

Abstract
Exosomes have been proposed as candidates for therapeutic immunization. The present study demonstrates that incorporation of the G protein of vesicular stomatitis virus (VSV-G) into exosome-like vesicles (ELVs) enhances their uptake and induces the maturation of dendritic cells. Targeting of VSV-G and ovalbumin as a model antigen to the same ELVs increased the cross-presentation of ovalbumin via an endosomal acidification mechanism. Immunization of mice with VSV-G and ovalbumin containing ELVs led to an increased IgG2a antibody response, expansion of antigen-specific CD8 T cells, strong in vivo CTL responses, and protection from challenge with ovalbumin expressing tumor cells. Thus, incorporation of VSV-G and targeting of antigens to ELVs are attractive strategies to improve exosomal vaccines.
Keywords: Exosomes, VSV-G, Vaccines
1. Introduction
Exosomes released from dendritic cells (DC) or cancer cells have been proposed as vaccine candidates for immunotherapy of tumors [1], [2], [3], [4], [5]. Arising from multivesicular bodies, exosomes contain a restricted set of cellular proteins [6], [7]. Some exosomal hallmark proteins such as heat shock proteins and tetraspanins can modulate DC activity, which can lead to improved immunogenicity and efficacy of exosome-based cancer vaccines [8].
Although exosomes released from cancer cells constitutively contain some tumor antigens, the process of protein sorting into vesicles within the multivesicular bodies still remains unclear [6]. Poor results in clinical trials also raise concerns regarding the efficacy of exosomal-based cancer vaccines [5]. This suggests that new approaches to improve the efficacy of exosome-based vaccines are warranted.
Direct targeting of protein antigens to exosomes might improve specificity of exosomal vaccines. As one possible strategy, the exogenous loading of MHC-I molecules of DC-derived exosomes with peptides was reported [4]. We recently demonstrated that replacing the transmembrane and cytoplasmic domain of the severe acute respiratory syndrome-associated coronavirus (SARS-CoV) S protein with that of the G protein of vesicular stomatitis virus (VSV-G) resulted in enhanced shedding of the S protein in a vesicle-associated form into the supernatant of transfected 293T cells. The presence of proteins such as HSP90 and CD82 indicated an exosome-like nature of these vesicles [9].
Viral fusion proteins might provide an attractive strategy to enhance uptake of the exosomal vaccines by professional antigen presenting cells and thus improve the immunogenicity. Since the transmembrane and intracytoplasmic domains of VSV-G are sufficient to tether heterologous proteins to the exosome-like vesicle (ELV), VSV-G itself should also be incorporated into the ELVs. The G protein of the VSV is a type I transmembrane glycoprotein about 66 kDa in size and is composed of an ectodomain, a membrane spanning domain and a cytoplasmic domain. The VSV-G protein mediates attachment of the viral particle to the cell membrane and induces fusion of the viral envelope with that of the target cell at pH below 6.0 [10].
Buseyne et al. demonstrated that incorporation of fusion-competent VSV-G into HIV-based viral vectors leads to an efficient uptake of the particles by immature DC and presentation of Gag-derived peptides on MHC-I molecules [11]. Furthermore, VSV-G has been shown to enhance the immunogenicity of a HIV-gag DNA vaccine when co-expressed with the antigen [12]. We also previously demonstrated that incorporating a fusion-competent, but not a fusion-deficient, VSV-G into an HIV-1-derived viral-like particle (VLP) vaccine led to a more than 100-fold increase in antibody titers against the HIV-gag proteins of the VLP [13]. Given the immunostimulatory properties of VSV-G, its incorporation into ELV potentially could increase the immunogenicity of exosome-based vaccines.
In the present study, we analyzed the effects of VSV-G on ELV-uptake by DC and DC-function, and characterized immune responses to ELV-associated antigens. For this purpose, we tethered the model antigen ovalbumin (OVA) to the ELVs by fusing the OVA protein to the transmembrane and intracytoplasmic domains of VSV-G. Incorporation of VSV-G into the ELV provided enhanced uptake of the vesicles and presentation of exosomal antigens by DC, as well as phenotypic and functional DC maturation in vitro. Furthermore, in vivo studies demonstrated efficient induction of antigen-specific CTL responses. Thus, targeting of antigens and a viral fusion protein to the same ELVs can improve the immunogenicity of exosomal vaccines.
2. Material and methods
2.1. Cell culture
The human embryonic kidney cell derivative 293T [14], 293 cells (Quantum Biotechnologies, Montreal, Canada) and the mouse melanoma cell lines B16-OVA and B16 were maintained in DMEM supplemented with 10% FCS, l-glutamine, penicillin and streptomycin.
2.2. Plasmids
The pEGFP-C1 plasmid was purchased from Clontech, Heidelberg, Germany. The construction of VCGΔBH [15], Hgpsyn [16] and pCD-Gsyn [17] has been described. For the construction of pCD-Gmut, a glutamine to asparagine point mutation was introduced at position 117 in the codon-optimized VSV-G gene as described for the wild type VSV-G [18]. To construct the expression plasmid for the membrane-bound OVA protein the OVA gene was amplified using the forward primer 5′ agctggctagcaagcttccaccatgaagtgcctgctgtacctggccttcctgt tcatcggcgtgaactgcggatccatgggctccatcgg cgcagcaagcatgga 3′ (1) containing the VSV-G leader sequence and flanked with NheI and HindIII restriction enzymes, and the reverse primer 5′ ggatccagcgctaggggaaacacatctgccaaagaa gag 3′ (2) flanked with BamHI and Eco47III restriction enzymes. Then the transmembrane domain was amplified from the plasmid pCD-Gsyn using the forward primer 5′ tctcttctttggcagatgtgtttcccctagcgctggatccttcggcgacaccggcctga gcaagaac 3′ (3) and the reverse primers 5′ ctgcagaattctt atcacttgcccagcctg 3′(4) flanked with EcoRI restriction site. The products were purified using a PCR purification kit (Qiagen, Oldenburg, Germany) mixed and used as template for the subsequent PCR using the primers (1) and (4). The resulting PCR fragment was cloned into the NheI–EcoRI digested pCDNA3.1 plasmid (Invitrogen, Karlsruhe, Germany) to generate pExOVA. All DNA preparations for immunizations and ELV production were performed using Endofree plasmid preparation kits (Qiagen).
2.3. Production and characterization of ELV
Semi-confluent 293T cell monolayer was transfected in 25(or 175) cm2 tissue culture flasks with 10(80) μg pExOVA alone, or together with 10(40) μg of pCD-Gsyn or pCD-Gmut to produce O-ELV, O-ELV-G or O-ELV-Gmut vesicles, respectively. For the production of ELV-G and ELV-Gmut vesicles, 10(40) μg of pCD-Gsyn or pCD-Gmut were transfected together with 10(80) μg of Carrier DNA (Invitrogen). Eight hours after transfection the FCS-containing medium was changed into the serum-free CD293 medium (Invitrogen). The conditioned medium was harvested 24 h later and the ELV were concentrated by ultracentrifugation through 20% sucrose cushion as described previously [9]. For the step gradient ultracentrifugation, the pelleted ELVs were resuspended in PBS and loaded onto a sucrose gradient containing 60, 50, 40, 30, 20 and 10% of sucrose and centrifuged for 2 h at 30,000 rpm. The resulting protein fractions were concentrated fourfold using 10,000 MW cutoff Vivaspin filters. For the determination of OVA and VSV-G contents of ELVs, ELISA assays were performed using soluble OVA protein and VSV-G peptides (Sigma–Aldrich, Tauchkirchen, Germany) as standards. Serial dilutions of ELV preparations and standard proteins/peptides were incubated overnight at 4 °C and then blocked with fat-free milk for 1 h at room temperature. As primary antibodies, the rabbit anti-OVA (Biodesign International, Saco, Maine) and mouse anti-VSV-G (Sigma–Aldrich) were used. Secondary HRP-conjugated antibodies and stabilized TMB chromogen were obtained from Dako, Hamburg, Germany. The total protein concentration in the ELVs was determined by Bio-Rad protein assay and the endotoxin level by QCL-1000® Chromogenic LAL Endpoint Assay (Cambrex, Germany) according to the manufacturer's instructions. The residual dsDNA (double strain DNA) in the ELV preparations was measured with the Qubit Quantitation Platform (Invitrogen) according to the manufacturer's instructions.
2.4. Western blot analysis
Western blot analysis was performed as described previously [9]. Blots were incubated with a rabbit anti-OVA, mouse anti-VSV-G antiserum (Sigma–Aldrich) or mouse anti-HSP90 antibodies (Santa Cruz Biotechnology Inc., Heidelberg, Germany). Thereafter goat anti-rabbit or rabbit anti-mouse HRP-conjugated antiserum were used (Sigma–Aldrich). Protein bands were detected with the Western blot detection kit (Biozym Scientific GmBH, Oldendorf, Germany) according to the manufacturer's instructions.
2.5. Cell surface expression of OVA
Cells were transfected with pExOVA or pSolOVA as described above. For the surface staining rabbit anti-OVA and FITC-conjugated anti-rabbit IgG Ab were used. The expression analysis was performed as described [9].
2.6. Fusion assay
293T cells were transfected with either pCD-Gsyn or pCD-Gmut in the presence or not of the pEGFP-C1. Two days after transfection, cells were incubated for 1 min in the VSV-G fusion buffer (10 mM MES and 10 mM HEPES, pH 5.5 in 1× PBS). After removing the fusion buffer, cells were incubated for 1 h at 37 °C with fresh DMEM media. Syncytia formation was monitored under a fluorescence microscope.
2.7. The viral vector pseudotyping experiment
293T cells were transfected with VCGΔBH, Sgpsyn and either pCD-GSyn or pCD-Gmut. The experiment was performed as described in Ref. [9]. For the titration 293A cells were used. GFP-positive cells were counted under a fluorescence microscope and the vector titers were calculated from the number of GFP-positive cells/well.
2.8. Mouse cells purification
Mice were housed at the animal facility of the faculty of medicine, Ruhr University Bochum, Germany and were handled according to the Federation of European Laboratory Animal Science Association. Six- to 8-week-old C57BL/6 mice (Charles River Wiga GmbH, Sulzfeld, Germany) were used. The TCR-transgenic OT-1 mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). For the purification of DC, single cell suspension of C57BL/6 mice splenocytes were prepared as previously described [19]. CD11c+ DC were enriched by positive selection with anti-CD11c magnetic beads (Miltenyi Biotec GmbH, Germany) according to the manufacture's instruction. The resulting cells were routinely >85% pure. CD8+ T cells from TCR-transgenic mice were purified from single cell splenocyte suspension by negative selection using magnetic beads (CD8+ T cell isolation kit, Miltenyi Biotec). For in vitro proliferation assay, cells were labeled with 3 μM of CFDA (Vybrant CFDA Cell Tracer Kit, Molecular Probes-Invitrogen) according to the manufacture's instruction.
2.9. Uptake experiment
ELVs were labeled with 20 μM CFDA for 4 h at room temperature. The labeled ELV were dialyzed against PBS/0.5 M NaCl overnight at 4 °C. DC were incubated with 10 μg (total protein content) ELVs for 120 min either at 37 °C or on ice. As control 1 μg/ml of FITC-dextran (FITC-dx) was added. At selected time points cells were collected, washed three times with ice-cold medium, incubated first with Fc-block and then stained with anti-CD11c-APC antibodies. The fluorescence of CD11c-positive and -negative cells was measured by flow cytometry.
2.10. Activation assay
DC were seeded at the density of 2 × 106 cells/well in 24-well plates and incubated with 1 or 10 μg/ml of ELV or with 1 μg/ml of LPS (E. coli, 26:b6, Sigma–Aldrich) as positive control. The conditioned media of cultured cells were harvested 24 h later for the determination of IL-12p70 production using a commercial available ELISA kit (BioSource Europe S.A., Nivelles, Belgium) according to manufacturer's instruction. The cells were collected and stained with anti-MHC II, anti-CD11c, anti-CD40, anti-CD80 and anti-CD86 antibodies (BD PharMingen, Heidelberg, Germany) to analyze the level of cell surface markers by flow cytometry.
2.11. Cross-priming assay for OT-1 CD8+ T cells
DC were plated in flat-bottom 96-well plates at the density of 2 × 105 cells/well and incubated with different concentrations of sOVA or ELV preparations for 2 h at 37 °C. After intensive washing with pre-warmed medium the cells were transferred into U-bottom 96-well plate. A total of 2 × 105 naïve pooled CFDA-labeled CD8+ T cells from spleens of OT-1 mice were then added to each well and incubated for 96 h at 37 °C. Thereafter the cells were incubated with APC-conjugated H-2Kb/OVA257–264 tetramers (Sanquin Reagents, Amsterdam, The Netherlands). The proliferation of primed OVA-specific tetramer+ cells was assessed by flow cytometric analysis of CFDA fluorescence intensity of the labeled cells.
2.12. Inhibition of cross-presentation
DC were first incubated for 1 h at 37 °C with either 10 μg/ml of Brefeldin A or 10 μM of lactacystin or 40 nM of ammonium chloride and pulsed thereafter for 2 h with 1 μg/ml sOVA, 1 μg/ml of OVA257–264 or 0.01 μg/ml O-ELV-G, in the presence of the same drugs. Then the cells were washed and co-cultured with CFDA-labeled OT-1 CD8+ T cells as described above.
2.13. Immunization and challenge experiment
C57BL/6 mice (n = 4 mice per group) were immunized subcutaneously twice at weeks 0 and 5 with 2 μg of ELV's (O-ELV, O-ELV-G or O-ELV-Gmut). The control groups were immunized either with 10 μg of soluble OVA or 50 μg of plasmid DNA (pSolOVA). For the challenge experiment, 2 × 106 B16 or B16-OVA cells were inoculated at the right flank of the mice. The volume of the tumor was determined every 3 days and mice were sacrificed when the volume reached 3500 mm3.
2.14. Measurement of serum antibody levels
One week after the second immunization mice were bled and the anti-OVA antibody production was determined by ELISA. Briefly, flat-bottom 96-well plates were coated overnight with 5 μg of OVA (Sigma–Aldrich), blocked with 5% fat-free milk and incubated with 1:100 or 1:10 diluted mice serum for the detection of OVA anti-IgG1 and -IgG2a antibodies, respectively. Corresponding HRP-conjugated secondary antibodies were obtained from Sigma–Aldrich.
2.15. CTL assay and tetramer binding
The CTL assay was performed as described previously [20] using 10 μg/ml OVA257–264 peptide to prime CFDAhi cells. Donor cells were prepared and transferred into recipient mice at day 7 after the second immunization. After 16 h, the recipient mice were euthanized, spleens and lymph nodes were obtained and CFDAhi and CFDAlo populations were identified by flow cytometry. Percent specific lysis was calculated by using the following formulae: percent specific lysis = [1 − (ratio vaccinated/ratio control)] × 100, where “ratio” = (percent CFDAhi/percent CFDAlo). For determination of TCR-positive cells, spleen cells were incubated subsequently with APC-conjugated H-2Kb/OVA257–264 tetramers and anti-CD8-FITC antibody and analyzed by flow cytometry.
2.16. Statistical analysis
The statistical analyses were performed using GraphPad Prism 4 Software Inc. (San Diego, CA, USA). For the comparison of different experimental groups, one-way ANOVA analysis was performed. Differences were considered significant at p < 0.05.
3. Results
3.1. Construction and expression of membrane-bound OVA and a fusion-defective VSV-G mutant
In order to target antigens to exosomes we fused the open reading frame of OVA to the N-terminus of the transmembrane and cytoplasmic domain of VSV-G (Fig. 1A). In addition, sequences coding for the leader peptide of the VSV-G protein were cloned in frame upstream of the OVA cDNA resulting in plasmid pExOVA. The FACS analysis of 293T cells transiently transfected with pExOVA revealed the presence of the OVA protein on the cell surface in contrast to cells transfected with pSolOVA, encoding secreted OVA (Fig. 1B).
Fig. 1.

Characterization of membrane-anchored OVA and fusion-defective VSV-G proteins. (A) Schematic drawing of chimeric OVA/VSV-G protein. Open box represents OVA; gray boxes, VSV-G. LP: leader peptide; TM: transmembrane domain; CD: cytoplasmic domain. (B) FACS analysis of 293T cells transfected with the expression plasmid for either pExOVA or pSolOVA and stained 2 days later for the presence of the OVA protein at the cell surface. Untransfected cells were used as negative control. (C) The 293T cells were transfected with either the pCD-Gsyn plasmid encoding VSV-G or the pCD-Gmut plasmid encoding fusion-defective VSV-G. The cell lysates were analyzed by Western blot for the presence of VSV-G. (D) The pCD-Gsyn or pCD-Gmut plasmids were co-transfected with a GFP expression plasmid. Two days later transfected cells were incubated with a low-pH fusion buffer and monitored over time under a fluorescence microscope for the presence of syncytia formation. The pictures were taken 1 h after addition of the buffer. Cells transfected with the GFP expression plasmid only served as negative control (Mock).
A fusion-defective mutant of VSV-G was constructed by introduction of a single Q-to-N point mutation at position 117 of the codon-optimized VSV-G expression plasmid [17] as previously reported for wild type VSV-G [21]. The expression of the recombinant mutant protein (VSV-Gmut) was confirmed by Western blot analysis (Fig. 1C). In a low-pH fusion buffer, the cells expressing VSV-G formed large syncytia, which were easily detectable when VSV-G was co-transfected with a GFP expression plasmid (Fig. 1D, right). In contrast, cells transfected with an expression plasmid encoding the VSV-Gmut showed no detectable syncytia (Fig. 1D, middle), indicating loss of fusion activity. Furthermore, a lentiviral vector pseudotyping assay was also performed. Lentiviral vectors pseudotyped with VSV-G reached titers of up to 1.2 × 106 infectious units/ml, while pseudotyping with the fusion-defective mutant reduced the titer to background levels of 0.4 × 103 infectious units/ml (data not shown).
3.2. Production and characterization of exosome-like vesicles
Transient transfection of 293T cells with pExOVA and pSolOVA showed that the OVA was readily detectable in lysates of cells transfected with both plasmids (Fig. 2A), but it could be pelleted by ultracentrifugation from the supernatant of pExOVA-transfected cells only, suggesting an association with cellular vesicles (Fig. 2A). In addition to the expected 65 kDa OVA band in the Western blot analysis, an additional band of about 40 kDa was observed in pExOVA-transfected cells, probably representing a proteolytic cleavage product of the chimeric OVA protein (Fig. 2A). The ultracentrifugation of the supernatant of pExOVA-transfected cells on a 20–60% sucrose step gradient revealed that most of the OVA protein was detectable in the 40% and 50% gradient fractions (Fig. 2B, upper panel), which also contained most the exosomal marker protein HSP90 (Fig. 2B, lower panel). This indicates a comparable density distribution of exosomes and OVA containing vesicles and is consistent with an exosomal localization of the chimeric OVA protein.
Fig. 2.

Characterization of the ELVs. (A) 293T cells were transfected with either pExOVA or pSolOVA plasmids. Cells lysate (Lysate) and the pellet obtained by ultracentrifugation of the conditioned media (Pellet) were analyzed for the presence of the OVA proteins by Western blot analysis. Mock-transfected cells were used as negative controls. (B) The conditioned media from 293T cells transfected with the pExOVA plasmid was loaded onto a step gradient of sucrose with the indicated sucrose concentration and ultracentrifuged. The different sucrose fractions were collected and analyzed by Western blot for the presence of the OVA protein (upper panel) and the HSP90 protein (lower panel).
For all further experiment, ELVs were prepared from the supernatant of transfected 293T cells by pelleting them through a 20% sucrose cushion. Transfection of different VSV-G and/or OVA expression plasmids resulted in various ELVs (Table 1 ). The concentrations of OVA and VSV-G in the different ELV preparations were determined by ELISA. The mean of the endotoxin levels were 1.49 ± 0.39 EU/ml. To further exclude potential effects of these low levels of endotoxin, exosomal preparations with equal endotoxin concentrations were compared side by side.
Table 1.
Exosome-like vesicles preparations used in this study
| Abbreviation | Transfected plasmidsa |
||
|---|---|---|---|
| pCD-Gsyn | pCD-Gmut | pExOVA | |
| ELV-G | + | − | − |
| ELV-Gmut | − | + | − |
| O-ELV | − | − | + |
| O-ELV-G | + | − | + |
| O-ELV-Gmut | − | + | + |
293T cells were co-transfected with “+” marked plasmids.
3.3. Uptake of exosome-like vesicles by dendritic cells in vitro
Binding and subsequent internalization of particulate antigens by DC may be the initial events, resulting in a cascade of signal transduction and cellular activation. First, we explored how efficiently different ELVs containing the fusion-competent VSV-G, its fusion-defective mutant or an irrelevant non-viral protein (OVA) are taken up by DC in vitro. The ELVs were labeled with the carboxyfluorescein diacetate (CFDA) dye, which becomes highly fluorescent upon internalization, while FITC-dextran was used as positive control. For the standardization of the data from several experiments and to consider the background fluorescence, the “uptake index” was calculated as the ratio of fluorescence intensity of the cells incubated at 37 °C to that of the cells incubated on ice. As compared to O-ELV and ELV-Gmut, ELV-G showed consistently significant higher level of uptake by CD11c+ DC after exposure times of 90 and 120 min (Fig. 3A). In contrast, the uptake indexes by CD11c− splenocytes for both FITC-dx and all ELV types were at the same level and only slightly above the background (Fig. 3B), consistent with a lower uptake activity of CD11c− cells. Thus, incorporation of VSV-G into ELVs significantly increased their internalization by DC.
Fig. 3.

Uptake experiment. Ten micrograms of different CFDA-labeled ELVs (ELV-G, ELV-Gmut or O-ELV) were incubated with cell suspension enriched for CD11c+ cells. Cells were incubated for the indicated time at 37 °C or on ice, washed, and stained with anti-CD11c antibody. The results are presented as an uptake index = (MFI at 37 °C/MFI ice) for CD11c-positive (A) and CD11c-negative (B) cells. The experiment was performed three times; data represent the mean ± standard deviation. Statistically significant difference (*p < 0.05) to the ELV-G group.
3.4. VSV-G containing exosome-like vesicles induce acute phenotypic and functional maturation of dendritic cells
The maturation of DC after antigen internalization is the next step that is required for the stimulation of T cell responses. Therefore, freshly isolated splenic CD11c+ DC were incubated with ELV-G, ELV-Gmut or O-ELV and expression of DC activation markers were determined to characterize phenotypic DC maturation induced by the ELVs. ELV-G at a concentration of 10 μg total protein/ml and 1 μg LPS/ml (positive control) led to a similar DC maturation as assessed by CD40 and CD86. Control ELVs (O-ELV, ELV-Gmut) showed lower levels of up-regulation that could reflect general ELVs features (Fig. 4A). The observed effect was dose-dependent and became undetectable at ELV concentrations of 1 μg/ml (data not shown).
Fig. 4.
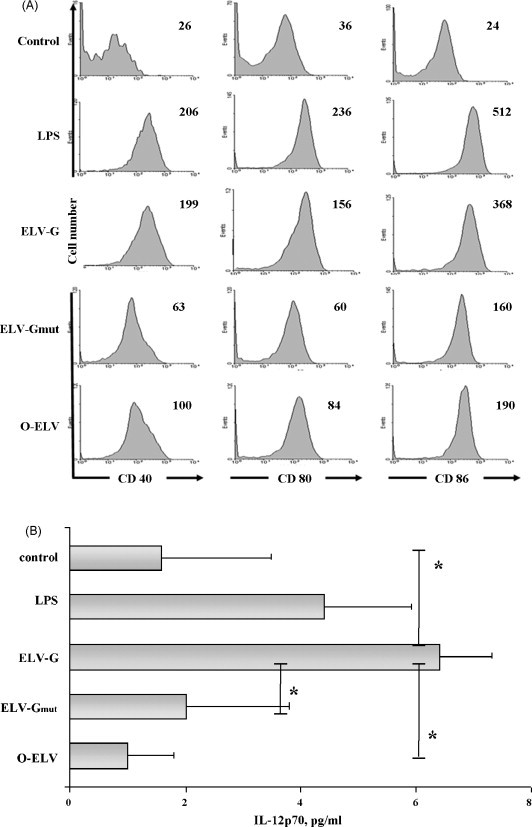
Activation of splenic DC by ELVs. (A) Freshly isolated splenic DC were incubated with LPS (1 μg/ml) or with 10 μg/ml of ELVs (ELV-G, ELV-Gmut or O-ELV) for 24 h. Cells were collected and stained for MHC II, CD11c, CD40, CD80, and CD86. CD11c+MHCII+ cells were analyzed for CD40, CD80, and CD86 expression levels by flow cytometry. Numbers indicated in the upper right of the histograms represent the Geometric log MFI. One representative experiment of three is shown. (B) Cell-free supernatants were analyzed in triplicate for the presence of IL-12p70 (pg/ml) by ELISA. Data present the mean from three independent experiments ± standard deviation. *p < 0.05.
For the induction of effective T cell responses, the production and secretion of cytokines (functional maturation of DC) is critical. DC incubated with ELV-G secreted similar high levels of IL-12p70 as DC stimulated with LPS. On the other hand, only low levels of IL-12p70 were detected in supernatants from untreated DC or those exposed to O-ELV or ELV-Gmut (Fig. 4B).
The final endotoxin concentration in the culture medium was at the same level between the various experimental ELV groups and did not exceed 0.15 EU/ml. In addition, the residual amounts of dsDNA from ELV preparations did not correlate with the DC maturation levels and the highest level in culture medium (up to 0.28 ng/ml) was reached in the ELV-Gmut group. Thus, neither LPS nor residual plasmid DNA can explain the strong DC maturation effect of ELV-G.
3.5. Cross-presentation of exosomal proteins by DC is enhanced by VSV-G incorporation and requires endosomal acidification
Dendritic cells are known to be the main cell type that is able to present exogenously derived peptides in the context of MHC class I molecules to CD8+ T cells. Since VSV-G increased uptake of the ELVs and stimulated DC maturation, it might also improve subsequent activation of antigen-specific, MHC class I-restricted T cells. O-ELV and O-ELV-Gmut did not enhance proliferation of TCR-transgenic CD8+ T cells specific for the immunodominant OVA epitope (OVA257–264) in comparison to the same amount of soluble OVA (Fig. 5C, D and A, respectively). In contrast, incorporation of fusion-competent VSV-G into ELVs (O-ELV-G) increased presentation approximately 100-fold, since the number of cell divisions after pulsing cells with O-ELV-G at a final concentration of 0.01 μg OVA/ml was comparable to that obtained after pulsing DC with 1 μg/ml of soluble OVA (Fig. 5E and B, respectively). Importantly, preparing ELV-G and O-ELV by separate transfections and mixing them just prior to incubations with DC, did not show any stimulatory effect. Similarly, mixing of soluble OVA with ELV-G did not enhance T cell proliferation either (Fig. 5F–I). This indicates, that the co-localization of fusion-competent VSV-G and the protein antigen of interest on the same ELV is necessary for enhancement of cross-presentation.
Fig. 5.

Cross-presentation of ELV targeted OVA to OVA-specific CD8+ cells. DC were incubated with soluble OVA at a concentration of 0.01 μg/ml (A) and 1 μg/ml (B) or with O-ELV (C), O-ELV-Gmut (D), or O-EVL-G (E) vesicles each containing 0.01 μg/ml of OVA. DC were also incubated with mixtures of separately prepared ELV-G or ELV-Gmut and O-ELV containing 0.01 μg/ml of OVA (F/H, respectively), or ELV-G or ELV-Gmut and 0.01 μg/μl of soluble OVA (G/I, respectively). After 2 h of incubation, DC were washed and mixed with CFDA-labeled CD8+ T cells from spleens of OT-1 mice. Ninety-six hours later, cells were washed and stained with APC-conjugated H-2Kb/OVA257–264 tetramers. The proliferation of tetramer-positive OT-1 CD8+ T cells was evaluated by flow cytometry analysis for CFDA fluorescence intensity. The percentages of cells that have undergone more than two divisions are given. One representative out of three independent experiments is shown.
Processing of internalized protein antigens can occur either by proteasomal degradation and loading onto MHC-I in a TAP-dependent manner (the classical MHC-I pathway) [22], [23] or by alternative pathways in endolysosomes or in phagosomes [24]. The processing pathway of O-ELV-G in DC was examined using various inhibitors. Brefeldin A was used as an inhibitor of exocytosis, lactacystin as an irreversible proteasomal inhibitor and ammonium chloride as an inhibitor of endosomal acidification, which also delays endosomal/lysosomal maturation. The processing of the Kb-restricted OVA257–264 has been reported to be proteasome-dependent, as determined by activation of a T cell hybridoma [25] and expression of cell surface OVA257–264-MHC class I complexes [26]. In our experiments, pre-treatment of DC with lactacystin inhibited the presentation of OVA257–264 peptide derived from soluble OVA. However, the same drug did not abrogate the proliferation of OT-1 cells if OVA was taken up by DC as O-ELV-G (Fig. 6 ). In contrast, ammonium chloride strongly inhibited cross-presentation of O-ELV-G even at a low dose of 40 nM, but it was absolutely ineffective for soluble OVA (Fig. 6). BFA dramatically blocked the proliferation of specific T cells in both cases (Fig. 6). The presentation of exogenous OVA257–264 peptide, which does not require any processing, was not affected by any of the treatments and could serve as an internal control for the proliferative features of the CD8+ T cells (data not shown). Therefore it can be concluded that DC process ELVs containing fusion-competent VSV-G through a degradation mechanism that involves endosomal acidification. The classical proteasomal degradation pathway does not seem to be the main source for the immunodominant peptides loaded onto MHC class I molecules.
Fig. 6.

Intracellular processing pathway of O-ELV-G. Splenic DC were first pre-incubated for 1 h with Brefeldin A, lactacystin or ammonium chloride at the indicated concentrations and then pulsed for 2 h with 1 μg/ml of soluble OVA (sOVA1) or 0.01 μg/ml of OVA in O-ELV-G in the presence of the drugs mentioned above. Cells were then washed and co-cultured with CFDA-labeled OT-1 CD8+ T cells during 96 h. Histograms represent CFDA fluorescence intensity of H-2Kb/OVA257–264 tetramer-positive cells. The percentages of cells that have undergone more than two divisions are given. The results are representative of three independent experiments.
3.6. Immunogenicity of exosome-like vesicles
The immunogenicity of ELVs containing fusion-competent VSV-G were compared in mice to the immunogenicity of ELVs lacking VSV-G and ELVs containing fusion-defective VSV-G in order to correlate the in vitro findings with the in vivo situation. In addition, mice also received a standard DNA vaccine encoding soluble OVA to be able to compare the immunogenicity of exosomal vaccines to other vaccination strategies.
Mice immunized with O-DNA exhibited high serum levels of anti-OVA antibodies of the IgG1 and IgG2a subclass (Fig. 7 ). ELV immunization preferentially induced an anti-OVA IgG2a response (Fig. 7B), whereas soluble OVA induced a predominant IgG1 antibody response (Fig. 7A). Incorporation of the fusion-competent VSV-G into the ELV selectively enhanced the IgG2a antibody levels (Fig. 7B).
Fig. 7.

Characterization of the humoral immune responses. Serum samples were collected 1 week after the second immunization, analyzed by ELISA for the presence of anti-OVA IgG1 (A) or IgG2a (B) immunoglobulin subclasses at a 1:1000 and 1:10 dilution, respectively. The histograms represent the mean from two independent experiments ± standard deviation (n = 8). Statistically significant difference (*p < 0.05) to the control group; statistically significant difference (#p < 0.05) to sOVA group; statistically significant difference (§p < 0.005) to O-DNA group; statistically significant difference (†p < 0.005) to O-ELV-Gmut group.
The ovalbumin-specific cellular immune response was assessed in both fluorescent tetramer-binding and in vivo cytotoxic T-lymphocyte (CTL) assays. The O-DNA vaccine induced moderate levels of tetramer+ cells (Fig. 8A). The soluble OVA protein immunization did not lead to Ag-specific CD8+ T cells levels exceeding the background. However, after immunization with OVA in the context of ELVs the numbers of tetramer+ cells were comparable to the O-DNA group. Incorporation of VSV-G further increased the levels of Ag-specific CD8+ T cells (Fig. 8A). The Ag-specific cells observed after ELV immunizations were also functionally active: the strongest specific lysis in the vivo CTL assay was obtained in mice immunized with O-ELV-G (59.2 ± 11%) compared to O-ELV-Gmut (25.2 ± 5.1%) or O-ELV immunized animals (26.3 ± 6.1%) (Fig. 8B).
Fig. 8.

Characterization of the cellular immune responses. (A) Tetramer staining. One week after the second immunization, spleen cells were analyzed by FACS for the percentage of H-2Kb/OVA257–264 tetramer-positive CD8+ T cells. The histogram represents the mean from two independent experiments ± standard deviation (n = 8). Statistically significant difference (*p < 0.05) to control and sOVA groups; statistically significant difference (#p < 0.05) to O-DNA, O-ELV, and O-ELV-Gmut groups. (B) In vivo CTL assay. Representative FACS histograms of CFDAlo and OVA257–264 loaded CFDAhi populations in the spleen 16 h after cell transfer on day 7 after second immunization. The percent of specific killing is shown.
The therapeutic potential of ELV vaccines was further analyzed in a tumor rejection model. Immunized and naïve animals were inoculated with B16 melanoma cells stably expressing OVA, and the rate of tumor growth was monitored over time by measuring the volume of the tumor. In the control mice and those immunized with sOVA, the tumor rapidly developed within 1–2 weeks after challenge. In contrast, the tumor growth was delayed in all groups of animals immunized with both ELV and O-DNA vaccines (Fig. 9 ). The rapid tumor growth in mice, immunized with O-ELV-G and challenged with B16 cells not expressing OVA, confirmed the specificity of the protective immune responses (Fig. 9).
Fig. 9.

Tumor challenge. (A) Mice immunized with the indicated vaccines were challenged with B16-OVA cells 1 week after the second immunization. As an antigen-specific control, mice immunized with O-ELV-G were also challenged with B16 cells not expressing OVA (B16). The tumor growth was monitored daily and the volume was determined at 3-day interval. (B) The histogram represents the size of the tumor at day 18 after challenge as the mean from two independent experiments ± standard deviation. Statistically significant difference (*p < 0.01) to control, B16 and sOVA groups.
4. Discussion
DC's are the most potent antigen presenting cells and their specific targeting and activation might advance vaccine design. Exosomes secreted by tumor cells were shown to be stable and safe immunogenic vehicles of tumor antigens; however, their function requires activated (or mature) DC [3], [4], [6]. In the present study we have shown that incorporation of VSV-G into exosome-like vesicles not only enhances uptake and presentation of exosome-associated antigens but also provides important maturation signals for DC.
It is known that VSV-G mediates binding of the virus to the cell surface and efficient entry into the target cells via endocytosis [10]. Indeed, although control exosomes can be effectively internalized by freshly isolated murine splenic DC, incorporation of fusogenic form of VSV-G improved the delivery of these ELVs to DC (Fig. 3). However, to stimulate immune responses, rather than tolerance, antigen internalization by DC should be accompanied by the innate immune system signals, which induce phenotypic and functional maturation of DC. The protein composition of exosomes was reported to have immunostimulatory effects [6], which could explain phenotypic activation of DC by control ELV in vitro (Fig. 4A). Another plausible explanation may be the xenogenic origin of the vesicles. However, in similar experiments with human monocyte-derived DC some degree of activation was also observed (unpublished observations). On the other hand, unlike VSV-G containing ELVs, control ELVs failed to up-regulate the activation markers on murine bone marrow-derived DC (data not shown) and to stimulate production of IL-12p70 by splenic DC (functional maturation) (Fig. 4B). Together this suggests that VSV-G containing exosomes provide a maturation signal for immature and not fully matured DC.
The molecular mechanism of the signal is under discussion. Using lentiviral vectors pseudotyped with VSV-G, Pichlmair et al. recently showed that murine bone marrow-derived plasmacytoid DC could be activated not due to the virus-like particle itself but rather due to specific shaped structures that carry residual amounts of plasmid DNA and act as agonists for TLR9 [27]. Although we could detect trace amounts of dsDNA in our ELV preparations ranging from 2.1 to 162 ng/ml (mean: 49.9 ± 50.6 ng/ml), the dsDNA was present in all ELV types independently of co-expression of VSV-G and within the same co-transfection experiment only minor differences in dsDNA content were found between samples containing fusiogenic and mutant form of VSV-G (data not shown). Thus, our results are more consistent with a VSV-G triggered signaling pathway recently described to act downstream from TLR4 [28]. The in vitro cross-presentation experiments further hint to a specific, fusion-dependent mechanism for VSV-G-mediated enhancement of DC's stimulatory activity. Despite stimulation of DC maturation, VSV-G containing ELVs were not able to stimulate cross-priming by DC co-exposed to soluble OVA or ELVs containing OVA. Physical co-localization of VSV-G and the OVA protein on the same ELVs seems to be required for the VSV-G-mediated enhancement of cross-presentation (Fig. 6). For both, the parental VSV and VSV-G pseudotyped viral vectors, the route of viral genetic material into the cytoplasm is through a phagosomal–endolysosomal low-pH compartment [10], [29], known as alternative MHC class I pathway for processing of internalized protein antigens [23]. We could demonstrate that a low dose of NH4Cl, which blocks not only the intravesicular acidification but also the phagosome–lysosome fusion, strongly inhibited the processing of VSV-G containing ELVs. At the same time, blockade of classical MHC class I pathway was rather ineffective (Fig. 6). This suggests that the main part of VSV-G-dependent cross-presentation of OVA is due to ELV internalization into acidic endolysosomal compartment from which the antigen gains access to the retrograde transport pathway for MHC class I presentation. Interestingly, Blander and Medzhitov proposed a model where MHC class II presentation is dependent on the presence of TLR ligands within the phagocytosed cargo [30]. In an individual phagosome, the TLR ligands enhance the kinetics of the fusion reaction in the endocytic pathway and activate hydrolytic enzymes [31]. This may explain why OVA physically associated with VSV-G was shuttled to the alternative MHC class I pathway instead of terminal degradation in lysosomes.
In vivo, the VSV-G has been reported to stimulate immune responses against co-administered antigens [11], [12], [13], [27]. We previously demonstrated that VSV-G incorporated into HIV-based viral-like particles enhanced both humoral and cellular immune responses against HIV-gag [13]. At the same time, our attempt to incorporate VSV-G into exosomal vaccines containing the S protein of SARS-CoV did not significantly increase the immune responses against the SARS-S [9]. Since both proteins are fusion-competent transmembrane glycoproteins, the immunostimulatory effect of VSV-G might be masked by the fusion activity of the SARS-S. In the present study, we therefore used recombinant OVA protein as a membrane-bound model antigen to characterize immunostimulatory properties of VSV-G in the context of ELV vaccines.
Immunization with ELVs containing OVA, but lacking fusion-competent VSV-G, stimulated IgG2a antibody production suggesting a T-helper cell type I (Th1) immune response. A further increase of IgG2a levels was observed after immunization with O-ELV-G consistent with induction of IL-12 release by DC stimulated in vitro with VSV-G containing ELVs (Fig. 4B). The increase in frequency and cytolytic activity of CD8+ T cells specific for an exosomal antigen further supports the conclusion, that VSV-G enhances the Th1 responses induced by ELV (Fig. 8A and B). Although the ELVs are an exogenous protein vaccine, they stimulated substantial CD8+ T cell responses and the frequency of OVA-specific CD8+ T cells induced by the O-ELV-G was even higher than after a conventional DNA immunization (Fig. 8A).
In our tumor challenge experiment, all groups of mice immunized with ELVs were protected independent of the presence or absence of fusion-competent VSV-G. Most likely, the CTL response induced by ELVs in the absence of VSV-G was already sufficient to control growth of the aggressive B16-OVA tumor cells, thereby masking a potential enhancement of protection by incorporation of VSV-G. The B16-OVA cells are known to express very low levels of MHC class I [32] and the fact that immunized mice were able to control the tumor growth implies that high avidity CTLs were generated.
Taken together, we demonstrated that co-expression of VSV-G and antigen on the surface of exosomes modulates DC activity in a fusion-dependent manner by (1) enhancing DC's co-stimulatory functions (up-regulation of CD80, CD86, CD40), (2) induction of DC effector functions (production of IL-12p70), and (3) acceleration of antigen internalization and presentation by the MHC class I alternative pathway involving acid endolysosomes. In vivo studies demonstrated more efficient antigen-specific cytotoxic CD8+ T cell responses.
Thus, the present study serves as a proof of concept for using VSV-G to enhance immunogenicity of exosomal vaccines. At the same time, the study provides a more general approach for tethering non-exosomal antigens to exosomes by fusing the antigen to the transmembrane and intracytoplasmic domains of VSV-G. This could offer a number of advantages over current strategies of exosome-based vaccines being tested primarily in the cancer field by targeting selected tumor antigens to exosomes after gene-based immunization.
Such strategies could also be exploited for the development of vaccines against intracellular parasites and viruses by incorporation of microbial antigen into VSV-G containing ELVs.
Acknowledgements
This work was supported by the collaborative research projects TIP-VAC (LSHP-CT-2004-012116), DEC-VAC (LSHP-CT-2005-018685) and EUROPRISE (LSHP-CT-2006-037611) granted by the European Commission. We thank Dr. Thomas Grunwald (Department of Molecular and Medical Virology, Ruhr University Bochum, Germany) for his contribution with characterization of exosomes.
References
- 1.Zitvogel L., Fernandez N., Lozier A., Wolfers J., Regnault A., Raposo G. Dendritic cells or their exosomes are effective biotherapies of cancer. Eur J Cancer. 1999;35(Suppl 3):36–38. doi: 10.1016/s0959-8049(99)00090-8. [DOI] [PubMed] [Google Scholar]
- 2.Andre F., Schartz N.E., Chaput N., Flament C., Raposo G., Amigorena S. Tumor-derived exosomes: a new source of tumor rejection antigens. Vaccine. 2002;20(Suppl 4):A28–A31. doi: 10.1016/s0264-410x(02)00384-5. [DOI] [PubMed] [Google Scholar]
- 3.Chaput N., Schartz N.E., Andre F., Taieb J., Novault S., Bonnaventure P. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol. 2004;172:2137–2146. doi: 10.4049/jimmunol.172.4.2137. [DOI] [PubMed] [Google Scholar]
- 4.Taieb J., Chaput N., Zitvogel L. Dendritic cell-derived exosomes as cell-free peptide-based vaccines. Crit Rev Immunol. 2005;25:215–223. doi: 10.1615/critrevimmunol.v25.i3.30. [DOI] [PubMed] [Google Scholar]
- 5.Escudier B., Dorval T., Chaput N., André F., Caby M.P., Novault S. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med. 2005;3:10. doi: 10.1186/1479-5876-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mignot G., Roux S., Thery C., Segura E., Zitvogel L. Prospects for exosomes in immunotherapy of cancer. J Cell Mol Med. 2006;10:376–388. doi: 10.1111/j.1582-4934.2006.tb00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wubbolts R., Leckie R.S., Veenhuizen P.T., Schwarzmann G., Mobius W., Hoernschemeyer J. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J Biol Chem. 2003;278:10963–10972. doi: 10.1074/jbc.M207550200. [DOI] [PubMed] [Google Scholar]
- 8.Chen W., Wang J., Shao C., Liu S., Yu Y., Wang Q. Efficient induction of antitumor T cell immunity by exosomes derived from heat-shocked lymphoma cells. Eur J Immunol. 2006;36:1598–1608. doi: 10.1002/eji.200535501. [DOI] [PubMed] [Google Scholar]
- 9.Kuate S., Cinatl J., Doerr H.W., Uberla K. Exosomal vaccines containing the S protein of the SARS coronavirus induce high levels of neutralizing antibodies. Virology. 2007;362(1):26–37. doi: 10.1016/j.virol.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matlin K.S., Reggio H., Helenius A., Simons K. Pathway of vesicular stomatitis virus entry leading to infection. J Mol Biol. 1982;156:609–631. doi: 10.1016/0022-2836(82)90269-8. [DOI] [PubMed] [Google Scholar]
- 11.Buseyne F., Le Gall S., Boccaccio C., Abastado J.P., Lifson J.D., Arthur L.O. MHC-I-restricted presentation of HIV-1 virion antigens without viral replication. Nat Med. 2001;7:344–349. doi: 10.1038/85493. [DOI] [PubMed] [Google Scholar]
- 12.Marsac D., Loirat D., Petit C., Schwartz O., Michel M.L. Enhanced presentation of major histocompatibility complex class I-restricted human immunodeficiency virus type 1 (HIV-1) Gag-specific epitopes after DNA immunization with vectors coding for vesicular stomatitis virus glycoprotein-pseudotyped HIV-1 Gag particles. J Virol. 2002;76:7544–7553. doi: 10.1128/JVI.76.15.7544-7553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuate S., Stahl-Hennig C., Stoiber H., Nchinda G., Floto A., Franz M. Immunogenicity and efficacy of immunodeficiency virus-like particles pseudotyped with the G protein of vesicular stomatitis virus. Virology. 2006;351:133–144. doi: 10.1016/j.virol.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 14.DuBridge R.B., Tang P., Hsia H.C., Leong P.M., Miller J.H., Calos M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuate S., Stefanou D., Hoffmann D., Wildner O., Uberla K. Production of lentiviral vectors by transient expression of minimal packaging genes from recombinant adenoviruses. J Gene Med. 2004;6:1197–1205. doi: 10.1002/jgm.623. [DOI] [PubMed] [Google Scholar]
- 16.Wagner R., Graf M., Bieler K., Wolf H., Grunwald T., Foley P. Rev-independent expression of synthetic gag-pol genes of human immunodeficiency virus type 1 and simian immunodeficiency virus: implications for the safety of lentiviral vectors. Hum Gene Ther. 2000;11:2403–2413. doi: 10.1089/104303400750038507. [DOI] [PubMed] [Google Scholar]
- 17.Ternette N., Stefanou D., Kuate S., Uberla K., Grunwald T. Expression of RNA virus proteins by RNA polymerase II dependent expression plasmids is hindered at multiple steps. Virol J. 2007;4:51. doi: 10.1186/1743-422X-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitt M.A., Zagouras P., Crise B., Rose J.K. A fusion-defective mutant of the vesicular stomatitis virus glycoprotein. J Virol. 1990;64:4907–4913. doi: 10.1128/jvi.64.10.4907-4913.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz O., Edwards A.D., Schito M., Aliberti J., Manickasingham S., Sher A. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;13:453–462. doi: 10.1016/s1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 20.Coles R.M., Mueller S.N., Heath W.R., Carbone F.R., Brooks A.G. Progression of armed CTL from draining lymph node to spleen shortly after localized infection with herpes simplex virus 1. J Immunol. 2002;168:834–838. doi: 10.4049/jimmunol.168.2.834. [DOI] [PubMed] [Google Scholar]
- 21.Chapple S.D., Jones I.M. Non-polar distribution of green fluorescent protein on the surface of Autographa californica nucleopolyhedrovirus using a heterologous membrane anchor. J Biotechnol. 2002;95:269–275. doi: 10.1016/s0168-1656(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 22.Kovacsovics-Bankowski M., Rock K.L. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267:243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez A., Regnault A., Kleijmeer M., Ricciardi-Castagnoli P., Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat Cell Biol. 1999;1:362–386. doi: 10.1038/14058. [DOI] [PubMed] [Google Scholar]
- 24.Norbury C.C., Princiotta M.F., Bacik I., Brutkiewicz R.R., Wood P., Elliott T. Multiple antigen-specific processing pathways for activating naive CD8+ T cells in vivo. J Immunol. 2001;166:4355–4362. doi: 10.4049/jimmunol.166.7.4355. [DOI] [PubMed] [Google Scholar]
- 25.Rock K.L., Gramm C., Rothstein L., Clark K., Stein R., Dick L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 26.Anton L.C., Snyder H.L., Bennink J.R., Vinitsky A., Orlowski M., Porgador A. Dissociation of proteasomal degradation of biosynthesized viral proteins from generation of MHC class I-associated antigenic peptides. J Immunol. 1998;160:4859–4868. [PubMed] [Google Scholar]
- 27.Pichlmair A., Diebold S.S., Gschmeissner S., Takeuchi Y., Ikeda Y., Collins M.K. Tubulovesicular structures within vesicular stomatitis virus G protein-pseudotyped lentiviral vector preparations carry DNA and stimulate antiviral responses via Toll-like receptor 9. J Virol. 2007;81:539–547. doi: 10.1128/JVI.01818-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Georgel P., Jiang Z., Kunz S., Janssen E., Mols J., Hoebe K. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology. 2007;362:304–313. doi: 10.1016/j.virol.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 29.Granelli-Piperno A., Finkel V., Delgado E., Steinman R.M. Virus replication begins in dendritic cells during the transmission of HIV-1 from mature dendritic cells to T cells. Curr Biol. 1999;9:21–29. doi: 10.1016/s0960-9822(99)80043-8. [DOI] [PubMed] [Google Scholar]
- 30.Blander J.M., Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 31.Blander J.M., Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 32.Mattes J., Hulett M., Xie W., Hogan S., Rothenberg M.E., Foster P. Immunotherapy of cytotoxic T cell-resistant tumors by T helper 2 cells: an eotaxin and STAT6-dependent process. J Exp Med. 2003;197:387–393. doi: 10.1084/jem.20021683. [DOI] [PMC free article] [PubMed] [Google Scholar]