

Abstract
Numerous human infections with avian influenza viruses in Asia in recent years have raised the concern that the next influenza pandemic is imminent. The most effective way to combat influenza is through the vaccination of the public. However, a minimum of 3–6 months is needed to develop an influenza vaccine using the traditional egg-based vaccine approach. The influenza hemagglutinin protein (HA), the active ingredient in the current vaccine, can be expressed in insect cells using the baculovirus expression vector system and purified rapidly. An influenza vaccine based on such a recombinant antigen allows a more timely response to a potential influenza pandemic. Here, we report an innovative monitoring assay for recombinant HA (rHA) expression and a rapid purification process. Various biochemical analyses indicate that the purified rHA is properly folded and biologically active.
Keywords: Influenza, Vaccine, Hemagglutinin, Baculovirus, Recombinant protein, Purification, Chromatography
1. Introduction
Influenza is a highly contagious, acute viral respiratory disease, which causes significant morbidity and mortality worldwide each year [1], [2], [3]. Influenza viruses are single-stranded ribonucleic acid (RNA) viruses surrounded by a lipid containing envelope spiked with two glycoproteins: hemagglutinin (HA) and neuraminidase (NA). These glycoproteins, and HA, in particular, have been recognized as key antigens in the host response to influenza virus in both natural infection and vaccination [4], [5]. The viruses are well known for their ability to mutate to circumvent immunity and re-infect the host. An antigenic shift, a major antigenic change of the virus due to the genetic re-assortment of two subtype strains that co-infected a host, can cause an influenza pandemic since the population may have no inherent immunity against the new strain [6], [7]. The avian influenza A (H5N1) epizootic outbreak and numerous human infections with H5N1 in Asia in recent years, and events such as the infection of two nurses attending to avian influenza patients in Vietnam (WHO, March 14, 2005) and a possible person-to-person transmission in a family cluster of the disease in Thailand [8], continue to raise concern that the next influenza pandemic is imminent.
A proven, effective way to combat influenza is through vaccination of the public using the trivalent vaccine produced in embryonated chicken eggs. In the current process, three influenza strains selected by WHO/CDC are propagated in chicken eggs, chemically inactivated, and semi-purified. The egg-based technology, however, is unable to respond to a pandemic crisis. Vaccine development and production take several months following the identification of potential strains and typically requires the re-assortment with a high yield strain to obtained adequate growth properties [9], [10], [11], [12]. A minimum of 3–6 months is needed to develop an influenza vaccine using this approach. More importantly, the H5 avian influenza strains responsible for recent epizootic outbreak involving numerous human infections are lethal to chicken eggs used for vaccine production and to the chickens that lay the eggs. If the 2003 severe acute respiratory syndrome (SARS)-coronavirus outbreak serves as a guide, the next influenza pandemic will likely have global consequences spreading within weeks, if not within days. Thus, a system that can rapidly produce new influenza vaccine is needed to prevent or to effectively reduce the impact of pandemic influenza.
Two new approaches have shown great promise to replace the egg-based technique [9], [10], [11]. One is cell culture-based, and the other is recombinant protein (antigen)-based. The cell culture-based approach involves production of influenza viruses in cell culture followed by the current (egg-based) virus inactivation and purification for the down stream processing. The advantages are: cell cultures are easier to handle and can be scaled up in a short period of time, and the influenza vaccines produced with this approach have been tested in Phase I and Phase II clinical trials and were found to be safe and at least as effective as the vaccines produced in embryonated chicken eggs [13], [14], [15]. A limitation of the cell culture-based approach is that the process still requires the production of a high-yielding re-assorted virus. This process also may introduce cell line specific mutations in the genes that can lead to the selection of variants characterized by antigenic and structural changes in the HA protein, potentially resulting in less-efficacious vaccines [16], [17], [18]. Additional hurdles include: the production and handling of a dangerous virus requires the availability of a high containment facility; mammalian cells can harbor animal viruses that may lead to safety concerns; the residues from the expressing cells may cause some unknown side-effects since no thorough purification process has been introduced into the manufacturing process. On the other hand, the recombinant protein-based approach involves production of viral antigens such as HA and NA in cell culture with recombinant DNA technology and utilization of the purified antigens as the active ingredients in the vaccine. The rHA influenza vaccines developed using the baculovirus-insect cell expression system has been tested in several Phase I and Phase II human clinical trials involving over 1200 subjects that demonstrated safety, immunogenicity and efficacy [19], [20], [21], [22], [23]. In elderly adults, rHA vaccine is equally or more immunogenic than the egg-based vaccine (Treanor JJ, et al. Dose-related safety and immunogenicity of a trivalent baculovirus-expressed influenza virus HA vaccine in elderly adults, manuscript in preparation). Interestingly, two H1N1 rHA vaccines (derived from two strains of A/New Caledonia/20/99 or A/Texas/36/91) provided partial protection against the lethal challenge of a reconstructed highly lethal 1918 pandemic influenza virus (also a H1N1 strain) in mice, suggesting that cross protection against drifted strains is definitely feasible [24].
To meet the challenge of a potential influenza pandemic, however, a reliable expression system and a quick, efficient downstream purification process are needed. In this communication, we reported a rapid process capable to purify rHA (H1N1, A/New Caledonia/20/99) from the fermentation to 95% purity within 6 h with a 57% overall yield. Since all chromatographic media used here are chemically stable and commercially available, the process can be easily scaled up in a GMP facility. Various biochemical analyses indicated that the purified rHA is properly folded and biologically active. In addition, we also developed a quick, simple analytical assay to monitor the expression of rHA in the insect cell fermentation to ensure the rHA production.
2. Materials and methods
2.1. Cloning and expression of influenza HA
The influenza vaccine strain-A/New Caledonia/20/99 (H1N1)—was obtained from the CDC. The full-length HA gene (containing the HA1 and HA2 genes) from the influenza viruses was cloned using RT-PCR and inserted into a baculovirus transfer vector developed by Protein Sciences Corporation. This specialized vector contained the promoter from the baculovirus polyhedrin gene flanked by sequences naturally surrounding the polyhedrin locus. Next, the transfer vector was co-transfected into insect cells with the linearized baculovirus genomic DNA (Autographa Californica Nuclear Polyhedrosis Virus) depleted of the polyhedrin gene and part of an essential gene downstream of the polyhedrin locus. The homologous recombination between the transfer plasmid and the linearized viral DNA rescued the virus, resulting in recovery efficiencies of recombinant virus of nearly 100%. Recombinant viruses were then selected by plaque assay. The plaque-derived recombinant baculovirus was then used to create a virus stock by infecting increasingly larger cultures of the proprietary insect cells (expresSF+®, derived from Sf9 cells) in serum-free culture medium (Protein Sciences Fortified Medium). The virus stock was then used to infect insect cells (2.0 × 106 cells/ml) to produce rHA in a 15-liter Applikon bioreactor. The multiplicity of infection (MOI) of the virus stock was 1 for the experiment.
To monitor the infection process and the expression of HA, 4 ml samples were taken from the bioreactor at various times. One millilitre was used for analyzing the changes in cell density, cell viability and cell size distribution. Two millilitres were centrifuged at 1600 rpm. The supernatant and pellet were stored separately at −80 °C to be used for SRID, gel and blot analysis. One millilitre was used for hemadsorption analysis. The rest was used for protein purification.
2.2. Hemadsorption assay
To 0.5 ml fermentation samples (insect cells uninfected, infected with recombinant baculovirus containing HA genes, and infected with recombinant baculovirus containing a non-HA gene) in a 1.5 ml Eppendorf tube, 0.10 ml of 5% chicken red blood cells (Charles River–Spafas, North Franklin, CT) in PBS was added and shaken gently for 10 min at room temperature. At the end of incubation, the tube was flipped gently for five times to get a homogenous suspension. Then, 10 μl of the suspension was pipetted on a glass plate and observed under a microscope (CK2, Olympus Optical Co., Japan) in three representative view fields. On average, about 20–70 insect cells were counted in each field. To reduce the chance of false positives, only the insect cells attached by three or more RBCs were counted as the RBC-bound insect cell. The percentage of RBC-bound insect cells against the total insect cells in each time point was calculated from three fields.
2.3. Cell analysis
At each time point, 1.0 ml of fermentation sample was analyzed with an automated cell analyzer (Cedex AS20, Innovatis GmbH, Germany) for cell density, cell viability and cell size distribution using the procedure described by the manufacture.
2.4. Buffers and columns
Buffer A: 20 mM sodium phosphate, 1.0 mM EDTA, 0.01 % Tergitol-NP9, 5% glycerol, pH 5.89. Buffer B: 20 mM sodium phosphate, 0.03 % Tergitol, 5% glycerol, pH 7.02. Buffer C: 20 mM sodium phosphate, 150 mM NaCl, 0.03% Tergitol, 5% glycerol, pH 7.02. Buffer D: 40 mM sodium phosphate, 0.05% Tween-20, 5% glycerol, pH 7.20. Buffer E: 100 mM sodium phosphate, 0.05% Tween-20, 5% glycerol, pH 7.20. Buffer F: 500 mM sodium phosphate, 0.05% Tween-20, 5% glycerol, pH 7.20. Buffer G: 10 mM sodium phosphate, 150 mM NaCl, 0.01% Tween-20, pH 7.22. Sanitation buffer: 1.0 M NaCl, 0.5 M NaOH.
UNOsphere-Q (Bio-Rad, Hercules, CA) column, ø 1.6 cm × 10 cm, 20 ml; SP-Sepharose Fast Flow (GE/Amersham/Pharmacia, Piscataway, NJ) column, ø 1.6 cm × 10 cm, 20 ml; Hydroxyapatite Type I column (HX-I, Bio-Rad, Hercules, CA), ø 1.0 cm × 4.6 cm, 3.6 ml.
2.5. Purification procedure
The fermentations producing rHA were harvested by centrifugation at 56–65 h post-infection. The cell pellet (6.4 g) was extracted with 225 ml of 1% Tergitol NP-9 in buffer A by stirring on a magnetic stirrer at 4 °C for 30 min. The extract was clarified by centrifugation at 10,000 × g for 25 min. The supernatant was loaded on Q/SP columns (equilibrated with Buffer A) in tandem at 5 ml/min. After loading, the columns were washed with 140 ml of Buffer A. Then, the columns were disconnected. HA was eluted from the SP column with 140 ml of buffer B and 80 ml of buffer C, consecutively. The Q/SP columns can be regenerated by washing with 5 column volumes (CV) of sanitation buffer and 5 CV of water and equilibrated with 5 CV of buffer A.
The HA fraction in buffer B (40 ml) was loaded on a HX-I column at 2 ml/min. The column was washed with 18 ml of buffer B. The HA was eluted from the HX-I column with increasing phosphate concentration (buffers D, E and F). The HA preparation in buffer D was further purified and concentrated by ultrafiltration with a stir cell using a 100 kDa MWCO regenerated cellulose membrane with buffer G. The HX-I column can be regenerated by washing with 10 CV of water, and equilibrated with 10 CV of buffer C.
2.6. Single radial immunodiffusion (SRID)
The rHA contents in all preparations were determined with SRID assay as described by Williams [25] and Manchini et al. [26]. The assay is based on the diffusion of rHA into a 1% agarose gel containing antibodies against the HA. The interaction between antigen and antibody produced a precipitation ring of which the size was directly proportional to the amount of antigen applied. The diameters of the rings in the SRID assays were determined with a measuring magnifier (Baush/Lomb, 81-34-38). The diameters of the precipitate ring were used to determine the actual concentrations based on standards provided by the Center for Biologics Evaluation and Research of FDA.
2.7. Deglycosylation
For complete deglycosylation, 20 μg of purified rHA was deglycosylated with 5000 units of peptide-N-glycosidase F (PNGase F, New England BioLabs, Beverly, MA, USA) or Endoglycosidase H (Endo H, New England BioLabs) at 37 °C for 60 min as described previously [27]. For limited deglycosylation, 20 μg of rHA was treated with 0.2 μg of trypsin on ice for 30 min. The digestion was stopped by adding 10× denaturing buffer and boiling for 5 min. Then the trypsin treated rHAs were deglycosylated with 2, 20 or 200 units of PNGase F or Endo H on ice, at 25 °C or at 37 °C for 2–60 min. The reactions were stopped by adding 2× SDS sample buffer and boiling for 5 min. The protein species at various deglycosylation stages were resolved on SDS–PAGE.
2.8. Other biochemical analyses
Vaxigrip influenza vaccine 2004–2005 was purchased from Canada Drug Delivery (Nanaimo BC, Canada). The purity of rHA was measured on SDS–polyacrylamide gels stained with Coomassie blue using scanning laser densitometry (model 710, Bio-Rad, Hercules, CA, USA) and peak integration analysis. The total amino acid analysis was carried out with a Beckman amino acid analyzer at Keck Facility of Yale University. The N-terminal amino acid sequence analysis was executed at the Protein Core Facility of Columbia University. The molecular size of the purified rHA was analyzed on a size-exclusion columns (TSK-4000, 7.5 × 300 mm, TosaHaas, Japan) at a constant flow rate of 0.8 ml/min, using the protein molecular weight markers as the reference (Sigma, St. Louis, USA) as previously described [28]. Elution buffer: 50 mM sodium phosphate, 50 mM NaCl and 0.001% NaN3. A trypsin resistance assay was carried out by incubating rHA for 30 min at 0 °C without or with 50 μg/ml TPCK-treated trypsin as described by Copeland et al. [29]. For this assay, the denatured HA was produced by boiling rHA for 5 min. Hemagglutination activity assays were done essentially as described by Barrett and Inglis [30] with a 0.5% solution of fresh chicken red blood cells in a U-bottom 96-well microtiter plate.
3. Results
3.1. Monitoring the expression of rHA in insect cell
A critical issue in production of a therapeutic protein using recombinant DNA technology is determining when to harvest the fermentation [31], [32]. Too early, the yield may be suboptimal. Too late, the expressed protein may be degraded by a variety of proteases released during the lytic process of infected cells. Thus, a rapid and sensitive assay is needed to monitor protein expression and to choose the right harvest time. HA is well known for its ability to bind the sialic acid on the surfaces of red blood cells (RBCs) and agglutinate these cells [30]. This phenomenon has been successfully used to detect cells and tissues infected with influenza viruses. To determine whether the insect cells infected with the recombinant baculoviruses containing the HA gene can also agglutinate RBCs, both uninfected and infected insect cells were incubated with RBCs for 10 min and observed under a microscope. In the uninfected insect cell sample, the RBCs (the smaller cells) were scattered around on the slide, and no specific binding of RBCs to the insect cells (the larger cells, about 16 μm in diameter) was observed as shown in Fig. 1a. On the other hand, most of RBCs were bound to the insect cells infected with baculovirus containing the HA gene derived from influenza strain A/New Caledonia/20/99 (H1N1) (Fig. 1b). This method also works for expression of the HAs derived from other strains such as B/Jiangsu/10/2003 (Fig. 1c) and A/New York/55/2004 (H3N2) (data not shown). To verify that the observed hemadsorption is due to the HA genes and not to the other genes in the baculovirus, insect cells infected with recombinant baculovirus containing a non-HA gene were also incubated with RBCs. No binding of RBCs on insect cells was observed (Fig. 1d). These observations clearly demonstrate that RBC's binding to the surface of insect cells is HA expression dependent, not infection dependent. The data also support the conclusion that the HAs expressed in insect cells are properly folded and biologically active.
Fig. 1.
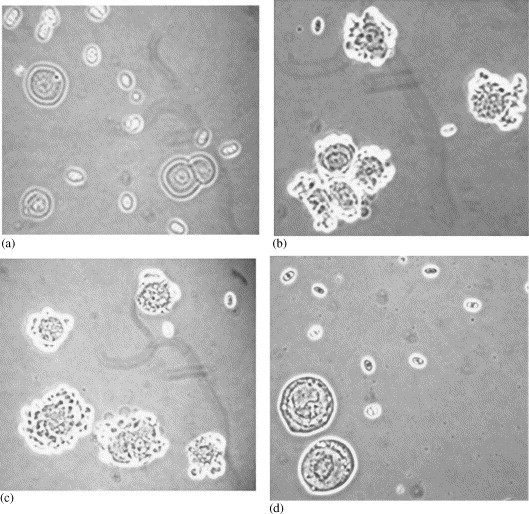
Monitoring rHA expression with hemadsorption. (a) Uninfected insect cells. (b) The insect cells infected with the baculovirus containing the HA gene from the A/New Caledonia/20/99 (H1N1) influenza virus. (c) The insect cells infected with the baculovirus containing the HA gene from the B/Jiangsu/10/2003 influenza virus. (d) The insect cells infected with the baculovirus containing a non-HA gene.
This hemadsorption assay was further optimized to monitor the expression of HA during fermentation. Even though the infection of insect cells can be seen by observing morphological changes as early as 6 hours post infection (HPI), the first binding of RBCs to insect cells was only observed around 23 HPI—about 20% of insect cells were bound by three to eight RBCs. Almost all insect cells were infected at 23 HPI according to cell morphology. The binding reached its peak around 45–55 HPI, when 70–80% of insect cells were bound by 8–20 RBCs (in some fermentations, almost all insect cells were bound by RBCs during this time), as shown in Table 1 . At the peak of hemadsorption, clusters of 20–200 insect cells agglutinated by RBCs have also been observed. Later, the binding gradually reduces to 30–40% of insect cells, most likely caused by the breakdown of some infected cells and the loss of cell membrane, as evidenced by the rapid decrease of cell viability. To determine the rHA levels in the fermentation at each sampling time, the single radial immunodiffusion (SRID) assay has been used, since it is a simple, reproducible technique, and relatively unaffected by other proteins in the crude extract [25], [26]. Consistently, the RBC binding to insect cells correlated well with the HA levels determined by SRID. Late HA expression is expected because the HA gene is regulated by the polyhedrin promoter, which is a late stage promoter in baculovirus infection. Similar results were also obtained for the expression of other HAs such as B/Jiangsu/10/2003 and A/New York/55/2004 (data not shown).
Table 1.
Possible correlation among HPI, hemadsorption, cell viability and HA yielda
| HPI | %Binding | Viability | HA (mg/L) |
|---|---|---|---|
| 0 | 0 | 92.9 | 0 |
| 6 | 0 | 92.9 | 0 |
| 23 | 20 | 87.6 | 4 |
| 30 | 30 | 88.2 | 7 |
| 46 | 65 | 74.7 | 18 |
| 50 | 80 | 52.4 | 20 |
| 54 | 70 | 50.1 | 20 |
| 70 | 40 | 21.8 | 11 |
| 75 | 30 | 16.1 | 11 |
HA yield was determined by SRID assay. The MOI of the virus stock was 1 for the experiment.
3.2. Designing and optimizing rHA purification process
A major challenge in the biotechnology industry is purification of biologically active recombinant proteins [33], [34]. An ideal purification process should be mild, efficient and capable of achieving high purity in a short period of time. Accordingly, each purification step has been carefully designed to optimize the whole process. As demonstrated in the hemadsorption studies, rHAs are expressed, folded and transported to the cell membrane at a late stage. To extract rHA from the cell membranes, several non-ionic detergents at various concentrations were tested for their efficiency. The best result was obtained using 1% Tergitol NP-9. To get a relatively clean extraction, a magnetic stirrer was used to avoid the disruption of cell nuclei and other organelles. The extract was clarified by centrifugation to remove cell nuclei and other debris.
The HA monomer of A/New Caledonia/20/99 influenza virus strain consists of 547 amino acids with a theoretical molecular weight of 63,156.43 and a pI of 6.30. Therefore, this rHA can be bound on a cation-exchange media like SP using a lower pH buffer and eluted with a higher pH buffer to achieve the primary purification and concentration. The supernatant of the extract was loaded on UNO Sphere Q/SP columns in tandem. The anion-exchange Q column acts as a scavenger by binding the negative-charged impurities that may foul the SP column. After loading and washing, the columns were detached and eluted separately. As shown in the chromatogram (Fig. 2a), about 50% of the proteins flow through Q/SP columns, a small amount of protein elutes from the SP with the pH 7 buffer and the rest bind tightly to either the SP or the Q column. As shown on the SDS–PAGE (Fig. 2b), the rHA captured on the SP at pH 6 was selectively eluted by a shift to pH 7. The pH shift resulted in a 14-fold increase in purity based on densitometry of the SDS–PAGE gel.
Fig. 2.
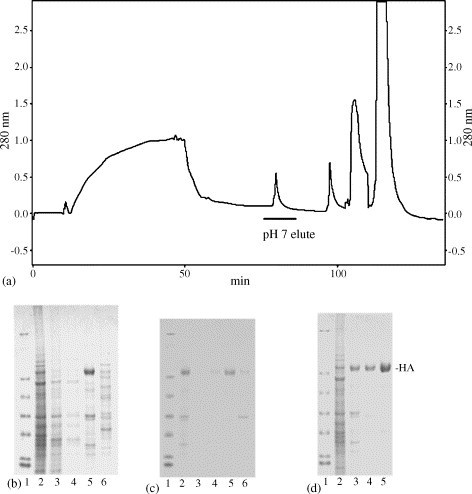
The purification process of rHA. (a) The chromatogram of primary purification and concentration on Q/SP columns. (b) The SDS–PAGE of Q/SP fractions, lanes 1–6 are molecular weight markers, applied sample, flow through, wash, pH 7 (buffer B) eluate, and buffer C eluate, respectively. (c) The SDS–PAGE of secondary purification on HX-I, lanes 1–6 are molecular weight markers, applied sample, flow through, wash, 40 mM phosphate eluate, and 100 mM phosphate eluate, respectively. (d) The SDS–PAGE of step-wise purification, lanes 1–5 are molecular weight markers, Q/SP applied sample, HA after the primary purification, HA after the secondary purification, HA after the final polish using ultrafiltration.
To further purify rHA, use of hydroxyapatite type I (HX-I) media was explored. The binding preference of HX-I media is significantly different from the ion-exchange media, and yet the binding and eluting conditions are relatively mild so as to preserve the biological activity of the target protein [35], [36]. Thus, the pH 7 SP column eluate was loaded on a HX-I column. After washing, rHA was eluted from the column with increased phosphate concentration. As shown in Fig. 2c, most rHA was eluted in 40 mM phosphate, and there was a small loss in the wash and in the 100 mM phosphate elution. The purity was increased from 52% to 91% according to the densitometry of the SDS–PAGE.
However, there was still a protein contaminant of about 36 kDa in the preparation as revealed on lane 5 of Fig. 2c. Since the size of rHA trimer is around 210 kDa, the difference between rHA size and this impurity could be explored to remove this impurity. Thus, the 40 mM phosphate eluate was further purified with ultrafiltration using a stir cell (100 kDa MWCO). As demonstrated in Fig. 2d, the 36 kDa band (lane 5) was selectively removed from the retentate. The purified rHA migrated on SDS–PAGE gel as a single polypeptide (rHA) with an apparent molecular weight of approximately 70 kDa. On the blot, a small amount of rHA1 and rHA2 (the cleavage products of rHA) were also observed, with apparent molecular weights of ≈50 and ≈28 kDa, respectively (data not shown). Trace amounts of rHA dimers and trimers were also detectable, with apparent molecular weights of ≈140 and ≈220 kDa, respectively.
As summarized in Table 2 , the process described here can purify rHA from the fermentation to 95% purity within 6 h with a 57% overall yield. The largest single step loss (27%) is on HX-1 column.
Table 2.
Stepwise mass balance of HA-NC purification by SRID assay
| HA (μg/ml) | Volume (ml) | HA (mg) | Puritya (%) | Step recovery (%) | Total recovery (%) | |
|---|---|---|---|---|---|---|
| Starting material | 53 | 225 | 12 | 3.6 | ||
| Q/SP | 240 | 40 | 9.6 | 52 | 80 | 80 |
| HX-1 | 280 | 25 | 7.0 | 91 | 73 | 58 |
| Ultrafiltration | 700 | 9.1 | 6.4 | 99 | 91 | 53 |
Purity was determined by the densitometry of SDS–PAGE.
3.3. Characterizing rHA protein
To confirm the authenticity of rHA, the purified protein was examined by N-terminal amino acid sequencing and total amino acid analysis (AAA). The N-terminal amino acid sequence matched the predicted one (10 cycles were used) and the signal sequence peptide (the first 17 amino acids) of the full-length HA gene was absent in rHA. The measured amino acid composition of the purified rHA was consistent with the theoretical one (data not shown). The authenticity of rHA was further verified by the Western blot using A/New Caledonia/20/99 antibody provided by the Food and Drug Administration (FDA).
The purified rHA in 0.005% Tween/PBS solution was analyzed using size-exclusion chromatography. It was eluted as a single peak at 9.1 min as shown in Fig. 3a, corresponding to a molecular weight around 800–1000 kDa, likely a complex of four to five HA trimers ((4–5) × 3 × 70 kDa). To test whether the purified rHA still retained its native structure, the purified protein was treated with trypsin on ice. As shown in the lane 3 of Fig. 3b, rHA was cleaved into only two bands, HA1-50.9 kDa and HA2-27.5 kDa. On the other hand, the heat-denatured rHA was digested into numerous small fragments. The trypsin-resistance data demonstrate that the rHA expressed in insect cells folded properly and retained its native structure after purification.
Fig. 3.
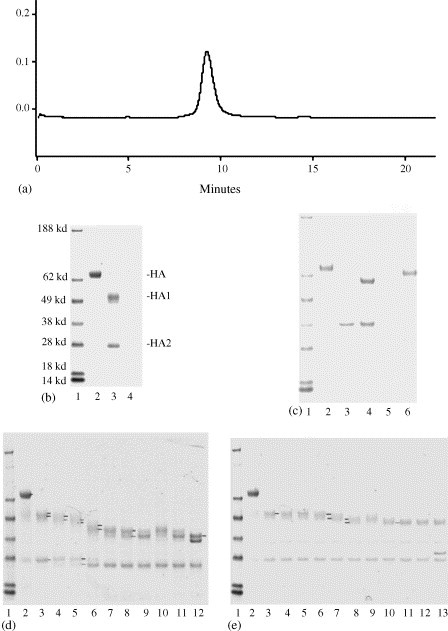
Characterization of rHA. (a) The SEC profile of the purified rHA. (b) The trypsin-resistant test analyzed on SDS–PAGE, lanes 1–4 are molecular weight markers, native rHA, native rHA treated with trypsin, and heat-denatured rHA treated with trypsin, respectively. (c) Deglycosylation of rHA with PNGase F and Endo H, lanes 1–6 are molecular weight markers, rHA, PNGase F, rHA treated with PNGase F, Endo H, and rHA treated with Endo H, respectively. (d) Limited deglycosylation of rHA with PNGase F. Lane 1, molecular weight markers; lane 2, rHA; lane 3, trypsin treated rHA; lanes 4–8, the trypsin treated rHAs were deglycosylated with 2 units of PNGase F on ice, at 25 °C or at 37 °C for 2–60 min; lanes 9–11, deglycosylated with 20 units of PNGase F; lane 12, deglycosylated with 200 units of PNGase F. (e) Limited deglycosylation of rHA with Endo H. Lane 1, molecular weight markers; lane 2, rHA; lane 3, trypsin treated rHA; lanes 4–8, the trypsin treated rHAs were deglycosylated with 2 units of Endo H on ice, at 25 °C or at 37 °C for 2–60 min; lanes 9–12, deglycosylated with 20 units of Endo H; lane 13, deglycosylated with 200 units of Endo H.
Since glycosylation may play an important role in the biological function of HA [37], [38], [39], it is of interest to explore whether the rHA produced in insect cells is properly glycosylated. Thus, rHA was deglycosylated with peptide-N-glycosidase F (PNGase F) or endoglycosidase H (Endo H) and resolved on SDS–PAGE. As shown in Fig. 3c, the untreated HA migrated at 70.4 kDa, PNGase F deglycosylated HA at 57.9 kDa, and Endo H treated HA at 64.8 kDa, respectively. These data indicate that the rHA produced in insect cells is indeed glycosylated with N-linked oligosaccharide side chains. About 5.6 kDa of oligosaccharide chains have high mannose content susceptible to Endo H, and 6.9 kDa of oligosaccharide chains have low mannose residues resistant to Endo H. To assess the number of N-linked oligosaccharide chains, the trypsin treated rHA was subjected to limited deglycosylation with PNGase F or Endo H under a variety of conditions. On the SDS–PAGE of PNGase F treated samples (Fig. 3d), there are seven distinguishable HA1 bands, 50.9, 49.0, 46.8, 45.2, 43.3, 42.0 and 39.8 kDa, and 2 HA2 bands, 27.1 and 25.4 kDa, respectively. On the SDS–PAGE of Endo H treated samples (Fig. 3e), there are three distinguishable HA1 bands, 51.2, 49.4 and 46.5 kDa, and one HA2 band, 27.1 kDa. The data suggest that there are six N-linked oligosaccharide chains in the HA1 region, and two of them have high mannose content. There is only one N-linked oligosaccharide chain in the HA2 region, which is likely a hybrid or a complex oligosaccharide. Consistently, there are six predicted N-linked glycosylation sites in the HA1 region and one predicted N-linked glycosylation site in the HA2 region of HA/A/New Caledonia/20/99, according to the sequence analysis (NetNGlyc) at Technical University of Denmark (http://www.cbs.dtu.dk/services/NetNGlyc/).
To directly compare with the A/New Caledonia antigen present in the egg-based vaccine, the purified rHA/A/New Caledonia was formulated either alone into a 15 μg/0.5 ml solution or with 15 μg rHA/B/Jiangsu and 45 μg rHA/A/Wyoming in a 0.5 ml dosage (FluBlØk™, the expected trade name of Protein Sciences’ rHA vaccine). As judged by the hemagglutination assay, FluBlØk is as active as Vaxigrip (a licensed egg-based vaccine manufactured by Sanofi-Pasteur-Aventis) in agglutinating RBCs and preventing them from forming a tight pellet as shown in Fig. 4a. Antigen specific SRID assay is widely used to determine the concentration of active ingredients in a vaccine. On the SRID gel prepared with A/New Caledonia/20/99 antibody (Fig. 4b), the diffusion ring of FluBlØk is slightly larger than that of Vaxigrip, suggesting that FluBlØk is equivalent to Vaxigrip for the active ingredient of A/New Caledonia/20/99 strain. On the SDS–PAGE (Fig. 4c), the FluBlØk lane is clean, and only three broad bands are visible, representing the HA, HA1 and HA2 of the three strains, respectively. The Vaxigrip lane is complicated with the major HA band around 59 kDa along with many minor bands. Similarly, numerous impurities have also been found in other egg-based vaccines [40].
Fig. 4.
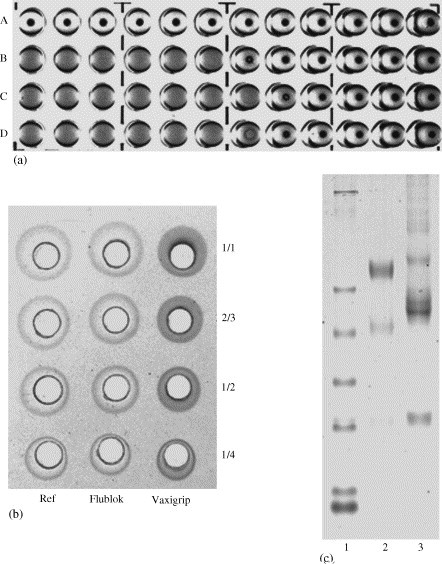
The comparison of FluBlØk with Vaxigrip. (a) The hemagglutination activities of 1.0 μg/ml rHA or equivalent amount from the vaccines. Row A, PBS as negative control; Row B, rHA; Row C, Vaxigrip; Row D, FluBlØk. (b) The evaluation of the effective antigen concentration with SRID assay. (c) The SDS–PAGE of the vaccines. Lane 1, molecular weight markers; lane 2, FluBlØk; lane 3, Vaxigrip.
All these data demonstrate that the purified rHA expressed in insect cells is correctly translated, properly glycosylated and folded and biologically active.
4. Discussion
The traditional egg-based vaccines have been successfully used for more than 50 years to prevent influenza. They are reliable, effective (if there is a good match), and affordable. However, the production cycle of the egg-based vaccines is lengthy and heavily dependent on egg supply and unable to be developed quickly in response to the urgent need in an influenza pandemic [9], [10], [11], [12]. To replace or supplement the egg-based vaccines, the new vaccine has to be equally effective, reliable, economical, and capable of being developed and delivered in a short period of time. The work reported here is some important progress toward an alternative influenza vaccine-the recombinant protein-based vaccine.
A new analytical method based on hemadsorption has been developed to closely monitor the expression of HA in insect cells. This method plays a critical role in ensuring optimal HA production and in determining the right harvest time, in addition to other harvest parameters such as HPI, cell's morphology and viability. It has also been successfully used at Protein Sciences Corporation to accelerate the screening process for the recombinant baculoviruses used for HA manufacturing.
A purification process has been developed to quickly purify the recombinant HA from the bulk harvested from the bioreactor while retaining its biological activity. Previously, HA purifications were heavily dependent on affinity chromatography using specific monoclonal antibodies or various lectins [41], [42]. Such methods are highly selective, but difficult to scale up to commercial levels due to a number of limiting factors: (1) some of the ligands will leach off the column during the purification and they must be removed from the final product; (2) it is difficult to regenerate an affinity column after use, and thus the performance declines after each use; (3) the batch to batch variations in the quality of affinity media make it almost impossible to have a robust purification process from time to time; (4) most affinity media are too expensive to be used at large scale. On the other hand, all chromatographic media used in the present process are chemically stable (can be regenerated repeatedly), commercially available and relatively inexpensive; thus more suitable to scale up in a GMP facility. If the reagents, columns and ancillary equipment are well prepared in advance, the whole purification can be completed in one full working day, avoiding the overnight storage of intermediate rHA preparation and possible inactivation of rHA. Several factors have made this rapid process possible. First, there is no sample manipulation between purification steps, which makes a quick, continuous purification process possible. Second, Q/SP is connected in tandem, combining two chromatographic processes into one. Third, a chemically different chromatographic media—HX-I is used to differentiate rHA from the remaining impurities. Unlike ion-exchange media, the adsorption of proteins to HX-I involves both anionic and cationic interaction. The calcium group can interact with carboxylate residues, whereas the phosphate group can bind the basic residues on the surface of the protein. The bound proteins can be eluted by an increasing phosphate gradient or a gradient of calcium, magnesium ions. It is worth to point out that the purification process described here needs to be further optimized for large-scale production, for example, a tangential flow filter should be used to replace a stir cell at the polish step. Nonetheless, we believe the strategies described here can also be used to develop a rapid purification process for other recombinant proteins.
Biologically, FluBlØk is as active as the egg-based vaccine—Vaxigrip as determined by the hemagglutination assay. Based on the SRID assay, FluBlØk is equivalent to Vaxigrip for the active ingredient of A/New Caledonia/20/99 strain of influenza. In challenge studies, chickens were effectively protected against the H5N1 virus infection after inoculation with the rHA of the virus [43]. Moreover, two distantly related H1N1 rHA influenza vaccines using the baculovirus-insect cell expression system have also been demonstrated to partially protect mice against the lethal challenge of a recombinant 1918 pandemic influenza virus [24]. In clinical trials, the trivalent rHA vaccine (FluBlØk) stimulates anti-HA antibody production at least as well as, and in the case of H3 rHA, superior to the traditional egg-based vaccine [19], [20], [21], [22], [23]. The 2004/05 influenza season Phase IIB field trial of FluBlØk that enrolled 460 healthy subjects aged 18–49 further showed that the 45/45/45 dose was 100% efficacious in preventing culture positive influenza illness compared to placebo (Press Release, Protein Sciences Corp., June 14, 2005).
Furthermore, a recombinant protein-based vaccine, such as the FluBlØk described here, also has other advantages over the traditional egg-based vaccines. It consists solely of three antigens (proteins) stored in sterile phosphate buffered-saline and without preservatives such as thimerosal (a mercury derivative currently used in the egg-based vaccine), antibiotics or adjuvants. Unlike the egg-based vaccines, no live influenza viruses, biocontainment facilities or harsh chemicals such as formaldehyde are used in manufacturing. This may explain why FluBlØk has shown lower side effects than the licensed vaccines in clinical trials [19], [20], [21], [22], [23]. Therefore, a reliable, effective, and affordable recombinant protein-based influenza vaccine can be and should be developed to meet the challenge of a potential influenza pandemic. For pandemic preparedness, developing and stockpiling rHA influenza vaccines against the present H5N1 strain may be a good option to provide some protection for the first response personnel and the population in the hard-hit areas in the case of a pandemic, and to win the precious time for manufacturing of a more specific influenza vaccine.
References
- 1.Hilleman M.R. Realities and enigmas of human viral influenza: pathogenesis, epidemiology and control. Vaccine. 2002;20:3068–3087. doi: 10.1016/s0264-410x(02)00254-2. [DOI] [PubMed] [Google Scholar]
- 2.Glezen W.P. Serious morbidity and mortality associated with influenza epidemics. Epidemiol Rev. 1982;4:24–44. doi: 10.1093/oxfordjournals.epirev.a036250. [DOI] [PubMed] [Google Scholar]
- 3.Cox N.J., Subbarao K. Influenza. Lancet. 1999;354:1277–1282. doi: 10.1016/S0140-6736(99)01241-6. [DOI] [PubMed] [Google Scholar]
- 4.Clements M.L., Betts R.L., Tierney R.L., Murphy B.R. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol. 1986;22:157–160. doi: 10.1128/jcm.24.1.157-160.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skehel J.J., Wiley D.C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 6.Wood J.M., Robertson J.S. From lethal virus to life-saving vaccine: developing inactivated vaccines for pandemic influenza. Nat Rev Microbiol. 2004;2:842–847. doi: 10.1038/nrmicro979. [DOI] [PubMed] [Google Scholar]
- 7.Stephenson I., Nicholson K.G., Wood J.M., Zambon M.C., Katz J.M. Confronting the avian influenza threat: vaccine development for a potential pandemic. Lancet Infect Dis. 2004;4:499–509. doi: 10.1016/S1473-3099(04)01105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ungchusak K., Auewarakul P., Dowell S.F., Kitphati R., Auwanit W., Puthavathana P. Probable person-to-person transmission of avian influenza A (H5N1) N Engl J Med. 2005;352:333–340. doi: 10.1056/NEJMoa044021. [DOI] [PubMed] [Google Scholar]
- 9.Cox M. Cell-based protein vaccines for influenza. Curr Opin Mol Ther. 2005;7:24–29. [PubMed] [Google Scholar]
- 10.Audsley J.M., Tannock G.A. The role of cell culture vaccines in the control of the next influenza pandemic. Exp Opin Biol Ther. 2004;4:709–717. doi: 10.1517/14712598.4.5.709. [DOI] [PubMed] [Google Scholar]
- 11.Sheridan C. Next generation flu vaccine boosted by Chiron debacle. Nat Biotechnol. 2004;22:1487–1488. doi: 10.1038/nbt1204-1487. [DOI] [PubMed] [Google Scholar]
- 12.Osterholm M.T. Preparing for the next pandemic. N Engl J Med. 2005;352:1839–1842. doi: 10.1056/NEJMp058068. [DOI] [PubMed] [Google Scholar]
- 13.Brands R., Visser J., Medema J., Palache A.M., van Scharrenburg G.J. Influvac: a safe Madin Darby Canine Kidney (MDCK) cell culture-based influenza vaccine. Dev Biol Stand. 1999;98:93–100. [PubMed] [Google Scholar]
- 14.Percheson P.B., Trepanier P., Dugre R., Mabrouk T. A Phase I, randomized controlled clinical trial to study the reactogenicity and immunogenicity of a new split influenza vaccine derived from a non-tumorigenic cell line. Dev Biol Stand. 1999;98:127–132. [PubMed] [Google Scholar]
- 15.Alymova I.V., Kodihalli S., Govorkova E.A., Fanget B., Gerdil C., Webster R.G. Immunogenicity and protective efficacy in mice of influenza B virus vaccines grown in mammalian cells or embryonated chicken eggs. J Virol. 1998;72:4472–4477. doi: 10.1128/jvi.72.5.4472-4477.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meiklejohn G., Eickhoff T.C., Graves P. Antigen drift and efficacy of influenza virus vaccines. J Infect Dis. 1987;138:618–624. doi: 10.1093/infdis/138.5.618. [DOI] [PubMed] [Google Scholar]
- 17.Robertson J.S., Naeve C.W., Webster R.G., Bootman J.S., Newman R., Schild G.C. Alterations in the hemagglutinin associated with adaptation of influenza B virus to growth in eggs. Virology. 1985;143:166–174. doi: 10.1016/0042-6822(85)90105-9. [DOI] [PubMed] [Google Scholar]
- 18.Schild G.C., Oxford J.S., de Jong J.C., Webster R.G. Evidence for host-cell selection of influenza virus antigenic variants. Nature. 1983;303:706–709. doi: 10.1038/303706a0. [DOI] [PubMed] [Google Scholar]
- 19.Lakey D.L., Treanor J.J., Betts R.F., Smith G.E., Thompson J., Sannella E. Recombinant baculovirus influenza A hemagglutinin vaccines are well tolerated and immunogenic in healthy adults. J Infect Dis. 1996;174:838–841. doi: 10.1093/infdis/174.4.838. [DOI] [PubMed] [Google Scholar]
- 20.Powers D.C., Smith G.E., Anderson E.L., Kennedy D.J., Hackett C.S., Wilkinson B.E. Influenza A virus vaccines containing purified recombinant H3 hemagglutinin are well-tolerated and induce protective immune responses in healthy adults. J Infect Dis. 1995;171:1595–1599. doi: 10.1093/infdis/171.6.1595. [DOI] [PubMed] [Google Scholar]
- 21.Powers D.C., McElhaney J.E., Florendo O.A., Jr., Manning M.C., Upshaw C.M., Bentley D.W. Humoral and cellular immune responses following vaccination with purified recombinant hemagglutinin from Influenza A (H3N2) virus. J Infect Dis. 1997;175:342–351. doi: 10.1093/infdis/175.2.342. [DOI] [PubMed] [Google Scholar]
- 22.Treanor J.J., Betts R.F., Smith G.E., Anderson E.L., Hackett C.S., Wilkinson B.E. Evaluation of a recombinant hemagglutinin expressed in insect cells as an influenza vaccine in young and elderly adults. J Infect Dis. 1996;173:1467–1470. doi: 10.1093/infdis/173.6.1467. [DOI] [PubMed] [Google Scholar]
- 23.Treanor J.J., Wilkinson B.E., Masseoud F., Hu-Primmer J., Battaglia R., O’Brien D. Safety and immunogenicity of a recombinant hemagglutinin vaccine for H5 influenza in human vaccine. Vaccine. 2001;19:1732–1737. doi: 10.1016/s0264-410x(00)00395-9. [DOI] [PubMed] [Google Scholar]
- 24.Tumpey T.M., Garcia-Sastre A., Taubenberger J.K., Palese P., Swayne D.E., Basler C.F. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc Natl Acad Sci USA. 2004;101:3166–3171. doi: 10.1073/pnas.0308391100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams M.S. Single-radial-immunodiffusion as an in vitro potency assay for human inactivated viral vaccines. Vet Microbiol. 1993;37:253–262. doi: 10.1016/0378-1135(93)90027-5. [DOI] [PubMed] [Google Scholar]
- 26.Mancini G., Carbonara A.O., Heremans J.F. Immunochemical quantitation of antigens by single radial immunodiffusion. Immunochemistry. 1965;2:235–254. doi: 10.1016/0019-2791(65)90004-2. [DOI] [PubMed] [Google Scholar]
- 27.Maley F., Trimble R.B., Tarentino A.L., Plummer T.H., Jr. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem. 1989;80:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- 28.Wang K., Spector A. The chaperone activity of bovine alpha crystalline. Interaction with other lens crystallins in native and denatured states. J Biol Chem. 1994;269:13601–13608. [PubMed] [Google Scholar]
- 29.Copeland C.S., Doms R.W., Bolzau E.M., Webster R.G., Helenius A. Assembly of influenza hemagglutinin trimers and its role in intracellular transport. J Cell Biol. 1986;103:1179–1191. doi: 10.1083/jcb.103.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett T., Inglis S.C. Growth, purification, and titration of influenza viruses. In: Mahy B.W.J., editor. Virology: A Practical Approach. IRL Press; Washington DC: 1991. pp. 119–150. [Google Scholar]
- 31.Palomares L.A., Estrada-Mondaca S., Ramirez O.T. Production of recombinant proteins: challenges and solutions. Methods Mol Biol. 2004;267:15–52. doi: 10.1385/1-59259-774-2:015. [DOI] [PubMed] [Google Scholar]
- 32.Molowa D.T., Mazanet R. The state of biopharmaceutical manufacturing. Biotechnol Annu Rev. 2003;9:285–302. doi: 10.1016/s1387-2656(03)09008-2. [DOI] [PubMed] [Google Scholar]
- 33.Ikonomou L., Schneider Y.J., Agathos S.N. Insect cell culture for industrial production of recombinant proteins. Appl Microbiol Biotechnol. 2003;62:1–20. doi: 10.1007/s00253-003-1223-9. [DOI] [PubMed] [Google Scholar]
- 34.Andersen D.C., Krummen L. Recombinant protein expression for therapeutic applications. Curr Opin Biotechnol. 2002;13:117–123. doi: 10.1016/s0958-1669(02)00300-2. [DOI] [PubMed] [Google Scholar]
- 35.Schroder E., Jonsson T., Poole L. Hydroxyapatite chromatography: altering the phosphate-dependent elution profile of protein as a function of pH. Anal Biochem. 2003;313:176–178. doi: 10.1016/s0003-2697(02)00567-5. [DOI] [PubMed] [Google Scholar]
- 36.Karlsson G., Winge S. Separation between the alpha and beta forms of human antithrombin by hydroxyapatite high-performance liquid chromatography. Protein Expr Purif. 2003;28:196–201. doi: 10.1016/s1046-5928(02)00678-2. [DOI] [PubMed] [Google Scholar]
- 37.Klenk H.D., Wagner R., Heuer D., Wolff T. Importance of hemagglutinin glycosylation for the biological functions of influenza virus. Virus Res. 2002;82:73–75. doi: 10.1016/s0168-1702(01)00389-6. [DOI] [PubMed] [Google Scholar]
- 38.Schulze I.T. Effects of glycosylation on the properties and functions of influenza virus hemagglutinin. J Infect Dis. 1997;176(Suppl. 1):S24–S28. doi: 10.1086/514170. [DOI] [PubMed] [Google Scholar]
- 39.Helenius A., Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 40.Renfrey S., Watts A. Morphological and biochemical characterization of influenza vaccines commercially available in the United Kingdom. Vaccine. 1994;12:747–752. doi: 10.1016/0264-410x(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 41.Holtz K.M., Anderson D.K., Cox M.M.J. Production of a recombinant influenza vaccine using the baculovirus expression vector system. Bioprocess J. 2005;2:65–73. [Google Scholar]
- 42.Mir-Shekari S.Y., Ashford D.A., Harvey D.J., Dwek R.A., Schulze I.T. The glycosylation of the influenza A virus hemagglutinin by mammalian cells. A site-specific study. J Biol Chem. 1997;272:4027–4036. doi: 10.1074/jbc.272.7.4027. [DOI] [PubMed] [Google Scholar]
- 43.Crawford J., Wilkinson B., Vosnesensky A., Smith G., Garcia M., Stone H. Baculovirus-derived rHA vaccines protect against lethal influenza infections by avian H5 & H7 subtypes. Vaccine. 1999;17:2265–2274. doi: 10.1016/s0264-410x(98)00494-0. [DOI] [PubMed] [Google Scholar]