

Abstract
We report the synthesis and antiviral activity of a new family of non-nucleoside antivirals, derived from the indole nucleus. Modifications of this template through Mannich and Friedel-Crafts reactions, coupled with nucleophilic displacement and reductive aminations led to 23 final derivatives, which were pharmacologically tested. Tryptamine derivative 17a was found to have a selective inhibitory activity against human varicella zoster virus (VZV) replication in vitro, being inactive against a variety of other DNA and RNA viruses. A structure-activity relationship (SAR) study showed that the presence of a biphenyl ethyl moiety and the acetylation at the amino group of tryptamine are a prerequisite for anti-VZV activity. The novel compound shows the same activity against thymidine kinase (TK)-competent (TK+) and TK-deficient (TK−) VZV strains, pointing to a novel mechanism of antiviral action.
Keywords: Indole derivatives, Antiviral activity, Varicella zoster virus, TK-deficient strains
Graphical abstract

Highlights
-
•
A library of indole-based derivatives has been designed and synthesized as potential antiviral agents.
-
•
Friedel-Crafts and Mannich reactions were used for the synthesis of different indole derivatives.
-
•
Tryptamine derivative 17a displays significant inhibitory activity against VZV replication.
-
•
A mechanism of action independent from the virus-encoded thymidine kinase is suggested.
1. Introduction
Varicella zoster virus (VZV) is a ubiquitous and highly infectious human virus that belongs to the herpesviridae family. It is classified within the group of α-herpesviruses which also includes herpes simplex virus (HSV) [1]. A VZV primary infection leads to acute varicella or “chickenpox”, while reactivation of latent virus, established in cranial nerve and dorsal root ganglia, causes herpes zoster (shingles). The course of varicella is generally benign in immune-competent children, but can cause severe morbidity and mortality in adults and in immune-compromised individuals [2]. Complications of herpes zoster in immune-competent hosts include post-herpetic neuralgia (PHN), a persistent pain syndrome, which is the most challenging complication particularly in older individuals [2], [3]. Central nervous system (CNS) complications can follow both primary infection and reactivation of VZV [4], [5]. The most serious manifestations arise when VZV invades the spinal cord or cerebral arteries after reactivation of the virus, causing diseases such as myelitis and focal vasculopathies [2], [4], [5]. Other neurological complications of herpes zoster include motor neuropathy, particularly in patients with zoster ophthalmicus [6], [7]. In patients with the acquired immune deficiency syndrome (AIDS), transplant recipients, and cancer patients, VZV infection can be associated with severe acute retinal necrosis (ARN), a disease with poor prognosis [8], [9]. The outcomes of varicella and herpes zoster have been dramatically improved by the development of safe and effective antiviral drugs with potent activity against VZV [10]. Three oral guanine-based antivirals are approved worldwide for the treatment of VZV-associated diseases: acyclovir, valacyclovir, and famciclovir [11]. The thymidine analog brivudin has been licensed for the therapy of herpes zoster in some European and Central American countries [12]. These drugs are (a) nucleoside analogs that after predominant phosphorylation by the virus-encoded thymidine kinases (TKs), act as competitive inhibitors of the viral DNA polymerase or alternate substrates to the natural triphosphates, inhibiting DNA replication [13]. Other anti-VZV nucleoside inhibitors such as the stearyl/valyl diester valomaciclovir and the valyl-ester prodrug of the bicyclic nucleoside analog (BCNA) FV100 are under clinical investigation [14], [15], [16], [17].
One of the limitations of the use of nucleoside derivatives is the emergence of single and multiple drug resistance which could be partially avoided with the use of non nucleoside compounds [18], [19]. A drug of choice for treatment of acyclovir-resistant VZV disease is foscarnet, a direct inhibitor of viral DNA polymerase that is not dependent on viral TK for activation [20], [21], [22]. A number of small molecules have been identified and reported as potent and selective VZV inhibitors with different mechanisms of action. Some examples are the 4-oxo-dihydroquinoline [23] and 4-oxo-dihydrothieno [2,3-b]pyridine derivatives [24] as inhibitors of the viral DNA polymerases, the oxadiazolephenyl derivative (ASP2151) as a helicase-primase inhibitor [25], and N-α-methylbenzyl-N′-arylthiourea derivatives that interfere with the function of the viral ORF54 protein, impairing morphogenesis of the capsid [26], [27]. Finally, a series of 4-benzyloxy-γ-sultone derivatives has been also reported as non-selective VZV inhibitors with unknown mechanism of action [28]. Given the difficulty of identifying initial hit compounds in this field, where the synthesized compounds are in primis subject to a cellular screening, we considered of interest to use a privileged scaffold as effective starting point in the search for anti-VZV ligands [29]. Indole and its bioisosteres, as privileged scaffolds, represent one of the most important structural motifs in drug discovery [30], [31], [32], and it is widely used in antiviral research [32], [33]. Arbidol [34] and delavirdine [35], are examples of marketed indole-containing antiviral drugs, whereas Panobinostat (LBH589) [36], being a HDAC (histone deacetylase) inhibitor, is actively undergoing clinical evaluation against human immunodeficiency virus (HIV) type 1 (See Fig. 1 ).
Fig. 1.

Indole-containing antiviral compounds. Structure of indole (A, B) and tryptamine (C) derivatives described in this work.
However the use of the indole-based structures in the research of anti-herpes virus agents is rather unusual. Hence we explored the minimum structural requirements for anti-VZV activity starting from this easily derivatizable scaffold. Two small libraries were synthesized based on substituted indoles (A, B) and tryptamines (C). Their cytotoxic and antiviral activity was then evaluated using cellular assays. Some interesting structure-activity relationships were evidenced regarding the N-1 and C-3 substituents.
2. Results and discussion
2.1. Chemistry
Compounds 4a-d were prepared starting from indole 1 according to Scheme 1 . N-1 alkylation of 1 with propyl iodide or 4-phenylbenzyl iodide in DCM/DMF using NaH as base, gave the corresponding intermediates 2 and 3, respectively. The 3-acyl derivative 4a was obtained from 2 by Friedel-Crafts acylation, using 4-chlorobenzoyl chloride and AlCl3 in acetonitrile (32% yield). Functionalization of 2 and 3 through a Mannich reaction, using formaldehyde and piperidine/or biphenyl ethyl amine, and TFA as catalyst, led to 3-methylamine derivatives 4b-d (25–38% yield).
Scheme 1.

Synthesis of N- substituted-3- acyl or 3-methylamine indole derivatives 4a-d. a Reagents and conditions, i: NaH, n-propyl iodide or 4-phenylbenzyl iodide, DCM/DMF, r.t, ii: AlCl3, 4-chlorobenzoyl chloride, CH3CN. iii: CH2O, TFA, RNH2, DCM, r.t.
The 5-substituted indole derivatives (8a-c) were synthesized using the indole-5-carboxyaldehyde (5) and the 5-aminoindole (9) as starting material and following the two-synthetic strategy indicated in Scheme 2 . 5 was first N-alkylated (6) and then subjected to a Mannich reaction to obtain the corresponding aldehydes 7a and 7b, as described above. Treatment of these intermediates with 4-chloro aniline and sodium triacetoxyborohydride in reductive amination conditions, led to final products 8a and 8b in 24% and 30% yield, respectively. On the other hand, reaction of 9 with benzoic acid using HOBt/HBTU as coupling agents gave N-(1H-indol-5-yl)benzamide 10 in 63% yield. N-alkylation of 10 with n-propyl iodide followed by Mannich reaction with formaldehyde and piperidine afforded final compound 8c.
Scheme 2.

Synthesis of 5-Substituted-3-methylamine indole derivatives 8a-c.a Reagents and conditions, i: NaH, n-propyl iodide, DCM/DMF, r.t; ii: CH2O, TFA, piperidine or biphenylethylamine, DCM, r.t; iii: 4-Cl aniline, DCM/CH3COOH, 1,5 h reflux, then (CH3COO)3BH4, 3–5 h, r.t iv: HOBt, HBTU, DIPEA, benzoic acid, DCM/DMF, v: CH2O, TFA, piperidine, DCM, r.t.
The tryptamine-based derivatives 15, 16a-i were prepared following Scheme 3 . N-alkylation of 3-(2-bromoethyl)-1H-indole (12) with methyl iodide or 4-phenylbenzyl iodide led to derivatives 13 and 14 in 67% and 61% yield, respectively. Nucleophilic displacement of the bromine atom in these intermediates by different commercially available amines was performed under microwave conditions, using palladium acetate as catalyst, obtaining the compounds 15, 16a-i in 38–75% yield [37].
Scheme 3.

Synthesis of tryptamine derivatives 15, 16 and 17. a Reagents and conditions, i: NaH, n-propyl iodide or 4-phenylbenzyl iodide, DCM/DMF, r.t; ii: methyl 4-(aminomethyl)benzoate, (CH3COO)2Pd, TEA, THF, μW 100 °C, 20′ iii: R'NH2 CH3COO)2Pd, TEA, THF, μW 100 °C, 20′, iv ClCOCH3, TEA, DCM, 30′, r.t.
Treatment of compounds 16a and 16d-i with acetyl chloride gave the corresponding acetyl derivatives 17a and 17d-i (42–58% yield).
Analogously, treatment of 16a with different aromatic and aliphatic acyl chlorides gave the corresponding acylated compounds 18a-c (48–57% yield). The carbamoyl derivative 18d was obtained in 58% yield by reaction of 16a with di-tert-butyl dicarbonate in DCM/TEA (See Scheme 4).
Scheme 4.

Synthesis of acyl derivatives 18a-c. a Reagents and conditions, i: ClCOR or Boc2O, TEA, DCM, 30′-24 h, r.t.
The acyl derivatives 17, except 17d and 17e, and 18 were obtained as a mixture of rotamers due to the atropisomerism of the amide function, as confirmed by ROESY experiments, which showed exchange cross-peaks between the hydrogen signals of the two conformers (as examples see NMR spectra of compounds 17f and 17 d in Supporting Information) [38], [39].
2.2. Antiviral activity
First, compounds 4, 8, and 15, 16 and 17a were evaluated for their ability to inhibit the replication of VZV in human embryonic lung (HEL) cells, and the results were compared to those obtained for the reference compounds acyclovir and brivudin (Table 1 ). The antiviral activity was expressed as EC50, being the compound concentration required to reduce virus-plaque formation (VZV) by 50%.
Table 1.
Activity of compounds 4, 8, 15, 16 and 17a against varicella-zoster virus (VZV) in human embryonic lung (HEL) cells.
| Comp. | R | R1 | EC50 (μM)a |
CC50 (μM)b | ||
|---|---|---|---|---|---|---|
| TK+ |
TK- |
|||||
| OKA | 07–1 | YS-R | ||||
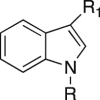 | ||||||
| 4a | Propyl | Phenylcarbonyl | >100 | >100 | nd | >100 |
| 4b | Propyl | Piperidinemethyl | >100 | >100 | nd | >100 |
| 4c | Propyl | Biphenylethylaminemethyl | >0.8 | >0.8 | nd | 1.7 |
| 4d | Biphenylmethyl | Piperidinemethyl | >4 | >4 | nd | 6.9 |
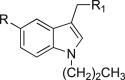 | ||||||
| 8a | 4-Ph-NHCH2 | Piperidine | >4 | >4 | nd | 15 |
| 8b | 4-Ph-NHCH2 | Biphenylethylamine | >4 | >4 | nd | 9.7 |
| 8c | Ph-CO-NH | Piperidine | >20 | >20 | nd | 31 |
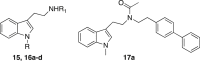 | ||||||
| 15 | Biphenyl methyl | Biphenyl ethyl | nd | >4 | >4 | 7.3 |
| 16a | Methyl | Biphenyl ethyl | >0.8 | >4 | nd | 20 |
| 16b | Methyl | 4-(OCH3)benzyl | >20 | >20 | nd | 28 |
| 16c | Methyl | 4-(Cl) benzyl | >4 | >4 | nd | 22 |
| 16d | Methyl | 2-naphtyl | >100 | >100 | nd | 41 |
| 17a (1)c | 3.6 | 3.1 | nd | 31 | ||
| 17a (2) | 2.5 | 1.7 | 2.1 | 39 | ||
| Acyclovir (1) | 2.1 | 26 | 224 | |||
| Acyclovir (2) | 1.9 | 165 | nd | 191 | ||
| Brivudin (1) | 0.019 | 33 | 300 | |||
| Brivudin (2) | 0.035 | 103 | nd | 160 | ||
Effective concentration required to reduce virus plaque formation by 50%. Virus input was 20 plaque forming units (PFU).
Cytotoxic concentration required to reduced cell growth by 50%.
Experiment number.
As shown in Table 1, the N-propyl-3-acyl (4a) and N-propyl-3-piperidine methyl (4b) derivatives do not displayed antiviral or cytotoxic activities at 100 μM, while the substitution of piperidine at the 3-position by a biphenylethylamine group and of the propyl group at the N-1 indole by a biphenylmethyl moiety dramatically increased the cytotoxic activity of resultant compounds 4c and 4d, with CC50 values of 1.7 and 6.9 μM, respectively. Compounds 8 a-c, derivatized at the C-5 indole position, proved less cytotoxic, CC50 ≈ 10–30 μM, but were also ineffective as inhibitors of VZV-induced plaque formation at similar concentrations.
The most interesting results were obtained for the tryptamine derivatives. In this series, the presence of a methyl group at the N-indole position lead to weaker cytotoxic derivatives (16a-d versus 15, CC50 20–41 versus 7.3 μM), while the acylation of the amine group (17a) also reduces cytotoxicity (CC50 ≈ 31–39 μM) but with an inhibitory effect on the anti-VZV activity. Tryptamine derivative 17a was indeed able to inhibit replication of TK+ and TK- VZV strains with EC50 values in the range of 1.7–3.6 μM (Table 1). The potency of 17a against OKA strain (EC50 = 2.5–3.6 μM) was found comparable to that of the reference drug acyclovir (EC50 = 1.9–2.1 μM) but two orders of magnitude lower than that of brivudin (EC50 = 0.02–0.03 μM). However, the activity of this compound against thymidine kinase-deficient VZV strains, both 07-1 and YS-R, was maintained in the micromolar range with EC50 = 1.7–3.1 μM being, at least, one order of magnitude more active than acyclovir (EC50 = 33–103 μM) and brivudin (EC50 = 26–165 μM).
The activity of these compounds against other members of the Herpesviridae family was tested in HEL cell cultures. As shown in Table 2 none of these compounds was found active either towards cytomegalovirus (HCMV, AD-169 strain and Davis strain) or against herpes simplex virus-1 (HSV-1; KOS and thymidine kinase-deficient acyclovir-resistant) (HSV-1/TK- KOS ACVr strains) or against herpes simplex virus-2 (HSV-2; G strain).
Table 2.
Activity of compounds 4, 8, 15, 16, and 17a against cytomegalovirus (HCMV), herpes simplex virus-1 (HSV-1) and herpes simplex virus-1 (HSV-2) in human embryonic lung (HEL) cell cultures.
| Comp. | Antiviral activity EC50 (μM)a |
Cytotoxicity (μM) |
||||
|---|---|---|---|---|---|---|
| HMCV |
HSV-1 |
HSV-2 |
MMCb | |||
| AD-169 | Davis | (KOS) | TK− KOS ACVr | (G) | ||
| 4a | nd | >100 | >100 | >100 | >100 | >100 |
| 4b | >4 | >4 | >100 | >100 | >100 | >100 |
| 4c | nd | >0.16 | >100 | >100 | >100 | 4 |
| 4d | nd | >4 | >20 | >20 | >20 | 20 |
| 8a | nd | >4 | >20 | >20 | >20 | 20 |
| 8b | nd | >4 | >4 | >4 | >4 | 20 |
| 8c | nd | >20 | >100 | >100 | >100 | 100 |
| 15 | >4 | >4 | >100 | >100 | >100 | 20 |
| 16a | >4 | >4 | >100 | >100 | >100 | >4 |
| 16b | >20 | >20 | >20 | >20 | >20 | 100 |
| 16c | >20 | >20 | >20 | >20 | >20 | 20 |
| 16d | 76 | 100 | >100 | >100 | >100 | >100 |
| 17a | >4 | >4 | >100 | >100 | >100 | 20 |
| Ganciclovir | 3.1 | 2.1 | 0.09 | 100 | 0.03 | >100 |
| Cidofovir | 0.38 | 0.51 | 0.4 | 1.2 | 0.7 | >250 |
| Acyclovir | 0.08 | 112 | 0.2 | >250 | ||
| Brivudin | 0.007 | 50 | 96 | >250 | ||
Effective concentration required to reduce virus plaque formation by 50%. Virus input was 20 plaque-forming units (PFU).
Maximum cytotoxic concentration that cause a microscopically detectable alteration of morphology.
The compounds were not active against other DNA viruses such as vaccinia virus, adenovirus-2, and feline herpesvirus. All these compounds also lacked inhibitory activity against a broad variety of RNA viruses, including HIV-1 and HIV-2, feline coronavirus, vesicular stomatitis virus, Coxsackie virus B4 and respiratory syncytial virus, para-influenza-3 virus, reovirus-1, Sindbis virus, Coxsackie virus B4, Punta Toro virus, influenza A virus (H1N1 and H3N2 subtypes) and influenza B virus (data not shown).
To further explore the SAR within the tryptamine series, we next prepared a second set of 17a analogs featuring a) a modified aromatic moiety linking the acetamide group (compounds 17 d-i) and b) a modified acyl group (compounds 18a-d).
As shown in Table 3 , substitution of biphenyl ethyl moiety by the more rigid naphthyl (compounds 17d, 17e) or their superior analog naphthylmethyl groups (17f, 17g), as well as, the introduction of a biphenylmethyl or phenyloxybenzyl moieties (17h, 17i) led to a complete loss of the antiviral activity. However, compounds containing 1-naphtyl (17e) or 1-naphtyl methyl (17g) groups or the inferior analog biphenylmethyl group (17h) were less cytotoxic. On the other hand, substitution of the acylmethyl group by bulkier alkyl groups such as cyclohexyl, morpholinethyl and tert-butyloxy groups (compound 18a, 18c, 18d) led to an annihilation of antiviral activity. In this series of compounds, only the benzoyl derivative 18b somewhat maintains the antiviral activity at an EC50 of 20 μM, displaying also a reduced cytotoxic activity (MMC >100 μM).
Table 3.
Activity of acyl analogs 17 and 18 against varicella-zoster virus (VZV) in human embryonic lung (HEL) cells..
| Compounds | R | R1 | EC50 (μM)a |
MCC (μM)b | |
|---|---|---|---|---|---|
| TK+ |
TK- |
||||
| OKA | 07–1 | ||||
| 17a | Biphenylethyl | Methyl | 2.6 | 2.0 | 35 |
| 17d | 2-naphtyl | >4 | >4 | 20 | |
| 17e | 1-naphtyl | >20 | >20 | 100 | |
| 17f | 2-naphtylmethyl | >4 | >4 | 20 | |
| 17g | 1-naphtylmethyl | >20 | >20 | 100 | |
| 17h | Biphenylmethyl | >20 | >20 | 100 | |
| 17i | Phenyloxybenzyl | >4 | >4 | 20 | |
| 18a | Cyclohexyl | >4 | >4 | 20 | |
| 18b | Phenyl | 20 | 20 | >100 | |
| 18c | Morpholinethyl | >4 | >4 | 20 | |
| 18d | t-butyloxy | >4 | >4 | 20 | |
| Acyclovir | 3.8 | 145 | >440 | ||
| Brivudin | 0.026 | 143 | >300 | ||

Effective concentration required to reduce virus plaque formation by 50%. Virus input was 20 plaque forming units (PFU).
Minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology.
3. Conclusions
The identification of a new family of non-nucleoside anti-VZV agents based upon the indole template has been reported. We have shown that the substitution on the tryptamine moiety strongly influences the activity/toxicity of these compounds. In particular, the concomitant presence of a biphenylethyl and a methyl(phenyl)carboxy group at the amino group of tryptamine is a requisite for antiviral activity. In fact, the N-biphenylethyl acetamide derivative 17a displayed an interesting inhibition against VZV and was endowed with a selectivity of 10–20 (ratio CC50/EC50). It must be also considered that 17a showed similar activity against TK+ and TK- VZV strains, thus indicating a mechanism of action independent from the virus-encoded thymidine kinase. Additionally, this compound is able to exert antiviral activity exclusively on VZV, being totally inactive against other members of Herpesviridae family as well as against a wide variety of RNA virus strains suggesting a selective action on a specific antiviral target untargeted by the currently existing compounds. The possible mechanism of action of this novel class of compounds will be further investigated.
4. Experimental section
4.1. Chemistry
Reagents, starting materials, and solvents were purchased from Sigma-Aldrich (Milan, Italy) and used as received. Reactions were carried out with magnetic stirring in round-bottomed flasks unless otherwise noted. Moisture-sensitive reactions were conducted in oven-dried glassware under a positive pressure of dry nitrogen, using pre-dried, freshly distilled solvents. Microwave assisted reactions were performed in a Biotage Initiator + reactor. Analytical thin layer chromatography (TLC) was performed on pre-coated glass silica gel plates 60 (F254, 0.25 mm, VWR International). Purifications were performed by flash column chromatography on silica gel (230–400 mesh, Merck Millipore). NMR spectra were recorded on Varian Mercury-400 apparatus. 1HNMR and 13C NMR spectra were recorded with a Varian-400 spectrometer, operating at 400 and 100 MHz, respectively. Chemical shifts are reported in δ values (ppm) relative to internal Me4Si, and J values are reported in hertz (Hz). The following abbreviations are used to describe peaks: s (singlet), d (doublet), dd (double double), t (triplet), q (quadruplet) and m (multiplet). ESI-MS experiments were performed on an Applied Biosystem API 2000 triple-quadrupole spectrometer. Combustion microanalyses were performed on a Carlo Erba CNH 1106 analyzer, and were within 0.4% of calculated values and confirmed >95% purity for the final products.
4.1.1. General procedure for the synthesis of N-alkylated indole intermediates (2, 3, 6, 11, 13 and 14)
Indole (1, 1.0 eq) or 1H-indole-5-carbaldehyde (5, 1.0 eq), or N-(1H-indol-5-yl)benzamide (10, 1.0 eq), or 3-(2-Bromoethyl)indole (13, 1.0 eq.) were dissolved in a mixture of anhydrous DCM/DMF (2/1 v/v) under magnetic stirring and the temperature was set to 0 °C. To this solution, 1.5 equivalents of NaH were added portionwise and the mixture was allowed to react for 30 min. Then, 1.5 equivalents of alkyl iodide [methyl iodide, or n-propyl iodide, or 4-(phenyl)iodomethylbenzene] in DCM were added dropwise and the reaction was warmed to room temperature and maintained under stirring for further 12 h. The reaction was then quenched by a 10% aqueous solution of citric acid and washed with brine. The organic layer was separated, dried over anhydrous Na2SO4, filtered and evaporated in vacuo. Crude products were purified by column chromatography using n-hexane/ethyl acetate (4:1 v:v) as mobile phase. N-alkylated compounds were obtained in 75% yield (derivative 2); 80% yield (derivatives 3 and 13); 67% yield (derivative 6); 55% yield (derivative 11) and 50% yield (derivative 14).
4.1.2. Synthesis of derivative 4a
To a solution of 1-propyl-1H-indole (2) in dry CH3CN was added aluminium trichloride (15 eq) and benzoyl chloride (10 eq), the reaction was stirred at room temperature overnight. The resulting mixture was dried in vacuo and reconstituted in DCM. The organic phase was washed with water (3 × 50 mL), dried over anhydrous Na2SO4, filtered, concentrated and purified by column chromatography using n-hexane/ethyl acetate (3/2) as mobile phase. Compound 4a was obtained in 32% yield.
4.1.3. Synthesis of derivatives 4b-d
A solution of formaldehyde (2 eq.), trifluoroacetic acid (2 eq.) and 2 equivalents of the proper amine (piperidine or bifenylethilamine) was added portionwise to a solution of the intermediate 2 or 3 (1 eq.) in DCM. The mixture was stirred for 3 h at room temperature, then was washed with water (3 × 50 mL), dried over anhydrous Na2SO4, filtered, concentrated and purified by column chromatography using DCM/MeOH (9:1 v/v) as mobile phase. Compounds 4b-d were obtained in 25%–38% yield.
4.1.4. Synthesis of derivatives 8a-b
The intermediate 7a or 7b (1.0 eq) was dissolved in a solution of DCM:CH3COOH (5:1 v/v) at room temperature. To this solution 2.0 equivalents of 4-chloroaniline was added and the mixture was warmed to reflux for 1.5 h. Then, 1.8 equivalents of sodium triacetoxyborohydride were added portionwise and the mixture was allowed to reflux for further 3–5 h. After cooling to room temperature, NaOH 1 N was added. The organic phase was separated and extracted one more time with the alkaline solution. Then it was dried over Na2SO4, filtered and concentrated in vacuo. The crude products were purified by column chromatography using mixtures of DCM/MeOH as eluent giving desired compounds 8a and 8b in 24% and 30% yield respectively.
4.1.5. Synthesis of derivative 8c
To a solution of 5-amino indole (9, 1.0 eq) in dichloromethane/DMF (20 mL/5 mL) were successively added benzoic acid (1.1 eq), HBTU (1.2 eq), HOBt (1.2 eq), and DIEA (2.4 eq), and stirring was continued at room temperature for 24 h. Afterward, the reaction mixture was diluted with dichloromethane (20 mL), and the resulting solution was washed successively with 10% citric acid (2 × 25 mL), 10% NaHCO3 (2 × 25 mL), and water (2 × 25 mL), dried over Na2SO4, and evaporated to dryness. Intermediate 10 was obtained after flash chromatography using n-hexane/ethyl acetate 2/1 as ratio with 63% yield. Substitution with propyl group of indolic N1 was carried out using the same conditions described elsewhere, to give compound 11 with 44% yield. Mannich reaction performed on position 3 of intermediate 11 follows the procedure previously described yielded final compound 12 in 24% yield.
4.1.6. General procedure for the synthesis of derivatives 15, and 16a-i
One equivalent of intermediate 14a or 14b was dissolved in THF and 1.5 eq of the proper amine, 1.5 eq of TEA, 1.5 eq of NaI and 0.3 eq of (CH3COO)2Pd were added to this solution. The reaction was conducted under μW, at 100 °C, for 20 min. The resulting mixture was filtered through Celite, dried in vacuo and reconstituted in DCM. The organic phase was washed with water (3 × 50 mL), dried over anhydrous Na2SO4, filtered, concentrated and purified by column chromatography using DCM/MeOH (9:1 v/v) as mobile phase giving derivative 15 in 38% yield and intermediates 16a-i in 55–75% yield.
4.1.7. General procedure for the synthesis of derivatives 17a-i and 18a-c
One equivalent of intermediate 16a-i was dissolved in DCM and 1.5 eq of the proper acyl chloride and 1.5 eq of TEA, were added to the solution. The reaction was conducted for 20 min at room temperature. The organic phase was washed with water (3 × 50 mL), dried over anhydrous Na2SO4, filtered, concentrated and purified by column chromatography using n-hexane/ethyl acetate (2:1 v/v) as mobile phase. Final derivatives 17a-i and 18a-d were obtained as an atropisomer mixtures in 42–58% yield.
4.1.8. Synthesis of derivative 18d
To a mixture of 16a (1.0 eq.) in DCM, (Boc)2O (3.0 eq.) was added followed by TEA (1.5 eq.). The mixture was allowed to stir at room temperature for 12 h. Afterward, the reaction mixture was diluted with dichloromethane (20 mL), and the resulting solution was washed with 10% citric acid (2 × 25 mL). The organic phase was dried over Na2SO4, and evaporated to dryness. Compound 17 k was obtained as atropisomer mixture after flash chromatography using n-hexane/ethyl acetate 2/1 in 58% yield.
4.2. Antiviral assays
The compounds were evaluated against different herpes viruses, including varicella-zoster virus (VZV) strains Oka and YS, TK− VZV strains 07-1 and YS-R, herpes simplex virus type 1 (HSV-1) strain KOS, thymidine kinase-deficient (TK−) HSV-1 KOS strain resistant to ACV (ACVr), herpes simplex virus type 2 (HSV-2) strain G, human cytomegalovirus (HCMV) strains AD-169 and Davis as well as feline herpes virus (FHV), the poxvirus vaccinia virus (Lederle strain), adenovirus-2, parainfluenza-3 virus, reovirus-1, Sindbis virus, Coxsackie virus B4, Punta Toro virus, respiratory syncytial virus (RSV), feline coronavirus (FIPV) and influenza A virus subtypes H1N1 (A/PR/8), H3N2 (A/HK/7/87) and influenza B virus (B/HK/5/72) and human immune deficiency virus (HIV-1 (IIIB) and HIV-2 (ROD)). The antiviral assays were based on inhibition of virus-induced cytopathicity (CPE) or (VZV) plaque formation in human embryonic lung (HEL) fibroblasts, African green monkey kidney cells (Vero), human epithelial cervix carcinoma cells (HeLa), human CD4 + T-lymphocyte cells (CEM), Crandell-Rees feline kidney cells (CRFK), or Madin Darby canine kidney cells (MDCK). Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) or with 20 VZV plaque forming units (PFU) and the cell cultures were incubated in the presence of varying concentrations of the test compounds. Viral CPE or plaque formation (VZV) was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50, the concentration required to reduce virus-induced cytopathicity or viral plaque formation by 50%.
4.3. Cytotoxicity assays
Cytotoxicity measurements were based on the inhibition of HEL cell growth. HEL cells were seeded at a rate of 5 × 103 cells/well into 96-well microtiter plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37 °C, the cell number was determined with a Coulter counter. The 50% cytostatic concentration (CC50) was calculated as the compound concentration required to reduce cell growth by 50% relative to the number of cells in the untreated controls. CC50 values were estimated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds. Cytotoxicity was expressed as the minimum cytotoxic concentration (MCC) or the compound concentration that causes a microscopically detectable alteration of cell morphology.
Funding
This work was supported by grant from the Italian Ministry of Education (MIUR) (PRIN n°2010-11E61J12000210001). The biological part of this work was supported by the KU Leuven (GOA 15/19 TBA).
Acknowledgements
The authors wish to express their gratitude to Mrs. Leentje Persoons, Mrs. Lies Van Den Heurck and Mrs. Ellen De Waegenaere for excellent technical assistance.
Footnotes
Supplementary data associated with this article can be found in the online version, at http://dx.doi.org/10.1016/j.ejmech.2016.09.014. These data include MOL files and InChiKeys of the most important compounds described in this article.
Contributor Information
Alessia Bertamino, Email: abertamino@unisa.it.
Isabel M. Gomez-Monterrey, Email: imgomez@unina.it.
Appendix A. Supplementary data
NMR and mass spectrometry data for synthesized compounds. Mono and bidimensional NMR spectra for compounds 17d and 17f.
Mol Files
The following ZIP file contains the MOL files of the most important compounds referred to in this article.
MOL file. ZIP file containing the MOL files of the most important compounds in this article.
References
- 1.Rahaus M., Schunemann S., Wolff M.H. Morphological and biological characteristics of varicella-zoster virus. Contrib. Microbiol. 1999;3:1–9. doi: 10.1159/000060312. [DOI] [PubMed] [Google Scholar]
- 2.Mueller N.H., Gilden D.H., Cohrs R.J., Mahalingam R., Nagel M.A. Varicella zoster virus infection: clinical features, molecular pathogenesis of disease, and latency. Neurol. Clin. 2008;26:675–697. doi: 10.1016/j.ncl.2008.03.011. (viii) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson R.W. Herpes zoster and postherpetic neuralgia. Expert Rev. Vaccines. 2010;9:21–26. doi: 10.1586/erv.10.30. [DOI] [PubMed] [Google Scholar]
- 4.Gilden D. Varicella zoster virus and central nervous system syndromes. Herpes J. IHMF. 2004;11(Suppl 2):89A–94A. [PubMed] [Google Scholar]
- 5.Kennedy P.G. Varicella-zoster virus latency in human ganglia. Rev. Med. Virol. 2002;12:327–334. doi: 10.1002/rmv.362. [DOI] [PubMed] [Google Scholar]
- 6.Gilden D., Cohrs R.J., Mahalingam R., Nagel M.A. Varicella zoster virus vasculopathies: diverse clinical manifestations, laboratory features, pathogenesis, and treatment. Lancet Neurol. 2009;8:731–740. doi: 10.1016/S1474-4422(09)70134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilden D.H., Kleinschmidt-DeMasters B.K., LaGuardia J.J., Mahalingam R., Cohrs R.J. Neurologic complications of the reactivation of varicella-zoster virus. N. Engl. J. Med. 2000;342:635–645. doi: 10.1056/NEJM200003023420906. [DOI] [PubMed] [Google Scholar]
- 8.Hillenkamp J., Nolle B., Rautenberg P., Fickenscher H., Roider J. Acute retinal necrosis: clinical features and therapy options. Ophthalmol. Z. Dtsch. Ophthalmol. Ges. 2009;106:1058–1064. doi: 10.1007/s00347-009-2047-5. [DOI] [PubMed] [Google Scholar]
- 9.Vafai A., Berger M. Zoster in patients infected with HIV: a review. Am. J. Med. Sci. 2001;321:372–380. doi: 10.1097/00000441-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Gnann J.W., Jr. Antiviral therapy of varicella-zoster virus infections. In: Arvin A., Campadelli-Fiume G., Mocarski E., Moore P.S., Roizman B., Whitley R., Yamanishi K., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. 2007. Cambridge. [Google Scholar]
- 11.Snoeck R., Andrei G., De Clercq E. Current pharmacological approaches to the therapy of varicella zoster virus infections: a guide to treatment. Drugs. 1999;57:187–206. doi: 10.2165/00003495-199957020-00005. [DOI] [PubMed] [Google Scholar]
- 12.Breuer J., Whitley R. Varicella zoster virus: natural history and current therapies of varicella and herpes zoster. Herpes J. IHMF. 2007;14(Suppl 2):25–29. [PubMed] [Google Scholar]
- 13.De Clercq E. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin. Microbiol. Rev. 2003;16:569–596. doi: 10.1128/CMR.16.4.569-596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrei G., Snoeck R. Advances in the treatment of varicella-zoster virus infections. Adv. Pharmacol. 2013;67:107–168. doi: 10.1016/B978-0-12-405880-4.00004-4. [DOI] [PubMed] [Google Scholar]
- 15.Balzarini J., McGuigan C. Chemotherapy of varicella-zoster virus by a novel class of highly specific anti-VZV bicyclic pyrimidine nucleosides. Biochim. Biophy. Acta. 2002;1587:287–295. doi: 10.1016/s0925-4439(02)00091-1. [DOI] [PubMed] [Google Scholar]
- 16.Pentikis H.S., Matson M., Atiee G., Boehlecke B., Hutchins J.T., Patti J.M., Henson G.W., Morris A. Pharmacokinetics and safety of FV-100, a novel oral anti-herpes zoster nucleoside analogue, administered in single and multiple doses to healthy young adult and elderly adult volunteers. Antimicrob. Agents Chemother. 2011;55:2847–2854. doi: 10.1128/AAC.01446-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor S.L., Kinchington P.R., Brooks A., Moffat J.F. Roscovitine, a cyclin-dependent kinase inhibitor, prevents replication of varicella-zoster virus. J. Virol. 2004;78:2853–2862. doi: 10.1128/JVI.78.6.2853-2862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert C., Bestman-Smith J., Boivin G. Resistance of herpesviruses to antiviral drugs: clinical impacts and molecular mechanisms. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2002;5:88–114. doi: 10.1016/s1368-7646(02)00021-3. [DOI] [PubMed] [Google Scholar]
- 19.Kimberlin D.W., Whitley R.J. Antiviral resistance: mechanisms, clinical significance, and future implications. J. Antimicrob. Chemother. 1996;37:403–421. doi: 10.1093/jac/37.3.403. [DOI] [PubMed] [Google Scholar]
- 20.Breton G., Fillet A.M., Katlama C., Bricaire F., Caumes E. Acyclovir-resistant herpes zoster in human immunodeficiency virus-infected patients: results of foscarnet therapy. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1998;27:1525–1527. doi: 10.1086/515045. [DOI] [PubMed] [Google Scholar]
- 21.De Clercq E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wathen M.W. Non-nucleoside inhibitors of herpesviruses. Rev. Med. Virol. 2002;12:167–178. doi: 10.1002/rmv.354. [DOI] [PubMed] [Google Scholar]
- 23.Hartline C.B., Harden E.A., Williams-Aziz S.L., Kushner N.L., Brideau R.J., Kern E.R. Inhibition of herpesvirus replication by a series of 4-oxo-dihydroquinolines with viral polymerase activity. Antivir. Res. 2005;65:97–105. doi: 10.1016/j.antiviral.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Schnute M.E., Cudahy M.M., Brideau R.J., Homa F.L., Hopkins T.A., Knechtel M.L., Oien N.L., Pitts T.W., Poorman R.A., Wathen M.W., Wieber J.L. 4-Oxo-4,7-dihydrothieno[2,3-b]pyridines as non-nucleoside inhibitors of human cytomegalovirus and related herpesvirus polymerases. J. Med. Chem. 2005;48:5794–5804. doi: 10.1021/jm050162b. [DOI] [PubMed] [Google Scholar]
- 25.Chono K., Katsumata K., Kontani T., Kobayashi M., Sudo K., Yokota T., Konno K., Shimizu Y., Suzuki H. ASP2151, a novel helicase-primase inhibitor, possesses antiviral activity against varicella-zoster virus and herpes simplex virus types 1 and 2. J. Antimicrob. Chemother. 2010;65:1733–1741. doi: 10.1093/jac/dkq198. [DOI] [PubMed] [Google Scholar]
- 26.Di Grandi M.J., Curran K.J., Feigelson G., Prashad A., Ross A.A., Visalli R., Fairhurst J., Feld B., Bloom J.D. Thiourea inhibitors of herpesviruses. Part 3: inhibitors of varicella zoster virus. Bioorg. Med. Chem. Lett. 2004;14:4157–4160. doi: 10.1016/j.bmcl.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 27.Visalli R.J., Fairhurst J., Srinivas S., Hu W., Feld B., DiGrandi M., Curran K., Ross A., Bloom J.D., van Zeijl M., Jones T.R., O'Connell J., Cohen J.I. Identification of small molecule compounds that selectively inhibit varicella-zoster virus replication. J. Virol. 2003;77:2349–2358. doi: 10.1128/JVI.77.4.2349-2358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Castro S., Garcia-Aparicio C., Andrei G., Snoeck R., Balzarini J., Camarasa M.J., Velazquez S. 4-Benzyloxy-gamma-sultone derivatives: discovery of a novel family of non-nucleoside inhibitors of human cytomegalovirus and varicella zoster virus. J. Med. Chem. 2009;52:1582–1591. doi: 10.1021/jm8014662. [DOI] [PubMed] [Google Scholar]
- 29.Welsch M.E., Snyder S.A., Stockwell B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cichero E., Ligresti A., Allara M., di Marzo V., Lazzati Z., D'Ursi P., Marabotti A., Milanesi L., Spallarossa A., Ranise A., Fossa P. Homology modeling in tandem with 3D-QSAR analyses: a computational approach to depict the agonist binding site of the human CB2 receptor. Eur. J. Med. Chem. 2011;46:4489–4505. doi: 10.1016/j.ejmech.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 31.Cichero E., Cesarini S., Mosti L., Fossa P. CoMFA and CoMSIA analyses on 1,2,3,4-tetrahydropyrrolo[3,4-b]indole and benzimidazole derivatives as selective CB2 receptor agonists. J. Mol. Model. 2010;16:1481–1498. doi: 10.1007/s00894-010-0664-1. [DOI] [PubMed] [Google Scholar]
- 32.Sharma V., Kumar P., Pathak D. Biological importance of the indole nucleus in recent years: a comprehensive review. J. Heterocycl. Chem. 2010;47:491–502. [Google Scholar]
- 33.Tonelli M., Novelli F., Tasso B., Vazzana I., Sparatore A., Boido V., Sparatore F., La Colla P., Sanna G., Giliberti G., Busonera B., Farci P., Ibba C., Loddo R. Antiviral activity of benzimidazole derivatives. III. Novel anti-CVB-5, anti-RSV and anti-Sb-1 agents. Bioorg. Med. Chem. 2014;22:4893–4909. doi: 10.1016/j.bmc.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 34.Boriskin Y.S., Leneva I.A., Pecheur E.I., Polyak S.J. Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008;15:997–1005. doi: 10.2174/092986708784049658. [DOI] [PubMed] [Google Scholar]
- 35.Freimuth W.W. Delavirdine mesylate, a potent non-nucleoside HIV-1 reverse transcriptase inhibitor. Adv. Exp. Med. Biol. 1996;394:279–289. doi: 10.1007/978-1-4757-9209-6_25. [DOI] [PubMed] [Google Scholar]
- 36.Rasmussen T.A., Schmeltz Sogaard O., Brinkmann C., Wightman F., Lewin S.R., Melchjorsen J., Dinarello C., Ostergaard L., Tolstrup M. Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccines Immunother. 2013;9:993–1001. doi: 10.4161/hv.23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertamino A., Ostacolo C., Ambrosino P., Musella S., Di Sarno V., Ciaglia T., Soldovieri M.V., Iraci N., Fernandez Carvajal A., de la Torre-Martinez R., Ferrer-Montiel A., Gonzalez Muniz R., Novellino E., Taglialatela M., Campiglia P., Gomez-Monterrey I. Tryptamine-based derivatives as transient receptor potential melastatin type 8 (TRPM8) channel modulators. J. Med. Chem. 2016;59:2179–2191. doi: 10.1021/acs.jmedchem.5b01914. [DOI] [PubMed] [Google Scholar]
- 38.Neuhaus D., Williamson M.P. second ed. Wiley; New York: 2000. The Nuclear Overhauser Effect in Structural and Conformational Analysis. [Google Scholar]
- 39.Bertamino A., Soprano M., Musella S., Rusciano M.R., Sala M., Vernieri E., Di Sarno V., Limatola A., Carotenuto A., Cosconati S., Grieco P., Novellino E., Illario M., Campiglia P., Gomez-Monterrey I. Synthesis, in vitro, and in cell studies of a new series of [indoline-3,2′-thiazolidine]-based p53 modulators. J. Med. Chem. 2013;56:5407–5421. doi: 10.1021/jm400311n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MOL file. ZIP file containing the MOL files of the most important compounds in this article.