

Highlights
-
•
Immunization with VSV-based vaccine is able to generate strong mucosal immunity.
-
•
VSV-based vaccine can induce significant higher polyfunctional T cells.
-
•
VSV-based vaccine significantly alleviates CVB3-induced viral myocarditis.
Keywords: Coxsackievirus B3, Vesicular stomatitis virus, Viral myocarditis, Dendritic cell, Polyfunctional T cells, Mucosal vaccine
Abstract
Recombinant vesicular stomatitis virus (VSV) is widely used as a vaccine platform. However, the capacity of VSV-based vaccines to induce mucosal immunity has not been fully investigated. In the present study, a recombinant VSV expressing coxsackievirus B3 (CVB3) major immunogen VP1 has been generated and the immune protection elicited by VSV-VP1 was evaluated. We demonstrated that intranasal delivery of VSV-VP1 can induce a potent antigen-specific mucosal immune response as well as a systemic immune response, particularly the induction of polyfunctional T cells. Importantly, mice immunized with VSV-VP1 were better protected against CVB3-induced viral myocarditis than those receiving a chitosan-formulated DNA vaccine. Increased dendritic cell (DC) maturation in the mesenteric lymph node (MLN) was observed in the mice vaccinated with VSV-VP1, which could be a potential mechanism for the protective immune response. These findings support VSV as a viral delivery vector that can induce robust mucosal immunity that should be considered for further vaccine development.
1. Introduction
Viral myocarditis (VMC) is induced by viral infection and often progresses to chronic myocarditis with persisting myocardial inflammation [1], [2]. Patients with severe cardiac dysfunction may also develop dilated cardiomyopathy [3], which is one of the most frequent reasons for heart transplantation. Although many different infectious agents have been attributed as the cause of viral myocarditis, the enterovirus coxsackievirus B3 (CVB3) is considered to be the most common cause of heart disease in adults and children [4], [5].
To date, many potential vaccines to prevent acute infection of myocarditis have been tested in animal models, including inactivated virus vaccines, attenuated virus vaccines, recombinant protein vaccines, DNA vaccines, and virus-like particle (VLP) vaccines [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16]. Intramuscular immunization of mice with a DNA vaccine was reported to induce variable humoral and cellular immunities, resulting in the protection of mice against lethal CVB3 infection [6], [7], [8]. Although these studies are very promising, no effective clinical vaccine against CVB3 is currently available. Hence, there is still an urgent need to develop new vaccines to protect against CVB3 infection.
Previous studies have demonstrated the importance of both humoral and cellular immune responses for protection from CVB3. Agammaglobulinemic individuals are particularly susceptible to CVB3-associated encephalitis, and mice lacking B cells develop a chronic infection and remain viremic for at least 2 months; viremia can be alleviated by the adoptive transfer of B cells from CVB3-immune wild-type mice [17], [18], [19]. Reports have also highlighted the importance of cellular immunity against CVB3-induced viral myocarditis. Comparison of C57BL/6 mice (resistant to chronic CVB3 myocarditis) and A.BY/SnJ mice (susceptible to chronic CVB3 myocarditis) found that a failure to clear the virus may be attributed to defects in dendritic cell (DC) maturation/activation and MHC class I antigen processing [20]. The neutralization of interleukin 17 (IL-17) increases both CD4+ and CD8+ T cells, which ameliorates the pathological cardiac changes and reduces viral replication [21].
As a member of enteroviruses, CVB3 invades host through gastrointestinal tract [5]. Thus, it is important for a CVB3 vaccine to produce mucosal immunity at the sites of viral transmission to block the initial infection as well as the induction of potent systemic immune responses. A multitude of vehicles, including microparticles, liposomes, and attenuated pathogens, have been developed as alternative platforms for mucosal delivery. Vesicular stomatitis virus (VSV), a viral vaccine vector, has been shown to be an excellent vector to deliver foreign antigens [22], [23]. Using reverse genetics, an exogenous gene can be inserted into the VSV genome and recombinant VSV can be recovered entirely from an infectious cDNA clone [24], [25]. The exogenous antigen is expressed continuously in vivo once the recombinant viruses are inoculated into animals, and thus it triggers specific immune responses. Recombinant VSV has been successfully developed for a number of vaccine candidates, such as human immunodeficiency virus [26], [27], [28], [29], severe acute respiratory syndrome virus [30], [31], hepatitis C virus [32], [33], hepatitis B virus [34], influenza virus [35], papillomavirus [36], human respiratory syncytial virus [37], poxvirus [38], Ebola virus, and Marburg virus [39], [40]. The VSV-based HIV vaccine candidate has been so successful in early studies that it has recently been moved to clinical study through the HIV Vaccine Design and Development Teams program [41]. Collectively, these studies have demonstrated the great potential of VSV-based vaccines to trigger strong immunity even after a single immunizing dose. In spite of this, the capacity of VSV-based vaccines to induce mucosal immunity has not yet been fully characterized.
We previously reported that a mucosal vaccine chito-pcDNA3.1-VP1, a DNA plasmid pcDNA3.1-VP1 encapsulated in a chitosan particle, protected mice against CVB3-induced viral myocarditis [14]. A moderate level of mucosal IgA and an IFN-γ+ T cell immune response were induced by intranasal immunization with chito-pcDNA3.1-VP1. To develop a more efficient vaccine, in the present study we have applied a VSV viral vector as a mucosal delivery system to express the main antigenic protein for CVB3 VP1. We evaluated the potential of this VSV-based vaccine to induce antigen-specific mucosal and systemic immunity in a viral myocarditis mouse model.
2. Materials and methods
The coding region of CVB3 (Nancy strain) major capsid protein VP1 was amplified by PCR from pcDNA3.1-VP1 [14] and cloned into the G–L junction of the pVSVXN2 plasmid. Recombinant VSV containing VP1 (VSV-VP1) was recovered as previously described [22]. Six-week-old male BALB/c mice were purchased from the experimental animal center of Chinese Academy of Science (Shanghai, China) and remained in pathogen-free conditions and housed at the Soochow University School of Medicine animal facilities. All animal experiments were performed according to the guidelines for the Care and Use of Laboratory Animals (Ministry of Health, China, 1998) and the guidelines of the Laboratory Animal Ethical Commission of Soochow University. Mice were lightly anesthetized by injecting 0.75% pentobarbital sodium into cavum abdominis (30 mg/kg) prior to all immunizations. Each group contained 6 mice. For VSV-VP1 or VSV–GFP immunization, single dose of 106 plaque-forming units (PFU) viruses in a 25 μl volume was intranasally administered. The group of mice receiving PBS alone was used as control. For chitosan–DNA immunization, each group of mice were mildly intranasally immunized with chitosan encapsulated 50 μg pcDNA3.1-VP1 (chito-pcDNA3.1-VP1) or 50 μg pcDNA3.1 (chito-pcDNA3.1) for 4 times biweekly. The detection of antibody response, lymphocyte proliferation assay, CTL activity assay, measurement of dendirtic cell maturation et al. as described previously [42]. All data are given as mean ± SD. Statistical analysis of the data was performed with the two-tailed independent Student's t-test using GraphPad Prism version 4.01 (GraphPad Software Incorporated). P < 0.05 was considered statistically significant. Full detailed methods are provided in Supplemental materials and methods.
3. Results
3.1. Construction and identification of the recombinant VSV based vaccines
The coding region of CVB3 main antigenic protein VP1 was amplified by PCR and cloned into pVSVXN2 plasmid at the G–L junction in the VSV genome by XhoI and NheI restriction enzymes as indicated in Fig. 1A. A VSV–GFP recombinant virus was generated as a control. We then recovered the recombinant viruses in BHK-21 cells as previously reported [24] and the recombinant viruses were purified by plaque assay. Immunofluorescence staining of VP1 indicated that VP1 was efficiently expressed in the cytoplasmic region of VSV-VP1-infected BHK-21 cells (Fig. 1B). We also detected the expected green fluorescence in VSV–GFP infected cells. A western blot further confirmed the expression of VP1 as well as the expression of VSV glycoprotein VSVG in infected cells (Fig. 1C). These results demonstrate the successful generation of recombinant virus VSV-VP1 expressing CVB3 antigen VP1.
Fig. 1.
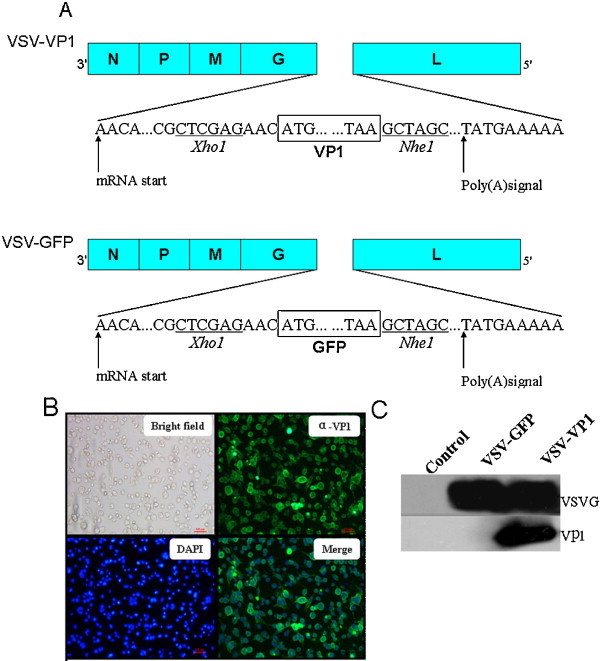
Characterization of VP1 expression from recombinant VSV. (A) The constructions of VSV-VP1 and VSV–GFP. Gene encoding CVB3 major antigen VP1 or GFP was inserted in the fifth position of the VSV genome. (B) The immunofluorescence detection of recombinant VSV infected BHK-21 cells. Six hours post infection, BHK-21 cells were fixed with 4% paraformaldehyde prior to permeabilization and staining. VP1 localization was detected using primary anti-CVB3 Vp1 antibody and secondary Alexa Fluor 488-labeled goat anti-mouse antibodies. (C) BHK-21 cells were harvested 6 h post infection with VSV–GFP or VSV-VP1 and assayed for Vp1 or VSV-G expression by Western blot using anti-CVB3 VP1 monoclonal antibody or anti-VSV-G polyclonal antibody.
3.2. The humoral response generated by VSV-VP1 vaccination
Administration of vaccines at mucosal surfaces has been shown to elicit an adequate local immune response at the administration site and at distant mucosal sites, as well as a systemic immune response [43]. Thus, groups of mice were intranasally immunized with VSV-VP1, VSV–GFP, chito-pcDNA3.1-VP1, chito-pcDNA3.1 and PBS described in Section 2. We monitored the weight change of immunized mice to assess the effects of viral infection. As previously reported [44], mice inoculated with 106 PFU of either VSV-VP1 or VSV–GFP demonstrated severe weight loss as well as other clinical symptoms such as tremors, paralysis, and circling within the 6 days post inoculation but recovery was noted thereafter (data not shown). Meanwhile, the weight of mice immunized with chito-pcDNA3.1-VP1, chito-pcDNA3.1 and PBS steadily increased, and were not significantly different from those of VSV-VP1 and VSV–GFP groups 2 weeks post inoculation.
To analyze the antibody response in the immunized mice, blood samples were collected from each mouse and the serum IgG antibody response was determined by ELISA at indicated time points. As shown in Fig. 2A mice immunized with a single dose of VSV-VP1 exhibited a high serum IgG response compared with that of mice immunized with VSV–GFP and it peaked at week 4 post inoculation with a drop in the following weeks. The serum IgG level in mice receiving chito-pcDNA3.1-VP1 gradually increased with three additional boosts and reached a peak at week 8. Interestingly, although the trend of fecal secretory IgA (sIgA) generated by VSV-VP1 was similar to that for serum IgG, its production was faster and higher than that in chito-pcDNA3.1-VP1 immunized mice (Fig. 2B). These results indicate that intranasal immunization with VSV-VP1 can efficiently induce both mucosal and systemic antibody responses.
Fig. 2.
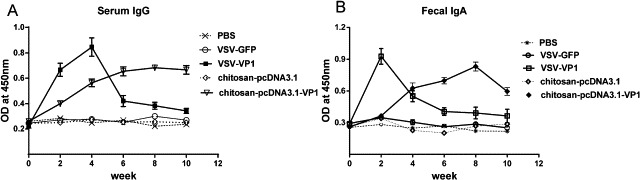
CVB3-specific antibody responses elicited by intranasal immunization with various groups of VSV-VP1, VSV–GFP, chito-pcDNA3.1-VP1, chito-pcDNA3.1 or PBS administration. Mice were immunized with four doses of various DNA vaccines at 2 weeks intervals, while the two VSV-based vaccine groups were immunized only with a single dose of 106 PFU viruses. The levels of CVB3-specific serum IgG(A) and fecal sIgA (B) at 2-week intervals. The results are expressed as the mean ± SD from three separate experiments.
3.3. Vaccination of mice with VSV-VP1 elicits strong cellular immunity
We next investigated the CVB3-specific T cell immune responses in the spleen and mesenteric lymph node (MLN) induced by VSV-VP1 immunization. As shown in Fig. 3A, compared with mice given PBS or VSV–GFP, mice immunized with VSV-VP1 displayed a significant increase in specific T cell proliferation in both the spleen and MLN as measured by BrdU assay (P < 0.001). This proliferation level was even higher than that of mice immunized with chito-pcDNA3.1-VP1 (P < 0.05).
Fig. 3.
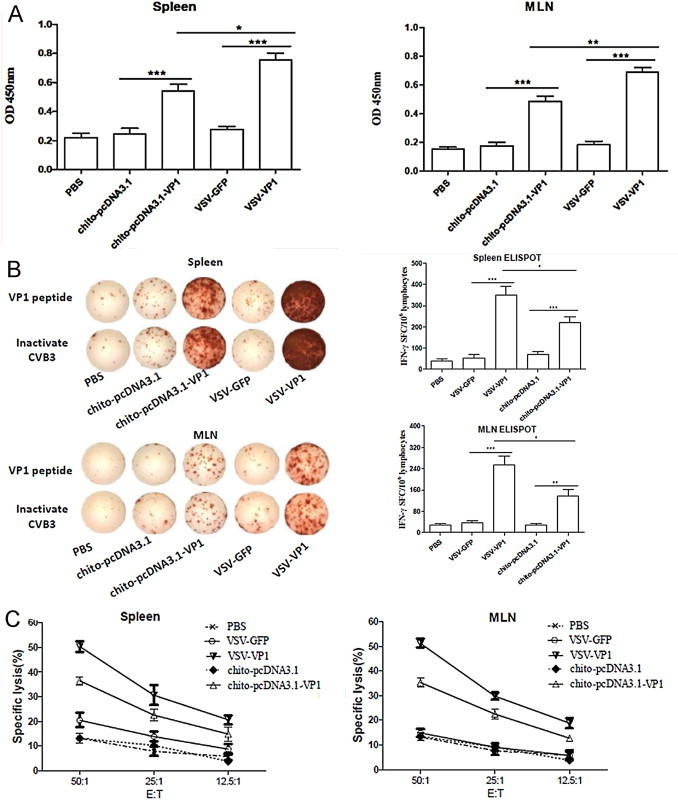
CVB3-specific T cell response elicited by intranasal immunization with VSV-based vaccine or DNA in chitosan formulations. Two weeks after final DNA immunization, spleen and MLN cells were collected from immunized mice and expanded in vitro with VP1237–249 peptide. (A) CVB3-specific T cell proliferation was assessed by Roche BrdU-Kit after stimulation with 20 μg/ml VP1237–249 peptide in the culture of 20 U/ml IL-2 for 72 h. (B) CVB3-specific cytokine-secreting lymphocytes were quantified by ELISPOT assay in response to CVB3 VP1237–249 peptide or inactivated CVB3 for 48 h. (C) CVB3-specific CTL activity of splenic and mesenteric cells was evaluated by lactate dehydrogenase assays using pcDNA3.1-VP1 stable-transfected autologous SP2/0 cells as target cells. The effector/target cell ratio was between 50:1 and 12.5:1. Results are represented as the mean ± SD (n = 6) of three separate experiments.*P < 0.05, **P < 0.01, ***P < 0.001.
An IFN-γ ELISPOT analysis indicated that mice immunized with VSV-VP1 developed the most IFN-γ-secreting T cells in the spleen and MLN, with frequencies reaching 351 and 255 spot-forming cells (SFC)/106 cells, respectively (Fig. 3B; P < 0.05). Mice immunized with chito-pcDNA3.1-VP1 produced a modest amount of IFN-γ secreting T cells compared with control groups. In agreement with the ELISPOT data, the strongest CVB3-specific cytotoxicity responses were also detected in the splenic (50.3%) and mesenteric lymphocytes (51.1%) derived from mice immunized with VSV-VP1 at an E:T ratio of 50:1 (Fig. 3C). Taken together, these data indicate that in addition to the previously reported VSV-induced specific systemic T cell immune responses against other viral pathogens, VSV-based vaccines can also induce a strong mucosal antigen-specific T cell immune response.
3.4. VSV-VP1 vaccination significantly alleviates CVB3-induced viral myocarditis
To determine if VSV-VP1 immunization has protective effects, 2 weeks after the final DNA vaccine immunization, all of groups mice were infected with a 50% lethal dose (LD50) of 3 of CVB3 to induce acute myocarditis. Seven days post-viral infection, mice in the groups vaccinated with PBS, VSV–GFP or chito-pcDNA3.1 suffered from loss of body weight, up to approximately 17.9% of total weight (Fig. 4A), indicating the development of myocarditis in these mice. Detection of serum creatine kinase (CK) and CK-MB activity reflects the amount of cardiac damage. Immunization with VSV-VP1 significantly reduced the serum CK and CK-MB activity (Fig. 4B and C). For example, CK-MB in the VSV-VP1 immunized group was only 631.3 IU/L, while it was 1420.0 IU/L in the VSV–GFP immunized group. Additionally, the viral titer of heart tissue in the VSV-VP1 immunized group was at least 250-fold lower than that of the VSV–GFP immunized group (Fig. 4D). Histology analysis of hematoxylin and eosin (HE)-stained heart sections showed a large amount of myocyte necrosis and immune-infiltration in the control (PBS), VSV–GFP and chito-pcDNA3.1 immunized groups. Only a tiny amount of inflammation was seen in the VSV-VP1 immunized group, suggesting that this vaccination prevented the development of myocarditis (Fig. 4E). Similar results were also observed in HE-stained pancreas sections (Fig. S2).
Fig. 4.
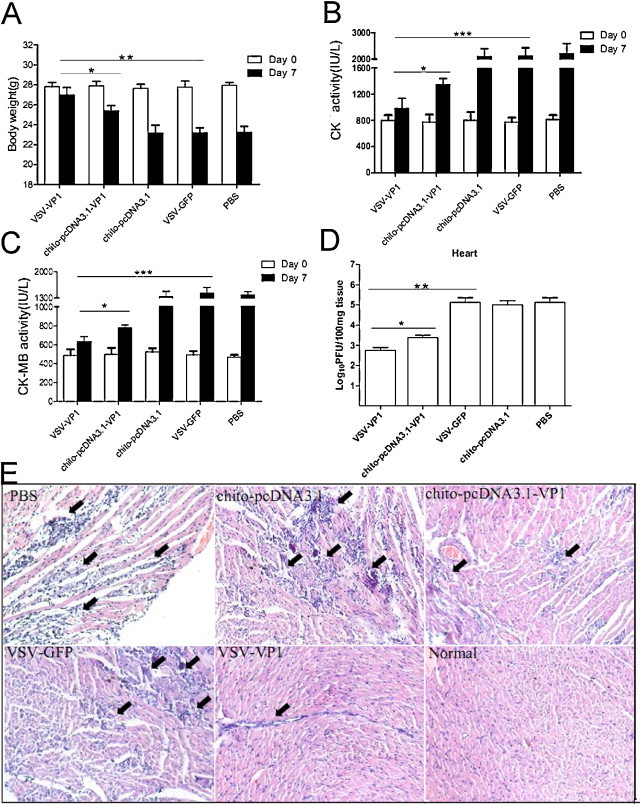
Prevention of CVB3 myocarditis and protection against CVB3 challenge by intranasal immunization with VSV-based vaccine or DNA in chitosan formulations. Seven days after 3LD50 CVB3 challenge, (A) body weight loss, (B) serum CK, (C) CK-MB, (D) Viral titers in heart and (E) inflammation of HE-stained sections were measured as indicated by the arrows. The results are represented as the mean ± SD (n = 6) of three separate experiments.*P < 0.05; **P < 0.01; ***P < 0.001.
In vivo ventricular systolic function was measured by fractional shortening (FS) and ejection fraction (EF) using an echocardiography assay (Fig. 5A). The left ventricular FS (LVFS) in chito-pcDNA3.1-VP1 immunized mice was ∼12% lower compared with that in the VSV-VP1 immunized group. When left ventricular EF (LVEF) was calculated, it was ∼10% lower in chito-pcDNA3.1-VP1 immunized mice compared with that in the VSV-VP1 immunized group (Fig. 5B), indicating that in vivo ventricular function after infection was improved by prior immunization with VSV-VP1.
Fig. 5.
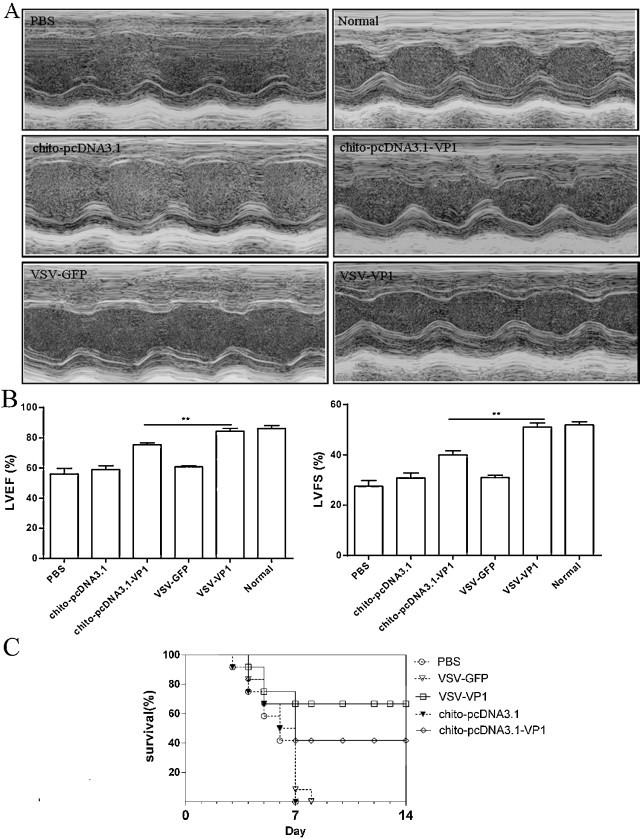
Cardiac function was detected by echocardiography using a 2-dimensional guided M-mode ultrasound system in each group by 3LD50 CVB3 challenge 7 days after infection. (A) Representative M-mode echocardiograms for each group (B) VSV-VP1 immunization significantly improved the cardiac systolic function against CVB3 induced myocarditis as evidenced by reduced left ventricular fraction shortening and ejection fraction from echocardiographic data. Values were presented as mean ± SD; n = 6 for each group; **P < 0.01. (C) The survival rate of mice was observed for 14 days following 5LD50 infection. Individual experiment was conducted three times with 12 mice per group and one representative data was shown.
To further evaluate the protective abilities of the vaccine candidates, mice were challenged with 5 LD50 of CVB3 2 weeks after the final DNA immunization. All mock-immunized mice succumbed to severe illness and died within 8 days, while 41.67% (5/12) of the chito-pcDNA3.1-VP1 immunized mice and 66.67% (8/12) of the VSV-VP1 immunized mice survived to 14 days with no signs of illness (Fig. 5C), indicating that nasal immunization with VSV-VP1 provides some protection against CVB3-induced myocarditis.
3.5. VSV-VP1 immunization promotes dendritic cell (DC) maturation and increases frequency of polyfunctional T cells
As DC play an important role in the induction of the adaptive immune response, we further examined the maturation of MLN DC in the immunized mice using flow cytometry. Two weeks after final immunization, MLN cells were isolated and stained by maturation makers MHCII, CD40, CD80 or CD86 together with CD11c, which is a characteristic marker of myeloid-derived DC. Few DC fully maturated in the VSV–GFP, chito-pCDNA3.1 and PBS immunized groups. Intranasal immunization with chito-pcDNA3.1-VP1 mildly promoted DC maturation compared with the levels of maturation seen for the VSV-VP1 immunized group, in which the cell surface expressions of MHCII, CD40, CD80 or CD86 molecules were efficiently up-regulated (Fig. 6A; P < 0.05).
Fig. 6.
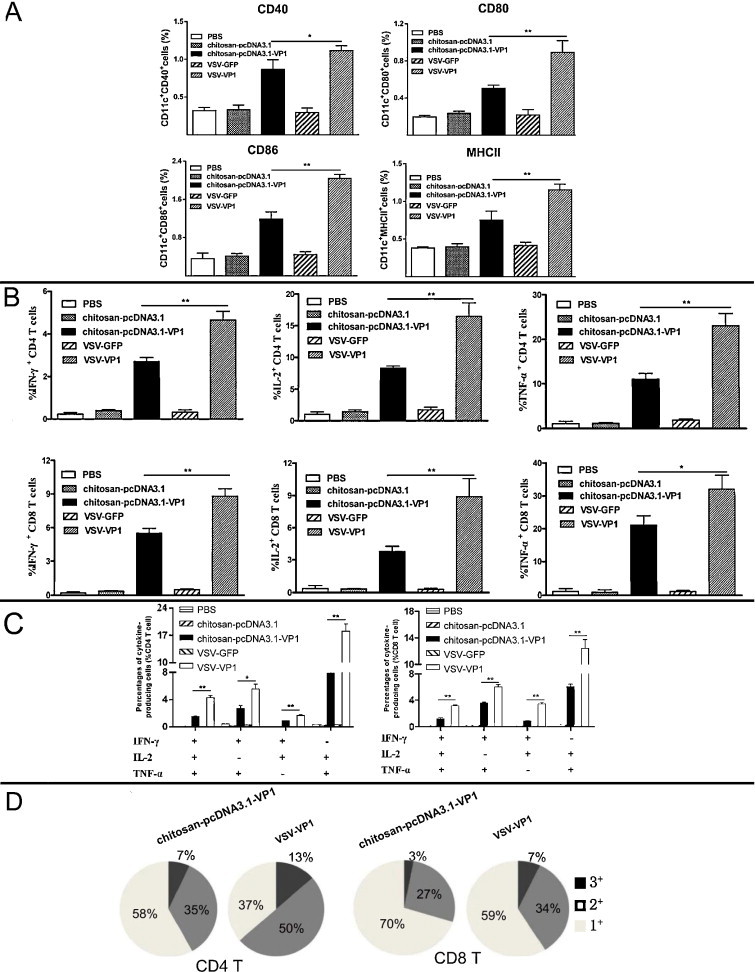
VSV-VP1 immunization promoted MLN DCs maturation and increased proportion of polyfunctional T cells. (A) DC maturation markers CD40, CD80, CD86 and MHCII were stained and determined by flow cytometry from MLN. (B) The frequency of CD4+ and CD8+ T cells producing IFN-γ and IL-2 or TNF-α alone was analyzed after stimulation for 6 h. (C) Frequency of CD4+ and CD8+ T cells expressing two or three of IFN-γ, IL-2 and TNF-α was analyzed. (D) Proportion of total triple, double and single positive IFN-γ, IL-2 and TNF-α producing CD4+ and CD8+ T cells was shown in pie charts. The results were expressed as the mean ± SD (n = 6) from three independent experiments. *P < 0.05, **P < 0.01.
To further explore the consequences of DC maturation, we next measured the development of polyfunctional T cells. Previous reports have shown that T cells with multiple effector functions, such as the concomitant production of IFN-γ, TNF-α, and IL-2, are functionally superior to their single cytokine-producing counterparts for the clearance of viral pathogens [45], [46]. Therefore, we evaluated the phenotype and proportion of polyfunctional T cells in immunized mice using intracellular cytokine staining.
Multiple T cell populations could be delineated based on the expression of IFN-γ, IL-2 and TNF-α. CD4+ or CD8+ T population was first gated, and the expression levels of IFN-γ, TNF-α, and IL-2 were further analyzed. Single-positive CD4+ or CD8+ T cells expressing IFN-γ, TNF-α, or IL-2 was significantly increased in the VSV-VP1 immunization group (4.64% for CD4+IFN-γ+, 16.48% for CD4+IL-2+ and 23.04% for CD4+TNF-α+) upon VP1 peptide stimulation and was significantly higher than those in the chito-pcDNA3.1-VP1 immunization group. T cells expressing these cytokines were barely detectable in the control immunization groups: VSV–GFP, chito-pcDNA3.1 and PBS (Fig. 6B; P < 0.05). To assess whether VSV-VP1-induced T cells were producing more than one of these cytokines, we next examined the co-expression patterns of IFN-γ, TNF-α, and IL-2. Analysis of IFN-γ+TNF-α+, IFN-γ+IL-2+, and TNF-α+IL-2+ double-positive CD4+ T cells revealed that the proportion of IFN-γ+IL-2+ cells was the lowest (1.69% for the VSV-VP1 immunized group and 0.88% for the chito-pcDNA3.1-VP1immunized group). TNF-α+IL-2+ double-positive T cells were the most abundant, reaching 18.10% for the VSV-VP1 immunized group and 7.92% for the chito-pcDNA3.1-VP1 immunized group. The percentage of the IFN-γ+TNF-α+IL-2+ triple-positive polyfunctional cells was higher than that of the IFN-γ+TNF-α+ double-positive cells (4.28% for the VSV-VP1 immunized group and 1.52% for the chito-pcDNA3.1-VP1 immunized group). Similar levels of IFN-γ, TNF-α, and IL-2 expression were seen in CD8+ T cells (Fig. 6C). Analysis of the proportions of the single-, double-, or triple-positive T cells in the VSV-VP1 and chito-pcDNA3.1-VP1 immunization groups indicated that VSV-VP1 immunization can enhance the magnitude of polyfunctional CD4+ and CD8+ T cells as detected by increased double-, or triple-positive populations (Fig. 6D).
4. Discussion
In the present study, we generated a VSV-based vaccine encoding CVB3 major antigenic protein VP1. Although VSV-based vectors have been widely used as vaccine platforms, few studies have characterized the capacity of VSV-based vaccines to induce potent mucosal immunity. We compared the antigen-specific mucosal immunity elicited by intranasal administration of a VSV-VP1 vaccine with that of a chitosan-based mucosal vaccine, chitosan-pcDNA3.1-VP1 [14]. With a single-dose intranasal delivery of VSV-VP1, mice exhibited significantly higher levels of both mucosal and systemic immune responses than mice vaccinated with chito-pcDNA3.1-VP1. This improvement in multiple aspects of immunity is one of the most important virtues of mucosal vaccination [43]. While the peak levels of antigen-specific fecal sIgA in the VSV-VP1 and chito-pcDNA3.1-VP1 groups were similar, the induction of antigen-specific sIgA in the VSV-VP1 immunized group was much faster than that in the chito-pcDNA3.1-VP1 immunized group. Since the same antigen, VP1, was used for both delivery systems in this study, these results demonstrate that the VSV-based vector is more potent at inducing mucosal immunity than the chitosan-based delivery system. The increased protection against viral myocarditis in VSV-VP1 immunized mice may result from the elevated humoral response as well as the cellular immunity comparing to that of chito-pcDNA3.1-VP1.
Oral delivery is a simple alternate way to induce mucosal immunity. Ma et al. found that a VSV-based vaccine against human norovirus administrated with the combination of intranasal and oral delivery could induce antigen-specific IgA in both the gastrointestinal tract and vagina [47]. Based on their success with oral delivery, it would likely be worthwhile to investigate the capacity of VSV-VP1 vaccination to induce mucosal and systemic immunity by oral delivery, alone or in combination with intranasal delivery.
Several features make VSV an ideal CVB3 vaccine delivery mechanism. First, VSV can induce strong mucosal immunity by intranasal or oral immunization. Because the gastrointestinal tract is the main route through which CVB3 invades the host, it is critical to induce mucosal immune responses that can block virus infection at the initial stage and the primary site. Second, being a virus, VSV can induce strong cellular immunity to compensate the impaired cellular immunity after CVB3 infection. Kemball et al. found that CVB3 infection rapidly induces phenotypic changes in conventional DC, and markedly impairs specific CD8+ T cell antigen presentation through MHCI pathway in vivo [48], [49]. Third, the VSV reverse genetics system is well characterized and easy to manipulate [24], [25]. High titers of recombinant VSV can be achieved by propagating virus in mammalian cell lines approved for vaccine production. Compared with other potential vaccine platforms such as baculovirus, the VSV system is faster and less expensive. Finally, VSV vaccines can be administered needle-free. This is especially valuable for people who live in rural regions that are short of health care.
The polyfunctional T cells induced by immunization with VSV-VP1 are an important component of the robust immune response induced by this vaccine. Numerous studies have shown that the quality, rather than the level, of T cell responses is crucial for determining the outcome of various infections [45], [46], [50]. For HCV, it was reported that the resolution obtained under early interferon therapy correlated with the development of polyfunctional IFN-γ/IL-2/CD107α+ specific CD8+ T cells [51]. While the chito-pcDNA3.1-VP1 vaccine could induce some polyfunctional T cells (1.52% triple-positive IFN-γ+TNF-α+IL-2+ CD4+ T cells), immunization with VSV-VP1 resulted in the striking increase of this population up to 4.28%. In addition, the VSV-VP1 vaccine induced up to 18.10% double-positive TNF-α+IL-2+ CD4+ T cells. Based on previous studies, we speculate that these polyfunctional T cells could establish long-lasting immunological memory and will likely give rise to a strong and robust secondary response upon challenge with CVB3.
DC play a central role in priming immune responses and are therefore of interest for vaccine capacity. The immunological paradigm holds that DC capture antigens, while inflammatory signals trigger their maturation and migration to the draining lymph nodes to initiate an antigen-specific immune response. Viral vectors, such as recombinant adenovirus 35, that can efficiently infect and cause maturation of DC in vivo induce optimal vaccine responses [52]. As previously described in vitro for BMDC [53], [54], we observed that VSV infection significantly improved the maturation of MLN DC through the up-regulation of different molecules 2 weeks post-final immunization. One reason for this might be that the stimulation caused by VSV infection is stronger and lasts longer than that caused by chitosan-incorporated DNA at the sites of immunization. The increased maturation of MLN DC in VSV-VP1-immunized mice likely results from the sustained migration and activation of peripheral DC from the inoculation site. Additional experiments are ongoing to address this hypothesis.
This study characterized the potency of a VSV-based vaccine vector for the induction of mucosal immunity. Vaccination with VSV-VP1 promoted mucosal DC maturation, enhanced antigen-specific mucosal immunity as well as systemic immunity, and could alleviate CVB3-induced viral myocarditis. Our study might also provide insight into the importance of mucosal immunity for the further development of VSV-based vaccines.
Acknowledgments
The authors would like to thank Prof. John K Rose at Yale University for providing the VSV reverse genetic system. The authors also thank Dr. Bernard Moss at NIH for providing vTF7-3 vaccinia virus expressing T7 RNA polymerase and Dr. Renjing Liu for critical proof reading. This work was supported by grants from Major State Basic Research Development Program of China, Ministry of Science and Technology of China (2013CB531502, 2013CB530501), the National Natural Science Foundation of China (31270977, 81072413, 81101257, 31170878), The Scientific Research Foundation for Returned Overseas Chinese Scholars from State Education Ministry to CD, Jiangsu “Pan-Deng” Project, Department of Science of Technology of Jiangsu province (BK2010004), Jiangsu “333” project of cultivation of high-level talents, Qing Lan Project of the Jiangsu higher education institutions, Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Jiangsu Provincial Innovative Research Team.
Conflict of interest statement: The authors declare that there are no conflicting financial interests.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.vaccine.2014.05.052.
Contributor Information
Sidong Xiong, Email: sdxiongfd@126.com.
Chunsheng Dong, Email: chunshengdong@suda.edu.cn.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- 1.Feldman A.M., McNamara D. Myocarditis. N Engl J Med. 2000;343:1388–1398. doi: 10.1056/NEJM200011093431908. [DOI] [PubMed] [Google Scholar]
- 2.Woodruff J.F. Viral myocarditis: a review. Am J Pathol. 1980;101:425–484. [PMC free article] [PubMed] [Google Scholar]
- 3.Pauschinger M., Phan M.D., Doerner A., Kuehl U., Schwimmbeck P.L., Poller W. Enteroviral RNA replication in the myocardium of patients with left ventricular dysfunction and clinically suspected myocarditis. Circulation. 1999;99:889–895. doi: 10.1161/01.cir.99.7.889. [DOI] [PubMed] [Google Scholar]
- 4.Tam P.E. Coxsackievirus myocarditis: interplay between virus and host in the pathogenesis of heart disease. Viral Immunol. 2006;19:133–146. doi: 10.1089/vim.2006.19.133. [DOI] [PubMed] [Google Scholar]
- 5.Racaniello V.R. Picornaviridae: the virus and their replication. In: Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Straus S.E., Martin M.A., Roizman B., editors. Fields virology. 5th ed. Lippincott Williams &Wilkins; Philadelphia, PA: 2007. [Google Scholar]
- 6.Henke A., Wagner E., Whitton J.L., Zell R., Stelzner A. Protection of mice against lethal coxsackievirus B3 infection by using DNA immunization. J Virol. 1998;72:8327–8331. doi: 10.1128/jvi.72.10.8327-8331.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henke A., Zell R., Stelzner A. DNA vaccine-mediated immune responses in Coxsackie virus B3-infected mice. Antiviral Res. 2001;49:49–54. doi: 10.1016/s0166-3542(00)00132-7. [DOI] [PubMed] [Google Scholar]
- 8.Kim J.Y., Jeon E.S., Lim B.K., Kim S.M., Chung S.K., Kim J.M. Immunogenicity of a DNA vaccine for coxsackievirus B3 in mice: protective effects of capsid proteins against viral challenge. Vaccine. 2005;23:1672–1679. doi: 10.1016/j.vaccine.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Werner S., Klump W.M., Schonke H., Hofschneider P.H., Kandolf R. Expression of coxsackievirus B3 capsid proteins in Escherichia coli and generation of virus-specific antisera. DNA. 1988;7:307–316. doi: 10.1089/dna.1.1988.7.307. [DOI] [PubMed] [Google Scholar]
- 10.Beck M.A., Chapman N.M., McManus B.M., Mullican J.C., Tracy S. Secondary enterovirus infection in the murine model of myocarditis. Pathologic and immunologic aspects. Am J Pathol. 1990;136:669–681. [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman N.M., Ragland A., Leser J.S., Hofling K., Willian S., Semler B.L. A group B coxsackievirus/poliovirus 5′ nontranslated region chimera can act as an attenuated vaccine strain in mice. J Virol. 2000;74:4047–4056. doi: 10.1128/jvi.74.9.4047-4056.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fohlman J., Ilback N.G., Friman G., Morein B. Vaccination of Balb/c mice against enteroviral mediated myocarditis. Vaccine. 1990;8:381–384. doi: 10.1016/0264-410x(90)90098-7. [DOI] [PubMed] [Google Scholar]
- 13.See D.M., Tilles J.G. Efficacy of a polyvalent inactivated-virus vaccine in protecting mice from infection with clinical strains of group B coxsackieviruses. Scand J Infect Dis. 1994;26:739–747. doi: 10.3109/00365549409008644. [DOI] [PubMed] [Google Scholar]
- 14.Xu W., Shen Y., Jiang Z., Wang Y., Chu Y., Xiong S. Intranasal delivery of chitosan–DNA vaccine generates mucosal SIgA and anti-CVB3 protection. Vaccine. 2004;22:3603–3612. doi: 10.1016/j.vaccine.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H., Morgan-Capner P., Latif N., Pandolfino Y.A., Fan W., Dunn M.J. Coxsackievirus B3-induced myocarditis. Characterization of stable attenuated variants that protect against infection with the cardiovirulent wild-type strain. Am J Pathol. 1997;150:2197–2207. [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L., Parham N.J., Zhang F., Aasa-Chapman M., Gould E.A., Zhang H. Vaccination with coxsackievirus B3 virus-like particles elicits humoral immune response and protects mice against myocarditis. Vaccine. 2012;30:2301–2308. doi: 10.1016/j.vaccine.2012.01.061. [DOI] [PubMed] [Google Scholar]
- 17.Hertel N.T., Pedersen F.K., Heilmann C. Coxsackie B3 virus encephalitis in a patient with agammaglobulinaemia. Eur J Pediatr. 1989;148:642–643. doi: 10.1007/BF00441520. [DOI] [PubMed] [Google Scholar]
- 18.Mena I., Perry C.M., Harkins S., Rodriguez F., Gebhard J., Whitton J.L. The role of B lymphocytes in coxsackievirus B3 infection. Am J Pathol. 1999;155:1205–1215. doi: 10.1016/S0002-9440(10)65223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geller T.J., Condie D. A case of protracted coxsackie virus meningoencephalitis in a marginally immunodeficient child treated successfully with intravenous immunoglobulin. J Neurol Sci. 1995;129:131–133. doi: 10.1016/0022-510x(94)00261-l. [DOI] [PubMed] [Google Scholar]
- 20.Rahnefeld A., Ebstein F., Albrecht N., Opitz E., Kuckelkorn U., Stangl K. Antigen-presentation capacity of dendritic cells is impaired in ongoing enterovirus myocarditis. Eur J Immunol. 2011;41:2774–2781. doi: 10.1002/eji.201041039. [DOI] [PubMed] [Google Scholar]
- 21.Yuan J., Yu M., Lin Q.W., Cao A.L., Yu X., Dong J.H. Th17 cells contribute to viral replication in coxsackievirus B3-induced acute viral myocarditis. J Immunol. 2010;185:4004–4010. doi: 10.4049/jimmunol.1001718. [DOI] [PubMed] [Google Scholar]
- 22.Lichty B.D., Power A.T., Stojdl D.F., Bell J.C. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10:210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Roberts A., Buonocore L., Price R., Forman J., Rose J.K. Attenuated vesicular stomatitis viruses as vaccine vectors. J Virol. 1999;73:3723–3732. doi: 10.1128/jvi.73.5.3723-3732.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawson N.D., Stillman E.A., Whitt M.A., Rose J.K. Recombinant vesicular stomatitis viruses from DNA. Proc Nat Acad Sci USA. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whelan S.P., Ball L.A., Barr J.N., Wertz G.T. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Nat Acad Sci USA. 1995;92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haglund K., Leiner I., Kerksiek K., Buonocore L., Pamer E., Rose J.K. High-level primary CD8(+) T-cell response to human immunodeficiency virus type 1 gag and env generated by vaccination with recombinant vesicular stomatitis viruses. J Virol. 2002;76:2730–2738. doi: 10.1128/JVI.76.6.2730-2738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson J.E., Schnell M.J., Buonocore L., Rose J.K. Specific targeting to CD4+ cells of recombinant vesicular stomatitis viruses encoding human immunodeficiency virus envelope proteins. J Virol. 1997;71:5060–5068. doi: 10.1128/jvi.71.7.5060-5068.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose N.F., Marx P.A., Luckay A., Nixon D.F., Moretto W.J., Donahoe S.M. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell. 2001;106:539–549. doi: 10.1016/s0092-8674(01)00482-2. [DOI] [PubMed] [Google Scholar]
- 29.Tan G.S., McKenna P.M., Koser M.L., McLinden R., Kim J.H., McGettigan J.P. Strong cellular and humoral anti-HIV Env immune responses induced by a heterologous rhabdoviral prime-boost approach. Virology. 2005;331:82–93. doi: 10.1016/j.virol.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Faber M., Lamirande E.W., Roberts A., Rice A.B., Koprowski H., Dietzschold B. A single immunization with a rhabdovirus-based vector expressing severe acute respiratory syndrome coronavirus (SARS-CoV) S protein results in the production of high levels of SARS-CoV-neutralizing antibodies. J Gen Virol. 2005;86:1435–1440. doi: 10.1099/vir.0.80844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapadia S.U., Rose J.K., Lamirande E., Vogel L., Subbarao K., Roberts A. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology. 2005;340:174–182. doi: 10.1016/j.virol.2005.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buonocore L., Blight K.J., Rice C.M., Rose J.K. Characterization of vesicular stomatitis virus recombinants that express and incorporate high levels of hepatitis C virus glycoproteins. J Virol. 2002;76:6865–6872. doi: 10.1128/JVI.76.14.6865-6872.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ezelle H.J., Markovic D., Barber G.N. Generation of hepatitis C virus-like particles by use of a recombinant vesicular stomatitis virus vector. J Virol. 2002;76:12325–12334. doi: 10.1128/JVI.76.23.12325-12334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cobleigh M.A., Wei X., Robek M.D. A vesicular stomatitis virus-based therapeutic vaccine generates a functional CD8 T cell response to hepatitis B virus in transgenic mice. J Virol. 2013;87:2969–2973. doi: 10.1128/JVI.02111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts A., Kretzschmar E., Perkins A.S., Forman J., Price R., Buonocore L. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J Virol. 1998;72:4704–4711. doi: 10.1128/jvi.72.6.4704-4711.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reuter J.D., Vivas-Gonzalez B.E., Gomez D., Wilson J.H., Brandsma J.L., Greenstone H.L. Intranasal vaccination with a recombinant vesicular stomatitis virus expressing cottontail rabbit papillomavirus L1 protein provides complete protection against papillomavirus-induced disease. J Virol. 2002;76:8900–8909. doi: 10.1128/JVI.76.17.8900-8909.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kahn J.S., Roberts A., Weibel C., Buonocore L., Rose J.K. Replication-competent or attenuated, nonpropagating vesicular stomatitis viruses expressing respiratory syncytial virus (RSV) antigens protect mice against RSV challenge. J Virol. 2001;75:11079–11087. doi: 10.1128/JVI.75.22.11079-11087.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braxton C.L., Puckett S.H., Mizel S.B., Lyles D.S. Protection against lethal vaccinia virus challenge by using an attenuated matrix protein mutant vesicular stomatitis virus vaccine vector expressing poxvirus antigens. J Virol. 2012;84:3552–3561. doi: 10.1128/JVI.01572-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geisbert T.W., Daddario-Dicaprio K.M., Geisbert J.B., Reed D.S., Feldmann F., Grolla A. Vesicular stomatitis virus-based vaccines protect nonhuman primates against aerosol challenge with Ebola and Marburg viruses. Vaccine. 2008;26:6894–6900. doi: 10.1016/j.vaccine.2008.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones S.M., Feldmann H., Stroher U., Geisbert J.B., Fernando L., Grolla A. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med. 2005;11:786–790. doi: 10.1038/nm1258. [DOI] [PubMed] [Google Scholar]
- 41.Spearman P. HIV vaccine development: lessons from the past and promise for the future. Curr HIV Res. 2003;1:101–120. doi: 10.2174/1570162033352093. [DOI] [PubMed] [Google Scholar]
- 42.Wang M., Yue Y., Dong C., Li X., Xu W., Xiong S. Mucosal immunization with high-mobility group box 1 in chitosan enhances DNA vaccine-induced protection against coxsackievirus B3-induced myocarditis. Clin Vaccine Immunol. 2013;20:1743–1751. doi: 10.1128/CVI.00466-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neutra M.R., Kozlowski P.A. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 44.Ball L.A., Pringle C.R., Flanagan B., Perepelitsa V.P., Wertz G.W. Phenotypic consequences of rearranging the P, M, and G genes of vesicular stomatitis virus. J Virol. 1999;73:4705–4712. doi: 10.1128/jvi.73.6.4705-4712.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Betts M.R., Nason M.C., West S.M., De Rosa S.C., Migueles S.A., Abraham J. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ciuffreda D., Comte D., Cavassini M., Giostra E., Buhler L., Perruchoud M. Polyfunctional HCV-specific T-cell responses are associated with effective control of HCV replication. Eur J Immunol. 2008;38:2665–2677. doi: 10.1002/eji.200838336. [DOI] [PubMed] [Google Scholar]
- 47.Ma Y., Li J. Vesicular stomatitis virus as a vector to deliver virus-like particles of human norovirus: a new vaccine candidate against an important noncultivable virus. J Virol. 2011;85:2942–2952. doi: 10.1128/JVI.02332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kemball C.C., Flynn C.T., Hosking M.P., Botten J., Whitton J.L. Wild-type coxsackievirus infection dramatically alters the abundance, heterogeneity, and immunostimulatory capacity of conventional dendritic cells in vivo. Virology. 2012;429:74–90. doi: 10.1016/j.virol.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kemball C.C., Harkins S., Whitmire J.K., Flynn C.T., Feuer R., Whitton J.L. Coxsackievirus B3 inhibits antigen presentation in vivo, exerting a profound and selective effect on the MHC class I pathway. PLoS Pathog. 2009;5:e1000618. doi: 10.1371/journal.ppat.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dalton D.K., Haynes L., Chu C.Q., Swain S.L., Wittmer S. Interferon gamma eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J Exp Med. 2000;192:117–122. doi: 10.1084/jem.192.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Badr G., Bedard N., Abdel-Hakeem M.S., Trautmann L., Willems B., Villeneuve J.P. Early interferon therapy for hepatitis C virus infection rescues polyfunctional, long-lived CD8+ memory T cells. J Virol. 2008;82:10017–10031. doi: 10.1128/JVI.01083-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lore K., Adams W.C., Havenga M.J., Precopio M.L., Holterman L., Goudsmit J. Myeloid and plasmacytoid dendritic cells are susceptible to recombinant adenovirus vectors and stimulate polyfunctional memory T cell responses. J Immunol. 2007;179:1721–1729. doi: 10.4049/jimmunol.179.3.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed M., Brzoza K.L., Hiltbold E.M. Matrix protein mutant of vesicular stomatitis virus stimulates maturation of myeloid dendritic cells. J Virol. 2006;80:2194–2205. doi: 10.1128/JVI.80.5.2194-2205.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed M., Mitchell L.M., Puckett S., Brzoza-Lewis K.L., Lyles D.S., Hiltbold E.M. Vesicular stomatitis virus M protein mutant stimulates maturation of Toll-like receptor 7 (TLR7)-positive dendritic cells through TLR-dependent and -independent mechanisms. J Virol. 2009;83:2962–2975. doi: 10.1128/JVI.02030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.