

Abstract
Two series of 6-(1,2,3-triazolyl)-2,3-dibenzyl-l-ascorbic acid derivatives with the hydroxyethylene (8a−8u) and ethylidene linkers (10c−10p) were synthesized and evaluated for their antiproliferative activity against seven malignant tumor cell lines and antiviral activity against a broad range of viruses. Conformationally unrestricted spacer between the lactone and 1,2,3-triazole units in 8a−8u series had a profound effect on antitumor activity. Besides, the introduction of a long side chain at C-4 of 1,2,3-triazole that led to the synthesis of decyl-substituted 2,3-dibenzyl-l-ascorbic acid 8m accounted for a selective and potent antiproliferative activity on breast cancer MCF-7 cells cells in the nM range. Further analysis showed that compound 8m strongly enhanced expression of hypoxia inducible transcription factor 1 α (HIF-1α) and to some extent decreased expression of nitric oxide synthase 2 (NOS2) suggesting its role in regulating HIF-1α signalling pathway. The p-methoxyphenyl-substituted derivative 10g displayed specific anti-cytomegalovirus (CMV) potential, whereas aliphatic-substituted derivatives 8l and 8m had the most potent, yet relatively non-specific, anti-varicella-zoster (VZV) activity.
Keywords: Vitamin C; Butenolide; 1,2,3-Triazole; HIF-1; NOS2; Antitumoral; Antiviral activity
Graphical abstract

Highlights
-
•
Two series of 1,2,3-triazolyl 2,3-dibenzyl-l-ascorbic acid conjugates were synthesized.
-
•
Conformationally unrestricted spacer had a major effect on antitumor activities.
-
•
Decyl-substituted l-ascorbic acid 8m caused inhibition of breast cancer MCF-7 cells in the nM range.
-
•
8m increased the expression of hypoxia inducible transcription factor HIF-1α.
-
•
p-Methoxyphenyl-substituted derivative 10g had specific anti-CMV activity.
1. Introduction
Even though l-ascorbic acid (L-ASA) cancer therapy has a controversial history [1], interest in L-ASA has been renewed due to its recently demonstrated ability to selectively kill cancer cells [[2], [3], [4], [5]]. It was shown that L-ASA inhibits the growth of KRAS and BRAF mutant colorectal cancer cells by causing oxidative stress and accumulation of reactive oxygen species (ROS), the latter leading to inactivation of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), an energy crisis and consequently cell death [6]. l-ascorbic acid is an established activator of non-heme iron α-ketoglutarate (αKG) dioxygenases, such as hypoxia inducible factor prolyl hydroxylases (HIF PHD), and epigenetic ten-eleven translocation (TET) enzymes, which are directly relevant to cancer susceptibility and progression [7,8]. HIF-1 is a critical mediator of the cellular response to hypoxia, making it an attractive molecular target for anticancer therapy. Hydroxylation of proline in HIF-1α under normoxia tags it for subsequent ubiquitinylation and proteasomal degradation [9]. Activation of the enzyme HIF prolyl hydroxylase inhibits the HIF-triggered antihypoxic program, the latter being the adaptive response of malignant tumors to hypoxic stress. Such activation was recently observed by l-ascorbic acid and its 2-phosphate derivative that promoted tumor cells death [10,11]. Furthermore, recent progress in the epigenetics identified a new role of ascorbate in the regulation of the demethylation of deoxyribonucleic acid (DNA) and histone proteins [12]. It was found that L-ASA acts as a direct regulator of TET activity and DNA methylation in mouse embryonic stem (ES) cells, thus indicating its potential application for treating cancers driven by aberrant DNA methylation in the clinic [13]. l-ascorbate also showed a selective killing effect against various human breast cancer cell lines which was dependent on the sodium-vitamin C transporter 2 (SVCT-2) expression responsible for l-ascorbate uptake [14]. Some 1,2,4-triazole and imidazole L-ASA derivatives exerted a pronounced cytostatic effect in human T-cell acute lymphoblastic leukemia cells [15]. Moreover, pyrimidine and purine derivatives of 6-deoxy-l-ascorbic acid and 4,5-didehydro-5,6-dideoxy-l-ascorbic acid derivatives showed antiproliferative effect in a panel of tumor cells and antiviral potency against VZV and CMV (Fig. 1 ) [[16], [17], [18], [19]]. It was observed that 2,3-O,O-dibenzyl-l-ascorbic acid derivatives exhibited better growth inhibition of malignant tumor cells and antiviral activity than their 2,3-dihydroxy analogues.
Fig. 1.

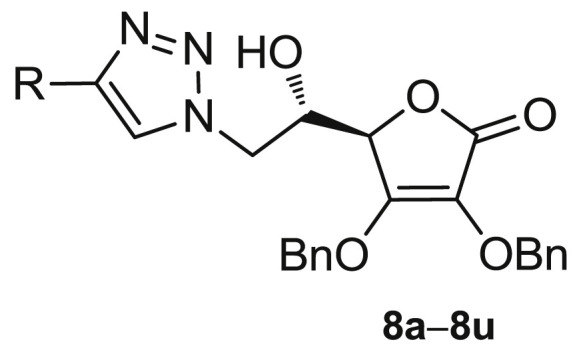
Purine and pyrimidine derivatives of l-ascorbic acid with antiproliferative and antiviral activities and 4-substituted 1,2,3-triazolyl-2,3-dibenzyl-l-ascorbic acid derivatives (8a−8u and 10c−10p).
1,2,3-Triazole has found increased application in the drug discovery because it has been recognized as good amide bioisostere that readily associates with biological targets through hydrogen bonding and dipole−dipole interactions [20]. Accordingly, a series of molecules containing a pentacyclic triterpene moiety connected to L-ASA via 1,2,3-triazole were shown to exhibit anti-influenza A/WSN/33 virus activity, showing that 2,3-O,O-dibenzylated L-ASA skeleton was crucial for their antiviral potency, regardless of the triterpene fragment [21]. Additionally, several aspulvinone compounds containing γ-butenolide moiety showed significant anti-influenza A (H1N1) virus activity [22].
Having in mind these effects and potential synergy of the antitumor and antiviral activity of L-ASA and 1,2,3-triazole moiety, we designed and synthesized l-ascorbic acid derivatives and their C4=C5 unsaturated analogues with a 1,4-disubstituted 1,2,3-triazole core on the C-6 atom hypothesizing that new chemical entities might exhibit a pronounced and selective antitumor and/or antiviral activity. To investigate the influence of substituents at the C-4 of 1,2,3-triazole conjugated with the L-ASA on biological activity, straight and branched alkyl chain of different lengths, cyclopropyl, variously substituted aromatic, benzenesulfonamide and dithiocarbamate moieties were introduced and their biological activity was compared to the unsubstituted triazole l-ascorbic acid conjugate (Fig. 1).
2. Results and discussion
2.1. chemistry
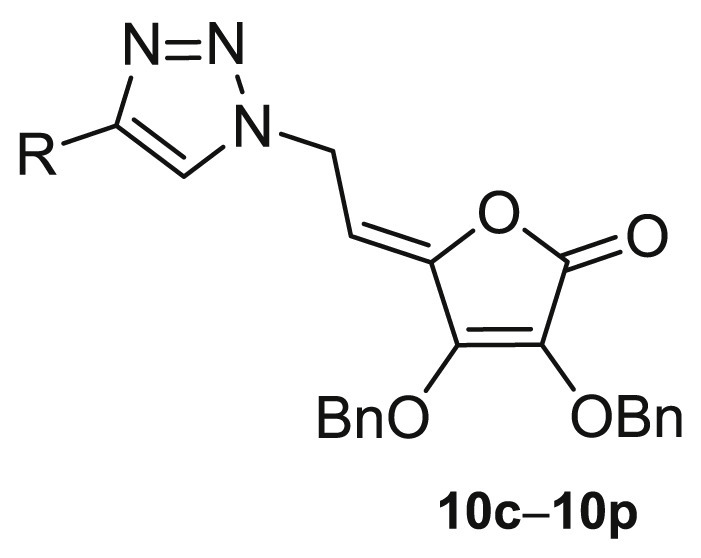
To evaluate the impact of substituents at the C-4 of 1,2,3-triazole and the hydroxyethylene and conformationally restricted ethylidene spacer between the lactone and 1,2,3-triazole moieties on biological activity, two series of 6-(1,2,3-triazolyl)-6-deoxy- (8a−8u) and 6-(1,2,3-triazolyl)-4,5-didehydro-5,6-dideoxy-l-ascorbic acid (10c−10p) derivatives were synthesized as outlined in Scheme 1 .
Scheme 1.

Reagents and conditions: i) acetyl chloride, acetone; ii) benzyl chloride, potassium carbonate (K2CO)3; iii) acetic acid, methanol; iv) tosyl chloride, pyridine, dichloromethane (CH2Cl2); v) sodium azide (NaN3), dimethylformamide (DMF), water; vi) Cu, 1 M copper (II) sulphate (CuSO4), tert-butanol, DMF, water, microwave reactor; vii) NaN3, Cu, 1 M CuSO4, DMF, water, microwave reactor; viii) NaN3, acetonitrile; ix) copper (II) acetate (Cu(OAc)2), methanol.
The isopropylidene protection of 5,6-hydroxyl groups, 2,3-dibenzylation, and subsequent removal of 5,6-isopropylidene in l-ascorbic acid to afford compound 3, were performed according to procedures described in the literature [18,23,24]. The tosylation of the 5,6-hydroxyl groups in compound 3 using tosyl chloride in pyridine afforded the 6-monotosylated 2,3-dibenzyl-l-ascorbic acid derivative 4 and the 5,6-ditosylated compound 5 in the same reaction, which were then used as parent molecules for the synthesis of the corresponding C-6 azido derivatives of 6-deoxy- (6) and 4,5-didehydro-5,6-dideoxy-l-ascorbic acid (9). The azido derivative 6 was synthesized by the reaction of the monotosylated derivative 4 with sodium azide in a yield of 77%. The 1,2,3-triazole was introduced at the C-6 position of the 2,3-dibenzyl-l-ascorbic acid via copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition (CuAAC) of C-6-azido-6-deoxy-2,3-dibenzyl-l-ascorbic acid 6 and the corresponding terminal alkynes 7b−7u (method A, Scheme 1) in yields from 16% to 72%.
A Cu(II) salt in the presence of a reducing agent, metallic copper, was employed to generate the required Cu(I) catalyst in situ. Besides diverse aliphatic and aromatic substituents at C-4, benzenesulfonamide [[25], [26], [27], [28]] and dithiocarbamate [[29], [30], [31]], recognized as important pharmacophores in the design of anticancer agents, were also introduced. Benzenesulfonamide-based terminal alkynes 7q−7t were prepared in a reaction of propargylamine and corresponding benzenesulfonyl chlorides [32], while tert-butyl 4-((prop-2-ynylthio)carbonothioyl)piperazine-1-carboxylate (7u) was synthesized via reaction of piperazine with carbon disulfide (CS2) and propargyl bromide in the presence of a base [33]. Following method A (Scheme 1), the best yields of 72% and 71% were obtained in the synthesis of compounds 8n and 8o with branched 4-tert-butyl and 4-(3-chloropropyl) side chains, respectively. Optimization of reactions was performed using another synthetic pathway (Scheme 1, method B). Method B represents a convenient one-pot two-step procedure to obtain the target compounds by the in situ generation of azide 6 via reaction of the monotosylated l-ascorbic acid derivative 4 with sodium azide, and followed by CuAAC reaction of azide 4 and corresponding alkyne to afford 1,4-disubstituted 1,2,3-triazole derivatives of l-ascorbic acid 8a−8u. For environmental reasons, the microwave-assisted reactions were performed in both methods over a period of 1 h for completion of the reaction. However, comparing the yields of the two synthetic pathways, it can be concluded that the method A, that included the isolation of the C-6 azido derivative 6, was generally more successful than the one-pot procedure (method B).
4-Substituted 1,2,3-triazolyl-4,5-didehydro-5,6-dideoxy-l-ascorbic acid (10c−10p) derivatives containing C-4=C-5 double bond were prepared starting from the ditosylated compound 5 which, by elimination and subsequent nucleophilic substitution with NaN3, gave the 4,5-unsaturated azido derivative 9 [34]. The target compounds 10c−10p were then obtained in a click reaction of the azido derivative 9 and the corresponding terminal alkynes 7c−7p using Cu(OAc)2,- as a catalytic Cu(I) source, under both micro-flow and batch conditions following a previously described procedure [34].
2.2. Biological profiling
2.2.1. In vitro antiproliferative evaluations and in silico physicochemical profiling
A series of 4-substituted 1,2,3-triazolyl 2,3-dibenzyl-l-ascorbic acid conjugates (8a−8u and 10c−10p) were evaluated against seven human tumor cell lines: lung adenocarcinoma (A549), ductal pancreatic adenocarcinoma (CFPAC-1), colorectal carcinoma (HCT-116), cervical carcinoma (HeLa), hepatocellular carcinoma (HepG2), breast adenocarcinoma (MCF-7) and colorectal adenocarcinoma, metastatic (SW620) cell lines as well as normal skin fibroblasts (HFF) (Table 1, Table 2, Table 3 ).
Table 1.
The growth-inhibition effects in vitro of l-ascorbic acid and compounds 8a−8u on selected tumor cell lines and normal fibroblasts.
| Compd. | R | IC50a/μM |
clogPb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A549 | CFPAC-1 | HCT-116 | HeLa | HepG2 | MCF-7 | SW620 | HFF | |||
| 8a | H | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 0.61± 0.42 |
0.80 |
| 8b | Si(CH3)3 | 19.18± 12.63 |
52.56± 6.35 |
31.81± 0.36 |
22.30± 0.36 |
24.11± 0.37 |
12.13± 10.51 |
56.03± 11.09 |
20.02± 1.07 |
3.16 |
| 8c | 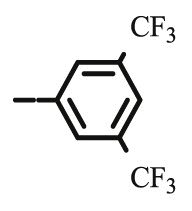 |
28.89± 15.94 |
66.47± 14.92 |
57.19± 43.37 |
35.53± 17.15 |
33.78± 15.04 |
45.89± 21.84 |
49.20± 4.19 |
80.69± 45.52 |
4.25 |
| 8d | 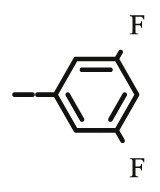 |
41.24± 22.18 |
62.37± 52.22 |
48.24± 61.60 |
40.77± 1.34 |
11.61± 5.52 |
27.73± 30.59 |
>100 | >100 | 2.75 |
| 8e | 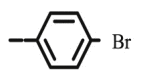 |
0.75± 4.26 |
5.83± 0.06 |
9.42± 1.93 |
3.91± 3.04 |
5.07± 2.61 |
5.62± 2.26 |
3.76± 4.64 |
5.10± 6.76 |
3.28 |
| 8f | 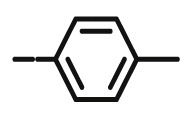 |
8.69± 0.05 |
>100 | >100 | 25.47± 66.03 |
8.77± 0.90 |
>100 | >100 | >100 | 2.89 |
| 8g | 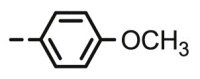 |
28.27± 0.24 |
26.02± 9.79 |
>100 | 16.39± 21.05 |
46.43± 17.89 |
99.73± 0.77 |
58.22± 59.57 |
<0.01 | 2.48 |
| 8h | 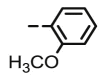 |
>100 | >100 | >100 | >100 | >100 | >100 | >100 | 0.05± 70.14 |
2.48 |
| 8i | 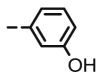 |
59.94± 7.82 |
54.08± 5.17 |
42.87± 19.40 |
39.26± 17.70 |
62.66± 32.48 |
51.84± 11.29 |
>100 | 39.04± 27.59 |
2.20 |
| 8j | 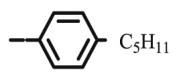 |
6.22± 1.73 |
9.89± 30.48 |
13.83± 31.97 |
8.39± 1.16 |
7.46± 0.43 |
9.01± 12.40 |
12.18± 8.62 |
>100 | 4.67 |
| 8k | 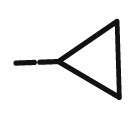 |
43.85± 9.99 |
47.67± 12.21 |
30.36± 3.18 |
26.52± 19.26 |
38.09± 19.04 |
25.91± 1.51 |
54.22± 17.58 |
24.31± 7.04 |
1.97 |
| 8l |  |
28.56± 34.71 |
73.22± 12.44 |
10.36± 17.35 |
18.07± 21.40 |
32.83± 22.34 |
7.91± 11.42 |
47.53± 1.32 |
26.27± 0.40 |
2.52 |
| 8m |  |
6.75± 24.43 |
40.27± 41.70 |
28.83± 32.21 |
22.89± 20.12 |
12.24± 22.17 |
0.08± 55.51 |
45.15± 2.86 |
>100 | 5.25 |
| 8n | 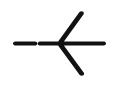 |
33.97± 13.52 |
54.88± 2.03 |
28.26± 2.22 |
18.33± 11.76 |
29.21± 9.51 |
0.73± 8.48 |
35.78± 6.38 |
13.33± 5.15 |
2.44 |
| 8o |  |
29.00± 20.77 |
58.78± 23.78 |
36.08± 19.22 |
36.66± 11.34 |
34.99± 5.45 |
24.53± 18.57 |
68.50± 4.37 |
27.07± 10.95 |
2.30 |
| 8p |  |
>100 | >100 | 72.69± 7.35 |
68.77± 43.87 |
68.06± 1.73 |
63.37± 2.61 |
>100 | 58.53± 41.44 |
0.69 |
| 8q | 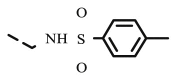 |
16.76± 50.14 |
68.01± 20.79 |
13.09± 22.76 |
14.36± 0.63 |
24.94± 7.80 |
6.40± 11.19 |
45.79± 6.49 |
0.05± 5.63 |
1.74 |
| 8r | 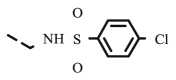 |
2.87± 29.22 |
38.87± 32.64 |
8.38± 0.66 |
13.63± 11.70 |
7.51± 1.60 |
4.17± 24.27 |
18.00± 3.71 |
0.02± 0.03 |
2.00 |
| 8s |  |
39.06± 59.22 |
58.16± 9.96 |
21.61± 13.87 |
20.71± 4.18 |
25.46± 7.57 |
3.39± 14.43 |
42.39± 2.36 |
0.02± 0.02 |
2.10 |
| 8t | 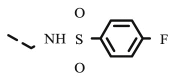 |
22.51± 29.96 |
34.68± 8.04 |
22.90± 2.18 |
24.97± 9.16 |
18.34± 8.31 |
26.81± 11.19 |
41.05± 12.13 |
0.03± 0.04 |
1.50 |
| 8u | 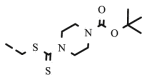 |
>100 | >100 | >100 | >100 | >100 | >100 | >100 | 0.06± 0.14 |
2.84 |
| L-ASA | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | ||
50% inhibitory concentration or compound concentration required to inhibit tumor cell proliferation by 50%. NA = not active.
Values of clog P were calculated by DataWarrior [36].
Table 2.
The growth-inhibition effects in vitro of 1,2,3-triazolyl 4,5-unsaturated l-ascorbic acid derivatives (10c−10p) on selected tumor cell lines and normal fibroblasts.
| Compd. | R | IC50a/μM |
cLogPb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A549 | CFPAC-1 | HCT-116 | HeLa | HepG2 | MCF-7 | SW620 | HFF | |||
| 10c | 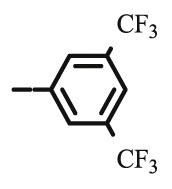 |
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 5.36 |
| 10d |  |
>100 | NA | >100 | 17.69± 58.20 |
>100 | >100 | >100 | >100 | 3.86 |
| 10e | 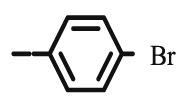 |
>100 | >100 | >100 | 24.31± 53.52 |
>100 | >100 | >100 | >100 | 4.39 |
| 10f |  |
>100 | >100 | >100 | 7.48± 70.70 |
>100 | >100 | >100 | >100 | 4.0 |
| 10g |  |
>100 | >100 | >100 | >100 | >100 | 39.14± 70.11 |
>100 | >100 | 3.59 |
| 10h | 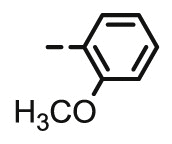 |
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 3.59 |
| 10i | 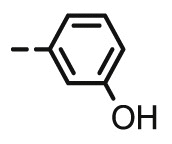 |
23.3± 0.26 |
33.69± 0.43 |
63.58± 0.41 |
74.80± 0.40 |
61.68± 0.65 |
27.77± 0.35 |
69.79± 1.66 |
42.57± 0.16 |
3.32 |
| 10j | 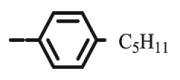 |
>100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 5.78 |
| 10k | 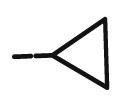 |
>100 | 97.11± 0.37 |
32.43± 0.96 |
76.75± 0.30 |
53.12± 0.69 |
9.27± 0.25 |
29.49± 1.24 |
17.03± 0.49 |
3.08 |
| 10l | >100 | 72.49± 0.35 |
61.19± 0.53 |
67.70± 0.37 |
77.07± 0.56 |
57.24± 0.80 |
49.45± 1.04 |
49.51± 0.38 |
3.63 | |
| 10m |  |
>100 | >100 | 98.83± 0.31 |
>100 | >100 | 72.02± 0.35 |
91.69± 0.42 |
>100 | 6.36 |
| 10n | 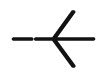 |
5.25± 0.13 |
>100 | 47.29± 0.54 |
>100 | 62.19± 0.60 |
4.93± 0.17 |
20.67± 0.58 |
49.85± 0.23 |
3.55 |
| 10o | 96.52± 0.17 |
>100 | 66.84± 0.56 |
66.84± 0.25 |
81.12± 0.49 |
23.55± 0.42 |
21.61± 0.53 |
94.07± 0.13 |
3.41 | |
| 10p |  |
>100 | 43.38± 0.51 |
27.90± 0.89 |
27.90± 1.10 |
39.03± 1.0 |
37.18± 1.24 |
41.91± 1.3 |
25.61± 0.80 |
1.80 |
| L-ASA | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | −2.47 | |
50% inhibitory concentration or compound concentration required to inhibit tumor cell proliferation by 50%. NA = not active.
Values of n-octanol/water partition coefficients clog P were calculated by DataWarrior [36].
Table 3.
Summary of cytostatic activities on seven tumor cell lines presented as median antiproliferative IC50 values and derivatives with IC50 value within the given range.
| A549 |
CFPAC-1 |
SW620 |
HCT-116 |
HeLa |
HepG2 |
MCF-7 |
|
|---|---|---|---|---|---|---|---|
| 8a−8u | |||||||
| Median IC50 | 28.9 | 58.2 | 54.2 | 31.8 | 25.0 | 29.2 | 25.9 |
| IC50 ≥ 100 | 4 | 5 | 7 | 5 | 3 | 3 | 4 |
| IC50 < 30 | 12 | 3 | 3 | 9 | 13 | 11 | 13 |
| IC50 < 1 |
1 |
0 |
0 |
0 |
0 |
0 |
2 |
|
10c−10p |
|||||||
| Median IC50 | 100.0 | 100.0 | 95.8 | 99.4 | 75.8 | 100.0 | 64.6 |
| IC50 ≥ 100 | 11 | 109 | 7 | 7 | 6 | 8 | 6 |
| IC50 < 30 | 2 | 0 | 3 | 1 | 4 | 0 | 4 |
| IC50 < 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
By varying the substituent at position C-4 of the 1,2,3-triazole ring, the library of 35 compounds has been created (Scheme 1, Fig. 2 ).
Fig. 2.

Groupings of 35 synthesized compounds from 8a−8u and 10c−10p series according to their structural similarity (more blue/red-more similar/dissimilar compounds). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Structural similarity in aromatic and aliphatic fragments in 8a−8u and 10c−10p series has been recognized, whereas the introduction of a sulfonamide unit (8q−8t) and formation of C4=C5 double bond (10c−10p) boosted the highest structural diversification (Fig. 2). The synthetized compounds have spanned a wide range of lipophilicity (Table 1, Table 2). The n-octanol/water partition coefficients (clog P) of the dibenzylated derivatives 8a−8u and 10c−10p are within the range of 0.687−6.359. The compounds 10c−10p with a C4=C5 double bond showed to be more lipophilic than their C-5-OH analogues 8c−8p. In addition, although the dibenzylated derivatives are quite large molecules with molecular weight (MW) range from 407.4 to 665.8, only six of them (8q−8u and 10c) break the Lipinski rule of five filter for drug-like molecules (Table S1 in Supplementary information) [35].
In general, it can be observed that the 2,3-dibenzyl-l-ascorbic acid derivative 8a, with an unsubstituted 1,2,3-triazole ring, did not exhibit any cytostatic activity against evaluated tumor cell lines indicating that the nature of C-4 substituent had a crucial effect on antiproliferative activity (Table 1). L-ASA itself was also lacking any cytostatic effect. Among l-ascorbic acid derivatives 8c−8j containing 4-aryl-substituted 1,2,3-triazole, the p-bromophenyl substituent in 8e caused a strong inhibitory effect on all tumor cell lines, particularly against lung adenocarcinoma cells (A549: IC50 = 0.75 μM). While the electron-withdrawing trifluoromethyl-in 8c and fluoro-substituted aromatic moiety in 8d had a small influence on the antiproliferative effect, the electron-donating methyl group in 8f caused selectivity in the cytostatic activity. This compound strongly inhibited the growth of lung adenocarcinoma (A549: IC50 = 8.69 μM) and hepatocellular carcinoma (HepG2: IC50 = 8.77 μM) cell lines. A longer alkyl substituent, such as 4-pentyl, at the phenyl moiety in 8j also contributed to marked albeit non-specific cytostatic activity. On the contrary, p-methoxy and o-methoxy, as well as p-hydroxy substituents at the phenyl in 8g, 8h and 8i, respectively, did not have a significant impact on the antiproliferative effect on tumor cell lines. However, both p-methoxy and o-methoxy substituents had a pronounced cytotoxicity on normal fibroblasts (HFF). From compounds with a 4-aliphatic-substituted 1,2,3-triazole, long straight 4-decyl and branched 4-tert-butyl in 8m and 8n, respectively, had a strong effect on cytostatic activity, particularly on breast adenocarcinoma cell line MCF-7. Thus, these compounds exhibited antitumor activity against MCF-7 cells in nM range (8m: IC50 = 0.08 μM; 8n: IC50 = 0.73 μM). However, compound 8n was also cytotoxic to normal fibroblasts (IC50 = 13.33 μM), while 8m showed to be nontoxic (IC50 > 100 μM).
Among compounds containing a sulfonamide-substituted 1,2,3-triazole moiety, p-methylphenyl- (8q), p-chlorophenyl- (8r) and 4-fluoro-2-chlorophenylsulfonamide (8s) moieties in 1,2,3-triazolyl-2,3-dibenzyl-l-ascorbic acid derivatives affected marked inhibitory activities on MCF-7 cells, in the range from 3.39 μM to 6.4 μM. Compound 8r exerted also a strong antiproliferative activity on A549 cells. It is noteworthy that sulfonamide-substituted 1,2,3-triazolyl-2,3-dibenzyl-l-ascorbic acid derivatives (8q−8t) exhibited non-specific inhibitory effects and high cytotoxicity to normal fibroblasts (HFF). (Methylthiocarbothionyl)piperazine 2,3-dibenzyl-l-ascorbic acid derivative 8u did not have any impact on antiproliferative activity on evaluated tumor cell lines.
Some relationship between lipophilicity and biological activity of 4-substituted 6-(1,2,3-triazolyl)-6-deoxy-2,3-dibenzyl-l-ascorbic acid derivatives (8a−8u) has been observed, suggesting that compounds 8a and 8p with the lowest lipophilic properties (clogP ≤ 0.8) had a lack of antiproliferative activity on tumor cells. The most lipophilic compounds 8j (clogP = 4.67) and 8m (clogP = 5.25) showed cytostatic activity on tumor cells without being cytotoxic to normal skin fibroblasts.
By replacing the C-5-OH group with stereochemically constrained C4=C5 double bond in Z-configuration [34], cytostatic effect of compounds from the 10c−10p series were decreased against the majority of cancer cells lines (Table 2, Table 3). However, 4,5-unsaturated analogues 10c−10p showed a higher cell specificity with the best activity against the growth of cells MCF-7 and HeLa, derived from female tumors (Table 3).
In addition, aliphatic-substituted compounds 10l−10p exhibited better antiproliferative effect compared to their aromatic-substituted analogues 10c−10j. Among 4-aryl-substituted unsaturated 2,3-dibenzyl-l-ascorbic acid derivatives, compounds 10d, 10e and 10f with di(trifluoromethyl), p-bromo and p-methyl substituents, respectively, displayed a selective cytotoxic effect towards cervical carcinoma cell lines HeLa (Table 2). Besides, these compounds were not cytotoxic (IC50 > 100 μM) to other tumor cell lines and normal fibroblasts HFF. 4,5-Unsaturated 2,3-dibenzyl-l-ascorbic acid derivatives 10k, with a cyclopropyl and 10n, with tert-butyl side chain, exerted strong antiproliferative effect (10k: IC50 = 9.27 μM; 10n: IC50 = 4.93 μM) on breast adenocarcinoma cells MCF-7. Compound 10n exerted an approximate 7-fold reduced antiproliferative effect on MCF-7 cells compared to its saturated analogue 8n. This compound exhibited also a strong cytostatic activity on A549 cells and moderate cytotoxicity to normal fibroblasts. Within the 4,5-unsaturated series, 4-cyclopropyl-1,2,3-triazole derivative 10k had a three-fold more pronounced inhibitory activity on MCF-7 cells than its saturated 2,3-dibenzyl-l-ascorbic acid analogue 8k. On the opposite, 4-aliphatic-substituted 1,2,3-triazolyl 2,3-dibenzyl-l-ascorbic acid 10m had only marginal or no antiproliferative effect, whereas its analogue 8m, from 6-deoxy-2,3-dibenzyl-l-ascorbic acid series, had a strong antiproliferative effect on MCF-7 in nM range.
The antiproliferative activity of synthetized compounds particularly for the saturated derivatives 8a−8u, against seven tested tumor cell lines were mutually quite linearly correlated, as it is illustrated by the PCA analysis (Fig. 3 ).
Fig. 3.

PCA biplot visualization of synthesized compounds according to experimental IC50 values (Table 1, Table 2). Coluoring denotes aromatic (red)/nonaromatic (blue) character of the R substituent (Scheme 1). More active compounds are grouped towards the left side of the plot. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The linear correlations coefficients between IC50 values of various pairs of tumor cell lines for the compounds 8a−8u spanned range 0.632–0.968, with median coefficient value of 0.749. The greatest similarity in antiproliferative activity was observed between tumor cell lines A549, HeLa and HepG2 (Table 3). The most growth inhibiting compounds for all tumor cell lines were 8e, 8j, and 8r, while 8m and 8n were particularly antiproliferative against tumor cell line MCF-7.
Lipophilicity comparison of the synthesized compounds from two series, 8 and 10, revealed that introduction of C-4=C-5 double bond increased the lipophilicity of compounds 10c−10p in comparison to their analogues 8a−8u. Within the 4,5-unsaturated series, compound 10n with the best antiproliferative activity, has a clogP value of 3.55. The most hydrophilic compound within the 4,5-unsaturated series 10p, containing 2-hydroxyethyl side chain at C-4, has a clogP value of 1.80 and showed a low to moderate inhibitory activity, while compounds exhibiting clogP >4.0 did not show appreciable antitumor activity.
Overall, the structure–activity relationship of 4-substituted 1,2,3-triazolyl appended 2,3-dibenzyl-l-ascorbic acid derivatives (8a−8u and 10c−10p) indicates that both type of substituents at C-4 of 1,2,3-triazole scaffolds and the hydroxyethylene and ethylidene spacer connecting the lactone and 1,2,3-triazole moieties, had an impact on antiproliferative activity as presented in Fig. 4 .
Fig. 4.

Insights into the structure–activity relationship of 4-substituted 1,2,3-triazolyl appended 2,3-dibenzyl-l-ascorbic acid derivatives (8a−8u and 10c−10p).
2.2.2. Apoptosis detection and validation of HIF-1-α and NOS2 protein targets for compound 8m by Western blot analysis
Due to the excellent selective cytostatic activities observed on breast adenocarcinoma MCF-7 cells, 4-decyl-substituted 1,2,3-triazolyl 6-deoxy-2,3-dibenzyl-l-ascorbic acid 8m was chosen for further evaluation of its mechanisms of action in vitro. The mode of cell death induced by 8m was assessed by Annexin V assay, performed as described previously [37] (Table 4 ).
Table 4.
Results of the Annexin V assaya for apoptosis/necrosis detection in MCF-7 cells treated with compound 8m.
| Cell line | Duration of treatment | Compd. | Early Apoptosis/% |
Late Apoptosis/% |
Necrosis/% |
|---|---|---|---|---|---|
| MCF-7 | 24 h | Control | 0 | 4.4 | 0 |
| 8m | 0 | 1.68 | 11.36 | ||
| 48 h | Control | 0 | 10.60 | 0.84 | |
| 8m | 13.4 | 35.3 | 8.76 |
Results are presented as percentages of early apoptotic cells, late apoptotic/primary necrotic cells, and secondary necrotic cells after 24 h and 48 h treatment at 2 × IC50 (0.16 μM).
Obtained results showed induction of apoptosis in MCF-7 cells treated with compound 8m. Compound 8m induced moderate increase in early and late apoptotic cell populations by 13.4% and 24.7% after 48 h, respectively (Table 4). A small increase of necrotic cell populations by 7.92% was also observed (Supplementary information, Fig. S22).
Since breast cancer and other solid tumors have long been known to be hypoxic, HIF-1α that is crucially involved in the response of mammalian cells to hypoxia has been recognized as a suitable target for development of antibreast cancer therapeutics [[38], [39], [40], [41]]. Besides having pro-carcinogenic effects, some reports indicated that stabilization of HIF-1 in breast cancer cells decreases cancer cells growth by promoting the cell cycle arrest and autophagy [[42], [43], [44]]. It was also found that several cellular responses to HIF-1 are mediated by nitric oxide (NO) and vice versa. A complex relationship between HIF-1, NO and cancer has been thus explored [44,45] where it was shown that a biphasic response to NO occurs in HIF-1 regulation. Indeed, NO initially inhibits HIF PHD activity that leads to HIF-1 accumulation in the cell which in turn results in increased HIF PHD levels that then prevent HIF-1 accumulation at later stages [45]. In breast cancer tissues, nitric oxide synthase (NOS) activity is higher, especially in highly invasive tumors [46]. Accordingly, the relative expression analysis of HIF-1α (hydroxylated form at position Pro564) and NOS2 proteins’ levels, as potential targets of the chosen candidate structure 8m, was performed (Fig. 5 ). Proline hydroxylation of HIF-1α is known to lead to von Hippel-Lindau protein-dependent ubiquitination and rapid proteasomal degradation, which means that measurement of increased levels of this HIF-1α form is indicative for regulation of the HIF-1α levels in tumor cells.
Fig. 5.

Representative Western blots of predicted protein targets for compound 8m. Cellular levels of selected proteins before and after treatment of MCF-7 cells with compound 8m at IC50 of 0.08 μM value for 24 and 48 h are presented. Approximate molecular weights (kDa) of primary antibodies are indicated. Relative protein expressions were determined by densitometric analysis of protein lines on the blots and normalized to the alpha-tubulin loading control. Data are presented as mean values ± SD. Statistically significant (p < 0.5) differences in the expression levels are marked with an asterisk; C = control cells.
In MCF-7 cells treated with 8m, a significant increase of hydroxylated HIF-1α levels along with decreased levels of NOS2 were observed, that suggest involvement of 8m in regulation of HIF-1α signalling pathway in MCF-7, what is in accord with its strong growth-inhibition effect.
2.2.3. In vitro antiviral evaluations
The 4-substituted-1,2,3-triazole-2,3-dibenzyl-l-ascorbic acid derivatives (8a−8u and 10c−10p) were evaluated against CMV, VZV, thymidine kinase deficient (TK−) and thymidine kinase positive (TK+) strains of VZV, herpes simplex virus type 1 and 2 (HSV-1 and HSV-2), vaccinia virus, adenovirus-2, vesicular stomatitis virus, human coronavirus, influenza A/H1N1, influenza A/H3N2, influenza B, Coxsackie virus B4, reovirus and Sindbis virus. Compounds that exhibited some antiviral activities are presented in Table 5 and their activity was compared with those of ganciclovir, cidofovir, acyclovir and brivudin. By comparison of the antiviral activities of two series of compounds (8a−8u and 10c−10p), we can infer that conformationally constrained 4,5-unsaturated derivatives from 10c−10p series showed a better antiviral potential against CMV and similar potential to VZV (Table 5).
Table 5.
Antiviral activity of selected compounds against CMV and VZV in human embryonic lung (HEL) cells..
| Antiviral activity EC50a/μM |
Cytotoxicity/μM |
||||||
|---|---|---|---|---|---|---|---|
| Compd. |
R |
CMV |
VZV |
||||
| AD-169 | Davis | TK+ VZV (OKA) | TK− VZV (07/1) | Cell morphology (MCC)b | CC50c | ||
| 8b | Si(CH3)3 | >100 | 12.12 | >20 | >20 | 100 | 22.27 |
| 8k | 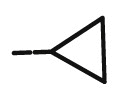 |
44.72 | >20 | >100 | >100 | 100 | NDd |
| 8l | >20 | >4 | >20 | 7.31 | 20 | NDd | |
| 8m |  |
>4 | >4 | 10.73 | 7.76 | 20 | NDd |
| 8n | 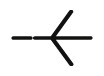 |
44.72 | >20 | >20 | 48.9 | 100 | NDd |
| 10d | 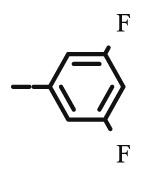 |
9.36 | 8.94 | >100 | 14.62 | 100 | >100 |
| 10e | 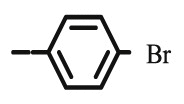 |
>100 | 8.94 | >100 | 44.72 | >100 | >100 |
| 10f | 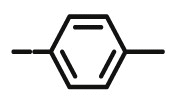 |
8.94 | 5.66 | 27.59 | >4 | 20 | >100 |
| 10g | 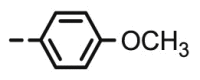 |
20 | 5.74 | >100 | >100 | >100 | >100 |
| 10h | 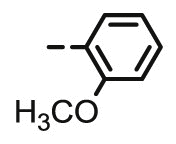 |
>100 | 10.79 | >20 | >100 | 100 | 4.29 |
| 10i | 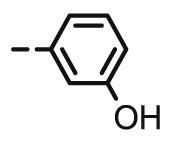 |
9.36 | 8.94 | 14.68 | 11.64 | 100 | 11.05 |
| 10n | 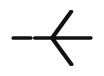 |
8.94 | 5.2 | 10.03 | 11.70 | 100 | 55.52 |
| 10p |  |
>100 | >100 | 78.06 | >100 | >100 | NDd |
| Ganciclovir | 3.15 | 2.3 | >350 | >350 | |||
| Cidofovir | 0.43 | 0.65 | >300 | 214 | |||
| Acyclovir | 1.56 | 96 | >440 | >440 | |||
| Brivudin | 0.0066 | 18.44 | >300 | 179 | |||

Effective concentration required to reduce virus plaque formation by 50%. Virus input was 100 plaque forming units (PFU).
Minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology.
Cytotoxic concentration required to reduce cell growth by 50%.
Not determined.
Among 6-(1,2,3-triazolyl)-6-deoxy-2,3-dibenzyl-l-ascorbic acid derivatives, only 8b, 8k, 8l, 8m and 8n exhibited antiviral activity. Compounds 8b and 8k, bearing 4-trimethylsilyl- and 4-cyclopropyl-substituted 1,2,3-triazole, respectively, displayed moderate antiviral potency against CMV (AD-169 and Davis strains). 2,3-Dibenzyl-l-ascorbic acid derivatives 8l−8n, with 4-aliphatic side chains, had antiviral activity against VZV (TK−), with the best value of EC50 = 7.31 μM for compound 8l. However, 8l and 8m produced an alteration of the cellular morphology at a concentration somewhat lower to that at which the compounds inhibited viral plaque formation (MCC = 20 μM).
Among 4,5-unsaturated structural analogues, it can be observed that the majority of 4-aromatic-substituted 2,3-dibenzyl-l-ascorbic acid 10d−10i were active against CMV, showing that 10g with p-methoxyphenyl substituent expressed the highest activity (EC50 = 5.74 μM, Davis) with no cytotoxic effect. However, o-methoxyphenyl in 10h and m-hydroxyphenyl in 10i caused cytostatic effect for HEL cells (CC50 = 4.29 μM and 11.05 μM). Some 4-aromatic-substituted derivatives (10d, 10f, 10g and 10i) were able to inhibit the replication of both laboratory CMV strains. While 8n, with branched 4-tert-butyl side chain, had marginal activity against CMV (AD-169) and VZV (07/1), its 4,5-unsaturated analogue 10n expressed anti-VZV activity (07/1) (EC50 = 11.70 μM), albeit not highly specific, within the same order of magnitude as that of brivudine (EC50 = 18.44 μM).
Compounds did not show appreciable antiviral activity against HSV-1 and HSV-2, vaccinia virus, adenovirus-2, vesicular stomatitis virus, human coronavirus, influenza A/H1N1, influenza A/H3N2, influenza B, Coxsackievirus B4, reovirus and Sindbis virus. Only exceptions were 4-unsubstituted 1,2,3-triazolyl-2,3-dibenzyl-l-ascorbic acid derivative 8a that exhibited slight activity against influenza A subtype H1N1 and H3N2 (Supplementary information, Tables S2) and 4,5-unsaturated 4-butyl-substituted 2,3-dibenzyl-l-ascorbic acid derivative 10l that displayed modest activity against HSV-2, adenovirus-2, and coronavirus (Supplementary information, Table S3).
3. Conclusions
Synthesis of different 4-substituted 6-(1,2,3-triazolyl)-2,3-dibenzyl-l-ascorbic acid derivatives (8a−8u and 10c−10p) was performed by microwave-assisted copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition (CuAAC) of C-6-azido 2,3-O,O-dibenzyl-l-ascorbic acid derivatives and terminal alkynes. Antiproliferative evaluations on a panel of tumor cell lines showed that both type of substituents at C-4 of 1,2,3-triazole and the linker connecting the lactone and 1,2,3-triazole units had an impact on growth inhibitory activity. The p-bromophenyl substituent in 8e caused a strong, albeit not highly specific inhibitory effect on all tested cell lines, where the strongest effect (IC50 = 0.75 μM) was observed on lung adenocarcinoma (A549) cells. Long straight 4-decyl in 8m and branched 4-tert-butyl in 8n contributed to marked cytostatic activity (8m: IC50 = 0.08 μM; 8n: IC50 = 0.73 μM) on breast adenocarcinoma cells (MCF-7). However, compound 8n was cytotoxic to normal fibroblasts, while 8m showed to be nontoxic and the most lipophilic compound from the 8a−8u series, with clogP 5.25. Within the 4,5-unsaturated 10c−10p series, 4-tert-butyl-substituted structural analogue 10n had the most potent inhibitory activity on A549 and MCF-7 cells. However, it exhibited considerably reduced antiproliferative activity on MCF-7 compared to its analogue 8n. A more detailed investigation of the mechanism of action for the selected candidate 8m showed that this compound induced cell death of breast cancer MCF-7 cells, partially due to apoptosis induction. Moreover, 8m was shown to affect the HIF-1α signalling pathway, as it enhanced expression of the hydroxylated HIF-1α form, tagged for degradation and concomitantly decreasing expression of NOS2.
Additionally, the specific antiviral effect against CMV (Davis) was observed for p-methoxyphenyl-substituted 4,5-unsaturated derivative 10g and its activity was 3-fold reduced (EC50 = 5.74 μM) in comparison with ganciclovir. Moreover, compounds 8l, 8m, 10i and 10n exhibited antiviral potential against VZV (07/1) with EC values similar to that of brivudin. However, they showed cytotoxicity at concentrations close to antiviral active concentrations.
In summary, our findings encourage further development of new structurally related conformationally unrestricted l-ascorbic acid analogues targeting the HIF-1α signalling pathway for breast cancer therapy. Also, additional efforts should be devoted to increase anti-CMV and anti-VZV potency and/or selectivity, along with a more detailed investigation of these compounds mechanisms of antiviral action that may be different from those of antiviral drugs ganciclovir and brivudin.
4. Experimental part
4.1. chemistry
4.1.1. General
Melting points (uncorrected) were determined with a Kofler micro hot-stage (Reichert, Wien, Austria). Precoated Merck (Darmstadt, Germany) silica gel 60F-254 plates were used for thin layer chromatography and the spots were detected under UV light (254 nm). Column chromatography was performed using Fluka (Buchs, Switzerland) silica gel (0.063–0.2 mm) and Merck (Darmstadt, Germany) aluminium oxide (Al2O3, 0.063–0.2 mm); glass columns were slurry-packed under gravity. 1H and 13C NMR spectra were acquired on a Bruker 300 and 600 MHz NMR spectrometer (Bruker Biospin, Rheinstetten, Germany). All data were recorded in DMSO‑d 6 at 298 K. Chemical shifts were referenced to the residual solvent signal of DMSO at δ 2.50 ppm for 1H and δ 39.50 ppm for 13C. Individual resonances were assigned on the basis of their chemical shifts, signal intensities, multiplicity of resonances and H–H coupling constants. Mass spectra were recorded on an Agilent 6410 instrument (Agilent Technologies, Wilmington, USA) equipped with an electrospray interface and triple quadrupole analyzer (LC/MS/MS). Microwave-assisted synthesis was performed in a Milestone start S microwave oven using quartz cuvette (1 cm) under the pressure of 40 bar. High-resolution mass spectra were recorded on Applied Biosystems 4800 Plus Maldi TOF/TOF Analyzer. All elemental compositions were within the 0.4% of the calculated values.
4.1.2. Synthetic procedures
5,6-O,O-Isopropylidene-l-ascorbic acid (1) [23], 2,3-O,O-dibenzyl-5,6-O,O-isopropylidene-l-ascorbic acid (2) [23], 2,3-O,O-dibenzyl-l-ascorbic acid (3) [23], 2,3-O,O-dibenzyl-6-tosyl-l-ascorbic acid (4) [24], 2,3-O,O-dibenzyl-4,5-didehydro-5,6-O,O-ditosyl-l-ascorbic acid (5) [18] (Z)-C-6-azido-2,3-O,O-dibenzyl-4,5-didehydro-5,6-dideoxy-l-ascorbic acid (9) [34] and unsaturated 1,2,3-triazole-2,3-dibenzyl-l-ascorbic acid derivatives (10c–10p) [34,47] were synthesized in accordance with procedures given in the literature.
4.1.2.1. 6-Azido-2,3-O,O-dibenzyl-6-deoxy-l-ascorbic acid (6)
To a solution of the monotosylated compound 4 (1.05 g; 2.057 mmol) in DMF (20 ml), NaN3 (334.3 mg; 5.143 mmol) dissolved in water (2 ml) was added. The reaction mixture was stirred at 70 °C for 3 h and was then left overnight at 50 °C. The solvent was evaporated and the product was dissolved in ethyl acetate (30 ml) and washed with water (2 × 20 ml). The organic layer was dried over magnesium sulphate (MgSO4), filtered and evaporated. The product 6 was isolated as yellow oil (600 mg; 77%). 1H NMR (600 MHz, DMSO) δ 7.50–7.26 (m, 10H, CH2 Ph), 5.71 (d, J = 6.6 Hz, 1H, OH), 5.24 (dd, J = 29.7 Hz; 11.7 Hz, 2H, CH 2Ph), 4.98 (q, J = 11.2 Hz, 2H, CH 2Ph), 4.87 (d, J = 1.7 Hz, 1H, H-4), 3.97–3.79 (m, 1H, H-5), 3.43 (dd, J = 12.6 Hz, 8.5 Hz, 1H, H-6), 3.34 (dd, J = 12.6 Hz, 4.3 Hz, 1H, H-6) ppm. 13C NMR (151 MHz, DMSO) δ 169.0 (C-1), 157.4 (C-3), 136.1 (Ph-q), 135.6 (Ph-q), 128.4 − 127.7 (Ph), 120.9 (C-2), 75.7 (C-4), 73.6 (CH2Ph), 72.8 (CH2Ph), 67.7 (C-5), 52.7 (C-6) ppm.
4.1.2.2. General procedure for the preparation of 1,2,3-triazolyl-2,3-dibenzyl-L-ascorbic acid derivatives (8a−8u)
4.1.2.2.1. Method A
To the solution of 6-azido-2,3-O,O-dibenzyl-6-deoxy-l-ascorbic acid (6) (200–300 mg) in DMF (3 ml); tert-butanol (3 ml) and H2O (3 ml), Cu (0.8 eq), 1 M CuSO4 solution (0.3 eq) and the corresponding alkyne (1.2 eq) were added. The reaction mixture was heated for 1 h using microwave radiation (300 W). The solvent was evaporated and the product was purified by column chromatography on silica gel and aluminium oxide. The obtained oily compound was further triturated using n-hexane to obtain white powder.
4.1.2.2.2. Method B
To the solution of C-6 monotosylated l-ascorbic acid derivative 4 (200–350 mg) in DMF (8 ml), Cu (0.8 eq), 1 M CuSO4 solution (0.3 eq), NaN3 (2.5 eq) dissolved in H2O (2 ml) and the corresponding alkyne (1.2 eq) were added. The reaction mixture was heated for 1 h using microwave radiation (300 W). The solvent was evaporated and the product was purified with column chromatography on silica gel and aluminium oxide. The obtained oily compound was further triturated using n-hexane to obtain white powder.
4.1.2.3. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-(1,2,3-triazol-1-yl)-L-ascorbic acid (8a) and 2,3-O,O-dibenzyl-6-deoxy-6-(4-trimethylsilyl-1,2,3-triazol-1-yl)-L-ascorbic (8b) acid
4.1.2.3.1. Method A
Compounds 8a and 8b were synthesized following general procedure using compound 6 (200 mg; 0.524 mmol); 1 M CuSO4 (0.16 ml); Cu (27.6 mg; 0.434 mmol); trimethylsilylacetylene (0.09 ml; 0.629 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8a (34.2 mg; 16%; m.p. = 120–121 °C) was isolated as white powder and 8b (66.1 mg; 26%) as colorless oil.
4.1.2.3.2. Method B
Compounds 8a and 8b were synthesized following general procedure using compound 4 (200 mg; 0.392 mmol); NaN3 (63.8 mg; 0.98 mmol); 1 M CuSO4 (0.12 ml); Cu (20 mg; 0.32 mmol); trimethylsilylacetylene (0.07 ml; 0.470 mmol); DMF (7 ml) and H2O (3 ml). Compound 8a (46.4 mg; 29%) was isolated as white powder and 8b (9 mg; 5%) as colorless oil. 8a: 1H NMR (300 MHz, DMSO) δ 8.14 (s, 1H, H-7), 7.72 (s, 1H, H-8), 7.47–7.27 (m, 10H, CH2 Ph), 5.66 (bs, 1H, OH), 5.22 (q, J = 11.7 Hz, 2H, CH 2Ph), 5.10–4.84 (m, 3H, CH 2Ph, H-4), 4.68 (dd, J = 13.8 Hz, 3.6 Hz, 1H, H-6), 4.43 (dd, J = 13.7Hz, 9.5 Hz, 1H, H-6), 4.21–4.10 (m, 1H, H-5) ppm. 13C NMR (151 MHz, DMSO) δ 169.0 (C-1), 157.2 (C-3), 136.1 (Ph-q), 135.6 (Ph-q), 133.0 (C-8), 128.7 − 127.8 (CH2 Ph), 125.8 (C-7), 121.0 (C-2), 75.6 (C-4), 73.6 (CH2Ph), 72.8 (CH2Ph), 67.4 (C-5), 52.0 (C-6) ppm. Calcd for C22H21N3O5: C, 64.86; H, 5.20; N, 10.31 . Found: C, 64.93; H, 5.19; N, 10.35.
8b: 1H NMR (600 MHz, CD3CN) δ 8.14 (s, 1H, H-7), 7.53–7.22 (m, 10H, CH2 Ph), 5.65 (d, J = 7.0 Hz, 1H, OH), 5.22 (dd, J = 49.3 Hz, 11.6 Hz, 2H, CH 2Ph), 5.07–4.89 (m, 3H, CH 2Ph, H-4), 4.66 (dd, J = 13.7 Hz, 3.6 Hz, 1H, H-6), 4.41 (dd, J = 13.8 Hz, 9.6 Hz, 1H, H-6), 4.20–4.13 (m, 1H, H-5), 0.25 (s, 9H, 3 x CH3) ppm. 13C NMR (151 MHz, DMSO) δ 169.0 (C-1), 157.2 (C-3), 144.4 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 131.5 (C-7), 128.7 − 127.8 (CH2 Ph), 120.9 (C-2), 75.6 (C-4), 73.7 (CH2Ph), 72.8 (CH2Ph), 67.3 (C-5), 51.7 (C-6), −1.0 (3 x CH3) ppm.
4.1.2.4. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-(3,5-bis(trifluoromethyl)phenyl)-1,2,3-triazol-1-yl]-L-ascorbic acid (8c)
4.1.2.4.1. Method A
Compound 8c was synthesized following general procedure using compound 6 (210.6 mg; 0.552 mmol); 1 M CuSO4 (0.17 ml); Cu (14.7 mg; 0.231 mmol); 3,5-bis(trifluoromethyl)phenylacetylene (0.12 ml; 0.663 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8c (150.5 mg; 44%; m.p. = 174–175 °C) was isolated as white powder.
4.1.2.4.2. Method B
Compound 8c was synthesized following general procedure using compound 4 (300 mg; 0.588 mmol); NaN3 (95.7 mg; 1.470 mmol); 1 M CuSO4 (0.18 ml); Cu (30.56 mg; 0.481 mmol); 3,5-bis(trifluoromethyl)phenylacetylene (0.13 ml; 0.706 mmol); DMF (7 ml) and H2O (3 ml). Compound 8c (173 mg; 48%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 9.02 (s, 1H, Ph-4′), 8.52 (s, 2H, Ph-2′, Ph-6′), 8.07 (s, 1H, H-7), 7.45–7.30 (m, 10H, CH2 Ph), 5.81 (d, J = 6.6 Hz, 1H, OH), 5.22 (dd, J = 25.4 Hz, 11.6 Hz, 2H, CH 2Ph), 5.05 (s, 1H, H-4), 5.04–4.92 (m, 2H, CH 2Ph), 4.77 (dd, J = 13.8 Hz, 3.5 Hz, 1H, H-6), 4.47 (dd, J = 13.7, 9.5 Hz, 1H, H-6), 4.28–4.13 (m, 1H, H-5) ppm. 13C NMR (75 MHz, DMSO) δ 169.0 (C-1), 157.2 (C-3), 143.5 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 133.4 (Ph-q), 131.7; 131.3; 130.8; 130.4 (q, J CF = 32.9 Hz, Ph-3′, Ph-5′), 128.7–127.8 (Ph), 125.3 (Ph-4′), 124.5 (Ph’), 125.1; 121.4 (d, J CF = 272.8 Hz, CF3), 124.5 (C-7), 121.0 (C-2), 75.5 (C-4), 73.7 (CH2Ph), 72.9 (CH2Ph), 67.2 (C-5), 52.7 (C-6) ppm. Calcd for C30H23F6N3O5: C, 58.16; H, 3.74; N, 6.78. Found: C, 58.32; H, 3.75; N, 6.78.
4.1.2.5. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-(3,5-difluorophenyl)-1,2,3-triazol-1-yl]-L-ascorbic acid (8d)
4.1.2.5.1. Method A
Compound 8d was synthesized following general procedure using compound 6 (209.4 mg; 0.549 mmol); 1 M CuSO4 (0.17 ml); Cu (29.6 mg; 0.466 mmol); 3,5-difluorophenylacetylene (0.08 ml; 0.659 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8d (160.7 mg; 57%; m.p. = 189–191 °C) was isolated as white powder.
4.1.2.5.2. Method B
Compound 8d was synthesized following general procedure using compound 4 (300 mg; 0.588 mmol); NaN3 (95.7 mg; 1.470 mmol); 1 M CuSO4 (0.18 ml); Cu (30.56 mg; 0.481 mmol); 3,5-difluorophenylacetylene (0.08 ml; 0.706 mmol); DMF (7 ml) and H2O (3 ml). Compound 8d (75.8 mg; 25%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 8.75 (s, 1H, H-7), 7.65–7.49 (m, 2H, Ph-2′, Ph-6′), 7.47–7.28 (m, 10H, CH2 Ph), 7.21 (tt, J = 9.3 Hz, 2.3 Hz, 1H, Ph-4′), 5.79 (d, J = 6.7 Hz, 1H, OH), 5.22 (dd, J = 25.2 Hz, 11.6 Hz, 2H, CH 2Ph), 5.05 (d, J = 1.1 Hz, 1H, H-4), 5.04–4.93 (m, 2H, CH 2Ph), 4.74 (dd, J = 13.7 Hz, 3.4 Hz, 1H, H-6), 4.44 (dd, J = 13.8 Hz, 9.6 Hz, 1H, H-6), 4.27–4.01 (m, 1H, H-5) ppm. 13C NMR (151 MHz, DMSO) δ 169.6 (C-1), 162.8 (dd, J CF = 245.0 Hz; J CF = 13.4 Hz, Ph-3′, Ph-5′), 157.8 (C-3), 144.6 (C-8), 136.4 (Ph-q), 135.8 (Ph-q), 134.5 (t, J CF = 10.7 Hz, Ph-1′), 129.2 − 128.2 (Ph), 124.2 (C-7), 121.3 (C-2), 108.5 (d, J CF = 26.7 Hz, Ph-2′, Ph-6’), 103.5 (t, J CF = 25.9 Hz, Ph-4′), 76.0 (C-4), 74.1 (CH2Ph), 73.3 (CH2Ph), 67.6 (C-5), 53.1 (C-6) ppm. Calcd for C28H23F2N3O5: C, 64.74; H, 4.46; N, 8.09. Found: C, 64.81; H, 4.47; N, 8.10.
4.1.2.6. Preparation of 2,3-O,O-dibenzyl-6-[4-(4-bromophenyl)-1,2,3-triazol-1-yl]-6-deoxy-L-ascorbic acid (8e)
4.1.2.6.1. Method A
Compound 8e was synthesized following general procedure using compound 6 (300 mg; 0.787 mmol); 1 M CuSO4 (0.24 ml); Cu (40.9 mg; 0.644 mmol); 4-bromophenylacetylene (178 mg; 0.944 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8e (306.1 mg; 69%; m.p. = 203–205 °C) was isolated as white powder.
4.1.2.6.2. Method B
Compound 8e was synthesized following general procedure using compound 4 (350 mg; 0.686 mmol); NaN3 (111.6 mg; 1.720 mmol); 1 M CuSO4 (0.21 ml); Cu (35.66 mg; 0.560 mmol); 4-bromophenylacetylene (152 mg; 0.823 mmol); DMF (7 ml) and H2O (3 ml). Compound 8e (83 mg; 22%) was isolated as white powder. 1H NMR (600 MHz, DMSO) δ 8.65 (s, 1H, H-7), 7.80 (d, J = 8.5 Hz, 2H, Ph-’), 7.65 (d, J = 8.5 Hz, 2H, Ph’), 7.44–7.28 (m, 10H, CH2 Ph), 5.75 (d, J = 6.8 Hz, 1H, OH), 5.22 (dd, J = 45.3 Hz, 11.6 Hz, 2H, CH 2Ph), 5.03 (d, J = 1.1 Hz, 1H, H-4), 4.99 (q, J = 11.2 Hz, 2H, CH 2Ph), 4.71 (dd, J = 13.8 Hz, 3.6 Hz, 1H, H-6), 4.44 (dd, J = 13.8 Hz, 9.6 Hz, 1H, H-6), 4.35–4.12 (m, 1H, H-5) ppm. 13C NMR (75 MHz, DMSO) δ 169.0 (C-1), 157.3 (C-3), 145.0 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 131.9 (Ph), 130.1 (Ph-q), 128.7 − 127.1 (Ph), 122.9 (C-7), 121.0 (C-2), 120.7 (Ph-q), 75.6 (C-4), 73.7 (CH2Ph), 72.8 (CH2Ph), 67.2 (C-5), 52.6 (C-6) ppm. Calcd for C28H24BrN3O5: C, 59.80; H, 4.30; N, 7.47. Found: C, 59.99; H, 4.30; N, 7.49.
4.1.2.7. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-tolyl-1,2,3-triazol-1-yl]-L-ascorbic acid (8f)
4.1.2.7.1. Method A
Compound 8f was synthesized following general procedure using compound 6 (150 mg; 0.393 mmol); 1 M CuSO4 (0.12 ml); Cu (20.45 mg; 0.320 mmol); 4-tolylacetylene (0.06 ml; 0.472 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8f (92 mg; 47%; m.p. = 190–191 °C) was isolated as white powder.
4.1.2.7.2. Method B
Compound 8f was synthesized following general procedure using compound 4 (300 mg; 0.588 mmol); NaN3 (95.4 mg; 1.470 mmol); 1 M CuSO4 (0.18 ml); Cu (30.48 mg; 0.48 mmol); 4-tolylacetylene (0.09 ml; 0.706 mmol); DMF (7 ml) and H2O (3 ml). Compound 8f (97.2 mg; 33%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 8.52 (s, 1H, H-7), 7.72 (d, J = 8.1 Hz, 2H, Ph’), 7.43 − 7.20 (m, 12H, CH2 Ph, Ph’), 5.73 (d, J = 6.8 Hz, 1H, OH), 5.23 (dd, J = 24.3 Hz, 11.6 Hz, 2H, CH 2Ph), 5.03 − 4.97 (m, 3H, CH 2Ph, H-4), 4.69 (dd, J = 13.7 Hz, 3.6 Hz, 1H, H-6), 4.43 (dd, J = 13.7 Hz, 9.5 Hz, 1H, H-6), 4.28–4.17 (m, 1H, H-5), 2.33 (s, 3H, CH3) ppm. 13C NMR (151 MHz, DMSO) δ 169.0 (C-1), 157.3 (C-3), 146.2 (C-8), 137.1 (Ph-q), 136.1 (Ph-q), 135.6 (Ph-q), 129.4 − 128.4 (Ph), 128.0 (Ph-q), 127.8 (Ph), 125.0 (Ph), 122.1 (C-7), 121.0 (C-2), 75.6 (C-4), 73.7 (CH2Ph), 72.8 (CH2Ph), 67.3 (C-5), 52.5 (C-6), 20.8 (CH3) ppm. Calcd for C29H27N3O5: C, 70.01; H, 5.47; N, 8.45. Found: C, 7.22; H, 5.48; N, 8.46.
4.1.2.8. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-(4-methoxyphenyl)-1,2,3-triazol-1-yl]-L-ascorbic acid (8g)
4.1.2.8.1. Method A
Compound 8g was synthesized following general procedure using compound 6 (250 mg; 0.656 mmol); 1 M CuSO4 (0.2 ml); Cu (34 mg; 0.535 mmol); 4-methoxyphenylacetylene (0.1 ml; 0.787 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8g (222.2 mg; 66%; m.p. = 190–192°) was isolated as white powder.
4.1.2.8.2. Method B
Compound 8g was synthesized following general procedure using compound 4 (250 mg; 0.490 mmol); NaN3 (79.7 mg; 1.226 mmol); 1 M CuSO4 (0.15 ml); Cu (25.5 mg; 0.401 mmol); 4-methoxyphenylacetylene (0.08 ml; 0.588 mmol); DMF (7 ml) and H2O (3 ml). Compound 8g (125.2 mg; 50%) was isolated as white powder. 1H NMR (600 MHz, DMSO) δ 8.54 (s, 1H, H-7), 7.82 (d, J = 8.8 Hz, 2H, Ph’), 7.52–7.33 (m, 10H, CH2 Ph), 7.07 (d, J = 8.8 Hz, 2H, Ph’), 5.81 (d, J = 6.8 Hz, 1H, OH), 5.28 (dd, J = 43.8 Hz, 11.6 Hz, 2H, CH 2Ph), 5.11–4.96 (m, 3H, H-4, CH 2Ph), 4.75 (dd, J = 13.8 Hz, 3.7 Hz, 1H, H-6), 4.48 (dd, J = 13.8 Hz, 9.6 Hz, 1H, H-6), 4.31–4.23 (m, 1H, H-5), 3.85 (s, 3H, OCH3) ppm. 13C NMR (151 MHz, DMSO) δ 169.0 (C-1), 158.9 (Ph-q), 157.2 (C-3), 146.0 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 128.7 − 126.4 (Ph), 123.4 (Ph-q), 121.5 (C-7), 121.0 (C-2), 114.3 (Ph’), 75.6 (C-4), 73.6 (CH2Ph), 72.8 (CH2Ph), 67.3 (C-5), 55.1 (OCH3), 52.4 (C-6) ppm. Calcd for C29H27N3O6: C, 67.83; H, 5.30; N, 8.18. Found: C, 68.00; H, 5.32; N, 8.20.
4.1.2.9. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-(2-methoxyphenyl)-1,2,3-triazol-1-yl]-L-ascorbic acid (8h)
4.1.2.9.1. Method A
Compound 8h was synthesized following general procedure using compound 6 (250 mg; 0.656 mmol); 1 M CuSO4 (0.2 ml); Cu (34 mg; 0.535 mmol); 2-methoxyphenylacetylene (0.1 ml; 0.787 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8h (201 mg; 60%; m.p. = 215–217 °C) was isolated as white powder.
4.1.2.9.2. Method B
Compound 8h was synthesized following general procedure using compound 4 (300 mg; 0.588 mmol); NaN3 (95.7 mg; 1.470 mmol); CuSO4 1 M (0.18 ml); Cu (30.56 mg; 0.481 mmol); 2-methoxyphenylacetylene (0.1 ml; 0.705 mmol); DMF (7 ml) and H2O (3 ml). Compound 8h (153.4 mg; 51%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 8.44 (s, 1H, H-7), 8.15 (dd, J = 7.7, 1.6 Hz, 1H, Ph’), 7.65–7.25 (m, 11H, Ph, Ph’), 7.13 (d, J = 8.0 Hz, 1H, Ph’), 7.05 (t, J = 7.5 Hz, 1H, Ph’), 5.71 (d, J = 7.0 Hz, 1H, OH), 5.23 (dd, J = 23.9 Hz, 11.6 Hz, 2H, CH 2Ph), 5.05 − 4.93 (m, 3H, H-4, CH 2Ph), 4.72 (dd, J = 13.7 Hz, 3.4 Hz, 1H, H-6), 4.47 (dd, J = 13.7 Hz, 9.7 Hz, 1H, H-6), 4.28–4.15 (m, 1H, H-5), 3.91 (s, 3H, OCH3) ppm. 13C NMR (151 MHz, DMSO) δ 169.0 (C-1), 157.2 (Ph-q), 155.3 (C-3), 141.5 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 128.8 − 125.0 (Ph), 120.9 (C-2), 120.6 (C-7), 119.2 (Ph-q), 111.5 (Ph’), 75.6 (C-4), 73.6 (CH2Ph), 72.8 (CH2Ph), 67.4 (C-5), 55.4 (OCH3), 52.3 (C-6) ppm. Calcd for C29H27N3O6: C, 67.83; H, 5.30; N, 8.18. Found: C, 67.91; H, 5.29; N, 8.19.
4.1.2.10. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-(3-hydroxyphenyl)-1,2,3-triazol-1-yl]-L-ascorbic acid (8i)
4.1.2.10.1. Method A
Compound 8i was synthesized following general procedure using compound 6 (250 mg; 0.656 mmol); 1 M CuSO4 (0.2 ml); Cu (34 mg; 0.535 mmol); 3-hydroxyphenylacetylene (0.1 ml; 0.787 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8i (209.1 mg; 64%; m.p. = 194–195 °C) was isolated as white powder.
4.1.2.10.2. Method B
Compound 8i was synthesized following general procedure using compound 4 (250 mg; 0.490 mmol); NaN3 (79.7 mg; 1.226 mmol); 1 M CuSO4 (0.15 ml); Cu (25.5 mg; 0.401 mmol); 3-hydroxyphenylacetylene (0.07 ml; 0.588 mmol); DMF (7 ml) and H2O (3 ml). Compound 8i (126.4 mg; 52%) was isolated as white powder. 1H NMR (600 MHz, DMSO) δ 9.51 (s, 1H, OH), 8.51 (s, 1H, H-7), 7.49–7.19 (m, 13H, CH2 Ph, Ph’), 6.74–6.70 (m, 1H, Ph’), 5.73 (d, J = 6.8 Hz, 1H, OH), 5.22 (dd, J = 43.7 Hz, 11.7 Hz, 2H, CH 2Ph), 5.08–4.89 (m, 3H, H-4, CH 2Ph), 4.69 (dd, J = 13.8 Hz, 3.6 Hz, 1H, H-6), 4.43 (dd, J = 13.8 Hz, 9.6 Hz, 1H, H-6), 4.25–4.18 (m, 1H, H-5) ppm. 13C NMR (75 MHz, DMSO) δ 169.0 (C-1), 157.7 (C-3), 157.3 (Ph-q), 146.1 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 132.0 (Ph-q), 129.9 − 127.8 (Ph), 122.4 (C-7), 121.0 (C-2), 116.0 (Ph’), 114.8 (Ph’), 111.8 (Ph’), 75.6 (C-4), 73.7 (CH2Ph), 72.8 (CH2Ph), 67.3 (C-5), 52.4 (C-6) ppm. Calcd for C28H25N3O6: C, 67.33; H, 5.04; N, 8.41. Found: C, 67.44; H, 5.05; N, 8.39.
4.1.2.11. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-(4-pentylphenyl)-1,2,3-triazol-1-yl]-L-ascorbic acid (8j)
4.1.2.11.1. Method A
Compound 8j was synthesized following general procedure using compound 6 (75 mg; 0.197 mmol); 4-pentylphenylacetylene (0.05 ml; 0.236 mmol); 1 M CuSO4 (0.06 ml); Cu (10.2 mg; 0.16 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8j (63.9 mg; 59%; m.p. = 140–141 °C) was isolated as white powder.
4.1.2.11.2. Method B
Compound 8j was synthesized following general procedure using compound 4 (300 mg; 0.588 mmol); NaN3 (95.4 mg; 1.470 mmol); 1 M CuSO4 (0.18 ml); Cu (30.48 mg; 0.480 mmol); 4-pentylphenylacetylene (0.14 ml; 0.706 mmol); DMF (7 ml) and H2O (3 ml). Compound 8j (111.1 mg; 34%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 8.53 (s, 1H, H-7), 7.74 (d, J = 8.1 Hz, 2H, Ph’), 7.51–7.11 (m, 12H, CH2 Ph, Ph’), 5.75 (d, J = 6.8 Hz, 1H, OH), 5.22 (dd, J = 24.1 Hz, 11.6 Hz, 2H, CH 2Ph), 5.04 − 4.95 (m, 3H, CH 2Ph, H-4), 4.70 (dd, J = 13.7 Hz, 3.6 Hz, 1H, H-6), 4.43 (dd, J = 13.7 Hz, 9.5 Hz, 1H, H-6), 4.27–4.17 (m, 1H, H-5), 2.67–2.52 (m, 2H, H-1′), 1.59 (quint, J = 7.5 Hz, 2H, H-2’), 1.35–1.15 (m, 4H, H-3′, H-4’), 0.86 (t, J = 6.8 Hz, 3H, CH3’) ppm. 13C NMR (151 MHz, DMSO) δ 168.9 (C-1), 157.1 (C-3), 146.1 (C-8), 142.0 (Ph-q), 136.1 (Ph-q), 135.5 (Ph-q), 128.7 − 128.3 (Ph), 128.2 (Ph-q), 127.7 (Ph), 125.0 (Ph), 122.0 (C-7), 121.0 (C-2), 75.5 (C-4), 73.6 (CH2Ph), 72.8 (CH2Ph), 67.3 (C-5), 52.4 (C-6), 34.8 (C-1′), 30.8 (CH2’), 30.4 (CH2’), 21.8 (C-4′), 13.8 (CH3’) ppm. Calcd for C33H35N3O5: C, 71.59; H, 6.37; N, 7.59. Found: C, 71.47; H, 6.36; N, 7.59.
4.1.2.12. Preparation of 2,3-O,O-dibenzyl-6-[4-cyclopropyl-1,2,3-triazol-1-yl]-6-deoxy-L-ascorbic acid (8k)
4.1.2.12.1. Method A
Compound 8k was synthesized following general procedure using compound 6 (200 mg; 0.524 mmol); 1 M CuSO4 (0.16 ml); Cu (27.6 mg; 0.434 mmol); cyclopropylacetylene (0.06 ml; 0.630 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8k (147.5 mg; 63%; m.p. = 103–105 °C) was isolated as white powder.
4.1.2.12.2. Method B
Compound 8k was synthesized following general procedure using compound 4 (300 mg; 0.588 mmol); NaN3 (95.7 mg; 1.470 mmol); CuSO4 1 M (0.18 ml); Cu (30.56 mg; 0.481 mmol); cyclopropylacetylene (0.06 ml; 0.705 mmol); DMF (7 ml) and H2O (3 ml). Compound 8k (101.8 mg; 39%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 7.84 (s, 1H, H-7), 7.49–7.21 (m, 10H, CH2 Ph), 5.66 (d, J = 6.9 Hz, 1H, OH), 5.21 (dd, J = 23.7 Hz, 11.6 Hz, 2H, CH 2Ph), 5.04–4.89 (m, 3H, CH 2Ph, H-4), 4.56 (dd, J = 13.7 Hz, 3.8 Hz, 1H, H-6), 4.30 (dd, J = 13.7 Hz, 9.4 Hz, 1H, H-6), 4.17–3.97 (m, 2H, H-5), 1.96–1.88 (m, 1H, H-1′), 0.91 − 0.83 (m, 2H, CH2 ’), 0.77–0.59 (m, 2H, CH2 ’). 13C NMR (151 MHz, DMSO) δ 169.3 (C-1), 157.5 (C-3), 148.8 (C-8), 136.2 (Ph-q), 135.7 (Ph-q), 128.9 − 127.9 (Ph), 122.0 (C-7), 121.1 (C-2), 75.8 (C-4), 73.8 (CH2Ph), 73.0 (CH2Ph), 67.5 (C-5), 52.3 (C-6), 7.7 (CH2’), 7.7 (CH2’), 6.6 (C-1′). Calcd for C25H25N3O5: C, 67.10; H, 5.63; N, 9.39. Found: C, 67.24; H, 5.64; N, 9.40.
4.1.2.13. Preparation of 2,3-O,O-dibenzyl-6-(4-butyl-1,2,3-triazol-1-yl)-6-deoxy-L-ascorbic acid (8l)
4.1.2.13.1. Method A
Compound 8l was synthesized following general procedure using compound 6 (208 mg; 0.545 mmol); 1 M CuSO4 (0.16 ml); Cu (28.4 mg; 0.447 mmol); 1-hexyne (0.08 ml; 0.655 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8l (150 mg; 59%; m.p. = 84 − 85 °C) was isolated as white powder.
4.1.2.13.2. Method B
Compound 8l was synthesized following general procedure using compound 4 (200 mg; 0.392 mmol); NaN3 (63.8 mg; 0.98 mmol); 1 M CuSO4 (0.12 ml); Cu (20 mg; 0.32 mmol); 1-hexyne (0.06 ml; 0.470 mmol); DMF (7 ml) and H2O (3 ml). Compound 8l (39.6 mg; 27%) was isolated as white powder. 1H NMR (600 MHz, DMSO) δ 7.84 (s, 1H, H-7), 7.50–7.23 (m, 10H, CH2 Ph), 5.63 (d, J = 7.0 Hz, 1H, OH), 5.22 (dd, J = 42.8 Hz, 11.6 Hz, 2H, CH 2Ph), 4.98 (q, J = 11.2 Hz, 1H, CH 2Ph), 4.92 (d, J = 1.2 Hz, 1H, H-4), 4.58 (dd, J = 13.8 Hz, 3.9 Hz, 1H, H-6), 4.34 (dd, J = 13.8 Hz, 9.3 Hz, 1H, H-6), 4.17–4.12 (m, 1H, H-5), 2.60 (t, J = 7.5 Hz, 2H, H-1′), 1.68–1.50 (m, 2H, CH2’), 1.37 − 1.28 (m, 2H, CH2’), 0.89 (t, J = 7.4 Hz, 3H, CH3’). 13C NMR (75 MHz, DMSO) δ 169.4 (C-1), 157.6 (C-3), 147.0 (C-8), 136.3 (Ph-q), 135.8 (Ph-q), 129.0 − 128.0 (Ph), 123.1 (C-7), 121.2 (C-2), 75.9 (C-4), 74.0 (CH2Ph), 73.1 (CH2Ph), 67.6 (C-5), 52.3 (C-6), 31.3 (C-1′), 24.8 (C-2′), 21.9 (C-3′), 13.9 (CH3’). Calcd for C26H29N3O5: C, 67.37; H, 6.31; N, 9.07. Found: C, 67.61; H, 6.30; N, 9.08.
4.1.2.14. Preparation of 2,3-O,O-dibenzyl-6-(4-decyl-1,2,3-triazol-1-yl)-6-deoxy-L-ascorbic acid (8m)
4.1.2.14.1. Method A
Compound 8m was synthesized following general procedure using compound 6 (210 mg; 0.551 mmol); 1 M CuSO4 (0.20 ml); Cu (29.1 mg; 0.458 mmol); 1-dodecyne (0.14 ml; 0.661 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8m (184 mg; 61%; m.p. = 72–74 °C) was isolated as white powder. HRMS (ESI Q-TOF): calcd. For C32H41N3O5 [M+H] = 548. 3124; found = 548.3126.
4.1.2.14.2. Method B
Compound 8m was synthesized following general procedure using compound 4 (300 mg; 0.588 mmol); NaN3 (95.7 mg; 1.470 mmol); CuSO4 1 M (0.18 ml); Cu (30.56 mg; 0.481 mmol); 1-dodecyne (0.15 ml; 0.705 mmol); DMF (7 ml) and H2O (3 ml). Compound 8m (110.7 mg; 34%) was isolated as white powder. 1H NMR (600 MHz, DMSO) δ 7.84 (s, 1H, H-7), 7.43-7.29 (m, 10H, CH2 Ph), 5.62 (d, J = 7.0 Hz, 1H, OH), 5.22 (dd, J = 42.3 Hz, 11.7 Hz, 2H, CH 2Ph), 4.98 (q, J = 11.2 Hz, 2H, CH 2Ph), 4.91 (d, J = 1.2 Hz, 1H, H-4), 4.58 (dd, J = 13.8 Hz, 3.9 Hz, 1H, H-6), 4.34 (dd, J = 13.8 Hz, 9.3 Hz, 1H, H-6), 4.18–4.07 (m, 1H, H-5), 2.59 (t, J = 7.6 Hz, 2H, H-1′), 1.68–1.49 (m, 2H, H-2’), 1.34–1.22 (m, 14H, CH2’ x 7), 0.85 (t, J = 7.0 Hz, 3H, CH3’) ppm. 13C NMR (151 MHz, DMSO) δ 169.0 (C-1), 157.2 (C-3), 146.6 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 128.7 − 127.8 (Ph), 122.8 (C-7), 121.0 (C-2), 75.6 (C-4), 73.7 (CH2Ph), 72.8 (CH2Ph), 67.4 (C-5), 52.1 (C-6), 31.3 (C-1′), 29.0 (CH2’), 29.0 (CH2’), 29.0 (CH2’). 28.8 (CH2’), 28.7 (CH2’), 28.6 (CH2’), 25.0 (CH2’), 22.1 (CH2’), 13.9 (CH3) ppm.
4.1.2.15. Preparation of 2,3-O,O-dibenzyl-6-(4-tert-butyl-1,2,3-triazol-1-yl)-6-deoxy-L-ascorbic acid (8n)
4.1.2.15.1. Method A
Compound 8n was synthesized following general procedure using compound 6 (279 mg; 0.732 mmol); 1 M CuSO4 (0.22 ml); Cu (38 mg; 0.598 mmol); 3,3-dimethylbutyne (0.11 ml; 0.877 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8n (245 mg; 72%; m.p. = 64–66 °C) was isolated as white powder.
4.1.2.15.2. Method B
Compound 8n was synthesized following general procedure using compound 4 (200 mg; 0.392 mmol); NaN3 (63.8 mg; 0.98 mmol); 1 M CuSO4 (0.12 ml); Cu (20 mg; 0.320 mmol); 3,3-dimethylbutyne (0.06 ml; 0.470 mmol); DMF (7 ml) and H2O (3 ml). Compound 8n (56 mg; 31%) was isolated as white powder. 1H NMR (600 MHz, DMSO) δ 7.85 (s, 1H, H-7), 7.44–7.28 (m, 10H, CH2 Ph), 5.64 (d, J = 6.9 Hz, 1H, OH), 5.23 (dd, J = 48.9 Hz, 11.6 Hz, 2H, CH 2Ph), 4.98 (q, J = 11.2 Hz, 2H, CH 2Ph), 4.94 (d, J = 1.3 Hz, 1H, H-4), 4.58 (dd, J = 13.8 Hz, 3.7 Hz, 1H, H-6), 4.33 (dd, J = 13.8 Hz, 9.4 Hz, 1H, H-6), 4.19–4.14 (m, 1H, H-5), 1.27 (s, 9H, 3 x CH3). 13C NMR (75 MHz, DMSO) δ 169.6 (C-1), 157.7 (C-3), 156. 4 (C-8), 136.5 (Ph-q), 136.3 (Ph-q), 135.8 (Ph-q), 129.0 − 128.1 (Ph), 121.2 (C-2), 121.2 (C-7), 76.0 (C-4), 74.0 (CH2Ph), 73.1 (CH2Ph), 67.6 (C-5), 52.4 (C-6), 30.5 (3 x CH3). Calcd for C26H29N3O5: C, 67.37; H, 6.31; N, 9.07. Found: C, 67.58; H, 6.33; N, 9.10.
4.1.2.16. Preparation of 2,3-O,O-dibenzyl-6-[4-(3-chloropropyl)-1,2,3-triazol-1-yl]-6-deoxy-L-ascorbic acid (8o)
4.1.2.16.1. Method A
Compound 8o was synthesized following general procedure using compound 6 (258.5 mg; 0.678 mmol); 1 M CuSO4 (0.2 ml); Cu (35.3 mg; 0.555 mmol); 5-chloropentyne (0.12 ml; 0.813 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8o (232.2 mg; 71%; m.p. = 104–106 °C) was isolated as white powder.
4.1.2.16.2. Method B
Compound 8o was synthesized following general procedure using compound 4 (200 mg; 0.392 mmol); NaN3 (63.8 mg; 0.98 mmol); 1 M CuSO4 (0.12 ml); Cu (20 mg; 0.320 mmol); 5-chloropentyne (0.05 ml; 0.470 mmol); DMF (7 ml) and H2O (3 ml). Compound 8o (65.2 mg; 35%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 7.91 (s, 1H, H-7), 7.53–6.95 (m, 10H, CH2 Ph), 5.65 (d, J = 6.9 Hz, 1H, OH), 5.22 (q, J = 11.7 Hz, 2H, CH 2Ph), 5.03–4.96 (m, 2H, CH 2Ph), 4.94 (d, J = 1.3 Hz, 1H, H-4), 4.60 (dd, J = 13.7 Hz, 3.8 Hz, 1H, H-6), 4.35 (dd, J = 13.7 Hz, 9.3 Hz, 1H, H-6), 4.22–4.02 (m, 1H, H-5), 3.68 (t, J = 6.5 Hz, 1H, H-3′), 2.75 (t, J = 7.4 Hz, 2H, H-1′), 2.12–1.92 (m, 2H, H-2′) ppm. 13C NMR (151 MHz, DMSO) δ 169.3 (C-1), 157.5 (C-3), 145.4 (C-8), 136.2 (Ph-q), 135.7 (Ph-q), 128.9 − 128.0 (Ph), 123.4 (C-7), 121.1 (C-2), 75.8 (C-4), 73.8 (CH2Ph), 73.0 (CH2Ph), 67.5 (C-5), 52.3 (C-6), 44.9 (C-3′), 31.9 (C-1′), 22.4 (C-2′) ppm. Calcd for C25H26ClN3O5: C, 62.05; H, 5.42; N, 8.68. Found: C, 62.21; H, 5.42; N, 8.70.
4.1.2.17. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-(2-hydroxyethyl)-1,2,3-triazol-1-yl]-L-ascorbic acid (8p)
4.1.2.17.1. Method A
Compound 8p was synthesized following general procedure using compound 6 (250 mg; 0.656 mmol); 1 M CuSO4 (0.2 ml); Cu (34 mg; 0.535 mmol); 3-butyne-1-ol (0.06 ml; 0.787 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8p (155.3 mg; 53%; m.p. = 105–107 °C) was isolated as white powder.
4.1.2.17.2. Method B
Compound 8p was synthesized following general procedure using compound 4 (200 mg; 0.392 mmol); NaN3 (63.8 mg; 0.98 mmol); 1 M CuSO4 (0.12 ml); Cu (20 mg; 0.32 mmol); 3-butyne-1-ol (0.04 ml; 0.470 mmol); DMF (7 ml) and H2O (3 ml). Compound 8p (60 mg; 34%) was isolated as white powder. HRMS (ESI Q-TOF): calcd. For C24H25N3O6 [M+H] = 452.1822; found = 452.1836. 1H NMR (300 MHz, DMSO) δ 7.87 (s, 1H, H-7), 7.56–7.19 (m, 10H, CH2 Ph), 5.64 (d, J = 6.9 Hz, 1H, OH), 5.22 (dd, J = 23.3 Hz, 11.7 Hz, 2H, CH 2Ph), 5.04 − 4.96 (m, 2H, CH 2Ph), 4.93 (d, J = 1.3 Hz, 1H, H-4), 4.63 (t, J = 5.3 Hz, 1H, OH), 4.61 − 4.55 (m, 1H, H-6), 4.35 (dd, J = 13.7 Hz, 9.3 Hz, 1H, H-6), 4.19–4.08 (m, 1H, H-5), 3.62 (dd, J = 12.3 Hz, 6.9 Hz, 2H, H-2′), 2.76 (t, J = 7.0 Hz, 2H, H-1′) ppm. 13C NMR (151 MHz, DMSO) δ 169.0 (C-1), 157.2 (C-3), 144.0 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 128.7 − 127.8 (Ph), 123.4 (C-7), 121.0 (C-2), 75.6 (C-4), 73.6 (CH2Ph), 72.8 (CH2Ph), 67.4 (C-5), 60.4 (C-2′), 52.1 (C-6), 29.1 (C-1′) ppm.
4.1.2.18. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[(4-(4-methylbenzenesulfonamide)methyl-1,2,3-triazol-1-yl]-L-ascorbic acid (8q)
4.1.2.18.1. Method A
Compound 8q was synthesized following general procedure using compound 6 (250 mg; 0.650 mmol); 1 M CuSO4 (0.20 ml); Cu (34 mg; 0.530 mmol); 4-methyl-N-(2-propynyl)benzenesulfonamide (164.6 mg; 0.790 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8q (196 mg; 51%; mp = 116–118 °C) was isolated as white powder.
4.1.2.18.2. Method B
Compound 8q was synthesized following general procedure using compound 4 (250 mg; 0.490 mmol); NaN3 (79.7 mg; 1.226 mmol); 1 M CuSO4 s(0.15 ml); Cu (25.5 mg; 0.401 mmol); 4-methyl-N-(2-propynyl)benzenesulphonamide (125.5 mg; 0.588 mmol); DMF (7 ml) and H2O (3 ml). Compound 8q (52.8 mg; 18%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 8.01 (bs, 1H, NH), 7.86 (s, 1H, H-7), 7.67 (d, J = 8.2 Hz, 2H, Ph’), 7.48–7.19 (m, 12H, CH2 Ph, Ph’), 5.65 (d, J = 7.0 Hz, 1 H, OH), 5.22 (q, J = 11,6 Hz, 2H, CH 2Ph), 5.04–4.94 (m, 2H, CH 2Ph), 4.89 (d, J = 1.3 Hz, 1H, H-4), 4.57 (dd, J = 13.7 Hz, 4.0 Hz, 1H, H-6), 4.34 (dd, J = 13.8, 9.3 Hz, 1H, H-6), 4.16–4.06 (m, 1H, H-5), 4.00 (d, J = 4.1 Hz, 2H, CH 2NH), 2.36 (s, 3H, CH3’) ppm. 13C NMR (151 MHz, DMSO) δ 169.3 (C-1), 157.4 (C-3), 143.5 (Ph-q), 143.0 (C-8), 137.5 (Ph-q), 136.2 (Ph-q), 135.7 (Ph-q), 129.8–126.8 (Ph), 124.4 (C-7), 121.1 (C-2), 75.7 (C-4), 73.9 (CH2Ph), 73.0 (CH2Ph), 67.5 (C-5), 52.3 (C-6), 38.2 (CH2NH), 21.1 (CH3’) ppm. Calcd for C30H30N4O7S: C, 61.01; H, 5.12; N, 9.49. Found: C, 61.20; H, 5.14; N, 9.50.
4.1.2.19. Preparation of 2,3-O,O-dibenzyl-6-[(4-(4-chlorobenzenesulfonamide)methyl-1,2,3-triazol-1-yl]-6-deoxy-L-ascorbic acid (8r)
4.1.2.19.1. Method A
Compound 8r was synthesized following general procedure using compound 6 (190 mg; 0.500 mmol); 1 M CuSO4 (0.20 ml); Cu (29.1 mg; 0.458 mmol); 4-chloro-N-(2-propynyl)benzenesulfonamide (137.5 mg; 0.600 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8r (154 mg; 51%; mp = 123–124 °C) was isolated as white powder.
4.1.2.19.2. Method B
Compound 8r was synthesized following general procedure using compound 4 (250 mg; 0.490 mmol); NaN3 (79.7 mg; 1.226 mmol); 1 M CuSO4 (0.15 ml); Cu (25.5 mg; 0.401 mmol); 4-chloro-N-(2-propynyl)benzenesulfonamide (137.7 mg; 0.601 mmol); DMF (7 ml) and H2O (3 ml). Compound 8r (70 mg; 23%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 8.21 (t, J = 5.1 Hz, 1H, NH), 7.89 (s, 1H, H-7), 7.75 (d, J = 8.6 Hz, 2H, Ph’), 7.61 (d, J = 8.6 Hz, 2H, Ph’), 7.45–7.22 (m, 10H, CH2 Ph), 5.63 (d, J = 7.1 Hz, 1H, OH), 5.21 (dd, J = 22.0 Hz, 11.7 Hz, 2H, CH 2Ph), 5.03–4.91 (m, 2H, CH 2Ph), 4.88 (d, J = 1.1 Hz, 1H, H-4), 4.56 (dd, J = 13.8 Hz, 3.9 Hz, 1H, H-6), 4.32 (dd, J = 13.8 Hz, 9.3 Hz, 1H, H-6), 4.15–4.06 (m, 1H, H-5), 4.04 (d, J = 5.4 Hz, 2H, CH 2NH) ppm. 13C NMR (75 MHz, DMSO) δ 169.1 (C-1), 157.4 (C-3), 143.1 (C-8), 139.4 (Ph-q), 137.4 (Ph-q), 136.2 (Ph-q), 135.6 (Ph-q), 129.3 − 128.0 (Ph), 124.5 (C-7), 121.0 (C-2), 75.6 (C-4), 73.8 (CH2Ph), 73.0 (CH2Ph), 67.5 (C-5), 52.2 (C-6), 38.1 (CH2NH) ppm. Calcd for C29H27ClN4O7S: C, 57.00; H, 4.45; N, 9.17. Found: C, 57.16; H, 4.47; N, 9.19.
4.1.2.20. Preparation of 2,3-O,O-dibenzyl-6-[(4-(2-chloro-4-fluorobenzenesulfonamide)methyl-1,2,3-triazol-1-yl]-6-deoxy-L-ascorbic acid (8s)
4.1.2.20.1. Method A
Compound 8s was synthesized following general procedure using compound 6 (250 mg; 0.650 mmol); 1 M CuSO4 (0.20 ml); Cu (34 mg; 0.530 mmol); 2-chloro-4-fluoro-N-(2-propynyl)benzenesulfonamide (194.8 mg; 0.787 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8s (239.5 mg; 59%; m.p. = 120–121 °C) was isolated as white powder.
4.1.2.20.2. Method B
Compound 8s was synthesized following general procedure using compound 4 (154.3 mg; 0.302 mmol); NaN3 (49.1 mg; 0.755 mmol); 1 M CuSO4 (0.1 ml); Cu (15.7 mg; 0.247 mmol); 2-chloro-4-fluoro-N-(2-propynyl)benzenesulfonamide (89.9 mg; 0.363 mmol); DMF (7 ml) and H2O (3 ml). Compound 8s (38.2 mg; 20%) was isolated as white powder. 1H NMR (600 MHz, DMSO) δ 8.42 (bs, 1H, NH), 7.95 (dd, J = 8.9 Hz, 6.0 Hz, 1H, Ph’), 7.86 (s, 1H, H-7), 7.56 (dd, J = 8.7 Hz, 2.5 Hz, 1H, Ph’), 7.45–7.29 (m, 11H, CH2 Ph, Ph’), 5.65 (d, J = 7.1 Hz, 1H, OH), 5.22 (dd, J = 34.6 Hz, 11.6 Hz, 2H, CH 2Ph), 4.99 (q, J = 11.2 Hz, 2H, CH 2Ph), 4.87 (d, J = 1.3 Hz, 1H, H-4), 4.55 (dd, J = 13.8 Hz, 4.0 Hz, 1H, H-6), 4.32 (dd, J = 13.8 Hz, 9.2 Hz, 1H, H-6), 4.16 (s, 2H, CH 2NH), 4.10–4.04 (m, 1H, H-5) ppm. 13C NMR (75 MHz, DMSO) δ 169.0 (C-1), 165.3; 161.9 (d, J CF = 254.1 Hz, Ph-4′), 157.2 (C-3), 143.0 (C-8), 136.1 (Ph-q), 135.5 (Ph-q), 134.8; 134.7 (d, J CF = 3.7 Hz, Ph-1′), 132.7 (d, J CF = 10.0 Hz, Ph-2′), 132.6; 132.39 (d, J CF = 11.5 Hz, Ph-6′), 128.7 − 127.9 (Ph), 124.2 (C-7), 120.9 (C-2), 119.1; 118.8 (d, J CF = 26.0 Hz, Ph’), 114.7; 114.4 (d, J CF = 21.7 Hz, Ph’), 75.5 (C-4), 73.7 (CH2Ph), 72.8 (CH2Ph), 67.4 (C-5), 52.0 (C-6), 37.8 (CH2NH) ppm. Calcd for C29H26ClFN4O7S: C, 55.37; H, 4.17; N, 8.91. Found: C, 55.48; H, 4.16; N, 8.90.
4.1.2.21. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[(4-(4-fluorobenzenesulfonamide)methyl-1,2,3-triazol-1-yl]-L-ascorbic acid (8t)
4.1.2.21.1. Method A
Compound 8t was synthesized following general procedure using compound 6 (100 mg; 0.461 mmol); 1 M CuSO4 (0.08 ml); Cu (20 mg; 0.315 mmol); 4-fluoro-N-(2-propynyl)benzenesulfonamide (81.9 mg; 0.384 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8t (62 mg; 38%; m.p. = 128–130 °C) was isolated as white powder.
4.1.2.21.2. Method B
Compound 8t was synthesized following general procedure using compound 4 (300 mg; 0.588 mmol); NaN3 (95.7 mg; 1.470 mmol); CuSO4 1 M (0.18 ml); Cu (30.56 mg; 0.481 mmol); 4-fluoro-N-(2-propynyl)benzenesulphonamide (150.3 mg; 0.705 mmol); DMF (7 ml) and H2O (3 ml). Compound 8t (115 mg; 33%) was isolated as white powder. 1H NMR (600 MHz, DMSO) δ 8.17 (bs, 1H, NH), 7.91 (s, 1H, H-7), 7.84–7.78 (m, 2H, Ph’), 7.47–7.27 (m, 12H, CH2 Ph, Ph’), 5.66 (d, J = 7.1 Hz, 1H, OH), 5.34–5.13 (m, 2H, CH 2Ph), 4.99 (q, J = 11.1 Hz, 2H, CH 2Ph), 4.90 (d, J = 1.3 Hz, 1H, H-4), 4.58 (dd, J = 13.8 Hz, 3.8 Hz, 1H, H-6), 4.34 (dd, J = 13.8 Hz, 9.4 Hz, 1H, H-6), 4.10 (dd, J = 10.4 Hz, 8.6 Hz, 1H, H-5), 4.05 (s, 2H, CH 2NH) ppm. 13C NMR (151 MHz, DMSO) δ 167.0 (C-1), 164.8; 164.2 (d, J CF = 250.7 Hz, Ph-4′), 157.1 (C-3), 143.0 (C-8), 136.8; 136.7 (d, J CF = 2.6 Hz, Ph-1′), 136.1 (Ph-q), 135.5 (Ph-q), 129.6 − 127.8 (Ph), 124.2 (C-7), 120.9 (C-2), 116.2; 116.1 (d, J CF = 22.7 Hz, Ph-3′, Ph-5′), 75.5 (C-4), 73.6 (CH2Ph), 72.8 (CH2Ph), 67.4 (C-5), 52.1 (C-6), 38.0 (CH2NH) ppm. Calcd for C29H27FN4O7S: C, 58.58; H, 4.58; N, 9.42. Found: C, 55.50; H, 4.57; N, 9.42.
4.1.2.22. Preparation of 2,3-O,O-dibenzyl-6-deoxy-6-[4-((tert-butyl 1-carboxylate)-4-(methylthiocarbothionyl)piperazine))-1,2,3-triazol-1-yl]-L-ascorbic acid (8u)
4.1.2.22.1. Method A
Compound 8u was synthesized following general procedure using compound 6 (240 mg; 0.629 mmol); 1 M CuSO4 (0.19 ml); Cu (32.7 mg; 0.515 mmol); tert butyl 4-((prop-2-ynylthio)carbonothioyl)piperazine-1-carboxylate (226.6 mg; 0.755 mmol); tert-butanol (3 ml); DMF (3 ml) and H2O (3 ml). Compound 8u (250 mg; 60%; m.p. = 104–106 °C) was isolated as white powder.
4.1.2.22.2. Method B
Compound 8u was synthesized following general procedure using compound 4 (250 mg; 0.490 mmol); NaN3 (79.7 mg; 1.226 mmol); 1 M CuSO4 (0.15 ml); Cu (25.5 mg; 0.401 mmol); tert butyl 4-((prop-2-ynylthio)carbonothioyl)piperazine-1-carboxylate (179.9 mg; 0.588 mmol); DMF (7 ml) and H2O (3 ml). Compound 8u (203.1 mg; 62%) was isolated as white powder. 1H NMR (300 MHz, DMSO) δ 8.09 (s, 1H, H-7), 7.68–7.05 (m, 10H, CH2 Ph), 5.67 (d, J = 7.0 Hz, 1H, OH), 5.21 (dd, J = 25.0 Hz, 11.6 Hz, 2H, CH 2Ph), 5.04–4.93 (m, 3H, CH 2Ph, H-4), 4.77–4.54 (m, 3H, CH2S, H-6), 4.37 (dd, J = 13.8 Hz, 9.7 Hz, 1H, H-6), 4.28–4.09 (m, 3H, H-5, CH2’), 3.91 (s, 2H, CH2’), 3.59–3.40 (m, 4H, 2 x CH2’), 1.41 (s, 9H, 3 x CH3) ppm. 13C NMR (75 MHz, DMSO) δ 194.6 (CS), 169.0 (C-1), 157.2 (C-3), 153.7 (CO), 141.5 (C-8), 136.1 (Ph-q), 135.6 (Ph-q), 128.7 − 127.8 (Ph), 124.8 (C-7), 121.0 (C-2), 79.4 (4 x CH2’), 75.6 (C-4), 73.7 (CH2Ph), 72.8 (CH2Ph), 67.4 (C-5), 52.3 (C-6), 31.5 (CH2S), 28.0 (3 x CH3’) ppm. Calcd for C33H39N5O6S2: C, 59.53; H, 5.90; N, 10.52. Found: C, 59.67; H, 5.92; N, 10.55.
4.2. In silico analysis
The MACCS 166-bit fingerprints generation, clustering based on structural similarity with using a binary (Tanimoto) distance as a similarity metric, as well as principle component analysis (PCA) were done by R packages rcdk, factoextra and princomp, respectively [48,49]. PCA analyses were performed in terms of measured IC50 values and 1D/2D molecular descriptors. Simple structural parameters (molecular weight (MW), number of hydrogen-bond accepting/donating atoms (HBA/HBD), total/polar surface area (TSA/PSA), molecular flexibility (low < 0.5 < high), shape index (spherical < 0.5 < linear), numbers of rotable bonds, rings and specifically aromatic rings) and physicochemical properties (lipophilicity clogP, aqueous solubility clogS) were calculated by algorithms of DataWarrior (Table S1 in Supplementary information) [36]. No significant groupings and patterns, in terms of the molecular descriptors used, were revealed. Inputs for all algorithms were SMILES (simplified molecular-input line-entry system) representation of the compounds (Supplementary information, Table S1).
4.3. Cell culturing
Human carcinoma cell lines A549, CFPAC-1, HCT-116, HeLa, HepG2, MCF-7, SW620 and normal fibroblasts HFF-1 were cultured in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10 FBS, 100 U/ml streptomycin and penicillin and 2 Mm l-glutamine (GIBCO, Invitrogen), at 37 C with 5 CO2 in humidified atmosphere. Cell lines were obtained from American Type Culture Collection (ATCC).
4.4. Proliferation assay
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed to evaluate anti-proliferative or proliferative effects on tumor and normal cell lines. Tumor cells and normal cells were seeded on 96-well microtiter plates at a seeding density of 5000 cells/well 24 h after seeding, cells were treated with freshly prepared compounds’dilutions in five concentrations (0.01–100 μM) and incubated for 72 h. Solvent DMSO, was tested for eventual cytotoxic effects and its concentration neverexceeded 0.1 μM. Following 72 h cells incubation with compounds, MTT measurment was performed. Measured absorbance of treated cells were transformed into a cell percentage growth (PG) using the formulas proposed by NIH and described previously [50]. Results of the test were obtained from two different experiments perfomed in quadruplicate and IC50 values were calculated using linear regression, and were further statistically analyzed by ANOVA, Tukey post-hoc test (at p < 0.5).
4.5. Apoptosis detection - Annexin V assay
Programmed cell death and necrosis induction were tested by Annexin V assay. Briefly, 2 x 104 of A549, MCF-7 and HFF-1 cells were seeded onto 8-well Nunc Lab-Tek II Chamber Slide system (Thermo Fischer Scientific) and treated with compound 8m at 1 x IC50 (0.08 μM) and 2 x IC50 (0.16 μM) for 24 and 48 h. Cells were stained by use of reagents provided in the Annexin-V-FLUOS Staining kit (Roche Applied Science) according to the manufacturer recommendations. Cells were analyzed by fluorescence microscopy (Zeiss Axio Observer Z1 Inverted Phase Contrast Fluorescent Microscope) and 100 cells were counted for each condition. Results were statistically analyzed by ANOVA, Tukey post-hoc test (at p < 0.5).
4.6. Western blot
MCF-7 cells were seeded in Petri dishes, at 1 x 106 cells/dish and treated with compound 8m at IC50 (0.08 μM) for 24 and 48 h. Compound 8m was assessed as effective and selective on MCF-7 cells with low IC50. Protein lysates were prepared using a buffer containing 50 mM Tris HCl (pH 8), 150 mM NaCl, 1 NP-40, 0.5 sodium deoxycholate, 0.1 SDS and a protease and phosphatase inhibitor cocktail (Roche, Switzerland). A total of 50 μg of proteins were resolved on 12 SDS polyacrylamide gels using the Mini-protean cell (Bio-Rad, USA). The membranes were incubated with primary antibodies against CDC25 (1:1000, rabbit mAb, Abcam, UK), NOS2 (1:500, mouse mAb, SantaCruzBiotechnology, USA) and hydroxy-HIF-1α (Pro564) (HIF-1α, 1:1000, rabbit mAb, Cell Signaling Technology, NL) at 4 °C overnight. Secondary antibody linked to anti-mouse (1:1000, Dako, USA) was used. The signal was visualized by Western Lightning Chemiluminescence Reagent Plus Kit (PerkinElmer, USA) on the ImageQuant LAS500 (GE Healthcare, USA) and α-tubulin (1:1000, mouse mAb, Sigma, USA) was used as a loading control. The signal intensities of particular protein lines on the blot were normalized with the intensity of the loading control and compared in Quantity One software (Bio-Rad, USA). The values are expressed as the average ± SD. Statistical analysis was performed in Microsoft Excel by using the ANOVA at p < 0.05.
4.7. Antiviral assays
Antiviral activity against CMV, VZV, HSV-1, HSV-2, vaccinia virus, vesicular stomatitis virus, adenovirus-2, vesicular stomatitis virus, human coronavirus, influenza A/H1N1, influenza A/H3N2, influenza B, Coxsackie virus B4, reovirus and Sindbis virus was determined essentially as described previously [50,51]. Confluent HEL fibroblasts were grown in 96-well microtiter plates and infected with the human CMV strains Davis and AD-169 at 100 PFU per well. After a 2 h incubation period, residual virus was removed and the infected cells were further incubated with the medium containing different concentrations of the compounds tested (in duplicate). After incubation for 7 days at 37 °C, virus-induced cytopathogenicity was monitored microscopically and after ethanol fixation and staining with Giemsa (for CMV and VZV). Antiviral activity was expressed as the EC50 or concentration required to reduce virus-induced cytopathogenicy by 50%. EC50 values were calculated from graphic plots of the percentage of cytopathogenicity as a function of the concentration of the compounds. Cytostatic measurements based on the inhibition of HEL cell growth were performed as follows: HEL cells were seeded at a rate of 5 x 103 cells/well into 96-well microtiter plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37 °C, the cell number was determined with a Coulter counter. The cytostatic concentration was calculated as the CC50, or the compound concentration required to reduce cell growth by 50% relative to the number of cells in the untreated controls. CC50 values were estimated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds. Cytotoxicity was expressed as minimum cytotoxic concentration (MCC) or the compound concentration that causes a microscopically detectable alteration of cell morphology of the confluent cell cultures that were exposed to the compounds.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgements
Financial support from the Croatian Science Foundation under the project HRZZ-IP-2018-01-4682 is gratefully acknowledged.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2019.111739.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Kuiper C., Vissers M.C.M. Ascorbate as a co-factor for fe- and 2-oxoglutarate dependent dioxygenases: physiological activity in tumor growth and progression. Front. Oncol. 2014;4:359. doi: 10.3389/fonc.2014.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carosio R., Zuccari G., Orienti I., Mangraviti S., Montaldo P.G. Sodium ascorbate induces apoptosis in neuroblastoma cell lines by interfering with iron uptake. Mol. Cancer. 2007;6:55. doi: 10.1186/1476-4598-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Q., Espey M.G., Krishna M.C., Mitchell J.B., Corpe C.P., Buettner G.R., Shacter E., Levine M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. 2005;102:13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Y., Chapman J., Levine M., Polireddy K., Drisko J., Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014;6:222ra18. doi: 10.1126/scitranslmed.3007154. [DOI] [PubMed] [Google Scholar]
- 5.Kim J., Lee S.-D., Chang B., Jin D.-H., Jung S.-I., Park M.-Y., Han Y., Yang Y., Il Kim K., Lim J.-S., Kang Y.-S., Lee M.-S. Enhanced antitumor activity of vitamin C via p53 in Cancer cells. Free Radic. Biol. Med. 2012;53:1607–1615. doi: 10.1016/j.freeradbiomed.2012.07.079. [DOI] [PubMed] [Google Scholar]
- 6.Yun J., Mullarky E., Lu C., Bosch K.N., Kavalier A., Rivera K., Roper J., Chio I.I.C., Giannopoulou E.G., Rago C., Muley A., Asara J.M., Paik J., Elemento O., Chen Z., Pappin D.J., Dow L.E., Papadopoulos N., Gross S.S., Cantley L.C. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–1396. doi: 10.1126/science.aaa5004. 80- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osipyants A.I., Poloznikov A.A., Smirnova N.A., Hushpulian D.M., Khristichenko A.Y., Chubar T.A., Zakhariants A.A., Ahuja M., Gaisina I.N., Thomas B., Brown A.M., Gazaryan I.G., Tishkov V.I. L-ascorbic acid: a true substrate for HIF prolyl hydroxylase? Biochimie. 2018;147:46–54. doi: 10.1016/j.biochi.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagle D.G., Zhou Y.-D. Natural product-derived small molecule activators of hypoxia-inducible factor-1 (HIF-1) Curr. Pharmaceut. Des. 2006;12:2673–2688. doi: 10.2174/138161206777698783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruick R.K., McKnight S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. (80-. ) [DOI] [PubMed] [Google Scholar]
- 10.Kuiper C., Dachs G.U., Currie M.J., Vissers M.C.M. Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Radic. Biol. Med. 2014;69:308–317. doi: 10.1016/j.freeradbiomed.2014.01.033. [DOI] [PubMed] [Google Scholar]
- 11.Miles S.L., Fischer A.P., Joshi S.J., Niles R.M. Ascorbic acid and ascorbate-2-phosphate decrease HIF activity and malignant properties of human melanoma cells. BMC Canc. 2015;15:867. doi: 10.1186/s12885-015-1878-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camarena V., Wang G. The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. 2016;73:1645–1658. doi: 10.1007/s00018-016-2145-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blaschke K., Ebata K.T., Karimi M.M., Zepeda-Martínez J.A., Goyal P., Mahapatra S., Tam A., Laird D.J., Hirst M., Rao A., Lorincz M.C., Ramalho-Santos M. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–226. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong S.-W., Lee S.-H., Moon J.-H., Hwang J.J., Kim D.E., Ko E., Kim H.-S., Cho I.J., Kang J.S., Kim D.J., Kim J.-E., Shin J.-S., Jung D.-J., Jeong Y.-J., Cho B.-J., Kim T.-W., Lee J.S., Kang J.-S., Hwang Y.-I., Noh D.-Y., Jin D.-H., Lee W.J. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene. 2013;32:1508–1517. doi: 10.1038/onc.2012.176. [DOI] [PubMed] [Google Scholar]
- 15.Wittine K., Stipković Babić M., Makuc D., Plavec J., Kraljević Pavelić S., Sedić M., Pavelić K., Leyssen P., Neyts J., Balzarini J., Mintas M. Novel 1,2,4-triazole and imidazole derivatives of l-ascorbic and imino-ascorbic acid: synthesis, anti-HCV and antitumor activity evaluations. Bioorg. Med. Chem. 2012;20:3675–3685. doi: 10.1016/j.bmc.2012.01.054. [DOI] [PubMed] [Google Scholar]
- 16.Gazivoda T., Plevnik M., Plavec J., Kraljević S., Kralj M., Pavelić K., Balzarini J., De Clercq E., Mintas M., Raić-Malić S. The novel pyrimidine and purine derivatives of l-ascorbic acid: synthesis, one- and two-dimensional 1H and 13C NMR study, cytostatic and antiviral evaluation. Bioorg. Med. Chem. 2005;13:131–139. doi: 10.1016/j.bmc.2004.09.052. [DOI] [PubMed] [Google Scholar]
- 17.Gazivoda T., Šokčević M., Kralj M., Šuman L., Pavelić K., De Clercq E., Andrei G., Snoeck R., Balzarini J., Mintas M., Raić-Malić S. Synthesis and antiviral and cytostatic evaluations of the new c-5 substituted pyrimidine and furo[2,3-d]pyrimidine 4′,5′-didehydro-L- ascorbic acid derivatives. J. Med. Chem. 2007;50:4105–4112. doi: 10.1021/jm070324z. [DOI] [PubMed] [Google Scholar]
- 18.Gazivoda T., Wittine K., Lovrić I., Makuc D., Plavec J., Cetina M., Mrvoš-Sermek D., Šuman L., Kralj M., Pavelić K., Mintas M., Raić-Malić S. Synthesis, structural studies, and cytostatic evaluation of 5,6-di-O-modified L-ascorbic acid derivatives. Carbohydr. Res. 2006;341:433–442. doi: 10.1016/j.carres.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Gazivoda T., Raić-Malić S., Marjanović M., Kralj M., Pavelić K., Balzarini J., De Clercq E., Mintas M. The novel C-5 aryl, alkenyl, and alkynyl substituted uracil derivatives of l-ascorbic acid: synthesis, cytostatic, and antiviral activity evaluations. Bioorg. Med. Chem. 2007;15:749–758. doi: 10.1016/j.bmc.2006.10.046. [DOI] [PubMed] [Google Scholar]
- 20.Kolb H.C., Sharpless K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today. 2003;8:1128–1137. doi: 10.1016/S1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 21.Wang H., Xu R., Shi Y., Si L., Jiao P., Fan Z., Han X., Wu X., Zhou X., Yu F., Zhang Y., Zhang L., Zhang L., Zhou D., Xiao S. Design, synthesis and biological evaluation of novel l-ascorbic acid-conjugated pentacyclic triterpene derivatives as potential influenza virus entry inhibitors. Eur. J. Med. Chem. 2016;110:376–388. doi: 10.1016/j.ejmech.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Gao H., Guo W., Wang Q., Zhang L., Zhu M., Zhu T., Gu Q., Wang W., Li D. Aspulvinones from a mangrove rhizosphere soil-derived fungus Aspergillus terreus Gwq-48 with anti-influenza A viral (H1N1) activity. Bioorg. Med. Chem. Lett. 2013;23:1776–1778. doi: 10.1016/j.bmcl.2013.01.051. [DOI] [PubMed] [Google Scholar]
- 23.Von Dallacker F., Sanders J. Derivate der L-Ascorbinsäure 1, Darstellung und Eigenschaften der O2,O3-Ethandiyl- und der O2,O3-Dibenzyl-L-Ascorbinsäuren. Chem. Ztg. 1985;109:197–202. [Google Scholar]
- 24.Von Dallacker F., Sanders J. Derivate der L-Ascorbinsäure 2, Darstellung von Deoxy-L-Ascorbinsäuren. Chem. Ztg. 1985;109:277–280. [Google Scholar]
- 25.Nocentini A., Trallori E., Singh S., Lomelino C.L., Bartolucci G., Di Cesare Mannelli L., Ghelardini C., McKenna R., Gratteri P., Supuran C.T. 4-Hydroxy-3-nitro-5-ureido-benzenesulfonamides selectively target the tumor-associated carbonic anhydrase isoforms IX and XII showing hypoxia-enhanced antiproliferative profiles. J. Med. Chem. 2018;61:10860–10874. doi: 10.1021/acs.jmedchem.8b01504. [DOI] [PubMed] [Google Scholar]
- 26.Kazokaitė J., Aspatwar A., Parkkila S., Matulis D. An update on anticancer drug development and delivery targeting carbonic anhydrase IX. PeerJ. 2017;5 doi: 10.7717/peerj.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kazokaitė J., Aspatwar A., Kairys V., Parkkila S., Matulis D. Fluorinated benzenesulfonamide anticancer inhibitors of carbonic anhydrase IX exhibit lower toxic effects on zebrafish embryonic development than ethoxzolamide. Drug Chem. Toxicol. 2017;40:309–319. doi: 10.1080/01480545.2016.1223095. [DOI] [PubMed] [Google Scholar]
- 28.Al-Dosari M., Ghorab M., Alsaid M., Nissan Y., Ahmed A. Synthesis and anticancer activity of some novel trifluoromethylquinolines carrying a biologically active benzenesulfonamide moiety. Eur. J. Med. Chem. 2013;69:373–383. doi: 10.1016/J.EJMECH.2013.08.048. [DOI] [PubMed] [Google Scholar]
- 29.Wang F., Zhai S., Liu X., Li L., Wu S., Dou Q.P., Yan B. A novel dithiocarbamate analogue with potentially decreased ALDH inhibition has copper-dependent proteasome-inhibitory and apoptosis-inducing activity in human breast cancer cells. Cancer Lett. 2011;300:87–95. doi: 10.1016/j.canlet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray S., Chaturvedi D. Application of organic carbamates in drug design. Part 1:anticancer agents - recent reports. Drugs Future. 2004;29:343–357. doi: 10.1358/dof.2004.029.04.787236. [DOI] [Google Scholar]
- 31.Ray S., Pathak S.R., Chaturvedi D. Organic carbamates in drug development. Part II: antimicrobial agents - recent reports. Drugs Future. 2005;30:161–180. doi: 10.1358/dof.2005.030.02.869228. [DOI] [Google Scholar]
- 32.Kraljević T.G., Harej A., Sedić M., Pavelić S.K., Stepanić V., Drenjančević D., Talapko J., Raić-Malić S. Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1,2,3-triazole–coumarin hybrids. Eur. J. Med. Chem. 2016;124:794–808. doi: 10.1016/j.ejmech.2016.08.062. [DOI] [PubMed] [Google Scholar]
- 33.Duan Y.-C., Ma Y.-C., Zhang E., Shi X.-J., Wang M.-M., Ye X.-W., Liu H.-M. Design and synthesis of novel 1,2,3-triazole-dithiocarbamate hybrids as potential anticancer agents. Eur. J. Med. Chem. 2013;62:11–19. doi: 10.1016/j.ejmech.2012.12.046. [DOI] [PubMed] [Google Scholar]
- 34.Meščić A., Šalić A., Gregorić T., Zelić B., Raić-Malić S. Continuous flow-ultrasonic synergy in click reactions for the synthesis of novel 1,2,3-triazolyl appended 4,5-unsaturated l -ascorbic acid derivatives. RSC Adv. 2017;7:791–800. doi: 10.1039/C6RA25244C. [DOI] [Google Scholar]
- 35.C.A. Lipinski, Drug-like properties and the causes of poor solubility and poor permeability, J. Pharmacol. Toxicol. Methods. 44 (n.d.) 235-249. doi:10.1016/S1056-8719(00)00107-6. [DOI] [PubMed]
- 36.Sander T., Freyss J., von Korff M., Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 37.Gregorić T., Sedić M., Grbčić P., Tomljenović Paravić A., Kraljević Pavelić S., Cetina M., Vianello R., Raić-Malić S. Novel pyrimidine-2,4-dione–1,2,3-triazole and furo[2,3-d]pyrimidine-2-one–1,2,3-triazole hybrids as potential anti-cancer agents: synthesis, computational and X-ray analysis and biological evaluation. Eur. J. Med. Chem. 2017;125:1247–1267. doi: 10.1016/j.ejmech.2016.11.028. [DOI] [PubMed] [Google Scholar]
- 38.Kimbro K.S., Simons J.W. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr. Relat. Cancer. 2006;13:739–749. doi: 10.1677/erc.1.00728. [DOI] [PubMed] [Google Scholar]
- 39.Bos R., Van Diest P.J., De Jong J.S., Van Der Groep P., Van Der Valk P., Van Der Wall E. Hypoxia-inducible factor-1α is associated with angiogenesis, and expression of bFGF, PDGF-BB, and EGFR in invasive breast cancer. Histopathology. 2005;46:31–36. doi: 10.1111/j.1365-2559.2005.02045.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhong H., De Marzo A.M., Laughner E., Lim M., Hilton D.A., Zagzag D., Buechler P., Isaacs W.B., Semenza G.L., Simons J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830–5835. [PubMed] [Google Scholar]
- 41.Bos R., van Diest P.J., van der Groep P., Shvarts A., Greijer A.E., van der Wall E. Expression of hypoxia-inducible factor-1α and cell cycle proteins in invasive breast cancer are estrogen receptor related. Breast Cancer Res. 2004;6:R450–R459. doi: 10.1186/bcr813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capparelli C., Whitaker-Menezes D., Guido C., Balliet R., Pestell T.G., Howell A., Sneddon S., Pestell R.G., Martinez-Outschoorn U., Lisanti M.P., Sotgia F. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11:2272–2284. doi: 10.4161/cc.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soussi T., Wiman K.G. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303–312. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Poyton R., Hendrickson M. Crosstalk between nitric oxide and hypoxia-inducible factor signaling pathways: an update. Res. Rep. Biochem. 2015;5:147–161. doi: 10.2147/RRBC.S58280. [DOI] [Google Scholar]
- 45.Olson N., van der Vliet A. Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide. 2011;25:125–137. doi: 10.1016/j.niox.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koshiji M., Kageyama Y., Pete E.A., Horikawa I., Barrett J.C., Huang L.E. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–1956. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devender N., Ramakrishna K.K.G., Hamidullah, Shukla S.K., Konwar R., Tripathi R.P. Synthesis and anticancer activity of γ-(triazolyl ethylidene)butenolides and polyfunctional pyrrolinones. Eur. J. Med. Chem. 2014;82:106–119. doi: 10.1016/j.ejmech.2014.05.042. [DOI] [PubMed] [Google Scholar]
- 48.Durant J.L., Leland B.A., Henry D.R., Nourse J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002;42:1273–1280. doi: 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- 49.R: the R project for statistical computing. https://www.r-project.org/ n.d.
- 50.Gazivoda T., Raić-Malić S., Krištafor V., Makuc D., Plavec J., Bratulić S., Kraljević-Pavelić S., Pavelić K., Naesens L., Andrei G., Snoeck R., Balzarini J., Mintas M. Synthesis, cytostatic and anti-HIV evaluations of the new unsaturated acyclic C-5 pyrimidine nucleoside analogues. Bioorg. Med. Chem. 2008;16:5624–5634. doi: 10.1016/j.bmc.2008.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Clercq E., Holý A., Rosenberg I., Sakuma T., Balzarini J., Maudgal P.C. A novel selective broad-spectrum anti-DNA virus agent. Nature. 1986;323:464–467. doi: 10.1038/323464a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.