

Abstract
A number of 5-arylisatin derivatives were synthesized in 5–6 steps from readily available starting materials. Their structures were confirmed by 1H NMR and 13C NMR as well as LC/MS. The cytotoxicity of these novel isatins against human leukemia K562 cells were evaluated by MTT assay in vitro. SAR studies indicated that the N-substituted benzyl and C-5 substituted phenyl groups greatly enhance their cytotoxic activity, whereas an intact carbonyl functionality on C-3 present in the parent ring is required to maintain such a potency. Particularly, N-(p-methoxybenzyl)-5-(p-methoxyphenyl)isatin (compound 2m) showed the highest antitumor activity against K562 cell lines (IC50 = 0.03 μM). Moreover, treatment with compound 2m significantly inhibited liver cancer HepG2 cells proliferation and migration, which could also reduce the human umbilical vein endothelial cells (HUVEC) tube formation. In conclusion, compound 2m exhibited very good cancer cells proliferation inhibition by angiogenesis responses in vitro, and 2m might be a promising angiogenesis inhibitor for cancer treatment.
Keywords: Synthesis, 5-phenylisatin, Proliferation, Migration, Angiogenesis
Graphical abstract
A series of 5-arylisatin derivatives were synthesized. The cytotoxicity evaluation results revealed that the most potent compound 2m inhibit tumor cells proliferation by decreasing migration and angiogenesis.

Highlights
-
•
The antitumor SAR studies of novel 5-phenylisatin derivatives were performed.
-
•
The methoxyl groups of C-5 and N-substitution may enhance their cytotoxicy.
-
•
Compound 2m displayed the most potent cytotoxic activity (IC50 = 0.03 μM) against K562 cell lines.
-
•
2m inhibited the proliferation of tumor cells by decreasing migration and angiogenesis.
1. Introduction
Isatins are a class of structurally versatile molecules and their core structure appears in many biologically active molecules and pharmaceutical agents. Isatin derivatives have been reported to possess a broad spectrum of activities such as anticancer [1,2], antidepressant [3], anticonvulsant [4], antifungal [5], anti-HIV [6]and anti-inflammatory [7], etc. In the last decade, N-substituted isatins have attracted increasing attention from both industry and academia. Vine et al. has reported that some N-benzyl isatin derivatives were more cytotoxic than their N-H counterparts toward some lymphoma cells as well as a series of human cancer cell lines, including human leukemic (K562, U937 and Jurkat), liver (HepG2), breast (MDA-MB-231 and MCF-7), prostate (PC-3) and colorectal (HCT-116) cell lines [8]. Chen's and Chiyanzu's groups reported that N-substituted isatin derivatives exhibited inhibition activities against SARS CoV 3CLpro and parasitic cysteine proteases [9,10]. Later on, Limpachayaporn and Liu found that 5-sulfonylisatin analogues could act as caspase-3,7 inhibitors and SARS-CoV 3C-like protease inhibitors [11,12]. In addition, Chinnasamy's group found that some N-1 and C-5 disubstituted molecules such as 1-(substituted benzylidene)-3-(1-(morpholino/piperidinomethyl)-2,3-dioxoindolin-5-yl)urea derivatives exhibited antiepileptic activity and neurotoxicity [13]. Previously, we reported some 5-(2-carboxyethenyl)isatin derivatives as anticancer agents, and found that the combination of a 5-[trans-2-(methoxycarbonyl)ethen-1-yl] group and a 1-(4-methoxybenzyl) group (compound 5–61, Fig. 1 ) in isatin significantly enhanced it's cytotoxic activity [14]. However, further research on this molecule indicated that the presence of a metabolically unstable acrylic ester moiety on the C-5 position is the key to its in vivo efficacy. In order to further improve the antitumor property of this type of 1,5-disubstituted isatin molecules, we have synthesized a serious of isatin derivatives and studied their cytotoxicity. The results showed that although the substituent of 5-methacrylate could increase the antitumor activity of the target compound, it is not stable in metabolic processes (unpublished data). Therefore, we report a series of new compounds in this paper, which can maintain good anti-tumor efficacy even without the use of 5-methacrylate substituents. Among them, compound 2m was shown as the most active compound against human leukemia K562 cells. Moreover, the molecular mechanism of the cytotoxic activity of compound 2m was explored on anti-migration and anti-angiogenesis.
Fig. 1.
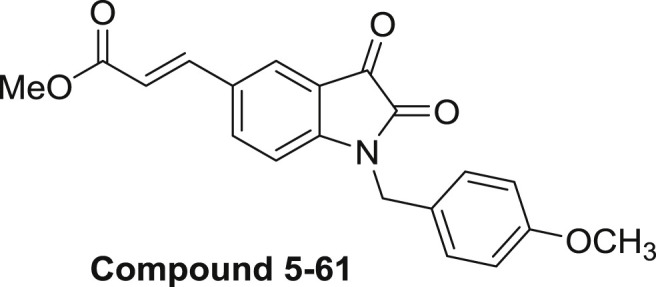
The structure of 1,5-disubstituted isatin derivative (compound 5–61).
2. Chemistry
Firstly, the target compounds were synthesized according to the procedures outlined in Scheme 1 . Monosubstituted isatins 1a-1g were obtained in two steps with a improved yield of 55–80% [15]. Moreover, 5-Bromoisatin (1d) was coupled with six different boronic acids by microwave-assistant Suzuki coupling reaction to afford compounds 1h-1k in 70–80% isolated yield [16]. Secondly, N-alkylation of 1i led to the synthesis of compounds 2a-2v in good to excellent yield (75–90%). Thirdly, compound 3a-3c were prepared from isatin derivatives by suzuki-coupling reaction (Scheme 2 ). Finally, compound 4a-4s were prepared from 5-bromo-1-(4-methoxybenzyl)indoline-2,3-dione by Suzuki-coupling (Scheme 3 ).
Scheme 1.

Synthesis of isatin derivatives (1a-1k, 2a-2v).
Scheme 2.

Synthesis of 3a-3c.
Scheme 3.

Synthesis of 4a-4r.
Compounds 5a-5k (Scheme 4 ) were prepared from 2m in good yield in one or two steps. Compound 2m was reduced with NaBH4 in methanol under room temperature to afford compound 5a. Compound 5b was prepared by treatment of the compound 2m with methylmagnesium bromide in tetrahydrofuran at −78 °C. Compound 2m was reduced with hydrazine monohydrate in ethanol under reflux to afford compound 5c. Compound 5d and 5e were prepared by treatment of the compound 2m with (triphenylphosphoranylidene)acetate in tetrahydrofuran at room temperature. Compound 5f was synthesized by treatment of the compound 2m with ethylene glycol in toluene at 110 °C. Compound 5g was prepared by treatment of the compound 2m with diethylaminosulfur trifluoride in DCM at reflux. Compound 5h was synthesized by treatment of the compound 2m with hydroxylamine hydrochloride in ethanol at reflux. The compounds 5i-5k were prepared by the treatment of 5c with benzaldehyde or substituted benzaldehyde and pyridine in ethanol.
Scheme 4.

C-3 modification of isatin derivatives (5a-5k).
The data obtained from 1H NMR, 13C NMR and Mass Spectrometry confirmed the proposed structures (Spectral data results are provided in supporting information).
3. Results and discussion
When contemplating a new class of 1,5-substituted isatin structure that could avoid the metabolically liabile acrylic acid ester group found in compound 5–61 while maintaining its biological potency, we envisaged a general structure compound 5–61 (Fig. 1) [14]. It can bear a various of aromatic groups at the C-5 position, that carry some structural similarity in terms of their size and conjugation property, whereas the in vivo Michael addition liability of the 5-acrylic acid ester group could be eliminated.
As illustrated in Fig. 2 , with the isatin as the core structure of our drug design, this strategy would allow us to start with the modification of the substitution pattern of the benzene ring, especially at C-5, followed by N-derivatization and then C-3 variation.
Fig. 2.

Structure modifications of isatin derivatives for SAR study.
The in vitro antitumor activities of the 5-substituted isatins 1a-1k against two human tumor cells K562 and HepG2 were first evaluated. As shown in Table 1 , the cytotoxic activities of the 5-phenyl substituted compounds are far better than the parent nucleus. Among them, compound 1i exhibited the highest cytotoxic activity against HepG2 cell lines (IC50 = 0.96 μM).
Table 1.
In vitro antitumor activities of compounds 1a-1k[14].
| Compd. | R; R1 | IC50(μM) |
|
|---|---|---|---|
| K562 | HepG2 | ||
| CPT | 0.07 ± 0.01 | 0.37 ± 0.06 | |
| 1a | H | >10 | >10 |
| 1b | F | >10 | >10 |
| 1c | Cl | >10 | >10 |
| 1d | Br | >10 | >10 |
| 1e | CH3 | >10 | >10 |
| 1f | OCH3 | >10 | >10 |
| 1g | (CH2)3CH3 | >10 | >10 |
| 1h | CH3 | >10 | 6.00 ± 2.16 |
| 1i | OCH3 | 6.92 ± 0.28 | 0.96 ± 0.32 |
| 1j | H | >10 | 5.33 ± 0.62 |
| 1k | CF3 | >10 | 5.43 ± 1.45 |

After compound 1i was identified as the potential lead, our SAR study was then focused on the N-derivatization based on the hypothesis that the combination of C-5 substitution with N-alkylation may further enhance their cytotoxic potency. A series of 5-(4-methoxyphenyl) N-substituted derivatives (Scheme 1) were screened for their in vitro cytotoxic activity.
As can be seen from the data in Table 2 , introduction of simple aliphatic groups at N-1 (2a-2c) did not significantly enhance their potency. Moreover, a p-methoxyphenyl group at N-1 significantly diminished the potency. However, the introduction of benzyl groups into the N-1 position (2d-2v) resulted in much better inhibitory activities, with 10–300 folds improvement compared to that of 1i, pointing to the tendency that the N-1 position needs a relatively sterically larger group with certain lipophilic property to increase the potency. Further examination of the structure difference and the cytotoxic data of the compounds 2d and 2f-2v against K562 showed that the substituted benzyl group did increase the potency. The improvement in potency does not seem to be related to the electron density of the substituent groups (2f, 2r and 2h, 2l); however, the substituents' size (2k, 2p) and location (2n and 2m, 2o) plays a more significant role. Moreover, the excellent cytotoxic activity for the compounds containing N-1 para or ortho mono methoxy benzyl group (2m, 2o), dimethoxy benzyl group (2q) as well as a piperonyl group (2v) indicated that hydrogen bond receptor in this direction of the molecule could significantly enhance the potency (8–25 folds, compared to that of 2d).
Table 2.
In vitro antitumor activities of compounds 2a-2v.
| Compd. | R2 | IC50(μΜ) |
|
|---|---|---|---|
| K562 | HepG2 | ||
| CPT | 0.07 ± 0.01 | 0.37 ± 0.06 | |
| 1i | H | 6.92 ± 0.28 | 0.96 ± 0.32 |
| 2a | CH3 | 4.98 ± 0.92 | 6.67 ± 2.82 |
| 2b | CH2CH3 | 2.20 ± 0.36 | 4.47 ± 0.23 |
| 2c | CH2CH=CH2 | 0.39 ± 0.05 | 7.09 ± 1.04 |
| 2d | CH2C6H5 | 0.57 ± 0.11 | 1.36 ± 0.51 |
| 2e | C6H4-4-OCH3 | >10 | >10 |
| 2f | CH2-4-C6H4F | 0.56 ± 0.08 | 1.12 ± 0.33 |
| 2g | CH2-3,4-C6H3Cl2 | 0.43 ± 0.15 | 0.99 ± 0.02 |
| 2h | CH2-4-C6H4Cl | 0.35 ± 0.13 | 0.25 ± 0.09 |
| 2i | CH2-3-C6H4Cl | 0.20 ± 0.12 | 0.32 ± 0.12 |
| 2j | CH2-2-C6H4Cl | 0.38 ± 0.20 | 0.60 ± 0.24 |
| 2k | CH2-4-C6H4Br | 0.10 ± 0.15 | 0.70 ± 0.03 |
| 2l | CH2-4-C6H4CH3 | 0.27 ± 0.03 | 0.33 ± 0.03 |
| 2m | CH2-4-C6H4OCH3 | 0.03 ± 0.01 | 0.05 ± 0.03 |
| 2n | CH2-3-C6H4OCH3 | 0.68 ± 0.27 | 0.70 ± 0.18 |
| 2o | CH2-2-C6H4OCH3 | 0.04 ± 0.01 | 0.13 ± 0.02 |
| 2p | CH2-3,4-C6H3(CH3)2 | 0.14 ± 0.03 | 0.41 ± 0.03 |
| 2q | CH2-3,4C6H3(OCH3)2 | 0.06 ± 0.01 | 1.35 ± 1.86 |
| 2r | CH2-4-C6H4CN | 0.38 ± 0.07 | 1.00 ± 0.29 |
| 2s | CH2-4-C6H4CF3 | 0.27 ± 0.03 | 0.38 ± 0.02 |
| 2t | CH2CH2OC6H5 | 0.40 ± 0.06 | 0.70 ± 0.25 |
| 2u | CH2-2-naphthyl | 0.40 ± 0.04 | 0.38 ± 0.05 |
| 2v | CH2-2-piperonyl | 0.03 ± 0.01 | 0.44 ± 0.19 |

With the identification of the p-methoxybenzyl group as an optimal substituent at N-1, our attention then turned back to the p-methoxyphenyl substitution on the benzene ring. Specifically, we were interested in finding out how the location would influence the cytotoxic potency. For this purpose, a series of isatins bearing p-methoxyphenyl group(s) at various position on the benzene ring were synthesized and their cytotoxic activities were examined. The compounds 3a-3c were synthesized following the procedure similar to that of 2m (Scheme 2) and their potency were listed in Table 3 . The 4-, 6- and 7-monosubstituted compounds 3a-3c exhibited much weaker potency than that of 2m (>220 folds), which may suggest that the N-1, C-5 disubstituted pattern in 2m possess a much favorable 2-dimensional orientation toward the biological targets.
Table 3.
In vitro antitumor activities of compounds 3a-3c.
| Compd. | Position | IC50(μM) |
|
|---|---|---|---|
| K562 | HepG2 | ||
| CPT | 0.07 ± 0.01 | 0.37 ± 0.06 | |
| 2m | 5 | 0.03 ± 0.01 | 0.05 ± 0.03 |
| 3a | 4 | >10 | >10 |
| 3b | 6 | 7.46 ± 1.69 | >10 |
| 3c | 7 | 4.43 ± 0.75 | 8.56 ± 0.06 |

The SAR analysis of the compounds in Table 3 revealed that the N-(p-methoxybenzyl) and 5-(p-substituted phenyl) disubstituted isatins are the most attractive candidates, with compound 2m exhibiting the highest potency. To further investigate the activity of the structural analogues of 2m, a number of 5-aryl N-(p-methoxyphenyl)satins were prepared (Scheme 3) and their inhibition against two human tumor cells were tested (Table 4 ).
Table 4.
In vitro antitumor activities of compounds 4a-4r.
| Compd. | R3 | IC50(μΜ) |
|
|---|---|---|---|
| K562 | HepG2 | ||
| CPT | 0.07 ± 0.01 | 0.37 ± 0.06 | |
| 2m | C6H4-4-OCH3 | 0.03 ± 0.01 | 0.05 ± 0.03 |
| 4a | C6H4-3-OCH3 | 0.10 ± 0.03 | 1.89 ± 1.48 |
| 4b | C6H4-2-OCH3 | 0.37 ± 0.09 | >10 |
| 4c | C6H4-3,5-(OCH3)2 | 0.51 ± 0.21 | 1.20 ± 0.58 |
| 4d | C6H4-3,4-(OCH3)2 | 0.26 ± 0.11 | >10 |
| 4e | C6H4-4-CC3H9 | 2.25 ± 0.73 | 4.58 ± 1.09 |
| 4f | C6H4-4-F | 0.24 ± 0.11 | 3.15 ± 2.31 |
| 4g | C6H4-4-Cl | 0.23 ± 0.05 | 0.38 ± 0.21 |
| 4h | C6H3-3-Cl-4-F | 0.38 ± 0.08 | >10 |
| 4i | C6H4-4-CH3a | 0.40 ± 0.12 | >10 |
| 4j | C6H4-4-(O-Pr) | 0.31 ± 0.08 | >10 |
| 4k | C6H5 | 0.15 ± 0.02 | 1.08 ± 0.60 |
| 4l | C6H4-4-CNa | 0.26 ± 0.04 | 0.89 ± 0.11 |
| 4m | C6H4-4-CF3a | 0.35 ± 0.08 | 0.82 ± 0.20 |
| 4n | C6H4-4-OCF3a | 0.06 ± 0.01 | 0.41 ± 0.32 |
| 4o | C6H4-4-OHa | 0.06 ± 0.02 | >10 |
| 4p | 2-naphthyl | 0.34 ± 0.10 | >10 |
| 4q | 4-pyridyl | 0.04 ± 0.02 | >10 |
| 4r | 2-thiophenyl | 0.08 ± 0.04 | 3.67 ± 1.40 |

As shown in Table 4, among all the 18 compounds examined, the cytotoxic activities of the phenyl analogue 4k, region-isomers 4a, 4b as well as the 3,5-dimethoxy-phenyl substituted compounds 4c exihibted a 5–25 folds decrease in potency compared to 2m, indicating that the para methoxy group is necessary and its linear orientation maybe a critical factor in maintaining its high potency.
The halogen-containing compounds (4f, 4g, 4h), compounds (4i, 4j, 4l, 4m) possess either electron-donating or electron-withdrawing groups, all exhibit very similar inhibition, indicating that variation of the electron density at this position did not significantly change their potency.
The excellent potency 5-(4-pyridyl) substituted compound 4q and 5-(2-thiophenyl) substituted compound 4r suggest that groups possessing a Lewis basic site could potentially improve the compound's activity. The hydroxyl group containing compound 4o did not exhibit any improved activity in comparison to compounds 2m, 4q and 4r.
There was no significant potency change when the 5-phenyl substitution (4k) was replaced by 5-(4-methyphenyl) group (4i) or 5-(2-naphthyl) group (4p). However, the cytotoxic activity of the 5-(4-n-butyl)phenyl compound 4e dropped 15 folds from that of the non-substituted counterpart (4k), which indicated the bulky aliphatic chain is detrimental.
Derivatization of the C-3 ketone group of 2m led to compounds 5a-5l (Scheme 4) and their cytotoxic activities were evaluated to establish the importance of this functionality. The testing result showed that all these derivatives are inferior candidates as inhibitors of K562 and HepG2 (Table 5 ), suggesting that the carbonyl functionality at C-3 is essential in order to maintain the observed high antitumor activity.
Table 5.
Antitumor activity of the C-3 derivatized compounds 5a-5k.
| Compd. | C-3 | IC50(μM) |
|
|---|---|---|---|
| K562 | HepG2 | ||
| CPT | 0.07 ± 0.01 | 0.37 ± 0.06 | |
| 2m | = O | 0.03 ± 0.01 | 0.05 ± 0.03 |
| 5a | -OH·H | 0.14 ± 0.09 | 0.70 ± 0.03 |
| 5b | -OH·CH3 | >10 | >10 |
| 5c | -H·H | >10 | >10 |
| 5d | = COOCH2CH3 | 1.24 ± 0.54 | 3.39 ± 0.27 |
| 5e | = COOCH3 | 1.90 ± 0.14 | 4.73 ± 1.24 |
| 5f | -CH2O(CH2)2OCH2- | >10 | >10 |
| 5g | -F·F | >10 | >10 |
| 5h | = NOH | >10 | >10 |
| 5i | = C6H5 | >10 | >10 |
| 5j | = C6H4-4-CF3 | 1.90 ± 0.26 | >10 |
| 5k | = C6H4-4-CH(CH3)2 | >10 | >10 |
Based on these SAR studies, compound 2m exhibited significant cancer inhibitory activity in vitro against the tested cancer cell lines. The IC50 of compounds 2m against K562 cells and HepG2 cells were 30 nM and 50 nM, respectively (Fig. 3 A). Meanwhile, a normal human cell line (umbilical vein endothelial cells, HUVEC) has been tested for 2m, the IC50 was 920 nM. Then the molecular mechanism by which compound 2m exerts its cytotoxic activity on cells proliferation, migration and angiogenesis was further investigated.
Fig. 3.

Morphological changes in compound 2m-treated K562 cells. (A) Structure and in vitro cell proliferation inhibitory activity of compound 2m. (B) Morphological changes induced by compound 2m treatment (30 nM) were observed.
Firstly, the morphological changes in compound 2m-treated K562 cells were observed. K562 cells treated by compound 2m showed the cells morphology change of G2/M phase arrest (such as elongation of the cells) and typical apoptotic morphology (such as cell shrinkage and/or blebbing) (Fig. 3B). As shown in Fig. 3B, the elongated cells increased significantly as early as 6 h after compound 2m treatment at a concentration of 30 nM as compared to DMSO-treated cells. The apoptotic cells were clearly observed in K562 cells after the treatment with compound 2m for 24 h. These results indicated that compound 2m might inhibit the cell proliferation of K562 cells through the cell cycle arrest and apoptosis.
To evaluate the cell cycle distribution of K562 cells with or without compound 2m treatment, we measured the DNA content by flow cytometry. As shown in Fig. 4 , after treatment with compound 2m (30 and 300 nM) for 48 h, the percentage of K562 cells in G2/M phase increased. Only 0.02% of cells were in G2/M phase in basic DMSO cultures. However, the exposure to compound 2m caused increased cell fraction in G2/M phase in a time-dependent and dose-dependent manner. These results demonstrated that compound 2m has cell proliferation inhibitory effect and can induce the cell cycle arrest of K562 cells in G2/M phase.
Fig. 4.

Compound 2m induced the cell cycle arrest of K562 cells in G2/M phase. K562 cells were treated with 30 nM and 300 nM compound 2m. At the time points indicated, cells were labeled with PI and their DNA content was determined using FACS analysis.
The presence of apoptotic K562 cells following compound 2m treatment was further revealed by flow cytometric analysis of cells double-labeled with Annexin V-FITC and PI (Fig. 5 ). After 48 h of treatment with compound 2m (30 and 300 nM), we observed that compound 2m showed significant apoptosis against K562 cells. The apoptotic rates of compound 2m-treated K562 cells increased to 43.4% and 95.2% of the total cells whereas only 4.1% and 4.3% apoptosis respectively at 30 nM and 300 nM. Result also indicated that the apoptosis was increased obviously in a time-dependent and dose-dependent manner. We have concluded that compound 2m inhibits the proliferation of K562 cells by inducing apoptosis in a time-dependent and dose-dependent manner.
Fig. 5.

Compound 2m induced apoptosis in K562 cells. K562 cells were treated with 30 nM and 300 nM compound 2m for different periods of time. K562 cells were labeled with Annexin V-FITC and PI and apoptosis was determined using FACS analysis.
To study whether compound 2m could inhibit migration and repair ability of tumor cells, wound healing assay and transwell assay were performed by HepG2 cells. After HepG2 cells were wounded, they were treated with the indicated concentrations of compound 2m (10 nM, 30 nM, 100 nM) for 0, 6, 12, 24, 36 and 48 h respectively. As shown in Fig. 6 A, after treatment by DMSO for 48 h, the migration distance was 184.89 μm. The migration distances of HepG2 cells treated with compound 2m were 162.89 μm, 135.22 μm and 96.24 μm respectively at 10 nM, 30 nM and 100 nM. By calculation, we found that the higher concentration of compound 2m, the higher the inhibition ratio. The inhibition ratios of HepG2 cells treated with compound 2m were 11.9%, 26.9% and 47.9% respectively at 10 nM, 30 nM and 100 nM.
Fig. 6.

Effect of compound 2m on HepG2 cells migration at 48 h by using a wound healing assay (A) and transwell assay (B).
Transwell assay is an experimental technique for studying the tendency of cells which is applied in cell culture, cytotaxis, cell migration and cell invasion. As shown in Fig. 6B, basic DMSO did not affect migration of HepG2 cells. While, the compound 2m could inhibit the HepG2 cells migration and have a dose-dependent inhibitory effect.
Tube formation plays an important role in the development of tumors. Angiogenesis is also the path which tumors transition from benign to malignant. Inhibiting this process can significantly prevent the development and spread of tumor tissue. As shown in Fig. 7 , basic DMSO did not affect tube forming of HUVEC cells. However, the compound 2m could inhibit the angiogenesis of HUVEC cells and have a dose-dependent inhibitory effect.
Fig. 7.

Suppressive effects of different concentrations compound 2m on tube formation of HUVEC cells. HUVECs cultured on matrigel coated plates were treated with the indicated concentrations of compound 2m (10 nM, 30 nM, 100 nM) for 12 h. Morphological changes in HUVECs were observed by microscopy and representative images are shown.
4. Conclusions
In conclusion, a series of novel N-1 benzyl and C-5 phenyl substituted isatin derivatives were synthesized and tested for their in vitro antitumor activity against two strains of cancer cell lines (human leukemia K562 cells and liver cancer HepG2 cells). Among them, compounds 2m exhibited excellent cancer inhibitory activity in vitro against K562 cell lines (IC50 = 0.03 μM) and HepG2 cell lines (IC50 = 0.05 μM). SAR analysis showed that the carbonyl group of the isatin, N-benzyl and C-5 phenyl substituted pattern, the para-methoxyl group of the benzyl played a significant role in the antitumor activity.
Both morphological results and flow cytometry analysis showed that compound 2m induce apoptosis and cause cell cycle arrest in a time-dependent and dose-dependent manner. Furthermore, wound healing and transwell experiments showed that compound 2m could inhibit the migration and repair ability of HepG2 cells. In addition, the results of cell tube formation showed that the compound 2m could inhibit the angiogenesis of HUVEC cells and has a dose-dependent inhibitory effect.
The effects of indole derivative compound 2m on tumor cell growth, migration and angiogenesis were studied, which provide a strong foundation for further study on the antitumor activity and mechanism of indole compounds.
5. Experimental section
5.1. Chemistry
All reagents and solvents used in this paper were of reagent grade, were obtained from commercial suppliers and used without further purification. 1H NMR and 13C NMR spectra were recorded on a Bruker AM-400 NMR spectrometer (Billerica, Middlesex, MA, USA) in CDCl3 or DMSO‑d 6. The chemical shifts are reported in δ(ppm) relative to tetramethylsilane as internal standard. (1H NMR: TMS at 0.00 ppm, CDCl3 at 7.26 ppm, DMSO‑d 6 at 2.50 ppm; 13C NMR: CDCl3 at 77.23 ppm, DMSO‑d 6 at 39.51 ppm). Mass spectra were obtained on a Q-TOF mass spectrometer (Agilent, Santa Clara, CA, USA). TLC analyses were carried out on silica gel F254, and the spots were examined with UV light. All final products had a purity of ≥95%. The purity of the final products was determined by HPLC (LabTech) on a Diamonsil C18 column (4.6 mm × 250 mm, 5 μm) with methanol/H2O (90/10 v/v) at 0.5 mL/min flow rate and 254 nm detector wavelength.
5.2. Synthesis of compounds 1a-1k and 2a-2v
The general procedure for the preparation of isatin derivatives was as follows. A mixture of 0.05 mol substituted anilines, 0.15 mol Hydroxylammonium chloride, 0.35 mol Na2SO4, 2 mol/L Hydrochloric acid at 5 mL and Chloral hydrate at 0.06 mol were stirred in 250 mL H2O at 95 °C. The progress of the reaction was monitored by TLC (petroleum ether/ethyl acetate). After the reaction was completed, the reaction mixture was cooled to room temperature and the precipitate was filtered and dried. The crude product was used directly for the next step without further purification.
To a flask (100 mL) which contained concentrated sulfuric acid (20 mL) was added N-2-(hydroxyimino)acetamide derivatives (7.0 g) in portions at 50 °C with vigorous stirring. The reaction temperature was maintained at 50 °C-75 °C during the addition. After the addition was completed, the mixture was heated to 80 °C and stir at 80 °C for 30 min. The reaction mixture was cooled to room temperature and then poured onto ice (250 g). The solid which resulted was filtered out and dried over air to yield the crude which was purified by dissolving in dilute sodium hydroxide (5%, 100 mL) followed by acidified with 4N hydrochloric acid (20 mL). The solid which formed was filtered out and dried over air to provide the purified compounds 1a-1g.
To a microwave reactor vial (5 mL) which contained the solution of 5-bromoindoline-2,3-dione (1.0 g, 4.4 mmol) in 1,4-dioxane (5 mL) were added PdCl2(dppf) (161 mg, 0.22 mmol), CH3COOK (0.6 g, 6.2 mmol) and substituted benzeneboronic acid (0.45 g, 5.3 mmol) under the atmosphere of argon. The microwave reactor vial was caped and placed into the microwave cavity. The reaction mixture was irradiated at high level for 30 min at 120 °C. The reaction mixture was cooled to room temperature, poured onto 100 mL ice-water and then extracted with dichloromethane (3 × 100 mL). The combined organic layers were washed with water (2 × 100 mL), brine (100 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under vacuum to afford the crude which was purified by flash column chromatography (silica gel, petroleum ether/ethyl acetate 3:1) to yield the desired compound 1h-1k.
To a flask (25 mL) which contained the solution of compound 1i (0.2 g, 0.79 mmol) in dry N,N-dimethylformamide (2 mL). The reaction temperature was maintained at 0 °C followed by addition K2CO3 (0.33 g, 2.37 mmol). Stirring 5 min, was added alkyl or benzyl (1.0 mmol) and the mixture allowed warm to room temperature. The reaction mixture was stirred at room temperature for 4 h. The orange solution was poured water (25 mL) and extracted with dichloromethane (3 × 100 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 10:1–3:1) afforded the title compounds 2a-2v.
5.2.1. 5-(p-tolyl)indoline-2,3-dione (1h)
Yield 54%; 1H NMR (400 MHz, DMSO‑d 6) δ 2.36 (s, 3H), 6.98 (d, 1H, J = 8.0 Hz), 7.25 (d, 2H, J = 8.0 Hz), 7.53 (d, 2H, J = 8.0 Hz), 7.72 (s, 1H), 7.87 (d, 1H, J = 8.0 Hz), 11.11 (s, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 21.10, 113.10, 118.84, 122.62, 126.50, 126.50, 130.05, 130.05, 135.34, 136.31, 136.64, 137.27, 150.17, 160.00, 184.86. ESI-MS: 238.1 [M+H]+, 260.1[M+Na]+.
5.2.2. 5-(4-methoxyphenyl)indoline-2,3-dione (1i)
Yield 55%; 1H NMR (400 MHz, DMSO‑d 6) δ 3.79 (s, 3H), 6.97 (d, 1H, J = 8.0 Hz), 7.00 (d, 2H, J = 8.0 Hz), 7.59 (d, 2H, J = 8.0 Hz), 7.70 (s, 1H), 7.84 (d, 1H, J = 8.0 Hz), 11.09 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 55.65, 113.08, 114.88, 114.88, 118.84, 122.38, 127.83, 127.83, 131.57, 135.18, 136.40, 149.84, 159.38, 160.01, 184.92. ESI-MS: 254.0 [M+H]+, 276.0[M+Na]+.
5.2.3. 5-phenylindoline-2,3-dione (1j)
Yield 43%; 1H NMR (400 MHz, DMSO‑d 6) δ 7.01 (d, 1H, J = 8.0 Hz), 7.36–7.38 (m, 1H), 7.44–7.47 (m, 2H), 7.64–7.66 (m, 2H), 7.76 (s, 1H), 7.89–7.92 (m, 1H), 11.13 (s, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 112.65, 118.37, 122.40, 126.18, 126.18, 127.44, 128.97, 128.97, 134.88, 136.44, 138.68, 149.91, 159.51, 184.32. ESI-MS: 224.1 [M+H]+, 246.1[M+Na]+.
5.2.4. 5-(4-(trifluoromethyl)phenyl)indoline-2,3-dione (1k)
Yield 44%; 1H NMR (400 MHz, DMSO‑d 6) δ 7.04 (d, 1H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.4 Hz), 7.87 (s, 1H), 7.90 (d, 2H, J = 8.0 Hz), 7.99 (d, 1H, J = 8.4 Hz), 11.20 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 102.01, 111.59, 113.26, 119.03, 123.40, 126.26, 126.29, 127.48, 127.48, 133.59, 137.24, 143.19, 151.15, 160.01, 184.63. ESI-MS: 292.0 [M+H]+.
5.2.5. 5-(4-methoxyphenyl)-1-methylindoline-2,3-dione (2a)
Yield 44%; 1H NMR (400 MHz, CDCl3) δ 3.28 (s, 3H), 3.85 (s, 3H), 6.94 (d, 1H, J = 8.0 Hz), 6.98 (d, 2H, J = 8.4 Hz), 7.46 (d, 2H, J = 8.4 Hz), 7.78 (d, 1H, J = 8.0 Hz), 7.79 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 26.33, 55.40, 110.23, 114.50, 114.50, 117.87, 123.26, 127.64, 127.64, 131.43, 136.28, 137.09, 149.96, 158.38, 159.59, 183.55. ESI-MS: 268.1 [M+H]+, 290.1 [M+Na]+.
5.2.6. 1-ethyl-5-(4-methoxyphenyl)indoline-2,3-dione (2b)
Yield 35%; 1H NMR (400 MHz, CDCl3) δ 1.32–1.35 (m, 3H), 3.78–3.84 (m, 2H), 3.85 (s, 3H), 6.95 (d, 1H, J = 8.0 Hz), 6.97 (d, 2H, J = 8.8 Hz), 7.45 (d, 2H, J = 8.8 Hz), 7.74 (s, 1H), 7.76 (d, 1H, J = 8.0 Hz). 13C NMR (100 MHz, CDCl3) δ 12.60, 35.08, 55.40, 110.33, 114.50, 114.50, 118.06, 123.47, 127.63, 127.63, 131.47, 136.24, 136.90, 149.16, 158.00, 159.57, 183.91. ESI-MS: 282.1 [M+H]+, 304.1 [M+Na]+.
5.2.7. 1-allyl-5-(4-methoxyphenyl)indoline-2,3-dione (2c)
Yield 42%; 1H NMR (400 MHz, CDCl3) δ 3.85 (s, 3H), 4.40 (d, 2H, J = 5.6 Hz), 5.30–5.37 (m, 2H), 5.82–5.96 (m, 1H), 6.94 (d, 1H, J = 8.0 Hz), 6.96 (d, 2H, J = 8.8 Hz), 7.45 (d, 2H, J = 8.8 Hz), 7.73 (d, 1H, J = 8.0 Hz), 7.80 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 42.62, 55.40, 111.17, 114.52, 114.52, 118.02, 118.70, 123.39, 127.67, 127.67, 130.41, 131.49, 136.24, 137.10, 149.34, 158.05, 159.61, 183.44. ESI-MS: 294.1 [M+H]+, 316.1 [M+Na]+.
5.2.8. 1-benzyl-5-(4-methoxyphenyl)indoline-2,3-dione (2d)
Yield 40%; 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H), 4.95 (s, 2H), 6.81 (d, 1H, J = 8.4 Hz), 6.95 (d, 2H, J = 8.4 Hz), 7.29–7.36 (m, 5H), 7.40 (d, 2H, J = 8.4 Hz), 7.64 (d, 1H, J = 8.4 Hz), 7.78 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 44.16, 55.38, 111.30, 114.50, 114.50, 118.12, 123.38, 127.47, 127.47, 127.63, 127.63, 128.20, 129.08, 129.08, 131.42, 134.57, 136.23, 137.14, 149.22, 158.41, 159.60, 183.44. ESI-MS: 344.1 [M+H]+, 366.1 [M+Na]+.
5.2.9. 1,5-bis(4-methoxyphenyl)indoline-2,3-dione (2e)
Yield 45%; 1H NMR (400 MHz, CDCl3) δ 3.85 (s, 3H), 3.87 (s, 3H), 6.88 (d, 1H, J = 8.0 Hz), 6.98 (d, 2H, J = 8.0 Hz), 7.07 (d, 2H, J = 8.0 Hz), 7.35 (d, 2H, J = 8.0 Hz), 7.46 (d, 2H, J = 8.0 Hz), 7.70 (d, 1H, J = 8.0 Hz), 7.85 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 55.41, 55.62, 111.50, 114.53, 114.53, 115.25, 115.25, 117.90, 123.40, 125.50, 127.39, 127.39, 127.69, 127.69, 131.45, 136.27, 137.45, 150.59, 157.76, 159.65, 159.72, 183.34. ESI-MS: 360.1 [M+H]+, 382.1 [M+Na]+.
5.2.10. 1-(4-fluorobenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2f)
Yield 42%; 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H), 4.92 (s, 2H), 6.81 (d, 1H, J = 8.4 Hz), 6.95 (d, 2H, J = 8.8 Hz), 7.02–7.07 (m, 2H), 7.32–7.36 (m, 2H), 7.40 (d, 2H, J = 8.8 Hz), 7.66 (d, 1H, J = 8.4 Hz), 7.78 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.47, 55.39, 111.12, 114.51, 114.51, 115.96, 116.17, 118.13, 123.49, 127.64, 127.64, 129.26, 129.35, 130.38, 130.42, 131.33, 136.24, 137.29, 148.95, 158.36, 159.63, 183.28. ESI-MS: 362.2 [M+H]+, 384.2 [M+Na]+.
5.2.11. 1-(3,4-dichlorobenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2g)
Yield 34%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 4.90 (s, 2H), 6.78 (d, 1H, J = 8.0 Hz), 6.97 (d, 2H, J = 8.4 Hz), 7.20 (d, 1H, J = 8.4 Hz), 7.42 (d, 2H, J = 8.0 Hz), 7.44 (d, 2H, J = 8.0 Hz), 7.69 (d, 1H, J = 8.0 Hz), 7.81 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.11, 55.39, 110.89, 114.56, 114.56, 118.19, 123.68, 126.74, 127.67, 127.67, 129.40, 131.13, 131.27, 132.58, 133.35, 134.86, 136.32, 137.60, 148.57, 158.33, 159.72, 182.85. ESI-MS: 412.1 [M+H]+.
5.2.12. 1-(4-chlorobenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2h)
Yield 43%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 4.92 (s, 2H), 6.78 (d, 1H, J = 8.0 Hz), 6.96 (d, 2H, J = 8.4 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.34 (d, 2H, J = 8.4 Hz), 7.41 (d, 2H, J = 8.4 Hz), 7.66 (d, 1H, J = 8.0 Hz), 7.79 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.52, 55.39, 111.11, 114.52, 114.52, 118.13, 123.52, 127.65, 127.65, 128.87, 128.87, 129.30, 129.30, 131.31, 133.10, 134.16, 136.26, 137.35, 148.86, 158.37, 159.64, 183.18. ESI-MS: 378.0 [M+H]+.
5.2.13. 1-(3-chlorobenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2i)
Yield 40%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 4.93 (s, 2H), 6.80 (d, 1H, J = 8.0 Hz), 6.96 (d, 2H, J = 8.8 Hz), 7.27 (d, 2H, J = 8.4 Hz), 7.32 (d, 2H, J = 8.4 Hz), 7.42 (d, 2H, J = 8.8 Hz), 7.68 (d, 1H, J = 8.0 Hz), 7.81 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.61, 55.39, 111.01, 114.52, 114.52, 118.13, 123.58, 125.56, 127.53, 127.67, 127.67, 128.51, 130.42, 131.32, 135.04, 136.34, 136.64, 137.41, 148.83, 158.37, 159.64, 183.11. ESI-MS: 378.1 [M+H]+.
5.2.14. 1-(2-chlorobenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2j)
Yield 44%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 5.08 (s, 2H), 6.81 (d, 1H, J = 8.4 Hz), 6.96 (d, 2H, J = 8.0 Hz), 7.23–7.26 (m, 2H), 7.41–7.45 (m, 4H), 7.66 (d, 1H, J = 8.0 Hz), 7.81 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 41.53, 55.39, 111.35, 114.51, 118.10, 123.31,127.50,127.61, 128.34, 129.43, 129.98, 131.26, 131.88, 133.03, 136.40, 137.23, 148.95, 158.56, 159.61, 183.16. ESI-MS: 378.2 [M+H]+.
5.2.15. 1-(4-bromobenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2k)
Yield 45%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 4.90 (s, 2H), 6.78 (d, 1H, J = 8.4 Hz), 6.96 (d, 2H, J = 8.8 Hz), 7.24 (d, 2H, J = 8.4 Hz), 7.41 (d, 2H, J = 8.8 Hz), 7.49 (d, 2H, J = 8.4 Hz), 7.66 (d, 1H, J = 8.0 Hz), 7.80 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.58, 55.39, 111.10, 114.52, 114.52, 118.14, 122.24, 123.55, 127.66, 127.66, 129.17, 129.17, 131.32, 132.26, 132.26, 133.61, 136.27, 137.39, 148.84, 158.36, 159.65, 183.15. ESI-MS: 422.1 [M+H]+.
5.2.16. 5-(4-methoxyphenyl)-1-(4-methylbenzyl)indoline-2,3-dione (2l)
Yield 43%; 1H NMR (400 MHz, CDCl3) δ 2.33 (s, 3H), 3.84 (s, 3H), 4.91 (s, 2H), 6.82 (d, 1H, J = 8.0 Hz), 6.95 (d, 2H, J = 8.8 Hz), 7.16 (d, 2H, J = 8.0 Hz), 7.25 (d, 2H, J = 8.0 Hz), 7.40 (d, 2H, J = 8.8 Hz), 7.64 (d, 1H, J = 8.0 Hz), 7.78 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 21.13, 43.94, 55.38, 111.33, 114.49, 114.49, 118.12, 122.44, 123.37, 127.50, 127.50, 127.63, 127.63, 129.73, 129.73, 131.49, 136.22, 137.08, 138.02, 149.29, 158.39, 159.57, 183.57. ESI-MS: 358.2 [M+H]+.
5.2.17. 1-(4-methoxybenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2m)
Yield 50%; 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 3H), 3.83 (s, 3H), 4.88 (s, 2H), 6.84 (d, 1H, J = 8.4 Hz), 6.87 (d, 2H, J = 8.4 Hz), 6.95 (d, 2H, J = 8.8 Hz), 7.28 (d, 2H, J = 8.4 Hz), 7.39 (d, 2H, J = 8.8 Hz), 7.64 (d, 1H, J = 8.4 Hz), 7.76 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.64, 55.30, 55.38, 111.32, 114.45, 114.45, 114.49, 114.49, 118.12, 123.32, 126.55, 127.61, 127.61, 128.96, 128.96, 131.43, 136.18, 137.04, 149.26, 158.37, 159.50, 159.58, 183.59; IR (KBr) 3435 (CAr-H), 2982, 2887, 1727 (C=O), 1618 (C=O), 1513 (CAr=CAr), 1480 (CAr=CAr), 1035 (C-O), 821 (CAr-H) cm−1; ESI-MS: 374.2 [M+H]+, 396.1 [M+Na]+. HRMS (ESI, m/z): calcd for C23 H19 N O4Na [M+Na]+ 396.1206, found 396.1195.
5.2.18. 1-(3-methoxybenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2n)
Yield 40%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 3.83 (s, 3H), 4.92 (s, 2H), 6.83 (d, 1H, J = 8.4 Hz), 6.87 (d, 2H, J = 8.4 Hz), 6.96 (d, 2H, J = 8.0 Hz), 6.97 (s, 1H), 7.28 (d, 1H, J = 8.0 Hz), 7.41 (d, 2H, J = 8.4 Hz), 7.65 (d, 1H, J = 8.0 Hz), 7.79 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 44.12, 55.31, 55.39, 111.34, 113.28, 113.38, 114.50, 114.50, 118.10, 119.68, 123.39, 127.64, 127.64, 130.14, 131.44, 136.12, 136.28, 137.16, 149.24, 158.40, 159.59, 160.16, 183.43. ESI-MS: 374.2 [M+H]+, 396.2 [M+Na]+.
5.2.19. 1-(2-methoxybenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2o)
Yield 42%; 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H), 3.90 (s, 3H), 4.97 (s, 2H), 6.90–6.96 (m, 5H), 7.29 (d, 2H, J = 8.4 Hz), 7.41 (d, 2H, J = 8.4 Hz),7.65 (d, 1H, J = 8.0 Hz), 7.69 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 38.98, 55.38, 55.46, 110.62, 111.54, 114.47, 114.47, 118.07, 120.92, 122.53, 123.14, 127.62, 127.62, 129.00, 129.39, 131.57, 136.27, 136.84, 149.71, 157.15, 158.56, 159.52, 183.82. ESI-MS: 374.2 [M+H]+, 396.2 [M+Na]+.
5.2.20. 1-(3,5-dimethylbenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2p)
Yield 46%; 1H NMR (400 MHz, CDCl3) δ 2.29 (s, 6H), 3.84 (s, 3H), 4.86 (s, 2H), 6.83 (d, 1H, J = 8.0 Hz), 6.93–6.96 (m, 5H), 7.41 (d, 2H, J = 8.8 Hz), 7.65 (d, 1H, J = 8.0 Hz), 7.78 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 21.29, 21.29, 44.13, 55.39, 111.37, 114.49, 114.49, 118.11, 123.34, 125.24, 125.24, 127.63, 127.63, 129.85, 131.49, 134.44, 136.28, 137.05, 138.74, 138.74, 149.39, 158.42, 159.57, 183.62. ESI-MS: 372.2 [M+H]+, 394.2 [M+Na]+.
5.2.21. 1-(3,4-dimethoxybenzyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2q)
Yield 40%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 3.86 (s, 3H), 3.86 (s, 3H), 4.89 (s, 2H), 6.83 (d, 1H, J = 8.4 Hz), 6.86 (d, 2H, J = 8.4 Hz), 6.92 (d, 1H, J = 8.0 Hz), 6.96 (d, 2H, J = 8.8 Hz), 7.41 (d, 2H, J = 8.8 Hz), 7.66 (d, 1H, J = 8.4 Hz), 7.79 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 44.10, 55.39, 55.93, 56.03, 110.75, 111.25, 111.32, 114.50, 114.50, 118.13, 120.09, 123.42, 127.01, 127.64, 127.64, 131.43, 136.21, 137.15, 149.05, 149.28, 149.60, 158.43, 159.60, 183.56. ESI-MS: 404.1 [M+H]+, 426.1 [M+Na]+.
5.2.22. 4-((5-(4-methoxyphenyl)-2,3-dioxoindolin-1-yl)methyl)benzonitrile (2r)
Yield 29%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 5.01 (s, 2H), 6.74 (d, 1H, J = 8.0 Hz), 6.97 (d, 2H, J = 8.4 Hz), 7.42 (d, 2H, J = 8.4 Hz), 7.47 (d, 2H, J = 8.4 Hz), 7.67 (d, 2H, J = 8.4 Hz), 7.68 (d, 1H, J = 8.0 Hz), 7.82 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.73, 55.40, 110.84, 112.36, 114.56, 114.56, 118.16, 118.25, 123.73, 127.67, 127.67, 128.06, 128.06, 131.16, 132.92, 132.92, 136.35, 137.68, 139.98, 148.49, 158.37, 159.73, 182.76. ESI-MS: 369.1 [M+H]+, 391.1 [M+Na]+.
5.2.23. 5-(4-methoxyphenyl)-1-(4-(trifluoromethyl)benzyl)indoline-2,3-dione (2s)
Yield 41%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 5.01 (s, 2H), 6.77 (d, 1H, J = 8.0 Hz), 6.96 (d, 2H, J = 8.4 Hz), 7.42 (d, 2H, J = 8.4 Hz), 7.48 (d, 2H, J = 8.0 Hz), 7.63 (d, 2H, J = 8.0 Hz), 7.65 (d, 1H, J = 8.0 Hz), 7.82 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.68, 55.40, 110.84, 112.36, 114.56, 114.56, 118.16, 118.25, 123.73, 127.67, 127.67, 128.06, 128.06, 131.16, 132.92, 132.92, 136.35, 137.68, 139.98, 148.49, 158.37, 159.73, 182.76. ESI-MS: 412.2 [M+H]+.
5.2.24. 5-(4-methoxyphenyl)-1-(2-phenoxyethyl)indoline-2,3-dione (2t)
Yield 41%; 1H NMR (400 MHz, CDCl3) δ 3.85 (s, 3H), 4.16 (t, 2H, J = 10.4 Hz), 4.27 (t, 2H, J = 10.4 Hz), 6.845 (d, 2H, J = 8.0 Hz), 6.95–6.99 (m, 3H), 7.21–7.28 (m, 4H), 7.46 (d, 2H, J = 8.0 Hz), 7.79 (d, 1H, J = 8.0 Hz), 7.80 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 40.31, 55.40, 65.72, 111.47, 111.50, 114.37, 114.40, 114.52, 118.03, 121.49, 123.21, 127.67, 127.67, 129.60, 129.60, 131.50, 136.15, 137.07, 149.97, 158.02, 158.63, 159.63, 183.28. ESI-MS: 374.2 [M+H]+, 396.2 [M+Na]+.
5.2.25. 5-(4-methoxyphenyl)-1-(naphthalen-2-ylmethyl)indoline-2,3-dione (2u)
Yield 48%; 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H), 5.12 (s, 2H), 6.84 (d, 1H, J = 8.0 Hz), 6.94 (d, 2H, J = 8.4 Hz), 7.39 (d, 2H, J = 8.8 Hz), 7.44 (d, 1H, J = 8.4 Hz), 7.44–7.50 (m, 2H), 7.59–7.62 (m, 1H), 7.80–7.83 (m, 5H). 13C NMR (100 MHz, CDCl3) δ 44.41, 55.37, 113.36, 114.50, 111.50, 118.17, 123.39, 125.04, 126.39, 126.46, 126.63, 127.62, 127.62, 127.77, 127.80, 129.16, 131.41, 131.99, 133.04, 133.32, 136.23, 137.18, 149.22, 158.51, 159.61, 183.42. ESI-MS: 394.2 [M+H]+.
5.2.26. 1-(benzo[d] [1,3]dioxol-5-ylmethyl)-5-(4-methoxyphenyl)indoline-2,3-dione (2v)
Yield 46%; 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 4.85 (s, 2H), 5.95 (s, 2H), 6.78 (d, 1H, J = 8.4 Hz), 6.82–6.86 (m, 3H), 6.96 (d, 2H, J = 8.4 Hz), 7.41 (d, 2H, J = 8.4 Hz), 7.66 (d, 1H, J = 8.0 Hz), 7.78 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 44.00, 55.39, 101.33, 107.98, 108.58, 111.29, 114.50, 114.50, 118.13, 121.11, 123.43, 127.65, 127.65, 128.27, 131.44, 136.23, 137.17, 147.61, 148.35, 149.15, 158.34, 159.60, 183.44. ESI-MS: 388.1 [M+H]+, 410.1 [M+Na]+.
5.3. Synthesis of compounds 3a-3c
To a microwave reactor vial (5 mL) which contained the solution of 4 or 5 or 6 or 7-bromoindoline-2,3-dione in 1,4-dioxane (5 mL) were added PdCl2(dppf) (155 mg, 0.22 mmol), CH3COOK (0.6 g, 6.2 mmol) and 4-Methoxyphenylboronic acid (5.3 mmol) under the atmosphere of argon. The microwave reactor vial was caped and placed into the microwave cavity. The reaction mixture was irradiated at high level for 30 min at 120 °C. The reaction mixture was cooled to room temperature, pourd onto 100 mL ice-water and then extracted with dichloromethane (3 × 100 mL). The combined organic layers were washed with water (2 × 100 mL), brine (100 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under vacuum to afford the crude which was purified by flash column chromatography (silica gel, petroleum ether/ethyl acetate 3:1) to yield the desired phenylisatin derivatives.
To a flask (25 mL) which contained the solution of phenylisatin derivatives (0.86 mmol) in dry N,N-dimethylformamide (2 mL). The reaction temperature was maintained at 0 °C followed by addition K2CO3 (0.35 g, 2.6 mmol). Stirring 5 min, was added 4-methoxybenzylchloride (0.16 g, 1.0 mmol) and the mixture allowed warm to room temperature. The reaction mixture was stirred at room temperature for 6 h. The orange solution was poured water (25 mL) and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 8:1–3:1) afforded the title compounds 3a-3c.
5.3.1. 1-(4-methoxybenzyl)-4-(4-methoxyphenyl)indoline-2,3-dione (3a)
Yield 51%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 3.87 (s, 3H), 4.89 (s, 2H), 6.72 (d, 1H, J = 7.6 Hz), 6.88 (d, 2H, J = 8.4 Hz), 6.97 (d, 2H, J = 8.8 Hz), 7.02 (d, 1H, J = 7.6 Hz), 7.29 (d, 2H, J = 8.8 Hz), 7.42–7.46 (m, 1H), 7.49 (d, 2H, J = 8.0 Hz). 13C NMR (100 MHz, CDCl3) δ 43.52, 55.31, 55.34, 108.96, 113.74, 113.83, 113.83, 114.41, 114.41, 125.61, 126.70, 128.33, 128.90, 128.90, 130.40, 130.40, 137.40, 143.22, 151.50, 158.09, 159.44, 160.53, 182.02. ESI-MS: 374.2 [M+H]+.
5.3.2. 1-(4-methoxybenzyl)-6-(4-methoxyphenyl)indoline-2,3-dione (3b)
Yield 46%; 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 3H), 3.86 (s, 3H), 4.91 (s, 2H), 6.88 (d, 2H, J = 8.4 Hz), 6.94 (s, 1H), 6.98 (d, 2H, J = 8.8 Hz), 7.24 (d, 1H, J = 8.0 Hz), 7.30 (d, 2H, J = 8.8 Hz), 7.45 (d, 2H, J = 8.4 Hz), 7.63 (d, 1H, J = 8.0 Hz). 13C NMR (100 MHz, CDCl3) δ 43.52, 55.31, 55.45, 108.87, 114.45, 114.45, 114.60, 114.60, 115.96, 121.92, 125.86, 126.71, 128.50, 128.50, 128.93, 128.93, 131.79, 151.12, 151.47, 158.95, 159.46, 160.77, 182.52. ESI-MS: 374.2 [M+H]+, 396.2 [M+Na]+.
5.3.3. 1-(4-methoxybenzyl)-7-(4-methoxyphenyl)indoline-2,3-dione (3c)
Yield 32%; 1H NMR (400 MHz, CDCl3) δ 3.73 (s, 3H), 3.86 (s, 3H), 4.65 (s, 2H), 6.52 (d, 2H, J = 8.4 Hz), 6.63 (d, 2H, J = 8.4 Hz), 6.82 (d, 2H, J = 8.4 Hz), 6.99 (d, 2H, J = 8.4 Hz), 7.07–7.11 (m, 1H), 7.27–7.30 (m, 1H), 7.63 (d, 1H, J = 8.4 Hz). 13C NMR (100 MHz, CDCl3) δ 44.71, 55.25, 55.44, 113.49, 113.49, 113.65, 113.65, 119.30, 123.54, 124.42, 126.99, 127.58, 127.58, 127.76, 129.23, 130.72, 130.72, 142.20, 147.24, 158.87, 159.61, 159.99, 183.62. ESI-MS: 374.1 [M+H]+, 396.1 [M+Na]+.
5.4. Synthesis of compounds 4a-4r
To a flask (25 mL) which contained the solution of 5-bromoindoline-2,3-dione (1 g, 4.4 mmol) in dry N,N-dimethylformamide (4 mL). The reaction temperature was maintained at 0 °C followed by addition K2CO3 (1.83 g, 13.2 mmol). Stirring 5 min, was added 4-methoxybenzylchloride (0.82 g, 5.3 mmol) and the mixture allowed warm to room temperature. The reaction mixture was stirred at room temperature for 8 h. The orange solution was poured water (25 mL) and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 10:1) afforded the title compounds 5-bromo-1-(4-methoxybenzyl)indoline-2,3-dione.
To a microwave reactor vial (2 mL) which contained the solution of 5-bromo-1-(4-methoxybenzyl)indoline-2,3-dione (0.2 g, 0.6 mmol) in 1,4-dioxane (2 mL) were added PdCl2(dppf) (21 mg, 0.03 mmol), CH3COOK (82 mg, 0.84 mmol) and substituted benzeneboronic acid (0.72 mmol) under the atmosphere of argon. The microwave reactor vial was caped and placed into the microwave cavity. The reaction mixture was irradiated at high level for 30 min at 120 °C. The reaction mixture was cooled to room temperature, poured onto 30 mL ice-water and then extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with water (2 × 100 mL), brine (100 mL) and dried over anhydrous magnesium sulfate. The solvent was removed under vacuum to afford the crude which was purified by flash column chromatography (silica gel, petroleum ether/ethyl acetate 3:1) to yield the desired compound 4a-4r.
5.4.1. 1-(4-methoxybenzyl)-5-(3-methoxyphenyl)indoline-2,3-dione (4a)
Yield 47%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 3.85 (s, 3H), 4.90 (s, 2H), 6.86 (d, 2H, J = 7.6 Hz), 6.91 (d, 2H, J = 8.0 Hz), 6.99 (t, 1H), 7.06 (d, 1H, J = 7.6 Hz), 7.30 (d, 2H, J = 8.0 Hz), 7.35 (d, 1H, J = 7.6 Hz), 7.70 (d, 1H, J = 8.0 Hz), 7.82 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.70, 55.32, 55.36, 111.33, 112.31, 113.29, 114.46, 114.46, 118.09, 118.98, 123.90, 126.45, 128.98, 128.98, 130.11, 136.77, 137.22, 140.42, 149.89, 158.35, 159.51, 160.14, 183.50. ESI-MS: 374.1 [M+H]+, 396.1 [M+Na]+.
5.4.2. 1-(4-methoxybenzyl)-5-(2-methoxyphenyl)indoline-2,3-dione (4b)
Yield 43%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 3.80 (s, 3H), 4.89 (s, 2H), 6.83 (d, 1H, J = 8.0 Hz), 6.88 (d, 2H, J = 8.4 Hz), 6.96–7.03 (m, 2H), 7.22 (d, 1H, J = 8.0 Hz), 7.29–7.33 (m, 3H), 7.63 (d, 1H, J = 8.0 Hz), 7.80 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.61, 55.32, 55.51, 110.68, 111.22, 114.42, 114.42, 117.49, 121.03, 126.59, 126.64, 128.23, 129.01, 129.01, 129.37, 130.18, 134.52, 139.26, 149.40, 156.26, 158.49, 159.47, 183.56. ESI-MS: 374.2 [M+H]+, 396.2 [M+Na]+.
5.4.3. 5-(3,5-dimethoxyphenyl)-1-(4-methoxybenzyl)indoline-2,3-dione (4c)
Yield 41%; 1H NMR (400 MHz, CDCl3) δ 3.81 (s, 3H), 3.85 (s, 6H), 4.92 (s, 2H), 6.48 (t, 1H, J = 4.4 Hz), 6.61 (s, 2H), 6.87–6.91 (m, 3H), 7.31 (d, 2H, J = 8.4 Hz), 7.71 (d, 1H, J = 8.4 Hz), 7.84 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.68, 55.32, 55.48, 55.48, 99.71, 104.83, 104.83, 111.32, 114.45, 114.45, 118.00, 123.84, 126.44, 128.99, 128.99, 136.78, 137.20, 141.02, 149.97, 158.35, 159.50, 161.28, 161.28, 183.50. ESI-MS: 404.2 [M+H]+, 426.2 [M+Na]+.
5.4.4. 5-(3,4-dimethoxyphenyl)-1-(4-methoxybenzyl)indoline-2,3-dione (4d)
Yield 38%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 3.91 (s, 3H), 3.93 (s, 3H), 4.90 (s, 2H), 6.85 (d, 1H, J = 8.0 Hz), 6.88 (d, 2H, J = 8.0 Hz), 6.92 (d, 1H, J = 8.4 Hz), 6.97 (s, 1H), 7.04 (d, 1H, J = 8.0 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.67 (d, 1H, J = 8.0 Hz), 7.80 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.67, 55.31, 56.01, 56.01, 109.71, 111.33, 111.60, 114.44, 114.44, 118.08, 118.90, 123.48, 126.47, 128.96, 128.96, 131.86, 136.35, 137.22, 149.08, 149.39, 149.39, 158.35, 159.49, 183.65. ESI-MS: 404.2 [M+H]+.
5.4.5. 5-(4-(tert-butyl)phenyl)-1-(4-methoxybenzyl)indoline-2,3-dione (4e)
Yield 34%; 1H NMR (400 MHz, CDCl3) δ 1.35 (s, 9H), 3.79 (s, 3H), 4.90 (s, 2H), 6.86 (d, 1H, J = 8.4 Hz), 6.89 (d, 2H, J = 8.4 Hz), 7.30 (d, 2H, J = 8.4 Hz), 7.41–7.47 (m, 4H), 7.70 (d, 1H, J = 8.4 Hz), 7.83 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 31.31, 31.31, 31.31, 34.61, 43.67, 55.31, 111.32, 114.45, 114.45, 118.12, 123.66, 126.04, 126.04, 126.19, 126.19, 126.52, 128.99, 128.99, 136.02, 136.54, 137.23, 149.58, 151.07, 158.38, 159.49, 183.58. ESI-MS: 400.1 [M+H]+.
5.4.6. 5-(4-fluorophenyl)-1-(4-methoxybenzyl)indoline-2,3-dione (4f)
Yield 41%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.90 (s, 2H), 6.86–6.90 (m, 3H), 7.10–7.15 (m, 2H), 7.29 (d, 2H, J = 8.0 Hz), 7.42–7.46 (m, 2H), 7.64 (d, 1H, J = 8.0 Hz), 7.78 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.69, 55.32, 111.41, 114.46, 114.46, 115.92, 116.13, 118.14, 123.69, 126.40, 128.18, 128.26, 128.97, 128.97, 135.16, 136.39, 136.54, 149.76, 158.27, 159.52, 161.47, 183.44. ESI-MS: 362.2 [M+H]+, 384.2 [M+Na]+.
5.4.7. 5-(4-chlorophenyl)-1-(4-methoxybenzyl)indoline-2,3-dione (4g)
Yield 48%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.90 (s, 2H), 6.87 (d, 1H, J = 8.4 Hz), 6.88 (d, 2H, J = 8.4 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.40 (m, 4H), 7.66 (d, 1H, J = 8.4 Hz), 7.79 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.71, 55.32, 111.46, 114.48, 114.48, 118.18, 123.65, 126.36, 127.79, 127.79, 128.96, 128.96, 129.25, 129.25, 134.09, 136.10, 136.50, 137.43, 149.99, 158.24, 159.54, 183.34. ESI-MS: 378.1 [M+H]+, 400.1 [M+Na]+.
5.4.8. 5-(3-chloro-4-fluorophenyl)-1-(4-methoxybenzyl)indoline-2,3-dione (4h)
Yield 40%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.90 (s, 2H), 6.88 (d, 1H, J = 8.4 Hz), 6.89 (d, 2H, J = 8.4 Hz), 7.21 (d, 1H, J = 8.4 Hz), 7.28–7.34 (m, 3H), 7.51 (d, 1H, J = 8.4 Hz), 7.63 (d, 1H, J = 8.4 Hz), 7.76 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.73, 55.32, 111.54, 114.49, 114.49, 117.09, 117.30, 118.17, 123.62, 126.23, 126.30, 128.78, 128.97, 128.97, 135.06, 136.28, 136.49, 150.15, 156.70, 158.17, 159.65, 183.23. ESI-MS: 396.1 [M+H]+, 418.1 [M+Na]+.
5.4.9. 1-(4-methoxybenzyl)-5-(p-tolyl)indoline-2,3-dione (4i)
Yield 46%; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H), 3.79 (s, 3H), 4.89 (s, 2H), 6.85 (d, 1H, J = 8.0 Hz), 6.88 (d, 2H, J = 8.4 Hz), 7.24 (d, 2H, J = 8.0 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.37 (d, 2H, J = 8.0 Hz), 7.68 (d, 1H, J = 8.4 Hz), 7.81 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 21.10, 43.66, 55.31, 111.30, 114.44, 114.44, 118.12, 123.66, 126.38, 126.38, 126.50, 128.96, 128.96, 129.77, 129.77, 136.08, 136.48, 137.35, 137.83, 149.55, 158.37, 159.49, 183.56. ESI-MS: 358.2 [M+H]+.
5.4.10. 5-(4-isopropoxyphenyl)-1-(4-methoxybenzyl)indoline-2,3-dione (4j)
Yield 43%; 1H NMR (400 MHz, CDCl3) δ 1.34 (s, 3H), 1.36 (s, 3H), 3.79 (s, 3H), 4.55–4.61 (m, 1H), 4.89 (s, 2H), 6.83 (d, 1H, J = 8.4 Hz), 6.88 (d, 2H, J = 8.4 Hz), 6.93 (d, 2H, J = 8.8 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.39 (d, 2H, J = 8.8 Hz), 7.64 (d, 1H, J = 8.4 Hz), 7.78 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 22.04, 22.04, 43.65, 55.31, 70.02, 111.28, 114.44, 114.44, 116.27, 116.27, 118.12, 123.35, 126.54, 127.63, 127.63, 128.96, 128.96, 131.15, 136.15, 137.15, 149.21, 157.92, 158.38, 159.48, 183.62. ESI-MS: 402.2 [M+H]+.
5.4.11. 1-(4-methoxybenzyl)-5-phenylindoline-2,3-dione (4k)
Yield 45%; 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 3H), 4.89 (s, 2H), 6.87 (d, 1H, J = 8.0 Hz), 6.88 (d, 2H, J = 8.0 Hz), 7.30 (d, 2H, J = 8.8 Hz), 7.36 (d, 1H, J = 8.0 Hz), 7.42 (d, 2H, J = 8.0 Hz), 7.45 (d, 2H, J = 8.8 Hz), 7.46–7.48 (d, 1H, J = 8.0 Hz), 7.71 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.68, 55.32, 111.36, 114.45, 114.45, 118.13, 123.85, 126.47, 126.55, 126.55, 127.90, 128.98, 128.98, 129.08, 129.08, 136.73, 137.37, 138.96, 149.79, 158.35, 159.50, 183.50. ESI-MS: 344.2 [M+H]+.
5.4.12. 4-(1-(4-methoxybenzyl)-2,3-dioxoindolin-5-yl)benzonitrile (4l)
Yield 29%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.91 (s, 2H), 6.89 (d, 2H, J = 8.8 Hz), 6.93 (d, 1H, J = 8.4 Hz), 7.29 (d, 2H, J = 8.8 Hz), 7.59 (d, 2H, J = 8.4 Hz), 7.71 (d, 2H, J = 8.4 Hz), 7.73 (d, 1H, J = 8.4 Hz), 7.84 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.77, 55.32, 111.60, 111.70, 114.50, 114.50, 118.27, 118.54, 123.88, 126.16, 127.13, 127.13, 128.98, 128.98, 132.89, 132.89, 135.10, 136.77, 143.34, 150.76, 158.11, 159.58, 183.06. ESI-MS: 369.2 [M+H]+.
5.4.13. 1-(4-methoxybenzyl)-5-(4-(trifluoromethyl)phenyl)indoline-2,3-dione (4m)
Yield 31%; 1H NMR (400 MHz, CDCl3) δ 3.81 (s, 3H), 4.94 (s, 2H), 6.91 (d, 2H, J = 8.8 Hz), 6.94 (d, 1H, J = 8.0 Hz), 7.32 (d, 2H, J = 8.8 Hz), 7.61 (d, 2H, J = 8.0 Hz), 7.72 (d, 2H, J = 8.8 Hz), 7.74 (d, 1H, J = 8.4 Hz), 7.86 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.76, 55.32, 111.57, 114.51, 114.51, 118.24, 123.92, 126.04, 126.08, 126.28, 126.85, 126.85, 128.98, 128.98, 129.84, 130.16, 135.74, 136.80, 142.46, 150.47, 158.19, 159.58, 183.20. ESI-MS: 412.1 [M+H]+, 434.1 [M+Na]+.
5.4.14. 1-(4-methoxybenzyl)-5-(4-(trifluoromethoxy)phenyl)indoline-2,3-dione (4n)
Yield 39%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.90 (s, 2H), 6.87–6.90 (m, 3H), 7.28–7.30 (m, 4H), 7.49 (d, 2H, J = 8.8 Hz), 7.66 (d, 1H, J = 8.0 Hz), 7.80 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.72, 55.31, 111.48, 114.48, 114.48, 118.19, 121.15, 121.15, 123.75, 126.35, 127.96, 127.96, 128.97, 128.97, 135.91, 136.62, 136.62, 137.71, 149.03, 150.08, 158.23, 159.55, 183.31. ESI-MS: 428.1 [M+H]+, 450.0 [M+Na]+.
5.4.15. 5-(4-hydroxyphenyl)-1-(4-methoxybenzyl)indoline-2,3-dione (4o)
Yield 28%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.89 (s, 2H), 6.84 (d, 1H, J = 8.4 Hz), 6.87–6.90 (m, 4H), 7.29 (d, 2H, J = 8.4 Hz), 7.36 (d, 2H, J = 8.4 Hz), 7.63 (d, 1H, J = 8.4 Hz), 7.77 (s, 1H). 13C NMR (100 MHz, DMSO) δ 42.91, 55.53, 112.03, 114.53, 114.53, 116.23, 116.23, 118.68, 121.99, 127.80, 127.92, 127.92, 129.33, 129.33, 129.76, 135.61, 136.16, 149.16, 157.73, 158.84, 159.18, 183.78. ESI-MS: 360.1 [M+H]+, 382.1 [M+Na]+.
5.4.16. 1-(4-methoxybenzyl)-5-(naphthalen-2-yl)indoline-2,3-dione (4p)
Yield 47%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.92 (s, 2H), 6.89 (d, 2H, J = 8.8 Hz), 6.91 (d, 1H, J = 8.4 Hz), 7.31 (d, 2H, J = 8.8 Hz), 7.49–7.53 (m, 2H), 7.61 (d, 1H, J = 8.4 Hz), 7.82–7.89 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 43.71, 55.32, 111.44, 114.48, 114.48, 118.24, 124.06, 124.57, 125.41, 126.39, 126.47, 126.68, 127.69, 128.16, 128.88, 128.98, 128.98, 132.76, 133.56, 136.22, 136.90, 137.26, 149.82, 158.35, 159.54, 183.49. ESI-MS: 394.1 [M+H]+, 416.1 [M+Na]+.
5.4.17. 1-(4-methoxybenzyl)-5-(pyridin-4-yl)indoline-2,3-dione (4q)
Yield 25%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.92 (s, 2H), 6.89 (d, 2H, J = 8.4 Hz), 6.94 (d, 1H, J = 8.4 Hz), 7.27 (d, 2H, J = 8.4 Hz), 7.45 (d, 2H, J = 6.0 Hz), 7.78 (d, 1H, J = 8.4 Hz), 7.90 (s, 1H), 8.68 (d, 2H, J = 6.0 Hz). 13C NMR (100 MHz, CDCl3) δ 43.79, 55.33, 111.73, 114.51, 114.51, 118.30, 120.92, 123.73, 126.15, 128.98, 128.98, 133.99, 136.63, 136.63, 146.25, 150.40, 150.40, 151.14, 158.12, 159.59, 183.01. ESI-MS: 345.2 [M+H]+, 367.2 [M+Na]+.
5.4.18. 1-(4-methoxybenzyl)-5-(thiophen-3-yl)indoline-2,3-dione (4r)
Yield 40%; 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 4.89 (s, 2H), 6.83 (d, 1H, J = 8.0 Hz), 6.88 (d, 2H, J = 8.8 Hz), 7.27–7.30 (m, 3H), 7.37–7.41 (m, 2H), 7.71 (d, 1H, J = 8.0 Hz), 7.82 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.69, 55.32, 111.38, 114.45, 114.45, 118.07, 120.52, 123.09, 125.68, 126.43, 127.01, 128.96, 128.96, 132.15, 135.87, 140.08, 149.43, 158.30, 159.50, 183.52. ESI-MS: 350.1 [M+H]+, 372.0 [M+Na]+.
5.5. Synthesis of compounds 5a-5k
A mixture of 0.4 mmol compound 2m, 0.8 mmol sodium borohydride were stirred in 2 mL dichloromethane and 8 mL ethanol at room temperature under an argon atmosphere. The progress of the reaction was monitored by TLC (petroleum ether/ethyl acetate). After the reaction finished, the reaction mixture was poured water (25 mL) and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 3:1) afforded the title compound 5a.
A mixture of 0.4 mmol compound 2m, 0.8 mmol methyl-magnesiubromide (1.0M solution in THF) were stirred in 2 mL dry THF at −78 °C under a argon atmosphere. The progress of the reaction was monitored by TLC (petroleum ether/ethyl acetate). After the reaction finished, the reaction mixture was poured ice water and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 3:1) afforded the title compound 5b.
A mixture of 0.4 mmol compounds 2m, 1.2 mmol Hydrazine hydrate were stirred in 5 mL ethanol at reflux under a argon atmosphere. The progress of the reaction was monitored by TLC (petroleum ether/ethyl acetate). After the reaction finished, the reaction mixture was poured water (25 mL) and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 6:1) afforded the title compounds 5c.
To a flask (25 mL) which contained the solution of compound 2m (0.15 g, 0.4 mmol) in dry THF (8 mL). The reaction temperature was maintained at 0 °C followed by the added triethyl phosphonoacetate (0.48 mmol) and the mixture allowed warm to room temperature. The reaction mixture was stirred at room temperature for 1 h. The orange solution was poured water (25 mL) and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 8:1) afforded the title compounds 5d and 5e.
A mixture of 0.4 mmol compounds 2m, 1.6 mmol ethylene glycol and 0.06 mmol toluene-p-sulfonic acid were stirred in 15 mL toluene at 125 °C under a argon atmosphere. The progress of the reaction was monitored by TLC (petroleum ether/ethyl acetate). After the reaction finished, the reaction mixture was poured water (25 mL) and extracted with ethyl acetate (3 × 40 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 10:1) afforded the title compounds 5f.
To a flask (25 mL) which contained the solution of compound 2m (0.15 g, 0.44 mmol) in dry dichloromethane (8 mL). The reaction temperature was maintained at 0 °C followed by the added DAST (77 mg, 0.48 mmol) and the mixture allowed warm to room temperature. The reaction mixture was stirred at room temperature for 2 h. The orange solution was poured water (25 mL) and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 5:1) afforded the title compounds 5g.
A mixture of 0.4 mmol compounds 2m, 0.44 mmol hydroxylamine hydrochloride and 0.2 mmol Na2CO3 were stirred in 10 mL ethanol at reflux under an argon atmosphere. The progress of the reaction was monitored by TLC (petroleum ether/ethyl acetate). After the reaction finished, the reaction mixture was filtered. The filtrate was concentrated to dryness and subjected to flash column chromatography (silica gel), eluting with petroleum ether/ethyl acetate, afforded compounds 5h.
To a flask (25 mL) which contained the solution of compound 5c (0.15 g, 0.42 mmol) in ethanol (8 mL) were added Piperidine (4 mg, 0.04 mmol) and substituted benzaldehyde (0.5 mmol) under the atmosphere of argon. The reaction mixture was stirred at reflux for 4 h. The orange solution was poured water (25 mL) and extracted with dichloromethane (3 × 50 mL). The combined extracts were dried over anhydrous magnesium sulfate, filtered and evaporated. Chromatography (petroleum ether/ethyl acetate 10:1–3:1) afforded the title compounds 5i-5k.
5.5.1. 3-hydroxy-1-(4-methoxybenzyl)-5-(4-methoxyphenyl)indolin-2-one (5a)
Yield 59%; 1H NMR (400 MHz, CDCl3) δ 3.76 (s, 3H), 3.83 (s, 3H), 4.77–4.90 (m, 2H), 5.20 (s, 1H), 6.78 (d, 1H, J = 8.4 Hz), 6.85 (d, 2H, J = 8.4 Hz), 6.94 (d, 2H, J = 8.8 Hz), 7.26 (d, 2H, J = 8.4 Hz), 7.39 (d, 1H, J = 8.4 Hz), 7.44 (d, 2H, J = 8.8 Hz), 7.65 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.48, 55.27, 55.35, 70.01, 109.74, 114.26, 114.26, 114.28, 114.28, 123.78, 127.36, 127.50, 127.81, 127.95, 127.95, 128.79, 128.79, 133.12, 136.38, 141.79, 159.05, 159.22, 176.97. ESI-MS: 376.2 [M+H]+.
5.5.2. 3-hydroxy-1-(4-methoxybenzyl)-5-(4-methoxyphenyl)-3-methylindolin-2-one (5b)
Yield 59%; 1H NMR (400 MHz, CDCl3) δ 1.68 (s, 3H), 3.77 (s, 3H), 3.83 (s, 3H), 4.77–4.93 (m, 2H), 6.78 (d, 1H, J = 8.4 Hz), 6.85 (d, 2H, J = 8.4 Hz), 6.94 (d, 2H, J = 8.8 Hz), 7.24 (d, 2H, J = 8.4 Hz), 7.37 (d, 1H, J = 8.4 Hz), 7.44 (d, 2H, J = 8.8 Hz), 7.59 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 25.14, 43.32, 55.26, 55.35, 73.87, 109.83, 114.26, 114.26, 114.26, 114.26, 122.09, 127.51, 127.78, 127.82, 127.82, 128.64, 128.64, 131.92, 133.19, 136.40, 140.71, 159.02, 159.16, 178.56. ESI-MS: 390.1 [M+H]+, 412.1 [M+Na]+.
5.5.3. 1-(4-methoxybenzyl)-5-(4-methoxyphenyl)indolin-2-one (5c)
Yield 54%; 1H NMR (400 MHz, CDCl3) δ 3.65 (s, 2H), 3.77 (s, 3H), 3.83 (s, 3H), 4.87 (s, 2H), 6.78 (d, 1H, J = 8.4 Hz), 6.85 (d, 2H, J = 8.4 Hz), 6.94 (d, 2H, J = 8.8 Hz), 7.27 (d, 2H, J = 8.4 Hz), 7.33 (d, 1H, J = 8.4 Hz), 7.42 (d, 2H, J = 8.8 Hz), 7.43 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 35.93, 43.33, 55.27, 55.35, 109.22, 114.18, 114.18, 114.26, 114.26, 123.04, 125.08, 126.20, 127.80, 127.80, 127.99, 128.80, 128.80, 133.50, 133.55, 143.22, 158.93, 159.11, 175.06. ESI-MS: 360.2 [M+H]+, 382.2 [M+Na]+.
5.5.4. (Z)-ethyl 2-(1-(4-methoxybenzyl)-5-(4-methoxyphenyl)-2-oxoindolin-3-ylidene)acetate (5d)
Yield 76%; 1H NMR (400 MHz, CDCl3) δ 1.35–1.39 (m, 3H), 3.77 (s, 3H), 3.84 (s, 3H), 4.31–4.37 (m, 2H), 4.90 (s, 2H), 6.75 (d, 1H, J = 8.0 Hz), 6.85 (d, 2H, J = 8.8 Hz), 6.96 (d, 2H, J = 8.4 Hz), 7.01 (s, 1H), 7.25 (d, 2H, J = 8.4 Hz), 7.44 (d, 1H, J = 8.0 Hz), 7.48 (d, 2H, J = 8.8 Hz), 8.81 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 14.24, 43.45, 55.28, 55.36, 61.26, 109.34, 114.25, 114.25, 114.28, 114.28, 120.49, 123.00, 127.27, 127.50, 127.81, 127.81, 128.70, 128.70, 130.53, 133.18, 135.88, 137.86, 143.89, 159.02, 159.20, 165.63, 167.73. ESI-MS: 466.3 [M+Na]+.
5.5.5. (Z)-methyl 2-(1-(4-methoxybenzyl)-5-(4-methoxyphenyl)-2-oxoindolin-3-ylidene)acetate (5e)
Yield 68%; 1H NMR (400 MHz, CDCl3) δ 3.77 (s, 3H), 3.84 (s, 3H), 3.88 (s, 3H), 4.89 (s, 2H), 6.75 (d, 1H, J = 8.0 Hz), 6.85 (d, 2H, J = 8.8 Hz), 6.96 (d, 2H, J = 8.8 Hz), 7.00 (s, 1H), 7.25 (d, 2H, J = 8.8 Hz), 7.44 (d, 1H, J = 8.0 Hz), 7.49 (d, 2H, J = 8.8 Hz), 8.82 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.74, 52.25, 55.28, 55.37, 109.37, 114.25, 114.25, 114.29, 114.29, 120.45, 122.34, 127.30, 127.47, 127.79, 127.79, 128.71, 128.71, 130.62, 133.13, 135.90, 138.23, 143.94, 159.04, 159.21, 166.07, 167.66. ESI-MS: 430.1 [M+H]+, 452.2 [M+Na]+.
5.5.6. 1'-(4-methoxybenzyl)-5'-(4-methoxyphenyl)spiro[ [1,3]dioxolane-2,3′-indolin]-2′-one (5f)
Yield 89%; 1H NMR (400 MHz, CDCl3) δ 3.77 (s, 3H), 3.83 (s, 3H), 4.35–4.38 (m, 2H), 4.63–4.66 (m, 2H), 4.78 (s, 2H), 6.72 (d, 1H, J = 8.0 Hz), 6.85 (d, 2H, J = 8.4 Hz), 6.93 (d, 2H, J = 8.8 Hz), 7.24 (d, 2H, J = 8.4 Hz), 7.41 (d, 1H, J = 8.0 Hz), 7.42 (d, 2H, J = 8.8 Hz), 7.55 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.04, 55.27, 55.34, 65.94, 65.94, 102.39, 109.96, 114.22, 114.22, 114.27, 114.27, 123.39, 124.53, 127.28, 127.80, 127.80, 128.64, 128.64, 129.75, 132.99, 136.55, 142.59, 159.06, 159.18, 173.46. ESI-MS: 418.2 [M+H]+, 440.2 [M+Na]+.
5.5.7. 3,3-difluoro-1-(4-methoxybenzyl)-5-(4-methoxyphenyl)indolin-2-one (5g)
Yield 81%; 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 3H), 3.84 (s, 3H), 4.85 (s, 2H), 6.83 (d, 1H, J = 8.4 Hz), 6.87 (d, 2H, J = 8.4 Hz), 6.96 (d, 2H, J = 8.4 Hz), 7.26 (d, 2H, J = 8.4 Hz), 7.42 (d, 2H, J = 8.4 Hz), 7.52 (d, 1H, J = 8.4 Hz), 7.72 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.61, 55.30, 55.38, 110.83, 114.44, 114.44, 114.44, 114.44, 120.75, 123.10, 123.10, 126.31, 127.83, 127.83, 128.81, 128.81, 131.48, 132.03, 137.27, 141.67, 159.46, 159.48, 165.46. ESI-MS: 396.1 [M+H]+, 418.1 [M+Na]+.
5.5.8. (Z)-3-(hydroxyimino)-1-(4-methoxybenzyl)-5-(4-methoxyphenyl)indolin-2-one (5h)
Yield 87%; 1H NMR (400 MHz, DMSO) δ 3.71 (s, 3H), 3.78 (s, 3H), 4.90 (s, 2H), 6.90 (d, 2H, J = 7.8 Hz), 7.00 (d, 2H, J = 7.8 Hz), 7.06 (d, 1H, J = 8.0 Hz), 7.30 (d, 2H, J = 8.0 Hz), 7.50 (d, 2H, J = 8.0 Hz), 7.59 (d, 1H, J = 8.0 Hz), 8.20 (s, 1H), 13.63 (s, 1H). 13C NMR (100 MHz, DMSO) δ 42.56, 55.52, 55.63, 110.51, 114.57, 114.57, 114.88, 114.88, 116.42, 125.15, 127.94, 127.94, 128.57, 129.19, 129.19, 130.26, 132.54, 135.32, 142.01, 143.96, 159.11, 159.19, 163.68. ESI-MS: 389.2 [M+H]+.
5.5.9. (Z)-3-benzylidene-1-(4-methoxybenzyl)-5-(4-methoxyphenyl)indolin-2-one (5i)
Yield 41%; 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 3H), 3.81 (s, 3H), 4.96 (s, 2H), 6.79 (d, 1H, J = 8.4 Hz), 6.85–6.91 (m, 4H), 7.28–7.35 (m, 5H), 7.43–7.45 (m, 3H), 7.47 (d, 2H, J = 8.4 Hz), 7.70 (s, 1H), 7.84 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 43.35, 55.28, 55.34, 109.37, 114.19, 114.19, 114.26, 114.26, 121.27, 121.82, 127.28, 127.56, 127.56, 128.09, 128.13, 128.65, 128.65, 128.76, 128.76, 129.34, 129.34, 129.73, 133.40, 134.82, 134.99, 137.73, 142.25, 158.88, 159.09, 168.60. ESI-MS: 448.2 [M+H]+.
5.5.10. (Z)-1-(4-methoxybenzyl)-5-(4-methoxyphenyl)-3-(4-(trifluoromethyl)benzylidene) indolin-2-one (5j)
Yield 39%; 1H NMR (400 MHz, CDCl3) δ 3.77 (s, 3H), 3.85 (s, 3H), 4.93 (s, 2H), 6.80 (d, 1H, J = 8.0 Hz), 6.85 (d, 2H, J = 8.4 Hz), 6.98 (d, 2H, J = 8.4 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.39 (d, 1H, J = 8.0 Hz), 7.48 (d, 2H, J = 8.4 Hz), 7.63 (s, 1H), 7.69–7.71 (m, 3H), 8.36 (d, 2H, J = 8.0 Hz). 13C NMR (100 MHz, CDCl3) δ 43.33, 55.28, 55.36, 109.37, 114.21, 114.21, 114.29, 114.29, 118.01, 121.60, 121.88, 127.28, 127.85, 127.85, 128.04, 128.09, 128.81, 128.81, 128.90, 128.90, 129.65, 129.65, 130.15, 133.50, 134.82, 134.99, 137.75, 142.31, 158.90, 159.06, 168.71. ESI-MS: 516.2 [M+H]+.
5.5.11. (Z)-3-(4-isopropylbenzylidene)-1-(4-methoxybenzyl)-5-(4-methoxyphenyl)indolin-2-one (5k)
Yield 37%; 1H NMR (400 MHz, CDCl3) δ 1.27 (s, 3H), 1.29 (s, 3H), 2.93–3.00 (m, 1H), 3.76 (s, 3H), 3.85 (s, 3H), 4.95 (s, 2H), 6.78 (d, 1H, J = 8.0 Hz), 6.84 (d, 2H, J = 8.4 Hz), 6.97 (d, 2H, J = 8.4 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.31 (d, 2H, J = 8.0 Hz), 7.33 (d, 1H, J = 8.0 Hz), 7.49 (d, 2H, J = 8.4 Hz), 7.63 (s, 1H), 7.68 (s, 1H), 8.29 (d, 2H, J = 8.0 Hz). 13C NMR (100 MHz, CDCl3) δ 23.87, 23.87, 34.20, 43.34, 55.28, 55.35, 109.30, 114.19, 114.19, 114.22, 114.22, 121.29, 122.03, 126.64, 126.75, 126.75, 127.59, 127.59, 127.81, 128.22, 128.78, 128.78, 129.61, 129.61, 132.49, 133.49, 134.69, 138.08, 142.14, 151.07, 158.87, 159.08, 168.75. ESI-MS: 490.2 [M+H]+.
5.6. Biology evaluation
5.6.1. Cell lines and culture conditions
The human leukemia K562 cell line was purchased from the Shanghai Institutes of Biological Sciences (Shanghai, China). Cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum, 2.05 mM glutamine and 1% penicillin/streptomycin. The medium was replaced once every third days. The umbilical vein endothelial cells (HUVECs) were purchased from ATCC (American type culture collection, USA) and cultured in Ham's F-12 medium supplemented with 10% fetal bovine serum, 2.05 mM glutamine and 1% penicillin/streptomycin. The liver cancer cells (HepG2) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2.05 mM glutamine and 1% penicillin/streptomycin. All cell lines were incubated in a humidified atmosphere of 5% CO2 at 37 °C.
5.6.2. Cytotoxic activity assay
Inhibition of cell proliferation by compound 2m was measured by the MTT assay. In brief, 100 μl of K562 cells were plated in 96-well plates at a density of 5 × 104 cells/ml for 2 h. Different concentrations of compound 2m (10–100,000 nM) were added to each well to culture for another 48 h. After 48 h of incubation, 10 μl MTT (5 mg/mL) was added to each well and the plates were further incubated for 4 h. MTT assay was performed using thermo microplate reader. The DMSO-treated controls were calculated as a cell viability value of 100%. The inhibitory concentrations (IC50) were obtained by nonlinear regression using GraphPad Prism 4.0. For each experiment, IC50 value was calculated from three independent assays.
5.6.3. Cell cycle analysis
For the DNA content analysis, 5 × 104 cells were cultured for 2 h and treated with compound 2m (30 nM, 300 nM) for 0, 6, 12, 24 and 48 h respectively. Cells were collected and fixed with 1 mL of 70% ice-cold ethanol at −20 °C overnight. Cells then were washed with PBS, incubated with propidium iodide (PI, 50 μg/mL), RNase A (100 μg/mL) and 0.2% Triton × 100 for 30 min in dark at room temperature and analyzed using a FACS Calibur system (version 2.0, BD) using the CELLQuest program (Becton Dickinson). Results were representatives of at least three independent experiments.
5.6.4. Flow cytometric analysis of apoptosis
Apoptotic cells were assayed by the Annexin-V-FITC Apoptosis Detection Kit (Tianjin Sungene Biotech Co., Ltd.) according to the manufacturer's instructions. K562 cells were seeded in six-well plates and treated with DMSO or compound 2m (30 nM, 300 nM) for 0, 6, 12, 24 and 48 h respectively. After the treatment, cells were harvested, washed twice with ice-cold PBS at 2500 rpm and resuspended in 1 × Binding buffer. Then, the cells were stained with 5 μl of Annexin-V-FITC for 10 min and 5 μl of PI (50 μg/mL) for 10 min at room temperature in dark and analyzed by flow cytometry. The fraction of cell population in different quadrants was analyzed using quadrant statistics.
5.6.5. Wound healing assay
Wound healing assay was performed to determine migration rate and repair ability of HepG2 cells. The HepG2 cells were plated in 6-well plates at a density of 2 × 105 cells/well and cultured in Dulbecco's modified Eagle's medium. After 24 h incubation, the cultured cells grew against the wall of 6-well plates. Then, the HepG2 cells were scratched with a 10 μl pipette tip to make a straight line on the HepG2 cells. The medium in the well was removed and the well of the HepG2 cells was washed for three times by 1 × PBS. The cells were treated with DMSO or compound 2m (10 nM, 30 nM, 100 nM) for 0, 6, 12, 24, 36 and 48 h respectively, and incubated at 37 °C and 5% CO2 atmosphere. Images of the HepG2 cells were got by a Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan).
5.6.6. Transwell assay
The transwell assay of HepG2 cells was performed using 24-well culture plates containing polycarbonate filter inserts (EMD Millipore corporation, Germany). The HepG2 cells were suspended in Dulbecco's modified Eagle's medium without fetal bovine serum and 200 μl (2 × 105 cells/well) were added to the upper compartment of the chamber. 600 μl Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum was added to the lower chamber. After 30 min, the cells were treated with DMSO or compound 2m (10 nM, 30 nM, 100 nM) for 24 h and incubated in 5% CO2 atmosphere at 37 °C. After 24 h, medium in the chamber was removed and the well of the HepG2 cells was washed for three times by 1 × PBS. Then, use the methanol as fixative and DAPI as stain. HepG2 cells on the upper side of the filter were removed using cotton swabs. The photographs of three horizons were taken in random. The migrated HepG2 cells to the lower side of the filter were counted.
5.6.7. Tube forming assay
The capillary-like network formation of HUVEC cells was investigated with Matrigel-coated 96-well culture plates [17]. Matrigel (13.9 mg/mL; BD Bioscience, San Jose, CA, USA) was thawed at 4 °C. Fifty microliters of the Matrigel was added to each well of the 24-well culture plates and incubated for 30 min at 37 °C for polymerization. The HUVEC cells (1 × 105 cells/well) were suspended in Ham's F-12 medium containing 10% FBS, and were then added to the Matrigel-coated wells. After 40 min incubation at 37 °C in 5% CO2 atmosphere, added DMSO or compound 2m (10 nM, 30 nM, 100 nM) to the cells. Then, the plates were incubated at 37 °C in 5% CO2 atmosphere. After 12 h incubation, the capillary-like tube formation in each well of the culture plates was photographed by a Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan).
Acknowledgments
The authors sincerely thank the financial support from National Natural Science Foundation of China (31601203), Undergraduate Training Program for Innovation and Entrepreneurship of Tianjin University of Science and Technology (201710057099).
The authors are thankful to the Research Centre of Modern Analytical Technology, Tianjin University of Science & Technology for NMR and HRMS analysis.
Footnotes
Supplementary data associated with this article can be found in the online version, at https://doi.org/10.1016/j.ejmech.2018.07.032. These data include MOL files and InChiKeys of the most important compounds described in this article.
Appendix A. Supplementary data
Mol Files
The following ZIP file contains the MOL files of the most important compounds referred to in this article.
MOL file. ZIP file containing the MOL files of the most important compounds in this article.
References
- 1.Yu B., Wang S.Q., Qi P.P., Yang D.X., Tang K., Liu H.M. Design and synthesis of isatin/triazole conjugates that induce apoptosis and inhibit migration of MGC-803 cells. Eur. J. Med. Chem. 2016;124:350–360. doi: 10.1016/j.ejmech.2016.08.065. [DOI] [PubMed] [Google Scholar]
- 2.V K.L., L J.M., Marie Ranson S.G.P., Bremner J.B. An investigation into the cytotoxicity and mode of action of some novel N-Alkyl-Substituted isatins. J. Med. Chem. 2007;50:5109–5117. doi: 10.1021/jm0704189. [DOI] [PubMed] [Google Scholar]
- 3.Pakravan P., Kashanian S., Khodaei M.M., Harding F.J. Biochemical and pharmacological characterization of isatin and its derivatives: from structure to activity. Pharmacol. Rep. 2013;65:313–335. doi: 10.1016/s1734-1140(13)71007-7. [DOI] [PubMed] [Google Scholar]
- 4.Shahid M., Subhan F., Ullah I., Ali G., Alam J., Shah R. Beneficial effects of Bacopa monnieri extract on opioid induced toxicity. Heliyon. 2016;2 doi: 10.1016/j.heliyon.2016.e00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pandeya S.N., Raja A.S., Stables J.P. Synthesis of isatin semicarbazones as novel anticonvulsants–role of hydrogen bonding, Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences. Societe canadienne des sciences pharmaceutiques. 2002;5:266. [PubMed] [Google Scholar]
- 6.Selvam P., Murugesh N., Chandramohan M., Debyser Z., Witvrouw M. Design, synthesis and antiHIV activity of novel isatine-sulphonamides. Indian J. Pharmaceut. Sci. 2008;70:779–782. doi: 10.4103/0250-474X.49121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Socca E.A., Luizferreira A., de Faria F.M., de Almeida A.C., Dunder R.J., Manzo L.P., Brito A.R. Inhibition of tumor necrosis factor-alpha and cyclooxigenase-2 by Isatin: a molecular mechanism of protection against TNBS-induced colitis in rats. Chem. Biol. Interact. 2014;209:48. doi: 10.1016/j.cbi.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Vine K.L., Locke J.M., Ranson M., Benkendorff K., Pyne S.G., Bremner J.B. Corrigendum to “In vitro cytotoxicity evaluation of some substituted isatin derivatives”. Bioorg. Med. Chem. 2007;15:931–938. doi: 10.1016/j.bmc.2006.10.035. [DOI] [PubMed] [Google Scholar]
- 9.Wang L., Bao B.B., Song G.Q., Chen C., Zhang X.M., Lu W., Wang Z., Cai Y., Li S., Fu S. Discovery of unsymmetrical aromatic disulfides as novel inhibitors of SARS-CoV main protease: chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. Eur. J. Med. Chem. 2017;137:450. doi: 10.1016/j.ejmech.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinezmayorga K., Byler K.G., Ramirezhernandez A.I., Terrazasalvares D.E. Cruzain inhibitors: efforts made, current leads and a structural outlook of new hits. Drug Discov. Today. 2015;20:890–898. doi: 10.1016/j.drudis.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Limpachayaporn P., Schã¤Fers M., Schober O., Kopka K., Haufe G. Synthesis of new fluorinated, 2-substituted 5-pyrrolidinylsulfonyl isatin derivatives as caspase-3 and caspase-7 inhibitors: nonradioactive counterparts of putative PET-compatible apoptosis imaging agents. Bioorg. Med. Chem. 2013;21:2025–2036. doi: 10.1016/j.bmc.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 12.Liu W., Zhu H.M., Niu G.J., Shi E.Z., Chen J., Sun B., Chen W.Q., Zhou H.G., Yang C. Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2014;22:292–302. doi: 10.1016/j.bmc.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prakash C.R., Raja S. Design, synthesis and antiepileptic properties of novel 1-(substituted benzylidene)-3-(1-(morpholino/piperidino methyl)-2,3-dioxoindolin-5-yl)urea derivatives. Eur. J. Med. Chem. 2011;46:6057–6065. doi: 10.1016/j.ejmech.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 14.Teng Y.O., Zhao H.Y., Jing W., Liu H., Gao M.L., Yao Z., Han K.L., Fan Z.C., Zhang Y.M., Hua S. Synthesis and anti-cancer activity evaluation of 5-(2-carboxyethenyl)-isatin derivatives. Eur. J. Med. Chem. 2016;112:145. doi: 10.1016/j.ejmech.2015.12.050. [DOI] [PubMed] [Google Scholar]
- 15.Kaila N., Janz K., Huang A., Moretto A., Debernardo S., Bedard P.W., Tam S., Clerin V., J K., Jr., Tsao D.H. 2-(4-Chlorobenzyl)-3-hydroxy-7,8,9,10-tetrahydrobenzo[H]quinoline-4-carboxylic acid (PSI-697): identification of a clinical candidate from the quinoline salicylic acid series of P-selectin antagonists. J. Med. Chem. 2007;50:40–64. doi: 10.1021/jm060631p. [DOI] [PubMed] [Google Scholar]
- 16.Gérard A.L., Lisowski V., Rault S. Direct synthesis of new arylanthranilic acids via a Suzuki cross-coupling reaction from iodoisatins. ChemInform. 2005;61:6082–6087. [Google Scholar]
- 17.Chung T.W., Kim S.J., Choi H.J., Kwak C.H., Song K.H., Suh S.J., Kim K.J., Ha K.T., Park Y.G., Chang Y.C. CAPE suppresses VEGFR-2 activation, and tumor neovascularization and growth. J. Mol. Med. (Berl.) 2013;91:271. doi: 10.1007/s00109-012-0952-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MOL file. ZIP file containing the MOL files of the most important compounds in this article.