in culture conditions. Toxicol. In Vitro. 2002;16:107–112. doi: 10.1016/s0887-2333(01)00115-1. [DOI] [PubMed] [Google Scholar]
- 30.Gul M., Atalay M., Gul H.I., Nakao C., Lappalainen J., Hänninen O. The effects of some Mannich bases on heat shock proteins HSC70 and GRP75, and thioredoxin and glutaredoxin levels in Jurkat cells. Toxicol. In Vitro. 2005;19:573–580. doi: 10.1016/j.tiv.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Dimmock J.R., Padmanilayam M.P., Das U., Zello G.A., Sharma R.K., Shrivastav A., Selvakumar P., Pasha M.K., Nienaber K.H., Lee J.S., Allen T.M., Santos C.L., Balzarini J., de Clercq E. Cytotoxic Mannich bases of 1-arylidene-2-tetralones. J. Enzyme Inhib. Med. Chem. 2003;18:313–324. doi: 10.1080/1475636031000121929. [DOI] [PubMed] [Google Scholar]
- 32.Dimmock J.R., Chamankhah M., Das U., Zello G.A., Quail J.W., Yang J., Nienaber K.H., Sharma R.K., Selvakumar P., Balzarini J., De Clercq E., Stables J.P. Cytotoxic and topographical properties of 6-arylidene-2-dimethylaminomethylcyclohexanone hydrochlorides and related compounds. J. Enzyme Inhib. Med. Chem. 2004;19:1–10. doi: 10.1080/14756360310001624975. [DOI] [PubMed] [Google Scholar]
- 33.Selvakumar P., Shrivastav A., Dimmock J.R., Sharma R.K. Potential role of N-myristoyltransferase in cancer. Prog. Lipid Res. 2007;46:1–36. doi: 10.1016/j.plipres.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Das U., Kumar S., Dimmock J.R., Sharma R.K. Inhibition of protein N-myristoylation: a therapeutic protocol in developing anticancer agents. Curr. Cancer Drug Targets. 2012;12:667–692. doi: 10.2174/156800912801784857. [DOI] [PubMed] [Google Scholar]
- 35.Zhao K., Wang Y., Li D., Tisdale M.J., Zhao L., Ji Z., Schwalbe C.H. Synthesis and cytotoxic and mechanistic studies of α-arylidenecyclohex(pent)anone or α-arylcyclohexanone α'-Mannich bases and their deoxo bisaryl cyclohex(pent)ene analogs. Pharm. Chem. J. 2004;38:229–238. [Google Scholar]
- 36.Wang J., Zhao L., Wang R., Lu M., Chen D., Jing Y. Synthesis and anticancer activity of 2-alkylaminomethyl-5-diaryl-methylenecyclopentanone hydrochlorides and related compounds. Bioorg. Med. Chem. 2005;13:1285–1291. doi: 10.1016/j.bmc.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Pati H.N., Das U., Ramirez-Erosa I.J., Dunlop D.M., Hickie R.A., Dimmock J.R. α-Substituted 1-aryl-3-dimethylaminopropanone hydrochlorides: potent cytotoxins towards human WiDr colon cancer cells. Chem. Pharm. Bull. 2007;55:511–515. doi: 10.1248/cpb.55.511. [DOI] [PubMed] [Google Scholar]
- 38.Pati H.N., Das U., Kawase M., Sakagami H., Balzarini J., De Clercq E., Dimmock J.R. 1-Aryl-2-dimethylaminomethyl-2-propen-1-one hydrochlorides and related adducts: a quest for selective cytotoxicity for malignant cells. Bioorg. Med. Chem. 2008;16:5747–5753. doi: 10.1016/j.bmc.2008.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gul H.I., Yerdelen K.O., Gul M., Das U., Pandit B., Li P.-K., Secen H., Sahin F. Synthesis of 4′-hydroxy-3′-piperidinomethylchalcone derivatives and their cytotoxicity against PC-3 cell lines. Arch. Pharm. (Weinheim) 2007;340:195–201. doi: 10.1002/ardp.200600072. [DOI] [PubMed] [Google Scholar]
- 40.Gul H.I., Yerdelen K.O., Das U., Gul M., Pandit B., Li P.-K., Dimmock J.R. Synthesis and cytotoxicity of novel 3-aryl-1-(3′-dibenzylaminomethyl-4′-hydroxyphenyl)-propenones and related compounds. Chem. Pharm. Bull. 2008;56:1675–1681. doi: 10.1248/cpb.56.1675. [DOI] [PubMed] [Google Scholar]
- 41.Hieu B.T., Thuy L.T., Thuy V.T., Tien H.X., Chinh L.V., Hoang V.D., Vu T.K. Design, synthesis and in vitro cytotoxic activity evaluation of new Mannich bases. Bull. Korean Chem. Soc. 2012;33:1586–1592. [Google Scholar]
- 42.Vijaya Bhaskar Reddy M., Su C.-R., Chiou W.-F., Liu Y.-N., Chen R.Y.-H., Bastow K.F., Lee K.-H., Wu T.-S. Design, synthesis, and biological evaluation of Mannich bases of heterocyclic chalcone analogs as cytotoxic agents. Bioorg. Med. Chem. 2008;16:7358–7370. doi: 10.1016/j.bmc.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Bilginer S., Gul H.I., Mete E., Das U., Sakagami H., Umemura N., Dimmock J.R. 1-(3-Aminomethyl-4-hydroxyphenyl)-3-pyridinyl-2-propen-1-ones: a novel group of tumour-selective cytotoxins. J. Enzyme Inhib. Med. Chem. 2013;28:974–980. doi: 10.3109/14756366.2012.700927. [DOI] [PubMed] [Google Scholar]
- 44.J.H. Van Der Westhuizen, A.E.M.N. Eljaleel, S.L. Bonnet, A. Wilhelm-Mouton, Preparation of Aminoalkyl Substituted Chalcones for Use in Anticancer and Antimalarial Pharmaceutical Compositions, PCT Int. Appl. WO 2011151789, 2011.
- 45.Vijaya Bhaskar Reddy M., Chen S.-S., Lin M.-L., Chan H.-H., Kuo P.-C., Wu T.-S. Preparation of a series of novel bichalcones linked with a 1,4-dimethylenepiperazine moiety and examination of their cytotoxicity. Chem. Pharm. Bull. 2011;59:1549–1554. doi: 10.1248/cpb.59.1549. [DOI] [PubMed] [Google Scholar]
- 46.Vijaya Bhaskar Reddy M., Shen Y.-C., Yang J.-S., Hwang T.-L., Bastow K.F., Qian K., Lee K.-H., Wua T.-S. New bichalcone analogs as NF-ĸB inhibitors and as cytotoxic agents inducing Fas/CD95-dependent apoptosis. Bioorg. Med. Chem. 2011;19:1895–1906. doi: 10.1016/j.bmc.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pereira J., Levy D., Ruiz J.L., Brocardo G.A., Ferreira K.A., Costa R.O., Queiroz R.G., Maria D.A., Neto A.E., Chamone D.A., Bydlowski S.P. Azidothymidine is effective against human multiple myeloma: a new use for an old drug. Anticancer Agents Med. Chem. 2013;13:186–192. [PubMed] [Google Scholar]
- 48.Celewicz L., Jóźwiak A., Ruszkowski P., Laskowska H., Olejnik A., Czarnecka A., Hoffmann M., Hładoń B. Synthesis and anticancer activity of 5′-chloromethylphosphonates of 3′-azido-3′-deoxythymidine (AZT) Bioorg. Med. Chem. 2011;19:6375–6382. doi: 10.1016/j.bmc.2011.08.069. [DOI] [PubMed] [Google Scholar]
- 49.Uckun F.M., Tai H.L., D'Cruz O.J. Antileukemic activity and cellular metabolism of the aryl phosphate derivative of bromo-methoxy zidovudine (compound WHI-07) Arzneimittelforschung. 2005;55:50–65. doi: 10.1055/s-0031-1296824. [DOI] [PubMed] [Google Scholar]
- 50.Vu T.K., Patil S.P., Park Y.J., Thao D.T. Synthesis and in vitro cytotoxic activity evaluation of novel Mannich bases and modified AZT derivatives possessing Mannich base moieties via click chemistry. Lett. Drug Des. Discov. 2013;10:585–593. [Google Scholar]
- 51.Chen H.-L., Chang C.-Y., Lee H.-T., Lin H.-H., Lu P.-J., Yang C.-N., Shiau C.-W., Shaw A.Y. Synthesis and pharmacological exploitation of clioquinol-derived copper-binding apoptosis inducers triggering reactive oxygen species generation and MAPK pathway activation. Bioorg. Med. Chem. 2009;17:7239–7247. doi: 10.1016/j.bmc.2009.08.054. [DOI] [PubMed] [Google Scholar]
- 52.Shen A.Y., Hwang M.H., Roffler S., Chen C.F. Cytotoxicity and antimicrobial activity of some naphthol derivatives. Arch. Pharm. (Weinheim) 1995;328:197–201. doi: 10.1002/ardp.19953280220. [DOI] [PubMed] [Google Scholar]
- 53.Shen A.-Y., Wu S.-N., Chiu C.-T. Synthesis and cytotoxicity evaluation of some 8-hydroxyquinoline derivatives. J. Pharm. Pharmacol. 1999;51:543–548. doi: 10.1211/0022357991772826. [DOI] [PubMed] [Google Scholar]
- 54.Shaw A.Y., Chang C.-Y., Hsu M.-Y., Lu P.-J., Yang C.-N., Chen H.-L., Lo C.-W., Shiau C.-W., Chern M.-K. Synthesis and structure-activity relationship study of 8-hydroxyquinoline-derived Mannich bases as anticancer agents. Eur. J. Med. Chem. 2010;45:2860–2867. doi: 10.1016/j.ejmech.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 55.Yadav Y., MacLean E.D., Bhattacharyya A., Parmar V.S., Balzarini J., Barden C.J., Too C.K.L., Jha A. Design, synthesis and bioevaluation of novel candidate selective estrogen receptor modulators. Eur. J. Med. Chem. 2011;46:3858–3866. doi: 10.1016/j.ejmech.2011.05.054. [DOI] [PubMed] [Google Scholar]
- 56.Jiao J., Xiang H., Liao Q. Recent advancement in nonsteroidal aromatase inhibitors for treatment of estrogen-dependent breast cancer. Curr. Med. Chem. 2010;17:3476–3487. doi: 10.2174/092986710792927877. [DOI] [PubMed] [Google Scholar]
- 57.Mohammed H.A., Ba L.A., Burkholz T., Schumann E., Diesel B., Zapp J., Kiemer A.K., Ries A.C., Hartmann R.W., Hosny M., Jacob C. Facile synthesis of chrysin-derivatives with promising activities as aromatase inhibitors. Nat. Prod. Commun. 2011;6:31–34. [PubMed] [Google Scholar]
- 58.Liu R., Zhao B., Wang D.-E., Yao T., Pang L., Tu Q., Ahmed S.M., Liu J.-J., Wang J. Nitrogen-containing apigenin analogs: preparation and biological activity. Molecules. 2012;17:14748–14764. doi: 10.3390/molecules171214748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Issa S., Walchshofer N., Kassab I., Termoss H., Chamat S., Geahchan A., Bouaziz Z. Synthesis and antiproliferative activity of oxazinocarbazole and N,N-bis(carbazolylmethyl)amine derivatives. Eur. J. Med. Chem. 2010;45:2567–2577. doi: 10.1016/j.ejmech.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 60.Asche C., Frank W., Albert A., Kucklaender U. Synthesis, antitumour activity and structure–activity relationships of 5H-benzo[b]carbazoles. Bioorg. Med. Chem. 2005;13:819–837. doi: 10.1016/j.bmc.2004.10.038. [DOI] [PubMed] [Google Scholar]
- 61.Paull K.D., Shoemaker R.H., Hodes L., Monks A., Scudiero D.A., Rubinstein L., Plowman J., Boyd M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 62.Schenck L.W., Kuna K., Frank W., Albert A., Asche C., Kucklaender U. 1,4,9,10-Anthradiquinone as precursor for antitumor compounds. Bioorg. Med. Chem. 2006;14:3599–3614. doi: 10.1016/j.bmc.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 63.Asche C. Antitumour quinones. Mini Rev. Med. Chem. 2005;5:449–467. doi: 10.2174/1389557053765556. [DOI] [PubMed] [Google Scholar]
- 64.Lee J.-H., Yeon J.-H., Kim H., Roh W., Chae J., Park H.-O., Kim D.-M. The natural anticancer agent plumbagin induces potent cytotoxicity in MCF-7 human breast cancer cells by inhibiting a PI-5 kinase for ROS generation. PLOS One. 2012;7:e45023. doi: 10.1371/journal.pone.0045023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawamura M., Kuriyama I., Maruo S., Kuramochi K., Tsubaki K., Yoshida H., Mizushina Y. Anti-tumor effects of novel 5-O-acyl plumbagins based on the inhibition of mammalian DNA replicative polymerase activity. PLOS One. 2014;9:e88736. doi: 10.1371/journal.pone.0088736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maruo S., Kuriyama I., Kuramochi K., Tsubaki K., Yoshida H., Mizushina Y. Inhibitory effect of novel 5-O-acyl juglones on mammalian DNA polymerase activity, cancer cell growth and inflammatory response. Bioorg. Med. Chem. 2011;19:5803–5812. doi: 10.1016/j.bmc.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 67.Montenegro R.C., Araújo A.J., Molina M.T., Marinho Filho J.D.B., Rocha D.D., Lopéz-Montero E., Goulart M.O.F., Bento E.S., Alves A.P.N.N., Pessoa C., de Moraes M.O., Costa-Lotufo L.V. Cytotoxic activity of naphthoquinones with special emphasis on juglone and its 5-O-methyl derivative. Chem. Biol. Interact. 2010;184:439–448. doi: 10.1016/j.cbi.2010.01.041. [DOI] [PubMed] [Google Scholar]
- 68.Bonifazi E.L., Ríos-Luci C., León L.G., Burton G., Padrón J.M., Misico R.I. Antiproliferative activity of synthetic naphthoquinones related to lapachol. First synthesis of 5-hydroxylapachol. Bioorg. Med. Chem. 2010;18:2621–2630. doi: 10.1016/j.bmc.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 69.Neves A.P., da Silva G.B., Vargas M.D., Pinheiro C.B., Visentin L. do C., Marinho Filho J.D.B., Araújo A.J., Costa-Lotufo L.V., Pessoa C., de Moraes M.O. Novel platinum(II) complexes of 3-(aminomethyl)naphthoquinone Mannich bases: synthesis, crystal structure and cytotoxic activities. Dalton Trans. 2010;39:10203–10216. doi: 10.1039/c0dt00572j. [DOI] [PubMed] [Google Scholar]
- 70.Neves A.P., Pereira M.X.G., Peterson E.J., Kipping R., Vargas M.D., Silva F.P., Jr., Carneiro J.W.M., Farrell N.P. Exploring the DNA binding/cleavage, cellular accumulation and topoisomerase inhibition of 2-hydroxy-3-(aminomethyl)-1,4-naphthoquinone Mannich bases and their platinum(II) complexes. J. Inorg. Biochem. 2013;119:54–64. doi: 10.1016/j.jinorgbio.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 71.da Silva G.B., Neves A.P., Vargas M.D., Alves W.A., Marinho-Filho J.D.B., Pessoa C., Moraes M.O., Costa-Lotufo L.V. Novel 3-(aminomethyl)naphthoquinone Mannich base-platinum(IV) complexes: synthesis, characterization, electrochemical and cytotoxic studies. J. Braz. Chem. Soc. 2013;24:675–684. [Google Scholar]
- 72.Huczyński A., Rutkowski J., Borowicz I., Wietrzyk J., Maj E., Brzezinski B. One-pot synthesis and cytotoxicity studies of new Mannich base derivatives of polyether antibiotic – Lasalocid acid. Bioorg. Med. Chem. Lett. 2013;23:5053–5056. doi: 10.1016/j.bmcl.2013.07.040. [DOI] [PubMed] [Google Scholar]
- 73.Wang S., Li Y., Liu Y., Lu A., You Q. Novel hexacyclic camptothecin derivatives. Part 1: synthesis and cytotoxicity of camptothecins with an A-ring fused 1,3-oxazine ring. Bioorg. Med. Chem. Lett. 2008;18:4095–4097. doi: 10.1016/j.bmcl.2008.05.103. [DOI] [PubMed] [Google Scholar]
- 74.Behery F.A., Akl M.R., Ananthula S., Parajuli P., Sylvester P.W., El Sayed K.A. Optimization of tocotrienols as antiproliferative and antimigratory leads. Eur. J. Med. Chem. 2013;59:329–341. doi: 10.1016/j.ejmech.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 75.Di Antonio M., Doria F., Richter S.N., Bertipaglia C., Mella M., Sissi C., Palumbo M., Freccero M. Quinone methides tethered to naphthalene diimides as selective G-quadruplex alkylating agents. J. Am. Chem. Soc. 2009;131:13132–13141. doi: 10.1021/ja904876q. [DOI] [PubMed] [Google Scholar]
- 76.Doria F., Nadai M., Folini M., Di Antonio M., Germani L., Percivalle C., Sissi C., Zaffaroni N., Alcaro S., Artese A., Richter S.N., Freccero M. Hybrid ligand-alkylating agents targeting telomeric G-quadruplex structures. Org. Biomol. Chem. 2012;10:2798–2806. doi: 10.1039/c2ob06816h. [DOI] [PubMed] [Google Scholar]
- 77.Zhang L., Ren L., Bai M., Weng L., Huang J., Wu L., Deng M., Zhou X. Synthesis and biological activities of quinazoline derivatives with ortho-phenol-quaternary ammonium salt groups. Bioorg. Med. Chem. 2007;15:6920–6926. doi: 10.1016/j.bmc.2007.07.053. [DOI] [PubMed] [Google Scholar]
- 78.Abdelhafez O.M., Abedelatif N.A., Badria F.A. DNA binding, antiviral activities and cytotoxicity of new furochromone and benzofuran derivatives. Arch. Pharm. Res. 2011;34:1623–1632. doi: 10.1007/s12272-011-1006-2. [DOI] [PubMed] [Google Scholar]
- 79.Lan W., Wei J., Qiu J., Yang Z., Su G., Dai Z. Synthesis and biological evaluation of 1,3-dihydroxyxanthone Mannich base derivatives as potential antitumor agents. Lett. Drug Des. Discov. 2013;10:689–698. [Google Scholar]
- 80.Yarim M., Koksal M., Schepmann D., Wünsch B. Synthesis and in vitro evaluation of novel indole-based sigma receptors ligands. Chem. Biol. Drug Des. 2011;78:869–875. doi: 10.1111/j.1747-0285.2011.01215.x. [DOI] [PubMed] [Google Scholar]
- 81.Koksal M., Yarim M., Durmaz I., Cetin-Atalay R. Synthesis and cytotoxic activity of novel 3-methyl-1-[(4-substituted-1-piperazinyl)methyl]-1H-indole derivatives. Arzneimittelforschung. 2012;62:389–394. doi: 10.1055/s-0032-1314868. [DOI] [PubMed] [Google Scholar]
- 82.Köksal Akkoç M., Yarim Yüksel M., Durmaz I., Çetin Atalay R. Design, synthesis, and biological evaluation of indole-based 1,4-disubstituted piperazines as cytotoxic agents. Turk. J. Chem. 2012;36:515–525. [Google Scholar]
- 83.Go M.-L., Leow J.L., Gorla S.K., Schüller A.P., Wang M., Casey P.J. Amino derivatives of indole as potent inhibitors of isoprenylcysteine carboxyl methyltransferase. J. Med. Chem. 2010;53:6838–6850. doi: 10.1021/jm1002843. [DOI] [PubMed] [Google Scholar]
- 84.Ramanujulu P.M., Yang T., Yap S.-Q., Wong F.-C., Casey P.J., Wang M., Go M.-L. Functionalized indoleamines as potent, drug-like inhibitors of isoprenylcysteine carboxyl methyltransferase (Icmt) Eur. J. Med. Chem. 2013;63:378–386. doi: 10.1016/j.ejmech.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 85.Shchekotikhin A.E., Shtil A.A., Luzikov Y.N., Bobrysheva T.V., Buyanov V.N., Preobrazhenskaya M.N. 3-Aminomethyl derivatives of 4,11-dihydroxynaphtho[2,3-f]indole-5,10-dione for circumvention of anticancer drug resistance. Bioorg. Med. Chem. 2005;13:2285–2291. doi: 10.1016/j.bmc.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 86.Shchekotikhin A.E., Glazunova V.A., Luzikov Y.N., Buyanov V.N., Susova O.Yu., Shtil A.A., Preobrazhenskaya M.N. Synthesis and structure–activity relationship studies of 4,11-diaminonaphtho[2,3-f]indole-5,10-diones. Bioorg. Med. Chem. 2006;14:5241–5251. doi: 10.1016/j.bmc.2006.03.052. [DOI] [PubMed] [Google Scholar]
- 87.Dezhenkova L.G., Susova O.Yu., Shchekotikhin A.E., Preobrazhenskaya M.N., Shtil A.A. Naphtho[2,3-f]indole-5,10-dione aminoalkyl derivatives: a new class of topoisomerase I inhibitors. Bull. Exp. Biol. Med. 2008;145:334–337. doi: 10.1007/s10517-008-0084-8. [DOI] [PubMed] [Google Scholar]
- 88.Simonov A.Yu., Lakatosh S.A., Luzikov Yu.N., Reznikova M.I., Susova O.Yu., Shtil' A.A., Elizarov S.M., Danilenko V.N., Preobrazhenskaya M.N. Synthesis of 4-substituted 3-[3-(dialkylaminomethyl)indol-1-yl]maleimides and study of their ability to inhibit protein kinase C-α, prevent development of multiple drug resistance of tumor cells and cytotoxicity. Russ. Chem. Bull. 2008;57:2011–2020. [Google Scholar]
- 89.Chen Z., Venkatesan A.M., Dehnhardt C.M., Ayral-Kaloustian S., Brooijmans N., Mallon R., Feldberg L., Hollander I., Lucas J., Yu K., Kong F., Mansour T.S. Synthesis and SAR of novel 4-morpholinopyrrolopyrimidine derivatives as potent phosphatidylinositol 3-kinase inhibitors. J. Med. Chem. 2010;53:3169–3182. doi: 10.1021/jm901783v. [DOI] [PubMed] [Google Scholar]
- 90.Vine K.L., Matesic L., Locke J.M., Ranson M., Skropeta D. Cytotoxic and anticancer activities of isatin and its derivatives: a comprehensive review from 2000-2008. Anti-Cancer Agents Med. Chem. 2009;9:397–414. doi: 10.2174/1871520610909040397. [DOI] [PubMed] [Google Scholar]
- 91.Yogeeswari P., Sriram D., Kavya R., Tiwari S. Synthesis and in-vitro cytotoxicity evaluation of gatifloxacin Mannich bases. Biomed. Pharmacother. 2005;59:501–510. doi: 10.1016/j.biopha.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 92.Raja Solomon V., Hu C., Lee H. Hybrid pharmacophore design and synthesis of isatin–benzothiazole analogs for their anti-breast cancer activity. Bioorg. Med. Chem. 2009;17:7585–7592. doi: 10.1016/j.bmc.2009.08.068. [DOI] [PubMed] [Google Scholar]
- 93.Taher A.T., Khalil N.A., Ahmed E.M. Synthesis of novel isatin-thiazoline and isatin-benzimidazole conjugates as anti-breast cancer agents. Arch. Pharm. Res. 2011;34:1615–1621. doi: 10.1007/s12272-011-1005-3. [DOI] [PubMed] [Google Scholar]
- 94.Raja Solomon V., Hu C., Lee H. Design and synthesis of anti-breast cancer agents from 4-piperazinylquinoline: a hybrid pharmacophore approach. Bioorg. Med. Chem. 2010;18:1563–1572. doi: 10.1016/j.bmc.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 95.Aboraia A.S., Abdel-Rahman H.M., Mahfouz N.M., El-Gendy M.A. Novel 5-(2-hydroxyphenyl)-3-substituted-2,3-dihydro-1,3,4-oxadiazole-2-thione derivatives: promising anticancer agents. Bioorg. Med. Chem. 2006;14:1236–1246. doi: 10.1016/j.bmc.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 96.Dash S., Kumar B.A., Singh J., Maiti B.C., Maity T.K. Synthesis of some novel 3,5-disubstituted 1,3,4-oxadiazole derivatives and anticancer activity on EAC animal model. Med. Chem. Res. 2011;20:1206–1213. [Google Scholar]
- 97.Rahman D.E.A. Synthesis, quantitative structure-activity relationship and biological evaluation of 1,3,4-oxadiazole derivatives possessing diphenylamine moiety as potential anticancer agents. Chem. Pharm. Bull. 2013;61:151–159. doi: 10.1248/cpb.c12-00637. [DOI] [PubMed] [Google Scholar]
- 98.Savariz F.C., Formagio A.S.N., Barbosa V.A., Foglio M.A., de Carvalho J.E., Duarte M.C.T., Dias Filho B.P., Sarragiotto M.H. Synthesis, antitumor and antimicrobial activity of novel 1-substituted phenyl-3-[3-alkylamino(methyl)-2-thioxo-1,3,4-oxadiazol-5-yl]-β-carboline derivatives. J. Braz. Chem. Soc. 2010;21:288–298. [Google Scholar]
- 99.Chen Y., Wang G., Duan N., Cao T., Wen X., Yin J., Wang W., Xie S., Huang W., Hu G. Synthesis and antitumor activity of fluoroquinolone C3-isostere derivatives: oxadiazole Mannich base derivatives (in Chinese) Yingyong Huaxue. 2012;29:1246–1250. [Google Scholar]
- 100.Sunil D., Isloor A.M., Shetty P., Chandrakantha B., Satyamoorthy K. Synthesis, characterization and in vitro cytotoxic properties of some new Schiff and Mannich bases in Hep G2 cells. Med. Chem. Res. 2011;20:1024–1032. [Google Scholar]
- 101.Hu G., Wang G., Duan N., Wen X., Cao T., Xie S., Huang W. Design, synthesis and antitumor activities of fluoroquinolone C-3 heterocycles (IV): s-triazole Schiff–Mannich bases derived from ofloxacin. Acta Pharm. Sin. B. 2012;2:312–317. [Google Scholar]
- 102.Hanna M.M., George R.F. Facile synthesis and quantitative structure–activity relationship study of antitumor active 2-(4-oxo-thiazolidin-2-ylidene)-3-oxo-propionitriles. Chem. Pharm. Bull. 2012;60:1195–1206. doi: 10.1248/cpb.c12-00498. [DOI] [PubMed] [Google Scholar]
- 103.Havrylyuk D., Zimenkovsky B., Vasylenko O., Day C.W., Smee D.F., Grellier P., Lesyk R. Synthesis and biological activity evaluation of 5-pyrazoline substituted 4-thiazolidinones. Eur. J. Med. Chem. 2013;66:228–237. doi: 10.1016/j.ejmech.2013.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jin L., Song B., Zhang G., Xu R., Zhang S., Gao X., Hu D., Yang S. Synthesis, X-ray crystallographic analysis, and antitumor activity of N-(benzothiazole-2-yl)-1-(fluorophenyl)-O,O-dialkyl-α-aminophosphonates. Bioorg. Med. Chem. Lett. 2006;16:1537–1543. doi: 10.1016/j.bmcl.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 105.Ma J., Zhu W., Li J., Ji P., Zhu Z., Liao X. Synthesis and biological activities of α-aminophosphonates derivatives containing thieno[3,2-c]pyridine (in Chinese) Chin. J. Org. Chem. 2013;33:1472–1477. [Google Scholar]
- 106.Abdel-Megeed M.F., Badr B.E., Azaam M.M., El-Hiti G.A. Synthesis, antimicrobial and anticancer activities of a novel series of diphenyl 1-(pyridin-3-yl)ethylphosphonates. Bioorg. Med. Chem. 2012;20:2252–2258. doi: 10.1016/j.bmc.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 107.Rezaei Z., Firouzabadi H., Iranpoor N., Ghaderi A., Jafari M.R., Jafari A.A., Zare H.R. Design and one-pot synthesis of α-aminophosphonates and bis(α-aminophosphonates) by iron(III) chloride and cytotoxic activity. Eur. J. Med. Chem. 2009;44:4266–4275. doi: 10.1016/j.ejmech.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 108.Euzébio F.P.G., dos Santos F.J.L., Piló-Veloso D., Alcântara A.F.C., Ruiz A.L.T.G., de Carvalho J.E., Foglio M.A., Ferreira-Alves D.L., de Fátima A. Synthesis, antiproliferative activity in cancer cells and theoretical studies of novel 6α,7β-dihydroxyvouacapan-17β-oic acid Mannich base derivatives. Bioorg. Med. Chem. 2010;18:8172–8177. doi: 10.1016/j.bmc.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 109.Zhao X.-L., Zhao Y.-F., Guo S.-C., Song H.-S., Wang D., Gong P. Synthesis and anti-tumor activities of novel [1,2,4]triazolo[1,5-a]pyrimidines. Molecules. 2007;12:1136–1146. doi: 10.3390/12051136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhai X., Zhao Y.-F., Liu Y.-J., Zhang Y., Xun F.-Q., Liu J., Gong P. Synthesis and cytotoxicity studies of novel [1,2,4]triazolo[1,5-a]pyrimidine-7-amines. Chem. Pharm. Bull. 2008;56:941–945. doi: 10.1248/cpb.56.941. [DOI] [PubMed] [Google Scholar]
- 111.Hickey T., Claffey J., Fitzpatrick E., Hogan M., Pampillón C., Tacke M. Dimethylamino-functionalised and N-heteroaryl-substituted titanocene anticancer drugs: synthesis and cytotoxicity studies. Investig. New Drugs. 2007;25:425–433. doi: 10.1007/s10637-007-9061-8. [DOI] [PubMed] [Google Scholar]
- 112.Pampillón C., Claffey J., Hogan M., Strohfeldt K., Tacke M. Synthesis and cytotoxicity studies of new dimethylamino-functionalised and indolyl-substituted titanocene anti-cancer drugs. Transit. Metal Chem. 2007;32:434–441. [Google Scholar]
- 113.Hogan M., Claffey J., Pampillón C., Watson R.W.G., Tacke M. Synthesis and cytotoxicity studies of new dimethylamino-functionalized and azole-substituted titanocene anticancer drugs. Organometallics. 2007;26:2501–2506. [Google Scholar]
- 114.Hogan M., Gleeson B., Tacke M. Synthesis and preliminary cytotoxicity studies of achiral indolyl-substituted titanocenes. Organometallics. 2010;29:1032–1040. [Google Scholar]
- 115.Bisi A., Meli M., Gobbi S., Rampa A., Tolomeo M., Dusonchet L. Multidrug resistance reverting activity and antitumor profile of new phenothiazine derivatives. Bioorg. Med. Chem. 2008;16:6474–6482. doi: 10.1016/j.bmc.2008.05.040. [DOI] [PubMed] [Google Scholar]
- 116.Csuk R., Nitsche C., Sczepek R., Schwarz S., Siewert B. Synthesis of antitumor-active betulinic acid-derived hydroxypropargylamines by copper-catalyzed Mannich reactions. Arch. Pharm. (Weinheim) 2013;346:232–246. doi: 10.1002/ardp.201200428. [DOI] [PubMed] [Google Scholar]
- 117.Csuk R., Sczepek R., Siewert B., Nitsche C. Cytotoxic betulin-derived hydroxypropargylamines trigger apoptosis. Bioorg. Med. Chem. 2013;21:425–435. doi: 10.1016/j.bmc.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 118.Fenick D.J., Taatjes D.J., Koch T.H. Doxoform and Daunoform: anthracycline-formaldehyde conjugates toxic to resistant tumor cells. J. Med. Chem. 1997;40:2452–2461. doi: 10.1021/jm970237e. [DOI] [PubMed] [Google Scholar]
- 119.Cogan P.S., Fowler C.R., Post G.C., Koch T.H. Doxsaliform: a novel N-Mannich base prodrug of a doxorubicin formaldehyde conjugate. Lett. Drug Des. Discov. 2004;1:247–255. [Google Scholar]
- 120.Burke P.J., Koch T.H. Design, synthesis, and biological evaluation of doxorubicin-formaldehyde conjugates targeted to breast cancer cells. J. Med. Chem. 2004;47:1193–1206. doi: 10.1021/jm030352r. [DOI] [PubMed] [Google Scholar]
- 121.Burke P.J., Kalet B.T., Koch T.H. Antiestrogen binding site and estrogen receptor mediate uptake and distribution of 4-hydroxytamoxifen-targeted doxorubicin-formaldehyde conjugate in breast cancer cells. J. Med. Chem. 2004;47:6509–6518. doi: 10.1021/jm049496b. [DOI] [PubMed] [Google Scholar]
- 122.Cogan P.S., Koch T.H. Rational design and synthesis of androgen receptor-targeted nonsteroidal anti-androgen ligands for the tumor-specific delivery of a doxorubicin-formaldehyde conjugate. J. Med. Chem. 2003;46:5258–5270. doi: 10.1021/jm0303305. [DOI] [PubMed] [Google Scholar]
- 123.Cogan P.S., Koch T.H. Studies of targeting and intracellular trafficking of an anti-androgen doxorubicin-formaldehyde conjugate in PC-3 prostate cancer cells bearing androgen receptor-GFP chimera. J. Med. Chem. 2004;47:5690–5699. doi: 10.1021/jm0495226. [DOI] [PubMed] [Google Scholar]
- 124.Burkhart D.J., Kalet B.T., Koch T.H. Doxorubicin-formaldehyde conjugates targeting αvβ3 integrin. Mol. Cancer Ther. 2004;3:1593–1604. [PubMed] [Google Scholar]
- 125.Zhao Y.-J., Wei W., Su Z.-G., Ma G.-H. Poly (ethylene glycol) prodrug for anthracyclines via N-Mannich base linker: design, synthesis and biological evaluation. Int. J. Pharm. 2009;379:90–99. doi: 10.1016/j.ijpharm.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 126.Chen J., Lou J., Liu T., Wu R., Dong X., He Q., Yang B., Hu Y. Synthesis and in-vitro antitumor activities of some Mannich bases of 9-alkyl-1,2,3,4-tetrahydrocarbazole-1-ones. Arch. Pharm. (Weinheim) 2009;342:165–172. doi: 10.1002/ardp.200800179. [DOI] [PubMed] [Google Scholar]
- 127.Kumbhare R.M., Kumar K.V., Ramaiah M.J., Dadmal T., Pushpavalli S.N.C.V.L., Mukhopadhyay D., Divya B., Anjana Devi T., Kosurkar U., Pal-Bhadra M. Synthesis and biological evaluation of novel Mannich bases of 2-arylimidazo[2,1-b]benzothiazoles as potential anti-cancer agents. Eur. J. Med. Chem. 2011;46:4258–4266. doi: 10.1016/j.ejmech.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 128.Ivanova Y., Momekov G., Petrov O., Karaivanova M., Kalcheva V. Cytotoxic Mannich bases of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones. Eur. J. Med. Chem. 2007;42:1382–1387. doi: 10.1016/j.ejmech.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 129.Petrov O., Ivanova Y., Momekov G., Kalcheva V. New synthetic chalcones: cytotoxic Mannich bases of 6-(4-chlorocinnamoyl)-2(3H)-benzoxazolone. Lett. Drug Des. Discov. 2008;5:358–361. [Google Scholar]
- 130.Pau A., Murineddu G., Asproni B., Murruzzu C., Grella G.E., Pinna G.A., Curzu M.M., Marchesi I., Bagella L. Synthesis and cytotoxicity of novel hexahydrothieno-cycloheptapyridazinone derivatives. Molecules. 2009;14:3494–3508. doi: 10.3390/molecules14093494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lóránd T., Kocsis B., Sohár P., Nagy G., Kispál G., Krane H.-G., Schmitt H., Weckert E. Synthesis and antibacterial study of unsaturated Mannich ketones. Eur. J. Med. Chem. 2001;36:705–717. doi: 10.1016/s0223-5234(01)01264-8. [DOI] [PubMed] [Google Scholar]
- 132.Lóránd T., Kocsis B., Sohár P., Nagy G., József P., Kispál G., László R., Prókai L. Synthesis and antibacterial activity of fused Mannich ketones. Eur. J. Med. Chem. 2002;37:803–812. doi: 10.1016/s0223-5234(02)01404-6. [DOI] [PubMed] [Google Scholar]
- 133.Srivastava B.K., Kapadnis P.B., Pandya P., Lohray V.B. Novel Mannich ketones of oxazolidinones as antibacterial agents. Eur. J. Med. Chem. 2004;39:989–992. doi: 10.1016/j.ejmech.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 134.Bandgar B.P., Patil S.A., Korbad B.L., Biradar S.C., Nile S.N., Khobragade C.N. Synthesis and biological evaluation of a novel series of 2,2-bisaminomethylated aurone analogues as anti-inflammatory and antimicrobial agents. Eur. J. Med. Chem. 2010;45:3223–3227. doi: 10.1016/j.ejmech.2010.03.045. [DOI] [PubMed] [Google Scholar]
- 135.Gul H.I., Sahin F., Gul M., Ozturk S., Yerdelen K.O. Evaluation of antimicrobial activities of several Mannich bases and their derivatives. Arch. Pharm. (Weinheim) 2005;338:335–338. doi: 10.1002/ardp.200400962. [DOI] [PubMed] [Google Scholar]
- 136.Sankappa Rai U., Isloor A.M., Shetty P., Isloor N., Malladi S., Fun H.-K. Synthesis and biological evaluation of aminoketones. Eur. J. Med. Chem. 2010;45:6090–6094. doi: 10.1016/j.ejmech.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 137.El-Bayouki Kh.A.M., Basyouni W.M., El-Sayed A.S., Tohamy W.M., El-Henawy A.A. Highly efficient one-pot synthesis, antimicrobial and docking studies of newer β-amino carbonyl derivatives catalyzed by silica sulfuric acid. Croat. Chem. Acta. 2012;85:255–268. [Google Scholar]
- 138.Kenchappa R., Bodke Y.D., Peethambar S.K., Telkar S., Bhovi V.K. Synthesis of β-amino carbonyl derivatives of coumarin and benzofuran and evaluation of their biological activity. Med. Chem. Res. 2013;22:4787–4797. [Google Scholar]
- 139.Damljanović I., Stevanović D., Pejović A., Vukićević M., Novaković S.B., Bogdanović G.A., Mihajlov-Krstev T., Radulović N., Vukićević R.D. Antibacterial 3-(arylamino)-1-ferrocenylpropan-1-ones: synthesis, spectral, electrochemical and structural characterization. J. Organomet. Chem. 2011;696:3703–3713. [Google Scholar]
- 140.Chouhan H.S., Singh S.K., Moorthy N.S.H.N. Synthesis and characterization of some Mannich bases as potential antimicrobial agents. Asian J. Chem. 2010;22:7903–7908. [Google Scholar]
- 141.Gres A., Loginova N., Koval'chuk T., Polosov G., Osipovich N., Strakha I., Azarko I., Zheldakova R. Synthesis, characterization and antimicrobial evaluation of copper(II) complexes of 5-tert-butyl-pyrocatechin-derived Mannich bases. Chemija. 2012;23:286–293. [Google Scholar]
- 142.Husain A., Maaz M., Ansari K.A., Ahmad A., Rashid M. Synthesis and microbiological evaluation of Mannich bases derived from 4,6-diacetylresorcinol. J. Chil. Chem. Soc. 2010;55:332–334. [Google Scholar]
- 143.Tambo-ong A., Chopra S., Glaser B.T., Matsuyama K., Tran T., Madrid P.B. Mannich reaction derivatives of novobiocin with modulated physiochemical properties and their antibacterial activities. Bioorg. Med. Chem. Lett. 2011;21:5697–5700. doi: 10.1016/j.bmcl.2011.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Neves A.P., Barbosa C.C., Greco S.J., Vargas M.D., Visentin L.C., Pinheiro C.B., Mangrich A.S., Barbosa J.P., da Costa G.L. Novel aminonaphthoquinone Mannich bases derived from lawsone and their copper(II) complexes: synthesis, characterization and antibacterial activity. J. Braz. Chem. Soc. 2009;20:712–727. [Google Scholar]
- 145.Akhter M., Husain A., Akhter N., Khan M.S.Y. Synthesis, antiinflammatory and antimicrobial activity of some new 1-(3-phenyl-3,4-dihydro-2H-1,3-benzoxazin-6-yl)ethanone derivatives. Indian J. Pharm. Sci. 2011;73:101–104. doi: 10.4103/0250-474X.89767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mathew B.P., Kumar A., Sharma S., Shukla P.K., Nath M. An eco-friendly synthesis and antimicrobial activities of dihydro-2H-benzo- and naphtho-1,3-oxazine derivatives. Eur. J. Med. Chem. 2010;45:1502–1507. doi: 10.1016/j.ejmech.2009.12.058. [DOI] [PubMed] [Google Scholar]
- 147.Prasad D., Rohilla R.K., Roy N., Nath M. Synthesis and antibacterial evaluation of benzazoles tethered dihydro[1,3]oxazines. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2012;51B:739–745. [Google Scholar]
- 148.Kategaonkar A.H., Sonar S.S., Pokalwar R.U., Kategaonkar A.H., Shingate B.B., Shingare M.S. An efficient synthesis of 3,4-dihydro-3-substituted-2H-naphtho[2,1-e][1,3]oxazine derivatives catalyzed by zirconyl chloride and evaluation of its biological activities. Bull. Korean Chem. Soc. 2010;31:1657–1660. [Google Scholar]
- 149.Negm N.A., Morsy S.M.I., Said M.M. Biocidal activity of some Mannich base cationic derivatives. Bioorg. Med. Chem. 2005;13:5921–5926. doi: 10.1016/j.bmc.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 150.Donawade D.S., Gadaginamath G.S. Some electrophilic substitution reactions on 1-substituted-3-acetyl/carbethoxy-5-hydroxy-2-methylindole and the antimicrobial activity of these new indole derivatives. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2005;44B:1679–1685. [Google Scholar]
- 151.Us D., Berk B., Gürdal E., Aytekin N., Kocagöz T., Çağlayan B., Aksan Kurnaz I., Demir Erol D. Mannich base derivatives of 3-hydroxy-6-methyl-4H-pyran-4-one with antimicrobial activity. Turk. J. Chem. 2010;34:447–456. [Google Scholar]
- 152.Aytemir M.D., Özçelik B. A study of cytotoxicity of novel chlorokojic acid derivatives with their antimicrobial and antiviral activities. Eur. J. Med. Chem. 2010;45:4089–4095. doi: 10.1016/j.ejmech.2010.05.069. [DOI] [PubMed] [Google Scholar]
- 153.Aytemir M.D., Özçelik B. Synthesis and biological activities of new Mannich bases of chlorokojic acid derivatives. Med. Chem. Res. 2011;20:443–452. [Google Scholar]
- 154.Karakaya G., Aytemir M.D., Özçelik B., Çaliş Ü. Design, synthesis and in vivo/in vitro screening of novel chlorokojic acid derivatives. J. Enzyme Inhib. Med. Chem. 2013;28:627–638. doi: 10.3109/14756366.2012.666538. [DOI] [PubMed] [Google Scholar]
- 155.Emami S., Ghafouri E., Faramarzi M.A., Samadi N., Irannejad H., Foroumadi A. Mannich bases of 7-piperazinylquinolones and kojic acid derivatives: synthesis, in vitro antibacterial activity and in silico study. Eur. J. Med. Chem. 2013;68:185–191. doi: 10.1016/j.ejmech.2013.07.032. [DOI] [PubMed] [Google Scholar]
- 156.Khan S.A., Siddiqui N., Imran M., Haque S.W. Synthesis and antimicrobial screening of novel Mannich bases of isatin derivative. Indian J. Pharm. Sci. 2004;66:830–834. [Google Scholar]
- 157.Jain S., Sharma A., Agrawal M., Sharma S., Dwivedi J., Kishore D. Synthesis and antimicrobial evaluation of some novel trisubstituted s-triazine derivatives based on isatinimino, sulphonamido and azacarbazole. J. Chem. 2013:925439. [Google Scholar]
- 158.Mogilaiah K., Sakram B. Synthesis and antimicrobial screening of certain novel Mannich bases bearing 1,8-naphthyridine moiety. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2004;43B:2724–2726. [Google Scholar]
- 159.Shetgiri N.P., Nayak B.K. Synthesis and antimicrobial activities of oxadiazoles, phthalazines and indolinones. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2005;44B:1267–1272. [Google Scholar]
- 160.Ravichandran V., Mohan S., Kumar K.S. Synthesis and antimicrobial activity of Mannich bases of isatin and its derivatives with 2-[(2,6-dichlorophenyl)amino]phenylacetic acid. ARKIVOC xiv. 2007:51–57. [Google Scholar]
- 161.Karki S.S., Kulkarni A.A., Thota S., Nikam S., Kamble A.S., Dhawale N.D. Synthesis, antimicrobial screening and beta lactamase inhibitory activity of 3-(3-chloro-4-fluorophenylimino)indolin-2-one and 5-chloroindolin-2-one derivatives. Turk. J. Pharm. Sci. 2012;9:353–358. [Google Scholar]
- 162.Sriram D., Bal T.R., Yogeeswari P. Synthesis, antiviral and antibacterial activities of isatin Mannich bases. Med. Chem. Res. 2005;14:211–228. [Google Scholar]
- 163.Murthy Y.L.N., Govindh B., Diwakar B.S., Nagalakshmi K., Rao K.V.R. Synthesis and bioevaluation of Schiff and Mannich bases of isatin derivatives with 4-amino-5-benzyl-2,4-dihydro-3H-1,2,4-triazole-3-thione. Med. Chem. Res. 2012;21:3104–3110. [Google Scholar]
- 164.Prakash C.R., Raja S. Synthesis, characterization and in vitro antimicrobial activity of some novel 5-substituted Schiff and Mannich base of isatin derivatives. J. Saudi Chem. Soc. 2013;17:337–344. [Google Scholar]
- 165.El-Emam A.A., Al-Tamimi A.-M.S., Al-Omar M.A., Alrashood K.A., Habib E.E. Synthesis and antimicrobial activity of novel 5-(1-adamantyl)-2-aminomethyl-4-substituted-1,2,4-triazoline-3-thiones. Eur. J. Med. Chem. 2013;68:96–102. doi: 10.1016/j.ejmech.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 166.Plech T., Wujec M., Majewska M., Kosikowska U., Malm A. Microbiologically active Mannich bases derived from 1,2,4-triazoles. The effect of C-5 substituent on antibacterial activity. Med. Chem. Res. 2013;22:2531–2537. doi: 10.1007/s00044-012-0248-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Plech T., Wujec M., Siwek A., Kosikowska U., Malm A. Synthesis and antimicrobial activity of thiosemicarbazides, s-triazoles and their Mannich bases bearing 3-chlorophenyl moiety. Eur. J. Med. Chem. 2011;46:241–248. doi: 10.1016/j.ejmech.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 168.Plech T., Wujec M., Kaproń B., Kosikowska U., Malm A. Synthesis and antibacterial activity of some novel N2-hydroxymethyl and N2-aminomethyl derivatives of 4-aryl-5-(3-chlorophenyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione. Heteroat. Chem. 2011;22:737–743. [Google Scholar]
- 169.Plech T., Wujec M., Kosikowska U., Malm A., Rajtar B., Polz-Dacewicz M. Synthesis and in vitro activity of 1,2,4-triazole-ciprofloxacin hybrids against drug-susceptible and drug-resistant bacteria. Eur. J. Med. Chem. 2013;60:128–134. doi: 10.1016/j.ejmech.2012.11.040. [DOI] [PubMed] [Google Scholar]
- 170.Almajan G.L., Barbuceanu S.-F., Almajan E.-R., Draghici C., Saramet G. Synthesis, characterization and antibacterial activity of some triazole Mannich bases carrying diphenylsulfone moieties. Eur. J. Med. Chem. 2009;44:3083–3089. doi: 10.1016/j.ejmech.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 171.Alam M.M., Nazreen S., Haider S., Shafi S., Yar M.S., Hamid H., Alam M.S. Synthesis of some new S-alkylated 1,2,4-triazoles, their Mannich bases and their biological activities. Arch. Pharm. (Weinheim) 2012;345:203–214. doi: 10.1002/ardp.201100128. [DOI] [PubMed] [Google Scholar]
- 172.Koparir M., Orek C., Parlak A.E., Soylemez A., Koparir P., Karatepe M., Dastan S.D. Synthesis and biological activities of some novel aminomethyl derivatives of 4-substituted-5-(2-thienyl)-2,4-dihydro-3H-1,2,4-triazole-3-thiones. Eur. J. Med. Chem. 2013;63:340–346. doi: 10.1016/j.ejmech.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 173.Basoglu S., Yolal M., Demirci S., Demirbas N., Bektas H., Karaoglu S.A. Design, synthesis and antimicrobial activities of some azole derivatives. Acta Pol. Pharm. 2013;70:229–236. [PubMed] [Google Scholar]
- 174.Bayrak H., Demirbas A., Karaoglu S.A., Demirbas N. Synthesis of some new 1,2,4-triazoles, their Mannich and Schiff bases and evaluation of their antimicrobial activities. Eur. J. Med. Chem. 2009;44:1057–1066. doi: 10.1016/j.ejmech.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 175.Ceylan S., Bektas H., Bayrak H., Demirbas N., Alpay-Karaoglu S., Ülker S. Syntheses and biological activities of new hybrid molecules containing different heterocyclic moieties. Arch. Pharm. (Weinheim) 2013;346:743–756. doi: 10.1002/ardp.201300161. [DOI] [PubMed] [Google Scholar]
- 176.Özyanik M., Demirci S., Bektaş H., Demirbaş N., Demirbaş A., Alpay Karaoğlu S. Preparation and antimicrobial activity evaluation of some quinoline derivatives containing an azole nucleus. Turk. J. Chem. 2012;36:233–246. [Google Scholar]
- 177.Arafa W.A.A., Mohamed A.S. Convenient one pot synthesis and antibacterial evaluation of some new Mannich bases carrying 1,2,4-triazolyl moiety. Chin. J. Chem. 2011;29:1661–1668. [Google Scholar]
- 178.Bektaş H., Demirbaş A., Demirbaş N., Bayrak H., Karaoğlu S.A. Synthesis and antimicrobial activities of some new biheterocyclic compounds containing 1,2,4-triazol-3-one and 1,3,4-thiadiazole moieties. Turk. J. Chem. 2010;34:517–527. [Google Scholar]
- 179.Demirbas A., Sahin D., Demirbas N., Karaoglu S.A. Synthesis of some new 1,3,4-thiadiazol-2-ylmethyl-1,2,4-triazole derivatives and investigation of their antimicrobial activities. Eur. J. Med. Chem. 2009;44:2896–2903. doi: 10.1016/j.ejmech.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 180.Uygun Y., Bayrak H., Özkan H. Synthesis and biological activities of methylenebis-4H-1,2,4-triazole derivatives. Turk. J. Chem. 2013;37:812–823. [Google Scholar]
- 181.Isloor A.M., Kalluraya B., Shetty P. Regioselective reaction: synthesis, characterization and pharmacological studies of some new Mannich bases derived from 1,2,4-triazoles. Eur. J. Med. Chem. 2009;44:3784–3787. doi: 10.1016/j.ejmech.2009.04.038. [DOI] [PubMed] [Google Scholar]
- 182.Al-Omar M.A., Al-Abdullah E.S., Shehata I.A., Habib E.E., Ibrahim T.M., El-Emam A.A. Synthesis, antimicrobial and anti-inflammatory activities of novel 5-(1-adamantyl)-4-[(arylidene)amino]-3-mercapto-1,2,4-triazoles and related derivatives. Molecules. 2010;15:2526–2550. doi: 10.3390/molecules15042526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ashok M., Holla B.S., Poojary B. Convenient one pot synthesis and antimicrobial evaluation of some new Mannich bases carrying 4-methylthiobenzyl moiety. Eur. J. Med. Chem. 2007;42:1095–1101. doi: 10.1016/j.ejmech.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 184.Karthikeyan M.S., Prasad D.J., Poojary B., Bhat K.S., Holla B.S., Kumari N.S. Synthesis and biological activity of Schiff and Mannich bases bearing 2,4-dichloro-5-fluorophenyl moiety. Bioorg. Med. Chem. 2006;14:7482–7489. doi: 10.1016/j.bmc.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 185.Bayrak H., Demirbas A., Demirbas N., Karaoglu S.A. Synthesis of some new 1,2,4-triazoles starting from isonicotinic acid hydrazide and evaluation of their antimicrobial activities. Eur. J. Med. Chem. 2009;44:4362–4366. doi: 10.1016/j.ejmech.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 186.Hussein M.A., Shaker R.M., Ameen M.A., Mohammed M.F. Synthesis, anti-inflammatory, analgesic, and antibacterial activities of some triazole, triazolothiadiazole, and triazolothiadiazine derivatives. Arch. Pharm. Res. 2011;34:1239–1250. doi: 10.1007/s12272-011-0802-z. [DOI] [PubMed] [Google Scholar]
- 187.Suresh Kumar G.V., Rajendra Prasad Y., Mallikarjuna B.P., Chandrashekar S.M. Synthesis and pharmacological evaluation of clubbed isopropylthiazole derived triazolothiadiazoles, triazolothiadiazines and Mannich bases as potential antimicrobial and antitubercular agents. Eur. J. Med. Chem. 2010;45:5120–5129. doi: 10.1016/j.ejmech.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 188.Lingappa B., Girisha K.S., Kalluraya B., Rai N.S., Kumari N.S. Regioselective reaction: synthesis of novel Mannich bases derived from 3-(4,6-disubstituted-2-thiomethylpyrimidyl)-4-amino-5-mercapto-1,2,4-triazoles and their antimicrobial properties. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2008;47B:1858–1864. [Google Scholar]
- 189.Maddila S., Jonnalagadda S.B. New class of triazole derivatives and their antimicrobial activity. Lett. Drug Des. Discov. 2012;9:687–693. [Google Scholar]
- 190.Yunus U., Bhatti M.H., Rahman N., Mussarat N., Asghar S., Masood B. Synthesis, characterization, and biological activity of novel Schiff and Mannich bases of 4-amino-3-(N-phthalimidomethyl)-1,2,4-triazole-5-thione. J. Chem. 2013:638520. [Google Scholar]
- 191.Fandakli S., Başoğlu S., Bektaş H., Yolal M., Demirbaş A., Alpay Karaoğlu S. Reduction, Mannich reaction and antimicrobial activity evaluation of some new 1,2,4-triazol-3-one derivatives. Turk. J. Chem. 2012;36:567–582. [Google Scholar]
- 192.Demirci S., Basoglu S., Bozdereci A., Demirbas N. Preparation and antimicrobial activity evaluation of some new bi- and triheterocyclic azoles. Med. Chem. Res. 2013;22:4930–4945. [Google Scholar]
- 193.Manjunatha K., Poojary B., Lobo P.L., Fernandes J., Kumari N.S. Synthesis and biological evaluation of some 1,3,4-oxadiazole derivatives. Eur. J. Med. Chem. 2010;45:5225–5233. doi: 10.1016/j.ejmech.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 194.Naveena C.S., Boja P., Kumari N.S. Synthesis, characterization and antimicrobial activity of some disubstituted 1,3,4-oxadiazoles carrying 2-(aryloxymethyl)phenyl moiety. Eur. J. Med. Chem. 2010;45:4708–4719. doi: 10.1016/j.ejmech.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 195.Al-Omar M.A. Synthesis and antimicrobial activity of new 5-(2-thienyl)-1,2,4-triazoles and 5-(2-thienyl)-1,3,4-oxadiazoles and related derivatives. Molecules. 2010;15:502–514. doi: 10.3390/molecules15010502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Desai N.C., Bhatt N., Somani H., Trivedi A. Synthesis, antimicrobial and cytotoxic activities of some novel thiazole clubbed 1,3,4-oxadiazoles. Eur. J. Med. Chem. 2013;67:54–59. doi: 10.1016/j.ejmech.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sanjeeva Reddy Ch., Sanjeeva Rao L., Vani Devi M., Nagaraj A. Synthesis and antimicrobial activity of linked heterocyclics containing pyrazole and oxadiazoles. J. Heterocycl. Chem. 2012;49:1038–1042. [Google Scholar]
- 198.Frank P.V., Manjunatha Poojary M., Damodara N., Chikkanna C. Synthesis and antimicrobial studies of some Mannich bases carrying imidazole moiety. Acta Pharm. (Zagreb) 2013;63:231–239. doi: 10.2478/acph-2013-0016. [DOI] [PubMed] [Google Scholar]
- 199.Naganagowda G., Petsom A. Synthesis and antimicrobial activity of some new 5-(3-chloro-1-benzothiophen-2-yl)-1,3,4-oxadiazole-2-thiol and their derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2011;186:2112–2121. [Google Scholar]
- 200.Ravindra K.C., Vagdevi H.M., Vaidya V.P., Padmashali B. Synthesis, antimicrobial and antiinflammatory activities of 1,3,4-oxadiazoles linked to naphtho[2,1-b]furan. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2006;45B:2506–2511. [Google Scholar]
- 201.Bektaş H., Ceylan Ş., Demirbaş N., Alpay-Karaoğlu Ş., Sökmen B.B. Antimicrobial and antiurease activities of newly synthesized morpholine derivatives containing an azole nucleus. Med. Chem. Res. 2013;22:3629–3639. doi: 10.1007/s00044-012-0318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bektaş H., Karaali N., Şahin D., Demirbaş A., Karaoglu S.A., Demirbaş N. Synthesis and antimicrobial activities of some new 1,2,4-triazole derivatives. Molecules. 2010;15:2427–2438. doi: 10.3390/molecules15042427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Aggarwal N., Kumar R., Dureja P., Khurana J.M. Synthesis of novel nalidixic acid-based 1,3,4-thiadiazole and 1,3,4-oxadiazole derivatives as potent antibacterial agents. Chem. Biol. Drug Des. 2012;79:384–397. doi: 10.1111/j.1747-0285.2011.01316.x. [DOI] [PubMed] [Google Scholar]
- 204.Ozkan-Dagliyan I., Sahin F., Koksal M. Synthesis, characterization and antimicrobial activity of novel 3,5-disubstituted-1,3,4-oxadiazole-2-ones. Rev. Chim. (Bucharest) 2013;64:534–539. [Google Scholar]
- 205.Altintaş H., Ateş Ö., Birteksöz S., Ötük G., Uzun M., Şatana D. Synthesis of Mannich bases of some 2,5-disubstituted 4-thiazolidinones and evaluation of their antimicrobial activities. Turk. J. Chem. 2005;29:425–435. doi: 10.1002/1521-4184(200102)334:2<35::aid-ardp35>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 206.Altintaş H., Ateş Ö., Kocabalkanli A., Birteksöz S., Ötük G. Synthesis, characterization and evaluation of antimicrobial activity of Mannich bases of some 2-[(4-carbethoxymethylthiazol-2-yl)imino]-4-thiazolidinones. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2005;44B:585–590. [Google Scholar]
- 207.Mohamed M.S., Kamel M.M., Kassem E.M.M., Abotaleb N., Abd El-moez S.I., Ahmed M.F. Novel 6,8-dibromo-4(3H)quinazolinone derivatives of anti-bacterial and anti-fungal activities. Eur. J. Med. Chem. 2010;45:3311–3319. doi: 10.1016/j.ejmech.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 208.Patel D., Kumari P., Patel N. Synthesis of 3-{4-[4-dimethylamino-6-(4-methyl-2-oxo-2H-chromen-7-yloxy)-[1,3,5]triazin-2-ylamino]-phenyl}-2-phenyl-5-(4-pyridin-2-yl-piperazin-1-ylmethyl)-thiazolidin-4-one and their biological evaluation. Med. Chem. Res. 2012;21:2926–2944. [Google Scholar]
- 209.Kulakov I.V. Synthesis and biological activity of 3,4-dihydropyrimidine-2-thione aminomethylene derivatives based on the alkaloid cytosine. Chem. Nat. Compd. 2011;47:597–599. [Google Scholar]
- 210.Shah T.B., Gupte A., Patel M.R., Chaudhari V.S., Patel H., Patel V.C. Synthesis and in vitro study of biological activity of heterocyclic N-Mannich bases of 3,4-dihydropyrimidine-2(1H)-thiones. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2010;49B:578–586. [Google Scholar]
- 211.Shah T.B., Gupte A., Patel M.R., Chaudhari V.S., Patel H., Patel V.C. Synthesis and in vitro study of biological activity of heterocyclic N-Mannich bases. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2009;48B:88–96. [Google Scholar]
- 212.Joshi S., Khosla N., Khare D., Sharda R. Synthesis and in vitro study of novel Mannich bases as antibacterial agents. Bioorg. Med. Chem. Lett. 2005;15:221–226. doi: 10.1016/j.bmcl.2004.09.084. [DOI] [PubMed] [Google Scholar]
- 213.Das Manikpuri A., Joshi S., Khadikar P.V. Synthetic, spectral, antimicrobial and QSAR studies on novel Mannich bases of glutarimides. J. Chil. Chem. Soc. 2010;55:283–292. [Google Scholar]
- 214.Das Manikpuri A., Joshi S. Evaluation of antimicrobial activity and QSAR study of a molecule library of the Mannich bases of glutarimides. Oxid. Commun. 2013;36:156–175. [Google Scholar]
- 215.Tamilvendan D., Rajeswari S., Ilavenil S., Chakkaravarthy K., Venkatesa Prabhu G. Syntheses, spectral, crystallographic, antimicrobial, and antioxidant studies of few Mannich bases. Med. Chem. Res. 2012;21:4129–4138. [Google Scholar]
- 216.Joshi S., Das Manikpuri A., Tiwari P. Synthesis and biological study of medicinally important Mannich bases derived from 4-(dimethylamino)-1,4,4a,5,5a,6,11,12a-octahydro-3,6,10,12,12a-pentahydroxynaphthacenecarboxamide. Bioorg. Med. Chem. Lett. 2007;17:645–648. doi: 10.1016/j.bmcl.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 217.Leadbetter M.R., Adams S.M., Bazzini B., Fatheree P.R., Karr D.E., Krause K.M., Lam B.M.T., Linsell M.S., Nodwell M.B., Pace J.L., Quast K., Shaw J.-P., Soriano E., Trapp S.G., Villena J.D., Wu T.X., Christensen B.G., Judice J.K. Hydrophobic vancomycin derivatives with improved ADME properties: discovery of telavancin (TD-6424) J. Antibiot. 2004;57:326–336. doi: 10.7164/antibiotics.57.326. [DOI] [PubMed] [Google Scholar]
- 218.Krause K.M., Renelli M., Difuntorum S., Wu T.X., Debabov D.V., Benton B.M. In vitro activity of telavancin against resistant Gram-positive bacteria. Antimicrob. Agents Chemother. 2008;52:2647–2652. doi: 10.1128/AAC.01398-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Higgins D.L., Chang R., Debabov D.V., Leung J., Wu T., Krause K.M., Sandvik E., Hubbard J.M., Kaniga K., Schmidt D.E., Jr., Gao Q., Cass R.T., Karr D.E., Benton B.M., Humphrey P.P. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2005;49:1127–1134. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Long D.D., Aggen J.B., Chinn J., Choi S.K., Christensen B.G., Fatheree P.R., Green D., Hegde S.S., Judice J.K., Kaniga K., Krause K.M., Leadbetter M., Linsell M.S., Marquess D.G., Moran E.J., Nodwell M.B., Pace J.L., Trapp S.G., Turner S.D. Exploring the positional attachment of glycopeptide/β-lactam heterodimers. J. Antibiot. 2008;61:603–614. doi: 10.1038/ja.2008.80. [DOI] [PubMed] [Google Scholar]
- 221.K.D. James, R.G. Sherrill, B. Radhakrishnan, Vancomycin Derivatives, PCT Int. Appl. WO 2012129493, 2012.
- 222.Kataki D., Bhattacharyya P., Deka M., Jha D.K., Phukan P. Iodine catalyzed three-component synthesis of β-amino-β-keto-esters and their antimicrobial activity. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2013;52B:1505–1512. [Google Scholar]
- 223.Malhotra M., Sharma S., Deep A. Synthesis, characterization and antimicrobial evaluation of novel derivatives of isoniazid. Med. Chem. Res. 2012;21:1237–1244. [Google Scholar]
- 224.Malhotra M., Arora M., Samad A., Sahu K., Phogat P., Deep A. Synthesis and evaluation of some novel derivatives of 2-propoxybenzylideneisonicotinohydrazide for their potential antimicrobial activity. J. Serb. Chem. Soc. 2012;77:589–597. [Google Scholar]
- 225.Desai N.C., Pandya M.R., Rajpara K.M., Joshi V.V., Vaghani H.V., Satodiya H.M. Synthesis and antimicrobial screening of novel series of imidazo[1,2-a]pyridine derivatives. Med. Chem. Res. 2012;21:4437–4446. [Google Scholar]
- 226.Joshi S.D., Manish K., Badiger A. Synthesis and evaluation of antibacterial and antitubercular activities of some novel imidazo[2,1-b][1,3,4]thiadiazole derivatives. Med. Chem. Res. 2013;22:869–878. [Google Scholar]
- 227.Fassihi A., Abedi D., Saghaie L., Sabet R., Fazeli H., Bostaki G., Deilami O., Sadinpour H. Synthesis, antimicrobial evaluation and QSAR study of some 3-hydroxypyridine-4-one and 3-hydroxypyran-4-one derivatives. Eur. J. Med. Chem. 2009;44:2145–2157. doi: 10.1016/j.ejmech.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 228.Asundaria S.T., Pannecouque C., De Clercq E., Supuran C.T., Patel K.C. Synthesis of novel biologically active methylene derivatives of sydnones. Med. Chem. Res. 2013;22:5752–5763. [Google Scholar]
- 229.Savaliya P.P., Akbari V.K., Modi J.A., Patel K.C. Synthesis, characterization, and antimicrobial screening of some Mannich base sydnone derivatives. Med. Chem. Res. 2013;22:5789–5797. [Google Scholar]
- 230.Taj T., Kamble R.R., Gireesh T.M., Hunnur R.K. Facile syntheses of Mannich bases of 3-[p-(5-arylpyrazolin-3-yl)phenyl]sydnones, as anti-tubercular and anti-microbial agents, under ionic liquid/tetrabutylammonium bromide catalytic conditions. J. Serb. Chem. Soc. 2011;76:1069–1079. [Google Scholar]
- 231.Mendoza Á., Mendoza-Díaz G., Pedraza-Reyes M., Bernès S. Copper(II) complex with the tridentate ligand N,N-bis(2-ethyl-4-methylimidazol-5-ylmethyl)phenylethylamine (biaq). X-ray crystal structure and biological activity on Bacillus subtilis of [Cu(biaq)Cl2] Z. Anorg. Allg. Chem. 2013;639:1455–1460. [Google Scholar]
- 232.Sivakumar K.K., Rajasekharan A., Rao R., Narasimhan B. Synthesis, SAR study and evaluation of Mannich and Schiff bases of pyrazol-5(4H)-one moiety containing 3-(hydrazinyl)-2-phenylquinazolin-4(3H)-one. Indian J. Pharm. Sci. 2013;75:463–475. doi: 10.4103/0250-474X.119832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Al-kaissi E.N., Al-Ghrary N.F., Al-Kaisi N.K., Al-Shamma A., Muhi-eldeen Z. Synthesis and antimicrobial evaluation of 4,5-diaryl-2-[4-(t-amino)-2-butynyl]-2,4-dihydro-3H-1,2,4-triazol-3-ones. Med. Chem. Res. 2012;21:3390–3395. [Google Scholar]
- 234.Abuo-Rahma G.E.-D.A.A., Sarhan H.A., Gad G.F.M. Design, synthesis, antibacterial activity and physicochemical parameters of novel N-4-piperazinyl derivatives of norfloxacin. Bioorg. Med. Chem. 2009;17:3879–3886. doi: 10.1016/j.bmc.2009.04.027. [DOI] [PubMed] [Google Scholar]
- 235.Joshi S., Khosla N., Tiwari P. In vitro study of some medicinally important Mannich bases derived from antitubercular agent. Bioorg. Med. Chem. 2004;12:571–576. doi: 10.1016/j.bmc.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 236.Bapna M., Parashar B., Sharma V.K., Chouhan L.S. Microwave-assisted synthesis of some novel and potent antibacterial and antifungal compounds with biological screening. Med. Chem. Res. 2012;21:1098–1106. [Google Scholar]
- 237.Hamama W.S., Zoorob H.H., Gouda M.A., Afsah E.M. Synthesis and antimicrobial and antioxidant activities of simple saccharin derivatives with N-basic side chains. Pharm. Chem. J. 2011;45:118–124. [Google Scholar]
- 238.Radadiya V.R., Purohit D.M., Patolia V.N. Synthesis and antimicrobial activity of 10-arylaminomethyl-3-methoxyphenothiazine-9-carboxylic acid. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2005;44B:1112–1114. [Google Scholar]
- 239.Ramalingam P., Ganapaty S., Babu Rao Ch. In vitro antitubercular and antimicrobial activities of 1-substituted quinoxaline-2,3(1H,4H)-diones. Bioorg. Med. Chem. Lett. 2010;20:406–408. doi: 10.1016/j.bmcl.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 240.Rajasekaran A., Rajamanickam V., Darlinquine S. Synthesis of some new thioxoquinazolinone derivatives and a study on their anticonvulsant and antimicrobial activities. Eur. Rev. Med. Pharmacol. Sci. 2013;17:95–104. [PubMed] [Google Scholar]
- 241.Kodhati V., Vanga M.R., Yellu N.R. Synthesis and anti bacterial and anti-ulcer evaluation of new S-Mannich bases of 4,6-diaryl-3,4-dihydropyrimidin-2(1H)-thiones. J. Korean Chem. Soc. 2013;57:234–240. [Google Scholar]
- 242.World Health Organization 2010/2011 Tuberculosis Global Facts. http://www.who.int/tb/publications/2010/factsheet_tb_2010.pdf (accessed 17.05.14.).
- 243.Dimmock J.R., Kandepu N.M., Das U., Zello G.A., Nienaber K.H. Antimycobacterial arylidenecyclohexanones and related Mannich bases. Pharmazie. 2004;59:502–505. doi: 10.1002/chin.200447216. [DOI] [PubMed] [Google Scholar]
- 244.Das S., Das U., Bandy B., Gorecki D.K.J., Dimmock J.R. 2-[4-(4-Methoxyphenylcarbonyloxy)benzylidene]-6-dimethylaminomethyl cyclohexanone hydrochloride: a Mannich base which inhibits the growth of some drug-resistant strains of Mycobacterium tuberculosis. Pharmazie. 2010;65:849–850. [PMC free article] [PubMed] [Google Scholar]
- 245.Dunn M.F., Ramírez-Trujillo J.A., Hernández-Lucas I. Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology. 2009;155:3166–3175. doi: 10.1099/mic.0.030858-0. [DOI] [PubMed] [Google Scholar]
- 246.Muñoz-Elías E.J., McKinney J.D. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sharma R., Das O., Damle S.G., Sharma A.K. Isocitrate lyase: a potential target for anti-tubercular drugs. Recent Pat. Inflamm. Allergy Drug Discov. 2013;7:114–123. doi: 10.2174/1872213x11307020003. [DOI] [PubMed] [Google Scholar]
- 248.Ji L., Long Q., Yang D., Xie J. Identification of Mannich base as a novel inhibitor of Mycobacterium tuberculosis isocitrate by high-throughput screening. Int. J. Biol. Sci. 2011;7:376–382. doi: 10.7150/ijbs.7.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sriram D., Yogeeswari P., Madhu K. Synthesis and in vitro and in vivo antimycobacterial activity of isonicotinoyl hydrazones. Bioorg. Med. Chem. Lett. 2005;15:4502–4505. doi: 10.1016/j.bmcl.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 250.Us D., Gürdal E., Berk B., Öktem S., Kocagöz T., Çağlayan B., Kurnaz I.A., Erol D.D. 4H-Pyran-4-one derivatives: leading molecule for preparation of compounds with antimycobacterial potential. Turk. J. Chem. 2009;33:803–812. [Google Scholar]
- 251.Berk B., Us D., Öktem S., Kocagöz Z.T., Çağlayan B., Aksan Kurnaz I., Demir Erol D. Molecular modeling and antimycobacterial studies of Mannich bases: 5-hydroxy-2-methyl-4H-pyran-4-ones. Turk. J. Chem. 2011;35:317–330. [Google Scholar]
- 252.Deidda D., Lampis G., Fioravanti R., Biava M., Porretta G.C., Zanetti S., Pompei R. Bactericidal activities of the pyrrole derivative BM212 against multidrug-resistant and intramacrophagic Mycobacterium tuberculosis strains. Antimicrob. Agents Chemother. 1998;42:3035–3037. doi: 10.1128/aac.42.11.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Biava M., Fioravanti R., Porretta G.C., Sleiter G., Deidda D., Lampis G., Pompei R. Synthesis and microbiological activities of pyrrole analogs of BM 212, a potent antitubercular agent. Med. Chem. Res. 1999;9:19–34. [Google Scholar]
- 254.Biava M., Fioravanti R., Porretta G.C., Deidda D., Maullu C., Pompei R. New pyrrole derivatives as antimycobacterial agents analogs of BM212. Bioorg. Med. Chem. Lett. 1999;9:2983–2988. doi: 10.1016/s0960-894x(99)00510-7. [DOI] [PubMed] [Google Scholar]
- 255.Biava M., Porretta G.C., Deidda D., Pompei R., Tafi A., Manetti F. Importance of the thiomorpholine introduction in new pyrrole derivatives as antimycobacterial agents analogues of BM 212. Bioorg. Med. Chem. 2003;11:515–520. doi: 10.1016/s0968-0896(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 256.Biava M., Porretta G.C., Deidda D., Pompei R., Tafi A., Manetti F. Antimycobacterial compounds. New pyrrole derivatives of BM212. Bioorg. Med. Chem. 2004;12:1453–1458. doi: 10.1016/j.bmc.2003.12.037. [DOI] [PubMed] [Google Scholar]
- 257.Biava M., Porretta G.C., Poce G., Deidda D., Pompei R., Tafi A., Manetti F. Antimycobacterial compounds. Optimization of the BM 212 structure, the lead compound for a new pyrrole derivative class. Bioorg. Med. Chem. 2005;13:1221–1230. doi: 10.1016/j.bmc.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 258.Biava M., Porretta G.C., Poce G., Supino S., Deidda D., Pompei R., Molicotti P., Manetti F., Botta M. Antimycobacterial agents. Novel diarylpyrrole derivatives of BM212 endowed with high activity toward Mycobacterium tuberculosis and low cytotoxicity. J. Med. Chem. 2006;49:4946–4952. doi: 10.1021/jm0602662. [DOI] [PubMed] [Google Scholar]
- 259.Biava M., Porretta G.C., Poce G., De Logu A., Saddi M., Meleddu R., Manetti F., De Rossi E., Botta M. 1,5-Diphenylpyrrole derivatives as antimycobacterial agents. Probing the influence on antimycobacterial activity of lipophilic substituents at the phenyl rings. J. Med. Chem. 2008;51:3644–3648. doi: 10.1021/jm701560p. [DOI] [PubMed] [Google Scholar]
- 260.Biava M., Porretta G.C., Poce G., De Logu A., Meleddu R., De Rossi E., Manetti F., Botta M. 1,5-Diaryl-2-ethyl pyrrole derivatives as antimycobacterial agents: design, synthesis, and microbiological evaluation. Eur. J. Med. Chem. 2009;44:4734–4738. doi: 10.1016/j.ejmech.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 261.Biava M., Porretta G.C., Poce G., Battilocchio C., Alfonso S., De Logu A., Serra N., Manetti F., Botta M. Identification of a novel pyrrole derivative endowed with antimycobacterial activity and protection index comparable to that of the current antitubercular drugs streptomycin and rifampin. Bioorg. Med. Chem. 2010;18:8076–8084. doi: 10.1016/j.bmc.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 262.Poce G., Bates R.H., Alfonso S., Cocozza M., Porretta G.C., Ballell L., Rullas J., Ortega F., De Logu A., Agus E., La Rosa V., Pasca M.R., De Rossi E., Wae B., Franzblau S.G., Manetti F., Botta M., Biava M. Improved BM212 MmpL3 inhibitor analogue shows efficacy in acute murine model of tuberculosis infection. PLOS One. 2013;8:e56980. doi: 10.1371/journal.pone.0056980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.La Rosa V., Poce G., Canseco J.O., Buroni S., Pasca M.R., Biava M., Raju R.M., Porretta G.C., Alfonso S., Battilocchio C., Javid B., Sorrentino F., Ioerger T.R., Sacchettini J.C., Manetti F., Botta M., De Logu A., Rubin E.J., De Rossi E. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 2012;56:324–331. doi: 10.1128/AAC.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Friggeri L., Ballante F., Ragno R., Musmuca I., De Vita D., Manetti F., Biava M., Scipione L., Di Santo R., Costi R., Feroci M., Tortorella S. Pharmacophore assessment through 3-D QSAR: evaluation of the predictive ability on new derivatives by the application on a series of antitubercular agents. J. Chem. Inf. Model. 2013;53:1463–1474. doi: 10.1021/ci400132q. [DOI] [PubMed] [Google Scholar]
- 265.Biava M. BM 212 and its derivatives as a new class of antimycobacterial active agents. Curr. Med. Chem. 2002;9:1859–1869. doi: 10.2174/0929867023368953. [DOI] [PubMed] [Google Scholar]
- 266.Biava M., Porretta G.C., Poce G., Supino S., Sleiter G. New pyrroles with potential antimycobacterial, antifungal and selective COX-2 inhibiting activities. Synthetic methodologies. Curr. Org. Chem. 2007;11:1092–1112. [Google Scholar]
- 267.Biava M., Porretta G.C., Manetti F. New derivatives of BM212: a class of antimycobacterial compounds based on the pyrrole ring as a scaffold. Mini Rev. Med. Chem. 2007;7:65–78. doi: 10.2174/138955707779317786. [DOI] [PubMed] [Google Scholar]
- 268.Biava M., Porretta G.C., Poce G., Battilocchio C., Alfonso S., de Logu A., Manetti F., Botta M. Developing pyrrole-derived antimycobacterial agents: a rational lead optimization approach. ChemMedChem. 2011;6:593–599. doi: 10.1002/cmdc.201000526. [DOI] [PubMed] [Google Scholar]
- 269.S.K. Arora, N. Sinha, S. Jain, R.S. Upadhayaya, G. Jana, S. Ajay, R. Sinha, Pyrrole Derivatives as Antimycobacterial Compounds, US Patent 7,691,837, 2010.
- 270.Kamal A., Swapna P., Shetti R.V.C.R.N.C., Shaik A.B., Narasimha Rao M.P., Sultana F., Khan I.A., Sharma S., Kalia N.P., Kumar S., Chandrakant B. Anti-tubercular agents. Part 7: a new class of diarylpyrrole–oxazolidinone conjugates as antimycobacterial agents. Eur. J. Med. Chem. 2013;64:239–251. doi: 10.1016/j.ejmech.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 271.He R., Zeng L.-F., He Y., Wu L., Gunawan A.M., Zhang Z.-Y. Organocatalytic multicomponent reaction for the acquisition of a selective inhibitor of mPTPB, a virulence factor of tuberculosis. Chem. Commun. 2013;49:2064–2066. doi: 10.1039/c3cc38961h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Raja S., Prakash C.R. Novel 1-(4-substituted benzylidene)-4-(1-(substituted methyl)-2,3-dioxoindolin-5-yl)semicarbazide derivatives for use against Mycobacterium tuberculosis H37Rv (ATCC 27294) and MDR-TB strain. Arch. Pharm. Res. 2013;39:411–422. doi: 10.1007/s12272-013-0062-1. [DOI] [PubMed] [Google Scholar]
- 273.Sriram D., Yogeeswari P., Gopal G. Synthesis, anti-HIV and antitubercular activities of lamivudine prodrugs. Eur. J. Med. Chem. 2005;40:1373–1376. doi: 10.1016/j.ejmech.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 274.Güzel Ö., Karali N., Salman A. Synthesis and antituberculosis activity of 5-methyl/trifluoromethoxy-1H-indole-2,3-dione 3-thiosemicarbazone derivatives. Bioorg. Med. Chem. 2008;16:8976–8987. doi: 10.1016/j.bmc.2008.08.050. [DOI] [PubMed] [Google Scholar]
- 275.Karali N., Gürsoy A., Kandemirli F., Shvets N., Betül Kaynak F., Özbey S., Kovalishyne V., Dimoglo A. Synthesis and structure–antituberculosis activity relationship of 1H-indole-2,3-dione derivatives. Bioorg. Med. Chem. 2007;15:5888–5904. doi: 10.1016/j.bmc.2007.05.063. [DOI] [PubMed] [Google Scholar]
- 276.Sriram D., Aubry A., Yogeeswari P., Fisher L.M. Gatifloxacin derivatives: synthesis, antimycobacterial activities, and inhibition of Mycobacterium tuberculosis DNA gyrase. Bioorg. Med. Chem. Lett. 2006;16:2982–2985. doi: 10.1016/j.bmcl.2006.02.065. [DOI] [PubMed] [Google Scholar]
- 277.Sriram D., Yogeeswari P., Basha J.S., Radha D.R., Nagaraja V. Synthesis and antimycobacterial evaluation of various 7-substituted ciprofloxacin derivatives. Bioorg. Med. Chem. 2005;13:5774–5778. doi: 10.1016/j.bmc.2005.05.063. [DOI] [PubMed] [Google Scholar]
- 278.Feng L.-S., Liu M.-L., Zhang S., Chai Y., Wang B., Zhang Y.-B., Lv K., Guan Y., Guo H.-Y., Xiao C.-L. Synthesis and in vitro antimycobacterial activity of 8-OCH3 ciprofloxacin methylene and ethylene isatin derivatives. Eur. J. Med. Chem. 2011;46:341–348. doi: 10.1016/j.ejmech.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 279.Feng L., Liu M., Wang S., Chai Y., Li S., Guo H. Synthesis and in vitro antimycobacterial activity of moxifloxacin methylene and ethylene isatin derivatives. Chem. Res. Chin. Univ. 2012;28:61–66. [Google Scholar]
- 280.Mamolo M.G., Zampieri D., Vio L., Fermeglia M., Ferrone M., Pricl S., Scialino G., Banfi E. Antimycobacterial activity of new 3-substituted 5-(pyridin-4-yl)-3H-1,3,4-oxadiazol-2-one and 2-thione derivatives. Preliminary molecular modeling investigations. Bioorg. Med. Chem. 2005;13:3797–3809. doi: 10.1016/j.bmc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 281.Zampieri D., Mamolo M.G., Laurini E., Fermeglia M., Posocco P., Pricl S., Banfi E., Scialino G., Vio L. Antimycobacterial activity of new 3,5-disubstituted 1,3,4-oxadiazol-2(3H)-one derivatives. Molecular modeling investigations. Bioorg. Med. Chem. 2009;17:4693–4707. doi: 10.1016/j.bmc.2009.04.055. [DOI] [PubMed] [Google Scholar]
- 282.Ali M.A., Shaharyar M. Oxadiazole Mannich bases: synthesis and antimycobacterial activity. Bioorg. Med. Chem. Lett. 2007;17:3314–3316. doi: 10.1016/j.bmcl.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 283.Foks H., Janowiec M., Zwolska Z., Augustynowicz-Kopeć E. Synthesis and tuberculostatic activity of some 2-piperazinmethylene derivatives 1,2,4-triazole-3-thiones. Phosphorus Sulfur Silicon Relat. Elem. 2005;180:537–543. [Google Scholar]
- 284.Kolavi G., Hegde V., Khazi I.A., Gadad P. Synthesis and evaluation of antitubercular activity of imidazo[2,1-b][1,3,4]thiadiazole derivatives. Bioorg. Med. Chem. 2006;14:3069–3080. doi: 10.1016/j.bmc.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 285.Hegde V.S., Kolavi G.D., Lamani R.S., Khazi I.A.M. Mannich bases and novel benzothiazole derivatives of imidazo[2,1-b]-1,3,4-thiadiazoles and their biological evaluation. J. Sulfur Chem. 2006;27:553–569. [Google Scholar]
- 286.Badiger N.P., Khazi I.M. Synthesis & evaluation of antitubercular activity of novel Mannich bases of imidazo[2,1-b][1,3,4]thiadiazoles. Adv. Mater. Res. 2013;816–817:1197–1201. [Google Scholar]
- 287.Sriram D., Yogeeswari P., Senchani G., Banerjee D. Newer tetracycline derivatives: synthesis, anti-HIV, antimycobacterial activities and inhibition of HIV-1 integrase. Bioorg. Med. Chem. Lett. 2007;17:2372–2375. doi: 10.1016/j.bmcl.2006.11.055. [DOI] [PubMed] [Google Scholar]
- 288.Sriram D., Yogeeswari P., Reddy S.P. Synthesis of pyrazinamide Mannich bases and their antitubercular properties. Bioorg. Med. Chem. Lett. 2006;16:2113–2116. doi: 10.1016/j.bmcl.2006.01.064. [DOI] [PubMed] [Google Scholar]
- 289.Sriram D., Banerjee D., Yogeeswari P. Efavirenz Mannich bases: synthesis, anti-HIV and antitubercular activities. J. Enzyme Inhib. Med. Chem. 2009;24:1–5. doi: 10.1080/14756360902784425 . [DOI] [PubMed] [Google Scholar]
- 290.Malinka W., Świątek P. Indole Mannich bases and their antimycobacterial effect. Acta Pol. Pharm. 2004;61:107–111. [PubMed] [Google Scholar]
- 291.Malinka W., Świątek P., Śliwińska M., Szponar B., Gamian A., Karczmarzyk Z., Fruziński A. Synthesis of novel isothiazolopyridines and their in vitro evaluation against Mycobacterium and Propionibacterium acnes. Bioorg. Med. Chem. 2013;21:5282–5291. doi: 10.1016/j.bmc.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 292.Sitarz M., Foks H., Janowiec M., Zwolska Z., Augustynowicz-Kopec E. Studies on pyrazine derivatives. 39. Synthesis, reactions, and tuberculostatic activity of 3-pyrazinyl-1,2,4-triazolo[4,3-a]-1,3-diazacycloalkanes. Chem. Heterocycl. Compd. 2005;41:200–207. [Google Scholar]
- 293.Pfaller M.A., Diekema D.J. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 2010;36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- 294.Gul H.I., Ojanen T., Vepsalainen J., Gul M., Erciyas E., Hanninen O. Antifungal activity of some mono, bis and quaternary Mannich bases derived from acetophenone. Arzneimittelforschung. 2001;51:72–75. doi: 10.1055/s-0031-1300005. [DOI] [PubMed] [Google Scholar]
- 295.Gul H.I., Denizci A.A., Erciyas E. Antimicrobial evaluation of some Mannich bases of acetophenones and representative quaternary derivatives. Arzneimittelforschung. 2002;52:773–777. doi: 10.1055/s-0031-1299965. [DOI] [PubMed] [Google Scholar]
- 296.Gul H.I., Ojanen T., Hanninen O. Antifungal evaluation of bis Mannich base derived from acetophenones and their corresponding piperidinols and stability studies. Biol. Pharm. Bull. 2002;25:1307–1310. doi: 10.1248/bpb.25.1307. [DOI] [PubMed] [Google Scholar]
- 297.Mete E., Ozelgul C., Kazaz C., Yurdakul D., Sahin F., Gul H.I. Synthesis and antifungal activity of 1-aryl-3-phenethylamino-1-propanone hydrochlorides and 3-aroyl-4-aryl-1-phenethyl-4-piperidinols. Arch. Pharm. (Weinheim) 2010;343:291–300. doi: 10.1002/ardp.200900136. [DOI] [PubMed] [Google Scholar]
- 298.Mete E., Gul H.I., Bilginer S., Algul O., Topaloglu M.E., Gulluce M., Kazaz C. Synthesis and antifungal evaluation of 1-aryl-2-[(dimethylamino)methyl]-2-propen-1-one hydrochlorides. Molecules. 2011;16:4660–4671. doi: 10.3390/molecules16064660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Kocsis B., Kustos I., Kilár F., Nyul A., Jakus P.B., Kerekes S., Villarreal V., Prókai L., Lóránd T. Antifungal unsaturated cyclic Mannich ketones and amino alcohols: study of mechanism of action. Eur. J. Med. Chem. 2009;44:1823–1829. doi: 10.1016/j.ejmech.2008.10.038. [DOI] [PubMed] [Google Scholar]
- 300.Rossignol T., Kocsis B., Bouquet O., Kustos I., Kilár F., Nyul A., Jakus P.B., Rajbhandari K., Prókai L., d'Enfert C., Lóránd T. Antifungal activity of fused Mannich ketones triggers an oxidative stress response and is Cap1-dependent in Candida albicans. PLOS One. 2013;8:e62142. doi: 10.1371/journal.pone.0062142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Liu Y., Luo W., Sun L., Guo C. Synthesis and antifungal activity of 3-substituted thiochromanones. Drug Discov. Ther. 2008;2:216–218. [PubMed] [Google Scholar]
- 302.Malhotra M., Sharma R., Sanduja M., Kumar R., Jain J., Deep A. Synthesis, characterization and evaluation of Mannich bases as potent antifungal and hydrogen peroxide scavenging agents. Acta Pol. Pharm. 2012;69:355–361. [PubMed] [Google Scholar]
- 303.Lal B., Gund V.G., Bhise N.B., Gangopadhyay A.K. Mannich reaction: an approach for the synthesis of water soluble mulundocandin analogues. Bioorg. Med. Chem. 2004;12:1751–1768. doi: 10.1016/j.bmc.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 304.Aytemir M.D., Özçelik B., Karakaya G. Evaluation of bioactivities of chlorokojic acid derivatives against dermatophytes couplet with cytotoxicity. Bioorg. Med. Chem. Lett. 2013;23:3646–3649. doi: 10.1016/j.bmcl.2013.03.098. [DOI] [PubMed] [Google Scholar]
- 305.Aytemir M.D., Çaliş Ü., Özalp M. Synthesis and evaluation of anticonvulsant and antimicrobial activities of 3-hydroxy-6-methyl-2-substituted 4H-pyran-4-one derivatives. Arch. Pharm. (Weinheim) 2004;337:281–288. doi: 10.1002/ardp.200200754. [DOI] [PubMed] [Google Scholar]
- 306.Mahmoud A.M., El-Sherif H.A.H., Habib O.M.A., Sarhan A.A.O. Synthesis and studies of triazolothiadiazines. An approach toward new biologically active heterocyclic compounds. Phosphorus, Sulfur Silicon Relat. Elem. 2007;182:1757–1766. [Google Scholar]
- 307.Kidwai M., Thakur R., Mohan R. Ecofriendly synthesis of novel antifungal (thio)barbituric acid derivatives. Acta Chim. Slov. 2005;52:88–92. [Google Scholar]
- 308.World Health Organization Malaria Report. 2012. http://www.who.int/malaria/publications/world_malaria_report_2013/report/en (accessed 08.06.04.) [Google Scholar]
- 309.O'Neill P.M., Mukhtar A., Stocks P.A., Randle L.E., Hindley S., Ward S.A., Storr R.C., Bickley J.F., O'Neil I.A., Maggs J.L., Hughes R.H., Winstanley P.A., Bray P.G., Park B.K. Isoquine and related amodiaquine analogues: a new generation of improved 4-aminoquinoline antimalarials. J. Med. Chem. 2003;46:4933–4945. doi: 10.1021/jm030796n. [DOI] [PubMed] [Google Scholar]
- 310.O'Neill P.M., Park B.K., Shone A.E., Maggs J.L., Roberts P., Stocks P.A., Biagini G.A., Bray P.G., Gibbons P., Berry N., Winstanley P.A., Mukhtar A., Bonar-Law R., Hindley S., Bambal R.B., Davis C.B., Bates M., Hart T.K., Gresham S.L., Lawrence R.M., Brigandi R.A., Gomez-delas-Heras F.M., Gargallo D.V., Ward S.A. Candidate selection and preclinical evaluation of N-tert-butyl isoquine (GSK369796), an affordable and effective 4-aminoquinoline antimalarial for the 21st century. J. Med. Chem. 2009;52:1408–1415. doi: 10.1021/jm8012618. [DOI] [PubMed] [Google Scholar]
- 311.Davis C.B., Bambal R., Moorthy G.S., Hugger E., Xiang H., Park B.K., Shone A.E., O'Neill P.M., Ward S.A. Comparative preclinical drug metabolism and pharmacokinetic evaluation of novel 4-aminoquinoline anti-malarials. J. Pharm. Sci. 2009;98:362–377. doi: 10.1002/jps.21469. [DOI] [PubMed] [Google Scholar]
- 312.Okombo J., Kiara S.M., Abdirahman A., Mwai L., Ohuma E., Borrmann S., Nzila A., Ward S. Antimalarial activity of isoquine against Kenyan Plasmodium falciparum clinical isolates and association with polymorphisms in pfcrt and pfmdr1 genes. J. Antimicrob. Chemother. 2013;68:786–788. doi: 10.1093/jac/dks471. [DOI] [PubMed] [Google Scholar]
- 313.Gemma S., Camodeca C., Brindisi M., Brogi S., Kukreja G., Kunjir S., Gabellieri E., Lucantoni L., Habluetzel A., Taramelli D., Basilico N., Gualdani R., Tadini-Buoninsegni F., Bartolommei G., Moncelli M.R., Martin R.E., Summers R.L., Lamponi S., Savini L., Fiorini I., Valoti M., Novellino E., Campiani G., Butini S. Mimicking the intramolecular hydrogen bond: synthesis, biological evaluation, and molecular modeling of benzoxazines and quinazolines as potential antimalarial agents. J. Med. Chem. 2012;55:10387–10404. doi: 10.1021/jm300831b. [DOI] [PubMed] [Google Scholar]
- 314.Singh B., Chetia D., Puri S.K., Srivastava K., Prakash A. Synthesis and in vitro and in vivo antimalarial activity of novel 4-anilinoquinoline Mannich base derivatives. Med. Chem. Res. 2011;20:1523–1529. [Google Scholar]
- 315.Bora S., Chetia D., Prakash A. Synthesis and antimalarial activity evaluation of some isoquine analogues. Med. Chem. Res. 2011;20:1632–1637. [Google Scholar]
- 316.Roy S., Chetia D., Rudrapal M., Prakash A. Synthesis and antimalarial activity study of some new Mannich bases of 7-chloro-4-aminoquinoline. Med. Chem. 2013;9:379–383. doi: 10.2174/1573406411309030008. [DOI] [PubMed] [Google Scholar]
- 317.Casagrande M., Basilico N., Parapini S., Romeo S., Taramelli D., Sparatore A. Novel amodiaquine congeners as potent antimalarial agents. Bioorg. Med. Chem. 2008;16:6813–6823. doi: 10.1016/j.bmc.2008.05.068. [DOI] [PubMed] [Google Scholar]
- 318.Miroshnikova O.V., Hudson T.H., Gerena L., Kyle D.E., Lin A.J. Synthesis and antimalarial activity of new isotebuquine analogues. J. Med. Chem. 2007;50:889–896. doi: 10.1021/jm061232x. [DOI] [PubMed] [Google Scholar]
- 319.Croft S.L., Duparc S., Arbe-Barnes S.J., Craft J.C., Shin C.S., Fleckenstein L., Borghini-Fuhrer I., Rim H.J. Review of pyronaridine anti-malarial properties and product characteristics. Malar. J. 2012;11:270. doi: 10.1186/1475-2875-11-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Adegoke O.A., Babalola C.P., Oshitade O.S., Famuyiwa A.A. Determination of the physicochemical properties of pyronaridine – a new antimalarial drug. Pak. J. Pharm. Sci. 2006;19:1–6. [PubMed] [Google Scholar]
- 321.Park S.H., Pradeep K. Absorption, distribution, excretion, and pharmacokinetics of 14C-pyronaridine tetraphosphate in male and female Sprague–Dawley rats. J. Biomed. Biotechnol. 2010:590707. doi: 10.1155/2010/590707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Davis T.M., Hamzah J., Ilett K.F., Karunajeewa H.A., Reeder J.C., Batty K.T., Hackett S., Barrett P.H. In vitro interactions between piperaquine, dihydroartemisinin, and other conventional and novel antimalarial drugs. Antimicrob. Agents Chemother. 2006;50:2883–2885. doi: 10.1128/AAC.00177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Vivas L., Rattray L., Stewart L., Bongard E., Robinson B., Peters W., Croft S.L. Anti-malarial efficacy of pyronaridine and artesunate in combination in vitro and in vivo. Acta Trop. 2008;10:222–228. doi: 10.1016/j.actatropica.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 324.Auparakkitanon S., Chapoomram S., Kuaha K., Chirachariyavej T., Wilairat P. Targeting of hematin by the antimalarial pyronaridine. Antimicrob. Agents Chemother. 2006;50:2197–2200. doi: 10.1128/AAC.00119-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Görlitzer K., Kramer C., Meyer H., Walter R.D., Jomaa H., Wiesner J. Pyrido[3,2-b]indol-4-yl-amine – synthese und Prüfung auf Wirksamkeit gegen Malaria. Pharmazie. 2004;59:243–250. [PubMed] [Google Scholar]
- 326.Görlitzer K., Meyer H., Jomaa H., Wiesner J. [1]Benzofuro[3,2-b]pyridin-4-yl-amines – synthese und Prüfung auf Wirksamkeit gegen Malaria. Pharmazie. 2004;59:443–445. [PubMed] [Google Scholar]
- 327.Görlitzer K., Meyer H., Walter R.D., Jomaa H., Wiesner J. [1]Benzothieno[3,2-b]pyridin-4-yl-amine – synthese und Prüfung auf Wirksamkeit gegen Malaria. Pharmazie. 2004;59:506–512. [PubMed] [Google Scholar]
- 328.Görlitzer K., Gabriel B., Frohberg P., Wobst I., Drutkowski G., Wiesner J., Jomaa H. Thieno[2,3-c]chinoline – synthese und biologische Prüfung. Pharmazie. 2004;59:439–442. [PubMed] [Google Scholar]
- 329.Görlitzer K., Gabriel B., Jomaa H., Wiesner J. Thieno[3,2-c]chinolin-4-yl-amine – synthese und Prüfung auf Wirksamkeit gegen Malaria. Pharmazie. 2006;61:278–284. [PubMed] [Google Scholar]
- 330.Görlitzer K., Gabriel B., Jomaa H., Wiesner J. Thieno[3,4-c]chinolin-4-yl-amine – synthese und Prüfung auf Wirksamkeit gegen Malaria. Pharmazie. 2006;61:901–907. [PubMed] [Google Scholar]
- 331.Görlitzer K., Enge C., Jones P.G., Jomaa H., Wiesner J. Benzo[c][2,7]naphthyridin-5-yl-arylamine – phenol-Mannich-basen vom Amodiaquin-, Cycloquin- und Pyronaridin-typ. Pharmazie. 2007;62:89–93. [PubMed] [Google Scholar]
- 332.Görlitzer K., Bode M., Jones P.G., Jomaa H., Wiesner J. Benzo[c][2,7]naphthyridin-5-yl-amine und benzo[h][1,6]naphthyridin-5-yl-amine – potenzielle antimalariamittel. Pharmazie. 2007;62:15–26. [PubMed] [Google Scholar]
- 333.Chadwick J., Amewu R.K., Marti F., Garah F.B., Sharma R., Berry N.G., Stocks P.A., Burrell-Saward H., Wittlin S., Rottmann M., Brun R., Taramelli D., Parapini S., Ward S.A., O'Neill P.M. Antimalarial mannoxanes: hybrid antimalarial drugs with outstanding oral activity profiles and a potential dual mechanism of action. ChemMedChem. 2011;6:1357–1361. doi: 10.1002/cmdc.201100196. [DOI] [PubMed] [Google Scholar]
- 334.Sriram D., Devakaram R.V., Dinakaran M., Yogeeswari P. Aromatic amino analogues of artemisinin: synthesis and in vivo antimalarial activity. Med. Chem. Res. 2010;19:524–532. [Google Scholar]
- 335.Li Y., Yang Z.-S., Zhang H., Cao B.-J., Wang F.-D., Zhang Y., Shi Y.-L., Yang J.-D., Wu B.-A. Artemisinin derivatives bearing Mannich base group: synthesis and antimalarial activity. Bioorg. Med. Chem. 2003;11:4363–4368. doi: 10.1016/s0968-0896(03)00499-1. [DOI] [PubMed] [Google Scholar]
- 336.Ciana C.-L., Siegrist R., Aissaoui H., Marx L., Racine S., Meyer S., Binkert C., de Kanter R., Fischli C., Wittlin S., Boss C. Novel in vivo active anti-malarials based on a hydroxy-ethyl-amine scaffold. Bioorg. Med. Chem. Lett. 2013;23:658–662. doi: 10.1016/j.bmcl.2012.11.118. [DOI] [PubMed] [Google Scholar]
- 337.Pacorel B., Leung S.C., Stachulski A.V., Davies J., Vivas L., Lander H., Ward S.A., Kaiser M., Brun R., O'Neill P.M. Modular synthesis and in vitro and in vivo antimalarial assessment of C-10 pyrrole Mannich base derivatives of artemisinin. J. Med. Chem. 2010;53:633–640. doi: 10.1021/jm901216v. [DOI] [PubMed] [Google Scholar]
- 338.Davioud-Charvet E., McLeish M.J., Veine D.M., Giegel D., Arscott L.D., Andricopulo A.D., Becker K., Müller S., Schirmer R.H., Williams C.H., Jr., Kenyon G.L. Mechanism-based inactivation of thioredoxin reductase from Plasmodium falciparum by Mannich bases. Implication for cytotoxicity. Biochemistry. 2003;42:13319–13330. doi: 10.1021/bi0353629. [DOI] [PubMed] [Google Scholar]
- 339.Chipeleme A., Gut J., Rosenthal P.J., Chibale K. Synthesis and biological evaluation of phenolic Mannich bases of benzaldehyde and (thio)semicarbazone derivatives against the cysteine protease falcipain-2 and a chloroquine resistant strain of Plasmodium falciparum. Bioorg. Med. Chem. 2007;15:273–282. doi: 10.1016/j.bmc.2006.09.055. [DOI] [PubMed] [Google Scholar]
- 340.Rodrigues T., Guedes R.C., dos Santos D.J.V.A., Carrasco M., Gut J., Rosenthal P.J., Moreira R., Lopes F. Design, synthesis and structure–activity relationships of (1H-pyridin-4-ylidene)amines as potential antimalarials. Bioorg. Med. Chem. Lett. 2009;19:3476–3480. doi: 10.1016/j.bmcl.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 341.Baramee A., Coppin A., Mortuaire M., Pelinski L., Tomavo S., Brocard J. Synthesis and in vitro activities of ferrocenic aminohydroxynaphthoquinones against Toxoplasma gondii and Plasmodium falciparum. Bioorg. Med. Chem. 2006;14:1294–1302. doi: 10.1016/j.bmc.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 342.F.A. Trofimov, N.G. Tsyshkova, N.S. Bogdanova, I.S. Nikolaeva, S.A. Zotova, Z.M. Sakhaschik, E.N. Padeiskaya, A.N. Fomina, E.A. Svirina, D.M. Zlydnikov, O.I. Kubar, E.G. Shvetsova, S.N. Kutchak, V.V. Peters, E.A. Bryantseva, A.G. Konoplyannikov, B.P. Surinov, V.A. Yadrovskaya, L.S. Safonova, N.A. Karpova, E.P. Savina, L.A. Savinova, A.N. Grinev, G.N. Pershin, G.V. Grineva, E.G. Pershina, Indole Derivative Having Antiviral, Interferon-inducing and Immunomodulatory Effects, US Patent 5,198,552, 1993.
- 343.Boriskin Y.S., Leneva I.A., Pécheur E.-I., Polyak S.J. Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008;15:997–1005. doi: 10.2174/092986708784049658. [DOI] [PubMed] [Google Scholar]
- 344.Teissier E., Loquet A., Lavillette D., Lavergne J.-P., Montserret R., Cosset F.-L., Böckmann A., Meier B.H., Penin F., Pécheur E.-I. Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PLOS One. 2011;6:e15874. doi: 10.1371/journal.pone.0015874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Chai H., Zhao Y., Zhao C., Gong P. Synthesis and in vitro anti-hepatitis B virus activities of some ethyl 6-bromo-5-hydroxy-1H-indole-3-carboxylates. Bioorg. Med. Chem. 2006;14:911–917. doi: 10.1016/j.bmc.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 346.Zhao Y., Feng R., Liu Y., Zhang Y., Gong P. Synthesis and in vitro anti-hepatitis B virus activity of some ethyl 5-hydroxy-4-substituted aminomethyl-2-sulfinylmethyl-1H-indole-3-carboxylates. Chem. Res. Chin. Univ. 2010;26:272–277. [Google Scholar]
- 347.Zhao C., Zhao Y., Chai H., Gong P. Synthesis and in vitro anti-hepatitis B virus activities of some ethyl 5-hydroxy-1H-indole-3-carboxylates. Bioorg. Med. Chem. 2006;14:2552–2558. doi: 10.1016/j.bmc.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 348.Zhao C., Zhao Y., Chai H., Gong P. Synthesis and in vitro-anti-hepatitis B virus activities of several ethyl 5-hydroxy-1H-indole-3-carboxylates. Chem. Res. Chin. Univ. 2006;22:577–583. doi: 10.1016/j.bmc.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 349.Liu Y., Zhao Y., Zhai X., Feng X., Wang J., Gong P. Synthesis and anti-hepatitis B virus evaluation of novel ethyl 6-hydroxyquinoline-3-carboxylates in vitro. Bioorg. Med. Chem. 2008;16:6522–6527. doi: 10.1016/j.bmc.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 350.Jia W., Zhao Y., Li R., Wu Y., Li Z., Gong P. Synthesis and in-vitro anti-hepatitis-B virus activity of 6H-[1]benzothiopyrano[4,3-b]quinolin-10-ols. Arch. Pharm. (Weinheim) 2009;342:507–512. doi: 10.1002/ardp.200900070. [DOI] [PubMed] [Google Scholar]
- 351.Jia W., Liu Y., Li W., Liu Y., Zhang D., Zhang P., Gong P. Synthesis and in vitro anti-hepatitis B virus activity of 6H-[1]benzothiopyrano[4,3-b]quinolin-9-ols. Bioorg. Med. Chem. 2009;17:4569–4574. doi: 10.1016/j.bmc.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 352.Chen D., Zhai X., Yuan Q.H., Luo J., Xie S.C., Gong P. Synthesis and in vitro anti-hepatitis B virus activity of 1H-benzimidazol-5-ol derivatives. Chin. Chem. Lett. 2010;21:1326–1329. [Google Scholar]
- 353.Zhao Y., Liu Y., Chen D., Wei Z., Liu W., Gong P. Synthesis and biological evaluation of 1H-benzimidazol-5-ols as potent HBV inhibitors. Bioorg. Med. Chem. Lett. 2010;20:7230–7233. doi: 10.1016/j.bmcl.2010.10.099. [DOI] [PubMed] [Google Scholar]
- 354.Chen D., Liu Y., Zhang S., Guo D., Liu C., Li S., Gong P. Synthesis and in-vitro anti-hepatitis B virus activity of ethyl 6-bromo-8-hydroxyimidazo[1,2-a]pyridine-3-carboxylates. Arch. Pharm. (Weinheim) 2011;344:158–164. doi: 10.1002/ardp.201000045. [DOI] [PubMed] [Google Scholar]
- 355.Mazzei M., Nieddu E., Miele M., Balbi A., Ferrone M., Fermeglia M., Mazzei M.T., Pricl S., La Colla P., Marongiu F., Ibba C., Loddo R. Activity of Mannich bases of 7-hydroxycoumarin against Flaviviridae. Bioorg. Med. Chem. 2008;16:2591–2605. doi: 10.1016/j.bmc.2007.11.045. [DOI] [PubMed] [Google Scholar]
- 356.Xing S., Bhat S., Shroff N.S., Zhang H., Lopez J.A., Margolick J.B., Liu J.O., Siliciano R.F. Novel structurally related compounds reactivate latent HIV-1 in a bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J. Antimicrob. Chemother. 2012;67:398–403. doi: 10.1093/jac/dkr496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Terzioğlu N., Karali N., Gürsoy A., Pannecouque C., Leysen P., Paeshuyse J., Neyts J., De Clercq E. Synthesis and primary antiviral activity evaluation of 3-hydrazono-5-nitro-2-indolinone derivatives. ARKIVOC i. 2006:109–118. [Google Scholar]
- 358.Pawar V., Lokwani D., Bhandari S., Mitra D., Sabde S., Bothara K., Madgulkar A. Design of potential reverse transcriptase inhibitor containing isatin nucleus using molecular modeling studies. Bioorg. Med. Chem. 2010;18:3198–3211. doi: 10.1016/j.bmc.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 359.Sriram D., Bal T.R., Yogeeswari P. Newer aminopyrimidinimino isatin analogues as non-nucleoside HIV-1 reverse transcriptase inhibitors for HIV and other opportunistic infections of AIDS: design, synthesis and biological evaluation. Farmaco. 2005;60:377–384. doi: 10.1016/j.farmac.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 360.Sebastian L., Desai A., Shampur M.N., Perumal Y., Sriram D., Vasanthapuram R. N-Methylisatin-beta-thiosemicarbazone derivative (SCH 16) is an inhibitor of Japanese encephalitis virus infection in vitro and in vivo. Virol. J. 2008;5:64. doi: 10.1186/1743-422X-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.S.G. Alvarez, J. Botyanszki, J. De Los Angeles, J. Fu, R. Fujimoto, J.M. Gralapp, R.C. Griffith, P. Lu, S.M. Pham, C.D. Roberts, F.U. Schmitz, M. Seep-Ersaud, R. Tommasi, A.C. Villa, S. Wattanasin, A. Yifru, R. Zheng, X. Zheng, Condensed Pentacyclic Derivatives for Use in the Treatment of Flaviviridae Infections, PCT Int. Appl. WO 2009086139, 2009.
- 362.Selvam P., Lakra D.R., Pannecouque C., De Clercq E. Synthesis, antiviral and cytotoxicity studies of novel N-substituted indophenazine derivatives. Indian J. Pharm. Sci. 2012;74:275–278. doi: 10.4103/0250-474X.106077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Gul H.I., Calis U., Vepsalainen J. Synthesis of some mono-Mannich bases and corresponding azine derivatives and evaluation of their anticonvulsant activity. Arzneimittelforschung. 2004;54:359–364. doi: 10.1055/s-0031-1296984. [DOI] [PubMed] [Google Scholar]
- 364.Gul H.I., Calis U., Ozturk Z., Tutar E., Calikiran L. Evaluation of anticonvulsant activities of bis(3-aryl-3-oxo-propyl)ethylamine hydrochlorides and 4-aryl-3-arylcarbonyl-1-ethyl-4-piperidinol hydrochlorides. Arzneimittelforschung. 2007;57:133–136. doi: 10.1055/s-0031-1296595. [DOI] [PubMed] [Google Scholar]
- 365.Gul H.I., Calis U., Vepsalainen J. Synthesis and evaluation of anticonvulsant activities of some bis Mannich bases and corresponding piperidinols. Arzneimittelforschung. 2002;52:863–869. doi: 10.1055/s-0031-1299982. [DOI] [PubMed] [Google Scholar]
- 366.Aytemir M.D., Çaliş Ü. Synthesis of some new hydroxypyranone derivatives and evaluation of their anticonvulsant activities. FABAD J. Pharm. Sci. 2006;31:23–29. [Google Scholar]
- 367.Aytemir M.D., Çaliş Ü. Synthesis of some novel Mannich bases derived from allomaltol and evaluation of their anticonvulsant activities. Hacet. Univ. J. Fac. Pharm. 2007;27:1–10. [Google Scholar]
- 368.Aytemir M.D., Çaliş Ü. Anticonvulsant and neurotoxicity evaluation of some novel kojic acids and allomaltol derivatives. Arch. Pharm. (Weinheim) 2010;343:173–181. doi: 10.1002/ardp.200900236. [DOI] [PubMed] [Google Scholar]
- 369.Aytemir M.D., Septioğlu E., Çaliş Ü. Synthesis and anticonvulsant activity of new kojic acid derivatives. Arzneimittelforschung. 2010;60:22–29. doi: 10.1055/s-0031-1296244. [DOI] [PubMed] [Google Scholar]
- 370.Stratford E.S., Curley R.W., Jr. Synthesis of aminomethyl-substituted cyclic imide derivatives for evaluation as anticonvulsants. J. Med. Chem. 1983;26:1463–1469. doi: 10.1021/jm00364a020. [DOI] [PubMed] [Google Scholar]
- 371.Byrtus H., Obniska J., Czopek A., Kamiński K. Synthesis and anticonvulsant activity of new N-Mannich bases derived from 5-cyclopropyl-5-phenyl-hydantoins. Arch. Pharm. (Weinheim) 2011;344:231–241. doi: 10.1002/ardp.201000241. [DOI] [PubMed] [Google Scholar]
- 372.Byrtus H., Obniska J., Czopek A., Kamiński K., Pawłowski M. Synthesis and anticonvulsant activity of new N-Mannich bases derived from 5-cyclopropyl-5-phenyl- and 5-cyclopropyl-5-(4-chlorophenyl)-imidazolidine-2,4-diones. Bioorg. Med. Chem. 2011;19:6149–6156. doi: 10.1016/j.bmc.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 373.Patel H.J., Sarra J., Caruso F., Rossi M., Doshi U., Stephani R.A. Synthesis and anticonvulsant activity of new N-1′,N-3′-disubstituted-2′H,3H,5′H-spiro-(2-benzofuran-1,4′-imidazolidine)-2′,3,5′-triones. Bioorg. Med. Chem. Lett. 2006;16:4644–4647. doi: 10.1016/j.bmcl.2006.05.102. [DOI] [PubMed] [Google Scholar]
- 374.Zhu Q., Pan Y., Xu Z., Li R., Qiu G., Xu W., Ke X., Wu L., Hu X. Synthesis and potential anticonvulsant activity of new N-3-substituted 5,5-cyclopropanespirohydantoins. Eur. J. Med. Chem. 2009;44:296–302. doi: 10.1016/j.ejmech.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 375.Madaiah M., Prashanth M.K., Revanasiddappa H.D., Veeresh B. Synthesis and pharmacological evaluation of novel 1′-[2-(difluoromethoxy)benzyl]-2′H,5′H-spiro[8-azabicyclo[3.2.1]octane-3,4′-imidazolidine]-2′,5′-diones and their derivatives. Arch. Pharm. (Weinheim) 2014;347:370–380. doi: 10.1002/ardp.201300289. [DOI] [PubMed] [Google Scholar]
- 376.Obniska J., Byrtus H., Kamiński K., Pawłowski M., Szczesio M., Karolak-Wojciechowska J. Design, synthesis, and anticonvulsant activity of new N-Mannich bases derived from spirosuccinimides and spirohydantoins. Bioorg. Med. Chem. 2010;18:6134–6142. doi: 10.1016/j.bmc.2010.06.064. [DOI] [PubMed] [Google Scholar]
- 377.Obniska J., Zagorska A. Synthesis and anticonvulsant properties of new N-[(4-arylpiperazin-1-yl)-methyl] derivatives of 3-aryl pyrrolidine-2,5-dione and 2-aza-spiro[4.4]nonane-1,3-dione. Farmaco. 2003;58:1227–1234. doi: 10.1016/S0014-827X(03)00187-3. [DOI] [PubMed] [Google Scholar]
- 378.Obniska J., Kaminski K., Skrzynska D., Pichor J. Synthesis and anticonvulsant activity of new N-[(4-arylpiperazin-1-yl)-alkyl] derivatives of 3-phenyl-pyrrolidine-2,5-dione. Eur. J. Med. Chem. 2009;44:2224–2233. doi: 10.1016/j.ejmech.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 379.Obniska J., Chlebek I., Pichór J., Kopytko M., Kamiński K. Synthesis, physicochemical and anticonvulsant properties of new N-[(4-aryl-1-piperazinyl)alkyl]-1-phenyl-2,5-pyrrolidinedione and N-[(4-aryl-1-piperazinyl)alkyl]-3-(3-methylphenyl)-2,5-pyrrolidinedione derivatives. Acta Pol. Pharm. 2009;66:639–647. [PubMed] [Google Scholar]
- 380.Obniska J., Kopytko M., Zagórska A., Chlebek I., Kamiński K. Synthesis and anticonvulsant properties of new Mannich bases derived from 3-aryl-pyrrolidine-2,5-diones. Part 1. Arch. Pharm. (Weinheim) 2010;343:333–341. doi: 10.1002/ardp.200900250. [DOI] [PubMed] [Google Scholar]
- 381.Obniska J., Rzepka S., Kamiński K. Synthesis and anticonvulsant activity of new N-Mannich bases derived from 3-(2-fluorophenyl)- and 3-(2-bromophenyl)-pyrrolidine-2,5-diones. Part II. Bioorg. Med. Chem. 2012;20:4872–4880. doi: 10.1016/j.bmc.2012.05.032. [DOI] [PubMed] [Google Scholar]
- 382.Kamiński K., Obniska J., Chlebek I., Wiklik B., Rzepka S. Design, synthesis and anticonvulsant properties of new N-Mannich bases derived from 3-phenylpyrrolidine-2,5-diones. Bioorg. Med. Chem. 2013;21:6821–6830. doi: 10.1016/j.bmc.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 383.Obniska J., Kolaczkowski M., Bojarski A.J., Duszyńska B. Synthesis, anticonvulsant activity and 5-HT1A, 5-HT2A receptor affinity of new N-[(4-arylpiperazin-1-yl)-alkyl] derivatives of 2-azaspiro[4.4]nonane and [4.5]decane-1,3-dione. Eur. J. Med. Chem. 2006;41:874–881. doi: 10.1016/j.ejmech.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 384.Obniska J., Chlebek I., Kamiński K. Synthesis and anticonvulsant properties of new Mannich bases derived from 3,3-disubstituted pyrrolidine-2,5-diones. Part IV. Arch. Pharm. (Weinheim) 2012;345:713–722. doi: 10.1002/ardp.201200092. [DOI] [PubMed] [Google Scholar]
- 385.Obniska J., Chlebek I., Kamiński K., Karolak-Wojciechowska J. Synthesis and anticonvulsant properties of new N-Mannich bases derived from 3,3-diphenyl- and 3-ethyl-3-methyl-pyrrolidine-2,5-diones. Part III. Arch. Pharm. (Weinheim) 2013;346:71–82. doi: 10.1002/ardp.201200265. [DOI] [PubMed] [Google Scholar]
- 386.Kamiński K., Obniska J., Chlebek I., Liana P., Pękala E. Synthesis and biological properties of new N-Mannich bases derived from 3-methyl-3-phenyl- and 3,3-dimethyl-succinimides. Part V. Eur. J. Med. Chem. 2013;66:12–21. doi: 10.1016/j.ejmech.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 387.Obniska J., Chlebek I., Kamiński K., Bojarski A.J., Satała G. Anticonvulsant activity and 5-HT1A/5-HT7 receptors affinity of piperazine derivatives of 3,3-diphenyl- and 3,3-dimethyl-succinimides. Lett. Drug Des. Discov. 2013;10:173–182. [Google Scholar]
- 388.Archana, Srivastava V.K., Kumar A. Synthesis of some newer derivatives of substituted quinazolinonyl-2-oxo/thiobarbituric acid as potent anticonvulsant agents. Bioorg. Med. Chem. 2004;12:1257–1264. doi: 10.1016/j.bmc.2003.08.035. [DOI] [PubMed] [Google Scholar]
- 389.Bajaj K., Archana, Kumar A. Synthesis and pharmacological evaluation of newer substituted benzoxazepine derivatives as potent anticonvulsant agents. Eur. J. Med. Chem. 2004;39:369–376. doi: 10.1016/j.ejmech.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 390.Garg N., Chandra T., Archana, Jain A.B., Kumar A. Synthesis and evaluation of some new substituted benzothiazepine and benzoxazepine derivatives as anticonvulsant agents. Eur. J. Med. Chem. 2010;45:1529–1535. doi: 10.1016/j.ejmech.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 391.Pandeya S.N., Rajput N. Synthesis and anticonvulsant activity of various Mannich and Schiff bases of 1,5-benzodiazepines. Int. J. Med. Chem. 2012:237965. doi: 10.1155/2012/237965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Liu D., Yu W., Li J., Pang C., Zhao L. Novel 2-(E)-substituted benzylidene-6-(N-substituted aminomethyl)cyclohexanones and cyclohexanols as analgesic and anti-inflammatory agents. Med. Chem. Res. 2013;22:3779–3786. [Google Scholar]
- 393.Popovici I., Popovici I., Mărculescu A.D. Biochemical effects of some new Mannich base from ortho-hydroxyaryl alkyl ketones on rats with chronic inflammation. Farmacia (Bucharest) 2008;56:221–228. [Google Scholar]
- 394.Agababyan A.G., Isakhanyan A.Y., Papoyan O.A., Tumadzhyan A.E., Balasanyan A.S., Gevorgyan G.A. Synthesis and pharmacological activity of N-[β-(para-substituted benzoyl)ethyl]isoleucine and -methionine. Pharm. Chem. J. 2005;39:364–366. [Google Scholar]
- 395.Agababyan A.G., Gevorgyan G.A., Tumadzhyan A.E., Akopyan R.A., Aristakesyan S.A. Synthesis and anti-inflammatory, analgesic, and antipyretic activities of N-[β-(p-substituted benzoyl)ethyl]amino acids. Pharm. Chem. J. 2009;43:13–15. [Google Scholar]
- 396.Chukicheva I.Yu., Fedorova I.V., Buravlev E.V., Lumpov A.E., Vikharev Yu.B., Anikina L.V., Grishko V.V., Kuchin A.V. Anti-inflammatory activity of isobornylphenol derivatives. Chem. Nat. Compd. 2010;46:478–480. [Google Scholar]
- 397.Chen G., Shan W., Wu Y., Ren L., Dong J., Ji Z. Synthesis and anti-inflammatory activity of resveratrol analogs. Chem. Pharm. Bull. 2005;53:1587–1590. doi: 10.1248/cpb.53.1587. [DOI] [PubMed] [Google Scholar]
- 398.Khan M.S., Husain A., Sharma S. New 4,6-diacetylresorcinol Mannich bases: synthesis and biological evaluation. Acta Pol. Pharm. 2010;67:261–266. [PubMed] [Google Scholar]
- 399.Bandgar B.P., Patil S.A., Totre J.V., Korbad B.L., Gacche R.N., Hote B.S., Jalde S.S., Chavan H.V. Synthesis and biological evaluation of nitrogen-containing benzophenone analogues as TNF-α and IL-6 inhibitors with antioxidant activity. Bioorg. Med. Chem. Lett. 2010;20:2292–2296. doi: 10.1016/j.bmcl.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 400.Bandgar B.P., Patil S.A., Gacche R.N., Korbad B.L., Hote B.S., Kinkar S.N., Jalde S.S. Synthesis and biological evaluation of nitrogen-containing chalcones as possible anti-inflammatory and antioxidant agents. Bioorg. Med. Chem. Lett. 2010;20:730–733. doi: 10.1016/j.bmcl.2009.11.068. [DOI] [PubMed] [Google Scholar]
- 401.Kouskoura M., Hadjipavlou-Litina D., Giakoumakou M. Synthesis and anti-inflammatory activity of chalcones and related Mannich bases. Med. Chem. 2008;4:586–596. doi: 10.2174/157340608786242070. [DOI] [PubMed] [Google Scholar]
- 402.Vijaya Bhaskar Reddy M., Hwang T.-L., Leu Y.-L., Chiou W.-F., Wu T.-S. Inhibitory effects of Mannich bases of heterocyclic chalcones on NO production by activated RAW 264.7 macrophages and superoxide anion generation and elastase release by activated human neutrophils. Bioorg. Med. Chem. 2011;19:2751–2756. doi: 10.1016/j.bmc.2011.02.038. [DOI] [PubMed] [Google Scholar]
- 403.Akhter M., Habibullah S., Hasan S.M., Alam M.M., Akhter N., Shaquiquzzaman M. Synthesis of some new 3,4-dihydro-2H-1,3-benzoxazines under microwave irradiation in solvent-free conditions and their biological activity. Med. Chem. Res. 2011;20:1147–1153. [Google Scholar]
- 404.Kontogiorgis C.A., Hadjipavlou-Litina D.J. Synthesis and antiinflammatory activity of coumarin derivatives. J. Med. Chem. 2005;48:6400–6408. doi: 10.1021/jm0580149. [DOI] [PubMed] [Google Scholar]
- 405.Park J.-S., Woo M.-S., Kim D.-H., Hyun J.-W., Kim W.-K., Lee J.-C., Kim H.-S. Anti-inflammatory mechanisms of isoflavone metabolites in lipopolysaccharide-stimulated microglial cells. J. Pharmacol. Exp. Ther. 2007;320:1237–1245. doi: 10.1124/jpet.106.114322. [DOI] [PubMed] [Google Scholar]
- 406.Wang J., Zhong Y., Liu L., Ji X.Q., Zhang Z.G. Synthesis and NO production inhibition of novel Mannich base derivatives of irisolidone. Chin. Chem. Lett. 2008;19:387–389. [Google Scholar]
- 407.Radwan A.A., elTahir K.E.H. Synthesis and in-silico studies of some diaryltriazole derivatives as potential cyclooxygenase inhibitors. Arch. Pharm. Res. 2013;36:553–563. doi: 10.1007/s12272-013-0078-6. [DOI] [PubMed] [Google Scholar]
- 408.Sujith K.V., Rao J.N., Shetty P., Kalluraya B. Regioselective reaction: synthesis and pharmacological study of Mannich bases containing ibuprofen moiety. Eur. J. Med. Chem. 2009;44:3697–3702. doi: 10.1016/j.ejmech.2009.03.044. [DOI] [PubMed] [Google Scholar]
- 409.Nithinchandra, Kalluraya B., Aamir S., Shabaraya A.R. Regioselective reaction: synthesis, characterization and pharmacological activity of some new Mannich and Schiff bases containing sydnone. Eur. J. Med. Chem. 2012;54:597–604. doi: 10.1016/j.ejmech.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 410.Gowda J., Khader A.M.A., Kalluraya B., Shree P., Shabaraya A.R. Synthesis, characterization and pharmacological activity of 4-{[1-substituted aminomethyl-4-arylideneamino-5-sulfanyl-4,5-dihydro-1H-1,2,4-triazol-3-yl]methyl}-2H-1,4-benzothiazin-3(4H)-ones. Eur. J. Med. Chem. 2011;46:4100–4106. doi: 10.1016/j.ejmech.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 411.Toma A., Hapău D., Naghi M., Vlasec L., Mogoşan C., Zaharia V. Heterocycles 34. Synthesis and anti-inflammatory activity of new polyheterocyclic Schiff bases and Mannich bases. Stud. UBB Chem. 2013;58:93–104. [Google Scholar]
- 412.Thore S.N., Gupta S.V., Baheti K.G. Docking, synthesis, and pharmacological investigation of novel substituted thiazole derivatives as non-carboxylic, anti-inflammatory, and analgesic agents. Med. Chem. Res. 2013;22:3802–3811. [Google Scholar]
- 413.Köksal M., Gökhan N., Küpeli E., Yesilada E., Erdoğan H. Synthesis, analgesic and antiinflammatory properties of certain 5-/6-acyl-3-(4-substituted-1-piperazinylmethyl)-2-benzoxazolinones derivatives. Arch. Pharm. (Weinheim) 2005;338:117–125. doi: 10.1002/ardp.200400937. [DOI] [PubMed] [Google Scholar]
- 414.Gökhan N., Erdoğan H., Durlu N.T., Demirdamar R., Özalp M. Synthesis and evaluation of analgesic, anti-inflammatory and antimicrobial activities of 6-acyl-3-piperazinomethyl-2-benzoxazolinones. Arzneimittelforschung. 2003;53:114–120. doi: 10.1055/s-0031-1297081. [DOI] [PubMed] [Google Scholar]
- 415.Gökhan N., Köksal M., Küpeli E., Yeşilada E., Erdoğan H. Some new Mannich bases of 5-methyl-2-benzoxazolinones with analgesic and anti-inflammatory activities. Turk. J. Chem. 2005;29:445–454. [Google Scholar]
- 416.Köksal M., Gökhan N., Küpeli E., Yesilada E., Erdogan H. Analgesic and antiinflammatory activities of some new Mannich bases of 5-nitro-2-benzoxazolinones. Arch. Pharm. Res. 2007;30:419–424. doi: 10.1007/BF02980214. [DOI] [PubMed] [Google Scholar]
- 417.Muthukumar V.A., George S., Vaidhyalingam V. Synthesis and pharmacological evaluation of 1-(1-((substituted)methyl)-5-methyl-2-oxoindolin-3-ylidene)-4-(substituted pyridin-2-yl)thiosemicarbazide. Biol. Pharm. Bull. 2008;31:1461–1464. doi: 10.1248/bpb.31.1461. [DOI] [PubMed] [Google Scholar]
- 418.Saravanan G., Alagarsamy V., Prakash C.R. Synthesis, analgesic, antiinflammatory and ulcerogenic properties of some novel N′-((1-(substituted-amino)methyl)-2-oxoindolin-3-ylidene)-4-(2-(methyl/phenyl)-4-oxoquinazolin-3(4H)-yl)benzohydrazide derivatives. Drug Discov. Ther. 2012;6:78–87. [PubMed] [Google Scholar]
- 419.Jesudason E.P., Sridhar S.K., Malar E.J.P., Shanmugapandiyan P., Inayathullah M., Arul V., Selvaraj D., Jayakumar R. Synthesis, pharmacological screening, quantum chemical and in vitro permeability studies of N-Mannich bases of benzimidazoles through bovine cornea. Eur. J. Med. Chem. 2009;44:2307–2312. doi: 10.1016/j.ejmech.2008.03.043. [DOI] [PubMed] [Google Scholar]
- 420.Mariappan G., Bhuyan N.R., Kumar P., Kumar D., Murali K. Synthesis and biological evaluation of Mannich bases of benzimidazole derivatives. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2011;50B:1216–1219. [Google Scholar]
- 421.El-Sayed N.A., Awadallah F.M., Ibrahim N.A., El-Saadi M.T. Synthesis, anti-inflammatory and ulcerogenicity studies of some substituted pyrimido[1,6-a]azepine derivatives. Eur. J. Med. Chem. 2010;45:3147–3154. doi: 10.1016/j.ejmech.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 422.El-Sayed N.A., Awadallah F.M., Ibrahim N.A., El-Saadi M.T. Potential anti-inflammatory activity and ulcerogenicity study of some novel pyrimido[4′,5′:4,5]pyrimido[1,6-a]azepine derivatives. Med. Chem. Res. 2012;21:395–405. [Google Scholar]
- 423.Refaat H.M., Khalil O.M., Kadry H.H. Synthesis and anti-inflammatory activity of certain piperazinylthienylpyridazine derivatives. Arch. Pharm. Res. 2007;30:803–811. doi: 10.1007/BF02978828. [DOI] [PubMed] [Google Scholar]
- 424.Amir M., Javed S.A., Kumar H. Design and synthesis of 3-[3-(substituted phenyl)-4-piperidin-1-ylmethyl/-4-morpholin-4-ylmethyl-4,5-dihydro-isoxazol-5-yl]-1H-indoles as potent anti-inflammatory agents. Med. Chem. Res. 2010;19:299–310. [Google Scholar]
- 425.Vasilevsky S.F., Govdi A.I., Shults E.E., Shakirov M.M., Sorokina I.V., Tolstikova T.G., Baev D.S., Tolstikov G.A., Alabugin I.V. Efficient synthesis of the first betulonic acid–acetylene hybrids and their hepatoprotective and anti-inflammatory activity. Bioorg. Med. Chem. 2009;17:5164–5169. doi: 10.1016/j.bmc.2009.05.059. [DOI] [PubMed] [Google Scholar]
- 426.M. Gerlach, C. Maul, Substituted 1 and 2-naphthol Mannich Bases, US Patent 7,202,242, 2007.
- 427.Srivastava S., Pandeya S.N., Yadav M.K., Singh B.K. Synthesis and analgesic activity of novel derivatives of 1,2-substituted benzimidazoles. J. Chem. 2013:694295. [Google Scholar]
- 428.Malinka W., Karczmarzyk Z., Sieklucka-Dziuba M., Sadowski M., Kleinrok Z. Synthesis and in vivo pharmacology of new derivatives of isothiazolo[5,4-b]pyridine of Mannich base type. Farmaco. 2001;56:905–918. doi: 10.1016/s0014-827x(01)01157-0. [DOI] [PubMed] [Google Scholar]
- 429.Malinka W., Świątek P., Filipek B., Sapa J., Jezierska A., Koll A. Synthesis, analgesic activity and computational study of new isothiazolopyridines of Mannich base type. Farmaco. 2005;60:961–968. doi: 10.1016/j.farmac.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 430.Shen A.-Y., Huang M.-H., Liao L.-F., Wang T.-S. Thymol analogues with antioxidant and L-type calcium current inhibitory activity. Drug Dev. Res. 2005;64:195–202. [Google Scholar]
- 431.Fernández-Bachiller M.I., Pérez C., González-Muñoz G.C., Conde S., López M.G., Villarroya M., García A.G., Rodríguez-Franco M.I. Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer's disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010;53:4927–4937. doi: 10.1021/jm100329q. [DOI] [PubMed] [Google Scholar]
- 432.Li N.-G., Song S.-L., Shen M.-Z., Tang Y.-P., Shi Z.-H., Tang H., Shi Q.-P., Fu Y.-F., Duan J.-A. Mannich bases of scutellarein as thrombin-inhibitors: design, synthesis, biological activity and solubility. Bioorg. Med. Chem. 2012;20:6919–6923. doi: 10.1016/j.bmc.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 433.Joshi D., Field J., Murphy J., Abdelrahim M., Schonherr H., Sparrow J.R., Ellestad G., Nakanishi K., Zask A. Synthesis of antioxidants for prevention of age-related macular degeneration. J. Nat. Prod. 2013;76:450–454. doi: 10.1021/np300769c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Ma L., Xiao Y., Li C., Xie Z.-L., Li D.-D., Wang Y.-T., Mac H.-T., Zhu H.-L., Wang M.-H., Ye Y.-H. Synthesis and antioxidant activity of novel Mannich base of 1,3,4-oxadiazole derivatives possessing 1,4-benzodioxan. Bioorg. Med. Chem. 2013;21:6763–6770. doi: 10.1016/j.bmc.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 435.Jagadish P.C., Soni N., Verma A. Design, synthesis, and in vitro antioxidant activity of 1,3,5-trisubstituted-2-pyrazolines derivatives. J. Chem. 2013:765768. [Google Scholar]
- 436.Chernyshenko Yu.N., Mustafin A.G., Gimadieva A.R., Abdrakhmanov I.B., Gerchikov A.Ya., Safarova I.V. Synthesis and antioxidant activity of aminomethylated 6-methyluracil derivatives. Pharm. Chem. J. 2010;44:123–125. [Google Scholar]
- 437.Velázquez A.M., Martínez V., Abrego V., Balboa M.A., Torres L.A., Camacho B., Díaz-Barriga S., Romero A., López-Castañares R., Angeles E. Synthesis and antihypertensive effects of new methylthiomorpholinphenol derivatives. Eur. J. Med. Chem. 2008;43:486–500. doi: 10.1016/j.ejmech.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 438.Abrego V.H., Martínez-Pérez B., Torres L.A., Ángeles E., Martínez L., Marroquín-Pascual J.L., Moya-Hernández R., Amaro-Recillas H.A., Rueda-Jackson J.C., Rodríguez-Barrientos D., Rojas-Hernández A. Antihypertensive and antiarrhythmic properties of a para-hydroxy[bis(ortho-morpholinylmethyl)]phenyl-1,4-DHP compound: comparison with other compounds of the same kind and relationship with logP values. Eur. J. Med. Chem. 2010;45:4622–4630. doi: 10.1016/j.ejmech.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 439.Tandon V.K., Singh K.A., Goswamy G.K. 1- and 2-substituted naphthalenes: a new class of potential hypotensive agents. Bioorg. Med. Chem. Lett. 2004;14:2797–2800. doi: 10.1016/j.bmcl.2004.03.080. [DOI] [PubMed] [Google Scholar]
- 440.Shen A.Y., Teng C.-M., Wang J.-S. Studies on the cardiovascular action of TPY-β: an antihypertensive agent with antiplatelet activity. Life Sci. 1999;65:1817–1828. doi: 10.1016/s0024-3205(99)00433-6. [DOI] [PubMed] [Google Scholar]
- 441.Siddiqui A.A., Mishra R., Shaharyar M. Synthesis, characterization and antihypertensive activity of pyridazinone derivatives. Eur. J. Med. Chem. 2010;45:2283–2290. doi: 10.1016/j.ejmech.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 442.Anisimova V.A., Spasov A.A., Kovalev Yu.G., Kovalev S.G., Dudchenko G.P. Synthesis and pharmacological activity of 2- and 3-(aminomethyl)imidazo[1,2-a]benzimidazoles. Pharm. Chem. J. 2004;38:535–539. [Google Scholar]
- 443.El-Sabbagh O.I., Shabaan M.A., Kadry H.H., Al-Din E.S. New octahydroquinazoline derivatives: synthesis and hypotensive activity. Eur. J. Med. Chem. 2010;45:5390–5396. doi: 10.1016/j.ejmech.2010.08.064. [DOI] [PubMed] [Google Scholar]
- 444.Lee B., Bauer H., Melchers J., Ruppert T., Rattray L., Yardley V., Davioud-Charvet E., Krauth-Siegel R.L. Irreversible inactivation of trypanothione reductase by unsaturated Mannich bases: a divinyl ketone as key intermediate. J. Med. Chem. 2005;48:7400–7410. doi: 10.1021/jm0504860. [DOI] [PubMed] [Google Scholar]
- 445.Wenzel I.N., Wong P.E., Maes L., Mueller T.J.J., Krauth-Siegel R.L., Barrett M.P., Davioud-Charvet E. Unsaturated Mannich bases active against multidrug-resistant Trypanosoma brucei brucei strains. ChemMedChem. 2009;4:339–351. doi: 10.1002/cmdc.200800360. [DOI] [PubMed] [Google Scholar]
- 446.de Mello H., Echevarria A., Bernardino A.M., Canto-Cavalheiro M., Leon L.L. Antileishmanial pyrazolopyridine derivatives: synthesis and structure–activity relationship analysis. J. Med. Chem. 2004;47:5427–5432. doi: 10.1021/jm0401006. [DOI] [PubMed] [Google Scholar]
- 447.Siddiqui S.M., Salahuddin A., Azam A. Mannich base derivatives of 1,3,4-oxadiazole: synthesis and screening against Entamoeba histolytica. Med. Chem. Res. 2013;22:1313–1319. [Google Scholar]
- 448.Kamel M.M., Anwar M.M., Soliman A.M., Abdel-Hamid H.F. In vitro antischistosomal evaluation of some newly synthesized praziquantel derivatives. Res. Chem. Intermed. 2013;39:3417–3426. [Google Scholar]
- 449.Vijaya Bhaskar Reddy M., Tsai W.-J., Qian K., Lee K.-H., Wu T.-S. Structure–activity relationships of chalcone analogs as potential inhibitors of ADP- and collagen-induced platelet aggregation. Bioorg. Med. Chem. 2011;19:7711–7719. doi: 10.1016/j.bmc.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 450.Severino B., Fiorino F., Perissutti E., Frecentese F., Cirino G., Roviezzo F., Santagada V., Caliendo G. Synthesis and pharmacological evaluation of peptide-mimetic protease-activated receptor-1 antagonists containing novel heterocyclic scaffolds. Bioorg. Med. Chem. 2008;16:6009–6020. doi: 10.1016/j.bmc.2008.04.059. [DOI] [PubMed] [Google Scholar]
- 451.Zapata-Colindres J.C., Zepeda-Gómez S., Montaño-Loza A., Vázquez-Ballesteros E., de Jesús Villalobos J., Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal anti-inflammatory drugs in peptic ulcer disease. Can. J. Gastroenterol. 2006;20:277–280. doi: 10.1155/2006/175217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Subudhi B.B., Panda P.K., Bhatta D. Synthesis and antiulcer activity study of 1,4-dihydropyridines and their Mannich bases with sulfanilamide. Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem. 2009;48B:725–728. [Google Scholar]
- 453.Ragab F.A., Hassan G.S., Yossef H.A., Hashem H.A. Synthesis of 6- and 9-alkylaminomethyl furoflavones as gastroprotective agents. Eur. J. Med. Chem. 2007;42:1117–1127. doi: 10.1016/j.ejmech.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 454.World Health Organization . 2012. Report A65/10 on the Global Burden of Mental Disorders, Submitted to the 65th World Health Assembly.http://apps.who.int/gb/ebwha/pdf_files/WHA65/A65_10-en.pdf (accessed 02.07.14.) [Google Scholar]
- 455.Köksal M., Sirri Bilge S. Synthesis and antidepressant-like profile of novel 1-aryl-3-[(4-benzyl)piperidine-1-yl]propane derivatives. Arch. Pharm. (Weinheim) 2007;340:299–303. doi: 10.1002/ardp.200700028. [DOI] [PubMed] [Google Scholar]
- 456.Geronikaki A., Babaev E., Dearden J., Dehaen W., Filimonov D., Galaeva I., Krajneva V., Lagunin A., Macaev F., Molodavkin G., Poroikov V., Pogrebnoi S., Saloutin V., Stepanchikova A., Stingaci E., Tkach N., Vlad L., Voronina T. Design, synthesis, computational and biological evaluation of new anxiolytics. Bioorg. Med. Chem. 2004;12:6559–6568. doi: 10.1016/j.bmc.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 457.Abdel Gawad N.M., Hassan G.S., Georgey H.H., El-Zorba H.Y. Benzoxepin derivatives: design, synthesis, and pharmacological evaluation with sedative–hypnotic effect. Med. Chem. Res. 2012;21:747–759. [Google Scholar]
- 458.Dyubchenko O.I., Nikulina V.V., Markov A.F., Kandalintseva N.V., Prosenko A.E., Khoshchenko O.M., Shwarts Ya.Sh., Dushkin M.I. Synthesis and hepatoprotector activity of water-soluble derivatives of aminoalkylphenols. Pharm. Chem. J. 2006;40:243–247. [Google Scholar]
- 459.Romanelli M.N., Cerbai E., Dei S., Guandalini L., Martelli C., Martini E., Scapecchi S., Teodori E., Mugelli A. Design, synthesis and preliminary biological evaluation of zatebradine analogues as potential blockers of the hyperpolarization-activated current. Bioorg. Med. Chem. 2005;13:1211–1220. doi: 10.1016/j.bmc.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 460.Xu Q., Liu T., Tian R., Li Q., Ma D. Synthesis and antiemetic activity of 1,2,3,9-tetrahydro-9-methyl-3-(4-substituted-piperazin-1-ylmethyl)-4H-carbazol-4-one derivatives. Front. Chem. China. 2009;4:63–68. [Google Scholar]
- 461.Koufaki M., Theodorou E., Alexi X., Nikoloudaki F., Alexis M.N. Synthesis of tropolone derivatives and evaluation of their in vitro neuroprotective activity. Eur. J. Med. Chem. 2010;45:1107–1112. doi: 10.1016/j.ejmech.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 462.Zhao Q., Liu X., Zhang L., Shen X., Qi J., Wang J., Qian N., Deng L. Bone selective protective effect of a novel bone-seeking estrogen on trabecular bone in ovariectomized rats. Calcif. Tissue Int. 2013;93:172–183. doi: 10.1007/s00223-013-9739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Kasser J., Nazarov A.A., Hartinger C.G., Wdziekonski B., Dani C., Kuznetsov M.L., Arion V.B., Keppler B.K. A one step/one pot synthesis of N,N-bis(phosphonomethyl)amino acids and their effects on adipogenic and osteogenic differentiation of human mesenchymal stem cells. Bioorg. Med. Chem. 2009;17:3388–3393. doi: 10.1016/j.bmc.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 464.Stepkowski S.M., Kao J., Wang M.-E., Tejpal N., Podder H., Furian L., Dimmock J., Jha A., Das U., Kahan B.D., Kirken R.A. The Mannich base NC1153 promotes long-term allograft survival and spares the recipient from multiple toxicities. J. Immunol. 2005;175:4236–4246. doi: 10.4049/jimmunol.175.7.4236. [DOI] [PubMed] [Google Scholar]
- 465.Alonso D., Dorronsoro I., Rubio L., Muñoz P., García-Palomero E., Del Monte M., Bidon-Chanal A., Orozco M., Luque F.J., Castro A., Medina M., Martínez A. Donepezil–tacrine hybrid related derivatives as new dual binding site inhibitors of AchE. Bioorg. Med. Chem. 2005;13:6588–6597. doi: 10.1016/j.bmc.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 466.Soyer Z., Parlar S., Alptuzun V. Synthesis and acetylcholinesterase (AChE) inhibitory activity of some N-substituted-5-chloro-2(3H)-benzoxazolone derivatives. Marmara Pharm. J. 2013;17:15–20. [Google Scholar]
- 467.Qin J., Lan W., Liu Z., Huang J., Tang H., Wang H. Synthesis and biological evaluation of 1,3-dihydroxyxanthone Mannich base derivatives as anticholinesterase agents. Chem. Centr. J. 2013;7:78. doi: 10.1186/1752-153X-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Babu T.H., Rao V.R.S., Tiwari A.K., Babu K.S., Srinivas P.V., Ali A.Z., Rao J.M. Synthesis and biological evaluation of novel 8-aminomethylated oroxylin A analogues as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2008;18:1659–1662. doi: 10.1016/j.bmcl.2008.01.055. [DOI] [PubMed] [Google Scholar]
- 469.Han H.-Y., Yan J.-F., Jin X., Fei Y., Li F., Zhou G.-M., Yang D.-C. Synthesis and evaluation of 5-(6-methoxynaphthalen-2-yl)-1-aryl-1-(4-(trifluoromethyl)phenylamino)pentan-3-ones as potential antidiabetic agents. J. Chil. Chem. Soc. 2011;56:930–934. [Google Scholar]
- 470.Wang H., Yan J., Song X., Fan L., Xu J., Zhou G., Jiang L., Yang D. Synthesis and antidiabetic performance of β-amino ketone containing nabumetone moiety. Bioorg. Med. Chem. 2012;20:2119–2130. doi: 10.1016/j.bmc.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 471.Tang G.X., Yan J.F., Fan L., Xu J., Song X.L., Jiang L., Luo L.F., Yang D.C. Synthesis of novel β-amino ketones containing a p-aminobenzoic acid moiety and evaluation of their antidiabetic activities. Sci. China: Chem. 2013;56:490–504. [Google Scholar]
- 472.Evans G.B., Furneaux R.H., Lewandowicz A., Schramm V.L., Tyler P.C. Exploring structure–activity relationships of transition state analogues of human purine nucleoside phosphorylase. J. Med. Chem. 2003;46:3412–3423. doi: 10.1021/jm030145r. [DOI] [PubMed] [Google Scholar]
- 473.Evans G.B., Furneaux R.H., Lewandowicz A., Schramm V.L., Tyler P.C. Synthesis of second-generation transition state analogues of human purine nucleoside phosphorylase. J. Med. Chem. 2003;46:5271–5276. doi: 10.1021/jm030305z. [DOI] [PubMed] [Google Scholar]
- 474.Evans G.B., Furneaux R.H., Schramm V.L., Tyler P.C. Synthesis of a transition state analogue inhibitor of purine nucleoside phosphorylase via the Mannich reaction. Org. Lett. 2003;5:3639–3640. doi: 10.1021/ol035293q. [DOI] [PubMed] [Google Scholar]
- 475.Clinch K., Evans G.B., Fröhlich R.F.G., Furneaux R.H., Kelly P.M., Legentil L., Murkin A.S., Li L., Schramm V.L., Tyler P.C., Woolhouse A.D. Third-generation immucillins: syntheses and bioactivities of acyclic immucillin inhibitors of human purine nucleoside phosphorylase. J. Med. Chem. 2009;52:1126–1143. doi: 10.1021/jm801421q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Clinch K., Evans G.B., Fleet G.W.J., Furneaux R.H., Johnson S.W., Lenz D.H., Mee S.P.H., Rands P.R., Schramm V.L., Taylor Ringia E.A., Tyler P.C. Syntheses and bio-activities of the L-enantiomers of two potent transition state analogue inhibitors of purine nucleoside phosphorylases. Org. Biomol. Chem. 2006;4:1131–1139. doi: 10.1039/b517883e. [DOI] [PubMed] [Google Scholar]
- 477.Murkin A.S., Clinch K., Mason J.M., Tyler P.C., Schramm V.L. Immucillins in custom catalytic-site cavities. Bioorg. Med. Chem. Lett. 2008;18:5900–5903. doi: 10.1016/j.bmcl.2008.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Mason J.M., Murkin A.S., Li L., Schramm V.L., Gainsford G.J., Skelton B.W. A β-fluoroamine inhibitor of purine nucleoside phosphorylase. J. Med. Chem. 2008;51:5880–5884. doi: 10.1021/jm800792b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Evans G.B., Furneaux R.H., Greatrex B., Murkin A.S., Schramm V.L., Tyler P.C. Azetidine based transition state analogue inhibitors of N-ribosyl hydrolases and phosphorylases. J. Med. Chem. 2008;51:948–956. doi: 10.1021/jm701265n. [DOI] [PubMed] [Google Scholar]
- 480.Longshaw A.I., Adanitsch F., Gutierrez J.A., Evans G.B., Tyler P.C., Schramm V.L. Design and synthesis of potent “sulfur-free” transition state analogue inhibitors of 5′-methylthioadenosine nucleosidase and 5′-methylthioadenosine phosphorylase. J. Med. Chem. 2010;53:6730–6746. doi: 10.1021/jm100898v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Goeminne A., Berg M., McNaughton M., Bal G., Surpateanu G., Van der Veken P., De Prol S., Versées W., Steyaert J., Haemers A., Augustyns K. N-arylmethyl substituted iminoribitol derivatives as inhibitors of a purine specific nucleoside hydrolase. Bioorg. Med. Chem. 2008;16:6752–6763. doi: 10.1016/j.bmc.2008.05.056. [DOI] [PubMed] [Google Scholar]
- 482.Ozaki S., Ebisui E., Hamada K., Goto J.-I., Suzuki A.Z., Terauchi A., Mikoshiba K. Potent transglutaminase inhibitors, aryl β-aminoethyl ketones. Bioorg. Med. Chem. Lett. 2010;20:1141–1144. doi: 10.1016/j.bmcl.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 483.Ozaki S., Ebisui E., Hamada K., Suzuki A.Z., Terauchi A., Mikoshiba K. Potent transglutaminase inhibitors, dithio β-aminoethyl ketones. Bioorg. Med. Chem. Lett. 2011;21:377–379. doi: 10.1016/j.bmcl.2010.10.136. [DOI] [PubMed] [Google Scholar]
- 484.Büyükkidan N., Büyükkidan B., Bülbül M., Özer S., Gonca Yalçin H. Synthesis and characterization of phenolic Mannich bases and effects of these compounds on human carbonic anhydrase isozymes I and II. J. Enzyme Inhib. Med. Chem. 2013;28:337–342. doi: 10.3109/14756366.2012.693919. [DOI] [PubMed] [Google Scholar]
- 485.Zhang S., Ma J., Bao Y., Yang P., Zou L., Li K., Sun X. Nitrogen-containing flavonoid analogues as CDK1/cyclin B inhibitors: synthesis, SAR analysis, and biological activity. Bioorg. Med. Chem. 2008;16:7127–7133. doi: 10.1016/j.bmc.2008.06.055. [DOI] [PubMed] [Google Scholar]
- 486.Gozalbes R., Mosulén S., Ortí L., Rodríguez-Díaz J., Carbajo R.J., Melnyk P., Pineda-Lucena A. Hit identification of novel heparanase inhibitors by structure and ligand-based approaches. Bioorg. Med. Chem. 2013;21:1944–1951. doi: 10.1016/j.bmc.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 487.Wangtrakuldee P., Byrd M.S., Campos C.G., Henderson M.W., Zhang Z., Clare M., Masoudi A., Myler P.J., Horn J.R., Cotter P.A., Hagen T.J. Discovery of inhibitors of Burkholderia pseudomallei methionine aminopeptidase with antibacterial activity. ACS Med. Chem. Lett. 2013;4:699–703. doi: 10.1021/ml400034m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Danilenko V.N., Simonov A.Y., Lakatosh S.A., Kubbutat M.H.G., Totzke F., Schächtele C., Elizarov S.M., Bekker O.B., Printsevskaya S.S., Luzikov Y.N., Reznikova M.I., Shtil A.A., Preobrazhenskaya M.N. Search for inhibitors of bacterial and human protein kinases among derivatives of diazepines[1,4] annelated with maleimide and indole cycles. J. Med. Chem. 2008;51:7731–7736. doi: 10.1021/jm800758s. [DOI] [PubMed] [Google Scholar]
- 489.Cravotto G., Balliano G., Tagliapietra S., Palmisano G., Penoni A. Umbelliferone aminoalkyl derivatives, a new class of squalene-hopene cyclase inhibitors. Eur. J. Med. Chem. 2004;39:917–924. doi: 10.1016/j.ejmech.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 490.Bahgat M.M., Maghraby A.S., Heiba M.E., Ruppel A., Fathalla O.A.M. Synthesis of new 4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine derivatives with an incorporated thiazolidinone moiety and testing their possible serine protease and cercarial elastase inhibitory effects with a possible prospective to block penetration of Schistosoma mansoni cercariae into the mice skin. Arch. Pharm. Res. 2005;28:1002–1012. doi: 10.1007/BF02977392. [DOI] [PubMed] [Google Scholar]
- 491.Lam K.W., Syahida A., Ul-Haq Z., Abdul Rahman M.B., Lajis N.H. Synthesis and biological activity of oxadiazole and triazolothiadiazole derivatives as tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 2010;20:3755–3759. doi: 10.1016/j.bmcl.2010.04.067. [DOI] [PubMed] [Google Scholar]
- 492.Czopek A., Kolaczkowski M., Bucki A., Byrtus H., Pawlowski M., Siwek A., Bojarski A.J., Bednarski M., Wróbel D., Wesołowska A. Novel Mannich bases, 5-arylimidazolidine-2,4-dione derivatives with dual 5-HT1A receptor and serotonin transporter affinity. Arch. Pharm. (Weinheim) 2013;346:98–109. doi: 10.1002/ardp.201200378. [DOI] [PubMed] [Google Scholar]
- 493.Mahesh R., Perumal R.V., Pandi P.V. Pharmacophore based synthesis of 3-chloroquinoxaline-2-carboxamides as serotonin3 (5-HT3) receptor antagonist. Biol. Pharm. Bull. 2004;27:1403–1405. doi: 10.1248/bpb.27.1403. [DOI] [PubMed] [Google Scholar]
- 494.Perumal R.V., Mahesh R. Synthesis and biological evaluation of a novel structural type of serotonin 5-HT3 receptor antagonists. Bioorg. Med. Chem. Lett. 2006;16:2769–2772. doi: 10.1016/j.bmcl.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 495.Kulagowski J.J., Broughton H.B., Curtis N.R., Mawer I.M., Ridgill M.P., Baker R., Emms F., Freedman S.B., Marwood R., Patel S., Patel S., Ragan C.I., Lesson P.D. 3-[[4-(4-Chlorophenyl)piperazin-1-yl]methyl]-1H-pyrrolo[2,3-b]pyridine: an antagonist with high affinity and selectivity for the human dopamine D4 receptor. J. Med. Chem. 1996;39:1941–1942. doi: 10.1021/jm9600712. [DOI] [PubMed] [Google Scholar]
- 496.Löber S., Hübner H., Gmeiner P. Azaindole derivatives with high affinity for the dopamine D4 receptor: synthesis, ligand binding studies and comparison of molecular electrostatic potential maps. Bioorg. Med. Chem. Lett. 1999;9:97–102. doi: 10.1016/s0960-894x(98)00692-1. [DOI] [PubMed] [Google Scholar]
- 497.Abadi A.H. Phenylpiperazinylmethylheterocycle derivatives: synthesis and dopamine receptor binding profiles. Arch. Pharm. (Weinheim) 2004;337:383–390. doi: 10.1002/ardp.200300838. [DOI] [PubMed] [Google Scholar]
- 498.Linz S., Troschütz R. Synthesis of 5-[(4-phenylpiperazin-1-yl)methyl]pyrrolo[2,3-d]pyrimidine derivatives as potential dopamine D4 receptor ligands. J. Heterocycl. Chem. 2007;44:349–354. [Google Scholar]
- 499.Linz S., Müller J., Hübner H., Gmeiner P., Troschütz R. Design, synthesis and dopamine D4 receptor binding activities of new N-heteroaromatic 5/6-ring Mannich bases. Bioorg. Med. Chem. 2009;17:4448–4458. doi: 10.1016/j.bmc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 500.Abdelfattah M.A.O., Lehmann J., Abadi A.H. Discovery of highly potent and selective D4 ligands by interactive SAR study. Bioorg. Med. Chem. Lett. 2013;23:5077–5081. doi: 10.1016/j.bmcl.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 501.Sikazwe D.M.N., Nkansah N.T., Altundas R., Zhu X.Y., Roth B.L., Setola V., Ablordeppey S.Y. Synthesis and evaluation of ligands for D2-like receptors: the role of common pharmacophoric groups. Bioorg. Med. Chem. 2009;17:1716–1723. doi: 10.1016/j.bmc.2008.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Arnold L.A., Estebanez-Perpina E., Togashi M., Jouravel N., Shelat A., McReynolds A.C., Mar E., Nguyen P., Baxter J.D., Fletterick R.J., Webb P., Guy R.K. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J. Biol. Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 503.Arnold L.A., Kosinski A., Estebanez-Perpina E.R.J.F., Guy R.K. Inhibitors of the interaction of a thyroid hormone receptor and coactivators: preliminary structure–activity relationships. J. Med. Chem. 2007;50:5269–5280. doi: 10.1021/jm070556y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Estebanez-Perpina E., Arnold L.A., Jouravel N., Togashi M., Blethrow J., Mar E., Nguyen P., Phillips K.J., Baxter J.D., Webb P., Guy R.K., Fletterick R.J. Structural insight into the mode of action of a direct inhibitor of coregulator binding to the thyroid hormone receptor. Mol. Endocrinol. 2007;21:2919–2928. doi: 10.1210/me.2007-0174. [DOI] [PubMed] [Google Scholar]
- 505.Hwang J.Y., Arnold L.A., Zhu F., Kosinski A., Mangano T.J., Setola V., Roth B.L., Guy R.K. Improvement of pharmacological properties of irreversible thyroid receptor coactivator binding inhibitors. J. Med. Chem. 2009;52:3892–3901. doi: 10.1021/jm9002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Zhou C., Wu G., Feng Y., Li Q., Su H., Mais D.E., Zhu Y., Li N., Deng Y., Yang D., Wang M.-W. Discovery and biological characterization of a novel series of androgen receptor modulators. Br. J. Pharmacol. 2008;154:440–450. doi: 10.1038/bjp.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Du Y., Li Q., Xiong B., Hui X., Wang X., Feng Y., Meng T., Hu D., Zhang D., Wang M., Shen J. Aromatic β-amino-ketone derivatives as novel selective non-steroidal progesterone receptor antagonists. Bioorg. Med. Chem. 2010;18:4255–4268. doi: 10.1016/j.bmc.2010.04.092. [DOI] [PubMed] [Google Scholar]
- 508.Zlotos D.P., Tränkle C., Abdelrahman A., Gündisch D., Radacki K., Braunschweig H., Mohr K. 6H,13H-pyrazino[1,2-a;4,5-a′]diindole analogs: probing the pharmacophore for allosteric ligands of muscarinic M2 receptors. Bioorg. Med. Chem. Lett. 2006;16:1481–1485. doi: 10.1016/j.bmcl.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 509.Muhi-eldeen Z., Al-Kaissi E., Ghantous H. Synthesis and biological evaluation of N1,N2-bis-[4-(t-amino)-2-butynyl]phthalamides as oxotremorine and acetylcholine antagonists. Med. Chem. Res. 2013;22:1597–1603. [Google Scholar]
- 510.Costantino L., Gandolfi F., Sorbi C., Franchini S., Prezzavento O., Vittorio F., Ronsisvalle G., Leonardi A., Poggesi E., Brasili L. Synthesis and structure-activity relationships of 1-aralkyl-4-benzylpiperidine and 1-aralkyl-4-benzylpiperazine derivatives as potent σ ligands. J. Med. Chem. 2005;48:266–273. doi: 10.1021/jm049433t. [DOI] [PubMed] [Google Scholar]
- 511.Santillan A., Jr., McClure K.J., Allison B.D., Lord B., Boggs J.D., Morton K.L., Everson A.M., Nepomuceno D., Letavic M.A., Lee-Dutra A., Lovenberg T.W., Carruthers N.I., Grice C.A. Indole- and benzothiophene-based histamine H3 antagonists. Bioorg. Med. Chem. Lett. 2010;20:6226–6230. doi: 10.1016/j.bmcl.2010.08.103. [DOI] [PubMed] [Google Scholar]
- 512.Romeo G., Materia L., Pittalà V., Modica M., Salerno L., Siracusa M., Russo F., Minneman K.P. New 1,2,3,9-tetrahydro-4H-carbazol-4-one derivatives: analogues of HEAT as ligands for the α1-adrenergic receptor subtypes. Bioorg. Med. Chem. 2006;14:5211–5219. doi: 10.1016/j.bmc.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 513.Pégurier C., Collart P., Danhaive P., Defays S., Gillard M., Gilson F., Kogej T., Pasau P., Van Houtvin N., Van Thuyne M., van Keulen B. Pyrazolone methylamino piperidine derivatives as novel CCR3 antagonists. Bioorg. Med. Chem. Lett. 2007;17:4228–4231. doi: 10.1016/j.bmcl.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 514.Borioni A., Mustazza C., Sestili I., Sbraccia M., Turchetto L., Del Giudice M.R. Synthesis of new 4-heteroaryl-2-phenylquinolines and their pharmacological activity as NK-2/NK-3 receptor ligands. Arch. Pharm. (Weinheim) 2007;340:17–25. doi: 10.1002/ardp.200600113. [DOI] [PubMed] [Google Scholar]