

Abstract
A double-inactivated, candidate whole virus vaccine against severe acute respiratory syndrome associated coronavirus (SARS-CoV) was developed and manufactured at large scale using fermenter cultures of serum protein free Vero cells. A two step inactivation procedure involving sequential formaldehyde and U.V. inactivation was utilised in order to ensure an extremely high safety margin with respect to residual infectivity. The immunogenicity of this double-inactivated vaccine was characterised in the mouse model. Mice that were immunised twice with the candidate SARS-CoV vaccine developed high antibody titres against the SARS-CoV spike protein and high levels of neutralising antibodies. The use of the adjuvant Al(OH)3 had only a minor effect on the immunogenicity of the vaccine. In addition, cell mediated immunity as measured by interferon-γ and interleukin-4 stimulation, was elicited by vaccination. Moreover, the vaccine confers protective immunity as demonstrated by prevention of SARS-CoV replication in the respiratory tract of mice after intranasal challenge with SARS-CoV. Protection of mice was correlated to antibody titre against the SARS-CoV S protein and neutralising antibody titre.
Keywords: SARS coronavirus, Double-inactivated vaccine, Neutralisation, Protection
1. Introduction
Severe acute respiratory syndrome is a new disease which emerged in China in late 2002 and spread rapidly around the world. This epidemic, which resulted in over 8000 infections and almost 800 deaths, raised considerable concerns that this emerging agent constituted a serious threat for global public health. The identification of a novel coronavirus (SARS-CoV) associated with this syndrome [1], [2], [3] raised the possibility of development of a vaccine to prevent infection. Although there have no reports of community-acquired human SARS infections since January 2004, a re-emergence of the disease is still a possibility. The virus remains in animal reservoirs and there is a potential for further spread to humans [4]. Thus, despite the disappearance of the disease in humans, there is still an urgent need for rapid development of a safe and effective vaccine. It is generally considered that live attenuated viruses are the most effective viral vaccines because of their ability to induce strong cellular and humoral immune responses [5]. However, the need for extensive safety testing of such vaccines can delay entry of such candidate vaccines into clinical trials. Multiple studies have reported that antibodies directed against the spike (S) glycoprotein can neutralise SARS-CoV infectivity [6], [7], [8], [9]. The S protein is reported to bind to the human angiotensin converting enzyme-2 (hACE-2) protein to mediate viral entry to the cell [10]. Human monoclonal antibodies directed against the S protein have been reported to reduce replication of the virus in the lung of infected ferrets and to completely prevent the development of SARS Co-V induced lung pathology [11]. In addition, a number of candidate vaccines induced protection against challenge with live virus in animal models, and this protection was associated with high levels of neutralising antibody [12], [13], [14]. Thus, it is likely that an inactivated or recombinant candidate vaccine containing the S protein will be able to induce protective immunity based primarily on stimulation of a humoral immune response.
We have favoured the development of an inactivated candidate whole virus vaccine as we have a well-established technology for rapid development of such vaccines. We have previously reported on the use of large scale Vero cell fermenter cultures for the development of an inactivated whole virus influenza vaccine [15]. This technology has been adapted for the development of a double inactivated whole virus SARS Co-V candidate vaccine. We report here on the characterisation of this candidate vaccine with respect to safety and immunogenicity.
2. Material and methods
2.1. Virus strain
SARS-CoV CDC#200301157 (also referred to as strain Utah; GenBank accession number AY714217) was obtained from the CDC (Atlanta, USA). It was isolated from sputum of a SARS patient in the USA (specimen #809940) and passaged two times on GMP grade Vero cells.
2.2. Electrophoretic and Western blot analysis
Denatured samples of the purified SARS-CoV vaccine (about 1 μg protein/lane) were separated on a 10% Tris–HCl sodium dodecyl sulfate-polyacrylamide gel under reducing conditions and subsequently blotted onto a PVDF membrane. Protein staining was done with the AuroDye™ forte staining kit (Amersham Bioscience/GE Healthcare, UK) according to manufacturers instructions. Rabbit antibodies directed against the SARS-CoV nucleocapsid (N) protein (IMG-548) or S protein (IMG-557), respectively, were obtained from Imgenex (San Diego, CA, USA) and used for Western blot analysis. After blocking and application of the primary antibody overnight at 2–8 °C (working dilution: 1:1000), membranes were washed and incubated for 1 h at room temperature with horse radish peroxidase (HRP)-labelled goat anti-rabbit IgG (Accurate Chemical, Westbury, USA). Following final washing, blots were developed using 3,3′-diaminobenzidine tetrahydrochloride and H2O2.
2.3. Immunisation of mice
2.3.1. Immunisation of CD1 mice
Female CD1 mice (6–8 weeks old) were obtained from the Charles River Laboratories subsidiary in Sulzfeld, Germany. After sampling of pre-immune sera, mice were immunised with six different doses of the candidate vaccine ranging from 1 μg to 0.3 ng total protein (method according to Bradford [16]). To evaluate the effect of adjuvantation on the immunogenicity of the vaccine, a non-adjuvanted preparation and material adjuvanted with 0.05 and 0.2% aluminium hydroxide (alum) was tested at each antigen concentration. Groups of 10 mice were inoculated subcutaneously (s.c.) with 0.5 ml of each vaccine preparation; control groups received the same volume of buffer (Tris-buffered saline) with or without 0.2% aluminium hydroxide. Fourteen days post-immunisation, sera were drawn from each animal and a booster immunisation was carried out with the same formulation and dosage as inoculated during primary immunisation. Sera were then drawn from each animal 4 weeks after primary immunisation (animals treated with the 0.2% aluminium hydroxide containing vaccine were also bled 12 weeks after primary immunisation).
2.3.2. Immunisation of Balb/C mice
Balb/C mice (6–7 weeks old) were also obtained from the Charles River Laboratories subsidiary. For cytokine studies groups of 15 mice Balb/C mice were immunised s.c. with 1 μg of vaccine adjuvanted with 0.2% alum. Booster injections with the same formulation were given 14 and 28 days post primary immunisation. Mice were sacrificed by cervical dislocation 21 and 35 days after the primary immunisation (five mice per group). A single cell suspension was prepared from the pooled spleens of five animals by forcing minced tissue through 200 mesh stainless steel sieves. Red blood cells were depleted by incubation for 5 min at room temperature with lysis buffer containing 0.15 M NH4Cl, 10 mM KHCO3 and 0.1 M Na2-EDTA, pH 7.4 and the leukocytes were suspended in complete cell culture medium.
2.4. Challenge of CD1 mice
A subset of the immunised CD1 mice, i.e. four mice each of the groups that received vaccine doses of 0.2 μg–0.3 ng and the control groups (see Section 2.3.1), were challenged 5 weeks (groups that received the non-adjuvanted or the 0.05% aluminium hydroxide containing preparation) or 13 weeks (0.2% aluminium hydroxide) after primary immunisation. Prior to challenge, a blood sample was drawn for determination of neutralising antibody titres. For challenge, mice were anaesthetised with Isofluran and inoculated intranasally with a dose of 105 TCID50 SARS-CoV contained in a volume of 20 μl (10 μl per nostril). Non-cloned SARS-CoV CDC#200301157 that has been propagated five times on serum protein free Vero SF cells was used for homologous challenge. On day 3 post inoculation (p.i.), mice were euthanised with Isofluran followed by barbiturate, before lung and trachea were removed and frozen at ≤−60 °C. Prior to titration in the TCID50 assay, tissue samples were thawed and homogenised in 1 ml of Vero cell culture medium supplemented with antibiotics.
2.5. Enzyme linked immunosorbant assay (ELISA)
The IgG titre against the S protein of SARS-CoV was determined by an indirect ELISA. Briefly, 96-well microtiter plates were coated overnight at 2–8 °C with 100 ng of Baculovirus (BV)-expressed, full-length, His-tagged S protein (Protein Sciences, Meriden, CT, USA) in 50 mM carbonate buffer, pH 9.6 per well or left uncoated, respectively. Subsequent to washing and blocking for 1 h with 3% non-fat dry milk, serial fourfold dilutions of sera (starting with a 1:100 dilution) were applied for 1 h at room temperature, followed by three washes with phosphate-buffered saline (PBS) containing 0.1% Tween 20. Bound antibodies were detected by HRP-labelled goat anti-mouse IgG (Accurate Chemical, Westbury, NY, USA; working dilution of 1:5000) followed by washes and development using OPD/H2O2. Finally, colour development was stopped by the addition of 5 M H2SO4 and the plates were read at 490/620 nm by ELISA plate reader (BIO-TEK, Winooski, VT, USA). To determine the endpoint antibody titre, all absorbance readings equal or greater than the cut-off value (four times the mean absorbance value of a 1:100 dilution of a negative control serum) were considered positive.
2.6. Determination of neutralising antibody titres
Serum samples were serially diluted with cell culture medium in twofold steps (usually beginning at a dilution of 1:20 or 1:40). The serum dilutions were mixed at a ratio of 1:1 with a virus stock suspension adjusted to 103.5 TCID50/ml, incubated for 1 h at room temperature and transferred (eight replicates per dilution) to a 96-well tissue culture plate seeded with a Vero cells. The plates were incubated for 5 days at 37 °C in a CO2-incubator, before the cultures were inspected under a light microscope for the presence of a cytopathic effect (CPE) caused by SARS-CoV, i.e. cell rounding and detachment. The neutralising titre was calculated by the number of virus negative wells and the serum dilution according to the method of Spearman [17].
2.7. ELISPOT assay
The frequency of interferon-γ (IFN-γ) or interleukin-4 (IL-4) secreting cells was analysed using mouse IFN-γ and IL-4 ELISPOT kits (Mabtech AB, Nacka, Sweden) following the instructions of the manufacturer. Serial dilutions of freshly isolated spleen cells of Balb/C mice were added to the wells ranging from 5 × 104 to 2 × 105 cells per well of antibody-coated 96-well plates. For stimulation, SARS candidate vaccine and recombinant BV-expressed S protein were added at a concentration of 0.1–1 μg/ml. Wells containing no antigen or 1 μg/ml of Pokeweed mitogen (Sigma, St. Louis, USA) were used as negative and positive control, respectively. The plates were then incubated overnight at 37 °C and 5% CO2 before the cells were discarded and the plates were washed with PBS. Interferon-γ or IL-4, respectively, was detected by a biotinylated interferon-γ or IL-4 specific antibody followed by streptavidin-alkaline phosphatase and development with BCIP/NBT substrate solution. The number of spots was counted using an automated ELISPOT reader (AID, Strassberg, Germany). The number of spots observed in wells containing no antigen was subtracted from the number of spots observed in wells containing specific antigen and the results were expressed as spot forming cells (SFC) per 106 spleen cells.
2.8. TCID50 assay (tissue culture infectious dose 50%)
The infectious virus titre of SARS-CoV-containing samples was determined by a TCID50 assay. In brief, serial 10-fold dilutions of virus containing samples were inoculated onto 96-well microtiter plates seeded with Vero cells. After incubation for 5–7 days at 37 °C in a CO2-incubator, the plates were screened under a light microscope for the presence of a CPE. From the number of virus positive wells per dilution, the TCID50 was calculated according to the Poisson formula by means of an in-house calculation software program. This program provides estimation of TCID50 of titres, and its 95% confidence limits, based on the one-hit model [18].
3. Results
3.1. Development of fermentation, inactivation and purification process
We have previously reported on the development of a formalin inactivated, whole virus influenza vaccine which was produced using large-scale serum protein free Vero cell fermenter cultures [15]. This development of a candidate SARS-CoV vaccine was based on adaption of this well established technology, to establish optimal conditions for growth, inactivation and purification of the inactivated virus. The SARS-CoV had been reported to grow well on Vero cells [2], [3], so it was considered that this was probably the optimal cell matrix for rapid vaccine development. A primary virus seed was generated from a human isolate by five sequential plaque clonings. This primary seed was further amplified to generate a seed virus bank, a working virus bank and a production virus bank. The production virus was then used to infect serum protein free cultures using a fermenter volume of 100 l. Following an incubation period of 2–3 days, the virus containing supernatant was then harvested and inactivated by 0.05% formalin treatment for 48 h followed by U.V. inactivation with an U.V. dose of 20 mJ/cm2. Virus infection at a multiplicity of infection (m.o.i.) of 0.001 resulted in generation of high viral titres (∼108 TCID50/ml) in the supernatant and both the formalin and U.V. inactivation steps were independently capable of inactivating this titre with a large margin of safety (manuscript in preparation).
Following inactivation, the virus was then subjected to a two-step purification involving continuous flow zonal centrifugation over a 0–50% sucrose gradient followed by an ultra/diafiltration process. The results of this two-step purification procedure are demonstrated in Fig. 1 . Two lots of purified vaccine were subjected to Western blot analysis and stained with both a specific anti-S glycoprotein (Fig. 1A) and anti-N antibodies (Fig. 1B). In addition the protein composition was analysed by staining with colloidal gold (Fig. 1C). These figures demonstrate that the final purified bulk vaccine is very pure and contains S protein and N protein bands at the equivalent molecular weights to the infectious virus particle prior to inactivation and purification. Some cross-linking of these proteins was also seen in inactivated preparations. An electron micrograph of the purified inactivated preparation confirmed this data (Fig. 2 ) with virus particles being demonstrated to present well defined spikes on the virus membrane.
Fig. 1.

Electrophoretic and Western blot analysis of purified SARS-CoV candidate vaccine. Lysate of SARS-CoV infected Vero SF cells (lane 1) or two lots of purified vaccine (lanes 2 and 3) were separated by SDS-PAGE. Western blot analysis was performed with antibodies specific for the Spike-protein (A) or the Nucleocapsid-protein (B) of SARS-CoV; protein staining using colloidal gold is shown in (C).
Fig. 2.
Electron micrograph (187,000-fold magnification) of purified inactivated SARS-CoV candidate vaccine after staining with uranyl acetate. Spikes formed by S protein project from the viral surface.
3.2. Specific and neutralising antibody response
The immunogenicity of the candidate vaccine was initially investigated in dose-finding and adjuvant studies performed in CD1 mice. For this purpose, groups of 10 mice were immunised twice with decreasing doses of the purified non-adjuvanted or adjuvanted vaccine at doses ranging from 1 μg to 0.3 ng. To evaluate the effect of adjuvantation on the immunogenicity of the vaccine, a non-adjuvanted preparation and material adjuvanted with 0.05 and 0.2% aluminium hydroxide was tested at each antigen concentration. The S specific antibody titre and neutralising antibody titre for each individual serum was then determined as described in Sections 2.5, 2.6. The geometric mean titre (GMT) determined for each group of animals is presented in Fig. 3 . These data demonstrate that the candidate vaccine is highly immunogenic in CD1 mice. Following a single immunisation with 1 or 0.2 μg, S specific antibody titres with GMT up to approximately 1:5000 for the adjuvanted formulations and 1:1000 for the non-adjuvanted formulation were measured 2 weeks after the primary immunisation (data not shown). Neutralising antibody titres were not measured at this time point. Following the booster immunisation, ELISA titres were substantially increased and GMTs of up to 1:400,000 could be obtained with the 1 μg dosage adjuvanted with 0.2% aluminium hydroxide. Reduction of the dosage to as little as 0.3 ng in an adjuvanted formulation still resulted in the development of a low titre antibody response (Fig. 3A). High titre neutralising antibodies (approximately 1:1000) were also measured for the 0.2 μg adjuvanted and non-adjuvanted formulations 3 weeks after the booster immunisation (Fig. 3B). Groups of mice immunised with the 0.2% aluminium hydroxide formulations were bled 10 and 11 weeks after booster immunisation and it was demonstrated that high titre S specific and neutralising antibodies were still detected at this stage (Fig. 3C and D).
Fig. 3.
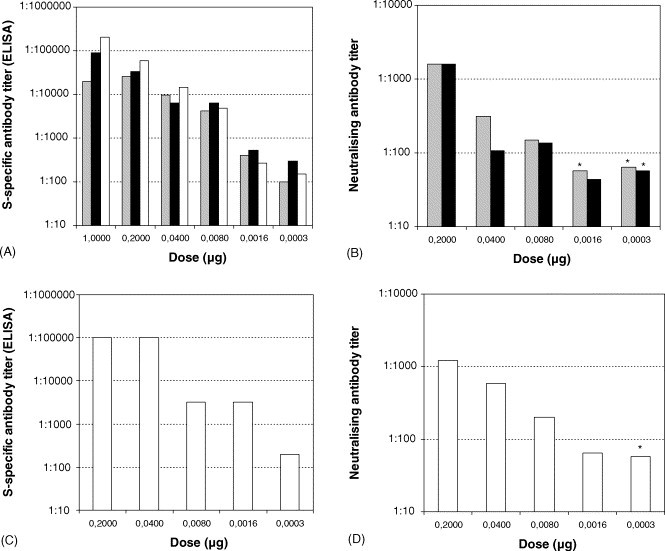
S-specific and neutralising antibody response against SARS-CoV vaccine of CD1 mice after booster immunisation. The S-specific antibody titre at week 4 (A) or 12 (C), respectively, was determined by ELISA. Neutralising antibody response of CD1 mice at week 5 (B) or week 13 (D), respectively, was analysed by a micro neutralisation assay. Geometric mean titres of groups that received the non-adjuvanted vaccine (grey bars), or vaccine adjuvanted with 0.05% (black bars) or 0.2% aluminium hydroxide (open bars) are presented (n = 10 for antibody titres; n = 4 for neutralising titres); ( ) no neutralising antibodies detectable (due to limited amount of serum in some instances the detection limit of the assay was at 1:57 or 1:71).
) no neutralising antibodies detectable (due to limited amount of serum in some instances the detection limit of the assay was at 1:57 or 1:71).
3.3. E.D.50 determination with and without adjuvant
The influence of adjuvant on S specific antibody responses was also determined by calculating the effective dose 50 (E.D.50), i.e. the minimum amount of antigen required for seroconversion in 50% of mice after two immunisations. The data in Table 1A demonstrates that following a booster immunisation the non-adjuvanted and the vaccine containing 0.2% alum induced a very similar antibody response; E.D.50 values of 1.1 and 0.8 ng, respectively, were obtained for non-adjuvanted and adjuvanted (0.2% alum) vaccines. The vaccine adjuvanted with 0.05% alum was somewhat more immunogenic. Seventy percent of the mice developed specific antibodies against the S protein even at the lowest dose applied, i.e. 0.3 ng. Due to the high proportion of seroconverters the E.D.50 could not be exactly calculated, but it was <0.1 ng. The proportion of animals which demonstrated a neutralising antibody titre was also analysed in a subset of immunised mice (n = 4, and the E.D.50 based on neutralising antibody response was also calculated (Table 1B ). This data was similar to that obtained for ELISA IgG determinations but differed somewhat in that the highest proportion of animals with neutralising antibodies were seen in the adjuvanted formulation with the highest concentration of alum (0.2%).
Table 1A.
Seroconversion (S-specific antibodies) of CD1 mice after immunisation with candidate SARS-CoV vaccine
| Vaccine group | Vaccine dose (μg) |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1.0 | 0.2 | 0.04 | 0.008 | 0.0016 | 0.0003 | Control | E.D.50 (ng) | |
| Number of seroconvertersa/number of animals tested (week 4) | ||||||||
| w/o adjuvant | 10/10 | 10/10 | 10/10 | 10/10 | 5/10 | 2/10 | 0/10 | 1.1 |
| 0.05% alum | 10/10 | 10/10 | 9/10 | 10/10 | 8/10 | 7/10 | 0/10 | <0.1 |
| 0.2% alum | 10/10 | 10/10 | 10/10 | 10/10 | 6/10 | 3/10 | 0/10 | 0.8 |
| Number of seroconvertersa/number of animals tested (week 12) | ||||||||
| 0.2% alum | n.d. | 4/4 | 4/4 | 3/4 | 3/4 | 1/4 | 0/4 | 0.9 |
Mice with S-specific antibody titres of ≥1:100 were rated as seroconverters; n.d.: not done.
Table 1B.
Seroconversion (neutralising antibodies) of CD1 mice after immunisation with candidate SARS-CoV vaccine
| Vaccine group | Vaccine dose (μg) |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 0.2 | 0.04 | 0.008 | 0.0016 | 0.0003 | Control | E.D.50 (ng) | |
| Number of seroconvertersa/number of animals tested (week 5) | ||||||||
| w/o adjuvant | n.d. | 4/4 | 4/4 | 4/4 | 0/4 | 0/4 | 0/4 | 3.6 |
| 0.05% alum | n.d. | 4/4 | 2/4 | 3/4 | 1/4 | 0/4 | 0/4 | 8 |
| Number of seroconvertersa/number of animals tested (week 13) | ||||||||
| 0.2% alum | n.d. | 4/4 | 4/4 | 4/4 | 2/4 | 0/4 | 0/4 | 1.6 |
Mice that developed neutralising antibodies were rated as seroconverters; n.d.: not done.
3.4. Characterisation of the cellular immune response to vaccination
Further characterisation of the immune responses in mice was carried out by investigating specific cytokine responses to vaccination. The type of immune response was determined by measurement of interferon-γ, a marker for Th-1 responses and interleukin-4 which is a marker for a Th-2 response. For cytokine studies Balb/C mice were immunised s.c. with 1 μg of antigen adjuvanted with 0.2% alum. Booster injections with the same formulation were given 14 and 28 days post primary immunisation. Mice were sacrificed 21 and 35 days after the primary immunisation. Spleen cells were isolated from Balb/C mice and stimulated with a purified recombinant S protein or candidate vaccine (see Sections 2.3.2, 2.7).
The data presented in Fig. 4 shows that substantial IFN-γ and IL-4 responses were obtained after two immunisations with whole virus vaccine. However, the responses to the whole virus vaccine were substantially higher than those obtained following stimulation with recombinant S protein, after two immunisations. Following a third immunisation, the IL-4 response to recombinant S protein was increased to the level obtained for whole virus antigen, whereas no significant IFN-γ response could still be detected upon stimulation with recombinant S protein. In addition, it could be demonstrated that as little as 0.1 μg/ml of whole virus antigen was sufficient to stimulate substantial Th-1 (IFN-γ) and Th-2 (IL-4) responses.
Fig. 4.

Cellular immune response to vaccination with SARS-CoV vaccine. Interferon-γ (A) and interleukin-4 responses (B) were determined by ELISPOT assays: spleen cells of naive Balb/C mice, or spleen cell obtained from of immunised Balb/C mice at day 21 or 35 after primary immunisation were stimulated with a SARS-CoV vaccine preparation, recombinant S protein or a Vero mock preparation.
3.5. Protection studies
Following the demonstration that the candidate vaccine was highly immunogenic, the protective efficacy of the vaccine was investigated in CD1 mice. A subset (i.e. four mice per group) of CD1 mice immunised during dose-finding and adjuvant studies (see above) were challenged 5 or 13 weeks post primary immunisation by intranasal delivery of 105 TCID50 of homologous virus strain. Virus replication in the respiratory tract was monitored as described in Section 2.4. The data presented in Table 2 demonstrates again that the non-adjuvanted vaccine and vaccine preparations adjuvanted with 0.05% aluminium hydroxide were highly effective in inducing specific anti-S antibodies and neutralising antibodies. An antigen dose as low 8 ng with or without adjuvant was effective in inducing neutralising antibodies in 3 of 4 or 4 of 4 immunised animals, respectively.
Table 2.
Immune response and protection of CD1 mice
| Dose (μg) | Animal | Non-adjuvanted |
Adjuvanted with 0.05% aluminium hydroxide |
||||
|---|---|---|---|---|---|---|---|
| ELISA titre (week 4) | NT (week 5) | Virus titre (day 3 p.i.) | ELISA titre (week 4) | NT (week 5) | Virus titre (day 3 p.i.) | ||
| 0.2 | 1 | 1:102,400 | 1:1522 | ≤1.7 | 1:25,600 | 1:1522 | ≤1.7 |
| 2 | 1:25,600 | 1:3620 | ≤1.7 | 1:25,600 | 1:1256 | ≤1.6 | |
| 3 | 1:25,600 | 1:1396 | ≤1.6 | 1:102,400 | 1:1974 | ≤1.5 | |
| 4 | 1:25,600 | 1:830 | ≤1.6 | 1:25,600 | 1:1810 | ≤1.7 | |
| 0.04 | 1 | 1:25,600 | 1:905 | ≤1.7 | 1:25,600 | 1:415 | ≤1.6 |
| 2 | 1:25,600 | 1:190 | ≤1.6 | 1:400 | ≤1:28 | ≤1.6 | |
| 3 | 1:6400 | 1:587 | ≤1.6 | 1:6400 | 1:349 | ≤1.6 | |
| 4 | 1:6400 | 1:95 | ≤1.7 | <1:100 | ≤1:33 | 2.2 | |
| 0.008 | 1 | 1:1600 | 1:113 | ≤1.5 | 1:1600 | 1:37 | ≤1.6 |
| 2 | 1:6400 | 1:320 | ≤1.7 | 1:1600 | ≤1:37 | ≤1.6 | |
| 3 | 1:25,600 | 1:415 | ≤1.6 | 1:25,600 | 1:587 | ≤1.6 | |
| 4 | 1:400 | 1:34 | 2.3 | 1:25,600 | 1:453 | ≤1.6 | |
| 0.0016 | 1 | 1:400 | ≤1:57 | 3.4 | 1:1600 | 1:160 | ≤1.5 |
| 2 | 1:1600 | ≤1:57 | ≤1.5 | 1:400 | ≤1:28 | ≤1.5 | |
| 3 | <1:100 | ≤1:57 | ≤1.6 | 1:1600 | ≤1:28 | ≤1.5 | |
| 4 | 1:1600 | ≤1:57 | 2.7 | 1:400 | ≤1:28 | 2.3 | |
| 0.0003 | 1 | 1:100 | ≤1:71 | ≤1.6 | 1:100 | ≤1:57 | 4.8 |
| 2 | <1:100 | ≤1:57 | 1.9 | <1:100 | ≤1:57 | 3.6 | |
| 3 | <1:100 | ≤1:71 | 5.6 | 1:100 | ≤1:57 | 3.4 | |
| 4 | 1:100 | ≤1:57 | 6.0 | 1:400 | ≤1:57 | ≤1.5 | |
| None | 1 | <1:100 (pool) | ≤1:57 | 4.5 | |||
| 2 | ≤1:57 | 4.8 | |||||
| 3 | ≤1:28 | 5.1 | |||||
| 4 | ≤1:57 | 3.4 | |||||
p.i.: post inoculation. S-specific antibody titres were measured by ELISA, neutralising antibody titres (NT) by a micro neutralisation assay and SARS-CoV titres of homogenised lung/trachea tissue were determined by a TCID50 assay and are given as log(TCID50/g lung tissue).
Challenge with 105 TCID50 of live virus resulted in virus replication in the lung of 100% of mock immunised mice. However, immunisation with candidate vaccine antigen doses as low as 8 ng resulted in a high degree of protection of vaccinated mice (3 of 4 and 4 of 4 animals protected). Reduced doses of antigen still resulted in partial protection with ≥50% of immunised mice being protected following immunisation with 1.6 ng of vaccine antigen. Similar data were obtained for vaccine preparations adjuvanted with 0.2% aluminium hydroxide (data not shown). The 50% protective dose (P.D.50) was calculated for all three vaccine formulations and were shown to be approximately equal with P.D.50's of 1.4, 0.7 and 1.0 ng being calculated for formulations without adjuvant, with 0.05% alum and 0.2% alum, respectively (Table 3 ).
Table 3.
Protection of CD1 mice after immunisation with candidate SARS-CoV vaccine
| Vaccine group | Vaccine dose (μg) |
||||||
|---|---|---|---|---|---|---|---|
| 0.2 | 0.04 | 0.008 | 0.0016 | 0.0003 | Control | P.D.50 (ng) | |
| Number of protected animalsa/number of challenged animals (week 5) | |||||||
| w/o adjuvant | 4/4 | 4/4 | 3/4 | 2/4 | 1/4 | } 0/4 | 1.4 |
| 0.05% alum | 4/4 | 3/4 | 4/4 | 3/4 | 1/4 | 0.7 | |
| Number of protected animalsa/number of challenged animals (week 13) | |||||||
| 0.2% alum | 4/4 | 4/4 | 4/4 | 3/4 | 0/4 | 1/4b | 1.0 |
Mice were considered protected if no infectious SARS-CoV was detected in lung/trachea.
One control animal became not infected after challenge.
3.6. Serological correlates of protection
The data presented in Table 2 indicated that there was a clear correlation between specific IgG titre against S protein, neutralising antibody titre and protection against intra-nasal challenge with live virus. This data was confirmed by summarising all protection data obtained in the mouse model. Table 4 summarises the data obtained for a total of 168 mice used in various immunisation studies during the course of this development. The data in Table 4A demonstrates that a specific anti-S IgG titre of ≥25,600 resulted in 100% protection of mice challenged with 105 TCID50 of live virus. It was also demonstrated that a specific neutralising titre ≥114 resulted in 100% protection against challenge (Table 4B). No virus replication was seen in mice with antibody titres above these levels. In contrast, 89% of the control animals were infected by intranasal challenge with 105 TCID50 of live virus.
Table 4.
Correlation of antibody titres and protection of mice
| (A) | ELISA antibody titre against the S protein (1:x) |
|||||||
|---|---|---|---|---|---|---|---|---|
| <100 | 100 | 400 | 1600 | 6,400 | 25,600 | 102,400 | 409,600 | |
| Total n of animals | 56 | 7 | 8 | 18 | 28 | 30 | 19 | 2 |
| N of infected animals | 47 | 3 | 5 | 3 | 1 | 0 | 0 | 0 |
| Infected animals (%) | 84 | 43 | 63 | 17 | 4 | 0 | 0 | 0 |
| (B) | Neutralising antibody titre (1:x)a |
|||||||
|
<57 |
58–113 |
114–227 |
228–455 |
456–911 |
912–1823 |
1824–3646 |
>3647 |
|
| Total n of animals | 84 | 9 | 12 | 15 | 7 | 22 | 7 | 2 |
| N of infected animals | 28 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Infected animals (%) | 69 | 11 | 0 | 0 | 0 | 0 | 0 | 0 |
In total 168 mice (Balb/C and CD1) were challenged with SARS-CoV. Of the 36 control animals 32, i.e. 89%, became infected.
Sera that contained no detectable neutralising antibodies are classified according to the detection limit of the respective assay, e.g. a serum with a titer of <1:71 is assigned to the category 114–227; n: number of animals.
4. Discussion
This report describes the extensive immunological characterisation of a candidate SARS coronavirus vaccine. A number of different strategies have previously been reported for the development of experimental and candidate human SARS vaccines. These include inactivated whole virus vaccines [19], [20], [21], attenuated viral vectors expressing SARS-CoV proteins [7], [8], [12], [13] and DNA vaccines [14], [22]. We have favoured the strategy of a whole virus vaccine development for a number of reasons, including speed of development. The virus grows to very high titres in Vero cells, a cell line accepted by most regulatory authorities. A similar technology used for this development has been utilised for development and industrial scale production of an influenza virus vaccine [15]. Manufacturing facilities are also in place at the appropriate biosafety level to utilise this technology for rapid large scale vaccine production. However, in addition to these technology and logistical reasons, it is also likely that an inactivated whole virus vaccine would be most efficient in inducing neutralising antibodies, which are possibly critical in preventing SARS-CoV infection.
Most processes for inactivated whole virus vaccines have utilised either formaldehyde or β-propiolactone as an inactivating agent. There have been reports of inactivation failures associated with both of these methodologies even in the recent past [23]. We have therefore developed a strategy involving a double inactivation process utilising formaldehyde and U.V. inactivations. Although formaldehyde is reported to react with protein and nucleic acid the reactions with nucleic acid, particularly when the nucleic acid is a component of a virus particle, are less well established. Therefore we have decided to introduce a second inactivation step in addition to formaldehyde, i.e. U.V. inactivation, directed primarily against nucleic acid. Both of these processes were independently demonstrated to be capable of inactivating >108 TCID50/ml with a large margin of safety (manuscript in preparation).
However, it was also essential to demonstrate that this combination of inactivation steps did not destroy the viral antigenic structure and immunogenicity. The data presented in Fig. 1 demonstrates that the major virus antigens, i.e. the S glycoprotein and N protein were not negatively effected. No additional breakdown products could be detected compared to the non-inactivated preparation. In addition, transmission electron microscopy analysis of uranyl acetate stained samples demonstrated that the inactivated virion presented well-defined spike structures on the virus particle with no apparent structural alterations resulting from the inactivation procedures.
These antigenic and structural analyses were then followed up with an extensive immunological characterisation of the candidate vaccine. Dose finding and adjuvant studies were carried out in mice with measurement of antibody responses by S specific ELISA and infectivity neutralising titres. The data presented in Fig. 3 demonstrated that the double inactivated preparation was highly immunogenic with S specific ELISA titres of up to 1:400,000 being obtained following two immunisations with 1 μg antigen adjuvanted with 0.2% aluminum hydroxide. High neutralising titres were also obtained following an immunisation and booster with 0.2 μg antigen, with titres of approximately 1:1000 being obtained. This immunogenicity appears to be higher than that reported for other whole virus vaccines subjected to a single inactivation step, although differences in methodologies make direct comparisons difficult [20], [21]. The double inactivated vaccine was also able to induce very high antibody responses even in the absence of adjuvant. This is similar to data reported for an U.V. inactivated vaccine [21] and for a β-propiolactone inactivated vaccine [20] where little difference in the level of neutralising antibodies in mice was reported. The minimal influence of adjuvantation on vaccine immunogenicity was also confirmed in these studies by calculation of the E.D.50. There was little difference in E.D.50 of the non-adjuvanted and 0.2% alum adjuvanted material although the 0.05% alum adjuvanted material appeared to be more immunogenic (Table 1A). However, this was not confirmed when the E.D.50 was calculated on the basis of neutralising antibody responses, with the non-adjuvanted material being more immunogenic than the preparation with 0.05% alum (Table 1B).
In addition to characterisation of humoral responses by antibody measurement, the Th-1 and Th-2 responses to immunisation were analysed by cytokine production. The data presented in Fig. 4 demonstrated that the double inactivated candidate vaccine was capable of inducing cytokine markers for both Th-1 and Th-2 responses. However, the responses to stimulation with recombinant S protein indicated that T cell responses to the S protein were predominantly Th-2 responses, which would be expected for an alum adjuvanted whole virus vaccine [24]. This is in agreement with data reported for immunisation with UV-inactivated SARS-CoV which demonstrated substantial IL-4 and IFN-γ responses to immunisation [21].
Following demonstration of the immunogenicity of the vaccine, investigations were then initiated to demonstrate the efficacy of the candidate vaccine in preventing infection in a small animal model. Balb/C mice have been established as a good rodent model for the replications of SARS Co-V [25]. We have demonstrated that the virus replicated very well in the respiratory tract of CD1 mice and this model was used to investigate the protective efficacy of the vaccine. The data in Table 2 demonstrates that both adjuvanted and non-adjuvanted vaccine preparations at an antigen concentration of 0.2 μg protected 100% of immunised mice from challenge with high titre (105 TCID50) virus by the intra-nasal route. The vaccine appeared to be effective at very low doses of antigen: calculation of the P.D.50 (Table 3) for non-adjuvanted and adjuvanted vaccines confirmed that very low antigen concentrations (approximately 1 ng) were sufficient to provide protection of 50% of immunised mice. This figure is even lower than that calculated for highly effective well-established Flavivirus vaccines such as tick-borne encephalitis which has a calculated P.D.50 of 32 ng in a similar model. ([26], unpublished data).
These protection studies were also used to establish a serological correlate of protection. The data in Table 4 demonstrate that induction of neutralising antibody titres ≥114 and a S specific ELISA titre of ≥25,600 resulted in 100% protection against intra-nasal challenge with 105 TCID50 of infectious virus.
These data clearly indicate that a double inactivated whole virus vaccine is a promising candidate for an effective human vaccine. The use of two inactivation steps based on different mechanisms of infection will ensure safety with respect to residual infectious virus. However, it has been reported that vaccination with a feline coronavirus vaccine resulted in antibody induced enhancement of infection [27], [28]. No suggestion of such a phenomenon could be induced from the challenge studies reported here or in other studies where protection was achieved by induction of neutralising antibodies with MVA [13] or parainfluenza [8] vectored spike protein live viral vaccines. However, vaccine induced enhancement of coronavirus infection will require further study as there have been reports that vaccination of ferrets with rMVA expressing SARS-CoV S protein was associated with enhanced hepatitis [29], [30].
Acknowledgements
This study was supported by the National Institutes of Health (NIH), Bethesda, USA (Contract no. N01-AI-30038). We thank Prof. P. Messner (Center for Ultrastructural Research, University of Natural Resources and Applied Life Sciences, Vienna, Austria) for performing the electron microscopic analysis and F. Cassels (NIH) for critical reading of the manuscript.
References
- 1.Peiris J.S.M., Lai S.T., Poon L.L.M., Guan Y., Yam L.Y.C., Lim W. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;318(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L. Isolation and characterization of virus related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 5.Ellis R.W. Technologies for making new vaccines. In: Plotkin S.A., Orenstein WA., editors. Vaccines. 4th ed. Elsevier; 2004. pp. 1177–1197. [Google Scholar]
- 6.Sui J., Li W., Murakami A., Tamin A., Matthews L.J., Wong S.K. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc Natl Acad Sci USA. 2004;101(8):2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao W., Tamin A., Soloff A., D’Aiuto L., Nwanegbo E., Robbins P.D. Effects of a SARS-associated coronavirus vaccine in monkeys. Lancet. 2003;362:1895–1896. doi: 10.1016/S0140-6736(03)14962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchholz U.J., Bukreyev A., Yang L., Lamirande E.W., Murphy B.R., Subbarao K. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc Natl Acad Sci USA. 2004;101:9804–9809. doi: 10.1073/pnas.0403492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann H., Hattermann K., Marzi A., Gramberg T., Geier M., Krumbiegel M. S protein of severe acute respiratory syndrome-associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralising antibodies in infected patients. J Virol. 2004;78:6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A. Angiostensin- converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ter Meulen J., Bakker A.B.H., van den Brink E.N., Weverling G.J., de Kruif J., Preiser W. Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet. 2004;363:2139–2141. doi: 10.1016/S0140-6736(04)16506-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bukreyev A., Lamirande E.W., Buchholz U.J., Vogel L.N., Elkins W.R., St Claire M. Mucosal immunisation of African green monkeys (Cercopithecus aethiops) with an attenuated parainfluenza virus expressing the SARS coronavirus spike protein for the prevention of SARS. Lancet. 2004;363:2122–2127. doi: 10.1016/S0140-6736(04)16501-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bisht H., Robert A., Vogel L., Bukreyev A., Collins P.L., Murphy B.R. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunises mice. Proc Natl Acad Sci USA. 2004;101:6641–6646. doi: 10.1073/pnas.0401939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z.Y., Kong W.P., Huang Y., Roberts A., Murphy B.R., Subbarao K., Nabel G.J. A DNA vaccine induces SARS coronavirus neutralisation and protective immunity in mice. Nature. 2004;428:561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kistner O., Barrett P.N., Mundt W., Reiter M., Schober-Bendixen S., Dorner F. Development of a mammalian cell (Vero) derived candidate influenza virus vaccine. Vaccine. 1998;16:960–968. doi: 10.1016/s0264-410x(97)00301-0. [DOI] [PubMed] [Google Scholar]
- 16.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 17.Spearman C. The method of “right or wrong cases” (constant stimuli) without Gausś formulae. Brit J Psychol. 1908;2:227–242. [Google Scholar]
- 18.Kundi M. One-hit models for virus inactivation studies. Antiviral Res. 1999;41(3):145–152. doi: 10.1016/s0166-3542(99)00008-x. [DOI] [PubMed] [Google Scholar]
- 19.Qu D., Zheng B., Yao X., Guan Y., Yuan Z.H., Zhong N.S. Intranasal immunisation with inactivated SARS-CoV (SARS-associated coronavirus) induced local and serum antibodies in mice. Vaccine. 2005;23:924–931. doi: 10.1016/j.vaccine.2004.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang L., Zhu Q., Qin E., Yu M., Ding Z., Shi H. Inactivated SARS-CoV Vaccine prepared from whole virus induces a high level of neutralising antibodies in BALB/c mice. DNA Cell Biol. 2004;23:391–394. doi: 10.1089/104454904323145272. [DOI] [PubMed] [Google Scholar]
- 21.Taksuka N., Fujii H., Takahashi Y., Kasai M., Morikawa S., Itamura S. A subcutaneoulsy injected UV-inactivated SARS coronavirus vaccine elicits systemic humoral immunity in mice. Int Immunol. 2004;16(10):1423–1430. doi: 10.1093/intimm/dxh143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada M., Takemoto Y., Okunobu Y., Hashimoto S., Yoshida S., Fukunaga Y. The development of vaccines against SARS corona virus in mice and SCID-PBL/hu mice. Vaccine. 2005;23:2269–2272. doi: 10.1016/j.vaccine.2005.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown F. Review of accidents caused by incomplete inactivation of viruses. Dev Biol Stand. 1993;81:103–107. [PubMed] [Google Scholar]
- 24.HogenEsch H. Mechanisms of stimulation of the immune response by aluminum adjuvants. Vaccine. 2002;20(Suppl. 3):34–39. doi: 10.1016/s0264-410x(02)00169-x. [DOI] [PubMed] [Google Scholar]
- 25.Subbarao K., McAuliffe J., Vogel L., Fahle G., Fischer S., Tatti K. Prior infection and passive transfer of neutralising antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J Virol. 2004;78:3572–3577. doi: 10.1128/JVI.78.7.3572-3577.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrett P.N., Dorner F., Ehrlich H., Plotkin S.A. Tick-borne encephalitis virus vaccine. In: Plotkin S.A., Orenstein WA., editors. Vaccines. 4th ed. Elsevier; 2004. pp. 1039–1055. [Google Scholar]
- 27.Vennema H., de Groot R.J., Harbour D.A., Dalderup M., Gruffydd-Jones T., Horzinek M.C. Early death after feline infectious peritonitis virus challenge due to recombinant vaccinia virus immunisation. J Virol. 1990;64(3):1407–1409. doi: 10.1128/jvi.64.3.1407-1409.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsen C.W. A review of feline infectious peritonitis virus: molecular biology, immunopathogenesis, clinical aspects, and vaccination. Vet Microbiol. 1993;361(1–2):1–37. doi: 10.1016/0378-1135(93)90126-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weingartl H., Czub M., Czub S., Neufeld J., Marszal P., Gren J. Immunisation with modified vaccinia virus Ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. J Virol. 2004;78(22):12672–12676. doi: 10.1128/JVI.78.22.12672-12676.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czub M., Weingartl H., Czub S., He R., Cao J. Evaluation of modified vaccinia Ankara based recombinant SARS vaccine in ferrets. Vaccine. 2005;23:2273–2279. doi: 10.1016/j.vaccine.2005.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]