

Abstract
Recombinant measles viruses (rMV) based on the live attenuated measles vaccine strain (MVb) expressing antigens of HIV-1 clade B were generated by reverse genetics. Recombinants expressing single or double antigens of HIV-1 (rMV-HIV) were genetically highly stable on human diploid cells. The production process of these viruses was essentially similar to the parental MV strain, yielding comparative end titers. Immunization of tg-mice by different regimens and formulations showed potent humoral and cellular immune responses against MV and HIV antigens. Recombinant MV-HIV expressing Gag protein conferred protective immunity in tg-mice after a high-dose pseudochallenge with recombinant vaccinia virus. In addition, rMV-HIV boosted anti-HIV antibodies, in the presence of pre-existing anti-vector antibodies.
Keywords: AIDS vaccine, Measles virus, Recombinant viral vectors
1. Introduction
Human immunodeficiency viruses (HIV) remain a global health problem of unprecedented dimensions. Since its discovery (>25 years ago), HIV has already caused an estimated 25 million deaths worldwide and has generated profound demographic changes in the most heavily affected countries. Globally, there is an estimated 40 million people living with HIV. An average of 2.5 million of newly infected people every year have been reported annually [1]. Despite all efforts of prevention and therapy programs, 2 million deaths are estimated every year.
While correlates of protection against HIV infection and persistence are not yet defined, the quest for a prophylactic vaccine remains a major goal in the fight against AIDS. Ideally an HIV vaccine should induce both neutralizing antibodies, to block establishment of an infection, and cell-mediated immunity to contain an escaped virus infection [2], [3]. In addition, the vaccine should be safe and feasible to manufacture on a large scale. Novel technologies were employed collectively to develop a vaccine, among these are the replication deficient viral vectors, like modified vaccinia Ankara, canarypox and adenoviruses and the live attenuated viral vectors, like MV. In addition, non-viral DNA and protein formulations were also employed [4]. However, the failure of Merck's clinical study, using a recombinant Ad5 expressing HIV-1 gag-pol-nef, led the scientific community to critically address the safety of such technologies [5]. While most of the vector systems did not originate from an existing vaccine, MV vector is derived from the live attenuated Moraten® vaccine (MVbv) (Zuniga et al., in preparation). MV vaccine has an excellent safety record as an injectable and as an aerosol vaccine [6], [7]. It strongly activates different arms of the immune system (cellular, humoral and mucosal) after a single or two low-dose injections [8], [9]. Persistence of antibodies and CTLs has been shown for as long as 25 years after vaccination [10]. Additionally, MV genome is not translocated into the nucleus which avoids viral genome integration into host cell genome, adding to the safety profile of the vaccine. A reverse genetics system for MV was established allowing the insertion and expression of additional transgenes by the viral genome [11], [12], [13]. In this paper, we show that rMVs expressing single and/or multiple HIV-1 genes remain stable and induce humoral and cellular immune responses against HIV-1 in transgenic mice, while retaining its efficacy against measles.
2. Materials and methods
2.1. Cells
MRC-5 cells were cultivated using BME/G13/1500 media supplemented with 10% of fetal bovine serum (FBS). The stably transfected rescue cell line 293-3-46 [14] was adapted to the media requirements of the MRC-5 cell line supplemented with 1.2 mg/ml of the antibiotic geneticin (G418, Invitrogen). Vero cells (African green monkey kidney) were maintained with MEM/880 NEAA media supplemented with 5% FBS, l-glutamine (0.58 mg/ml) and gentamycin (0.05 mg/ml).
2.2. Antigens
Plasmids encoding for HIV-1 clade B gp140dCFI, gp140dV1/2dCFI and the gag-pol genes were kindly obtained from Dr. G. Nabel of the Vaccine Research Center (VRC, NIH, USA) [15]. These genes were optimized for human codon usage. For gp140dCFI (e1), the cytoplasmic tail, the transmembrane domain, the cleavage site and the fusion domain were deleted to yield a secreted gp140 form. Moreover, to obtain a fusion intermediate-like configuration, a region encoding the interheptad space was additionally deleted. For the gp140dV1/2dCFI (e2) an additional sequence deletion in the hyper-variable loops 1 and 2 (dV1/2) was performed to enhance the exposure of conserved epitopes and therefore increase the chances to induce neutralizing antibodies. The gagpol construct (gp) represents a synthetic gene encoding a Gag-Pol fusion protein optimized to induce potent cellular immune responses [15]. Critical functions of Pol-derived enzymes were inactivated by mutagenesis for safety reasons. A pBC-gag plasmid coding for a codon-optimized HIV-1 clade B gag protein alone (gag) was kindly received from M. Feinberg, Emory University, Atlanta, USA.
2.3. Construction of antigenomic recombinant MV plasmids
The plasmid p(+)MVb2 is the cloned version of the Moraten®-Berna MV vaccine strain. Additional transcription units in position 2 between the P and the M genes or position 3 between H and L (Fig. 1A) were engineered.
Fig. 1.
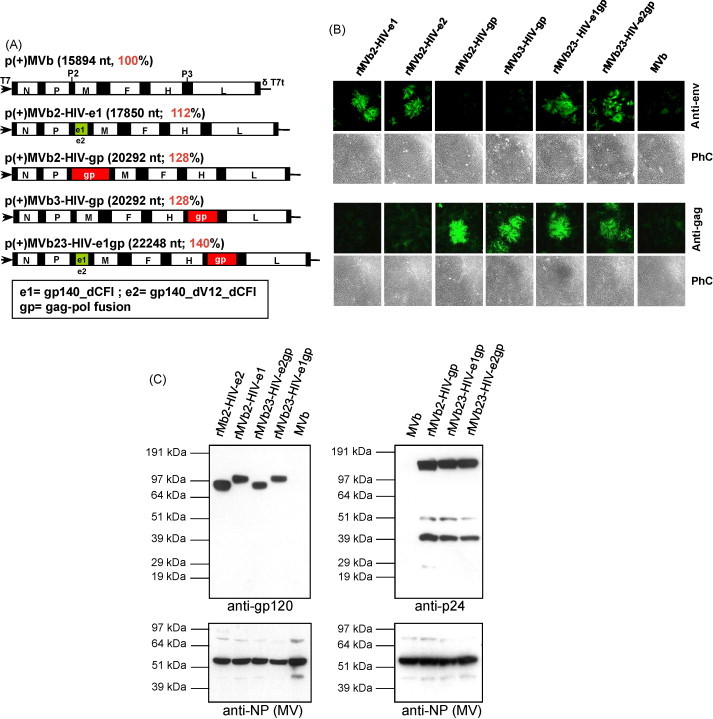
Recombinant MV constructs and transgenes expression. (A) p(+)MVb vector with ATUs at positions 2 and 3. The MV genes are indicated: N (nucleocapsid); P (phosphoprotein); M (matrix); F (fusion); H (hemagglutinin); L (large polymerase). Plasmid is flanked by 5′ T7 RNA polymerase promoter (T7) and 3′ hepatitis delta ribozyme (δ) and T7 terminater (T7t). HIV-transgenes coding for env derivatives (e1, e2) and gagpol fusion (gp) are inserted at position 2 or 3. The MV genome can be increased by 140% without affecting its biochemical and biophysical characteristics. (B) Transgenes expression by indirect immunofluorescence. Vero cells were cultured and inoculated with 200 pfu of different recombinant viruses. Expression of the transgenes was detected after 72 h p.i. by staining with specific antibodies. (C) Transgenes expression was analysed by Western Blots. Vero cells were infected with an MOI of 0.1 of different viruses harvested 72 h p.i. Cell lysates were analysed for the different transgenes expression.
The genes coding for e1, e2 and gp were amplified by PCR from the original plasmids. The PCR products were sub-cloned at unique sites MluI and AatII of p(+)MVb2 or p(+)MVb3, resulting with the plasmids p(+)MVb2-HIV-e1, p(+)MVb2-HIV-e2, p(+)MVb2-HIV-gag and p(+)MVb3-HIV-gp.
Similarly, plasmids containing two transgenes were generated by ligation of the fragment of p(+)MVb2-HIV-e1 or e2 containing e1 or e2 and the fragment of the p(+)MVb3-HIV-gp containing gp, resulting with the polyvalent plasmids p(+)MVb23HIVB-e1gp and p(+)MVb23HIVB-e2gp.
2.4. Rescue of recombinant MV-HIV viruses
Recombinant viruses were rescued from DNA using the 293-3-46 helper cell line as described previously [14].
2.5. Virus expansion and titration
Recombinant MVs were expanded on MRC-5 cells in roller bottles (1750 cm2 surface area, Corning) at 37 °C/5% CO2, according to standard protocols. Viral harvests were collected at different time points post-inoculation. Virus aliquots were stored at −80 °C after adding FBS as cryoprotector to a concentration of 0.02% (v/v). Viral titers of virus stocks were determined by standard MV plaque titration assays.
2.6. Indirect immunofluorescence and Western blots
Vero cells cultured on glass cover-slips were infected with rMVb-HIVs. Detection was essentially described previously [16]. Briefly, cells were washed with PBS, fixed with 4% paraformaldehyde and permeabilized with ice-cold methanol, and then blocked with 0.5% BSA. HIV antigens were detected by specific primary antibodies followed by Alexa 488-conjugated secondary antibodies.
Proteins were also detected by Western blot. Briefly, 3 days post-infection, cells were washed and subsequently lysed in 70 μl of lysis buffer (1% NP-40/150 mM NaCl/50 mM Tris–HCl pH 8) containing a protease inhibitor cocktail (Complete Mini, Roche Diagnostics GmbH).
Total cell extract was resolved on NuPAGE 4–12% Bis–Tris gels (Invitrogen) in the presence of MOPS-containing running buffer. Proteins were transferred on nitrocellulose membranes by semi-dry blotting. Clade B HIV antigens were detected using antisera directed against HIV gp120 or p24. Additionally, the MV nucleocapsid (N) and phosphoprotein (P) were detected with a specific antiserum.
2.7. Immunocytochemistry assay
Infected cells monolayers were fixed with 4% paraformaldehyde, permeabilized with methanol and stained with specific antibody. After extensive washes horseradish peroxidase-conjugated secondary antibody followed by SeramunBlue TMB/Substrate solution incubation (Seramun Diagnostics) was added. Syncytia were finally analysed under the microscope and counted for statistical relevance.
2.8. Antibodies
Antibodies against HIV antigens were kindly obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Goat HIV-1 gp120SF2 antiserum, Item No. 387 and Rabbit HIV-1 SF2 p24 antiserum, Item No. 4250). Additionally, a mouse anti-MV (N, P) serum (kindly obtained from Dr. F Wild, Inserm, France) was applied. The following secondary antibodies were used: anti-goat IgG Alexa 488-conjugated antibody (Invitrogen), anti-rabbit IgG Alexa 488-labelled antibody (Invitrogen) and anti-mouse, anti-goat and anti-rabbit HRP (Dako).
2.9. Growth kinetics
Growth of rMVs was evaluated on confluent MRC-5 cells. After infection, medium was collected at a daily interval and the dynamics of virus shed was determined using standard MV plaque titration assay [12].
2.10. IFNAR-CD46 transgenic mice and immunization
Transgenic mice lacking the interferon-α/β receptors expressing the human CD46 receptor with human-like tissue specificity (IFNAR−/−CD46) were employed for MV infection [17]. For humoral immune response analysis, mice (N = 6) were intraperitoneally (i.p.) immunized at the age of 6–12 weeks with parental or recombinant rMVb-HIV clade B, at a dosage of 1 × 104 or 1 × 105 pfu or as indicated. Blood was collected at weeks 4, 8, 12 and 25. Serum samples were heat inactivated (55 °C/15 min) and stored at 4 °C until use. For the analysis of cellular immune response a single immunization of 1 × 105 pfu per animal was performed. The animals were kept in optimal hygiene conditions, according to the Swiss animal welfare regulations, at Berna Biotech Ltd. animal facility.
2.11. Immunological assays
ELISA assays: Anti-MV antibody titers were determined according to standard protocols [11]. Anti-HIVenv clade B IgG titers were determined using a commercial ELISA assay kit (Genscreen HIV-1/2 ELISA Kit, Bio-Rad). Values above the cut-off background level (mean value of sera from control mice multiplied by a factor of three) were considered positive. Titers were depicted as log10 of the reciprocal end-dilutions.
ELISPOT assay: IFN-γ ELISPOT assay was performed according to the manufacturer instructions (ELISPOT set, BD Biosciences). Duplicates of splenocyte cultures were subsequently re-stimulated with either an equal volume of medium (negative control), medium containing Modified Vaccinia Ankara (MVA, MOI of 2) or medium supplemented with MVA-gag (MOI 2) kindly given by Dr. D. Garber (Emory, Atlanta, USA). The MVA and MVA-gag viruses were Cells were incubated for 18 h at 37 °C in 5% CO2. After a washing step, the detection antibody (biotinylated anti-mouse IFN-γ antibody, BD Biosciences) was incubated for 2 h at room temperature. Peroxidase-conjugated streptavidin (Streptavidin-HRP, BD Biosciences) was used prior spots development with the AEC substrate reagent set (BD Bioscience). The spots were counted under a dissection microscope.
Pseudochallenge: Two groups of 6 mice were pre-immunized with 1 × 104 pfu of MVb2-HIV-gag. Two groups of 4 mice were kept as naive controls. At day 10 post-immunization two groups of mice (one pre-immunized and one naive) were challenged with 5 × 106 pfu of rVac-gag administered intranasally (i.n.). The other two groups were challenged with Vac/wt virus. Weight loss was monitored daily.
3. Results
3.1. Generation of recombinant MVb-HIV-1 viruses
Transgenes coding for env, gag and gagpol polyprotein were inserted either in position 2 (between the measles P and M genes) or position 3 (between the measles H and L genes) to generate a set of rMV-HIV-1 antigenomic plasmids, p(+)MVb2-HIV-e1, p(+)MVb2-HIV-e2, p(+)MVb2-HIV-gag and p(+)MVb2-HIV-gp. Plasmids harboring two transgenes p(+)MVb23-HIV-e1gp and p(+)MVb23-HIV-e2gp were also constructed (Fig. 1A).
Constructed plasmids were successfully rescued by calcium-phosphate co-transfection in 293-3-46 cell line [14], resulting with rMVb2-HIV-e1, rMVb2-HIV-e2, rMVb2-HIV-gag, rMVb2-HIV-gp and recombinants expressing two transgenes rMVb23-HIV-e1gp and rMVb23-HIV-e2gp viruses. The successful rescue of rMVb-HIVs, expressing multiple HIV genes, env, gag and pol (increasing the viral genome by 40%) suggests that MV genome has the capacity to tolerate large gene segments. Viruses were purified on human diploid cells (MRC-5) by three rounds of serial end-dilutions and expanded using the roller flasks technique. Recombinant viruses were produced at approximately equal end titers (Table 1 ). The results suggest that insertion of single or double transgenes into the MV genome does not markedly affect the propagation efficiency of the virus.
Table 1.
Up-scaling of the recombinant MV-HIV on MRC-5 cells.
| Recombinant virus | End titer (pfu/ml) |
|---|---|
| MVb2-HIV-e1 | 2.9e+06 |
| MVb2-HIV-e2 | 2.5e+06 |
| MVb2-HIV-gp | 2.2e+06 |
| MVb3-HIV-gp | 2.2e+06 |
| MVb23-HIV-e1gp | 1.0e+06 |
| MVbv | 2.7e+06 |
Viruses were inoculated on MRC5 cells in 0.5 l roller bottles. Several harvests were performed. End titers were determined by plaque assays.
3.2. Expression of recombinant proteins by MVb-HIV-1 viruses
HIV-1 transgenes expression was assayed by immunofluorescence. HIV-env (e1 or e2), gag and gagpol (gp) were monitored using specific antibodies. The results show that all newly generated viruses specifically express their corresponding HIV-1 transgenes (Fig. 1B). These proteins were also detected by Western blots (Fig. 1C). As expected, the migrations of the proteins match with their expected molecular weights. The secreted e2 derivative antigen, lacking the hyper-variable loops V1 and V2 (gp140dV12dCFI), migrated at lower molecular wt than the e1 (gp140dCFI) that contains the variable loops V1 and V2. In the context of the bivalent viruses (rMVb23-HIVe1gp and rMVb23-HIV-e2gp), the env proteins (e1 and e2) were also expressed, but at slightly lower level, which is likely due to the slight delay of virus propagation.
The estimated molecular weights of the p24 (24 kDa), the p17–p24 precursor (41 kDa) and the unprocessed gagpol polyprotein (estimated molecular weight: 158 kDa) were experimentally confirmed, indicating proper expression of the entire gagpol polyprotein. No crossreactivity was observed in MVb infected cell extract (Fig. 1C). Probing of equivalent blots with an antiserum that recognizes the MV nucleocapsid (N) and phosphoprotein (P) confirmed similar degrees of replication of the diverse rMV-HIV-1 viruses, including parental MVb, in cell culture (Fig. 1C, lower panels).
3.3. Growth of recombinant MVs expressing single and double transgenes
Recombinant MVb-HIVs expressing either one transgene showed similar growth compared to parental MVbv on MRC-5 cells (Fig. 2A). The growth kinetics of recombinant virus expressing simultaneously the env derivative (e1 or e2) and gp displayed a slightly slower growth (approximately by 12–24 h), which is likely due to the overall increase in the genome size (approximately 40%). Similarly, the growth kinetics of the double recombinant expressing env and gagpol (e1,gp) in roller bottles showed only slight growth delay in the kinetics, but the end titers of the final harvests were comparable (Table 1). For possible large-scale production of the vaccine and adjustment of the harvest times would be enough to yield comparable titers.
Fig. 2.
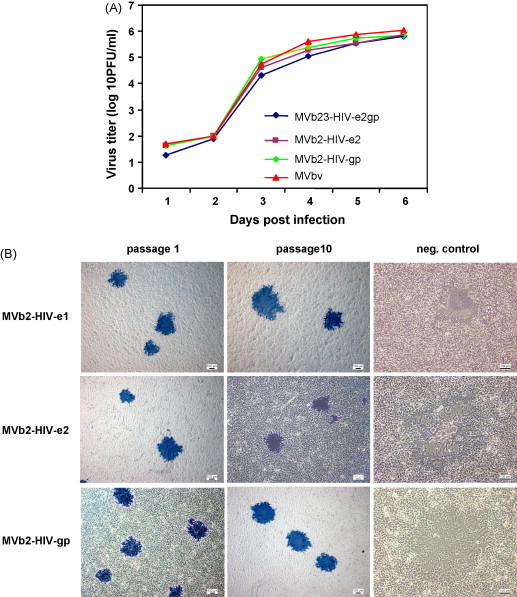
Potency and stability of recombinant MVb-HIVs. (A) Growth kinetics of different rMVb-HIV-1 compared with the parental vaccine. Viruses were harvested daily from infected cell cultures, and virus shed was determined by plaque assays. (B) Stability of protein expression was determined by immunocytochemistry. Serially passaged viruses were inoculated onto cell monolayers and syncytia were stained with specific monoclonal antibodies and HRP.
3.4. Expression of HIV-1 transgenes by rMVb is genetically stable
The rMVb-HIV-1 viruses are very stable, as shown by immunofluorescence and Western blots (Fig. 1C). To determine whether the stability of protein expression is confirmed genetically, recombinant rMVb2-HIV-e1, rMVb2-HIV-e2 and rMVb2-HIV-gp were serially passaged 10 times on MRC5 cells, with an amplification factor of 1020. Ten single syncytia were picked from passages 1, 5 and 10 and genomic viral RNA was sequenced after an RT-PCR. The results showed that genomes from all ten syncytia of each passage revealed identical nucleotide sequence to that of the original seed virus (not shown). Immunocytochemistry also confirmed stability of HIV-transgene expression after each passage (Fig. 2B). We found that titers of rMV-HIVs, after each passage, did not dramatically differ, yet remained comparable. Nevertheless, growth phenotypes were not observed.
3.5. Induction of humoral anti-HIV-1 and MV antibodies in transgenic mice
To test the ability of rMVb-HIV vectors to elicit immune responses, transgenic mice susceptible to MV infection were intraperitoneally (i.p.) immunized in a prime–boost schedule at week 0 and week 4 respectively. Mice were immunized with single rMV-HIV (rMVb2-HIV-e1, rMVb2-HIV-e2 rMVb2-HIV-gag and rMVb2-HIV-gp), with a mix of two different recombinants (rMVb2-HIV-e2 + rMVb2-HIV-gp) and with recombinants expressing double transgenes (rMVb23-HIVe1gp or rMVb23-HIV-e2gp). Standard MVb vaccine was used as a control. Blood was drawn at weeks 4, 8 and 12 and sera were assayed by ELISA for antibody responses against MV or HIV-1 clade B. The results show that all mouse groups (immunized with different rMV-HIVs) induced similar high-titers of anti-measles antibodies, and these were comparable to the antibody titers induced by standard MVb vaccine (Fig. 3A). Interestingly, probing the same sera for anti-HIV antibodies, comparable high titers of anti-HIV env antibodies were found in different mouse groups (whether they were immunized with a single recombinant expressing e1 or e2 or from mouse group immunized with rMV-HIV expressing double antigens (e1 and gp), or from a group immunized with a mixture of two single recombinants (rMVb2-HIV-e1 + rMVb2-HIV-gp) (Fig. 3A). Four weeks after the prime, considerable titers of anti HIV antibodies were readily measured (Fig. 3A, right panel, red bars). Four weeks after the boost, a 100-fold increase in HIV antibody titers were measured in all mouse groups, except the MVbv immunized animals and the negative controls. These results indicate that rMV vector is immunogenic and, in the presence of anti-MV antibodies (previously induced after the prime, left panel, red bars), rMVs expressing HIV antigens boosted anti HIV antibodies (Fig. 3A, right panel, blue and green bars). UV inactivated MVb2-HIV-e1 did not induce detectable antibodies against env, indicating that in vivo replication is central for the presentation of the env antigen and the induction of immune responses (Table 2 ). Similarly, anti-vector response was found severely affected after UV inactivation. This indicates that virus replication is required for an efficient humoral immune response.
Fig. 3.
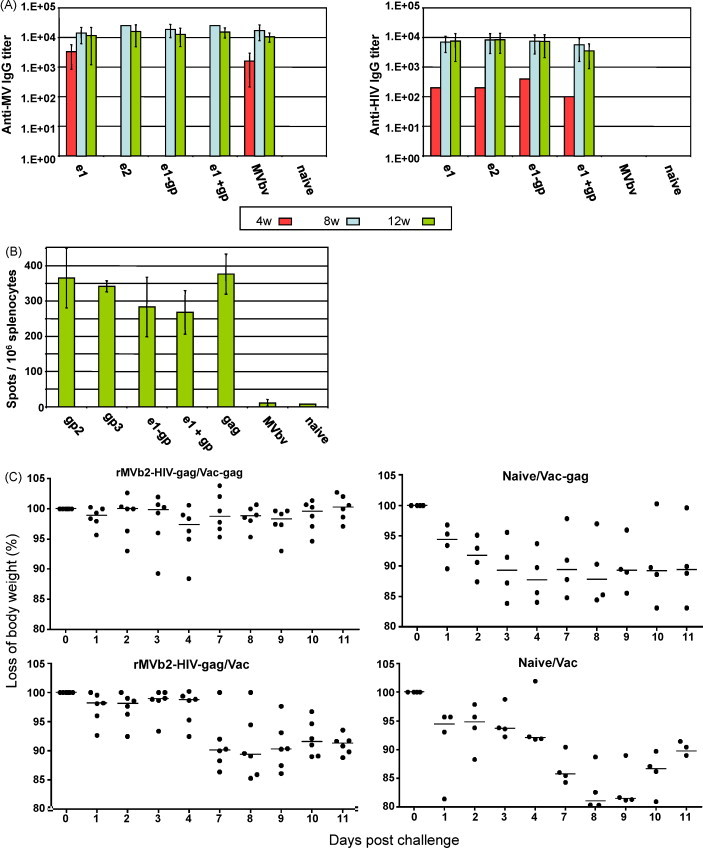
Induction of humoral and cellular immune response against the MV and HIV-1. (A) Humoral immune responses: Mice were intraperitoneally (i.p.) primed and then boosted at week 0 and week 4 respectively with of 1 × 104 pfu of different rMV-HIV-1 viruses. Blood was collected at weeks 4, 8, 12 and 25. ELISAs against MV or HIV-env were performed. (B) Determination of cellular immune responses by ELISPOT assays. Transgenic mice susceptible MV were immunized with 1 × 105 pfu and ELISPOTs were performed 3-week post-immunization. (C) Pseudochallenge of rMVb2-HIV-gag pre-immunization IFNAR−/−CD46 tg-mice. Mice were immunized with MVb2-HIV-gag or not (Naive) and each group was split to groups and 10 days later two groups were challenged with wild-type vaccinia (Vac-wt) or recombinant Vaccinia expressing HIV-1-gag (Vac-gag). Each dot represents an animal (e1 = rMVb2-HIV-e1; e2 = rMVb2-HIV-e2; e1-gp = rMVb23-HIV-e1gp; e1 + gp = rMVb2-HIV-e1 + rMVb2-HIV-gp; gp2 = rMVb2-HIV-gag; gp3 = rMVb3-HIV-gag).
Table 2.
Antibody responses to MV and HIV-env 4 and 8 weeks after immunization with active or UV-inactivated viruses.
| Virus | N | Anti-MV titer (ELISA)a |
Anti-HIV titer (ELISA) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Week 4b |
Week 8c |
Week 4 |
Week 8 |
||||||
| Titer log10 | SD (±) | Titer log10 | SD (±) | Titer log10 | SD (±) | Titer log10 | SD (±) | ||
| MVbvd | 4 | 3.00 | (0.35) | 4.21 | (0.17) | <0.10 | <0.10 | <0.10 | <0.10 |
| MVb2-HIV-e1 | 4 | 3.35 | (0.52) | 4.11 | (0.25) | 2.30 | (0.25) | 3.81 | (0.25) |
| MVb2-HIV-e1e (UV inactivated) | 4 | 0.50 | (1.00) | 0.50 | (1.00) | <0.10 | <0.10 | <0.10 | <0.10 |
| Naivef | 4 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 |
ELISA titers are optical density values measured at 492 nm. OD values are 2.1-fold above the background.
Immunization was performed on day 0 and measured at 4 weeks.
Boost was performed at 4 weeks after the prime and measured at 8 weeks.
Prime and boost were done with homologous viruses by injecting 104 pfu/mouse.
Immunization with a single injection of 104 pfu/mouse.
Pool of pre-immune sera of animals before immunization.
3.6. Induction of cellular immune responses
An IFN-γ ELISPOT assay was performed to measure gag-specific T cell activation from MVb-HIV-1 immunized mice (Fig. 3B). Splenocytes were extracted 3 weeks after a single immunization with 1 × 105 pfu/animal using diverse virus preparations expressing gag or gagpol recombinant protein. Splenocytes were then re-stimulated either with medium (negative control) or MVA-gag clade B. The MVA-gag re-stimulated splenocytes revealed significant T cell immune response activation. Mice immunized with single recombinant viruses which express the transgene from position 2 (MVb2-HIV-gag or MVb2-HIV-gp) induced the best responses. This result also shows that independently of the subgenomic site of gagpol transgene expression (position 2 or 3 within the MV genome; gp2 or gp3 respectively) similar cellular immune response could be induced. Moreover, the double recombinant viruses, MVb23-HIV-e1gp, or the mix of two recombinant viruses (MVb2-HIV-e1 + MVb2-HIV-gp) displayed comparable cellular immune response. Unexpectedly, the mix of MVb2-HIVe1 and MVb2-HIV-gp (e1 + gp) and the double recombinant (e1 − gp) gave a lower number of spot-forming cells (SFC) compared with the single recombinants, yet animals were immunized with the same amounts of viruses. It is likely that viruses in a heterogeneous mixture may give rise to unpredictable competition between the two viruses. Additionally, the delayed replication of the double recombinant may also explain the obtained lower values. The MVbv vaccinated mice, as expected, did not induce any gag-specific cellular immune response.
3.7. Pseudochallenge and protection of tg-mice
We determined the quality of cellular immune response induced by the rMVb2-HIV-gag by challenging immunized mice with rVac-gag or Vac-wt. IFNAR−/−CD46 tg-mice are optimal because they are sensitive to recombinant vaccinia challenge, characterized by a weight loss upon infection with either rVac-gag or Vac/wt viruses. Groups of 4 (naive) or 6 (pre-immunized) mice were challenged with rVac-gag or Vac/wt on day 10 post-immunization with rMVb2-HIV-gag. Animal weight loss was monitored daily. The results shown in Fig. 3C indicate that the rMVb2-HIV-gag pre-immunized mice were less susceptible to weight loss than the naïve mice indicating controlled replication of the challenge virus in one group but not the other. Animals pre-immunized with MVb2-HIV-gag but challenged with Vac/wt displayed a significant weight loss (10–15%) at 7 days post-challenge. Naive mice challenged with either rVac-gag or Vac/wt displayed a fast weight loss, 15% and 20% respectively. The results obtained suggest that MVb2-HIV-gag is induced functional T cells immunity that protected immunized animals against recombinant Vac-gag.
4. Discussion
We generated recombinant MVs expressing HIV-1 antigens from independent open reading frames inserted either between the P and M genes or between the H and L genes of the MV genome. We demonstrated previously the effectiveness of rMVb vector against SIV and SARS-CoV [11], [18]. In this manuscript we demonstrate that expression of HIV antigens remains genetically stable, after serial passages in MCR5 cells as shown by immunocytochemistry and sequencing. The replication kinetics of the rMV-HIVs followed the same trend as the parental MVb vaccine. Such behaviour is particularly favourable for rMVb vaccine candidates over other members of the Mononegavirales [19], [20], suggesting a high fidelity of MV RNA polymerase. The plasticity of MV and its ability to stably express foreign genes (HIV-1 genes in this study) after multiple passages, suggest that MV is a robust and superior delivery system in comparison to other RNA virus vectors. Unlike MV, many RNA viruses are unable to accommodate large foreign sequence in their icosahedral capsids and often rapidly lose even small inserts due to their dependence on RNA secondary and tertiary structures as well as high levels of recombination.
The MVb-based system has several advantages as a live viral vector. It uses an MV strain which is already in use as a safe and efficacious vaccine [13]. The production cost of standard MV vaccines is very low and the current MV vaccine is well-known to induce a life-long immunity. Here we demonstrated that rMV-HIV candidate vaccine production is up-scalable and that the end virus titers were comparable to those of the standard MV vaccine. Furthermore, parental and recombinant MVb viruses were shown to induce comparable anti-MV immune responses. These features are particularly important if mass immunization is envisaged for developing countries. In this regard, the use of MV vector cocktails delivering simultaneously several additional antigens could be envisaged instead of the routine MV vaccination in early childhood.
The development of an effective HIV vaccine remains a challenging endeavour; however, this complexity may not be due to the delivery systems per se, but due to the heterogeneity of HIV-1 itself. We demonstrate that MV vector induces humoral and cellular immune responses in experimental animals. The induction of cellular immune responses is shown to protect animals from a high dose or recombinant vaccinia virus challenge, indicating that the induced cellular immune responses are functionally protective, at least in a small animal model system.
Acknowledgments
The cloning of the recombinant vectors was performed at the institute of Molecular Biology, University of Zurich. This work was supported by the National Institute of Health (NIH AI46007) to HYN at the University of Zurich and the NIH contract No. N01-AI-60018. Thanks to Drs David Garber, Emory, for supplying the rMVAs, Fabian Wild for supplying the MV antibodies, the NIH AIDS research and reference reagent program for supplying all HIV reagents. Thanks to Ms Sara Weibel for her excellent technical support, and Dr Martin A. Billeter, University of Zurich, for his continued support.
References
- 1.United Nations Program on HIV/AIDs (UNAIDS). Report on the global HIV/AIDS epidemic. Geneva, Switzerland: UNAIDS; 2008.
- 2.Gandhi R.T., Walker B.D. Immunologic control of HIV-1. Annu Rev Med. 2002;53:149–172. doi: 10.1146/annurev.med.53.082901.104011. [review] [DOI] [PubMed] [Google Scholar]
- 3.Letvin N.L., Walker B.D. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat Med. 2003;9(July (7)):861–866. doi: 10.1038/nm0703-861. [review] [DOI] [PubMed] [Google Scholar]
- 4.Liniger M., Zuniga A., Naim H.Y. Use of viral vectors for the development of vaccines. Exp Rev Vaccines. 2007;6(2):255–266. doi: 10.1586/14760584.6.2.255. [DOI] [PubMed] [Google Scholar]
- 5.Sekaly R.P. The failed HIV Merck vaccine study: a step back or a launching point for future vaccine development? J Exp Med. 2008;205(January (1)):7–12. doi: 10.1084/jem.20072681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett J.V., Fernandez D.C., Valdespino-Gomez J.L., Garcia-Garcia Mde L., Islas-Romero R., Echaniz-Aviles G. Aerosolized measles and measles-rubella vaccines induce better measles antibody booster responses than injected vaccines: randomized trials in Mexican schoolchildren. Bull World Health Organ. 2002;80:806–812. [PMC free article] [PubMed] [Google Scholar]
- 7.Dilraj A., Sukhoo R., Cutts F.T., Bennett J.V. Aerosol and subcutaneous measles vaccine: measles antibody responses 6 years after re-vaccination. Vaccine. 2007;25(21):4170–4174. doi: 10.1016/j.vaccine.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Griffin D. 4th ed. Lippincott Williams & Wilkins; Philadelphia: 2001. Measles virus. [Google Scholar]
- 9.Hilleman M.R. Current overview of the pathogenesis and prophylaxis of measles with focus on practical implications. Vaccine. 2002;20:651–665. doi: 10.1016/s0264-410x(01)00384-x. [DOI] [PubMed] [Google Scholar]
- 10.Ovsyannikova I.G., Dhiman N., Jacobson R.M., Vierkant R.A., Poland G.A. Frequency of MV-specific CD4+ and CD8+ T cells in subjects seronegative or highly seropositive for measles vaccine. Clin Diagn Lab Immunol. 2003;10:411–416. doi: 10.1128/CDLI.10.3.411-416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuniga A., Wang Z., Liniger M., Hangartner L., Caballero M., Pavlovic J. Attenuated measles virus as a vaccine vector. Vaccine. 2007;25(16):2974–2983. doi: 10.1016/j.vaccine.2007.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z., Hangartner L., Martin L., Zuniga A., Billeter M.A., Naim H.Y. Recombinant measles viruses expressing heterologous antigens of mumps and simian immunodeficiency viruses. Vaccine. 2001;19:2319–2336. doi: 10.1016/s0264-410x(00)00523-5. [DOI] [PubMed] [Google Scholar]
- 13.Tangy F., Naim H.Y. Live attenuated measles vaccine as a potential multivalent pediatric vaccination vector. Viral Immunol. 2005;18:317–326. doi: 10.1089/vim.2005.18.317. [DOI] [PubMed] [Google Scholar]
- 14.Radecke F., Spielhofer P., Schneider H., Kaelin K., Huber M., Billeter M.A. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham B.S., Koup R.A., Roederer M., Bailer R.T., Enama M.E., Moodie Z. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 DNA candidate vaccine. J Infect Dis. 2006;194(December (12)):1650–1660. doi: 10.1086/509259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naim H.Y., Ehler E., Billeter M.A. Measles virus matrix protein specifies virus budding and envelope glycoprotein sorting in polarized epithelial cells. EMBO J. 2000;19:3576–3585. doi: 10.1093/emboj/19.14.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mrkic B., Pavlovic J., Rulicke T., Atkinson J.P., Aguzzi A., Cattaneo R. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72:420–7427. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liniger M., Zuniga A., Tamin A., Azzouz-Morin T.N., Knuchel M., Marty R. Induction of neutralizing and cellular immune responses against SARS coronavirus by recombinant measles viruses. Vaccine. 2008;26:2164–2174. doi: 10.1016/j.vaccine.2008.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spielhofer P., Bächi T., Fehr T., Kälin K., Billeter M.A., Naim H.Y. Chimeric measles viruses with a foreign envelope. J Virol. 1998;72:2150–2159. doi: 10.1128/jvi.72.3.2150-2159.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Billeter M.A., Naim H.Y., Udem S.A. Reverse genetics of measles virus and resulting multivalent recombinant vaccines. Curr Topics Microbiol Immun. 2008;329:129–162. doi: 10.1007/978-3-540-70523-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]